

Abstract
Background:
Calorie restriction (CR) during daily nutrition has been shown to affect the prognosis of many chronic diseases such as metabolic syndrome, diabetes, and aging. As an alternative nutrition model, prolonged intermittent fasting (PF) in humans is defined by the absence of food for more than 12 h. In our previous human studies, CR and PF models were compared and it was concluded that the two models might have differences in signal transduction mechanisms. We have investigated the effects of these models on neurons at the molecular level in this study.
Methods:
Neurons (SH-SY5Y) were incubated with normal medium (N), calorie-restricted medium (CR), fasting medium (PF), and glucose-free medium (G0) for 16 h. Simultaneously, ketone (beta-hydroxybutyrate; bOHB) was added to other experiment flasks containing the same media. Concentrations of lactate, lactate dehydrogenase (LDH), bOHB, and glucose were measured to demonstrate the changes in the energy metabolism together with the mitochondrial functions of cells. Citrate synthase activity and flow cytometric mitochondrial functions were investigated.
Results:
At the end of incubations, lactate and LDH levels were decreased and mitochondrial activity was increased in all ketone-added groups (P < .01) regardless of the glucose concentration in the environment. In the fasting model, these differences were more prominent.
Conclusion:
Our results demonstrated that neurons use ketones regardless of the amount of glucose, and bOHB-treated cells had positive changes in mitochondrial function. We conclude that the presence of bOHB might reverse neuron damage and that exogenous ketone treatment may be beneficial in the treatment of neurological diseases in the future.
Keywords: Calorie restriction, Citrate synthase, Fasting, Ketone bodies, Ketone bodies, Neuron cultures, Mitochondrial function
Introduction
In recent years, researchers are working hard on genetics and metabolic pathways to get insights into aging and increasing lifespan. CR is the only proven path to increased healthy longevity and resistance to disease.1, 2 Moreover, CR is a well-known treatment for obesity, metabolic syndrome, and prediabetes, and has shown to be beneficial in Parkinson’s disease and Alzheimer's.1, 3 However, daily CR requires continuous compliance and most patients have difficulties in changing their lifestyle. Currently, an alternative nutrition model is defined as intermittent fasting (IF) and has been used in animal studies. It has been shown that the lifespan of rats that are fed once every 24 or 48 h without CR had increased, and the disease course had slowed down in Alzheimer's models.4, 5
Prolonged intermittent fasting (PF) in humans is defined as fasting more than 12 h which increases fat catabolism and production of ketones in the liver, while physiologically gluconeogenesis tries to stabilize the blood glucose. Aside from CR, in PF it is thought that fasting stress may change energy metabolism pathways in the cells.6, 7 A number of mechanisms have been proposed, especially to ketone bodies, during PF that include alterations in antioxidant status, changes in brain neurotransmitter levels, such as GABA, and the metabolic consequences on cellular energy metabolism together with anti-inflammatory effects on neuronal cells.8, 9 The benefits of ketone therapy have been demonstrated in drug-resistant cases, especially in chronic neurological diseases such as epilepsy. 3 Ketogenic diet (KD) and exogenous ketone applications were first used in refractory epilepsy in 1920, and only with this treatment patients' incurable attacks could be stopped. 10 With the replacement of antiepileptic drugs in the following years, neuroprotective effects of ketones have been underestimated for many years. In the 1990s, with the new applications, KD therapy came back to the protocols of pharmaco-resistant epilepsy, and since then, it has been included in the treatment of pediatric patients. However, the exact mechanism of action of ketone bodies is still unknown today.
Although PF is practiced by many people all around the world, apart from the studies performed with a few volunteers, human studies with a large sample size are limited.5, 11, 12 In our previous study, we have compared CR to PF in obese and healthy individuals and showed that PF may enhance health and cellular resistance to disease by different mechanisms, even if the fasting period is followed by a period of overeating and overall caloric intake is not decreased. 6 Mechanism of action of PF is not fully understood; the most important finding is that, unlike normal, body is exposed to abundant ketone bodies as a result of fat breakdown during PF.
This study aims to investigate the changes in energy metabolism and contribution of ketone bodies to these changes during long-term (16 h) nutrient deprivation in neuronal cell cultures and try to provide further insight into the mechanisms whereby ketones may exert their beneficial effects on neuronal mitochondrial function.
Methods
Cell Cultures
SH-SY5Y cells were purchased from ATCC (human neuroblastoma cells-ATCC Cat# CRL-2266, RRID: CVCL_0019) and stored frozen. When required, cells were thawed and quickly seeded at 2 × 10 5 cells/cm2 density into 25 cm2 flasks and grown in media containing Dulbecco’s modified Eagle’s Medium/Ham’s F-12 Nutrient Mixture (DMEM/F-12; 1:1) containing L-glutamine and 10% fetal bovine serum (FBS) at 37°C. Growth medium was replaced every two days and doubling time studies showed up to be approximately 29 h. Cells were passaged at 80% to 90% confluency to 75 cm2 flasks and the total media volume was made up to 10 mL before incubating at 37°C and 5% CO2 + 95% air atmosphere. Experiments were performed in 75 cm2 flasks and early passages of cells (P2-5 after purchase) were used.
Experimental Cell Treatment Conditions
Eight groups were organized: Control group (N), cells were incubated with normal medium: This medium contained 300 g/dL glucose (DMEM/F12 + 10% FBS). Calorie restriction (CR) model: Earl's balanced salt solution (EBSS) + 10% FBS medium was used in this group during the experiments. The amount of glucose was 100 g/dL in the medium (EBSS). Prolonged fasting model (PF): Glucose amount was reduced to 50 g/dL in the medium of these cells with the dilution of EBSS with glucose-free medium (Dulbecco's Modified Eagle Medium, DMEM 0) still containing 10% FBS. A fourth group (negative control, G0) was added to the experiment in which glucose was completely withdrawn; the medium of this group was DMEM0 containing 10% FBS. Subgroups were formed by adding 5 mM ketone (βOHB) to all these aforementioned groups. For the ketone-added groups, the abbreviations “K” have been added: NK, CRK, PFK, G0K. G0 and G0K group of cells in the experiment do not reflect the physiological conditions because normally our cells never remain completely glucose-free during CR and fasting. These groups were designed specifically to observe the behaviors of cells when only ketone is found in the environment. DMEM/F12 (Cat. number: 11330032), DMEM0 (Cat. number: A1443001), and FBS (Cat. number: 10500064) were purchased from Gibco Life Technologies and EBSS was purchased from Pan Biotech (Cat. number: P04-31500).
Experiments
When the cells were 80% confluent before the experiments, normal media were withdrawn, and each flask was washed with 2 mL of sterile phosphate-buffered saline (PBS) and experiment media were added. About 1 mL of the unused media was allotted for the measurements of energy metabolites (glucose, lactate, βOHB, and LDH) before the experiments (initial values, i). Experiments were started the day before, and 16 h incubation ended the next morning. Experiment media were removed and 1 mL of them were allotted for the metabolic measurements again (end values, e). Cells were removed with Trypsin/EDTA and the viability rates were controlled with trypan blue. Then they were homogenized and divided into two for citrate synthase activity and flow-cytometric mitochondrial function assessments. In addition, microscopic pictures of the cells for morphological examination were taken before and after the experiments. These experiments were repeated seven times under the same conditions.
Beta-Hydroxybutyrate (βOHB) Application
Sodium βOHB was purchased from Sigma (Cat. number: 54965-50G). To test the effect of βOHB, cultures were treated with βOHB and dose-dependent (0.5, 2, 5, 8, and 10 mM) cell viabilities were performed to establish the optimum experiment concentrations (data not shown). A 5 mM concentration was chosen for incubation during the experiment periods, which was consistent with physiologic doses in a fasting state and other molecular studies.13–15 Calculations were made from βOHB with a molecular weight of 126.09 g to a total of 5 mM in a 10 mL medium. A 0.630 mg was weighed and dissolved in 50 mL sterile PBS, filtered with a 0.22 µm syringe to prepare the sterile stock solution. All the ketone-added media were prepared to have a final concentration of 5 mM βOHB.
Cell Viability
The 3-(4, 5-dimethylthiazol2-yl)-2, 5-diphenyltetrazolium-bromide (MTT) assay is a cytotoxicity experiment that is used to demonstrate the viability of cells under certain conditions numerically. While MTT dye is metabolized by living cells and gives color at 595 nm wavelength, color formation is not observed in dead cells. Color formation intensity is directly proportional to cell viability. MTT kit (Cat. number 11465007001-Roche-Germany) was used in the experiments. Cells were seeded onto PLL-coated 96-well plates at a density of 2 × 10 4 cells/well. Cell viability was estimated by measuring the metabolism of MTT under each experimental media. MTT solution (5 mg/mL) was added to each well in a 96-well plate, and cells were incubated for 4 h at 37°C. Then, solubilization solution (SDS) was added to each well. After overnight incubation, absorbance at 570 nm was measured. Cell viability was expressed as the percentage of viable cells relative to the control (N group).
Analysis of Metabolic Parameters
With the Abbot precision-extra POCT device, ketone (βOHB) concentrations of the media were measured before and after the experiments. About 10 µL of each medium was introduced to the ketone probe and individual measurements were made. Glucose, lactate, and LDH levels in the culture media were measured in ADVIA 1800 (Siemens) biochemistry autoanalyzer. Briefly, hexokinase and ATP transform glucose-to-glucose 6-phosphate plus ADP; glucose 6-phosphate is then reacted with NADP and glucose 6-phosphate dehydrogenase to form NADP which is measured spectrophotometrically at 340/410 nm. The analytic range of the glucose measurement method was 1 to 700 mg/dL. For lactate measurements, briefly, LDH oxidizes lactate to pyruvate with simultaneous reduction of NAD+ to NADH, which is monitored spectrophotometrically at 340 nm/383 nm. The analytic range of the lactate measurement method was 2 to 50 mg/L. LDH activity was assessed by measuring the decrease in the optical density resulting from the oxidation of NADH at 340 nm using pyruvate as a substrate. The analytic range of the LDH activity measurement was 5 to 750 U/L.
Citrate Synthase Activity Measurement
Citrate synthase is the first enzyme of the tricyclic acid cycle in the mitochondrial matrix. The activity of citrate synthase is frequently used as a biochemical marker for the mitochondrial content and functions of a cellular homogenate. A citrate synthase activity measurement kit (ab119692-Abcam, USA) was used for the enzyme activities in accordance with the experimental plan. Briefly, cells were collected in a centrifuge tube with Trypsin/EDTA after 16 h of incubation and centrifuged at 4°C with 500 g for 10 min. The supernatant was discarded and the pellet was washed twice with PBS. After the washings, 3 mL of extraction buffer provided by the kit were added to the tubes and incubated in ice for 20 min. After the incubation, tubes were centrifuged at 4°C with 16 g for 20 min. The amount of protein in the supernatant was measured. According to the protein concentrations, homogenates were diluted with the buffer solution to the desired protein concentration. Samples were added to a 96-well plate in a volume of 100 µL and incubated for 3 h at room temperature. After 3 h, wells were washed twice with the washing buffer. Absorbances were measured in the ELISA reader and color changes were recorded at 412 nm for 30 min. Activity graphs are drawn and the time for Vmax of the enzymes was calculated as minutes for each group.
Assessment of Mitochondrial Functions by Flow-Cytometry
Flow-cytometric techniques are conveniently used to simultaneously investigate the effects of many physiological and pathological conditions on mitochondrial functions (Robinson et al.). They are mainly based on the use of laser beams capable of measuring at a specific wavelength with the help of metabolite-specific fluorescent dyes. This method allows the metabolic changes in the cell to be measured quantitatively and expressed as mean fluorescence intensity (MFI). The MitoTracker Red dye is used for measuring the mitochondrial membrane potential selectively. It enters the mitochondria in the cell and, as the membrane potential increases, its color changes from green to orange-red and allows the flow-cytometric measurement at 599 nm wavelength. In this study, we have used this specific dye in cells incubated in different experimental media on mitochondrial membrane potential. For mitochondrial reactive oxygen species (ROS) formation, MitoSOX Red (ex: 510 nm, em: 580 nm) dye was used and for mitochondria amount of cells, MitoTracker Green (ex: 490 nm, em: 516 nm) dye was used. Cells were removed by trypsin/EDTA and divided into three (3 × 105 cells for each dye) after counting. Cells were once washed with sterile PBS and stained for 20 min in the dark at 37°C, with 100 nM MitoTracker Red, 5 µM MitoSOX, and 50 nM MitoTracker Green. After centrifugation at 500 g for 5 min and washing once with PBS again, they were scanned in the flow-cytometry (BD, FACS Verse) device. Flow-cytometric mitochondrial dyes were purchased from Thermo Fischer Scientific-USA, MitoTracker Red (CMXRos) Cat No: M7512, MitoSOX (Red Mitochondrial Superoxide Indicator-M36008) M36008, MitoTracker Green (GREEN FM) Cat No: M7514.
Data Analysis
All our data are presented as mean ± standard deviation (SD) from seven independent experiments. Our results were evaluated statistically by one-way ANOVA, followed by Tukey’s multiple-comparison test. Differences with a probability (P) less than .05 were considered statistically significant.
Results
Calorie Restriction and Fasting Model Optimization Studies
To test and decide the incubation period for CR and PF model, 105 cells were planted in 12-well flasks, each with an area of 4 cm2 in the second passage and when the density was 80% normal, growth media were removed and experiment media were added in four different groups: Normal (N), CR, PF, and G0. These media were added in a total volume of 2 mL according to the flask plan and cells were incubated in the aforementioned media for 4, 8, 12, 16, and 20 h. Media were collected from the wells at the beginning and end of each incubation, and the amount of glucose, lactate, and LDH were analyzed. Cells were removed with 200 µL trypsin/EDTA and homogenized in 1 mL medium and cell viability (Figure 1) was checked by trypan blue application. This experiment was repeated three times and it was decided that the most ideal incubation time was 16 h by comparing all the results obtained. This decision was based on the LDH levels of the media, viability of the cells, and the state of fasting for more than 12 h to be simulated.
Figure 1. Cell Viabilities in the Calorie Restriction and Fasting Model Optimization Studies (n = 3).
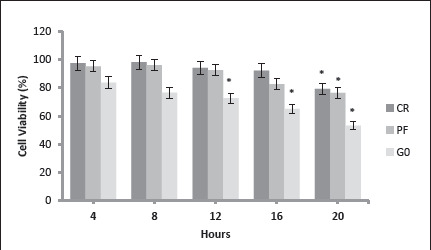
Note: CR, calorie restriction model (EBSS + 10% FBS, 100 mg/dL glucose containing medium); PF, prolonged fasting model (EBSS: DMEM0 1:1 + 10% FBS, 50 mg/dL glucose containing medium); G0 (DMEM0 + 10% FBS, glucose-free medium); * P < .01 when compared to the first 4 h of the same group.
Metabolic Parameters
All cells consumed glucose during experiments; cells incubated with ketone-added media also used βOHB simultaneously, and their glucose utilization decreased compared to other cells (Table 1). In addition, lactate formation was reduced in ketone-added cells compared to other media. Especially CRK and PFK media seemed to help the cells use their mitochondria to get energy rather than the anaerobic glycolysis. LDH enzyme activity measurements are one of the indicators of cell damage16 and LDH levels of cells that were incubated with ketone-added media were lower. Cell damage was the highest in cells with G0 and G0K media as expected, but surprisingly it was lower in G0K medium compared to G0 although both media was glucose deprived (Table 1). As mentioned before, the G0 and G0K group of cells in the experiment do not reflect the physiological conditions because normally our cells never remain completely glucose-free during CR and fasting. These groups were created specifically to observe the behaviors of cells only when ketone is found in the environment.
Table 1. Metabolites Before and After the Experiments in the Cell Media.
| Glucose | Glucose | Lactate | Lactate | βOHB | βOHB | LDH | LDH | |
| mg/dL | mg/dL | mg/dL | mg/dL | mmol/L | mmol/L | U/L | U/L | |
| N | 305 ± 16 | 231 ± 14† | 14,8 ± 0,9 | 24,6 ± 1,6† | 0 | 0 | 17 ± 2,1 | 25 ± 2,7† |
| NK | 311 ± 18 | 270 ± 28*† | 13,1 ± 1,2 | 14,5 ± 1,2* | 4,9 ± 0,3 | 3,7 ± 0,5† | 16 ± 3,6 | 19 ± 2*† |
| CR | 99 ± 13 | 83 ± 9*† | 14,7 ± 3,1 | 30,6 ± 2,8*† | 0 | 0 | 18 ± 1,4 | 26 ± 0,9† |
| CRK | 102 ± 9 | 87 ± 7*# | 13,9 ± 2,2 | 17,3 ± 1,7*# | 4,8 ± 0,8 | 3,1 ± 0,9† | 18 ± 2,3 | 21 ± 1,7† |
| PF | 47 ± 6 | 37 ± 6*† | 14,9 ± 2,9 | 41,2 ± 0,9*† | 0 | 0 | 19 ± 1,9 | 31 ± 2,3*† |
| PFK | 48 ± 4 | 39 ± 3*#† | 14,8 ± 1,9 | 30,2 ± 1,7*#† | 4,7 ± 0,7 | 2,9 ± 0,6† | 17 ± 1,4 | 22 ± 1,7*#† |
| G0 | 3 ± 0,2 | 3 ± 0,3* | 14,4 ± 2,1 | 23,6 ± 2,6*† | 0 | 0 | 18 ± 1,8 | 41 ± 1,6*† |
| G0K | 3 ± 0,6 | 1 ± 0,2* | 14,9 ± 2,3 | 14,3 ± 1,4*,# | 5,1 ± 0,4 | 1,9 ± 0,2† | 17 ± 2,7 | 29 ± 0,9*#† |
Note: Values are shown with "i" before the experiment and with "e" after the experiment. Data are expressed as mean ± SD (n = 7). N, (normal) control group (DMEM F12 + 10% FBS-300 mg/dL glucose containing medium); NK, ketone added to normal medium (DMEM F12 + 10% FBS + 5 mM bOHB); CR, calorie restriction model (EBSS + 10% FBS, 100 mg/dL glucose containing medium); CRK, ketone added to CR medium (EBSS + 10% FBS + 5 mM bOHB); PF, prolonged fasting model (EBSS: DMEM0 1:1 + 10% FBS, 50 mg/dL glucose containing medium); PFK, ketone added to PF medium (EBSS: DMEM0 1: 1 + 10% FBS + 5 mM bOHB), G0 (DMEM0 + 10% FBS, no-glucose containing medium); G0K, ketone added to G0 medium (DMEM0 + 10% FBS + 5 mM bOHB). * P < .01 when compared to N; # P < .01 when compared to the same group without bOHB; †P < .01 when compared to the values at the beginning.
Cell Viability
Cellular viability was assessed by MTT assay. Viability rates of the ketone-treated cells were higher than the cells kept in other media. In the case of ketone body, βOHB protected against cell death in neurons treated with decreased glucose media (CRK and PFK; Figure 2). Cell survival was expressed as percentage neuroprotection compared with the normal set at 100%. Even in glucose-free media (G0), cells survived to a certain degree and cells in glucose-free plus ketone-added media (G0K) were more viable (Figure 2).
Figure 2. Cell Viability Assessed with MTT Assay.
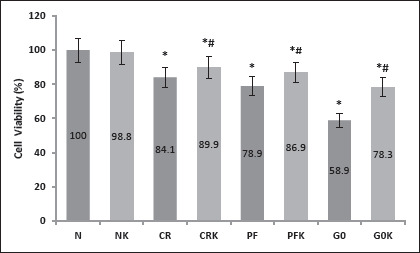
Note: Data are expressed as mean ± SD (n = 7). N, (normal) control group (DMEM F12 + 10% FBS-300 mg/dL glucose containing medium); NK, ketone added to normal medium (DMEM F12 + 10% FBS + 5 mM bOHB); CR, calorie restriction model (EBSS + 10% FBS, 100 mg/dL glucose containing medium); CRK, ketone added to CR medium (EBSS + 10% FBS + 5 mM bOHB); PF, prolonged fasting model (EBSS: DMEM0 1:1 + 10% FBS, 50 mg/dL glucose containing medium); PFK, ketone added to PF medium (EBSS: DMEM0 1: 1 + 10% FBS + 5 mM bOHB), G0 (DMEM0 + 10% FBS, no-glucose containing medium); G0K, ketone added to G0 medium (DMEM0 + 10% FBS + 5 mM bOHB). The viability of the cells in wells containing normal medium was considered 100%, and the viability rates of other groups were calculated accordingly. Values are given as mean ± SD. * P < .01 when compared to N, # P < .01 when compared to the same group without bOHB.
Citrate Synthase Activity
In our experiments, citrate synthase activity was measured in cells after 16 h of incubation with the experiment media. From the activity graphs, the time when the enzyme reaches the highest speed (Vmax) was calculated in minutes and is given in Figure 3. Cells incubated with a normal medium reached the Vmax value fastest. It was observed that the enzyme was working slowly as medium glucose content was decreased. Enzyme velocity was found to be statistically increased in media containing ketone when compared to no ketone-containing media of the same groups (Figure 3). As mentioned before, the G0 and G0K group of cells in the experiment do not reflect the physiological conditions because normally our cells never remain completely glucose-free during CR and fasting. These groups were created specifically to observe the behaviors of cells only when ketone is found in the environment. However, in the G0K group, the enzyme activity was observed to be faster than the G0 group, which suggests in the absence of glucose, cells use ketone vigorously (Table 1) and try to restore mitochondrial energy pathways.
Figure 3. Citrate synthase activity.
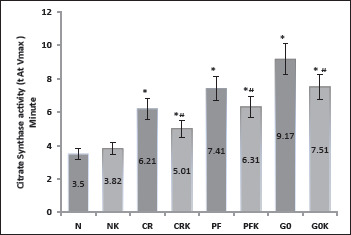
Note: Data are expressed as mean ± SD (n = 7). Graph drawn according to the minute measured when the enzyme reaches its highest velocity (Vmax). Values are mean ± SD. N, (normal) control group (DMEM F12 + 10% FBS-300 mg/dL glucose containing medium); NK, ketone added to normal medium (DMEM F12 + 10% FBS + 5 mM bOHB); CR, calorie restriction model (EBSS + 10% FBS, 100 mg/dL glucose containing medium); CRK, ketone added to CR medium (EBSS + 10% FBS + 5 mM bOHB); PF, prolonged fasting model (EBSS: DMEM0 1:1 + 10% FBS, 50 mg/dL glucose containing medium); PFK ketone added to PF medium (EBSS: DMEM0 1: 1 + 10% FBS + 5 mM bOHB); G0 (DMEM0 + 10% FBS, no-glucose containing medium); G0K, ketone added to G0 medium (DMEM0 + 10% FBS + 5 mM bOHB). * P < .01 when compared to N, # P < .01 when compared to the same group without bOHB.
Flow Cytometric Mitochondrial Functions
The SH-SY5Y human neuronal cell line is widely used in neurological research and in the study of mitochondrial function. In this study, SH-SY5Y cells were analyzed for mitochondrial measurements by staining with Mito Red, Mito Green, and MitoSOX. Flow-cytometric mitochondrial membrane potential, mitochondrial mass, and mitochondrial oxidant load were measured (Figures 4A, B, and C, respectively). During our experiments, it was observed that the 16 h incubation did not make a significant change in the mitochondria mass in all cell groups (Figure 4B), except for the decrease in cells in the G0 and G0K medium, which was expected as the media had no glucose. The mitochondrial membrane potential is the most important indicator of the electron transport chain and ATP formation, and therefore the mitochondrial energy function.17, 18 In the independent experiments, mitochondrial membrane potential was found to be increased especially in cells incubated with medium with ketone and reduced glucose (CRK and PFK; Figure 4C). In cells that were incubated with medium CR, ROS amounts were lowest, meanwhile, ROS was the highest in the cells incubated with PFK medium (Figure 4A). This finding was interesting because we were not expecting an increased ROS content in the cells with βOHB so we have repeated the experiments more than seven times to confirm this data. Data in Figure 4A were acquired from nine independent experiments. The decrease in ROS in the G0K group of cells was attributed to decreased viable cell number.
Figure 4. Flow-Cytometric Measurements, Cells Analyzed for Mitochondrial Functions.
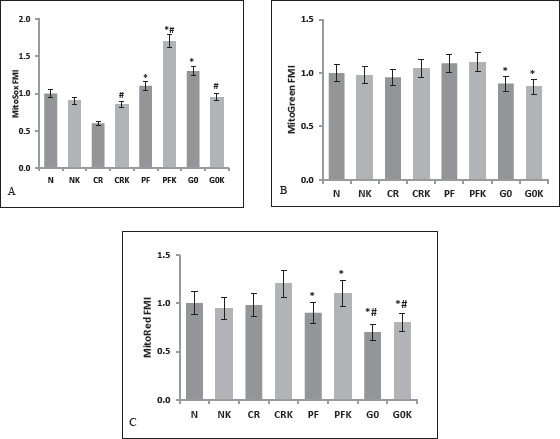
Note: Data are expressed as mean ± SD (n = 7). A, MitoSOX (mitochondrial oxidant load-ROS) (n = 9) B, Mito Green (mitochondrial mass) (n = 7) and C, MitoRed (mitochondrial membrane potential) were measured (n = 7). N, (normal) control group (DMEM F12 + 10% FBS-300 mg/dL glucose containing medium); NK, ketone added to normal medium (DMEM F12 + 10% FBS + 5 mM b OHB); CR, calorie restriction model (EBSS + 10% FBS, 100 mg/dL glucose containing medium); CRK, ketone added to CR medium (EBSS + 10% FBS + 5 mM bOHB); PF, prolonged fasting model (EBSS: DMEM0 1:1 + 10% FBS, 50 mg/dL glucose containing medium); PFK, ketone added to PF medium (EBSS: DMEM0 1: 1 + 10% FBS + 5 mM bOHB), G0 (DMEM0 + 10% FBS, no-glucose containing medium); G0K, ketone added to G0 medium (DMEM0 + 10% FBS + 5 mM bOHB). * P < .01 when compared to N, # P < 0,01 when compared to the same group without bOHB.
Microscopic pictures of cells were taken before and after the experiments. When examined microscopically, cells that were incubated with a normal medium appeared healthy and their axons were extended (Figures 5A and 5B), while 16 h later, there were noticeable drawbacks in the axons of the cells incubated with medium (CR, PF) with reduced glucose levels (Figure 5C and 5D). However, cells treated with 5 mM βOHB had longer axons (Figure 5E and 5F; CRK and PFK, respectively). Cells incubated with glucose-free medium (G0), pulled their axons back and were completely rounded (Figure 5G) but in the ketone-added medium (G0K) of the same cells, the shape of the cells was different with minimally extended axons (Figure 5H).
Figure 5. Microscopic Images of Cells Incubated with Different Media.
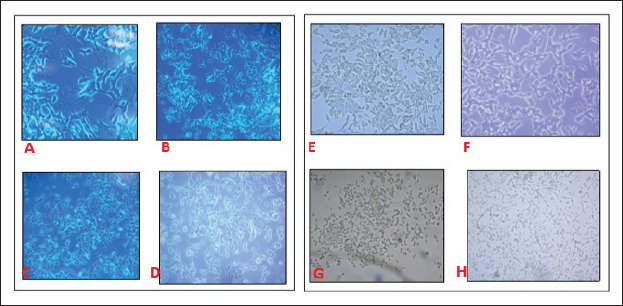
Note: Cells incubated with normal medium. A, 20X and B, 10X. Cells incubated with CR and PF medium, C and D respectively, there were withdrawls of the axons. Cells treated with βOHB had longer axons in CRK and PFK medium, E and F, respectively. G, Cells, incubated in G0 medium were completely rounded. H, only keton added medium (G0K) cell shapes were changed with minimally extended axons.
Discussion
In this study, our aim was to investigate the effects of CR and PF patterns on human neuron cells at the molecular level. We studied the potential of βOHB to substitute for glucose as an energy substrate during glucose depletion and to prevent the death of neurons as a result.
Although glucose is the main energy source, the brain can utilize other energy sources, such as ketone bodies βOHB. Under normal conditions, blood levels of ketone bodies are maintained below 0.5 mM. During prolonged fasting or a high-fat and low-carbohydrate diet, blood levels of ketone bodies are increased and used by peripheral tissues together with the brain.14, 19 Prolonged fasting is defined as fasting more than 12 h 3 and in these conditions, while glucose is provided from glycogen, free fatty acids are mobilized from the adipose tissue. The major signal here is the secretion of glucagon from the pancreas, and as a result, mobilized free fatty acids are directed to the liver. Fatty acids in the liver increase the production of acetyl-CoA in mitochondria, and while liver energy pathways are kept active, plenty of acetyl-CoA is produced and directed to ketone body synthesis. Ketone bodies, synthesized abundantly in the liver, are delivered to blood and used by peripheral tissues as a rapid energy source. With these pathways, while glucose concentrations are low, tissues can keep energy metabolism active. 7
Ketone bodies are in acid character, but do not affect blood pH levels at physiological doses (<6 mmol/L). 15 Especially in the case of diabetic ketoacidosis mostly seen in type I diabetes, the decrease in body pH and loss of consciousness is caused by the abruptly secreted huge volume of ketone bodies (>10 mmol/L). 15 This pathologic condition causes many scientists to overlook the beneficial effects of ketones. Because the main pathology in type I diabetes is insulin deficiency, glucose cannot enter the cells. Despite there being abundant glucose in the blood, cells send continuous low-energy signals to the liver, and glucagon stimulates the liver to produce both ketone bodies and glucose constantly. Hence ketone bodies, uncontrollably produced in large quantities, acidify the blood. However, because the insulin secretion in healthy individuals is normal, endogenously synthesized glucose enters the cell and ketone elevation in physiological doses does not harm the body. 20 Moreover, increasing the amount of ketone in healthy individuals is thought to be protective against aging-related dementia and other neurodegenerative diseases 21 , and the benefits of experimental ketone administration have been investigated in many neurodegenerative diseases. 22 Ketones have been shown to inhibit neuronal cell damage, in particular by inhibiting inflammatory promarkers, tumor necrosis factor-alpha (TNFα), interleukin-6 (IL-6), and interleukin-8 (IL-8).23, 24. Moreover, it has been recently shown KD expands metabolically protective γδ T cells that restrain inflammation. 25 Prolonged fasting model can increase the concentrations of ketones endogenously without CR and has been shown as an alternative to CR.
In this study, the CR model and PF models were simulated in human neuronal cell cultures. Following these conditions, cells were treated with βOHB and energy metabolites were measured in the media. Citrate synthase activity and flow-cytometric mitochondrial function assessments were analyzed in cell homogenates. In our optimization studies, it was observed that in the first 16 h, cell losses were not high (Figure 1), but there was significantly decreased cell viability after 16 h (P < .01), especially in the flasks containing glucose-free media (Figure 1). Because more than 12 h of fasting simulation was planned to be performed in our study, 16 h experimental incubations were applied. Consistent with us, there are recent rodent studies reporting that 16 h fasting is the most effective form of PF. 26
During the experiments, lactate formation was reduced in ketone-treated cells compared to other media (Table 1). These findings suggest that in the presence of ketones, neurons reduce anaerobic glycolysis and use mitochondrial pathways to produce energy. This may exert a positive effect in preserving membrane potential in epilepsy and degenerative neuron diseases. It has been shown that KD and βOHB stabilize hypoxia-induced factor (HIF), which is known to regulate the expression of genes involved in energy metabolism and antiapoptotic proteins such as Bcl-2. 27 Moreover, cells used glucose in glucose-containing media and also used ketones in ketone-added media simultaneously; their glucose utilization was decreased compared to other cells (Table 1). LDH activity measurements showed that cell damage was higher in cells incubated with reduced glucose media (CR and PF) compared to ketone-treated cells (CRK and PFK; Table 1). These results suggest that the presence of ketone in the environment decreases cell damage and ketones direct cell energy metabolism to mitochondria to ensure ATP production is kept constant when sufficient glucose is not available. Changes in glucose and lipid metabolism have been observed in rats given exogenous ketone (βOHB) with no further dietary changes, and these effects were linked to the increase of the metabolism of ketone bodies in the mitochondria.28, 29 Moreover, it has been observed that hypoglycemic animals, treated with glucose and D-beta-hydroxybutyrate (D-BHB) showed decreased cell death and ROS production in the cortex and hippocampus as compared to animals rescued with glucose alone. 3 Our findings agree with recent reports suggesting that βOHB can be used as an alternative energy substrate30, 31 even in the presence of glucose.
In this study, citrate synthase activity was measured in cells after 16 h of incubation with different concentrations of glucose and media containing 5 mM βOHB. Citrate synthase is known to be the first enzyme of the tricyclic acid cycle in the mitochondria matrix and measurements of citrate synthase activity in cell extracts are accepted as an index of mitochondrial activity. 17 Cells incubated with a normal medium reached the Vmax fastest (Figure 3). As the glucose in the media decreased, the enzyme worked slowly; however, enzyme velocity was found to be statistically increased in media containing ketone together with glucose. When ketone (5 mM βOHB) was added to a normal medium (300 g/dL glucose), there was no significant change in enzyme speed. This finding suggests that when excess glucose is found in the environment, mitochondrial enzyme activity does not change with the ketone presence. However, when we look at the ketone amounts in Table 1, we can see that the cells incubated with a normal medium also use ketone simultaneously. To our knowledge, our study shows the effects of ketone bodies on the relative Vmax of citrate synthase for the first time. Because it is observed that neurons treated with βOHB exhibited increased oxygen consumption and ATP production and an elevated NAD+/NADH ratio, 32 our results suggest cellular signaling mechanisms by which βOHB may mediate the adaptive responses of neurons to fasting or KD. KD has been shown to cause activation of type II nuclear transcription factor, peroxisome proliferator-activated receptors (PPAR), which regulate lipid and energy metabolism in neurons. 13
During the experiments, mitochondrial membrane potential, mitochondrial mass, and mitochondrial oxidant load were measured by flow cytometry. It was observed that the 16 h incubation did not make a statistically significant change in the mitochondrial mass in all cell groups (Figure 4B), It is shown that the number/mass of the mitochondria changed during incubation periods at least two to seven days. 17 We can conclude that 16 h of fasting or caloric restriction does not make a difference in the mitochondrial mass but the mitochondrial activity significantly changed with different nutrition models such as PF and CR (Figure 3 and Figure 4B) in our study. The mitochondrial membrane potential is the most important indicator of the electron transport chain and ATP formation, and thus the mitochondrial energy function.17, 18 In our experiments, mitochondrial membrane potential was found to be increased especially in cells incubated with glucose-reduced media containing ketone (CRK and PFK; Figure 4C). This suggests that ATP production is regulated in ketone environments, so that seizures can be prevented by maintaining membrane threshold values, especially in cases where uncontrolled cell discharges occur such as in epilepsy. Studies have shown that the rate of mitochondrial NADH and coenzyme Q oxidation and ATP production increase in rats treated with exogenous ketone. 33 Moreover, in addition to being a source of acetyl coenzyme A for neuronal energy metabolism, βOHB is suggested to influence certain cellular signaling pathways such as it can inhibit or activate protein deacetylases and can inhibit mitochondrial membrane permeability transition pore opening.34–36
Examination of mitochondrial reactive oxygen derivatives (ROS) content revealed different results than expected in the study. The mitochondrial oxidative load was the lowest in the CR group (100 mg/dL glucose and no ketone) and the highest in the PFK group (50 mg/dL glucose with 5 mM βOHB; Figure 4A). Mitochondria are a source of ROS and consequently, mitochondrial proliferation could lead to an increase in oxidative stress. However, because the mitochondrial masses did not change in our experiments (Figure 4B), differences here cannot be attributed to the number of mitochondria in the cells. In some studies, it is reported that pretreatment with ketone bodies caused ROS production and depleted glutathione (GSH) levels in primary rat hepatocytes. 37 This contradictory effect of ketones could be because of increased mitochondrial respiration in cells treated with βOHB in our study. Marosi et al. showed that βOHB metabolism increases mitochondrial respiration which drives changes in the expression of brain-derived neurotrophic factor (BDNF) in cultured cerebral cortical neurons. They concluded that increased mitochondrial ROS generation appears to mediate βOHB-induced BDNF expression resulting in neurogenesis and synaptic plasticity in hippocampus32, 38 Additionally, autophagy, a key survival mechanism, is regulated by several genes, such as AMPK, mTOR, and sirtuins, which are also known to regulate aging and stress resistance.39–42 Increased ROS in the presence of βOHB can activate adaptive compensatory mechanisms such as autophagy by generating mitochondrial and cellular stress. It has been thought that cellular fasting stress can activate autophagy and mitohormesis. 43 However, in contrary, Sullivan et al. reported that ketosis, induced by KD, significantly decreased ROS production in the hippocampus, presumably by increasing the expression of mitochondrial uncoupling protein activity. 44
Finally, electron microscope pictures of the cells showed us that cells incubated with a normal medium appear healthy and their axons were extended (Figures 5A and B), while 16 h later, there were noticeable drawbacks in the axons of the cells incubated with reduced glucose medium (CR and PF; Figures 5C and D). In the cells that were incubated under the same conditions plus added ketone (CRK and PFK) media, axon withdrawals were seen to be less (Figures 5E and F). These results suggest that as the glucose amount decreases, axon extensions of the neuronal cells decrease and thus their transmissions may slow down, but the extensions heal when ketone is found in the same environment. Even in the cells in group G0K (5 mM βOHB and no glucose), axons were longer than the G0 group cells suggesting that ketones can be alternative energy substrates to glucose.
Aside from the ketone bodies, in humans, it is shown that insulin-like growth factor (IGF-I) levels decrease in response to short-term starvation despite an increase in GH secretion. 45 This is thought to be the result of an increase in the IGF-I inhibitory protein IGFPB-1, 46 which results in decreased IGF-I bioavailability and prevents the feedback inhibition of GH secretion by IGF-I. 47 In primary neurons, IGF-I is shown to sensitize cells to oxidative stress by a Ras/Erk-dependent mechanism, 48 and experiments in rat primary glia suggest IGF-I sensitizes against oxidative damage and chemotherapeutic drugs. 49 In our previous study, we showed that there was a difference in the fasting and CR models, especially in the GH and IGF-I axis, and in this study, when the CR and PF models were compared at a molecular level in neurons, differences in cell viability and mitochondrial functions were shown. 6 Our results suggest that during prolonged fasting, βOHB contributes to beneficial effects of fasting on neuron protection and that these effects might be through the changes in the mitochondrial functions. These positive effects may make βOHB not only a preventive agent but also a putative therapeutic molecule in cases of neuronal damage such as epilepsy, Alzheimer’s and Parkinson’s diseases, stroke, and traumatic brain injury. In the light of our findings, we think, the presence of ketone in the environment increases the mitochondrial activity of neurons, and even in the glucose presence, cells try to protect their energy metabolism by using ketones efficiently. Revealing the translational changes in these models will guide future ketone therapies in aging and neurodegenerative diseases.
Acknowledgments
This work was supported by the Scientific and Technological Research Council of Turkey (TUBITAK) under grant #17S816.
Declaration of Conflicting Interests: The authors declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Funding: The authors disclosed receipt of the following financial support for the research, authorship, and/or publication of this article: This work was supported by the Scientific and Technological Research Council of Turkey (TUBITAK) under grant #17S816.
ORCID iD: Fehime Benli Aksungar https://orcid.org/0000-0002-7954-7177
https://orcid.org/0000-0002-7954-7177
Authors’ Contribution
MP, SB, AP carried out all the experiments. DOA and FBA planned and organized the study. All authors together analyzed the data and wrote the manuscript.
Statement of Ethics
No ethical statement is needed since this is a cell culture study. We did not any use human or animal subjects.
References
- 1.Lopez-Otin C, Blasco MA, Partridge L, et al. The hallmarks of aging. Cell 2013; 53: 1194–1217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Armanios M and Blackburn EH. The telomere syndromes. Nat Rev Genet 2012; 13(10): 693–704. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Adam L and Hartman MD. Neuroprotection in metabolism-based therapy. Epilepsy Res 2012; 100: 286–294. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Perlmutter D and Loberg K. Grain Brain . Hodder & Stoughton, 2013. [Google Scholar]
- 5.Anson RM, Guo Z, Cabo R, et al. Intermittent fasting dissociates beneficial effects of dietary restriction on glucose metabolism and neuronal resistance to injury from calorie intake. Proc Natl Acad Sci 2003; 100: 6216–6220. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Aksungar FB, Sarıkaya M, Coskun A, et al. Comparison of intermittent fasting and caloric restriction according to the GH/IGF-1 axis and insulin resistance in obese subjects: A two year follow-up. J Nutr Health Aging 2016; 21(6): 681–685. [DOI] [PubMed] [Google Scholar]
- 7.Amilpas AJ, Montiel T, Tinoco ES, et al. Protection of hypoglycemia-induced neuronal death by β-hydroxybutyrate involves the preservation of energy levels and decreased production of reactive oxygen species. J Cereb Blood Flow Metab 2015; 35(5): 851–860. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Lindsay BB, Ryan PN, Asker EJ.. Acute effects of dietary constituents on motor skill and cognitive performance in athletes. Nutr Rev 2016; 72(12): 790–802. [DOI] [PubMed] [Google Scholar]
- 9.Colman RJ, Anderson RM, Johnson SC, et al. Caloric restriction delays disease onset and mortality in rhesus monkeys. Science 2009; 10: 201–204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Wilder RM, Winter MD.. The treshold of ketogenesis. Biol Chem 1992; 52: 393–401. [Google Scholar]
- 11.Aksungar FB, Eren A, Ure S, et al. Effects of ıntermittent fasting on serum lipid levels, coagulation status, and plasma homocysteine levels. Ann Nutr Metab 2005; 49: 77–82. [DOI] [PubMed] [Google Scholar]
- 12.Aksungar FB, Topkaya AE, Akyıldız M.. Interleukin-6, c-reactive protein and biochemical parameters during prolonged intermittent fasting. Ann Nutr Metab 2007; 51: 88–95. [DOI] [PubMed] [Google Scholar]
- 13.Simeone TA, Stephanie A, Matthews MS, et al. Regulation of brain PPAR gamma2 contributes to ketogenic diet antiseizure efficacy. Exp Neurol 2016; 287 (Pt 1): 54–64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Noh HS, Hah YS, Nilufar R, et al. Acetoacetate protects neuronal cells from oxidative glutamate toxicity. J Neurosci Res 2006; 83(4): 702–709. [DOI] [PubMed] [Google Scholar]
- 15.Laffel L. Ketone bodies: A review of physiology, pathophysiology and application of monitoring to diabetes. Diabetes Metab Res Rev 1999; 15: 412–426. [DOI] [PubMed] [Google Scholar]
- 16.Chan FK, Moriwaki K, De Rosa MJ.. Detection of necrosis by release of lactate dehydrogenase (LDH) activity. Methods Mol Biol 2014; 979: 65–70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Hughes SD, Kanabus M, Anderson G, et al. The ketogenic diet component deconaic acid increases mitochondrial citrate synthase and complex I activity in neuronal cells. J. Neurochem 2014; 129: 426–433. [DOI] [PubMed] [Google Scholar]
- 18.Robinson JP, Li N, Narayanan PK.. High throughput-based mitochondrial function assays by multi-parametric flow cytometry. Curr Protoc Cytom 2015; 73: 9.48.1–9.48.9. [DOI] [PubMed] [Google Scholar]
- 19.Nordli DR, Koenigsberger D, Schroeder J, et al. Ketogenic diets. In: Resor SR Jr, ed. The Medical Treatment of Epilepsy . Marcel Dekker, 1992, 455–471. [Google Scholar]
- 20.Hartman AL, McNally MA.. Ketone bodies in epilepsy. J Neurochem 2012; 121: 28–35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Jeong JH, Yu KS, Bak DH, et al. Intermittent fasting is neuroprotective in focal cerebral ischemia by minimizing autophagic flux disturbance and inhibiting apoptosis. Exp Theraeup Med 2016; 12: 3021–3028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Lima PA, Sampaio LPB, Damasceno NRT.. Neurobiochemical mechanisms of a ketogenic diet in refractory epilepsy. Clinics 2014; 69(10): 699–705. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Ruskin D, Kawamura M, Masino SA.. Reduced pain and inflammation in juvenile and adult rats fed a ketogenic diet. PLoS One 2009; 4(12): e8349. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Paoli AG, Moro T, Bosco G, et al. Effects of n-3 polyunsaturated fatty acids (ω-3) supplementation on some cardiovascular risk factors with a ketogenic mediterranean diet. Mar Drugs 2015; 13(2): 996–1009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Goldberg EL, Shchukina I, Asher JL, et al. Ketogenesis activates metabolically protective γδ T cells in visceral adipose tissue. Nat Metab 2020, https://doi.org/10.1038/s42255-019-0160-6. [DOI] [PMC free article] [PubMed]
- 26.Baik S, Rajeev V, Fann DY, et al. Intermittent fasting increases adult hippocampal neurogenesis. Brain Behav 2019; 10(1002): brb3–1444. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Puchowicz MA, Zechel JL, Valerio J, et al. Neuroprotection in diet-induced ketotic rat brain after focal ischemia. J Cereb Blood Flow Metab 2008; 28: 1907–1916. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Maalouf M, Rho JM, Mattson MP.. The neuroprotective properties of calorie restriction, the ketogenic diet, and ketone bodies. Brain Res Rev 2009; 59: 293–315. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Shannon LK, Poff AN, Ward NP, et al. Effects of exogenous ketone supplementation on blood ketone, glucose, triglyceride, and lipoprotein levels in Sprague–Dawley rats. Nutr Metab 2016; 2016: 13–19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.LaManna JC, Salem N, Puchowicz M, et al. Ketones suppress brain glucose consumption. Adv Exp Med Biol 2009; 301: 306–332. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Zhang Y, Kuang Y, Xu K, et al. Ketosis proportionately spares glucose utilization in brain. J Cereb Blood Flow Metab 2013; 33: 1307–1311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Marosi K, Kim SW, Moeh K, et al. 3-Hydroxybutyrate regulates energy metabolism and induces BDNF expression in cerebral cortical neurons. J Neurochem 2016; 139(5): 769–781. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Maaluf M, Sullivan P, Davis L, et al. Ketones inhibit mitochondrial production of reactive oxygen species production following glutamate excitotoxicity by increasing NADH oxidation. Neurosience 2017; 145(1): 256–264. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Newman JC, Verdin E.. Ketone bodies as signaling metabolites. Trend Endocrin Metab 2014; 25: 42–52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Scheibye-Knudsen M, Mitchell SJ, Fang EF.. A high-fat diet and NAD(+) activate Sirt1 to rescue premature aging in cockayne syndrome. Cell Metab 2014; 20: 840–855. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Kim DY, Simeone KA, Simeone TA, et al. Ketone bodies mediate antiseizure effects through mitochondrial permeability transition. Ann Neurol 2015; 78: 77–87. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Abdelmegeed MA, Kim SK, Woodcroft KJ, et al. Acetoacetate activation of extracellular signal-regulated kinase 1/2 and p38 mitogen-activated protein kinase in primary cultured rat hepatocytes: Role of oxidative stress. J Pharmacol Exp Ther 2004; 10: 728–736. [DOI] [PubMed] [Google Scholar]
- 38.Phillips M. Fasting as a therapy in neurological disease. Nutrients 2019; 17: 11–10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Giralt A and Villarroya F.. SIRT3, a pivotal actor in mitochondrial functions, metabolism, cell death and aging. Biochem J 2012; 444: 1–10. [DOI] [PubMed] [Google Scholar]
- 40.Morselli E, Galluzzi L, Kepp O, et al. Autophagy mediates pharmacological lifespan extension by spermidine and resveratrol. Aging 2009; 1(12): 961–970. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Marino G and Kroemer G.. Ammonia: A diffusible factor released by proliferating cells that induces autophagy. Sci Signaling 2010; 3(124): pe19. [DOI] [PubMed] [Google Scholar]
- 42.Lee C and Longo VD. Fasting vs. dietary restriction in cellular protection and cancer treatment: from model organisms to patients. Oncogene 2011; 30(30): 3305–3316. [DOI] [PubMed] [Google Scholar]
- 43.Rubinsztein DC, Codogno P, Levine B.. Autophagy modulation as a potential therapeutic target for diverse diseases. Nat Rev Drug Discov 2012; 11(9): 709–730. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Sullivan PG, Rippy NA, Dorenbos K.. The ketogenic diet increases mitochondrial uncoupling protein levels and activity. Ann Neurol 2004; 55: 576–580. [DOI] [PubMed] [Google Scholar]
- 45.Moller N and Jorgensen OL. Effects of growth hormone on glucose, lipid, and protein metabolism in human subjects. Endocr Rev 2008; 30(2): 152–177. [DOI] [PubMed] [Google Scholar]
- 46.Zapf J. Physiological role of the insulin-like growth factor binding proteins. Eur J Endocrinol 1955; 132: 645–654. [DOI] [PubMed] [Google Scholar]
- 47.Muller W, Saegerl W, Wellhausen L, et al. Markers of function and proliferation in noninvasive and invasive bi- and plurihormonal adenomas of patients with acromegaly: An immunohistochemical study. Pathol Res Pract 1999; 195(9): 595–603. [DOI] [PubMed] [Google Scholar]
- 48.Li Y, Xu W, Mc Burney MW, et al. SirT1 inhibition reduces IGF-I/IRS-2/Ras/ERK1/2 signaling and protects neurons. Cell Metab 2008; m8(1): 38–48. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Lee C, Safdie FM, Raffaghello L, et al. Reduced levels of IGF-I mediate differential protection of normal and cancer cells in response to fasting and improve chemotherapeutic index. Cancer Res 2010; 70: 1564–1572. [DOI] [PMC free article] [PubMed] [Google Scholar]