

Abstract
Background:
Human neurodegenerative diseases occur as a result of various factors. Regardless of the variety in the etiology of development, many of these diseases are characterized by the accumulation of pathological, misfolded proteins; hence, such diseases are considered as proteinopathies. While plenty of research study has been conducted in order to identify the pathophysiology of these proteinopathies, there is still a lack of understanding in terms of potential therapeutic targets.
Summary:
Molecular chaperones present the main workforce for cellular protection and stress response. Therefore, considering these functions, molecular chaperones present a promising target for research within the field of conformational diseases that arise from proteinopathies. Since the association between neurodegenerative disorders and their long-term consequences is well documented, the need for the development of new therapeutic strategies becomes even more critical
Key message:
In this review, we summarized the molecular function of heat shock proteins and recent progress on their role, involvement, and other mechanisms related to neurodegeneration caused by different etiological factors. Based on the relevant scientific data, we will highlight the functional classification of heat shock proteins, regulation, and their therapeutic potential for neurodegenerative disorders.
Keywords: Heat shock protein, HSP27, HSP70, HSP90, Neurodegeneration, Alzheimer disease, Parkinson disease, Guillain–Barré syndrome
Neurodegenerative Disorders
The global incidence of the development of neurodegenerative diseases is high. 1 With increased life expectancy, the neurodegenerative disorders represent the fourth biggest cause of mortality in affluent countries and are growing more common in poorer countries. 2
Even though considerable research has been carried out, the underlying mechanisms remain poorly understood. 3
Neurodegenerative Factors
Various types of neurodegenerative disorders can be generated by a variety of genetic and environmental factors. Scientific evidence suggests that some of the multifactorial conditions influencing these disorders are abnormal protein dynamics with defective protein degradation and aggregation, oxidative and nitrosative stress, and mitochondrial dysfunction. Compromised bioenergetics, metal toxicity, and pesticide exposure represent significant environmental factors that can lead to neurodegenerations too. 4 5 6
Neurodegenerative Proteinopathies
A common feature of a number of neurodegenerative disorders is misfolding and progressive polymerization of proteins. In fact, many neurodegenerative diseases that arise from a wide array of factors are protein misfolding disorders. Even though a huge amount of research has been performed in order to learn more about the pathophysiology of these proteinopathies, possible drug targets are still ambiguous. Protein quality control (PQC) presents mechanisms that imply the appropriate clearance of misfolded proteins and associated aggregates in order to maintain protein homeostasis. Because neurons have an exceptional cellular structure with long extensions, PQC in neurons is very challenging compared to other cell types. As a crucial component of PQC, molecular chaperones inhibit the formation of disease-causing aggregates. Therefore, neurons consist of a set of chaperones that identify misfolded proteins via exposed hydrophobic surfaces and facilitate their refolding.7–13
Chaperones are the main workforce involved in cellular homeostasis and stress response. Because heat shock proteins (HSP) are highly conserved proteins and because they have always been involved with stress sensing and stress resistance, it is not surprising that they also participate with the nervous system which is developed to sense environmental challenges and consequently respond to them.14–19
Therefore, comprehension of the multifaceted role of HSP in neurodegeneration, that will be discussed below, could help to develop new diagnostic or therapeutic targets in human neurodegenerative disorders.
What Are Molecular Chaperones?
HSP function as a PQC system. HSP were discovered by Ritossa in 1962. He observed a puffing pattern in Drosophila chromosomes after exposure to heat. Several studies since then have demonstrated the significance of HSP in clinical settings. 20
HSP's role is to detect proteins that have been incorrectly folded or denatured. In order to protect the cellular environment, these "misfolded" proteins are refolded or degraded.21, 22 Chaperones for some proteins are also responsible for the initial folding. These newly transcribed genes during translation require immediate chaperone interactions. Other proteins, across the entire term of existence, often interact with chaperones. Molecular chaperones, in general, do not provide structural information for folding but rather avoid undesirable intermolecular interactions. 23
HSP can be classified as stress repressible and stress inducible proteins. Stress-repressible HSP’s are primarily responsible for the proper folding of nascent polypeptides, while after protein denaturation, stress-inducible HSP’s play a major role in protein refolding. 24
Based on their molecular weight, HSP are categorized into several types such as: HSP10, HSP40, HSP60, HSP70, HSP90, and HSP110. 25 Various types of HSP appear to have distinct functions in protein folding and unfolding.23, 26
Therefore, HSPs have an essential role in the maintenance of protein structure and function in all types of cells.
Investigations through a variety of models of brain diseases, induced by drug overexpression, have observed protective effects of HSP. Moreover, it has been observed that HSP are induced in different pathological conditions of the nervous system including epilepsy, trauma, cerebral ischemia, and neurodegenerative diseases. 27 Their presence has been found in different cell types, such as neurons, glia, and endothelial cells. 28 HSP that are present in the extracellular environment are released throughout necrotic cell death and through physiological secretory mechanisms. In the extracellular environment, HSP can bind to stress-sensitive recipient cells such as neurons; therefore, they can increase stress resistance. 13
HSP27 in Neurodegenerative Diseases
HSP27 expression in neurons is low, although it can be increased by proteotoxic stress. In neurodegenerative diseases, increased levels of HSP27 in neurons and glia correlate with pathogenic deposition of abnormal proteins. 29 HSP27 act as a mediator in survival responses of the central nervous system (CNS) insults as a result of antioxidant activity and through the suppression of cell death pathways. 30 Consequently, describing the possible mechanisms of HSP27's participation in neuronal survival will contribute to identifying new approaches for preventing neuronal death (Figure 1).
Figure 1. Schematic Presentation of the HSP27, HSP70, and HSP90 Involvement in: (a) Neurodegeneration (ND): Alzheimer Disease (AD), Parkinson’s Disease, Cerebral Ischemia and Peripheral Nerve Degeneration; (b) Immune Mediated ND: Multiple Sclerosis (MS) and Guillain-Barré syndrome (GBS).
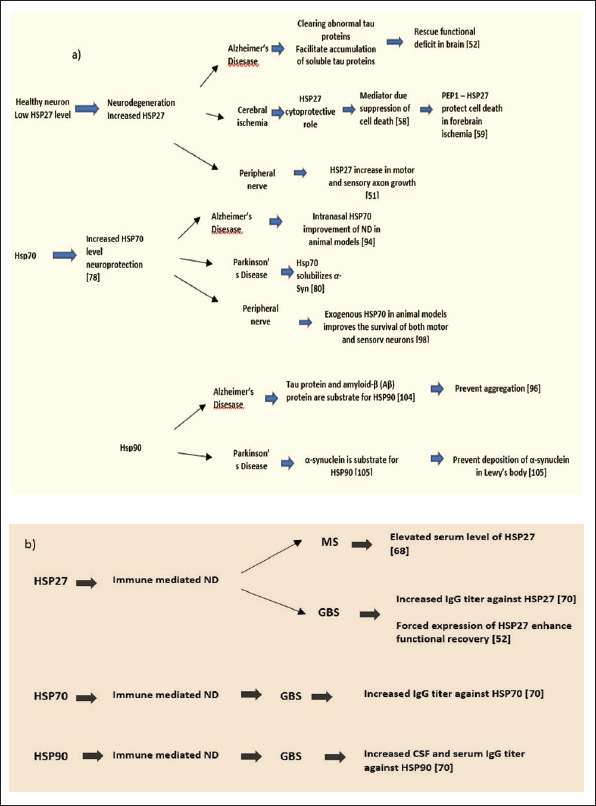
Wagstaff et al. 31 in an attempt to identify the HSP implicated in the apoptosis-protective effect constructed a Herpes Simplex Virus-based Vector in order to express individual HSP genes with high efficiency, which may be used to investigate their protective effects in vitro. Their results have shown the protective effect of overexpressing HSP27 against necrosis and programmed cell death. For the first time, such a protective effect of HSP27 was presented in neuronal cells. The possible proposed mechanisms of protection against apoptosis were through induction of the Fas/APO-1 pathway as well as by a protein kinase C inhibitor. 14
Later on, Abisambra et al. 18 investigated the role of HSP27 in the mechanism that develops Alzheimer's disease (AD). In their study, they investigated the ability of HSP27 to modulate tau dynamics. Tau presents the subunit protein that is one of the crucial hallmarks of AD and therefore it is of great importance in resolving disease mechanisms. 32 Because the mechanisms of HSP27 that are involved to facilitate the clearance of abnormal protein have been difficult to describe, they used a combined methodology of biophysical and physiological approaches that led to the discovery of an important aspect of HSP27 biology. While both mock phosphorylated HSP27 and wildtype HSP27 in vitro showed the ability to bind and prevent tau aggregation, only wildtype HSP27 had the ability to clear tau from the brain and therefore protecting from functional deficits. The mock-phosphorylated form of HSP27 in fact stabilized tau in the brain. This may point to the possibility that HSP27 may be capable to switch between phosphorylated and nonphosphorylated states in order to achieve the clearance of abnormal tau proteins. Otherwise, HSP27 might enable the accumulation of soluble tau intermediates. According to the findings of this study, HSP27 might assist in tau clearance from the brain and rescue from persistent increase in synaptic strength between hippocampal neurons, which is linked to memory and learning. Though, when this ability is conceded, HSP27 may potentially assist in the generation of soluble tau intermediates. 18
Additionally, the development of AD results from the extracellular accumulation of Aβ-rich plaques, as well. Amyloid β (Aβ) peptides present cleavage products of the amyloid precursor protein that are likely to aggregate into amorphous aggregates, toxic oligomers, and fibrils. 33 It has been reported in in vitro studies that HSP27 affect Aβ that is associated with senile plaques in AD brain tissue. 34 Even though a connection between HSP27 and Aβ has been reported, molecular mechanisms of this interaction remain not fully understood. 35 Although it is shown that in cellular systems, extracellular Aβ1-42 generates the expression of HSP27, yet, extracellular HSP27 exogenous exposure enhanced Aβ1-42 cellular toxicity. 36 Studies that involve recombinant protein have suggested that the role of HSP27 is to affect Aβ quaternary structure, isolating toxic oligomers into less toxic, 36 although other studies have demonstrated a role of HSP27 in the reduction of Aβ aggregation.34, 37
Recombinant protein studies have revealed that the role of HSP27 is to modify Aβ quaternary structure, separating toxic oligomers into less toxic ones. Furthermore, some data suggest that HSP27 might be involved in proteinopathies associated with Parkinson’s disease (PD) and dementia with Lewy Bodies. 33
α-Synuclein (α-syn) is a disordered neuronal protein involved in the control of synaptic vesicles.33, 38 Additionally, it is the principal structural component in large protein aggregates known as Lewy Bodies. These protein aggregates are formed in neurodegenerative synucleinopathies, such as PD. 39 It has been reported that HSP27 is involved in the reduction of α-syn aggregation. A recent study describes that the reduction of the disulfide bond in HSP27 increased its chaperone activity toward multiple client proteins, including α-syn.40, 41
Moreover, in order to fully understand the relationship that implicates the involvement of HSP27 in synucleinopathies, further in vivo studies are required to be performed in the future.
Furthermore, some data present the role of HSP27 in cerebral ischemia. There is also a growing evidence that overexpression of HSP27 provides a strong cytoprotective role in cerebral ischemia. 42 However, the actual mechanism underlying this protection is yet unknown. It has been suggested that in ischemic neurons HSP27-mediated neuroprotection is due to a direct or indirect inhibition of the mitochondrial cell death pathway. 43 While HSP27 has been proposed as a possible therapeutic protein in forebrain ischemia, its inability to penetrate cells limits its use for this purpose. Thus, in an endeavor to deliver HSP27 protein to cells and tissues, An et al. in 2008 examined the possibility of protein transduction using PEP-1 peptide, which was designed to enhance the biological activity of transduced proteins in cells. 44 To achieve this, they mixed HSP27 with PEP-1 peptide, using HSP27 as a target protein for direct transduction in vitro and in vivo. According to their findings, protection against cell death resulted when the PEP-1–HSP27 fusion protein was directly transduced into neuronal cells and across the blood–brain barrier. Therefore, they suggested that the PEP-1 peptide in combination with HSP27 could be effective as a possible therapeutic agent used to treat temporary forebrain ischemia.
While it is widely established that the CNS does not normally regenerate, peripheral nerves after injury regenerate spontaneously because of activation of the inherent growing ability of neurons and a conducive environment. 45 Even though peripheral nerves have the ability to regenerate after injury, motor functional repair in human damaged proximal nerves is minimal. One of the potential explanations is that induced axonal growth after the injury is too slow.46–48
Concerning the possible involvement of HSP27 in peripheral nerve injuries, several studies in animal models were conducted. Ma et al. 17 conducted a study in mice about the role of HSP27 after peripheral nerve injury. They reported that HSP27 increased axonal growth after peripheral nerve damage that stimulates motor recovery. They applied an unbiased bioinformatics technique to uncover genes that play a vital role in regenerative response following nerve transaction or crush injuries in dorsal root ganglion (DRG) neurons of mouse models. The authors identified HSP27 as a possible candidate to increase neuron regeneration. 49
Lindsay 50 reported that in adult rats, sensory neurons survive axotomy and Neuron Growth Factor (NGF) withdrawal, while in general neonatal sensory neurons do not. The potential explanation for this is that injured adult DRG neurons themselves develop into the source of the survival-promoting growth factors usually provided by the target tissue. Considering that Lewis et al. 16 investigated the involvement of HSP27 in neonatal sensory neurons following peripheral nerve damage and NGF withdrawal. Their findings suggested that HSP27 contributes substantially to the survival of sensory neurons and is possible to be a significant factor for the survival of adult sensory neurons, as well. From the reported results it can be indicated that the regulation of the HSP27 is associated with sensory neuronal survival and that the overexpression of human HSP27 in neonatal rat sensory neurons is necessary to decrease apoptosis after NGF withdrawal in vitro. Therefore, HSP27 can act as an inherent survival factor that has a crucial role of decreasing cell death in sensory neurons. But, from the in vitro studies, it has been observed that overexpression of HSP27 can also save sympathetic neurons from NGF withdrawal; hence, it can be proposed that this role for HSP27 is not limited only to sensory neurons. The mechanism that might be responsible for the antiapoptotic role of HSP27 in neurons can be explained through stabilization of actin microfilaments, increment of glutathione levels and reduction of generation of reactive oxygen species, enhancement of protein refolding, and association with protease complexes, or maybe any other novel function.16, 51
As a conclusion, HSP27 has been found to have a stronger protective impact on the neurological system, though there is a lack of knowledge regarding the role of HSP27 in immune-mediated neurological diseases.
Multiple sclerosis is the most common chronic inflammatory disease that affects the CNS. It presents the most diffuse chronic inflammatory disease of the CNS. The pathophysiology of this disease is clearly influenced by both immune-mediated and neurodegenerative mechanisms. 52
Both immune-mediated and neurodegenerative processes obviously play role in the pathogenesis of this disease. During the relapse phase of multiple sclerosis, patients' serum, and cerebral fluid have been shown to have increased levels of HSP27.27, 53, 54 Based on these findings, it can be suggested that through MS attacks, HSP27 may be overexpressed in order to protect neuronal cells by reducing misfolding of proteins and aggregation.
Concerning Guillain–Barré syndrome (GBS) an immune-mediated polyneuropathy, Yonekura et al. 55 and Helgeland et al. 56 examined antibodies against HSP in cerebrospinal fluids (CSF) and sera from patients with GBS. Considerably higher IgG antibody titers against HSP27 were found in CSF from GBS patients. 56
Recently, one promising study performed by Asthana et al. 57 in mouse models reported that HSP27 is a main therapeutic target on nerve repair in GBS immune-mediated polyneuropathy. According to their findings, in a clinically appropriate GBS animal model that was established with purified antibodies against gangliosides, forced expression of HSP27 increased functional recovery. Neurobehavioral, electrophysiological, and histological techniques revealed that HSP27 has a protective role on GBS during nerve regeneration and motor endplate reinnervation. They suggest that more research is needed in order to find the possibilities of discovering small compounds that can stimulate HSP27 under physiological conditions as a potential therapeutic agent for nerve regeneration. 57
Eventually, the therapeutic potential of HSP27 in most neurological disorders is yet not fully clarified, although their role cannot be neglected. So far, not much attention has been placed to these compounds; nonetheless, it is possible that these small molecules may shift the conventional paradigm and provide novel insights into the creation of innovative therapeutics for neurological disorders. It has been shown that HSP27 might be involved in multiple diseases modifications. Therefore, it is obvious that HSP27 may be a promising new therapeutic target in treating neurodegeneration diseases.
HSP70 in Neurodegeneration Diseases
Neurodegenerative diseases, such as AD and PD, share a similar feature: the aggregation and deposition of misfolded proteins inside and outside neurons of the brain, as well as selective neuronal loss in the CNS.58–60
In recent years, several studies have demonstrated that the activation of the heat shock response, and in particular the elevation of HSP70 levels, has a neuroprotective effect in several animal neurodegeneration models (Figure 1).13, 61
PD is characterized primarily by the accelerated and selective loss of dopaminergic neurons in the substantia nigra pars compacta, followed by a dopamine decrease in the nigrostriatal pathway, and the appearance of intracytoplasmic fibrillar α-syn protein aggregates (Lewy Bodies) in the remaining nigral neurons. α-syn is a 140-amino acid neuronal protein that is considered to regulate cell differentiation, synaptic plasticity, and dopaminergic neurotransmission. 61 Based on several reports, numerous results conducted on PD mechanism have indicated that HSP70 may solubilize α-syn and induce the degradation of its insoluble forms via chaperone-mediated autophagy and the proteasome.62, 63
Although PD neurodegenerative pathology and involved molecular etiopathogenesis are yet not fully understood, novel contributions have been achieved with the Drosophila model, as the first animal model in which the importance of molecular chaperones, specifically HSP70 alongside with α-syn-induced neurodegeneration, has been investigated. 64
Using a Drosophila model, it has been reported that controlled expression of the protein chaperone HSP70 inhibits dopaminergic neuronal death caused by α-syn expression. Furthermore, interfering with endogenous chaperone function increases α-syn toxicity. The author noticed that Lewy Bodies in human postmortem tissue are immunopositive for molecular chaperones which proves the significance of these findings to human diseases. These results suggest that aberrant protein-folding and -aggregation may be common processes in the pathophysiology of polyQ- and α-syn-related neurodegenerative disorders.65–67
In addition, Protein deglycase or Protein 7 (DJ-1) and α—syn have been associated with hereditary early onset forms of PD, as well. Based on this, Batelli et al. 68 explored the putative functional link between these two proteins in a cellular model of oxidative stress, in order to confirm if they belong to the same antioxidant pathway, which is a detrimental state that is thought to play a crucial role in neurodegenerative diseases.
From the reported results, they found that DJ-1 inactivation may promote aggregation of α-syn and lead to toxicity, while the HSP70 model may be implicated in the regulation of α-syn fibril formation and in the antioxidant response. They suggested that DJ-1 and HSP70 might be therapeutic targets in PD because of their role in neutralizing oxidative stress and their involvement in the solubility of α-syn aggregates. 68
It was observed that HSP70 might have a similar role in AD, as well. The pathological characteristic and etiological component of AD, often known as the "protein-folding" disease, is caused by extraneuronal amyloid-beta (A) deposits and intracellular neurofibrillary tangles formed of the protein tau. 29
Several studies indicate that in the brain of AD patients, the neuroprotective mechanism of HSP70 and other molecular chaperones fails. 69 It is apparent that the expression and functionality of the inducible form of HSP70 and other important HSPs are decreased in aged tissues. Neurons that are affected by high frequency in AD disease located in the hippocampus and entorhinal cortex show relatively low levels of HSP70. 70
So far, in numerous animal models of AD, the neuroprotective role of HSP70 has been demonstrated. A large body of data indicates that many symptoms resembling the main manifestations of AD are triggered in rats following olfactory bulb injury. 71 For this reason, olfactory bulbectomy (OBX) mice can be used as an animal model of AD.
Bobkova et al. 72 investigated endogenous inducible forms of HSP70 in different brain areas of mice after OBX. They found out that the surgical damage of the olfactory bulbs triggers the development of several pathologies, including amyloid-β accumulation and a strong decrease of neuron density in the cortex and hippocampus accompanied with significant disturbance of spatial memory. Following quantifying the endogenous inducible form of HSP70 in several brain locations of OBX mice, they discovered distinct oscillations in HSP70 levels depending on the time after the surgery and the age of mice. It is remarkable to mention that the maximal stimulation of HSP70 synthesis in the hippocampus was manifest in the recovery period of OBX animals. 72
Later on Bobkova et al. 73 studied the potential therapeutic effect of exogenous HSP70 in mouse. They used two rodent models of AD-like neurodegeneration: olfactory bulbectomized mice (OBX) and 5XFAD transgenic mice. The functional, morphological, and biochemical features of OBX mice were comparable to those described for AD patients, including significant memory loss, a decreased utilization of cerebral glucose, and an elevated amount of brain amyloid β protein precursor and Aβ.72, 74, 75 In OBX mice, it has reported a significant loss of neurons in the hippocampus and temporal cortex, 74 which are the brain areas most damaged in AD patients, 75 whereas 5XFAD mice shown an elevated amount of amyloid plaques that form early in their lifetime in diverse brain locations. 76
Full-length recombinant human HSP70 was administered intranasally to these two complimentary AD models: OBX and 5XFAD mice. Memory improvement following the HSP70 therapy is linked with a drop in Aβ levels, while in 5XFAD mice, with a reduction in the density of amyloid plaques. According to the findings of these studies, the HSP70 therapy is extremely successful in alleviating all main indications of neurodegeneration generated in OBX and 5XFAD mice at the histological and cognitive levels.75, 76
Additionally, the huge amount of work suggests that intracellular accumulation of amyloid β is increasingly established as an initial occurrence in the pathogenesis of AD. 76 HSP70 overexpression efficiently protects neurons against intracellular amyloid deposition in a cellular model of AD.77, 78
One of the objectives of this review is to explore the roles of HSP70 in neurodegeneration. As a summary, it can be suggested that HSP70 has a neuroprotective role in diseases that affect CNS. Therefore, future strategies can be suggested for targeting HSP70 in the development of neurodegenerative disease treatments.
Considering the role of HSP70 in peripheral nerve diminishing disorders, Tidwell et al. 79 reported that in neonatal mouse after exogenous, HSP70 showed very promising results in reducing the loss of neurons.
The authors investigated the modulation of the constitutive and stress-induced 70-kD HSP in the neonatal mouse following sciatic nerve axotomy. According to the findings, endogenous levels of HSP 70 in lumbar motor neurons and DRG sensory neurons did not increase substantially up to 24 h after axotomy.
Although after the administration of HSP 70 preparations to the sciatic nerve stump after axotomy, the survival of both motor and sensory neurons was significantly improved.
These findings support the concept that the rapid administration of exogenous HSC70 or HSP70, or both, may improve the survival of injured neurons by stimulating the production of endogenous HSP70.
A huge amount of data shows that HSP70 plays important roles in immune responses. The specific physiological environment substantially impacts the immune functions of HSP70. The first factor is the localization of HSP70, whether it is intracellular, on the cell surface, or in circulation. Intracellular HSP70 protects the cell and inhibits cytokine production, whereas extracellular HSP70 stimulates cytokine production and tags cells for destruction. The second factor is the type of receptors on the target cells that bind to HSP70. Toll-like receptors (TLRs) provide HSP70 the potential to trigger cytokine production and promote the innate response, whereas scavenger receptors assist HSP70 transport antigens to antigen-presenting cells and therefore stimulate the adaptive response. The third component is the condition under which HSP70 is synthesized and released from the cell. HSP70s, for example, are implicated in the creation of antigen-dependent immunological memory in the case of microbial invasion, and in the establishment of antigen-independent immune memory in the case of other stressors.80–82 Because HSP70 is involved in different ways in mechanisms that trigger an immune response, it is expected that HSP70 plays a significant role in neuroimmunological diseases, as well.
Helgeland et al. 56 and Yonekura et al. 55 reported that in CSF and sera from GBS patients increased titer of IgG autoantibodies against HSP70 has been found. It has been also reported that TLR2-, CD14-, or HSP70-based immunomodulation might be potentially implicated in the control of the undesirable effect of innate immune system activation for inflammatory neuropathies. 82 Loshaj-Shala et al. 83 in their study reported that Campylobacter jejuni chaperone proteins DnaK (∼70 kDa) and the different forms of HSP70 extracted and identified from human peripheral nerve have significant primary sequence homology and conservation of known epitopes.
Based on their findings, they concluded that C. jejuni chaperone proteins (70 kDa) can be suggested as potential antigens regulating the induction of immune-mediated disorders, such as GBS, or at least play a substantial role in this disease, presumably via a molecular mimicry mechanism. 83
To sum up, various studies suggested the neuroprotective effect of HSP70 through the reduction of cell death and recovery improvements in neurodegenerative disorders. Hence, additional studies of HSP70 might show alternative, “druggable” therapeutic targets for these conditions.
HSP90 in Neurodegeneration
HSP90 has been linked with a variety of neurodegenerative diseases characterized by protein aggregation including AD and PD. The hyperphosphorylated tau protein and the fibril- and plaque-forming protein amyloid-β (Aβ), which are involved in AD, present a substrate for HSP90 clients. 84 Evans et al., 77 based on the results from their investigation, proposed that in order to avoid aggregation, HSP90 can target abnormal folds in both Aβ and tau. In PD, deposits of α-synuclein in Lewy Bodies are HSP90 substrate. 85 Therefore, the upregulation of HSP90 function may be effective in treating certain neurodegenerative disorders in these circumstances. In AD, however, HSP90 suppression may reduce the activity of kinases that hyperphosphorylate tau, thus decreasing aggregation. Additionally, as a result of HSP90 suppression, abnormal disease-associated proteins may be targeted for degradation (Figure 1). 86
Several data recognize that HSP90 plays important role in immune responses. The impact of HSP90 in the immune system depends on the physiological environment—extracellular, intracellular, or cell surface.
Primarily, it should be noted that HSP90 chaperones predominantly are intracellular molecules that are engaged in facilitating the maturation of several receptors and kinases. 87 In eucaryotes cells, HSP90 occurs in four forms, that is, as two cytosolic isoforms (one is inducible, and the other is constitutive). The other two isoforms are localized in the ER and the mitochondrion. The presence of cytosolic HSP90 in the extracellular matrix originates from exosome secretion. 88 The inducible cytosolic isoform of HSP90 is secreted into the extracellular matrix throughout phosphorylation of its Thr-90 residue and cell stress. 89
Several data propose that extracellular HSP90 might assist the folding and hence the activation of immune cells including natural killer cells and T lymphocytes via their receptors. 90 HSP90, which is surface exposed, acts as a signal for the molecular patterns that are associated with danger or damage, through the activation of the innate immune response, and eventually the adaptive immune system. 91
In contrast, both intracellular and extracellular HSP90 are implicated in antigen presentation. The intracellular type of HSP90 adheres to antigenic proteins, allowing them to be processed into antigenic peptides. The produced antigenic peptides are finally presented to MHC-I/II by HSP90. 90 While extracellular HSP90 interacts with cell surface receptors through binding to substrate peptide antigens, it facilitates endocytosis of the HSP90-antigen complex. 92 Thus, HSP90 participates in the cross-presentation of antigens to immune cells.
In addition, based on the involvement of HSP90 in mechanisms that trigger an immune response, it is expected that HSP90 might have an impact on neuroimmunological diseases, as well.
Yonekura et al. 55 reported that in CSF and serum from GBS patients, the significantly increased titer of IgG autoantibodies against HSP90 has been found as compared with motor neuron disease. 55
Taken altogether, the complex role of HSP90 on immune pathways and neurodegeneration mechanisms (Figure 1) needs to be further investigated. Such a multifaceted role could lead to knowledge to develop HSP90 as a promising therapeutical strategy for several diseases, which involve neurodegeneration and all other diseases that are linked with protein misfolding.
Therapeutical Approaches of Heat Shock Proteins
HSP presents remarkable therapeutic targets for a variety of diseases. The most promising approach was studied through the last two decades in developing HSP90 inhibitors HSP90 inhibitors for therapeutic applications as an anticancer therapy. However, for the time being, the chances for HSP90 inhibitors development as a cancer therapy did not result in acceptable efficacy/safety margins. 93
While these studies did not result yet with an approved compound, many approaches that can be used toward the future development of other HSP agents have been launched. Regardless of all that, during this time it was learned that is still not clear how to use these agents. This remains a major challenge that needs to be sorted out in order to ensure that these agents play a role in the treatment of cancer in the future.
However, the use of HSP agents is not limited to cancer treatment only. A wide range of neurodegenerative disorders are characterized by neuronal cell death after misfold, aggregation, and accumulation of abnormally processed or mutant proteins. Therefore, pharmacological targeting of HSP may have an application in neurodegenerative disorders. The application has been impeded because of the inability to create compounds that can cross the blood–brain barrier and reach therapeutic brain concentrations at nontoxic dosages.94, 95
In the following, several concepts associated with the specific targeting of chaperones in neurodegeneration tested on cell and animal models will be discussed.
Studies that tested the therapeutical potential of HSP27 as a therapeutical target were performed o animal models in different neurodegenerative conditions. The obtained results were promising, although further studies need to be performed.
Therapeutical Targeting/Inhibition
In 2004, Shimura et al. first showed the therapeutic potential of HSP27 in AD. The experiment was conducted in human cortical neuron 2A cultured cells, in which HS27 was delivered. It was noticed that the level of phosphorylated tau decreased. 96 Later, HSP27's potential therapeutic potential in in vitro and in vivo models was demonstrated, showing that HSP27, which is overexpressed, and its constituents phosphorylated mutant form interacted with tau and inhibited it from accumulating. 18 Additionally, Toth et al. in 2013 showed that in a mouse model of AD, HSP27 also reduced the amount of aggregated amyloid-β. 97
Further, it has been reported for the impact of HSP27 injection treatment in transient middle cerebral artery occlusion in mice, which is an ischemic stroke model. Based on the result that has been reported intravenously administration of HSP27 showed protection of the brain in different stroke rat models. 98 The authors claim that they did not observe any significant adverse effect of HSP27. Further studies are needed in order to develop HSP27 intravenous therapy for the treatment of stroke. It might open new perspectives for establishing therapy for ischemic stroke, which could be also useful for other diseases as well, including AD and PD.
In the search for neuroprotective drugs development, targeting HSP70 appears as a promising approach. HSP70 targeting is related to the development of inhibitors aimed at the adenosine triphosphate (ATP)-binding site, i.e., the allosteric sites in the nucleotide-binding domain, which also serves as the substrate-binding domain. 99 It has been shown that previously investigated compounds with for anticancer ability that consists of the skeleton (benzothiazolin-2-yliden)-4-oxothiazolidin-2-ylidene (rhodacyanine) interact to distinct allosteric sites of HSP70. 99 Among them, MKT-077 and YM-01 homologs targeting HSP70 were suggested as candidates in AD therapy because of their capacity to promptly and efficiently reduce Tau levels in vitro and ex vivo.100, 101 Despite their promising results in Tau dynamics, because of their low Blood-Brain-Barrier (BBB) penetration and renal toxicity, these homologs were not further considered.100, 102, 103
Additionally, Methylene Blue and Azure C present an important class of molecules, phenothiazines. Through the inhibition of HSP70 ATPase function, they are able to reduce levels of total Tau and phosphorylated-Tau, although with low selectivity.104, 105 This suggests that phenothiazine derivatives might be used as multimodal drug toward AD. Furthermore, the synergistic effect of HSP70 ATPase activity and Tau aggregation inhibition appears to be a promising approach, therefore during drug screening, these two targets should be combined. 103
Furthermore, a drug approved for ulcer therapy, geranylgeranylacetone, demonstrates the ability to stimulate HSP70 expression while maintaining a safe profile. It improved cognitive function in an AD mouse model, lowering Aβ levels, A βplaque formation, and synaptic loss. 106 The mechanism of action of geranylgeranylacetone was first unknown, however, it was later demonstrated that improvement in the AD model arises through modulation of the ERK/p38 Mitogen-activated protein kinase (MAPK) signaling pathway. 107
Moreover, a well-known traditional Chinese medication, the extract of Ginkgo biloba leaves, reduces neurotoxicity of the Aβ1–42 oligomer in human-derived cell line (SH-SY5Y cell), by increasing HSP70, among other proteins. 108
In general, targeting HSP70 appears to be a viable approach in the quest for neuroprotective medicines, particularly for the management of Tau in AD and other tauopathies. Further research in this area should focus on the selective targeting of constitutive proteins versus stress-induced proteins.
Regarding HSP90, a large body of evidence shows that inhibiting HSP90 may be effective in the treatment of AD by neutralizing Tau protein hyperphosphorylation and aggregation. 94 Inhibitors of HSP90 mostly interact with the nucleotide-binding pocket in the N-terminal domain, where they attach to the ATP-binding site, blocking the ADP- and ATP-bound conformational changes required for chaperone action. Inhibitors such as Geldanamycin (GA), 17-N-allylamino-17-demethoxygeldanamycin (17-AAG), and radicicol target this protein site. 109
GA was isolated from the Streptomyces species and was the first HSP90 inhibitor identified. Initially, it was investigated for antibacterial and anticancer action modes; however, toxicity concerns hampered further research. 94 Various GA analogs have been designed, with 17-AAG being especially considered as an effective HSP90 inhibitor with a safer profile and improved solubility. The findings from pharmacokinetic data collected during the study of 17-AAG as an antitumoral medication led to its repurposing as a therapeutic for AD and other neurodegenerative disorders. In vivo activity of 17-AGG was established in a rat. 110 Oral administration of 17-AAG stimulates overexpression at the cellular level of HSP’s (HSP27, HSP40, and especially HSP70), resulted with reduced brain damage and improved cognitive functioning. The impact of this inhibitor on Tau—a crucial target for AD—was studied in mice models. It was shown that a high dosage of 17-AAG reduced neurofibrillary tangles in transgenic mice. 111 Similar pathways were observed for radicicol, which has also been proposed as a therapy for neurodegenerative diseases. 112
Thus, acquiring further knowledge is vital as many issues should be explained. In summary, therapeutic targeting of HSP might provide a foundation for future therapies in the perspective of multitargeted drug development and a polypharmacological approach to complicated neurodegenerative diseases.
Conclusions and Perspectives
Over the past several years have realized a remarkable rise in the number of studies of the involvement of HSP in neurodegenerative diseases, expecting that these studies could lead to a new strategy for the prevention and treatment of neurodegenerative conditions. 113
In this review, we have briefly focused on some of the recent areas of research on the molecular role of HSP27, HSP70, and HSP90 in nervous system diseases.
Based on the huge amount of data that confirms the neuroprotective role of HSP27, therapeutic approaches that lead to modification of HSP27 levels and function might be effective insight for treating different neurological disorders.
In order to select the most appropriate strategy for each disease, the circumstantial environment of the neuron must be considered. Increasing or decreasing HSP27 levels in some circumstances may be valuable, but in others, it might be more suitable to regulate the phosphorylation state and therefore the oligomerization properties of HSP27. Thus, by understanding these mechanisms, approaches targeting the chaperone network can move toward more individualized and effective therapeutic strategies. 18
Considering HSP70, the present findings not only show exogenous HSP70 as a possible therapeutic agent for the treatment of numerous CNS neurodegenerative diseases but also highlight the unique function of mammalian HSP70. It is important to note that HSP70 may form complexes with tau and participate in its degradation, 114 suggesting that HSP70 therapy may be useful not just for AD but also for other neurodegenerative disorders related with tauopathies.
Besides neuroprotective role in the CNS, some data suggest that HSP70 in neonatal mouse may significantly increase the survival of both motor and sensory neurons of peripheral nerves. 84 These encouraging findings urged pharmacological research on active compounds that modulate HSP70. Still, more studies should be performed to confirm these protective mechanisms in humans.
Because of the complex role of HSP90 on immune pathways and neurodegeneration mechanisms, it could lead to new insight for the development of HSP90 as a promising therapeutical strategy for numerous diseases, which involve neurodegeneration and all other diseases related with protein misfolding.
Regarding future perspectives, the mechanisms by which HSP27, HSP70, and HSP90 are generally involved in life circles of the cell, and in particular their role in directly caused neurodegeneration processes or immune-mediated neurodisorders, their facilitation in neuroprotection is still poorly understood. Additional research is required to get a sight of the complete and very promising therapeutic potential of this HSP during neurodegenerative processes.
Declaration of Conflicting Interests: The authors declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Funding: The authors received no financial support for the research, authorship, and/or publication of this article.
ORCID iD: Aida Loshaj Shala https://orcid.org/0000-0003-2640-4356
https://orcid.org/0000-0003-2640-4356
Authors’ Contribution
Conceptualization, ALS, and GB; writing-review and editing, ALS; Supervision, GB. The published version of this manuscript ‘Impact of Heat Shock Proteins in Neurodegeneration: Possible Therapeutical targets’ has been read and agreed by all authors.
Statement of Ethics
Not applicable
References
- 1.Feigin VL, Nichols E, Alam T, et al. Global, regional, and national burden of neurological disorders, 1990–2016: A systematic analysis for the global burden of disease study 2016. Lancet Neurol 2019; 18(5): 459–480. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Zhou L, Miranda-Saksena M, Saksena NK.. Viruses and neurodegeneration. Virol J 2013; 10: 1–17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Gitler AD, Dhillon P, disease: Shorter J. Neurodegenerative. Models, mechanisms, and a new hope. Dis Model Mech 2017; 10(5): 499–502. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Cannon JR, Greenamyre JT.. The role of environmental exposures in neurodegeneration and neurodegenerative diseases. Toxicol Sci 2011; 124(2): 225–250. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Sheikh S, Haque E, Mir S.. Neurodegenerative diseases: Multifactorial conformational diseases and their therapeutic interventions. J Neurodegener Dis 2012; 2013(563481): 8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Uttara B, Singh AV, Zamboni P, et al. Oxidative stress and neurodegenerative diseases: a review of upstream and downstream antioxidant therapeutic options. Curr Neuropharmacol 2009; 7(1): 65–74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Rosenberger K, Dembny P, Derkow K, et al. Intrathecal heat shock protein 60 mediates neurodegeneration and demyelination in the CNS through a TLR4- and MyD88-dependent pathway. Mol Neurodegener 2015; 10: 5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Bross P, Magnoni R, Bie AS.. Molecular chaperone disorders: Defective Hsp60 in neurodegeneration. Curr Top Med Chem 2012; 12(22): 2491–2503. [DOI] [PubMed] [Google Scholar]
- 9.Hansen JJ, Dürr A, Cournu-Rebeix I, et al. Hereditary spastic paraplegia SPG13 is associated with a mutation in the gene encoding the mitochondrial chaperonin Hsp60. Am J Hum Genet 2002; 70(5): 1328–1332. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Cheng W, Li Y, Hou X, et al. HSP60 is involved in the neuroprotective effects of naloxone. Mol Med Rep 2014; 10(4): 2172–2176. [DOI] [PubMed] [Google Scholar]
- 11.Lehnardt S, Schott E, Trimbuch T, et al. A vicious cycle involving release of heat shock protein 60 from injured cells and activation of toll-like receptor 4 mediates neurodegeneration in the CNS. J Neurosci 2008; 28(10): 2320–2331. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Ciechanover A and Kwon YT.. Protein quality control by molecular chaperones in neurodegeneration. Front Neurosci 2017; 11: 1–18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Lyon MS, Milligan C.. Extracellular heat shock proteins in neurodegenerative diseases: New perspectives. Neurosci Lett 2019; 711(May): 134462. [DOI] [PubMed] [Google Scholar]
- 14.Mehlem P, Schulze-Osthoff K, Arrigo A.. Small stress proteins as novel regulators of apoptosis. J Biol Chem 1996; 271(28): 16510–16514. [DOI] [PubMed] [Google Scholar]
- 15.Shimada Y, Tanaka R, Shimura H, et al. Phosphorylation enhances recombinant HSP27 neuroprotection against focal cerebral ischemia in mice. Neuroscience 2014; 278: 113–121. [DOI] [PubMed] [Google Scholar]
- 16.Lewis SE, Mannion RJ, White FA, et al. A role for HSP27 in sensory neuron survival. J Neurosci 1999; 19(20): 8945–8953. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Ma CHE, Omura T, Cobos EJ, et al. Accelerating axonal growth promotes motor recovery after peripheral nerve injury in mice. J Clin Invest 2011; 121(11): 4332–4347. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Abisambra JF, Blair LJ, Hill SE, et al. Phosphorylation dynamics regulate Hsp27-mediated rescue of neuronal plasticity deficits in tau transgenic mice. J Neurosci 2010; 30(46): 15374–15382. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.An JJ, Lee YP, Kim SY, et al. Transduced human PEP-1-heat shock protein 27 efficiently protects against brain ischemic insult. FEBS J 2008; 275(6): 1296–1308. [DOI] [PubMed] [Google Scholar]
- 20.De Maio A, Santoro MG, Tanguay RM, et al. Ferruccio Ritossa’s scientific legacy 50 years after his discovery of the heat shock response: A new view of biology, a new society, and a new journal. Cell Stress Chaperones 2012; 17(2): 139–143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Arispe N, Doh M, De Maio A.. Lipid interaction differentiates the constitutive and stress-induced heat shock proteins Hsc70 and Hsp70. Cell Stress Chaperones 2002; 7: 330–338. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Kampinga HH, Craig EA.. The HSP70 chaperone machinery: J proteins as drivers of functional specificity. Nat Rev Mol Cell Biol 2010; 11(8): 579–592. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Xu Q, Metzler B, Jahangiri M, et al. Molecular chaperones and heat shock proteins in atherosclerosis. Am J Physiol Heart Circ Physiol 2012; 302(85): H506–514. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Seigneuric R, Mjahed H, Gobbo J, et al. Heat shock proteins as danger signals for cancer detection. Front Oncol 2011; 1(November): 37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Schmitt E, Gehrmann M, Brunet M, et al. Intracellular and extracellular functions of heat shock proteins: Repercussions in cancer therapy. J Leukoc Biol 2007; 81(1): 15–27. [DOI] [PubMed] [Google Scholar]
- 26.Hartl FU, Hayer-Hartl M.. Molecular chaperones in the cytosol: From nascent chain to folded protein. Science 2002; 295(5561): 1852–1858. [DOI] [PubMed] [Google Scholar]
- 27.Turturici G, Tinnirello R, Sconzo G, et al. Positive or negative involvement of heat shock proteins in multiple sclerosis pathogenesis: An overview. J Neuropathol Exp Neurol 2014; 73(12): 1092–1106. [DOI] [PubMed] [Google Scholar]
- 28.Foster JA, Brown IR.. Differential induction of heat shock mRNA in oligodendrocytes, microglia, and astrocytes following hyperthermia. Mol Brain Res 1997; 45(2): 207–218. [DOI] [PubMed] [Google Scholar]
- 29.Koren J, Jinwal UK, Lee DC, et al. Chaperone signalling complexes in Alzheimer’s disease. J Cell Mol Med 2009; 13(4): 619–630. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Zhou ZB, Huang GX, Lu JJ, et al. Up-regulation of heat shock protein 27 inhibits apoptosis in lumbosacral nerve root avulsion-induced neurons. Sci Rep 2019; 9(1): 1–10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Wagstaff MJD, Smith J, De JS, et al. Protection of neuronal cells from apoptosis by Hsp27 delivered with a herpes simplex virus-based vector. J Biol Chem 1999; 274(8): 5061–5069. [DOI] [PubMed] [Google Scholar]
- 32.Mandelkow EM, Mandelkow E.. Biochemistry and cell biology of Tau protein in neurofibrillary degeneration. Cold Spring Harb Perspect Biol 2011; 3(10): 1–25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Webster JM, Darling AL, Uversky VN, et al. Small heat shock proteins, big impact on protein aggregation in neurodegenerative disease. Front Pharmacol 2019; 10(September): 1–18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Wilhelmus MMM, Boelens WC, Otte-Höller I, et al. Small heat shock protein HspB8: Its distribution in Alzheimer’s disease brains and its inhibition of amyloid-β protein aggregation and cerebrovascular amyloid-β toxicity. Acta Neuropathol 2006; 111(2): 139–149. [DOI] [PubMed] [Google Scholar]
- 35.Westerheide SD, Morimoto RI.. Heat shock response modulators as therapeutic tools for diseases of protein conformation. J Biol Chem 2005; 280(39): 33097–33100. [DOI] [PubMed] [Google Scholar]
- 36.Ojha J, Karmegam RV, Masilamoni JG, et al. Behavioral defects in chaperone-deficient Alzheimer’s disease model mice. PLoS One 2011; 6(2): e1655. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Yerbury JJ, Ooi L, Dillin A, et al. Walking the tightrope: Proteostasis and neurodegenerative disease. J Neurochem 2016; 137(4): 489–505. [DOI] [PubMed] [Google Scholar]
- 38.Bukach OV, Glukhova AE, Seit-Nebi AS, et al. Heterooligomeric complexes formed by human small heat shock proteins HspB1 (Hsp27) and HspB6 (Hsp20). Biochim Biophys Acta-Proteins Proteomics 2009; 1794(3): 486–495. [DOI] [PubMed] [Google Scholar]
- 39.Marino M, Papa S, Crippa V, et al. Differences in protein quality control correlate with phenotype variability in 2 mouse models of familial amyotrophic lateral sclerosis. Neurobiol Aging 2015; 36(1): 492–504. [DOI] [PubMed] [Google Scholar]
- 40.Cox D, Carver JA, Ecroyd H.. Preventing α-synuclein aggregation: The role of the small heat-shock molecular chaperone proteins. Biochim Biophys Acta-Mol Basis Dis 2014; 1842(9): 1830–1843. [DOI] [PubMed] [Google Scholar]
- 41.Alderson TR, Roche J, Gastall HY, et al. Local unfolding of the HSP27 monomer regulates chaperone activity. Nat Commun 2019; 10(1): 1–16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Stetler RA, Cao G, Gao Y, et al. Hsp27 protects against ischemic brain injury via attenuation of a novel stress-response cascade upstream of mitochondrial cell death signaling. J Neurosci 2008; 28(49): 13038–13055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Shimada Y, Shimura H, Tanaka R, et al. Phosphorylated recombinant HSP27 protects the brain and attenuates blood-brain barrier disruption following stroke in mice receiving intravenous tissue-plasminogen activator. PLoS One 2018; 13(5): 1–18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Morris MC, Depollier J, Mery J, et al. A peptide carrier for the delivery of biologically active proteins into mammalian cells. Nat Biotechnol 2001; 19(12): 1173–1176. [DOI] [PubMed] [Google Scholar]
- 45.Chen ZL, Yu WM, and Strickland S.. Peripheral regeneration. Annu Rev Neurosci 2007; 30: 209–233. [DOI] [PubMed] [Google Scholar]
- 46.Höke A. A (heat) shock to the system promotes peripheral nerve regeneration. J Clin Invest 2011; 121(11): 4231–4234. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Hoke A and Brushart T.. Challenges and opportunities for regeneration in the peripheral nervous system. J Exp Neurol 2009; 223(1): 1–4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Cashman CR, Höke A.. Mechanisms of distal axonal degeneration in peripheral neuropathies. Neurosci Lett 2015; 1–18. Available from: http://linkinghub.elsevier.com/retrieve/pii/S0304394015000610 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Hunt D, Raivich G, Anderson PN.. Activating transcription factor 3 and the nervous system. Front Mol Neurosci 2012; 5(February): 7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Lindsay M. Nerve growth factors (NGF, BDNF) Enhance Axonal Regeneration but Are not Required for Survival of Adult Sensory Neurons. J Neurosci . 1988;8(7):2394–405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Venugopal A, Sundaramoorthy K, Vellingiri B.. Therapeutic potential of Hsp27 in neurological diseases. Egypt J Med Hum Genet 2019; 20(1): 2–8. [Google Scholar]
- 52.Amor S, Puentes F, Baker D, et al. Inflammation in neurodegenerative diseases. Immunology 2010; 129(2): 154–169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Li HF, Zhang HL.. Elevated HSP27 levels during attacks in patients with multiple sclerosis. Acta Neurol Scand 2012; 126(2): 2005–2006. [DOI] [PubMed] [Google Scholar]
- 54.Ce P, Erkizan O, Gedizlioglu M.. Elevated HSP27 levels during attacks in patients with multiple sclerosis. Acta Neurol Scand 2011; 124(5): 317–320. [DOI] [PubMed] [Google Scholar]
- 55.Yonekura K, Yokota SI, Tanaka S, et al. Prevalence of anti-heat shock protein antibodies in cerebrospinal fluids of patients with Guillain-Barr?? syndrome. J Neuroimmunol 2004; 156: 204–209. [DOI] [PubMed] [Google Scholar]
- 56.Helgeland G, Petzold A, Hoff JM, et al. Anti-heat shock protein 70 antibody levels are increased in myasthenia gravis and Guillain-Barr?? syndrome. J Neuroimmunol 2010; 225(1–2): 180–183. [DOI] [PubMed] [Google Scholar]
- 57.Asthana P, Zhang G, Sheikh KA, et al. Heat shock protein is a key therapeutic target for nerve repair in autoimmune peripheral neuropathy and severe peripheral nerve injury. Brain Behav Immun 2021; 91: 48–64. [DOI] [PubMed] [Google Scholar]
- 58.Chaudhuri TK, Paul S.. Protein-misfolding diseases and chaperone-based therapeutic approaches. FEBS J 2006; 273(7): 1331–1349. [DOI] [PubMed] [Google Scholar]
- 59.Valastyan JS, Lindquist S.. Mechanisms of protein-folding diseases at a glance. Dis Model Mech . 2014; 7(1): 9–14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Soto C. Protein misfolding in neurodegenerative diseases: The key pending questions. J Neurol Transl Neurosci . 2013; 1(10): 19–22. [Google Scholar]
- 61.Turturici G, Sconzo G, Geraci F.. Hsp70 and its molecular role in nervous system diseases. Biochem Res Int 2011; 2011: 1–18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Labrador-garrido A, Bertoncini CW, Roodveldt C.. The Hsp70 chaperone system in Parkinson’s disease. In: Rana AQ (ed) Etiology and pathophysiology of Parkinson’s disease [Internet] . 1st ed. In Tech; 2011, pp. 221–46. http://www.intechopen.com/books/etiology-and-pathophysiology-of-parkinson-s-disease/the-hsp70-chaperone-system-in-parkinson-s-disease
- 63.Witt SN. Molecular chaperones, alpha-synuclein and neurodegeneration. Mol Neurobiol 2013; 47(2): 552–560. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Chan HY, Bonini NM.. Drosophila models of human neurodegenerative disease. Cell Death Differ. 2000; 7(11): 1075–1080. [DOI] [PubMed] [Google Scholar]
- 65.Auluck PK, Chan HYE, Trojanowski JQ, et al. Chaperone suppression of α-synuclein toxicity in a Drosophila model for Parkinson’s disease. Science 2002; 295(5556): 865–868. [DOI] [PubMed] [Google Scholar]
- 66.Lu B and Vogel H.. Drosophila models of neurodegenerative diseases. Annu Rev Pathol Mech Dis 2009; 4: 315–342. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Jellinger KA. Interaction between pathogenic proteins in neurodegenerative disorders. J Cell Mol Med 2012; 16(6): 1166–1183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Batelli S, Albani D, Rametta R, et al. DJ-1 modulates α-synuclein aggregation state in a cellular model of oxidative stress: Relevance for Parkinson’s disease and involvement of HSP70. PLoS One 2008; 3(4):e1884. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 69.Franklin TB, Krueger-Naug AM, Clarke DB, et al. The role of heat shock proteins Hsp70 and Hsp27 in cellular protection of the central nervous system. Int J Hyperthermia 2005; 21(5): 379–392. [DOI] [PubMed] [Google Scholar]
- 70.Heydari AR, Wu B, Takahashi R, et al. Expression of heat shock protein 70 is altered by age and diet at the level of transcription. Mol Cell Biol 1993; 13(5): 2909–2918. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Bobkova N V, Nesterova I V, Medvinskaya NI, Aleksandrova IY, Samokhin A N, Gershovich JG, et al. Possible role of the olfactory system in Alzheimer's disease genesis. In: Fisher A, Hanin I and Memo M. SF (eds) New trends in Alzheimer and parkinson disorders ADPD . 1st ed. International Proceedings Division, MEDIMOND s.r.l.; 2005, pp. 91–95. [Google Scholar]
- 72.Bobkova N, Guzhova I, Margulis B, et al. Dynamics of endogenous Hsp70 synthesis in the brain of olfactory bulbectomized mice. Cell Stress Chaperones 2013; 18(1): 109–118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Bobkova NV, Garbuz DG, Nesterova I, et al. Therapeutic effect of exogenous Hsp70 in mouse models of Alzheimer’s disease. J Alzheimer’s Dis 2014; 38(2): 425–435. [DOI] [PubMed] [Google Scholar]
- 74.Wesson DW, Levy E, Nixon RA, et al. Olfactory dysfunction correlates with amyloid-beta burden in an Alzheimer’s disease mouse model. J Neurosci 2010; 30(2): 505–514. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Aleksandrova Y, Kuvichkin VV, Kashparov IA, et al. Increased level of β-amyloid in the brain of bulbectomized mice. Biochemistry 2004; 69: 176–180. [DOI] [PubMed] [Google Scholar]
- 76.Takahashi RH, Nam EE, Edgar M, et al. Alzheimer beta-amyloid peptides: Normal and abnormal localization. Histol Histopathol 2002; 17(1): 239–246. [DOI] [PubMed] [Google Scholar]
- 77.Evans CG, Wisén S, Gestwicki JE.. Heat shock proteins 70 and 90 inhibit early stages of amyloid β-(1-42) aggregation in vitro. J Biol Chem 2006; 281(44): 33182–33191. [DOI] [PubMed] [Google Scholar]
- 78.Magrane J. Heat shock protein 70 participates in the neuroprotective response to intracellularly expressed-amyloid in neurons. J Neurosci 2004; 24(7): 1700–1706. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Tidwell JL, Houenou LJ, Tytell M.. Administration of Hsp70 in vivo inhibits motor and sensory neuron degeneration. Cell Stress Chaperones 2004; 9: 88–98. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Giffard RG, Macario AJL, De Macario EC.. The future of molecular chaperones and beyond. J Clin Invest 2013; 123(8): 3206–3208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Malyshev I. HSP70 in the Immune Responses. In: Immunity, tumors and aging: The role of HSP70 . 2013th ed. Moscow: Springer, 2013, pp. 63–82. [Google Scholar]
- 82.Zhang ZY, Zhang Z, Schluesener HJ.. Toll-like receptor-2, CD14 and heat-shock protein 70 in inflammatory lesions of rat experimental autoimmune neuritis. Neuroscience 2009; 159(1): 136–142. [DOI] [PubMed] [Google Scholar]
- 83.Loshaj-Shala A, Regazzoni L, Daci A, et al. Guillain Barré syndrome (GBS): New insights in the molecular mimicry between C. jejuni and human peripheral nerve (HPN) proteins. J Neuroimmunol 2015; 289: 168–176. [DOI] [PubMed] [Google Scholar]
- 84.Dickey CA, Kamal A, Lundgren K, et al. The high-affinity HSP90-CHIP complex recognizes and selectively degrades phosphorylated tau client proteins. J Clin Invest 2007; 117(3): 648–658. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.McLean PJ, Klucken J, Shin Y, et al. Geldanamycin induces Hsp70 and prevents α-synuclein aggregation and toxicity in vitro. Biochem Biophys Res Commun 2004; 321(3): 665–669. [DOI] [PubMed] [Google Scholar]
- 86.Luo W, Sun W, Taldone T, et al. Heat shock protein 90 in neurodegenerative diseases. Mol Neurodegener 2010; 5(1): 1–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Sreedhar AS, É Kalmár, Csermely P, et al. Hsp90 isoforms: Functions, expression and clinical importance. FEBS Lett 2004; 562(1–3): 11–15. [DOI] [PubMed] [Google Scholar]
- 88.Epple LM, Griffiths SG, Dechkovskaia AM, et al. Medulloblastoma exosome proteomics yield functional roles for extracellular vesicles. PLoS One 2012; 7(7): e42064. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Wang WB, Feng LX, Yue QX, et al. Paraptosis accompanied by autophagy and apoptosis was induced by celastrol, a natural compound with influence on proteasome, ER stress and Hsp90. J Cell Physiol 2012; 227(5): 2196–2206. [DOI] [PubMed] [Google Scholar]
- 90.Graner MW. HSP90 and immune modulation in cancer. Adv Cancer Res 2016; 129: 191–224. [DOI] [PubMed] [Google Scholar]
- 91.Multhoff G, Pockley AG, Streffer C, et al. Dual role of heat shock proteins (HSPs) in anti-tumor immunity. Curr Mol Med 2012; 12(9): 1174–1182. [DOI] [PubMed] [Google Scholar]
- 92.Oura J, Tamura Y, Kamiguchi K, et al. Extracellular heat shock protein 90 plays a role in translocating chaperoned antigen from endosome to proteasome for generating antigenic peptide to be cross-presented by dendritic cells. Int Immunol 2011; 23(4): 223–237. [DOI] [PubMed] [Google Scholar]
- 93.Biamonte MA, Van De Water R, Arndt JW, et al. Heat shock protein 90: Inhibitors in clinical trials. J Med Chem 2010; 53(1): 3–17. [DOI] [PubMed] [Google Scholar]
- 94.Waza M, Adachi H, Katsuno M, et al. Alleviating neurodegeneration by an anticancer agent: An Hsp90 inhibitor (17-AAG). Ann N Y Acad Sci 2006; 1086: 21–34. [DOI] [PubMed] [Google Scholar]
- 95.Shrestha L, Bolaender A, Patel HJ, et al. Heat shock protein (HSP) drug discovery and development: Targeting heat shock proteins in disease. Curr Top Med Chem 2016; 16(25): 2753–2764. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Shimura H, Miura-Shimura Y, Kosik KS.. Binding of tau to heat shock protein 27 leads to decreased concentration of hyperphosphorylated tau and enhanced cell survival. J Biol Chem 2004; 279(17): 17957–17962. [DOI] [PubMed] [Google Scholar]
- 97.Tóth ME, Szegedi V, Varga E, et al. Overexpression of Hsp27 ameliorates symptoms of Alzheimer’s disease in APP/PS1 mice. Cell Stress Chaperones 2013; 18(6): 759–771. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Shimura H, Tanaka R, Urabe T, et al. Heat shock protein 27 (HSP27) as a therapeutic target in ischemic stroke and neurodegenerative disorders. Juntendo Med J 2017; 63(1): 17–21. [Google Scholar]
- 99.Taldone T, Ochiana SO, Patel PD, et al. Selective targeting of the stress chaperome as a therapeutic strategy. Trends Pharmacol Sci 2014; 35(11): 592–603. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Rousaki A, Miyata Y, Jinwal UK, et al. Allosteric drugs: The interaction of antitumor compound MKT-077 with human Hsp70 chaperones. J Mol Biol 2011; 411(3): 614–632. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Abisambra J, Jinwal UK, Miyata Y, et al. Allosteric heat shock protein 70 inhibitors rapidly rescue synaptic plasticity deficits by reducing aberrant tau. Biol Psychiatry 2013; 74(5): 367–374. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Lectins: Sharon N. Carbohydrate-specific reagents and biological recognition molecules. J Biol Chem 2007; 282(5): 2753–2764. [DOI] [PubMed] [Google Scholar]
- 103.Martin MD, Baker JD, Suntharalingam A, et al. Inhibition of both Hsp70 activity and tau aggregation in vitro best predicts tau lowering activity of small molecules. ACS Chem Biol 2016; 11(7): 2041–2048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Congdon EE, Wu JW, Myeku N, et al. Methylthioninium chloride (methylene blue) induces autophagy and attenuates tauopathy in vitro and in vivo. Autophagy 2012; 8(4): 609–622. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Jinwal UK, Koren J, O’Leary JC, et al. Hsp70 ATPase modulators as therapeutics for Alzheimer’s and other Neurodegenerative diseases. Mol Cell Pharmacol 2010; 2(2): 43–46. [PMC free article] [PubMed] [Google Scholar]
- 106.Hoshino T, Suzuki K, Matsushima T, et al. Suppression of Alzheimer’s disease-related phenotypes by geranylgeranylacetone in mice. PLoS One 2013; 8(10): 2–11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Sun Y, Zhang JR, Chen S.. Suppression of alzheimer’s disease-related phenotypes by the heat shock protein 70 inducer, geranylgeranylacetone, in APP/PS1 transgenic mice via the ERK/p38 MAPK signaling pathway. Exp Ther Med 2017; 14(6): 5267–5274. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Liu L, Zhang C, Kalionis B, et al. EGb761 protects against Aβ1-42 oligomer-induced cell damage via endoplasmic reticulum stress activation andHsp70 protein expression increase in SH-SY5Y cells. Exp Gerontol 2016; 75: 56–63. [DOI] [PubMed] [Google Scholar]
- 109.Campanella C, Pace A, Bavisotto CC, et al. Heat shock proteins in Alzheimer’s disease: Role and targeting. Int J Mol Sci 2018; 19(9): 1–22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Ortega L, Calvillo M, Luna F, et al. 17-AAG improves cognitive process and increases heat shock protein response in a model lesion with Aβ25-35. Neuropeptides; 2014; 48(4): 221–232. [DOI] [PubMed] [Google Scholar]
- 111.Ho SW, Tsui YTC, Wong TT, et al. Effects of 17-allylamino-17-demethoxygeldanamycin (17-AAG) in transgenic mouse models of frontotemporal lobar degeneration and Alzheimer’s disease. Transl Neurodegener 2013; 2(1): 1–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Pillay I, Nakano H, Sharma SV.. Radicicol inhibits tyrosine phosphorylation of the mitotic Src substrate sam68 and retards subsequent exit from mitosis of Src-transformed cells. Cell Growth Differ 1996; 7(11): 1487–1499. [PubMed] [Google Scholar]
- 113.Ali YO, Kitay BM, Zhai RG.. Dealing with misfolded proteins: Examining the neuroprotective role of molecular chaperones in neurodegeneration. Molecules 2010; 15(10): 6859–6887. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Shimura H, Schwartz D, Gygi SP, et al. CHIP-Hsc70 complex ubiquitinates phosphorylated tau and enhances cell survival. J Biol Chem 2004; 279(6): 4869–4876. [DOI] [PubMed] [Google Scholar]