

Abstract
One catalyst, two reaction set‐ups, three monomers and unlimited macromolecular microstructural designs: The iron guanidine complex [FeCl2(TMG5NMe2asme)] (1) polymerizes lactide faster than the industrially used Sn(Oct)2 and shows high activity towards glycolide and ϵ‐caprolactone. Its distinguished features enable the synthesis of both block and random‐like copolymers in the melt by a simple change of the polymerization set‐up. Sequential addition of monomers yields highly ordered block copolymers including the symmetrical PLA‐b‐PGA‐b‐PCL‐b‐PGA‐b‐PLA pentablock copolymers, while polymerizations of monomer mixtures feature enhanced transesterifications and pave the way to di‐ and terpolymers with highly dispersed repeating unit distributions. A robust catalyst active under industrially applicable conditions and producing copolymers with desired microstructures is a major step towards biocompatible polymers with tailor‐made properties as alternatives for traditional plastics on the way towards a sustainable, circular material flow.
Keywords: Block Copolymers, Caprolactone, Glycolide, Iron, Lactide, Random Copolymers
Opposite microstructures accessible by a single catalyst: An iron guanidine complex produces defined block copolymers of lactide, ϵ‐caprolactone and glycolide under solvent‐free conditions at 150 °C. Increasing the reaction temperature leads to enhanced transesterifications producing random‐like copolymers. ϵ‐Caprolactone was identified as crucial driver to obtain polymer chains with highly dispersed repeating unit distributions.

Introduction
After 100 years of macromolecular science, the overwhelming innovations plastics brought are clouded by the approximately 6.3 billion metric tons of plastic waste found contaminating our planet. [1] To address this problem, a variety of monomers derived from renewables or waste streams leading to advanced, biocompatible materials are currently under investigation. [2] In addition to monomers being independent of depleting mineral oil, end‐of‐life options and recycling strategies have to be developed. [3] A bioplastic which has already reached a certain maturity is polylactide (PLA). It is produced from renewable feedstocks and shows high potential as circular material due to its several end‐of‐life options: biodegradation as well as mechanical or chemical recycling. [4] Production capacities are predicted to rise in the coming years and PLA is expected to replace conventional petroleum‐based polymers in a number of applications. [5] To end the application of the currently used toxic heavy metal catalyst in industry, benign catalysts meeting the principles of sustainable chemistry have been developed for polymerization under industrial conditions. [6] However, the mechanical properties of PLA face some limitations like a high rigidity or unsuited degradation time, which are necessary to overcome in order to make the bioplastic widely applicable. [7] The properties can be manipulated in some measure by copolymerization with the l‐lactide stereoisomers d‐ or meso‐lactide. [8] To adjust the mechanical properties further, other comonomers are needed. For lactide (LL), ϵ‐caprolactone (in this publication: Cap) has proven to be a suitable counterpart. [9] Cap is traditionally derived from oil, but recent developments show that it can be obtained efficiently from biomass feedstock. [10] In comparison to PLA, polycaprolactone (PCL) is more flexible due to a lower glass transition temperature and shows prolonged degradation times. [11] Especially PLA‐b‐PCL‐b‐PLA copolymers, resulting in a soft block surrounded by hard ones, are interesting due to their properties as thermoplastic elastomers. [12] The set of comonomers is completed by glycolide (GG), which forms the hydrophilic polymer polyglycolide (PGA) leading e.g., to a faster degradation. [13] Combining the three monomers LL, Cap and GG in one copolymer allows the tuning of the polymer properties to a large extent. [14] The characteristics of a copolymer depend on the ratios of the monomers as well as the microstructure which can vary from blocky via gradient and statistical to random. [15] Desirable microstructures are either blocky or random, since the building block distribution of the other microstructures are not distinctly defined and thus the properties of a likewise called polymer can vary significantly. Random copolymers are defined by following Bernoullian statistics: the probability of finding a certain repeating unit at any given site is independent of the preceding unit. [15] LL, GG and Cap are all lactones but they show very different reactivities in the ring‐opening polymerization. [16] Many catalysts have been reported to copolymerize these monomers, however, the resulting microstructures differ enormously. The reactivity ratios in the copolymerization are for most catalysts in the order r GG>r LL≫r Cap and it was reported that Cap cannot be polymerized after LL. [17] But the opposite was reported as well with catalysts preferably consuming Cap. [18] The relative reactivity towards the monomers is important for the formation of block copolymers which are typically produced by sequential addition. [19] To synthesize random copolymers, the reactivity ratios should best be equal which remains a challenge for most catalysts. Aluminum complexes with elaborated, bulky ligands are performing best in this discipline.[ 17c , 20 ] Another and much less extensive process to obtain random copolymers is by means of transesterification. The occurrence of transesterifications typically depends on the reaction temperature and normally they are avoided to preserve a controlled polymerization.[ 18a , 21 ] However, by increasing the reaction temperature, some highly active catalysts are capable of reaching a uniform distribution of the building units by transesterification. [22] Other catalysts promote transesterifications only to a lesser extent and rather produce statistical copolymers with a blocky character. [23] There are few examples of catalysts which produce copolymers of Cap and LL and/or GG with different microstructures by the change of the reaction conditions. Especially, defined block copolymers produced under melt conditions as well as highly randomized copolymers synthesized by a single catalyst remain a challenge.[ 17k , 23a , 24 ] The industrially employed tin octoate (Sn(Oct)2) was intensively studied regarding its capability to form copolymers but could not tackle those two challenges. Block copolymers of PCL and PLA were approached by sequential addition, however, a sharp transition between the blocks in the resulting copolymers could only be attained if the PCL prepolymer was purified before the addition of the second monomer.[ 23b , 24b ] The synthesis of random copolymers was attempted by transesterification. It was reported that, when using Sn(Oct)2 as catalyst, the temperature influences the occurrence of transesterifications and the average sequence length, but to a lesser extent than for other catalysts. [25] Therefore, the synthesis of random copolymers of LL and Cap with Sn(Oct)2 was only successful for very limited monomer‐to‐monomer ratios. [24b] Otherwise, only low degrees of randomness and the formation of micro blocks was observed.[ 23b , 26 ] Substituting Sn(Oct)2 having toxic features with a biocompatible catalyst highly active under solvent‐free conditions which overcomes its limitations in the synthesis of defined block copolymers or random copolymers, would lift the industrial process of lactone copolymer synthesis to a whole new level. Recently we have presented an iron guanidine complex as the first robust, biocompatible catalyst which supersedes the polymerization activity for lactide of Sn(Oct)2 significantly under industrially relevant melt conditions. [27] The catalyst marks a major milestone on the way towards the replacement of the toxic tin compound in the industrial production of PLA. In addition, iron guanidine catalysts conquer new areas of polymer chemistry e.g., by simultaneously copolymerizing lactide and styrene via ROP and ATRP in a one‐pot synthesis. [28]
In this report we are tackling two challenges with one iron guanidine catalyst: 1) The synthesis of highly defined block copolymers of LL, Cap and GG in a solvent‐free process and 2) the synthesis aiming for random copolymers of LL, Cap and GG despite their very different polymerization rates. At first, preliminary experiments are described which were necessary to gain basic knowledge on the catalysts behavior and find appropriate reaction conditions. Afterwards, findings regarding the synthesis and characterization of block copolymers are presented. Then, the process aiming for random copolymers is discussed and finally, mechanistic aspects are investigated and evaluated regarding the capability of this catalyst to produce such different microstructures with only small adjustments in the reaction conditions.
Results and Discussion
The iron guanidine complex [FeCl2(TMG5NMe2asme)] (1) was synthesized according to literature. [27] Its polymerization properties for lactide in bulk at 150 °C was already investigated, a k p of (0.554±0.024) L mol−1 s−1 was determined and it was shown that the catalyst follows the coordination–insertion mechanism. [27] For this study, the k p of the homopolymerization of Cap with 1 as catalyst under solvent‐free conditions at 150 °C was determined to be (5.97±1.55)×10−3 L mol−1 s−1 and thus two orders of magnitude lower in comparison with the k p for LL (see Figure S2). [27] Since the polymerization preferences of a catalyst can change if both monomers are present in a reaction mixture, the reactivity ratios for the copolymerization of LL and Cap were estimated via the Mayo–Lewis plot (see Figure S3). [29] Herefore, experiments were performed by polymerization in Schlenk tubes tempered by an oil bath. At a total monomer‐to‐initiator (M/I) ratio of 1000 : 1, LL‐to‐Cap ratios between 1 : 1 and 1 : 9 were employed, the polymerizations were stopped at low conversions and the monomer conversions were determined by 1H NMR spectroscopy. The resulting values were applied to the Mayo–Lewis equation resulting in straight lines intercepting in one point when plotting r LL versus r Cap. The intercept gives the reactivity ratios for the two monomers. In this case r LL≫r Cap is found reflecting the difference in reactivity found for the homopolymerizations of each monomer. 1 is therefore a catalyst having a strong preference for the incorporation of LL in comparison to Cap. Two polymerization set‐ups were afterwards tested and the microstructure of the corresponding copolymers was investigated. When LL and Cap were polymerized as a monomer mixture with a LL‐to‐Cap‐to‐co‐initiator‐to‐catalyst (LL+Cap:CoI:Cat) ratio of 500+500 : 1 : 1 at 150 °C with benzyl alcohol (BnOH) as co‐initiator, LL was converted within 10 min indicated by a fast increase in viscosity of the reaction mixture. The conversion of Cap did not exceed 45 % even after prolonged reaction times of up to 24 h (see Table S3, run 3). The resulting microstructure determined by 13C{1H} NMR spectroscopy was not blocky as one would expect, but statistical with only very short sequences of Cap (see Figure S19), which indicates the occurrence of transesterifications. The role of transesterification was investigated further and is reported in the second part of this publication (see below). Secondly, a sequential addition approach was tested where Cap was polymerized first and LL was added afterwards. Polymerization in melt is typically limited by diffusion and especially for block copolymerization it has to be assured that the newly added monomer can reach the active chain end without diffusion limitation. However, it was observed that the highly viscous PCL melt becomes liquid again because it dissolves in the added LL and therefore the mixing of active chains with the added monomer is ensured. With a monomer‐to‐coinitiator‐to‐catalyst (M/CoI/Cat) ratio of 500+500 : 1 : 1 (LL+Cap:BnOH:1) adding LL after 4 h of Cap polymerization, high conversions of both monomers were reached (see Table S3, run 1). The carbonyl region of the 13C{1H} NMR spectrum only displays peaks that are typical of a PLA‐PCL blocky microstructure, namely, long sequences of each of the repeat units (i.e., CapCapCap and LLLLLL), but no peaks of isolated repeat units in between the repeat units of the opposite monomer that would signify transesterification (see Figure 1A). However, the signals in the 13C{1H} NMR spectrum do not give information on whether two homopolymers or a block copolymer are formed. 1 is known to act as a single site catalyst [27] therefore, the formation of two unconnected homopolymers or species with different compositions produced by ambiguous initiation cannot be excluded. Immortal conditions were tested to inhibit the initiation by 1. To handle the low amounts of catalyst necessary for polymerizing under immortal conditions, a scale up to a steel reactor was performed which allows at the same time the monitoring of the polymerization by in situ Raman spectroscopy. We found that the catalyst is active even in the presence of 500 equiv. of BnOH as co‐initiator with 10 000 equiv. of LL supporting once again the high stability of this complex towards protic compounds. MALDI‐ToF‐MS end group analysis revealed that with a high excess of co‐initiator, an unambiguous initiation can be achieved with BnOH as the only initiating group (see Figure S33). The molar masses of the polymers produced under immortal conditions fit the theoretical ones very well and show dispersities below 1.10 (see Table S2). A M/CoI/Cat ratio of in total 10 000 : 200 : 1 is suited for the synthesis of block copolymers by sequential addition at 150 °C under solvent‐free conditions.
Figure 1.
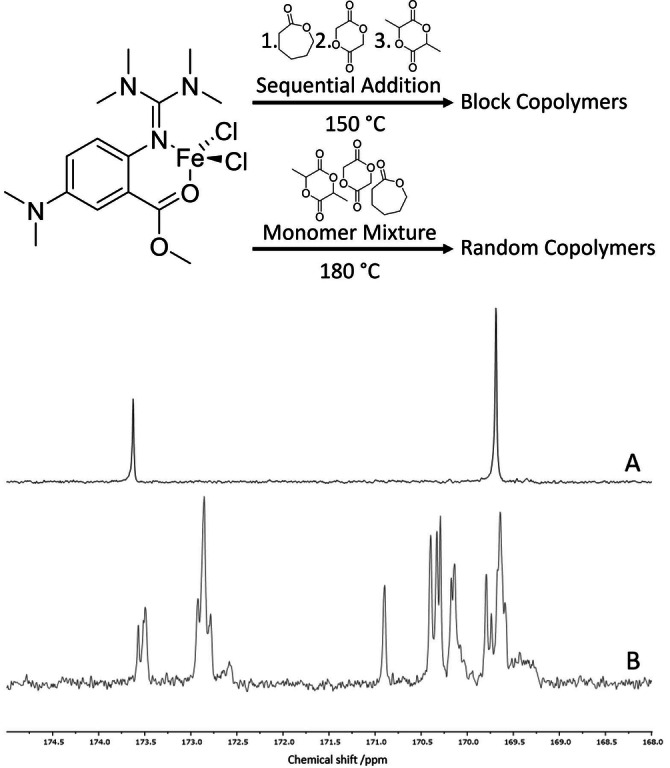
Top: Synthesis scheme of block and random copolymers. Bottom: Carbonyl region of the 13C{1H} NMR spectrum of A) PCL‐b‐PLA synthesized by sequential addition, M/CoI/Cat ratio 500+500 : 1 : 1 at 150 °C (Table S3, run 1) and B) PLA‐ran‐PCL synthesized from the monomer mixture, M/CoI/Cat ratio 500+500 : 1 : 1 at 180 °C (Table 2, run 1, CoI=co‐initiator).
Under the optimized reaction conditions block copolymers of LL and Cap were synthesized. In addition, GG was introduced as third monomer to expand the scope of the copolymers. Block copolymers were prepared with BnOH as monofunctional or 1,4‐benzenedimethanol (BDM) as bifunctional co‐initiator resulting in AB(C) or (C)BAB(C) block copolymers. Table 1 gives an overview of the monomer combinations employed. To decrease the total reaction time, polymerization of Cap was performed at 170 °C, then the polymerization temperature was reduced to 150 °C to minimize transesterifications. An exemplary spectrum of a copolymerization monitored by in situ Raman spectroscopy and the referring semilogarithmic plot of the integrated monomer bands are displayed in Figure 2 showing that a distinct peak appears for each monomer with the addition to the reaction mixture. The conversion of LL appears to proceed the slowest, however, it has to be considered that Cap is polymerized at a higher temperature and a deactivation of the catalyst could occur during the polymerizations of Cap and GG. It is remarkable that even after having polymerized two batches of monomers before, the polymerization of LL still follows first order kinetics and reaches high conversions supporting a truly living polymerization. Different monomer combinations and ratios were applied and for all runs high conversions of above 80 % were recorded. Since the polymerizations were performed under solvent‐free conditions, full conversion is hardly achievable due to diffusion limitation.
Table 1.
Polymerizations by sequential monomer addition leading to block copolymers.
|
|
Monomers |
M/CoI/Cat ratio (Cap(+GG)+LL:CoI:1) |
t |
p LL [e] |
p Cap [e] |
p GG [e] |
Feed ratio |
Incorp. ratio[f] |
M n,theo [g] [kg mol−1] |
M n [h] [kg mol−1] |
Đ [h] |
|---|---|---|---|---|---|---|---|---|---|---|---|
|
1 |
Cap+LL |
5000+5000 : 200 : 1[a] |
105 min[c]+5 h[d] |
0.82 |
0.92 |
– |
1 : 1 |
0.99 : 1.00 |
5.5 |
4.8 |
1.19 |
|
2 |
Cap+LL |
3333+6666 : 200 : 1[b] |
105 min[c]+10 h[d] |
0.92 |
0.96 |
– |
2 : 1 |
1.96 : 1.00 |
6.4 |
6.1 |
1.14 |
|
3 |
Cap+GG+LL |
4500+1000+4500 : 200 : 1[a] |
105 min[c]+1 h[d]+10 h[d] |
0.83 |
0.94 |
>0.99 |
1 : 1 : 0.22 |
1.08 : 1.00 : 0.28 |
5.7 |
5.1 |
1.23 |
|
4 |
Cap+GG+LL |
4500+1000+4500 : 200 : 1[b] |
105 min[c]+1 h[d]+10 h[d] |
0.94 |
0.99 |
>0.99 |
1 : 1 : 0.22 |
1.02 : 1.00 : 0.23 |
6.2 |
6.2 |
1.23 |
|
5 |
Cap+GG |
9000+1000 : 200 : 1[b] |
165 min[c]+1 h[d] |
– |
0.98 |
>0.99 |
1 : 0.11 |
1.00 : 0.09 |
5.6 |
6.1 |
1.33 |
[a] Benzyl alcohol as co‐initiator. [b] Benzenedimethanol as co‐initiator. [c] 170 °C. [d] 150 °C. [e] Conversion of the individual monomers determined by 1H NMR spectroscopy of the crude polymer. [f] Incorporated ratio of the monomers determined by 1H NMR spectroscopy of the precipitated polymer. [g] Calculated by the sum for all monomers of molar mass x conversion x monomer ratio of each monomer, respectively. [h] Determined by GPC in THF.
Figure 2.
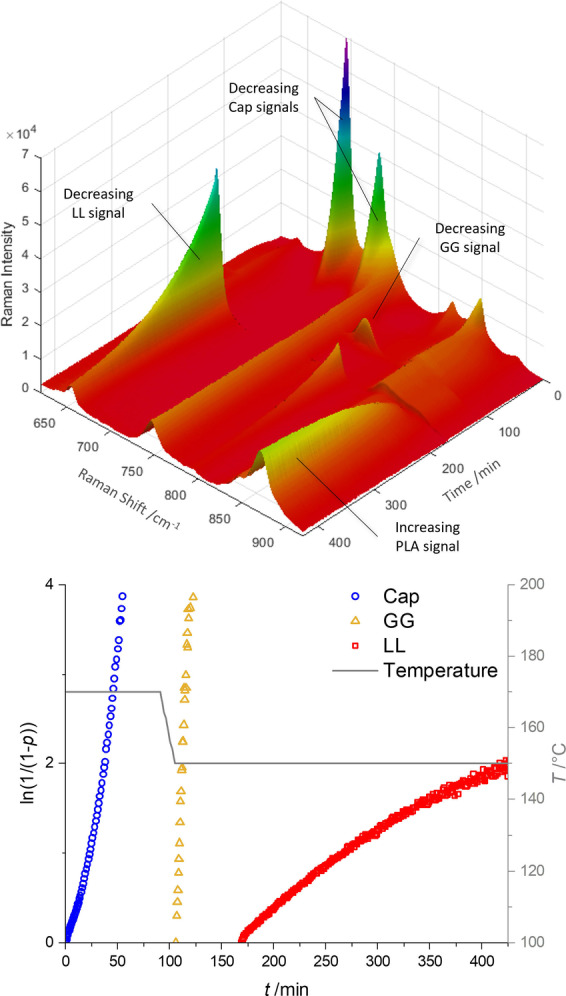
Top: Synthesis of PLA‐b‐PGA‐b‐PCL‐b‐PGA‐b‐PLA by sequential monomer addition to BDM monitored by in situ Raman spectroscopy. Bottom: Semilogarithmic plot in accordance with a polymerization of pseudo first order. Time‐resolved conversions determined by integration of decreasing monomer signals. The polymerization was continued after recording ended (Table 1, run 4).
The monomer ratio in the copolymers was determined by 1H NMR spectroscopy of the isolated product and matches the feed ratio accurately for all tested combinations. The variation of the block lengths of LL and Cap works without drawbacks. The share of GG was limited to 10 % since higher ratios would lead to the insolubility of the copolymer in common organic solvents. However, from a catalytic point of view there is no indication that higher ratios of GG would be problematic. To the best of our knowledge these are the first examples of triblock copolymers of LL, GG and Cap with homopolymeric blocks. In literature, rarely any examples of terpolymers of LL, GG and Cap with separated blocks can be found, while block copolymers of two of these monomers are vastly present. When all three monomers were employed and a blocky microstructure was obtained, one block consists of a statistical copolymer of two monomers while the other block is formed by the third monomer. [30] The presented copolymers therefore fill a gap in the already quite mature field of lactone polymerization.
The carbonyl regions in the 13C{1H} NMR spectra of the produced copolymer indicate the formation of long blocks of each repeat unit (see Figure 3 and Figures S6–S16). Additionally to the homotriads, small signals of the transition between the blocks are visible. These triads do not signal transesterification because for, e.g., the PLA‐b‐PCL‐b‐PLA (Figure 3A) the CapCapLL and CapLLLL triads matching the transition between the blocks are exclusively found. If a PGA block is located between PCL and PLA (Figure 3B) the CapLLLL signal disappears as expected since LL is no longer connected to Cap. Since the spectra do not only show the formation of long blocks but also the connection between them, they strongly indicate the formation of copolymers. It is further supported by diffusion ordered NMR spectroscopy (DOSY) spectra that reveal a single diffusion coefficient for all produced polymers (see Figure S7–S17). If two homopolymers would have formed, two different diffusion coefficients should have been observed. [31] The molar masses were determined by gel permeation chromatography (GPC). All copolymers show a monomodal molar mass distribution independently of the monomer‐to‐monomer ratio employed. The average molar masses match the theoretical values very well and the dispersities are with a maximum of 1.33 low. The values reveal the high degree of control maintained during the polymerization. 1 is therefore not only capable of homopolymerizing lactide under immortal conditions to highly defined PLA (see Table S2), but shows the same accuracy when several monomers are added throughout the polymerization. The thermal properties of the polymers were investigated with differential scanning calorimetry (DSC). Depending on the copolymer composition different melting events are found. For PCL‐b‐PLA (Table 1, run 1), consisting of equal shares of the monomers, two melting events referring to the microphase separated blocks are found slightly below the melting points of the homopolymers, respectively.[ 5b , 32 ] For the other di‐ or tercopolymers only one melting event is recorded in the region of the homopolymer of the major repeating unit. Further details can be found in Table S4 and Figure S34.
Figure 3.
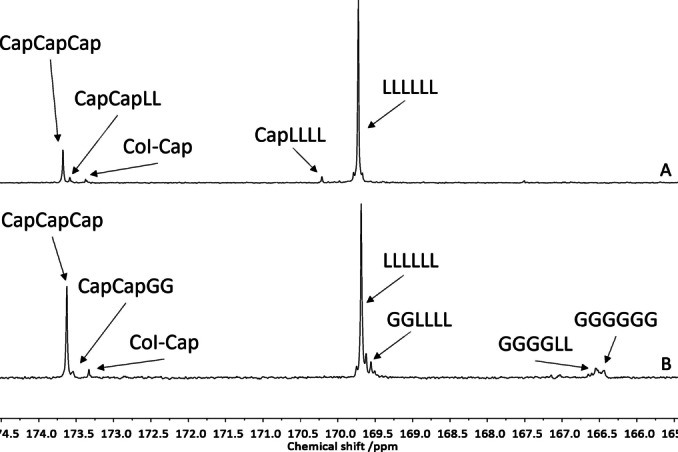
Carbonyl region of the 13C{1H} NMR spectra of A) PLA‐b‐PCL‐b‐PLA (Table 1, run 2) and B) PLA‐b‐PGA‐b‐PCL‐b‐PGA‐b‐PLA (Table 1, run 4).
In this first part, we were able to show that the synthesis of block copolymers containing LL, Cap and GG is possible in melt by sequential addition facilitated by a highly active catalyst performing a living ROP. Following, the synthesis procedure aiming for random copolymers is presented.
Monomers with different copolymerization reactivity ratios like LL, GG and Cap are a challenge when random copolymers are desired. However, if transesterifications occur, random copolymers are accessible due to scrambling of the formed polymer chain. The challenge is to facilitate such a high degree of transesterifications, that a uniformly random copolymer is formed. Here, a catalyst fulfilling special criteria is necessary: It has to be stable at high temperatures since transesterifications are enhanced by an increased polymerization temperature. Furthermore, the interplay between the polymerization preference and the tendency for transesterification has to be balanced.
1 is known to be especially active and stable in melt polymerizations at high temperatures. [27] Therefore, the polymerization temperature was risen to 180 °C in order to maximize its activity. Polymerizations were conducted for 24 h in Schlenk tubes tempered by an oil bath. In a first experiment LL and Cap were copolymerized in a LL+Cap:BnOH:1 ratio of 500+500 : 1 : 1. High conversions were found for both monomers and the 13C{1H} NMR spectrum of the isolated copolymer (see Figure 1B) revealed an approximately even distribution between the triads indicating the formation of a highly distributed copolymer. Further monomer ratios and combinations were tested with a total M/CoI/Cat ratio of 1000 : 1 : 1 with BnOH as co‐initiator. The results are summarized in Table 2. High conversions are found for all monomers in the copolymerization experiment, nevertheless, lowest values are around 70 % when Cap or LL is the minor monomer, while GG reaches full conversion throughout the experiments. The incorporation ratio of the monomers in the copolymer reflects the feed ratio well which shows that a target‐oriented copolymer design is possible. Figure 4 exhibits the carbonyl region of the 13C{1H} NMR spectra of the isolated copolymers of LL, Cap and GG. The different ratio of GG incorporated is visible by growing signal in the GG area with higher GG contents. When analyzing the produced copolymers with GPC, it was found for all copolymers that the average chain lengths are much lower than the theoretical ones and show quite high dispersities of up to Đ=1.72 indicating a deviation from controlled molar mass growth and the occurrence of transesterifications. The copolymers were analyzed with DSC to investigate their thermal behavior (see Table S4 and Figure S35). However, most of the polymers are completely amorphous neither exhibiting melting nor crystallization events. Only the copolymer containing a dominating content of PCL shows a signal referring to its crystalline domain at the expected temperature. The absence of melting events for most copolymers support that the copolymers consist of short sequences which cannot form crystalline domains.
Table 2.
Polymerizations of monomer mixtures aiming for random copolymers.
|
|
Monomers |
M/CoI/Cat ratio[a] (LL+Cap(+GG): CoI:1) |
p LL [b] |
p Cap [b] |
p GG [b] |
Feed |
Incorp.[c] |
M n,theo [d] [kg mol−1] |
M n [e] [kg mol−1] |
Đ [e] |
Seq. length (LL/Cap/ GG)[f] |
R (ref. LL)[g] |
T II (CapLCap/ CapGCap)[f] |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
|
1 |
LL+Cap |
500+500 : 1 : 1 |
0.90 |
0.85 |
– |
1 : 1 |
1.00 : 0.94 |
113.4 |
24.3 |
1.57 |
1.2/1.1/– |
1.30 |
0.80/– |
|
2 |
LL+Cap |
750+250 : 1 : 1 |
0.94 |
0.68 |
– |
3 : 1 |
3.90 : 1.00 |
121.0 |
42.8 |
1.35 |
3.7/1.2/– |
1.19 |
0.64/– |
|
3 |
LL+Cap |
250+750 : 1 : 1 |
0.68 |
0.89 |
– |
1 : 3 |
1.00 : 3.12 |
100.7 |
27.6 |
1.61 |
0.8/1.5/– |
1.03 |
0.90/– |
|
4 |
LL+Cap+GG |
475+475+50 : 1 : 1 |
0.91 |
0.81 |
>0.99 |
1 : 1 : 0.10 |
1.07 : 1.00 : 0.12 |
112.0 |
36.8 |
1.37 |
1.4/1.1/0.9 |
– |
0.47/1.85 |
|
5 |
LL+Cap+GG |
450+450+100 : 1 : 1 |
0.92 |
0.75 |
>0.99 |
1 : 1 : 0.22 |
1.37 : 1.00 : 0.33 |
109.8 |
39.7 |
1.27 |
1.7/1.1/1.3 |
– |
0.47/1.67 |
|
6 |
LL+Cap+GG |
425+425+150 : 1 : 1 |
0.93 |
0.75 |
>0.99 |
1 : 1 : 0.35 |
1.20 : 1.00 : 0.46 |
110.0 |
32.6 |
1.36 |
1.5/1.2/1.1 |
– |
0.47/2.12 |
|
7 |
LL+GG |
900+100 : 1 : 1 |
0.90 |
– |
>0.99 |
1 : 0.11 |
1.00 : 0.12 |
128.4 |
42.2 |
1.32 |
3.0/–/1.5 |
– |
– |
|
8 |
Cap+GG |
900+100 : 1 : 1 |
– |
0.93 |
>0.99 |
1 : 0.11 |
1.00 : 0.09 |
107.1 |
40.4 |
1.72 |
–/2.2/0.5 |
– |
– |
[a] BnOH as co‐initiator. [b] Conversions of the individual monomers determined by 1H NMR spectroscopy of the crude polymer after 24 h of polymerization at 180 °C. [c] Incorporated ratio of the monomers determined by 1H NMR spectroscopy of the precipitated polymer. [d] Calculated by the sum for all monomers of molar mass x conversion x monomer ratio of each monomer, respectively. [e] Determined by GPC in THF. [f] Calculated in accordance with ref. [21c]. [g] Randomness factor of LL calculated in accordance with ref. [33].
Figure 4.
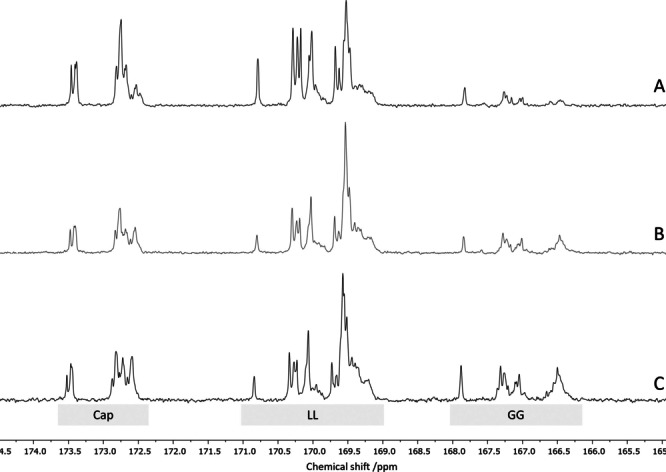
13C{1H} NMR carbonyl region of PCL‐ran‐PGA‐ran‐PLA with A) 5 % GG (Table 2, run 4), B) 12 % GG (Table 2, run 5), and C) 17 % GG (Table 2, run 6).
The microstructure of the copolymers was analyzed on the basis of the carbonyl region of their 13C{1H} NMR spectra (see Figure 1B, Figure 4 and Figure S21–S32). The peaks were assigned to their building blocks according to literature precedents.[ 20b , 21c ] In all spectra a variety of signals is found: The homotriads referring to LLLLLL, GGGGGG (if applicable) and CapCapCap as well as signals indicating the distribution of a repeating unit between the others. Furthermore, the two signals at 167.8 and 170.8 ppm which correspond to the CapGCap and the CapLCap triads, respectively, have to be highlighted. Both building blocks cannot form by traditional addition of a monomer but are the result of a so‐called second mode transesterification which splits a monomer consisting of two repeating units like LL or GG and leaves a single repeating unit surrounded by two others. [21c] The peaks for CapGCap and CapLCap are shifted from the others and therefore well visible in comparison with other triads formed by second mode transesterification like GGLGG and LLGLL. The occurrence of those signals proves that the microstructure of the copolymers is the result of transesterifications. The polymerization experiments were repeated under immortal conditions with an M/CoI/Cat ratio of in total 1000 : 5 : 1 and the produced copolymers do not differ significantly from the ones presented before and therefore confirm that the transesterification processes are not relying on a high concentration of the catalyst (see Table S3, run 4–6).
The reaction conditions were hence successfully changed from obtaining block towards statistical copolymers. The question remains to which degree the polymer was rearranged by transesterification and if a Bernoullian distribution of the repeating units was reached differentiating an undefined statistical copolymer from a random one. Since this differentiation is seldomly done in literature disregarding an evaluation based on the visual distribution of the signals in the 13C{1H} NMR carbonyl region, no holistic, sophisticated parameter is defined for tricopolymers of LL, Cap and GG. Herefore, an evaluation on the basis of different parameters is attempted. The average sequence lengths is defined for all copolymers and therefore accordingly calculated. [21c] The second mode transesterification yield is defined for copolymers of Cap in combination with LL or GG.[ 21c , 33 ] The randomness factor R is reported for dicopolymers of LL and Cap. [33] All calculated parameters are based on the integration of certain peaks of the 13C{1H} NMR carbonyl region and are listed in Table 2.
The copolymers exhibit very short average sequence lengths of around 1 for all three repeating units. The average sequence length increases for LL and Cap if they are present in excess and decrease if they are the minor component. Regardless, LL shows as a trend slightly higher sequence lengths in comparison with Cap under analogue conditions. The sequence lengths of LL and GG can be below 1 with a minimum of 0.5, if all repeating units are divided by second mode transesterification. This is the case for PCL‐co‐PGA (Table 2, run 8). The short sequences show the high degree of distribution the repeating units have in the copolymer. The randomness factor R is the quotient of the sequence length of a perfectly random copolymer with a Bernoullian distribution and the experimentally found average sequence lengths in the copolymer. [33] The LL sequence length is chosen as referring parameter. If R≥1, a truly random copolymer is at hand. And indeed, with R>1 for the dicopolymers of LL and Cap (Table 2, run 9–11), they fulfill the criteria for a random copolymer. The second mode transesterification yield T II describes to which degree the single unit (e.g. CapLCap or CapGCap) is found in the polymer in comparison with a perfectly random copolymer having a Bernoullian distribution (T II=1).[ 21c , 33 ] The parameter allows a comparison of the degree of distribution for different monomers. T II(CapLCap) shows values between 0.47 and 0.90 and is therefore less dispersed than necessary for a random copolymer. T II(CapGCap) on the other hand lies significantly above 1 for all copolymers indicating a higher dispersion than necessary for a random copolymer. These values reveal that the transesterifications do not proceed evenly in the copolymer and that LL is less dispersed in the polymer chain in comparison with GG. The short sequence lengths and randomness factors close to 1 would classify the copolymers as random, the T II values especially for the tercopolymers on the other hand show deviations from the Bernoullian statistics for LL. This contradiction points out the need for a holistic parameter to classify random copolymers. Nevertheless, it can be concluded that the repeating units are highly dispersed along the copolymer chain in a random‐like manner with some curtailments for LL.
The second part of this report demonstrates that it is possible to obtain two completely different microstructures with one catalyst. At a high enough temperature, full conversions can be reached when polymerizing a monomer mixture independently of monomer combination. Due to transesterifications the repeating units are highly distributed along the polymer chain within short sequences. However, not all kinds of repeating units are evenly highly dispersed along the polymer chain, which raises the question what influences the process of transesterification. This will be evaluated subsequently.
To gain more information on the incorporation of the monomers, a scaled‐up polymerization experiment aiming for random copolymers was performed with a M/CoI/Cat ratio of 450+450+100 : 1 : 1 (LL+Cap+GG:BnOH:1) in a steel reactor at 180 °C. Aliquots were removed from the reaction mixture after defined reaction times and the conversion of the three monomers was determined by 1H NMR spectroscopy. In the PCL region of the 1H NMR spectrum, the signals for Cap next to Cap and Cap next to either LL or GG (CapX) are clearly separated and therefore, it is possible to monitor whether the Cap active chain end attacks the already existing polymer chain and Cap is found next to GG or LL or if the Cap active chain end goes for further available Cap monomer. Whether Cap is located next to GG or LL cannot be withdrawn from the 1H NMR spectrum, therefore the experiment was repeated only with LL and Cap. The development of the conversion with time for both experiments is displayed in Figure 5. The graph of the terpolymer shows that GG is the first monomer to be polymerized. After only 10 min full conversion is reached. Within the first hour the conversion of LL rises above 90 %. During this time very small amounts of Cap are converted, significant conversion is only found after reaction times of more than one hour. Independently of the total conversion of Cap, the share of CapX represents approximately 70 %. It is peculiar that a clear majority of the incorporated Cap is found next to G or L while these monomers are already incorporated in the polymer. This experimental finding implies a revision of the mechanism of the synthesis of random copolymers by transesterification: The reorganization of the polymer chains does not take place between two polymer strands exchanging chains, instead Cap as monomer induces transesterification as active chain end reorganizing the polymer chains nearly every time it is incorporated. The Cap active chain end has a strong preference for the building blocks in the already existing polymer chain in comparison with the available Cap monomer. Due to the slow coordination and/or insertion of Cap with a Cap active chain end, the faster route of coordination and transesterification of the existing polymer chain is mainly preferred resulting in a scrambling of the polymer chain redistributing the repeating units. The proposed kinetic correlations are shown in Scheme 1.
Figure 5.
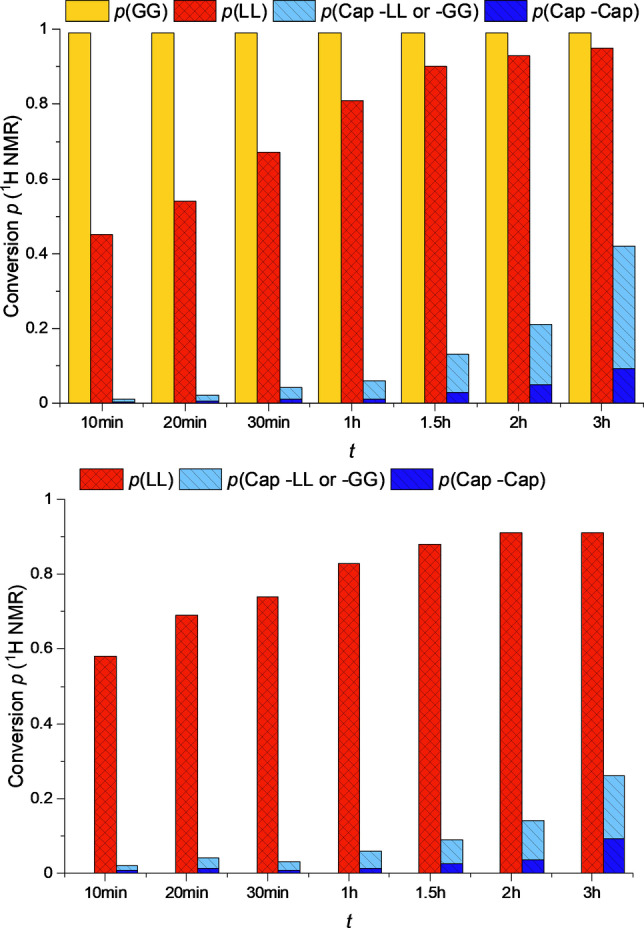
Top: Time dependent conversions of the polymerization of LL+Cap+GG:BnOH:1 450+450+100 : 1 : 1. Bottom: Time dependent conversions of the polymerization of LL+Cap:BnOH:1 500+500+100 : 1 : 1.
Scheme 1.

Proposed kinetic correlations for the synthesis aiming for random copolymers via transesterifications. At the described stage LL and GG monomers have been consumed completely due to their high polymerization rate and the only monomer remaining in the polymerization mixture is Cap.
When only LL and Cap are copolymerized the same ratio of CapX and CapCap is observed, however, the total conversion of Cap is lower (see Figure 5 bottom). Even though the total M/CoI/Cat ratio is maintained and GG constitutes only 10 % of the monomer mixture, the conversion is nearly twice as high in its presence. Since the T II values in Table 2 showed that the transesterifications preferably take place at GG in comparison with LL, the transesterification with GG might be kinetically favored leading to the observed faster conversion of Cap.
With the interactions summarized in Scheme 1 the formation of block copolymers becomes possible by polymerizing Cap first as described in the first part of this report. Once the PCL block is formed the caproyl units are fixed in the polymer chain and do not lead to transesterification anymore. The insertion of GG and LL monomer added after full conversion of Cap on the other hand occurs so fast, that the existing polymer chain is of no or only low interest as transesterification partner. When all monomers are consumed, the catalyst does not show a strong tendency for transesterification. When a block copolymer (PCL‐b‐PLA, Table 1, run 1) is heated with freshly added catalyst in a ratio of 1000 : 1 (referring to the monomers incorporated) for 24 h at 180 °C, the blocky structure is maintained and only very small peaks resulting from a low degree of transesterification could be detected (see Figure S4). Therefore, the active chain end must be responsible for the degree of transesterifications leading to random‐like copolymers.
The increase of transesterifications if Cap is present in the copolymerization mixture was reported for some other catalysts but never investigated in detail.[ 21b , 23a ] The tendency of an active chain end to promote transesterification with the existing polymer chain and the preferred attack of a certain monomer, was shown to depend on the nature of the catalyst.[ 21c , 33 ] The presented catalyst is therefore an exceptional example containing the perfect features to promote the synthesis of PCL‐PLA‐PGA copolymers with desired microstructures. It strongly supports transesterifications during the insertion of Cap on the one hand and on the other hand adds GG and LL sequentially in a controlled manner as well as it does not scramble two already formed polymer chains significantly by transesterification. Its high activity leads to a fast consumption of all three monomers while the sequential addition experiments revealed a truly living polymerization character. Both microstructures, blocky as well as highly dispersed, are obtained in a solvent‐free process which is only possible due to the high robustness of the complex. The immortal conditions employed, which reduce the amount of catalyst necessary for the polymerization, round up the picture and characterize [FeCl2(TMG5NMe2asme)] (1) as an outstanding catalyst for the ring‐opening polymerization of lactones.
Conclusion
The iron guanidine catalyst presented herein exhibits a remarkable control over the copolymer microstructure, taming or using the transesterification bias of Cap—as desired. It is therefore not only the first biocompatible, robust catalyst with a higher polymerization activity than Sn(Oct)2 under industrially relevant melt conditions, it also shows an amazingly versatile control in copolymerizations. Having one catalyst for the formation of neat block copolymers as well as random‐like copolymers gives a glimpse of the opportunities still to be discovered to reduce the intricacy of industrial copolymerization processes. Understanding the role of Cap as transesterification agent is one example of crucial steps necessary to gain knowledge on copolymerization procedures. Only with those details revealed, complex copolymerization systems can be controlled to form materials with tailor‐made properties meeting the high requirements necessary to be the sustainable substitute for plastics that currently endanger our environment.
Conflict of interest
The authors declare no conflict of interest.
Supporting information
As a service to our authors and readers, this journal provides supporting information supplied by the authors. Such materials are peer reviewed and may be re‐organized for online delivery, but are not copy‐edited or typeset. Technical support issues arising from supporting information (other than missing files) should be addressed to the authors.
Supporting Information
Acknowledgements
R.D.R. thanks the DBU (Deutsche Bundesstiftung Umwelt) and DAAD (German Academic Exchange Service) for generous funding. M.K. thanks the ISF (Israel Science Foundation) for generous funding. The authors thank S. Buschmann for DSC measurements, Dr. O. Kuzmich and M. Connolly for MALDI‐ToF‐MS measurements and acknowledge Total Corbion PLA for lactide donations. S.H.‐P. and A.P. thank the Deutsche Forschungsgemeinschaft for generous support in the framework of the SFB985 (project C6). Open Access funding enabled and organized by Projekt DEAL.
R. D. Rittinghaus, J. Zenner, A. Pich, M. Kol, S. Herres-Pawlis, Angew. Chem. Int. Ed. 2022, 61, e202112853; Angew. Chem. 2022, 134, e202112853.
The raw data have been deposited under https://doi.org/10.5281/zenodo.5872326
Contributor Information
Prof. Moshe Kol, Email: moshekol@tauex.tau.ac.il.
Prof. Dr. Sonja Herres‐Pawlis, Email: sonja.herres-pawlis@ac.rwth-aachen.de.
References
- 1. Fagnani D. E., Tami J. L., Copley G., Clemons M. N., Getzler Y. D., McNeil A. J., ACS Macro Lett. 2021, 10, 41–53. [DOI] [PubMed] [Google Scholar]
- 2.
- 2a. Diment W. T., Stößer T., Kerr R. W. F., Phanopoulos A., Durr C. B., Williams C. K., Catal. Sci. Technol. 2021, 11, 1737–1745; [Google Scholar]
- 2b. Carrodeguas L. P., Chen T. T. D., Gregory G. L., Sulley G. S., Williams C. K., Green Chem. 2020, 22, 8298–8307; [Google Scholar]
- 2c. Chen T. T. D., Carrodeguas L. P., Sulley G. S., Gregory G. L., Williams C. K., Angew. Chem. Int. Ed. 2020, 59, 23450–23455; [DOI] [PMC free article] [PubMed] [Google Scholar]; Angew. Chem. 2020, 132, 23656–23661; [Google Scholar]
- 2d. McGuire T. M., Bowles J., Deane E., Farrar E. H. E., Grayson M. N., Buchard A., Angew. Chem. Int. Ed. 2021, 60, 4524–4528; [DOI] [PMC free article] [PubMed] [Google Scholar]; Angew. Chem. 2021, 133, 4574–4578; [Google Scholar]
- 2e. Piccini M., Leak D. J., Chuck C. J., Buchard A., Polym. Chem. 2020, 11, 2681–2691; [Google Scholar]
- 2f. Sulley G. S., Gregory G. L., Chen T. T. D., Peña Carrodeguas L., Trott G., Santmarti A., Lee K.-Y., Terrill N. J., Williams C. K., J. Am. Chem. Soc. 2020, 142, 4367–4378. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.
- 3a. Zhu Y., Romain C., Williams C. K., Nature 2016, 540, 354–362; [DOI] [PubMed] [Google Scholar]
- 3b. Zhang X., Fevre M., Jones G. O., Waymouth R. M., Chem. Rev. 2018, 118, 839–885; [DOI] [PubMed] [Google Scholar]
- 3c. Payne J., McKeown P., Jones M. D., Polym. Degrad. Stab. 2019, 165, 170–181. [Google Scholar]
- 4.
- 4a. McKeown P., Jones M. D., Sustain. Chem. 2020, 1, 1–22, [Google Scholar]; 10.3390/suschem1010001; [DOI]
- 4b. Drumright R. E., Gruber P. R., Henton D. E., Adv. Mater. 2000, 12, 1841–1846; [Google Scholar]
- 4c. Auras R., Harte B., Selke S., Macromol. Biosci. 2004, 4, 835–864; [DOI] [PubMed] [Google Scholar]
- 4d. Sintim H. Y., Bary A. I., Hayes D. G., Wadsworth L. C., Anunciado M. B., English M. E., Bandopadhyay S., Schaeffer S. M., DeBruyn J. M., Miles C. A., Sci. Total Environ. 2020, 727, 138668. [DOI] [PubMed] [Google Scholar]
- 5.
- 5a. Madhavan Nampoothiri K., Nair N. R., John R. P., Bioresour. Technol. 2010, 101, 8493–8501; [DOI] [PubMed] [Google Scholar]
- 5b. Farah S., Anderson D. G., Langer R., Adv. Drug Delivery Rev. 2016, 107, 367–392; [DOI] [PubMed] [Google Scholar]
- 5c. Endres H., Siebert-Raths A., Behnsen H., Schulz C., Biopolymers—facts and statistics, Hochschule Hannover, Hannover, 2019, ISSN 2363–8559. [Google Scholar]
- 6.
- 6a. Schäfer P. M., Fuchs M., Ohligschläger A., Rittinghaus R., McKeown P., Akin E., Schmidt M., Hoffmann A., Liauw M. A., Jones M. D., Herres-Pawlis S., ChemSusChem 2017, 10, 3547–3556; [DOI] [PubMed] [Google Scholar]
- 6b. Schäfer P. M., McKeown P., Fuchs M., Rittinghaus R. D., Hermann A., Henkel J., Seidel S., Roitzheim C., Ksiazkiewicz A. N., Hoffmann A., Dalton Trans. 2019, 48, 6071–6082; [DOI] [PubMed] [Google Scholar]
- 6c. Schäfer P. M., Herres-Pawlis S., ChemPlusChem 2020, 85, 1044–1052; [DOI] [PubMed] [Google Scholar]
- 6d. Rittinghaus R. D., Tremmel J., Růžička A., Conrads C., Albrecht P., Hoffmann A., Ksiazkiewicz A. N., Pich A., Jambor R., Herres-Pawlis S., Chem. Eur. J. 2020, 26, 212–221; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6e. Jones M. D., Wu X., Chaudhuri J., Davidson M. G., Ellis M. J., Mater. Sci. Eng. C 2017, 80, 69–74. [DOI] [PubMed] [Google Scholar]
- 7.
- 7a. Castro-Aguirre E., Iniguez-Franco F., Samsudin H., Fang X., Auras R., Adv. Drug Delivery Rev. 2016, 107, 333–366; [DOI] [PubMed] [Google Scholar]
- 7b. Haider T. P., Völker C., Kramm J., Landfester K., Wurm F. R., Angew. Chem. Int. Ed. 2019, 58, 50–62; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2019, 131, 50–63. [Google Scholar]
- 8.
- 8a. Hador R., Botta A., Venditto V., Lipstman S., Goldberg I., Kol M., Angew. Chem. Int. Ed. 2019, 58, 14679–14685; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2019, 131, 14821–14827; [Google Scholar]
- 8b. Hador R., Lipstman S., Rescigno R., Venditto V., Kol M., Chem. Commun. 2020, 56, 13528–13531; [DOI] [PubMed] [Google Scholar]
- 8c. Rosen T., Rajpurohit J., Lipstman S., Venditto V., Kol M., Chem. Eur. J. 2020, 26, 17183–17189; [DOI] [PubMed] [Google Scholar]
- 8d. Rosen T., Goldberg I., Venditto V., Kol M., J. Am. Chem. Soc. 2016, 138, 12041–12044; [DOI] [PubMed] [Google Scholar]
- 8e. Yuntawattana N., McGuire T. M., Durr C. B., Buchard A., Williams C. K., Catal. Sci. Technol. 2020, 10, 7226–7239; [Google Scholar]
- 8f. Stopper A., Rosen T., Venditto V., Goldberg I., Kol M., Chem. Eur. J. 2017, 23, 11540–11548. [DOI] [PubMed] [Google Scholar]
- 9.
- 9a. Garkhal K., Verma S., Jonnalagadda S., Kumar N., J. Polym. Sci. Part A 2007, 45, 2755–2764; [Google Scholar]
- 9b. Zhang C., Zhai T., Turng L.-S., Dan Y., Ind. Eng. Chem. Res. 2015, 54, 9505–9511; [Google Scholar]
- 9c. Simic V., Spassky N., Hubert-Pfalzgraf L. G., Macromolecules 1997, 30, 7338–7340; [Google Scholar]
- 9d. Wang Y., Niu J., Jiang L., Niu Y., Zhang L., J. Polym. Sci. Part A 2016, 53, 374–381. [Google Scholar]
- 10.
- 10a. Vries J. G. D., Phua P. H., Cabrera I. V. M., Heeres H. J., US009199961B2, 2015;
- 10b. Thaore V., Chadwick D., Shah N., Chem. Eng. Res. Des. 2018, 135, 140–152. [Google Scholar]
- 11.
- 11a. Brode G., Koleske J., J. Polym. Sci. Part A 1972, 6, 1109–1144; [Google Scholar]
- 11b. Sinha V., Bansal K., Kaushik R., Kumria R., Trehan A., Int. J. Pharm. 2004, 278, 1–23; [DOI] [PubMed] [Google Scholar]
- 11c. Ng K. W., Achuth H. N., Moochhala S., Lim T. C., Hutmacher D. W., J. Biomater. Sci. Polym. Ed. 2007, 18, 925–938. [DOI] [PubMed] [Google Scholar]
- 12.
- 12a. Watts A., Kurokawa N., Hillmyer M. A., Biomacromolecules 2017, 18, 1845–1854; [DOI] [PubMed] [Google Scholar]
- 12b. Nakayama Y., Aihara K., Yamanishi H., Fukuoka H., Tanaka R., Cai Z., Shiono T., J. Polym. Sci. Part A 2015, 53, 489–495. [Google Scholar]
- 13.
- 13a. Montes de Oca H., Ward I., Polymer 2006, 47, 7070–7077; [Google Scholar]
- 13b. Hakkarainen M., Albertsson A.-C., Karlsson S., Polym. Degrad. Stab. 1996, 52, 283–291; [Google Scholar]
- 13c. Chu C., J. Appl. Polym. Sci. 1981, 26, 1727–1734. [Google Scholar]
- 14. Cai Q., Bei J., Wang S., J. Biomater. Sci. Polym. Ed. 2000, 11, 273–288. [DOI] [PubMed] [Google Scholar]
- 15. Jones R. G., Kitayama T., Hellwich K.-H., Hess M., Jenkins A. D., Kahovec J., Kratochvíl P., Mita I., Mormann W., Ober C. K., Pure Appl. Chem. 2016, 88, 1073–1100. [Google Scholar]
- 16.
- 16a. Duda A., Biela T., Libiszowski J., Penczek S., Dubois P., Mecerreyes D., Jérôme R., Polym. Degrad. Stab. 1998, 59, 215–222; [Google Scholar]
- 16b. Kaler S., McKeown P., Ward B. D., Jones M. D., Inorg. Chem. Front. 2021, 8, 711–719; [Google Scholar]
- 16c. Quilter H. C., Hutchby M., Davidson M. G., Jones M. D., Polym. Chem. 2017, 8, 833–837; [Google Scholar]
- 16d. Hermann A., Hill S., Metz A., Heck J., Hoffmann A., Hartmann L., Herres-Pawlis S., Angew. Chem. Int. Ed. 2020, 59, 21778–21784; [DOI] [PMC free article] [PubMed] [Google Scholar]; Angew. Chem. 2020, 132, 21962–21968. [Google Scholar]
- 17.
- 17a. Pensec S., Leroy M., Akkouche H., Spassky N., Polym. Bull. 2000, 45, 373–380; [Google Scholar]
- 17b. Jacobs C., Dubois P., Jérôme R., Teyssié P., Macromolecules 1991, 24, 3027–3034; [Google Scholar]
- 17c. Stirling E., Champouret Y., Visseaux M., Polym. Chem. 2018, 9, 2517–2531; [Google Scholar]
- 17d. Simic V., Pensec S., Spassky N., Macromol. Symp. 2000, 153, 109–121; [Google Scholar]
- 17e. Kreiser-Saunders I., Kricheldorf H. R., Macromol. Chem. Phys. 1998, 199, 1081–1087; [Google Scholar]
- 17f. Darensbourg D. J., Karroonnirun O., Macromolecules 2010, 43, 8880–8886; [Google Scholar]
- 17g. Stevels W. M., Ankoné M. J. K., Dijkstra P. J., Feijen J., Macromol. Chem. Phys. 1995, 196, 1153–1161; [Google Scholar]
- 17h. Deng X., Zhu Z., Xiong C., Zhang L., J. Polym. Sci. Part A 1997, 35, 703–708; [Google Scholar]
- 17i. Mandal M., Chakraborty D., Ramkumar V., RSC Adv. 2015, 5, 28536–28553; [Google Scholar]
- 17j. Kricheldorf H. R., Rost S., Polymer 2005, 46, 3248–3256; [Google Scholar]
- 17k. D'Auria I., Ferrara V., Tedesco C., Kretschmer W., Kempe R., Pellecchia C., ACS Appl. Polym. Mater. 2021, 3, 4035–4043. [Google Scholar]
- 18.
- 18a. Kricheldorf H. R., Mang T., Jonte J. M., Macromolecules 1984, 17, 2173–2181; [Google Scholar]
- 18b. Zhong Z., Yu D., Meng F., Gan Z., Jing X., Polym. J. 1999, 31, 633–636; [Google Scholar]
- 18c. Florczak M., Duda A., Angew. Chem. Int. Ed. 2008, 47, 9088–9091; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2008, 120, 9228–9231; [Google Scholar]
- 18d. Wyrębiak R., Oledzka E., Figat R., Sobczak M., Molecules 2019, 24, 4168; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18e. Florczak M., Libiszowski J., Mosnacek J., Duda A., Penczek S., Macromol. Rapid Commun. 2007, 28, 1385–1391. [Google Scholar]
- 19.
- 19a. Rosen T., Goldberg I., Navarra W., Venditto V., Kol M., Angew. Chem. Int. Ed. 2018, 57, 7191–7195; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2018, 130, 7309–7313; [Google Scholar]
- 19b. Contreras J., Pestana J., López-Carrasquero F., Torres C., Polym. Bull. 2014, 71, 1661–1674; [Google Scholar]
- 19c. Ayyoob M., Lee S., Kim Y. J., J. Polym. Res. 2020, 27, 109. [Google Scholar]
- 20.
- 20a. Nomura N., Akita A., Ishii R., Mizuno M., J. Am. Chem. Soc. 2010, 132, 1750–1751; [DOI] [PubMed] [Google Scholar]
- 20b. Kan C., Ma H., RSC Adv. 2016, 6, 47402–47409; [Google Scholar]
- 20c. Pappalardo D., Annunziata L., Pellecchia C., Macromolecules 2009, 42, 6056–6062; [Google Scholar]
- 20d. Li G., Lamberti M., Pappalardo D., Pellecchia C., Macromolecules 2012, 45, 8614–8620. [Google Scholar]
- 21.
- 21a. Dobrzynski P., Kasperczyk J., Janeczek H., Bero M., Macromolecules 2001, 34, 5090–5098; [Google Scholar]
- 21b. Dobrzynski P., J. Polym. Sci. Part A 2002, 40, 1379–1394; [Google Scholar]
- 21c. Dobrzynski P., J. Polym. Sci. Part A 2002, 40, 3129–3143. [Google Scholar]
- 22.
- 22a. Bero M., Kasperczyk J., Adamus G., Makromol. Chem. 1993, 194, 907–912; [Google Scholar]
- 22b. Ouyang H., Nie K., Yuan D., Zhang Y., Cui D., Yao Y., Sci. China Chem. 2018, 61, 708–714. [Google Scholar]
- 23.
- 23a. Dakshinamoorthy D., Peruch F., J. Polym. Sci. Part A 2012, 50, 2161–2171; [Google Scholar]
- 23b. Nalampang K., Molloy R., Punyodom W., Polym. Adv. Technol. 2007, 18, 240–248; [Google Scholar]
- 23c. Wei Z., Liu L., Qu C., Qi M., Polymer 2009, 50, 1423–1429; [Google Scholar]
- 23d. Dobrzyński P., Kasperczyk J., Jelonek K., Ryba M., Walski M., Bero M., J. Biomed. Mater. Res. Part A 2006, 79, 865–873. [DOI] [PubMed] [Google Scholar]
- 24.
- 24a. Qin L., Bai J., Zhang Y., Chen X., J. Organomet. Chem. 2018, 871, 40–47; [Google Scholar]
- 24b. In't Veld P. J., Velner E. M., Van De Witte P., Hamhuis J., Dijkstra P. J., Feijen J., J. Polym. Sci. Part A 1997, 35, 219–226; [Google Scholar]
- 24c. Lapenta R., Mazzeo M., Grisi F., RSC Adv. 2015, 5, 87635–87644. [Google Scholar]
- 25.
- 25a. Grijpma D., Pennings A., Polym. Bull. 1991, 25, 335–341; [Google Scholar]
- 25b. Choi E. J., Park J. K., Chang H. N., J. Polym. Sci. Part B 1994, 32, 2481–2489; [Google Scholar]
- 25c. Kricheldorf H. R., Boettcher C., Tönnes K.-U., Polymer 1992, 33, 2817–2824; [Google Scholar]
- 25d. Kricheldorf H. R., Hachmann-Thiessen H., J. Polym. Sci. Part A 2005, 43, 3268–3277. [Google Scholar]
- 26.
- 26a. Bero M., Czapla B., Dobrzyński P., Janeczek H., Kasperczyk J., Macromol. Chem. Phys. 1999, 200, 911–916; [Google Scholar]
- 26b. Baimark Y., Molloy R., ScienceAsia 2004, 30, 327; [Google Scholar]
- 26c. Fernández J., Etxeberria A., Sarasua J.-R., J. Mech. Behav. Biomed. 2012, 9, 100–112; [DOI] [PubMed] [Google Scholar]
- 26d. Hiljanen-Vainio M., Karjalainen T., Seppälä J., J. Appl. Polym. Sci. 1996, 59, 1281–1288; [Google Scholar]
- 26e. Fernández J., Meaurio E., Chaos A., Etxeberria A., Alonso-Varona A., Sarasua J., Polymer 2013, 54, 2621–2631; [Google Scholar]
- 26f. Weng F., Li X., Wang Y., Wang W. J., Severtson S. J., Macromol. React. Eng. 2015, 9, 535–544. [Google Scholar]
- 27. Rittinghaus R. D., Schäfer P. M., Albrecht P., Conrads C., Hoffmann A., Ksiazkiewicz A. N., Bienemann O., Pich A., Herres-Pawlis S., ChemSusChem 2019, 12, 2161–2165. [DOI] [PubMed] [Google Scholar]
- 28. Rittinghaus R. D., Karabulut A., Hoffmann A., Herres-Pawlis S., Angew. Chem. Int. Ed. 2021, 60, 21795–21800; [DOI] [PMC free article] [PubMed] [Google Scholar]; Angew. Chem. 2021, 133, 21965–21971. [Google Scholar]
- 29. Mayo F. R., Lewis F. M., J. Am. Chem. Soc. 1944, 66, 1594–1601. [Google Scholar]
- 30.
- 30a. Yuan W., Tang X., Huang X., Zheng S., Polymer 2005, 46, 1701–1707; [Google Scholar]
- 30b. Channuan W., Siripitayananon J., Molloy R., Sriyai M., Davis F. J., Mitchell G. R., Polymer 2005, 46, 6411–6428. [Google Scholar]
- 31. Stößer T., Chen T., Zhu Y., Williams C., Philos. Trans. R. Soc. A 2018, 376, 20170066. [Google Scholar]
- 32.
- 32a. Bittiger H., Marchessault R., Niegisch W., Acta Crystallogr. 1970, 26, 1923–1927; [Google Scholar]
- 32b. Van de Velde K., Kiekens P., Polym. Test. 2002, 21, 433–442. [Google Scholar]
- 33. Kasperczyk J., Bero M., Makromol. Chem. 1993, 194, 913–925. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
As a service to our authors and readers, this journal provides supporting information supplied by the authors. Such materials are peer reviewed and may be re‐organized for online delivery, but are not copy‐edited or typeset. Technical support issues arising from supporting information (other than missing files) should be addressed to the authors.
Supporting Information