

Abstract
Schistosoma mansoni, a snail‐borne, blood fluke that infects humans, was introduced into the Americas from Africa during the Trans‐Atlantic slave trade. As this parasite shows strong specificity to the snail intermediate host, we expected that adaptation to South American Biomphalaria spp. snails would result in population bottlenecks and strong signatures of selection. We scored 475,081 single nucleotide variants in 143 S. mansoni from the Americas (Brazil, Guadeloupe and Puerto Rico) and Africa (Cameroon, Niger, Senegal, Tanzania, and Uganda), and used these data to ask: (i) Was there a population bottleneck during colonization? (ii) Can we identify signatures of selection associated with colonization? (iii) What were the source populations for colonizing parasites? We found a 2.4‐ to 2.9‐fold reduction in diversity and much slower decay in linkage disequilibrium (LD) in parasites from East to West Africa. However, we observed similar nuclear diversity and LD in West Africa and Brazil, suggesting no strong bottlenecks and limited barriers to colonization. We identified five genome regions showing selection in the Americas, compared with three in West Africa and none in East Africa, which we speculate may reflect adaptation during colonization. Finally, we infer that unsampled populations from central African regions between Benin and Angola, with contributions from Niger, are probably the major source(s) for Brazilian S. mansoni. The absence of a bottleneck suggests that this is a rare case of a serendipitous invasion, where S. mansoni parasites were pre‐adapted to the Americas and able to establish with relative ease.
Keywords: Africa, Brazil, codispersal, exome, human parasite, migration
1. INTRODUCTION
Genomic characterization of parasites and pathogens is increasingly being used as an aid to traditional epidemiological methods in reconstructing transmission patterns (de Oliveira et al., 2020; Nadeau et al., 2021). On a longer timescale, genomic data can be used to understand biological invasions of pathogens into new continents, just as these methods are used for investigating biological invasions in free‐living organisms (Rius et al., 2015; Sherpa & Després, 2021). Such methods can determine the colonization route, source population, number of colonization events, whether diversity is reduced during colonization and evidence for adaptation in colonizing populations. Examining the consequences of historical invasions can inform our understanding of extant invasions.
The Trans‐Atlantic slave trade lasted from 1502 to 1888 (Bergad, 2007). During this time, more than 12 million people were trafficked from Africa to slave ports in the Americas, representing one of the largest forced migration events in human history (Eltis, 2001). Along with the human cargo, a number of human pathogens were introduced into the Americas. For example, parvovirus B19 (Primate erythroparvovirus 1) and Hepatitis B virus were successfully introduced into the Americas, leading to large‐scale outbreaks (Guzmán‐Solís et al., 2021). Today viable populations of pathogens including herpes simplex virus 2 (Human alphaherpesvirus 2; Forni et al., 2020), Yellow fever virus (Bryant et al., 2007), the parasitic nematode Wuchereria bancrofti (Small et al., 2019), among others (Steverding, 2020), are all a direct result of introductions during the Trans‐Atlantic slave trade. In some cases, the genetic signatures of the introduction are still visible. For example, genetic diversity in South American Leishmania chagasi populations is halved and the effective population size (N e) is reduced from 43.6 million to 15.5 thousand compared to source populations in Africa (Leblois et al., 2011; Schwabl et al., 2021). Here, we focus on another successful invasion by a human‐parasitic trematode, Schistosoma mansoni.
S. mansoni is distributed from Oman, through sub‐Saharan Africa, to the Caribbean and countries along the eastern coast of South America. Phylogenetic evidence indicates that S. mansoni in West Africa and the Americas are closely related (Crellen et al., 2016; Desprès et al., 1993; Fletcher et al., 1981; Morgan et al., 2005; Webster et al., 2013) and these observations, along with demographic reconstructions (Crellen et al., 2016), indicate a recent origin of S. mansoni in the Americas. As a result, there is strong evidence that S. mansoni comigrated to the Americas during the forced, human migrations of the Trans‐Atlantic slave trade (Files, 1951). Furthermore, reduced diversity in mitochondrial haplotypes (Desprès et al., 1993; Fletcher et al., 1981; Morgan et al., 2005; Webster et al., 2013) in South American S. mansoni suggests the presence of a bottleneck during parasite establishment.
Our central goal here was to use parasite genomic data to investigate this human‐mediated, biological invasion and the impacts of a relatively recent, transcontinental, migration event. Parasites in the genus Schistosoma have a complex life cycle involving human definitive hosts and aquatic snail intermediate hosts (reviewed in Anderson & Enabulele, 2021). Eggs are expelled in human faeces (S. mansoni and S. japonicum) or urine (S. haematobium). Larvae (miracidia) hatch in fresh water and infect receptive snails. Inside the snail host, the schistosomes reproduce asexually, and second‐stage larvae (cercariae) are released back into the water where they infect humans, mature into adult worms and restart their life cycle. S. mansoni is diploid, with a well‐characterized 363‐Mb genome (Berriman et al., 2009; International Helminth Genomes Consortium, 2019; Protasio et al., 2012), ZW sex determination, obligate sexual reproduction of adult worms, and a relatively long lifespan (5–10 years) (Fulford et al., 1995).
The distribution of the intermediate snail host is a major driver of schistosome distribution. S. mansoni shows strong specificity for species and even strains of snails in the genus Biomphalaria (Webster & Woolhouse, 1998). However, Biomphalaria pfeifferi, B. sudanica and B. alexandrina are the primary intermediate hosts in Africa (DeJong et al., 2001), while B. glabrata, B. tenagophila and B. straminea are the known snail hosts in South America (Vidigal et al., 2000). S. mansoni infections can impact the reproductive viability of their snail hosts, and there are strong co‐evolutionary interactions driving resistance to infection in snails and for infectivity in parasites (Davies et al., 2001; Theron et al., 2014; Webster et al., 2004). Several schistosome resistance genes have been localized within the snail genome (Tennessen et al., 2015, 2020) and polymorphic loci in snails and parasites are thought to determine compatibility (Mitta et al., 2017; Webster & Woolhouse, 1998; Woolhouse & Webster, 2000). Based on these observations, we hypothesize that the adaptation to novel Biomphalaria spp. hosts would place strong selective pressures on S. mansoni as it became established in the Americas.
Adult schistosomes live in the blood vessels, making them difficult to sample. Genome and exome sequencing of schistosomes is now possible using whole genome amplification of miracidia larvae isolated from faeces or urine (Doyle et al., 2019; Le Clec'h et al., 2018; Shortt et al., 2017), and several genome‐scale population analyses have recently been published (Berger et al., 2021; Platt et al., 2019; Shortt et al., 2017). Our goal here was to address the following questions with the available sequence data from both Africa (Niger, Senegal, Uganda, Tanzania) and the Americas (Caribbean, Brazil): (i) Are the genomic data consistent with a West African origin of colonizing schistosome populations? (ii) Is there evidence for genetic bottlenecks during colonization? (iii) Are there genomic signatures suggesting adaptation of colonizing parasites to the Americas? (iv) Can we determine the source country, or countries, for parasite populations in the Americas?
2. MATERIALS AND METHODS
2.1. Data and sample information
We examined published exomic and genomic data from 178 individual Schistosoma samples/isolates, from multiple geographical locations, available from three studies (Berriman et al., 2009; Chevalier et al., 2019; Crellen et al., 2016). Exome data are from Chevalier et al. (2016) and Chevalier et al. (2019). These data were generated from individual larval miracidia hatched from Schistosoma mansoni eggs and preserved on Flinders Technology Associates® (FTA) cards. Exome libraries were generated via whole genome amplification followed by targeted capture of the exome (Le Clec'h et al., 2018). This method specifically targets 95% (14.81 Mb) of the exome with 2× tiled probes. The whole genome sequence data came from adult worms cultured through laboratory rodents and snails for two or more generations before whole genome library preparation and sequencing (Berriman et al., 2009; Crellen et al., 2016; International Helminth Genomes Consortium, 2019). Sample origins are shown in Figure 1. Detailed metadata are available for each sample in Table S1 including country of origin, species identification and NCBI Short Read Archive.
FIGURE 1.
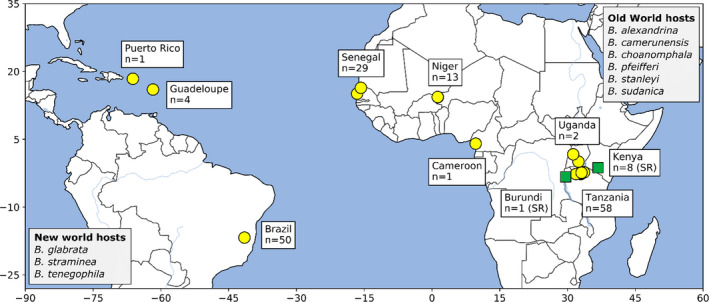
Sampling localities. Location and number of samples for Schistosoma mansoni (yellow circles) and S. rodhaini (green squares) samples included in this study. Members of the genus Biomphalaria are the predominant intermediate hosts with specific distributions of the different species involved in transmission varying across Africa and South America. Intermediate snail vectors are listed in greyed boxes
2.2. Computational environment
We used conda version 4.8.3 to manage computational, virtual environments for all analyses. Sequence read filtering through genotyping steps were documented in a snakemake version 5.18.1 (Köster & Rahmann, 2012) workflow and all other analyses were performed in a series of jupyter version 1.0.0 notebooks. The code for this project, including shell scripts, snakemake workflows notebooks, and environmental yaml files are available at https://github.com/nealplatt/sch_man_nwinvasion/releases/tag/v0.2 (last accessed October 21, 2021) and accessioned at https://doi.org/10.5281/zenodo.5590460 (last accessed October 21, 2021).
2.3. Genotyping
Paired‐end reads were quality filtered with trimmomatic version 0.39 (Bolger et al., 2014) with the following parameters: “LEADING:10TRAILING:10 SLIDINGWINDOW:4:15MINLEN:36.” Filtered reads were mapped to the S. mansoni genome (GenBank Assembly accession: GCA_000237925.3) using bwa version 0.7.17‐r1188 (Li & Durbin, 2010). We allowed up to 15 mismatches per 100‐bp read (‐n 15) to account for divergence between S. mansoni and S. rodhaini. Mapped single and paired reads were merged into a single file and all optical/PCR (polymerase chain reaction) duplicates were removed with gatk version 4.1.2.0’s (McKenna et al., 2010) MarkDuplicates. Single nucleotide variants (SNVs) were called with HaplotypeCaller and GenotypeGVCFs on a contig‐by‐contig basis, combined into a gvcf per individual, and finally merged into a single gvcf for the entire data set. We used high‐quality, SNV data from Le Clec’h et al. (2021) as a training data set for variant recalibration and scored SNV quality using “‐an SOR ‐an MQ ‐an MQRankSum ‐an ReadPosRankSum.” Sensitivity Tranches (‐‐truth‐sensitivity‐tranche) were set at 100, 99.5, 99, 97.5, 95 and 90. We recalibrated SNVs using the 97.5 sensitivity tranche and filtered low‐confidence sites with the following set of filters (‐‐filter‐expression): QD < 2.0, MQ < 30.0, FS > 60.0, SOR > 3.0, MQRankSum < −12.5 and ReadPosRankSum < −8.0." All genotyping steps from read filtering through variant recalibration were contained within a single snakemake version 5.18.1 (Köster & Rahmann, 2018) script.
We used vcftools version 0.1.16 (Danecek et al., 2011) for additional rounds of filtering. First, we removed low‐quality sites with quality score <25, read depth <12 and nonbiallelic sites. Second, we removed sites and individuals with a genotyping rate <50%. Third, we removed all sites that were on unresolved haplotigs by retaining only those SNVs that were on one of seven autosomal scaffolds (GenBank Nucleotide accessions: HE601624.2‐30.2), the sex‐linked ZW scaffold (HE601631.2) or the mitochondria (HE601612.2). Finally, for analyses requiring unlinked SNVs, we filtered linked sites within 250‐kb windows using plink version 1.90b4 (Purcell et al., 2007) with the following parameters: “—indep‐pairwise 250 kb 1 0.20.”
2.4. Summary statistics
We quantified read depth per probed‐exome region with mosdepth version 0.2.5 (Pedersen & Quinlan, 2018) and calculated genome‐wide summary statistics for each population, including F 3, F ST, Tajima's D, π, and the Watterson estimator (Θ) with scikit‐allele version 1.2.1 (Miles et al., 2019). We examined genome regions that were targeted by the Le Clec'h et al. (2018) probe set for these calculations; nontarget regions (i.e., nonexomic) were ignored because most samples lacked information from these regions. F ST between populations was calculated from the average Weir–Cockerham F ST (Weir & Cockerham, 1984) in windows of 100 SNVs. Effective population size (N e) was estimated from Θ and the mutation rate (μ = 8.1e‐9 per base per generation; Crellen et al., 2016) with:
We examined linkage disequilibrium (LD) within each population by calculating r 2 (‐‐r2) values with plink version 1.90b6.18 (Purcell et al., 2007). We excluded invariant sites from the analyses. Intra‐autosomal, pairwise comparisons between SNVs within 1 Mb of one another were allowed by setting the following parameters: “‐‐ldwindow 1000000”, “‐‐ld‐window‐kb 1000” and “‐‐ld‐window‐r2 to 0.0.” r 2 values were then binned into 500‐bp windows and averaged for each population using the R version 3.6.1 stats.bin function in the fields version 11.6 (Nychka et al., 2017) library. We used local regression to smooth the binned r 2 values with the loessMod function in the base R version 3.6.1 package and a span size of 0.5.
We used a Pearson Mantel test to examine correlation between genetic and physical distance. Since we did not have exact collection coordinates from whole genome samples, or they were laboratory‐derived, we excluded them from the analyses and instead focused only on the S. mansoni exome samples. We calculated pairwise p‐distances with vcf2dis (https://github.com/BGI‐shenzhen/VCF2Dis; commit: b7684d3, accessed February 13, 2021) and physical distances between samples the Python haversine 2.3.0 module. Finally, we used the mantel() function in the scikit‐bio 0.2.1 Python library to conduct a Pearson Mantel test that included 1000 permutations.
2.5. Population structure and admixture
We examined population substructure using principal components analysis (PCA) and admixture with unlinked autosomal SNVs (described above). Two PCAs were calculated in plink version 1.90b6.18, with and without the S. rodhaini samples. Population ancestry was estimated with admixture version 1.3.0 (Alexander et al., 2009). We examined between k = 1 and k = 20 populations and used the vross‐validation scores and the Evanno et al. (2005) method to determine a range of viable k value. Q estimates were used as a proxy for ancestry fractions.
We examined D (Patterson et al., 2012), D 3 (Hahn & Hibbins, 2019) and F 3 (Patterson et al., 2012) to identify gene flow between S. rodhaini and S. mansoni with emphasis on the Tanzanian S. mansoni population since it and the S. rodhaini samples are from East Africa. For D 3, we calculated the mean‐pairwise (Euclidean) distances between populations using scikit‐allele’s allel.pairwise_distance() function. To determine significance, we used 1000 block bootstrap replicates of 1000 SNV blocks. We calculated the average F 3 across the genome in blocks of 100 variants. Here we ran multiple tests that included some combination of an African S. mansoni population (Niger, Senegal and Tanzania) as the test group and Brazilian S. mansoni and S. rodhaini as the potential source populations. D, or the ABBA‐BABBA statistic, was averaged over blocks of 1000 variants assuming a phylogeny of (((a, Tanzania), S. rodhaini), S. margrebowiei), where the a population was either Brazil, Niger or Senegal. D, D 3 and F 3 values were calculated using scikit‐allel.
2.6. Phylogenetics
We used three different phylogenetic methods to visualize relationships among sampled schistosomes: a mitochondrial haplotype network, a coalescent‐based species tree and a phylogenetic network.
2.6.1. Mitochondrial haplotype network
We extracted mitochondrial SNVs from all S. mansoni individuals with vcftools and converted the subsequent VCF file to Nexus format with vcf2phylip version 2.0 (Ortiz, 2019). A median joining network (ε = 0) was created from the mitochondrial haplotypes with popart version 1.7 (Leigh & Bryant, 2015).
2.6.2. Coalescent‐based species tree
We generated a coalescent‐based species tree with svdquartets (Chifman & Kubatko, 2015) packaged in paup* version 4.0.a.build166 (Swofford, 2003). We examined parsimony‐informative, autosomal SNVs by removing private alleles (singleton and doubletons). All samples were assigned to a population based on their country of origin (examples include Niger, Puerto Rico, Brazil, Cameroon, etc.) except for the laboratory‐derived, Caribbean samples. Each of these samples was considered to represent an individual population given their histories of extensive laboratory passage (exclusing Guadeloupe1, Guadeloupe2, Puerto Rico). We randomly evaluated 100,000 random quartets and bootstrapped the quartet tree with 1000 standard replicates. The tree was rooted on the single S. margrebowiei individual.
2.6.3. Phylogenetic network
We used a phylogenetic network to visualize and quantify migration among schistosome populations. We only included S. mansoni populations with more than four individuals, which excluded all whole genome samples from this analysis including those from the Caribbean and the S. rodhaini samples. We used autosomal SNVs after filtering linked sites in 250‐kb blocks with plink version 1.90b6.18 and then used treemix version 1.12 (Pickrell & Pritchard, 2012) to generate the phylogenetic network. This analysis used a covariance matrix generated from blocks of 500 SNVs without sample‐size correction (“‐‐noss”) and the number of migration events was limited to 3.
2.7. Selection
We scanned the genome to identify regions under selection using haplotype (h‐scan version 1.3; Schlamp et al., 2016), allele frequency (sweepfinder2 version 2.1; DeGiorgio et al., 2016) and PCA‐based (pcadapt version 4.3.3; Luu et al., 2017) methods. In addition, to avoid false positives, we used msprime version 0.7.4 (Kelleher et al., 2016) to conduct simulations to estimate the range of values expected under neutrality from h‐scan and sweepfinder2.
2.7.1. Neutral simulations
We used msprime version 0.7.4 (Kelleher et al., 2016) to simulate a set of neutrally evolving SNVs along a single chromosome for each population and then used the simulated data with h‐scan and sweepfinder2 to define the range of values expected in the absence of selection. For these simulations we used a mutation rate (μ = 8.1e‐9 per base per generation; Crellen et al., 2016) and recombination rate (3.4e‐8 per base per generation; Criscione et al., 2009) from previous work on S. mansoni. Population‐specific estimates of N e are described above. The simulated chromosome length was set to 88.9 Mb which is equal to chromosome 1 (HE601624.2) in the S. mansoni assembly. The number of chromosomes sampled per msprime run was equal to the number of samples available from each population. We used these parameters to perform 342 simulations for each population, roughly equivalent to 100 genomes of simulated data. Next, we downsampled the SNVs along the entire simulated chromosome so that they were comparable with our targeted sequencing approach (i.e., only SNVs from “exomic” regions). Since the simulated chromosomes were the same size as chromosome 1, we transposed the chr1 annotation onto the simulated chromosome and extracted only those SNVs occurring in regions accessible by our biotinylated probes with vcftools. The simulated data were run through h‐scan and sweepfinder2 in parallel with the actual SNV data to establish a range of neutral values for each test (described above).
2.7.2. h‐scan
This method measures the length of homozygous haplotypes to identify regions under selection. Strong selection will drive adaptive alleles linked SNVs to high frequency and reduce homozygosity in the surrounding region. For each population, we converted autosomal SNVs from VCF to h‐scan format using vcf2hscan.py script from vcf2phylip version 2.0 (Ortiz, 2019) and ran h‐scan on each chromosome. To avoid calculating H across large distances without variants we set a maximum gap length (‐g) of 10 kb. H values were smoothed (H smoothed) by median filtering values in 201 SNV windows (step size =1) using the medfilt function in scipy version 1.5.2 (Virtanen et al., 2020) for visualization purposes.
2.7.3. sweepfinder2
This method uses deviations in allele frequency from a neutral expectation to estimate positive selection while accounting for the possibility of background selection via a likelihood ratio (LR) test (DeGiorgio et al., 2016). Empirical site‐frequency spectra were calculated for each population, and within each population LR was estimated along each autosome individually. We examined grid points (“g”), or window sizes, of 1, 5, 10 and 20 kb to accommodate possible gaps caused by exome data. These options had minimal impact on the results. Downstream analyses are reported on the runs with “g” =1 kb.
2.7.4. pcadapt
We used the R version 4.0.5 package pcadapt version 4.3.3 (Luu et al., 2017) to identify highly differentiated loci among populations via variants associated with population structure as identified by PCA. We only included samples from Brazil, Niger and Senegal since our primary goal was to identify variants involved in adaptation to the Americas. Rare variants (MAF < 5%) were excluded with vcftools version 0.1.16. We identified the appropriate number of principal components from the data by running an initial pcadapt run with 20 populations (K = 20) and LD filtered variants (LD.clumping = list(size =100, thr =0.2)). The major break in the subsequent scree plot was used as the optimal K choice. We used a second pcadapt run with the optimal K and the same LD filtering parameters as the initial run to assign p values to each site. Finally, we adjusted p values for multiple tests with Bonferroni correction and an α = .05 to identify SNV outliers associated with population differentiation.
2.7.5. Identifying regions of selection
We identified regions potentially under positive selection using a three‐step process. First, we identified SNVs whose h‐scan and sweepfinder2 values were in the 99th percentile of and greater than the neutral thresholds established with msprime. These were SNVs with the strongest signal of selection. Then, we expanded from the SNV to a broader region by merging all variants within 333,333 bp whose h‐scan or sweepfinder2 values were greater than the neutral thresholds. Finally, we looked for pcadapt outliers in each region. These regions are referred to as “putative selected regions” or “putative regions of selection.” Gene names and functions were taken from UniProtKB (release 2020_06) or HHsearch (Steinegger et al., 2019) annotations from Le Clec'h et al. (2021). The entire process is summarized in Figure S1.
3. RESULTS
3.1. Summary of sequence data
After genotyping and filtering, we removed 25 of the 178 samples with low numbers of reads, poor coverage or low genotyping rates. The final data set included 135 Schistosoma mansoni (exome), eight S. mansoni (genome), eight S. rodhaini (exome), one S. rodhaini (genome) and one S. margrebowiei (genome). We genotyped 1,823,890 sites, which was reduced to 475,081 autosomal and 815 mitochondrial variants after quality filtering: one variant per 641 bases. Further, because we required that any site be genotyped in >50% of the samples and only 11% (18 of 153) of the samples were from whole genome data, all of the variants outside of the exome were removed. The final data set comprised 153 samples with mean read depths of 520.7× (range: 251.4–998.2×) and 66.0× (range: 14.8–726.2×) at mitochondrial and autosomal loci. Location and sequence coverage statistics for all samples in the final data set are listed in Table S1.
3.2. Summary statistics
Autosomal and mitochondrial summary statistics for π, H, Tajima's D, Θ and N e are shown in Table 1. π, Θ and N e are similar between the West African and Brazilian S. mansoni populations but are two to three times lower than observed in Tanzanian S. mansoni. Tajima's D values ranged from slightly positive to negative (Tajima's D = −1.417 to 0.034) in S. mansoni (Table 1; Table S2). All the African populations show negative Tajima's D, consistent with natural selection or population expansion. However, the Brazilian Tajima's D is positive, which is inconsistent with a bottleneck during colonization of South America. Mitochondrial diversity was quantified with π and haplotype diversity (H). H in all populations is very high (>0.978) indicating that all mitochondrial haplotypes are unique. Mitochondrial π follows the same pattern as autosomal π with the exception that the mitochondrial π in Brazil is lower than expected when compared to measures in Niger and Senegal. F ST values between S. mansoni populations are shown in Table 2 and were highest in pairwise comparisons that included the Tanzanian population. Mantel tests showed significant signs of isolation‐by‐distance within Africa (r = .64, p = .001) and between African and Brazilian (r = .77, p = .001) S. mansoni samples.
TABLE 1.
Whole genome summary statistics for Schistosoma mansoni populations and S. rodhaini
| n | π | π (mito) | H (mito) | Tajima's D | Θ | N e | π (PRS) | Tajima's D (PRS) | |
|---|---|---|---|---|---|---|---|---|---|
| S. rodhaini | 9 a | 5.61E‐04 | Na b | Na b | 0.479 | 4.29E‐04 | 13,226 | Na c | Na c |
| Sm (Brazil) | 45 | 6.93E‐04 | 2.10E‐03 | 0.984 | 0.034 | 5.84E‐04 | 18,032 | 2.22E‐04 | −1.218 |
| Sm (Niger) | 10 | 6.00E‐04 | 5.89E‐03 | 0.978 | −0.579 | 6.07E‐04 | 18,737 | 2.46E‐04 | −1.018 |
| Sm (Senegal) | 25 | 4.97E‐04 | 4.72E‐03 | 0.997 | −1.417 | −1.375 | 21,992 | 1.75E‐04 | −1.816 |
| Sm (Tanzania) | 55 | 1.45E‐03 | 7.25E‐03 | 1.0 | −0.729 | −0.739 | 51,508 | Na c | Na c |
"n"—number of samples; "π"—nucleotide diversity; "H"—haplotype diversity; "Θ"—Watterson estimator; "N e"—effective population size.
Abbreviations: PRS, putative region of selection; Sm, Schistosoma mansoni.
Eight of nine S. rodhaini samples came from a single laboratory population: population statistics are probably biased.
No S. rodhaini reads mapped to the S. mansoni mitochondria.
Not calculated.
TABLE 2.
F ST between Schistosoma species and populations
| Pop1 | Pop2 | F ST | SE |
|---|---|---|---|
| S. rodhaini | Sm (Caribbean) | 0.929 | 0.0012 |
| S. rodhaini | Sm (Tanzania) | 0.844 | 0.0016 |
| S. rodhaini | Sm (Senegal) | 0.937 | 0.0013 |
| S. rodhaini | Sm (Niger) | 0.931 | 0.0011 |
| S. rodhaini | Sm (Brazil) | 0.919 | 0.0013 |
| Sm (Caribbean) | Sm (Tanzania) | 0.279 | 0.0036 |
| Sm (Caribbean) | Sm (Senegal) | 0.323 | 0.0085 |
| Sm (Caribbean) | Sm (Niger) | 0.236 | 0.0071 |
| Sm (Caribbean) | Sm (Brazil) | 0.154 | 0.0067 |
| Sm (Tanzania) | Sm (Senegal) | 0.416 | 0.0032 |
| Sm (Tanzania) | Sm (Niger) | 0.348 | 0.0031 |
| Sm (Tanzania) | Sm (Brazil) | 0.379 | 0.0034 |
| Sm (Senegal) | Sm (Niger) | 0.135 | 0.0042 |
| Sm (Senegal) | Sm (Brazil) | 0.235 | 0.0047 |
| Sm (Niger) | Sm (Brazil) | 0.152 | 0.0036 |
Abbreviation: Sm, Schistosoma mansoni.
Diversity and N e values were lower in S. rodhaini than all S. mansoni populations but should be interpreted with caution. Eight of the nine S. rodhaini samples are from the same laboratory‐maintained population. F ST between S. rodhaini and individual S. mansoni populations ranged from 0.844 (S. rodhaini vs. S. mansoni Tanzania) to 0.937 (S. rodhaini vs. S. mansoni Senegal).
Figure 2 shows LD decay (binned and smoothed) from pairwise r 2 values. LD decays to r 2 ≤ .2 within 500 kb for all populations. LD was weakest in the Tanzania population with r 2 decaying to .5 in 28 bp. LD decay in the other three populations (Senegal, Niger and Brazil) was relatively consistent with LD decaying by half (r 2 = .5) between 15,150 bp (Niger) and 26,196 bp (Brazil). Both Senegal and Niger show high levels of LD even between SNVs on different chromosomes: this results from lower sample size (Senegal, n = 25; Niger, n = 10) in these two populations.
FIGURE 2.
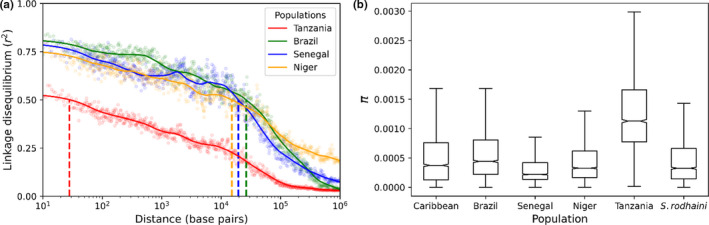
Linkage disequilibrium (LD) decay and diversity within populations—(a) LD between single nucleotide variants was quantified with r 2 values for each population. Mean r 2 values were taken in 500‐bp windows and loess smoothed. Vertical dotted lines indicate the distance where r 2 = .5 for each population. LD decayed to r 2 = .5 in 28 bp (Tanzania), 15,150 bp (Niger), 19,318 bp (Senegal) and 26,196 bp (Brazil). (b) Nucleotide diversity (π) varied between Schistosoma mansoni populations with the highest levels of diversity occurring in East Africa (Tanzania). π was measured in 100‐kb windows across the autosomal chromosomes. Outliers are not shown
3.3. Admixture with S. rodhaini
We asked whether hybridization with S. rodhaini, a closely related schistosome infecting rodents, might contribute to the high genetic diversity observed in East Africa vs. West Africa and South American S. mansoni. To investigate this, we used three statistics (D, D 3 and F 3) to test for admixture between S. mansoni and S. rodhaini, with particular emphasis on the Tanzanian populations of S. mansoni. Each of these statistics attempts to identify the presence of admixture in different ways. D and D 3 values ≠0 indicate admixture with positive and negative values determining the direction of introgression. F 3 values < 0 indicate admixture between the two source populations. None of the three statistics, or any of the population combinations, returned values containing significant signals of admixture (Table 3). These results suggest that hybridization/introgression between S. mansoni and S. rodhaini may make no detectable contribution to elevated diversity in East Africa.
TABLE 3.
Admixture statistics
| Comparison | Test Stat | SE | Z |
|---|---|---|---|
| D (Patterson et al., 2012) | |||
| (((Br, Tz)Sr), Smr) | −0.033 | 0.0404 | −0.807 |
| (((Ni, Tz)Sr), Smr) | 0.006 | 0.0437 | 0.129 |
| (((Se, Tz)Sr), Smr) | 0.051 | 0.0473 | 1.070 |
| F 3 (Patterson et al., 2012) | |||
| Br: Tz, Sr | 0.764 | 0.0207 | 36.910 |
| Ni: Tz, Sr | 0.993 | 0.0299 | 33.214 |
| Se: Tz, Sr | 1.265 | 0.0461 | 27.428 |
| D 3 (Hahn and Hibbens 2019) | |||
| (Br, Tz, Sr) | −0.0047 | 0.00036 | 0.418 |
| (Ni, Tz, Sr) | −0.0046 | 0.00034 | 0.419 |
| (Se, Tz, Sr) | −0.0054 | 0.00048 | 0.353 |
| (Cm, Tz, Sr) | 0.0005 | 0.00066 | −0.022 |
| (Cr, Tz, Sr) | −0.0048 | 0.00029 | 0.517 |
| (Ug, Tz, Sr) | 0.0034 | 0.00062 | −0.174 |
"Z"—Z‐score of the mean from 0.
Population abbreviations: Br, Brazil; Cm, Cameroon; Cr, Caribbean; Ni, Niger; Se, Senegal; Smr, Schistosoma margrebowiei; Sr, Schistosoma rodhaini; Tz, Tanzania; Ug, Uganda.
3.4. Population structure
We examined population structure using PCA and admixture with 38,197 unlinked autosomal SNVs. Two PCAs were generated, with and without the S. rodhaini outgroup (see Figure 4). The two species were differentiated along PC1 (34.7% variance) when S. rodhaini was included (Figure 3a). S. mansoni samples cluster into geographically defined groups when S. rodhaini is excluded (Figure 3b). East African samples were distinct from all other S. mansoni samples, except for one whole genome sample collected in Kenya (see below). Samples from the Americas, including those from the Caribbean and Brazil, showed a closer relationship with Cameroon and Nigerien samples than those from Senegal.
FIGURE 4.
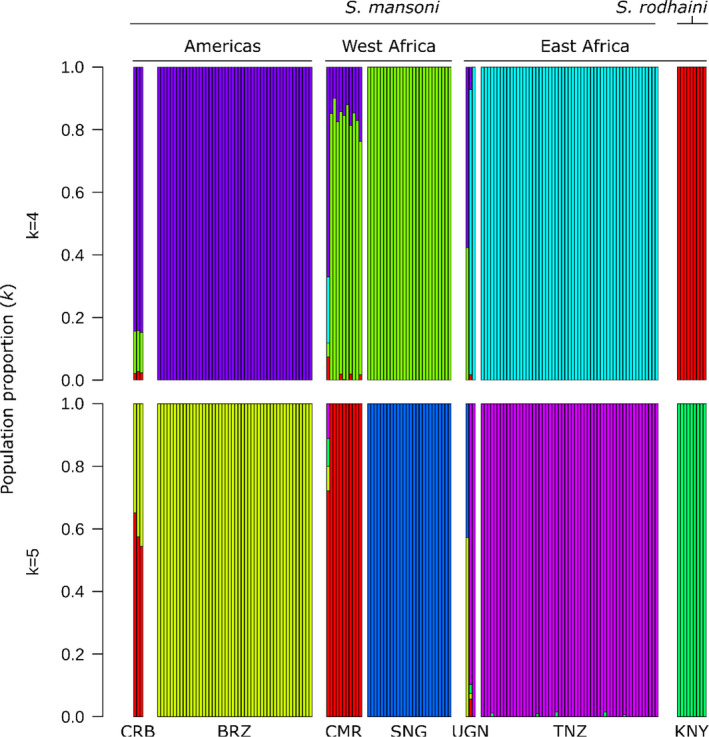
Population structure in Schistosoma mansoni. admixture analyses with k = 4 and k = 5 populations identified clear distinctions between each of the major sampling localities (the Americas, West Africa, East Africa and S. rodhaini). The population components in each of the whole genome samples from Uganda (UGN), Kenya (KNY), Cameroon (CMR) and the Caribbean (CRB) were more heterogeneous than samples with exome data. Cameroonian and Nigerien samples contain moderate proportions of the Brazilian population component. BRN, Burundi; BRZ, Brazil; NGR, Niger; SNG, Senegal; TNZ, Tanzania
FIGURE 3.
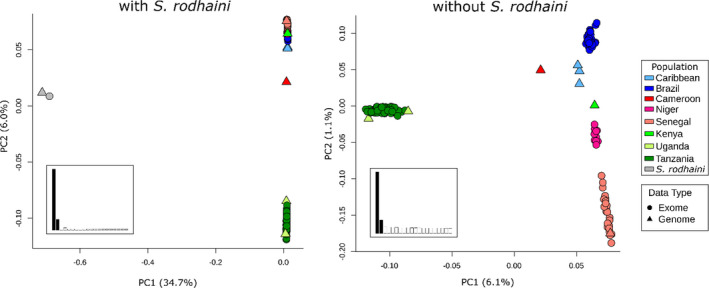
A principal component analysis (PCA) of unlinked autosomal single nucleotide variants. The PCA plot that included Schistosoma mansoni and S. rodhaini (left) clearly shows a large distinction between the two species with some variation within S. mansoni along PC2. A PCA with only S. mansoni (right) differentiates East African S. mansoni along PC1. The remaining S. mansoni samples fall along a continuum on PC2 that goes from samples in West Africa and transitions to the Americas. Inset bar charts represent the percentage variation explained along the first 20 PCs. Only PC1 and PC2 were examined (shaded)
We used admixture to assign individuals to one of k populations, where k is between 1 and 20 (Figure 4). Cross‐validation scores (Evanno et al., 2005) were minimized when k was 4 or 5. Both k = 4 and 5 split S. mansoni samples into geographically defined populations with two major differences. First, k = 4 showed that the allelic component primarily associated with Brazil was found at moderate levels in Cameroon and Nigerien individuals. Second k = 5 split the West African samples into a Senegalese and a Cameroonian + Nigerien population.
As observed in the PCA, the Kenyan, whole‐genome sample contained a large portion of alleles associated with samples from the Americas (~40%–60%) in the admixture analysis. Crellen et al. (2016) recovered similar results and hypothesized that the Kenyan sample may be reflecting human‐trafficking routes between Portuguese and Arab slave traders out of the port of Mombasa. Given that only a single sample is available from this region, and contamination with South American strains during laboratory passage is a possible alternative explanation, we chose to remove the Kenyan sample from downstream analyses.
3.5. Phylogenetics
We used three different phylogenetic methods to investigate the evolutionary relationships between populations (Figure 5). First, we generated a median‐joining network from 815 mitochondrial SNVs of which 477 where phylogenetically informative (Figure 5a). The haplotype network identified three major haplotypes roughly corresponding to the geographical partitioning of the samples. The haplogroups include an East African clade (Tanzania, Uganda), a Senegal group, and an intermediate haplogroup with samples from the Brazilian and Nigerien populations. Caribbean samples were not assigned to a single haplogroup. The single sample from Puerto Rico was associated with the major Brazilian and Nigerien haplotype, and the two Guadalupe samples tended to be more strongly associated with Senegalese haplotypes. Additionally, the sample from Cameroon was only a single step removed from the most common Brazilian haplotype.
FIGURE 5.
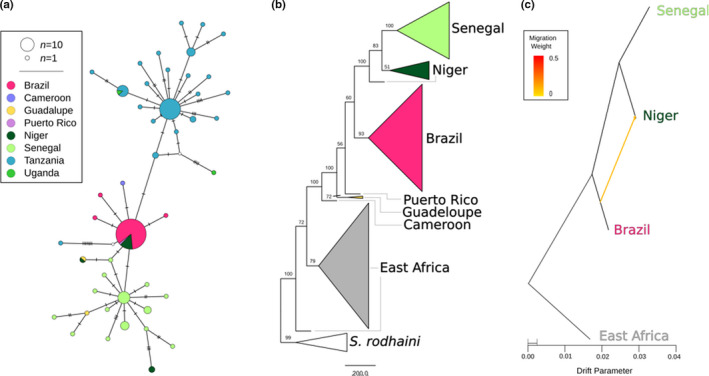
Phylogenetic relationships between Schistosoma mansoni populations. Multiple phylogenetic analyses and marker types were used to discern relationships between S. mansoni populations. (a) A median‐joining haplotype network was constructed from 815 variants across the mitochondria of all S. mansoni samples. (b) A coalescent‐based species tree from 100,819 parsimony‐informative single nucleotide variants with bootstrap values shown on each clade. Monophyletic populations are shown as a collapsed clade except in the case of East Africa which contains samples from Tanzania and Uganda. (c) A maximum‐likelihood phylogenetic network of autosomal variants identified a single, weak migration edge orientated from Brazil to Niger. All three analyses identify a relationship between Senegal, Niger and Brazil that excludes East African samples. The mitochondrial (a) and autosomal (c) networks both allow for direct relationship or allele sharing between Brazil and Niger. The species tree (b) indicates a strong relationship between Senegal and Niger that excludes Brazil (bootstrap percentage = 100)
A coalescent‐based species tree from 100,819 parsimony‐informative SNVs was generated with svd‐quartets (Figure 5b). Quartet sampling was limited to 100,00 quartets which sampled 0.43% of all distinct quartets present in the alignment. The final species tree was consistent with 84.7% of all the quartets sampled. Unlike the mitochondrial tree, samples fall into well‐supported clades corresponding to geography with two exceptions. Samples from East Africa and Niger formed independent paraphyletic clades. In both cases paraphyly was induced by a single individual. Bootstrap support was generally higher in the quartet species tree than in the mitochondrial tree. West African samples formed a well‐supported monophyletic clade with the Brazilian and Caribbean samples, indicating a shared origin for these parasites. Brazilian and Caribbean samples appear to have a polyphyletic origin within a larger clade containing West African parasites.
Finally, migration between populations was quantified with treemix (Figure 5c). We only examined populations with more than five individuals, which excluded Cameroonian and Caribbean samples from the analysis. The topologies linking the remaining populations (East Africa, Niger, Senegal and Brazil) in the treemix and species tree were identical. The species tree was slightly improved with the addition of a single migration edge from Brazil to Niger. This migration edge significantly improved the likelihood score of the topologies from 73.6026 to 73.7165 with an edge weight of 0.1 (p = .0081).
3.6. Selection
We used msprime to generate a set of neutrally evolving SNVs based on parameters specific to each of the sampled populations. These neutrally evolving SNVs were distributed across an 88.9‐Mb chromosome that was equal in size to S. mansoni chromosome 1 (HE601624.2). We then transposed the HE601624.2 exome annotation onto the simulated chromosome to extract “exome” data. This process was repeated 342 times per population to produce a set of neutrally evolving loci to use as controls when examining selection on actual samples. We used these neutral simulations to generate maximum (100%) threshold values for h‐scan and sweepfinder2 test statistics expected under neutrality (see below). The mean (x̅) simulated SNV count across all replicates was: x̅ Brazil =11,198 (range =10,874–11,552), x̅ Niger =8448 (range =8141–8706), x̅ Senegal =11,977 (range =11,642–12,309) and x̅ Tanzania =33,168 (range =32,575–33,741). The number of observed SNVs was between 72.2% and 89.7% of the mean number of simulated SNVs (Brazil =8947; Niger =6107; Senegal =9381; Tanzania =29,765). The reduced number of SNVs in the neutral data is probably due to the absence of selection that is presumed to be acting on the exome data sets.
The h‐scan and sweepfinder2 results are shown in Figure 6. pcadapt results are show in Figure S2. Each program uses a different methodology to detect sites under selection. h‐scan calculates H with homozygous tract length and the number of haplotypes to identify genome regions that have undergone selective sweeps from unphased variants. H values were highly variable for each population, even within windows smaller than 100 kb. In Tanzania only two of the 475,081 SNVs had H values higher than those generated from neutral simulations. sweepfinder2 calculates deviations from a neutral site frequency spectrum correcting for the possibility of background selection. An LR describes the probability of positive selection vs. neutral evolution and background selection within a designated window. sweepfinder2 was able to clearly define multiple peaks for each population when compared to h‐scan. Each of the four populations had regions greater than neutral expectations. pcadapt identifies SNVs significantly associated with population differentiation. In this analysis we found 442 SNV outliers, after multiple test correction, that were distributed across all seven autosomes at 127 loci. These regions were distributed across 280.2 Mb. F ST of the pcadapt outliers (F ST =0.544) was significantly higher than in the remaining population outliers (F ST =0.195).
FIGURE 6.
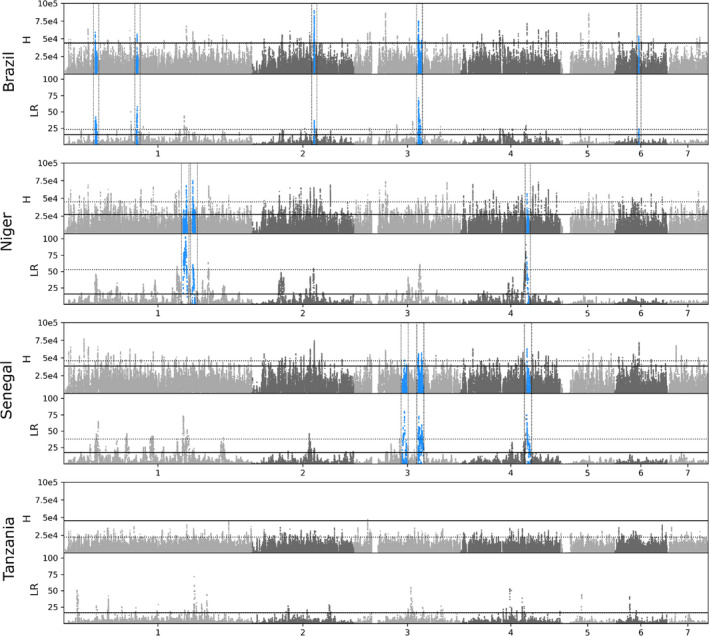
Positive selection across the Schistosoma mansoni genome. Selection across the S. mansoni genome was calculated with haplotype (H)‐ and allele frequency (likelihood ration; LR)‐based methods. Dotted lines represent positions with an H or LR value in the 99th percentile. The solid black line represents the maximum H or LR calculated from simulated data under neutral conditions. Regions of interest (blue, boxed) were identified by finding sites where the H and LR values were in the >99th percentile and were both greater than the maximum H or LR from simulated data. Once these sites were identified we combined variants within 333,333‐bp windows that showed signs of selection: an H or LR greater than the simulated threshold
We defined “putative regions of selection” as those that have most likely experienced positive selection. These regions contain variants (i) with both H and LR values in the 99th percentile, (ii) are greater than the neutral thresholds and (iii) have a signal of population‐specific directional selection. All SNVs meeting one or more of these criteria are listed in Table S4.
Our results recovered five, three and three putative selected regions in Brazil, Niger and Senegal respectively (Figure 6; Table S5). Information regarding the number of regions, SNVs and genes identified are presented in Tables 4 and 5. π (Figure S3) and Tajima's D (Figure S4) were depressed in these regions compared to genome‐wide values (Table S3), which is consistent with loci experiencing selection. On average the size of each region was relatively small (1,395,643 bp) and in two instances these sites were shared between populations. The Brazilian and Senegalese populations shared a site on chromosome 3 at HE601626.2:30,092,830:31,936,551, while the Nigerien and Senegalese populations shared a peak on chromosome 4 at HE601627.2:31,216,154:32,138,352. We did not recover any regions or sites of interest in the Tanzanian population in large part because only two of 475,081 sites had higher H values the largest H from neutral simulations (H = 45,532.9). These variants are on chromosome 3 at HE601626.2:6,500,422 and HE601626.2:6,503,809 and are adjacent to each other in our filtered SNV data set.
TABLE 4.
Number of SNPs and regions identified in genome‐wide scans for selection
| Brazil | Niger | Senegal | Tanzania | |
|---|---|---|---|---|
| Outlier SNVs (h‐scan) | 4366 | 44,024 | 9182 | 2 |
| Outlier SNVs (sweepfinder) | 12,871 | 41,525 | 23,379 | 4288 |
| Merged regions (1/3 Mb) | 113 | 250 | 168 | 46 |
| 99th Percentile SNPs | 703 | 239 | 176 | 0 |
| Outlier SNPs (pcadapt) a | 442 | 442 | 442 | Na |
| Putative regions under selection | 5 | 3 | 3 | 0 |
| Genes in selected regions | 116 | 112 | 157 | 0 |
| Genes w/99th percentile SNPs | 10 | 5 | 7 | 0 |
Abbreviations: SNP, single nucleotide polymorphism; SNV, single nucleotide variant.
Outlier SNPs are those that are greater than the maximum value derived from neutrally simulated data.
TABLE 5.
Genes in Schistosoma mansoni populations with the strongest signals of directional selection
| Gene ID | UniProtKB accession | UniProtKB description | HHsearch annotation |
|---|---|---|---|
| Brazil | |||
| Smp_060090 | G4VAJ9 | 40S ribosomal protein S12 | Ribosomal protein S12e ribosome |
| Smp_073680 | G4V701 | Putative TATA‐box binding protein | DNA‐directed RNA polymerase II subunit |
| Smp_123510 | A0A5K4EKW3 | Vacuolar protein sorting‐associated protein 16 homolog | Vps16_C |
| Smp_123520 a | A0A3Q0KKK4 | Putative RNA M5U methyltransferase | rRNA (Uracil‐5‐)‐methyltransferase RumA |
| Smp_123570 | A0A3Q0KKM5 | BHLH domain‐containing protein | Aryl hydrocarbon receptor nuclear translocator |
| Smp_123590 | A0A5K4EL22 | Uncharacterized protein | Swi5‐dependent recombination DNA repair protein |
| Smp_148460 | A0A3Q0KPP3 | Putative neurofibromin | GAP‐related domain of neurofibromin |
| Smp_162000 | A0A5K4ETP0 | UBR‐type domain‐containing protein | E3_UbLigase_R4 |
| Smp_246630 | A0A5K4F427 | UBC core domain‐containing protein | E2 Ubiquitin conjugating enzyme |
| Smp_341570 | A0A5K4FC62 | Uncharacterized protein | Uncharacterized protein family UPF0183 |
| Niger | |||
| Smp_008230 | G4V7H8 | Putative rab‐18 | di‐Ras2 |
| Smp_126620 | A0A3Q0KL42 | Uncharacterized protein | Ligand‐binding domain of low‐density lipoprotein receptor |
| Smp_165060 | A0A3Q0KRY3 | Uncharacterized protein | Uncharacterized protein |
| Smp_167890 b | Q6BC90 | Peptide‐methionine (R)‐S‐oxide reductase | C‐terminal MsrB domain of methionine sulphoxide reductase PilB |
| Smp_313490 b | A0A5K4F3H5 | Uncharacterized protein | Transforming protein RhoA, Rho‐associated, coiled‐coil |
| Senegal | |||
| Smp_070780 | G4VEM1 | UDP‐glucose 4‐epimerase | Uridine diphosphogalactose‐4‐epimerase |
| Smp_123440 | A0A3Q0KKL2 | Putative fad oxidoreductase | d‐amino‐acid oxidase |
| Smp_123520 a | A0A3Q0KKK4 | Putative RNA M5U methyltransferase | rRNA (Uracil‐5‐)‐methyltransferase RumA |
| Smp_164560 | A0A3Q0KRR1 | Uncharacterized protein | Na |
| Smp_167890 b | Q6BC90 | Peptide‐methionine (R)‐S‐oxide reductase | C‐terminal MsrB domain of methionine sulfoxide reductase PilB |
| Smp_213150 | A0A5K4EZI5 | Uncharacterized protein | Ribonucleases P/MRP protein subunit POP1 |
| Smp_313490 b | A0A5K4F3H5 | Uncharacterized protein | Transforming protein RhoA, Rho‐associated, coiled‐coil |
UniProtKB descriptions are from release 2020_06. HHsearch annotations are from Le Clec'h et al. (2021).
Shared between Brazil and Senegal.
Shared between Niger and Senegal.
We identified 116–157 genes within “putative selected regions” in the Brazilian, Nigerien and Senegalese populations (Table S6). Within these populations, 10, five and seven genes contain SNVs meeting the 99th percentile. Several genes identified in these regions were shared between populations. Brazil and Senegal shared 48 genes in target regions, and Senegal and Niger shared 22 genes in target regions. Three genes with 99th percentile SNVs were shared between populations: Smp_313490 (Uncharacterized protein) and Smp_167890 (Peptide‐methionine (R)‐S‐oxide reductase; Niger and Senegal), Smp_123520 (Putative RNA M5U methyltransferase; Brazil and Senegal).
4. DISCUSSION
We examined the impact of human‐mediated dispersal of Schistosoma mansoni during the Trans‐Atlantic slave trade. Previous analyses with S. mansoni used mitochondrial data or had limited sampling from the Americas (Crellen et al., 2016; Morgan et al., 2005; Webster et al., 2013). To build upon these previous studies, we included 135 exome sequences available from natural populations in Brazil (n = 45), Niger (n = 10), Senegal (n = 25) and Tanzania (n = 55) which we combined with the existing genome sequences from Cameroon (n = 1), the Caribbean (n = 4), Senegal (n = 1) and Uganda (n = 2) (Berriman et al., 2009; Chevalier et al., 2016; Crellen et al., 2016). We used these data to explore the S. mansoni expansion across Africa, examine parasite colonization of the Americas, quantify signatures of selection during colonization and detect hybridization with Schistosoma rodhaini, a closely related parasite utilizing a rodent host.
4.1. Elevated East African diversity and S. mansoni expansion across Africa
A striking result from this study is the dramatic reduction in genetic diversity between East and West Africa. Sequence summary statistics indicate that the East African population has two‐ to three‐fold greater nucleotide diversity (π), larger N e and greater mitochondrial diversity than the other populations (Table 1; Figure 5a). Phylogenetic analyses rooted with S. rodhaini clearly indicate that the East African S. mansoni samples from Tanzania and Uganda are sister to a clade containing all other S. mansoni, including those from the Caribbean, Brazil, Senegal, Niger and Cameroon. Our data support previous work from whole genomes, mitochondrial genes and microsatellites suggesting that S. mansoni emerged in East Africa (Crellen et al., 2016; Morgan et al., 2005; Webster et al., 2013). In addition, our estimates of N e are within a range book‐ended by other whole genome studies (Berger et al., 2021; Crellen et al., 2016).
It is possible that drug treatment differences between localities have an unseen impact on these results. Mass drug administration (MDA) has been shown to reduce, and in some cases even eliminate, transmission of schistosomes. However, it is not uncommon for schistosomes to persist in populations even in the midst of significant MDA efforts. In these areas, MDA has minimal impacts on the genetic diversity of schistosome populations (Berger et al., 2021; Faust et al., 2019; Gower et al., 2017; Huyse et al., 2013; Lelo et al., 2014).
The rapid decay in LD observed in East Africa compared with West African and American populations provides further evidence that East African S. mansoni populations are ancestral. Similar reductions in the rate of LD decay have been observed in humans and malaria parasites, outside of their ancestral Africa range (Anderson et al., 2000; Gurdasani et al., 2015; Neafsey et al., 2008). Rapid breakdown in LD also has important practical applications for genome‐wide association analyses (GWAS), because it allows mapping of phenotypic traits to very narrow regions of the genome (Mackay & Huang, 2018). There are multiple biomedically important traits of interest that vary in S. mansoni populations, including drug susceptibility or resistance, host specificity and cercarial production (Anderson et al., 2018). We recently used GWAS for mapping resistance to the first‐line drug (Praziquantel) in laboratory schistosome populations (Le Clec’h et al., 2021). Most laboratory S. mansoni populations tested were from South America, where LD decays relatively slowly: these are not ideal for GWAS. Establishment of laboratory S. mansoni populations from East Africa, or GWAS analyses using parasites directly from the field would be valuable for future GWAS with S. mansoni.
There is minimal allele sharing or migration between East African and other S. mansoni populations. F ST comparisons that include Tanzania are greater than other comparisons (with Tanzania F ST = 0.355; excluding Tanzania F ST = 0.206). East African populations are among the most strongly differentiated populations in the PCAs (Figure 3b) and the East African population component in admixture analyses is absent, or at minimal levels, in other S. mansoni populations. Finally, mitochondrial haplotypes in Uganda and Tanzania form a distinct haplogroup from other S. mansoni populations. These data indicate that while East Africa is the probable origin of S. mansoni, migration or allele sharing between East Africa and other populations is restricted.
We do not expect that human movement is a major barrier between East and West African schistosome populations. However, differences in snail–schistosome compatibility in East and West Africa may provide barriers to gene flow. This is seen in multiple host–parasite systems, including Daphnia–microsporidia (Ebert, 1994), and trematode infections of snails (Lively, 1989) and minnows (Ballabeni & Ward, 1993). Strong host‐specificity exists within the Biomphalaria and S. mansoni system (Mitta et al., 2017; Theron et al., 2014; Webster & Woolhouse, 1998) and a review shows that compatibility is greater between sympatric Biomphalaria–S. mansoni combinations (Morand et al., 1996). Sympatric schistosome–snail combinations result in rapid immune suppression and rapid parasite development, while allopatric schistosome–snail combinations result in a slower immune cell proliferation and a nonspecific generalized immune response which reduced parasite growth and establishment (Portet et al., 2019). There is a developing understanding of Biomphalaria phylogenetics (Jorgenson et al., 2007), phylogeography (Dejong et al., 2003) and compatibility relationships among East African snail species (B sudanica, B. pffeiferi and B. choanomphala) and S. mansoni (Mutuku et al., 2017, 2021), but further research is needed to understand the compatibility of allopatric snail–schistosome combinations from East and West Africa. We suggest that the presence of fine‐scale geographical structure of Biomphalaria populations (Webster et al., 2001) and local adaptation in sympatric Biomphalaria–schistosome combinations may limit parasite geneflow between East and West Africa.
PCAs differentiate S. mansoni populations on an East‐to‐West gradient along PC2 (Figure 3b). Mitochondrial haplotypes in Senegal and Cameroon are intermediate to those in Tanzania and Senegal (Figure 5a). The species tree from autosomal SNV data (Figure 5a) indicates that S. mansoni is in a series of nested, well‐supported clades from East Africa (Tanzania+Uganda), to Cameroon and to West Africa (Niger+Senegal). F ST and r (Mantel) values between Tanzania, Niger and Senegal reflect increasing isolation with distance across Africa. These observations combined with the origination of S. mansoni in East Africa confirms an East‐to‐West, stepwise expansion of S. mansoni from Tanzania and Uganda → Cameroon → Niger → Senegal (Crellen et al., 2016; Morgan et al., 2005; Webster et al., 2013).
4.2. Does hybridization between S. rodhaini and S. mansoni contribute to elevated East African diversity
Several closely related Schistosoma species are able to hybridize and produce viable offspring, as confirmed via experimental rodent infections. The potential for hybridization between animal and human Schistosoma species is a significant public health concern (Borlase et al., 2021; Leger & Webster, 2017; Léger et al., 2020; Stothard et al., 2020). Our group, and others, have recently shown that ancient hybridization and adaptive introgression has resulted in the transfer of genes from the livestock species Schistosoma bovis into S. haematobium: West African S. haematobium genomes contain 3%–8% introgressed S. bovis sequences and S. bovis alleles have reached a high frequency in some genome regions (Platt et al., 2019; Rey, Toulza, et al., 2021). The sister species of S. mansoni, S. rodhaini, parasitizes rodents and is primarily located in eastern Africa (Rey, Webster, et al., 2021). S. mansoni and S. rodhaini have been shown to readily hybridize in and produce fertile offspring in the laboratory (Théron, 1989). Natural hybrids have been reported in Kenya and Tanzania (Morgan et al., 2003; Steinauer, Hanelt, et al., 2008), although hybrids have only been detected from their snail intermediate host and never encountered in the mammalian hosts, humans and rodents (Rey, Webster, et al., 2021). We were unable to find evidence of hybridization in 55 samples collected from Tanzania. Both species are clearly separated in genotypic space with differences between the species accounting for the largest component in the PCA (Figure 3a; PC1 = 34.7% variation), and F ST between S. rodhaini and S. mansoni populations is very high (F ST = 0.912). admixture analyses also clearly differentiated S. rodhaini from all other S. mansoni populations (Figure 4) and we were unable to identify admixture signal between S. rodhaini and the Tanzanian population with genome‐wide statistics including D, D 3 and F 3 (Table 3).
Hybridization between these two species is thought to be rare (≤7.2%) (Morgan et al., 2003; Rey, Webster, et al., 2021; Steinauer, Hanelt, et al., 2008; Steinauer, Mwangi, et al., 2008). Our sample size may not be large enough to identify rare hybrids. Furthermore, we analysed exome (coding) data, which may underrepresent introgressed alleles if they are selected against. Finally, the S. rodhaini samples are primarily from a single laboratory population that may not be representative of natural populations. These caveats aside, our analyses clearly failed to identify recent hybridization between S. mansoni and S. rodhaini. We conclude that S. rodhaini introgression does not contribute to the high genetic diversity in our Tanzanian S. mansoni samples.
4.3. Expansion into the Americas
Previous work has shown that S. mansoni was exported from Africa to the Americas during the Trans‐Atlantic slave trade (Crellen et al., 2016; Desprès et al., 1993; Files, 1951; Fletcher et al., 1981; Morgan et al., 2005; Webster et al., 2013). Here, we use genomic data to investigate the probable source population(s), number of introductions, evidence for bottlenecks and parasite adaptation during colonization.
4.3.1. Source populations
Of the two West African populations sampled (Niger and Senegal), our results support stronger relationships between Brazil and Niger, than with Senegal. While the species tree (Figure 5b) appears to rule out Niger or Senegal as the direct source population for Brazilian S. mansoni, there is evidence of allele sharing between the Nigerian and Brazil populations. First, the dominant mitochondrial DNA (mtDNA) haplotype in Brazilian and Nigerien samples is shared (Figure 5a). Second, every Nigerien population contains at least 10.1% of the Brazilian component, as shown in the admixture analyses (mean 15.8%; Figure 4). Third, Niger and Brazil are more closely associated with each other along a genotypic continuum, represented by PC2, than Brazil is to other African populations (Figure 3b). Finally, treemix identified a single weak migration edge between Brazil and Niger (Figure 5c), confirming a relationship between these two populations.
A simple hypothesis from the data is that, assuming a general east‐to‐west expansion holds at finer geographical scales, the source population that was eventually exported to Brazil is probably located somewhere between Benin and Angola. These countries fell within the Bight of Benin, Bight of Biafra and West Central Africa slave trading regions (Figure 7). Our Brazilian samples were collected in Ponto dos Volantes in Minas Gerais, Brazil. This location is relatively equidistant from major slave ports in Bahia (527 km) and around Rio de Janeiro (706 km). In all, more than 3.5 million slaves were transported to Brazil (Table S7; Slave Voyages Database, 2009). Of all slaves taken to Brazil, 82% disembarked at ports in either Bahia (1.3 million) or Rio de Janerio (1.5 million). In Africa, slave‐exporting markets in the Bights of Benin and Biafra and West Central Africa were responsible for 53.6%, 5.0% and 33.8% of people exported to Bahia and 1.3%, 1.1% and 74.9% of those exported to southeast Brazilian ports (Slave Voyages Database, 2009). Taken together these data imply that the Brazilian population we sampled in Ponto dos Volantes probably originated from markets in the Bight of Benin or West Central Africa. In our phylogenetic analyses, the Brazilian population falls within a clade containing samples from Senegal, Niger and the Caribbean but excludes the single Cameroonian sample. If the Cameroonian sample is representative of the Cameroonian population, then it may be that Cameroon and Niger represent the eastern and western limits of the unrepresented source population. This area more closely aligns with the Bight of Benin, a region containing parts of Nigeria and Benin. Additional samples from these regions are needed to test this hypothesis. It is also important to note that the Brazilian samples here represent a single geographical location (Ponto dos Volantes) and that the source for this population may not extrapolate to larger regions, or even to other locations in Brazil.
FIGURE 7.
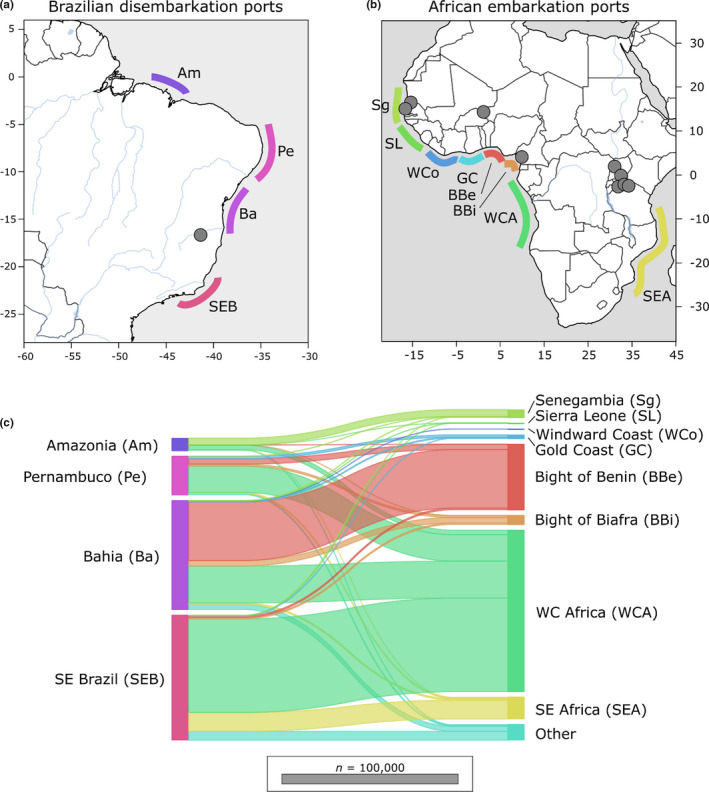
Export and importation of slaves between Africa and Brazil. During the Trans‐Atlantic slave trade, 12 million people were trafficked from Africa. More than 3.5 million were exported into Brazil at major ports along the east coast. Most slaves originated from regions along the West African coast from Benin to Angola. General locations of major embarkation and disembarkation ports are shown along the (a) Brazilian and (b) Africa coastlines. (c) The movement of people between Brazilian and African ports is shows with horizontal lines. Line width is proportional to the number of migrants. The Brazilian samples in this study were collected from Ponto dos Volantes in Minas Gerais, Brazil, between the Ports of Bahaia and South East Brazil. The primary embarkation points for Bahaia and South East Brazil were in West Central Africa and the Bight of Benin. These regions are a probable source for the S. mansoni collected in Ponto dos Volantes. Collection localities for S. mansoni are represented by grey dots. Slave exportation data are from the Slave Voyages Database (2009)
4.3.2. No evidence for population bottlenecks during colonization
Previous mtDNA analyses have shown reduced diversity in South American parasites (Desprès et al., 1993; Fletcher et al., 1981; Morgan et al., 2005; Webster et al., 2013), suggesting bottlenecks during colonization. Our data are consistent with this, showing a two‐ to three‐fold reduction in mtDNA in Brazil compared to West African parasites. However, genome‐wide summary statistics of autosomal sequence data tell a different story, and provide no evidence for population bottlenecks associated with S. mansoni introduction and establishment in Ponto dos Volantes, Brazil (Table 1). Nucleotide diversity, measured by π, was higher in Brazil than either of the West African populations (Niger or Senegal), perhaps because the Brazilian population is derived from multiple West African source populations. The Brazilian N e was roughly the same as for Niger and comparable to Senegal. Mean Tajima's D calculated from genome‐wide SNV data is negative in each of the African populations and highest in the Caribbean samples (mean Tajima's D = 0.929; Table S2). Tajima's D values in our study using exome SNV data from different regions of Niger, Senegal and Tanzania were comparable with values calculated using whole genome SNV data from Ugandan S. mansoni (Berger et al., 2021). By contrast, mean Tajima's D in Brazilian samples is close to 0 (mean Tajima's D = 0.034; 95% CI [−0.18 to −0.085]) and is significantly greater than mean Tajima's D in the African samples (t test p < .001; Table S2).
The nuclear genomic data clearly suggest that the establishment of S. mansoni in Brazil was not associated with a significant population bottleneck and had minimal impacts on genome‐wide levels of genetic diversity. The discrepancy between mtDNA and nuclear DNA may stem from two sources. First, mtDNA has an effective population size one‐quarter that of nuclear genes (Birky et al., 1983), and can potentially provide a more sensitive indicator of bottlenecks. Second, and perhaps more critical, mtDNA constitutes a single marker, so may poorly reflect population history (Anderson, 2001). Extensive laboratory passage may also result in bias in population summary statistics. For example, the Caribbean samples examined have undergone 2–15 generations of laboratory passage, which is probably responsible for the elevated Tajima's D in this population (Crellen et al., 2016).
4.3.3. Number of introductions
All Brazilian and Caribbean samples are paraphyletic and fall between the Cameroonian sample and West African clade in Figure 5b. The relationships among these samples are resolved but not supported outside of a monophyletic clade containing the Brazilian samples. As a result, the autosomal phylogeny by itself does not conclusively support one or multiple introductions into the Americas. The two Caribbean samples from Guadeloupe island contain unique mitochondrial haplotypes absent from Brazil (Figure 5a); however, this is, at best, only weak evidence for independent introductions into Brazil and the Caribbean from the data we have available. The higher autosomal diversity of Brazilian S. mansoni compared with two sampled West African populations provides additional indirect evidence for multiple origins.
Plasmodium falciparum and W. bancrofti became established in the Americas at the same time as S. mansoni, and without apparent bottlenecks (Small et al., 2019; Yalcindag et al., 2012). In both cases it is hypothesized that high levels of diversity were maintained by recurring introductions from multiple sources (Rodrigues et al., 2018; Small et al., 2019; Yalcindag et al., 2012). More than 3.5 million slaves were transported to Brazil from across the continent of Africa (Table S7; Slave Voyages Database, 2009). S. mansoni prevalence varies widely between sites in western and central Africa, with estimates ranging from 0.3% (Gambia; Sanneh et al., 2017) to 89% (Democratic Republic of the Congo; Kabongo et al., 2018). Assuming S. mansoni prevalence during the period of the Atlantic Slave Trade was comparable to current levels and that infected people contained multiple S. mansoni individuals (genotypes; Van den Broeck et al., 2014), it is plausible that hundreds of thousands, and more probably millions, of reproductively viable S. mansoni were introduced into Brazil from across Africa. As a result, individual genotypes that may have been separated by thousands of miles in Africa were brought into close contact in the Americas and would lead to higher levels of diversity there than in any single population in Africa. The lack of bottlenecks during establishment of S. mansoni in the New World is consistent with compatibility between West African S. mansoni and South American Biompahalaria species. We note that this scenario can be directly tested. It is possible to experimentally explore this biological invasion using experimental infection of laboratory‐maintained South American snail populations with miracidia derived from West Africa.
4.3.4. Adaptation during colonization
We hypothesized that S. mansoni introduced into the Americas would have been exposed to novel selective pressures as they adapted to new biotic and abiotic challenges. For example, the S. mansoni life cycle requires an intermediate snail host in which miracidia mature into cercariae that are capable of infecting humans. Previous work has shown the snail immune response is greater when exposed to sympatric parasite strains (Portet et al., 2019) and, in general, sympatric host/parasite combinations to be more compatible (Morand et al., 1996). In Africa, S. mansoni use Biomphalaria alexandrina, B. camerunensis, B. choanomphala, B. pfeifferi, B. stanleyi and/or B. sudanica as the snail host, none of which are present in the Americas (Figure 1). Instead, these parasites have adapted to using different Biomphalaria hosts, including B. glabrata, B. straminea and B. tenagophila (Hailegebriel et al., 2020; Kengne‐Fokam et al., 2018; Vidigal et al., 2000). We examined exomic SNV data to identify genes and larger regions of the genome under selection at a finer scale and identified zero to five putative regions of selection from each of the major populations (Table 4).
In the Brazilian samples, we identified five putative selected regions that contain 126 genes (Table S6). π and Tajima's D were significantly reduced in these five regions compared to genome‐wide averages, which is expected if these loci are, or have been, under selection (Table S3). One region is shared between the Brazilian and Senegalese populations. Forty‐six genes fall within this Senegal–Brazil overlapping region, leaving 80 genes and four loci that are probably experiencing population‐specific positive selection. Even within the group of 80 genes, there are nine with strong signals of selection (Table 5). These genes contained variants with H and LR values in the 99th percentile in addition to being greater than the threshold defined by neutral simulations. Several genes within this group are associated with housekeeping functions, including transcription and protein degradation (Smp_060090, 40S ribosomal protein S12; Smp_162000, UBR‐type domain‐containing protein; Smp_246630, UBC core domain‐containing protein). Two uncharacterized proteins were identified (Smp_341570 andSmp_123590) but we are not able to speculate on their function. Of particular interest are two possible transcription factors, an uncharacterized protein containing a helix, loop, helix domain (Smp_123570, BHLH domain‐containing protein) and a putative TATA‐box binding protein (Smp_073680, Putative TATA‐box binding protein). It is possible that adaptation to the Brazilian environment was driven by changes in gene expression, but more work is needed to understand the potential role of these loci in adaptation to the Americas.
4.3.5. Selection on African S. mansoni
We examined selection on African S. mansoni as part of the process to identify unique signals of selection in the Brazilian population. We identified 112 and 157 genes under selection in three regions each for the Nigerien and Senegalese populations (Table 4). One of the three regions, and 22 genes, was shared between Niger and Senegal. We failed to identify any regions of selection in Tanzania using our combined criteria, but results from individual tests of selection (h‐scan, sweepfinder2) did overlap at nine of 25 regions (Table S8) identified in a large Ugandan population (Berger et al., 2021). These regions were identified using a variety of within‐ (iHS) and between‐population (F ST, XP‐EHH) tests on miracidia isolated from two Ugandan locations with differing histories of praziquantel treatment.
5. CONCLUSIONS
Our analyses confirmed an east‐to‐west expansion of Schistosoma mansoni across Africa. Sometime during this expansion one or more Central African population(s), probably located between Angola and Benin, were transported to Brazil. Genome‐wide signatures of diversity, measures of allele frequencies (Tajima's D) and estimates of N e are comparable between S. mansoni in Brazil, Niger and Senegal and do not imply the presence of a bottleneck during the establishment of S. mansoni in Brazil. We did find five genome regions under selection in Brazil, four of which are population‐specific. In total, 80 genes fall within these regions and may be associated with S. mansoni's adaptation to novel selection pressures associated with the Americas. We identified nine genes with the strongest signals of selection that are candidates for future experimental work.
CONFLICT OF INTEREST
The authors declare no competing interests.
AUTHOR CONTRIBUTIONS
Study design: F.D.C., T.J.A. and W.L.C; formal analysis: F.D.C., R.N.P. and T.J.A.; writing, original draft: R.N.P. and T.J.A.; reviewing and editing: A.G., A.E., B.W., D.R., F.D.C., G.O., J.P.W., M.M, P.T.L., R.R.dA., R.N.P., S.K., T.J.A. and W.L.C.; investigationF.D.C., M.M. and W.L.C.; resources were provided by F.D.C. and T.J.A.
OPEN RESEARCH BADGES
This article has earned an Open Data Badge for making publicly available the digitally‐shareable data necessary to reproduce the reported results. The data is available at https://doi.org/10.5061/dryad.dv41ns209.
Supporting information
Fig S1
Table S1
ACKNOWLEDGMENTS
Sandra Smith and John M. Heaner (Texas Biomedical Research Institute) provided computational research support. Matthew Berriman provided the most recent S. mansoni genome sequence and annotation prior to publication. This research was funded by the National Institute of Allergy and Infectious Diseases (NIAD R01 AI097576‐01 and NIAD 5R21AI096277‐01), Texas Biomedical Research Institute Forum (award 0467). Sample acquisition was supported by funding from the Wellcome Trust (104958/Z/14/Z) for the SCAN project, the EU grant FP6 STREP contract no. 032203 for the CONTRAST project and the Gates Foundation coordinated by the University of Georgia Research Foundation Inc. (RR374‐053/5054146 and RR374‐053/4785426) for the SCORE project.
Platt, R. N. II , Le Clec'h, W. , Chevalier, F. D. , McDew‐White, M. , LoVerde, P. T. , Ramiro de Assis, R. , Oliveira, G. , Kinung'hi, S. , Djirmay, A. G. , Steinauer, M. L. , Gouvras, A. , Rabone, M. , Allan, F. , Webster, B. L. , Webster, J. P. , Emery, A. M. , Rollinson, D. , & Anderson, T. J. C. (2022). Genomic analysis of a parasite invasion: Colonization of the Americas by the blood fluke Schistosoma mansoni . Molecular Ecology, 31, 2242–2263. 10.1111/mec.16395
Roy N. Platt II, Winka Le Clec'h, and Frédéric D. Chevalier joint first authors.
Handling Editor: Sebastien Calvignac‐Spencer
DATA AVAILABILITY STATEMENT
Data used in this paper were previously published (Berriman et al., 2009; Chevalier et al., 2019; Crellen et al., 2016; International Helminth Genomes Consortium, 2019; Le Clec'h et al., 2021) under multiple NCBI BioProject (PRJNA439266, PRJNA560070, PRJEB522, PRJEB526, PRJNA743359 and PRJNA773498) and NCBI Short Read Archive (ERR046038, ERR103049, ERR103050, ERR119614, ERR119615, ERX284221, ERR310938, ERR539846, ERR539847, ERR539848, and ERR9974) accessions.
Code availability: Scripts, notebooks, and environmental yaml files are available at https://github.com/nealplatt/sch_man_nwinvasion/releases/tag/v0.2 (last accessed October 21, 2021) or https://doi.org/10.5281/zenodo.5590460 (last accessed October 21, 2021).
REFERENCES
- Alexander, D. H. , Novembre, J. , & Lange, K. (2009). Fast model‐based estimation of ancestry in unrelated individuals. Genome Research, 19(9), 1655–1664. 10.1101/gr.094052.109 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Anderson, T. (2001). The dangers of using single locus markers in parasite epidemiology: Ascaris as a case study. Trends in Parasitology, 17(4), 183–188. 10.1016/s1471-4922(00)01944-9 [DOI] [PubMed] [Google Scholar]
- Anderson, T. , & Enabulele, E. E. (2021). Schistosoma mansoni . Trends in Parasitology, 37(2), 176–177. 10.1016/j.pt.2020.06.003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Anderson, T. J. C. , Haubold, B. , Williams, J. T. , Estrada‐Franco§, J. G. , Richardson, L. , Mollinedo, R. , Bockarie, M. , Mokili, J. , Mharakurwa, S. , French, N. , Whitworth, J. , Velez, I. D. , Brockman, A. H. , Nosten, F. , Ferreira, M. U. , & Day, K. P. (2000). Microsatellite markers reveal a spectrum of population structures in the malaria parasite Plasmodium falciparum . Molecular Biology and Evolution, 17(10), 1467–1482. 10.1093/oxfordjournals.molbev.a026247 [DOI] [PubMed] [Google Scholar]
- Anderson, T. , LoVerde, P. T. , Le Clec'h, W. , & Chevalier, F. D. (2018). Genetic crosses and linkage mapping in schistosome parasites. Trends in Parasitology, 34(11), 982–996. 10.1016/j.pt.2018.08.001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ballabeni, P. , & Ward, P. I. (1993). Local Adaptation of the Tremadote Diplostomum phoxini to the European Minnow Phoxinus phoxinus, its Second Intermediate Host. Functional Ecology, 7(1), 84–90. 10.2307/2389870 [DOI] [Google Scholar]
- Bergad, L. W. (2007). The comparative histories of slavery in Brazil, Cuba, and the United States. Cambridge University Press. [Google Scholar]
- Berger, D. J. , Crellen, T. , Lamberton, P. H. L. , Allan, F. , Tracey, A. , Noonan, J. D. , Kabatereine, N. B. , Tukahebwa, E. M. , Adriko, M. , Holroyd, N. , Webster, J. P. , Berriman, M. , & Cotton, J. A. (2021). Whole‐genome sequencing of Schistosoma mansoni reveals extensive diversity with limited selection despite mass drug administration. Nature Communications, 12(1), 1–14. 10.1038/s41467-021-24958-0 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Berriman, M. , Haas, B. J. , LoVerde, P. T. , Wilson, R. A. , Dillon, G. P. , Cerqueira, G. C. , Mashiyama, S. T. , Al‐Lazikani, B. , Andrade, L. F. , Ashton, P. D. , Aslett, M. A. , Bartholomeu, D. C. , Blandin, G. , Caffrey, C. R. , Coghlan, A. , Coulson, R. , Day, T. A. , Delcher, A. , DeMarco, R. , … El‐Sayed, N. M. (2009). The genome of the blood fluke Schistosoma mansoni. Nature, 460(7253), 352–358. 10.1038/nature08160 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Birky, C. W. Jr , Maruyama, T. , & Fuerst, P. (1983). An approach to population and evolutionary genetic theory for genes in mitochondria and chloroplasts, and some results. Genetics, 103(3), 513–527. 10.1093/genetics/103.3.513 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bolger, A. M. , Lohse, M. , & Usadel, B. (2014). Trimmomatic: A flexible trimmer for Illumina sequence data. Bioinformatics, 30(15), 2114–2120. 10.1093/bioinformatics/btu170 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Borlase, A. , Rudge, J. W. , Léger, E. , Diouf, N. D. , Fall, C. B. , Diop, S. D. , Catalano, S. , Sène, M. , & Webster, J. P. (2021). Spillover, hybridization, and persistence in schistosome transmission dynamics at the human‐animal interface. Proceedings of the National Academy of Sciences of the United States of America, 118(41), 10.1073/pnas.2110711118 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bryant, J. E. , Holmes, E. C. , & Barrett, A. D. J. P. P. (2007). Out of Africa: A molecular perspective on the introduction of yellow fever virus into the Americas. PLoS Path, 3(5), e75. 10.1371/journal.ppat.0030075 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chevalier, F. D. , Le Clec’h, W. , Eng, N. , Rugel, A. R. , Assis, R. R. D. , Oliveira, G. , Holloway, S. P. , Cao, X. , Hart, P. J. , LoVerde, P. T. , & Anderson, T. J. C. (2016). Independent origins of loss‐of‐function mutations conferring oxamniquine resistance in a Brazilian schistosome population. International Journal for Parasitology, 46(7), 417–424. 10.1016/j.ijpara.2016.03.006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chevalier, F. D. , Le Clec’h, W. , McDew‐White, M. , Menon, V. , Guzman, M. A. , Holloway, S. P. , Cao, X. , Taylor, A. B. , Kinung'hi, S. , Gouvras, A. N. , Webster, B. L. , Webster, J. P. , Emery, A. M. , Rollinson, D. , Garba Djirmay, A. , Al Mashikhi, K. M. , Al Yafae, S. , Idris, M. A. , Moné, H. , … Anderson, T. J. C. (2019). Oxamniquine resistance alleles are widespread in Old World Schistosoma mansoni and predate drug deployment. PLoS Path, 15(10), e1007881. 10.1371/journal.ppat.1007881 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chifman, J. , & Kubatko, L. (2015). Identifiability of the unrooted species tree topology under the coalescent model with time‐reversible substitution processes, site‐specific rate variation, and invariable sites. Journal of Theoretical Biology, 374, 35–47. 10.1016/j.jtbi.2015.03.006 [DOI] [PubMed] [Google Scholar]
- Crellen, T. , Allan, F. , David, S. , Durrant, C. , Huckvale, T. , Holroyd, N. , Emery, A. M. , Rollinson, D. , Aanensen, D. M. , Berriman, M. , Webster, J. P. , & Cotton, J. A. (2016). Whole genome resequencing of the human parasite Schistosoma mansoni reveals population history and effects of selection. Scientific Reports, 6, 20954. 10.1038/srep20954 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Criscione, C. D. , Valentim, C. L. , Hirai, H. , LoVerde, P. T. , & Anderson, T. J. (2009). Genomic linkage map of the human blood fluke Schistosoma mansoni . Genome Biology, 10(6), R71. 10.1186/gb-2009-10-6-r71 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Danecek, P. , Auton, A. , Abecasis, G. , Albers, C. A. , Banks, E. , DePristo, M. A. , Handsaker, R. E. , Lunter, G. , Marth, G. T. , Sherry, S. T. , McVean, G. , & Durbin, R. (2011). The variant call format and VCFtools. Bioinformatics, 27(15), 2156–2158. 10.1093/bioinformatics/btr330 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Davies, C. M. , Webster, J. P. , & Woolhouse, M. E. (2001). Trade‐offs in the evolution of virulence in an indirectly transmitted macroparasite. Proceedings of the Royal Society of London. Series B: Biological Sciences, 268(1464), 251–257. 10.1098/rspb.2000.1367 [DOI] [PMC free article] [PubMed] [Google Scholar]
- de Oliveira, T. C. , Corder, R. M. , Early, A. , Rodrigues, P. T. , Ladeia‐Andrade, S. , Alves, J. M. P. , Neafsey, D. E. , & Ferreira, M. U. (2020). Population genomics reveals the expansion of highly inbred Plasmodium vivax lineages in the main malaria hotspot of Brazil. PLoS Neglected Tropical Diseases, 14(10), e0008808. 10.1371/journal.pntd.0008808 [DOI] [PMC free article] [PubMed] [Google Scholar]
- DeGiorgio, M. , Huber, C. D. , Hubisz, M. J. , Hellmann, I. , & Nielsen, R. (2016). SweepFinder2: Increased sensitivity, robustness and flexibility. Bioinformatics, 32(12), 1895–1897. 10.1093/bioinformatics/btw051 [DOI] [PubMed] [Google Scholar]
- DeJong, R. J. , Morgan, J. A. T. , Paraense, W. L. , Pointier, J.‐P. , Amarista, M. , Ayeh‐Kumi, P. F. K. , Babiker, A. , Barbosa, C. S. , Brémond, P. , Pedro Canese, A. , de Souza, C. P. , Dominguez, C. , File, S. , Gutierrez, A. , Incani, R. N. , Kawano, T. , Kazibwe, F. , Kpikpi, J. , Lwambo, N. J. S. , … Loker, E. S. (2001). Evolutionary relationships and biogeography of Biomphalaria (Gastropoda: Planorbidae) with implications regarding its role as host of the human bloodfluke, Schistosoma mansoni . Molecular Biology, 18(12), 2225–2239. 10.1093/oxfordjournals.molbev.a003769 [DOI] [PubMed] [Google Scholar]
- Dejong, R. J. , Morgan, J. A. , Wilson, W. D. , Al‐Jaser, M. H. , Appleton, C. C. , Coulibaly, G. , D’Andrea, S. P. , Doenhoff, M. J. , Haas, W. , Idris, M. A. , Magalhães, L. A. , Moné, H. , Mouahid, G. , Mubila, L. , Pointier, J.‐P. , Webster, J. P. , Zanotti‐Magalhães, E. M. , Paraense, W. L. , Mkoji, G. M. , & Loker, E. S. (2003). Phylogeography of Biomphalaria glabrat a and B. pfeifferi, important intermediate hosts of Schistosoma mansoni in the New and Old World tropics. Molecular Ecology, 12(11), 3041–3056. 10.1046/j.1365-294x.2003.01977.x [DOI] [PubMed] [Google Scholar]
- Desprès, L. , Imbert‐Establet, D. , & Monnerot, M. (1993). Molecular characterization of mitochondrial DNA provides evidence for the recent introduction of Schistosoma mansoni into America. Molecular and Biochemical Parasitology, 60(2), 221–229. 10.1016/0166-6851(93)90133-i [DOI] [PubMed] [Google Scholar]
- Doyle, S. R. , Sankaranarayanan, G. , Allan, F. , Berger, D. , Jimenez Castro, P. D. , Collins, J. B. , Crellen, T. , Duque‐Correa, M. A. , Ellis, P. , Jaleta, T. G. , Laing, R. , Maitland, K. , McCarthy, C. , Moundai, T. , Softley, B. , Thiele, E. , Ouakou, P. T. , Tushabe, J. V. , Webster, J. P. , … Holroyd, N. (2019). Evaluation of DNA extraction methods on individual helminth egg and larval stages for whole‐genome sequencing. Frontiers in Genetics, 10, 826. 10.3389/fgene.2019.00826 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ebert, D. (1994). Virulence and local adaptation of a horizontally transmitted parasite. Science, 265(5175), 1084–1086. 10.1126/science.265.5175.1084 [DOI] [PubMed] [Google Scholar]
- Eltis, D. (2001). The volume and structure of the transatlantic slave trade: A reassessment. The William and Mary Quarterly, 58(1), 17–46. 10.2307/2674417 [DOI] [PubMed] [Google Scholar]
- Evanno, G. , Regnaut, S. , & Goudet, J. (2005). Detecting the number of clusters of individuals using the software STRUCTURE: A simulation study. Molecular Ecology, 14(8), 2611–2620. 10.1111/j.1365-294X.2005.02553.x [DOI] [PubMed] [Google Scholar]
- Faust, C. L. , Crotti, M. , Moses, A. , Oguttu, D. , Wamboko, A. , Adriko, M. , Adekanle, E. K. , Kabatereine, N. , Tukahebwa, E. M. , Norton, A. J. , Gower, C. M. , Webster, J. P. , & Lamberton, P. H. L. (2019). Two‐year longitudinal survey reveals high genetic diversity of Schistosoma mansoni with adult worms surviving praziquantel treatment at the start of mass drug administration in Uganda. Parasites & Vectors, 12(1), 1–12. 10.1186/s13071-019-3860-6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Files, V. S. (1951). A study of the vector‐parasite relationships in Schistosoma mansoni . Parasitology, 41(3–4), 264–269. [DOI] [PubMed] [Google Scholar]
- Fletcher, M. , LoVerde, P. T. , & Woodruff, D. S. (1981). Genetic variation in Schistosoma mansoni: Enzyme polymorphisms in populations from Africa, Southwest Asia, South America, and the West Indies. American Journal of Tropical Medicine and Hygiene, 30(2), 406–421. 10.4269/ajtmh.1981.30.406 [DOI] [PubMed] [Google Scholar]
- Forni, D. , Pontremoli, C. , Clerici, M. , Pozzoli, U. , Cagliani, R. , & Sironi, M. (2020). Recent out‐of‐Africa migration of human herpes simplex viruses. Molecular Biology and Evolution, 37(5), 1259–1271. 10.1093/molbev/msaa001 [DOI] [PubMed] [Google Scholar]
- Fulford, A. J. , Butterworth, A. E. , Ouma, J. H. , & Sturrock, R. F. (1995). A statistical approach to schistosome population dynamics and estimation of the life‐span of Schistosoma mansoni in man. Parasitology, 110(Pt 3), 307–316. 10.1017/s0031182000080896 [DOI] [PubMed] [Google Scholar]
- Gower, C. M. , Gehre, F. , Marques, S. R. , Lamberton, P. H. , Lwambo, N. J. , & Webster, J. P. (2017). Phenotypic and genotypic monitoring of Schistosoma mansoni in Tanzanian schoolchildren five years into a preventative chemotherapy national control programme. Parasites & Vectors, 10(1), 1–12. 10.1186/s13071-017-2533-6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gurdasani, D. , Carstensen, T. , Tekola‐Ayele, F. , Pagani, L. , Tachmazidou, I. , Hatzikotoulas, K. , Karthikeyan, S. , Iles, L. , Pollard, M. O. , Choudhury, A. , Ritchie, G. R. S. , Xue, Y. , Asimit, J. , Nsubuga, R. N. , Young, E. H. , Pomilla, C. , Kivinen, K. , Rockett, K. , Kamali, A. , … Sandhu, M. S. (2015). The African genome variation project shapes medical genetics in Africa. Nature, 517(7534), 327–332. 10.1038/nature13997 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guzmán‐Solís, A. A. , Villa‐Islas, V. , Bravo‐López, M. J. , Sandoval‐Velasco, M. , Wesp, J. K. , Gómez‐Valdés, J. A. , Moreno‐Cabrera, M. D. L. L. , Meraz, A. , Solís‐Pichardo, G. , Schaaf, P. , TenOever, B. R. , Blanco‐Melo, D. , & Ávila Arcos, M. C. (2021). Ancient viral genomes reveal introduction of human pathogenic viruses into Mexico during the transatlantic slave trade. eLife, 10, 10.7554/eLife.68612 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hahn, M. W. , & Hibbins, M. S. (2019). A three‐sample test for introgression. Molecular Biology and Evolution, 36(12), 2878–2882. 10.1093/molbev/msz178 [DOI] [PubMed] [Google Scholar]
- Hailegebriel, T. , Nibret, E. , & Munshea, A. (2020). Prevalence of Schistosoma mansoni and S. haematobium in snail intermediate hosts in Africa: A systematic review and meta‐analysis. Journal of Tropical Medicine. 10.1155/2020/8850840 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huyse, T. , Van den Broeck, F. , Jombart, T. , Webster, B. L. , Diaw, O. , Volckaert, F. , Balloux, F. , Rollinson, D. , & Polman, K. (2013). Regular treatments of praziquantel do not impact on the genetic make‐up of Schistosoma mansoni in Northern Senegal. Infection, Genetics and Evolution, 18, 100–105. 10.1016/j.meegid.2013.05.007 [DOI] [PubMed] [Google Scholar]
- International Helminth Genomes Consortium . (2019). Comparative genomics of the major parasitic worms. Nature Genetics, 51(1), 163–174. 10.1038/s41588-018-0262-1 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jorgenson, A. , Kristensen, T. K. , & Stothard, J. R. (2007). Phylogeny and biogeography of African Biomphalaria (Gastropoda: Planorbidae), with emphasis on endemic species of the great East African lakes. Zoological Journal of the Linnean Society, 151(2), 337–349. 10.1111/j.1096-3642.2007.00330.x [DOI] [Google Scholar]
- Kabongo, M. M. , Linsuke, S. , Baloji, S. , Mukunda, F. , da Luz Raquel, I. , Stauber, C. , Geertruyden, J.‐P. , & Lutumba, P. (2018). Schistosoma mansoni infection and its association with nutrition and health outcomes: A household survey in school‐aged children living in Kasansa, Democratic Republic of the Congo. Pan African Medical Journal, 31, 197. 10.11604/pamj.2018.31.197.16364 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kelleher, J. , Etheridge, A. M. , & McVean, G. (2016). Efficient coalescent simulation and genealogical analysis for large sample sizes. PLoS Computational Biology, 12(5), e1004842. 10.1371/journal.pcbi.1004842 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kengne‐Fokam, A. C. , Nana‐Djeunga, H. C. , Bagayan, M. , & Njiokou, F. (2018). Biomphalaria camerunensis as a viable alternative intermediate host for Schistosoma mansoni in southern Cameroon. Parasites & Vectors, 11(1), 1–9. 10.1186/s13071-018-2763-2 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Köster, J. , & Rahmann, S. (2012). Snakemake—A scalable bioinformatics workflow engine. Bioinformatics, 28(19), 2520–2522. 10.1093/bioinformatics/bts480 [DOI] [PubMed] [Google Scholar]
- Köster, J. , & Rahmann, S. (2018). Snakemake‐a scalable bioinformatics workflow engine. Bioinformatics, 34(20), 3600. 10.1093/bioinformatics/bty350 [DOI] [PubMed] [Google Scholar]
- Le Clec’h, W. , Chevalier, F. D. , Ana, C. A. , Mattos, A. S. , Diaz, R. , McDew‐White, M. , Rohr, C. M. , Kinung’hi, S. , Allan, F. , Webster, B. L. , Webster, J. P. , Emery, A. M. , Rollinson, D. , Djirmay, A. G. , Al, K. M. , Mashikhi, S. A. , Yafae, M. A. , Idris, H. M. , Mouahid, G. , … Anderson, T. J. C. (2021). Genetic analysis of praziquantel response in schistosome parasites implicates a transient receptor potential channel. Science Translational Medicine. 10.1126/scitranslmed.abj9114 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Le Clec'h, W. , Chevalier, F. D. , McDew‐White, M. , Allan, F. , Webster, B. L. , Gouvras, A. N. , Kinunghi, S. , Tchuem Tchuenté, L.‐A. , Garba, A. , Mohammed, K. A. , Ame, S. M. , Webster, J. P. , Rollinson, D. , Emery, A. M. , & Anderson, T. J. C. (2018). Whole genome amplification and exome sequencing of archived schistosome miracidia. Parasitology, 145(13), 1739–1747. 10.1017/S0031182018000811 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Le Clec'h, W. , Chevalier, F. D. , McDew‐White, M. , Menon, V. , Arya, G. A. , & Anderson, T. J. C. (2021). Genetic architecture of transmission stage production and virulence in schistosome parasites. Virulence, 12(1), 1508–1526. 10.1080/21505594.2021.1932183 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leblois, R. , Kuhls, K. , François, O. , Schönian, G. , & Wirth, T. (2011). Guns, germs and dogs: On the origin of Leishmania chagasi . Infection, Genetics and Evolution, 11(5), 1091–1095. 10.1016/j.meegid.2011.04.004 [DOI] [PubMed] [Google Scholar]
- Léger, E. , Borlase, A. , Fall, C. B. , Diouf, N. D. , Diop, S. D. , Yasenev, L. , Catalano, S. , Thiam, C. T. , Ndiaye, A. , Emery, A. , Morrell, A. , Rabone, M. , Ndao, M. , Faye, B. , Rollinson, D. , Rudge, J. W. , Sène, M. , & Webster, J. P. (2020). Prevalence and distribution of schistosomiasis in human, livestock, and snail populations in northern Senegal: A One Health epidemiological study of a multi‐host system. Lancet Planet Health, 4(8), e330–e342. 10.1016/s2542-5196(20)30129-7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leger, E. , & Webster, J. P. (2017). Hybridizations within the genus Schistosoma: Implications for evolution, epidemiology and control. Parasitology, 144(1), 65–80. [DOI] [PubMed] [Google Scholar]
- Leigh, J. W. , & Bryant, D. (2015). POPART: Full‐feature software for haplotype network construction. Methods in Ecology Evolution, 6(9), 1110–1116. [Google Scholar]
- Lelo, A. E. , Mburu, D. N. , Magoma, G. N. , Mungai, B. N. , Kihara, J. H. , Mwangi, I. N. , Maina, G. M. , Kinuthia, J. M. , Mutuku, M. W. , Loker, E. S. , Mkoji, G. M. , & Steinauer, M. L. (2014). No apparent reduction in schistosome burden or genetic diversity following four years of school‐based mass drug administration in Mwea, central Kenya, a heavy transmission area. PLoS Neglected Tropical Diseases, 8(10), e3221. 10.1371/journal.pntd.0003221 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li, H. , & Durbin, R. (2010). Fast and accurate long‐read alignment with Burrows‐Wheeler transform. Bioinformatics, 26(5), 589–595. 10.1093/bioinformatics/btp698 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lively, C. M. (1989). Adaptation by a parasitic trematode to local populations of its snail host. Evolution, 43(8), 1663–1671. 10.1111/j.1558-5646.1989.tb02616.x [DOI] [PubMed] [Google Scholar]
- Luu, K. , Bazin, E. , & Blum, M. G. (2017). pcadapt: An R package to perform genome scans for selection based on principal component analysis. Molecular Ecology Resources, 17(1), 67–77. [DOI] [PubMed] [Google Scholar]
- Mackay, T. F. C. , & Huang, W. (2018). Charting the genotype‐phenotype map: Lessons from the Drosophila melanogaster genetic reference panel. Wiley Interdisciplinary Reviews: Developmental Biology, 7(1). 10.1002/wdev.289 [DOI] [PMC free article] [PubMed] [Google Scholar]
- McKenna, A. , Hanna, M. , Banks, E. , Sivachenko, A. , Cibulskis, K. , Kernytsky, A. , Garimella, K. , Altshuler, D. , Gabriel, S. , Daly, M. , & DePristo, M. A. (2010). The Genome Analysis Toolkit: A MapReduce framework for analyzing next‐generation DNA sequencing data. Genome Research, 20(9), 1297–1303. 10.1101/gr.107524.110 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miles, A. , Ralph, P. , Rae, S. , & Pisupati, R. (2019). scikit‐allel (Version 1.2.1)
- Mitta, G. , Gourbal, B. , Grunau, C. , Knight, M. , Bridger, J. , & Théron, A. (2017). The compatibility between Biomphalaria glabrata snails and Schistosoma mansoni: An increasingly complex puzzle. In Rollinson D., & Stothard J. R. (Eds.), Advances in parasitology (Vol. 97, pp. 111–145). Elsevier. [DOI] [PubMed] [Google Scholar]
- Morand, S. , Manning, S. D. , & Woolhouse, M. E. (1996). Parasite‐host coevolution and geographic patterns of parasite infectivity and host susceptibility. Proceedings of the Royal Society of London. Series B: Biological Sciences, 263(1366), 119–128. 10.1098/rspb.1996.0019 [DOI] [PubMed] [Google Scholar]
- Morgan, J. A. T. , Dejong, R. J. , Adeoye, G. O. , Ansa, E. D. O. , Barbosa, C. S. , Brémond, P. , Cesari, I. M. , Charbonnel, N. , Corrêa, L. R. , Coulibaly, G. , D’andrea, P. S. , De souza, C. P. , Doenhoff, M. J. , File, S. , Idris, M. A. , Incani, R. N. , Jarne, P. , Karanja, D. M. S. , Kazibwe, F. , … Loker, E. S. (2005). Origin and diversification of the human parasite Schistosoma mansoni . Molecular Ecology, 14(12), 3889–3902. 10.1111/j.1365-294X.2005.02709.x [DOI] [PubMed] [Google Scholar]
- Morgan, J. , DeJong, R. J. , Lwambo, N. J. , Mungai, B. N. , Mkoji, G. M. , & Loker, E. S. (2003). First report of a natural hybrid between Schistosoma mansoni and S. rodhaini . Journal of Parasitology, 89(2), 416–418. [DOI] [PubMed] [Google Scholar]
- Mutuku, M. W. , Laidemitt, M. R. , Spaan, J. M. , Mwangi, I. N. , Ochanda, H. , Steinauer, M. L. , Loker, E. S. , & Mkoji, G. M. (2021). Comparative vectorial competence of Biomphalaria sudanica and Biomphalaria choanomphala, snail hosts of Schistosoma mansoni, from transmission hotspots in lake Victoria, Western Kenya. Journal of Parasitology, 107(2), 349–357. 10.1645/20-138 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mutuku, M. W. , Lu, L. , Otiato, F. O. , Mwangi, I. N. , Kinuthia, J. M. , Maina, G. M. , Laidemitt, M. R. , Lelo, E. A. , Ochanda, H. , & Loker, E. S. , & Mkoji, G. M. (2017). A Comparison of Kenyan Biomphalaria pfe ifferi and B. sudanica as vectors for Schistosoma mansoni, including a discussion of the need to better understand the effects of snail breeding systems on transmission. Journal of Parasitology, 103(6), 669–676. 10.1645/17-72 [DOI] [PubMed] [Google Scholar]
- Nadeau, S. A. , Vaughan, T. G. , Scire, J. , Huisman, J. S. , & Stadler, T. (2021). The origin and early spread of SARS‐CoV‐2 in Europe. Proceedings of the National Academy of Sciences of the United States of America, 118(9), e2012008118. 10.1073/pnas.2012008118 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Neafsey, D. E. , Schaffner, S. F. , Volkman, S. K. , Park, D. , Montgomery, P. , Milner, D. A. , Lukens, A. , Rosen, D. , Daniels, R. , Houde, N. , Cortese, J. F. , Tyndall, E. , Gates, C. , Stange‐Thomann, N. , Sarr, O. , Ndiaye, D. , Ndir, O. , Mboup, S. , Ferreira, M. U. , … Wirth, D. F. (2008). Genome‐wide SNP genotyping highlights the role of natural selection in Plasmodium falciparum population divergence. Genome Biology, 9(12), R171. 10.1186/gb-2008-9-12-r171 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nychka, D. , Furrer, R. , Paige, J. , & Sain, S. (2017). fields: Tools for spatial data (Version 11.6). University Corporation for Atmospheric Research. https://github.com/NCAR/Fields [Google Scholar]
- Ortiz, E. M. (2019). vcf2phylip v2.0: Convert a VCF matrix into several matrix formats for phylogenetic analysis. 10.5281/zenodo.2540861 [DOI] [Google Scholar]
- Patterson, N. , Moorjani, P. , Luo, Y. , Mallick, S. , Rohland, N. , Zhan, Y. , Genschoreck, T. , Webster, T. , & Reich, D. (2012). Ancient admixture in human history. Genetics, 192(3), 1065–1093. 10.1534/genetics.112.145037 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pedersen, B. S. , & Quinlan, A. R. (2018). Mosdepth: Quick coverage calculation for genomes and exomes. Bioinformatics, 34(5), 867–868. 10.1093/bioinformatics/btx699 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pickrell, J. K. , & Pritchard, J. K. (2012). Inference of population splits and mixtures from genome‐wide allele frequency data. PLoS Genetics, 8(11), e1002967. 10.1371/journal.pgen.1002967 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Platt, R. N. , McDew‐White, M. , Le Clec’h, W. , Chevalier, F. D. , Allan, F. , Emery, A. M. , Garba, A. , Hamidou, A. A. , Ame, S. M. , Webster, J. P. , Rollinson, D. , Webster, B. L. , & Anderson, T. J. C. (2019). Ancient hybridization and adaptive introgression of an invadolysin gene in schistosome parasites. Molecular Biology and Evolution, 36(10), 2127–2142. 10.1093/molbev/msz154 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Portet, A. , Pinaud, S. , Chaparro, C. , Galinier, R. , Dheilly, N. M. , Portela, J. , Charriere, G. M. , Allienne, J.‐F. , Duval, D. , & Gourbal, B. (2019). Sympatric versus allopatric evolutionary contexts shape differential immune response in Biomphalaria/Schistosoma interaction. PLoS Path, 15(3), e1007647. 10.1371/journal.ppat.1007647 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Protasio, A. V. , Tsai, I. J. , Babbage, A. , Nichol, S. , Hunt, M. , Aslett, M. A. , De Silva, N. , Velarde, G. S. , Anderson, T. J. C. , Clark, R. C. , Davidson, C. , Dillon, G. P. , Holroyd, N. E. , LoVerde, P. T. , Lloyd, C. , McQuillan, J. , Oliveira, G. , Otto, T. D. , Parker‐Manuel, S. J. , … Berriman, M. (2012). A systematically improved high quality genome and transcriptome of the human blood fluke Schistosoma mansoni. PLoS Neglected Tropical Diseases, 6(1), e1455. 10.1371/journal.pntd.0001455 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Purcell, S. , Neale, B. , Todd‐Brown, K. , Thomas, L. , Ferreira, M. A. R. , Bender, D. , Maller, J. , Sklar, P. , de Bakker, P. I. W. , Daly, M. J. , & Sham, P. C. (2007). PLINK: A tool set for whole‐genome association and population‐based linkage analyses. American Journal of Human Genetics, 81(3), 559–575. 10.1086/519795 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rey, O. , Toulza, E. , Chaparro, C. , Allienne, J.‐F. , Kincaid‐Smith, J. , Mathieu‐Begné, E. , Allan, F. , Rollinson, D. , Webster, B. L. , & Boissier, J. (2021). Diverging patterns of introgression from Schistosoma bovis across S. haematobium African lineages. PLoS Path, 17(2), e1009313. 10.1371/journal.ppat.1009313 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rey, O. , Webster, B. L. , Huyse, T. , Rollinson, D. , Van den Broeck, F. , Kincaid‐Smith, J. , Onyekwere, A. , & Boissier, J. (2021). Population genetics of African Schistosoma species. Infection, Genetics and Evolution, 89, 104727. 10.1016/j.meegid.2021.104727 [DOI] [PubMed] [Google Scholar]
- Rius, M. , Bourne, S. , Hornsby, H. G. , & Chapman, M. A. (2015). Applications of next‐generation sequencing to the study of biological invasions. Current Zoology, 61(3), 488–504. 10.1093/czoolo/61.3.488 [DOI] [Google Scholar]
- Rodrigues, P. T. , Valdivia, H. O. , de Oliveira, T. C. , Alves, J. M. P. , Duarte, A. M. R. C. , Cerutti‐Junior, C. , Buery, J. C. , Brito, C. F. A. , de Souza, J. C. , Hirano, Z. M. B. , Bueno, M. G. , Catão‐Dias, J. L. , Malafronte, R. S. , Ladeia‐Andrade, S. , Mita, T. , Santamaria, A. M. , Calzada, J. E. , Tantular, I. S. , Kawamoto, F. , … Ferreira, M. U. (2018). Human migration and the spread of malaria parasites to the New World. Scientific Reports, 8(1), 1–13. 10.1038/s41598-018-19554-0 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sanneh, B. , Joof, E. , Sanyang, A. M. , Renneker, K. , Camara, Y. , Sey, A. P. , Jagne, S. , Baldeh, I. , Ceesay, S. J. , Sambou, S. M. , & Ogoussan, K. (2017). Field evaluation of a schistosome circulating cathodic antigen rapid test kit at point‐of‐care for mapping of schistosomiasis endemic districts in The Gambia. PLoS One, 12(8), e0182003. 10.1371/journal.pone.0182003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schlamp, F. , van der Made, J. , Stambler, R. , Chesebrough, L. , Boyko, A. R. , & Messer, P. W. (2016). Evaluating the performance of selection scans to detect selective sweeps in domestic dogs. Molecular Ecology, 25(1), 342–356. 10.1111/mec.13485 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schwabl, P. , Boité, M. C. , Bussotti, G. , Jacobs, A. , Andersson, B. , Moreira, O. , Freitas‐Mesquita, A. L. , Meyer‐Fernandes, J. R. , Telleria, E. L. , Traub‐Csekö, Y. , Vaselek, S. , Leštinová, T. , Volf, P. , Morgado, F. N. , Porrozzi, R. , Llewellyn, M. , Späth, G. F. , & Cupolillo, E. (2021). Colonization and genetic diversification processes of Leishmania infantum in the Americas. Communications Biology, 4(1), 1–13. 10.1038/s42003-021-01658-5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sherpa, S. , & Després, L. (2021). The evolutionary dynamics of biological invasions: A multi‐approach perspective. Evolutionary Applications, 14(6), 1463–1484. 10.1111/eva.13215 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shortt, J. A. , Card, D. C. , Schield, D. R. , Liu, Y. , Zhong, B. O. , Castoe, T. A. , Carlton, E. J. , & Pollock, D. D. (2017). Whole genome amplification and reduced‐representation genome sequencing of Schistosoma japonicum miracidia. PLoS Neglected Tropical Diseases, 11(1), e0005292. 10.1371/journal.pntd.0005292 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Slave Voyages Database . (2009). Voyages: The trans‐atlantic slave trade database. https://www.slavevoyages.org/ [Google Scholar]
- Small, S. T. , Labbe, F. , Coulibaly, Y. I. , Nutman, T. B. , King, C. L. , Serre, D. , & Zimmerman, P. A. (2019). Human migration and the spread of the nematode parasite Wuchereria bancrofti . Molecular Biology and Evolution, 36(9), 1931–1941. 10.1093/molbev/msz116 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Steinauer, M. L. , Hanelt, B. , Mwangi, I. N. , Maina, G. M. , Agola, L. E. , Kinuthia, J. M. , Mutuku, M. W. , Mungai, B. N. , Wilson, W. D. , Mkoji, G. M. , & Loker, E. S. (2008). Introgressive hybridization of human and rodent schistosome parasites in western Kenya. Molecular Ecology, 17(23), 5062–5074. 10.1111/j.1365-294X.2008.03957.x [DOI] [PubMed] [Google Scholar]
- Steinauer, M. L. , Mwangi, I. N. , Maina, G. M. , Kinuthia, J. M. , Mutuku, M. W. , Agola, E. L. , Mungai, B. , Mkoji, G. M. , & Loker, E. S. (2008). Interactions between natural populations of human and rodent schistosomes in the Lake Victoria region of Kenya: A molecular epidemiological approach. PLoS Neglected Tropical Diseases, 2(4), e222. 10.1371/journal.pntd.0000222 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Steinegger, M. , Meier, M. , Mirdita, M. , Vöhringer, H. , Haunsberger, S. J. , & Söding, J. (2019). HH‐suite3 for fast remote homology detection and deep protein annotation. BMC Bioinformatics, 20(1), 473. 10.1186/s12859-019-3019-7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Steverding, D. (2020). The spreading of parasites by human migratory activities. Virulence, 11(1), 1177–1191. 10.1080/21505594.2020.1809963 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stothard, J. R. , Kayuni, S. A. , Al‐Harbi, M. H. , Musaya, J. , & Webster, B. L. (2020). Future schistosome hybridizations: Will all Schistosoma haematobium hybrids please stand‐up! PLoS Neglected Tropical Diseases, 14(7), e0008201. 10.1371/journal.pntd.0008201 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Swofford, D. L. (2003). PAUP*. Phylogenetic Analysis Using Parsimony (*and other methods). Sinauer Associates. https://paup.phylosolutions.com/ [Google Scholar]
- Tennessen, J. A. , Bollmann, S. R. , Peremyslova, E. , Kronmiller, B. A. , Sergi, C. , Hamali, B. , & Blouin, M. S. (2020). Clusters of polymorphic transmembrane genes control resistance to schistosomes in snail vectors. eLife, 9, e59395. 10.7554/eLife.59395 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tennessen, J. A. , Theron, A. , Marine, M. , Yeh, J. Y. , Rognon, A. , & Blouin, M. S. (2015). Hyperdiverse gene cluster in snail host conveys resistance to human schistosome parasites. PLoS Genetics, 11(3), e1005067. 10.1371/journal.pgen.1005067 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Théron, A. (1989). Hybrids between Schistosoma mansoni and S. rodhaini: Characterization by cercarial emergence rhythms. Parasitology, 99(Pt 2), 225–228. 10.1017/s0031182000058674 [DOI] [PubMed] [Google Scholar]
- Theron, A. , Rognon, A. , Gourbal, B. , & Mitta, G. (2014). Multi‐parasite host susceptibility and multi‐host parasite infectivity: A new approach of the Biomphalaria glabrata/Schistosoma mansoni compatibility polymorphism. Infection, Genetics and Evolution, 26, 80–88. 10.1016/j.meegid.2014.04.025 [DOI] [PubMed] [Google Scholar]
- Van den Broeck, F. , Meurs, L. , Raeymaekers, J. A. M. , Boon, N. , Dieye, T. N. , Volckaert, F. A. M. , Polman, K. , & Huyse, T. (2014). Inbreeding within human Schistosoma mansoni: Do host‐specific factors shape the genetic composition of parasite populations? Heredity, 113(1), 32–41. 10.1038/hdy.2014.13 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vidigal, T. , Kissinger, J. , Caldeira, R. L. , Pires, E. , Monteiro, E. , Simpson, A. , & Carvalho, O. (2000). Phylogenetic relationships among Brazilian Biomphalaria species (Mollusca: Planorbidae) based upon analysis of ribosomal ITS2 sequences. Parasitology, 121(6), 611–620. 10.1017/S0031182000006831 [DOI] [PubMed] [Google Scholar]
- Virtanen, P. , Gommers, R. , Oliphant, T. E. , Haberland, M. , Reddy, T. , Cournapeau, D. , Burovski, E. , Peterson, P. , Weckesser, W. , Bright, J. , van der Walt, S. J. , Brett, M. , Joshua Wilson, K. , Millman, J. , Mayorov, N. , Nelson, A. R. J. , Jones, E. , Kern, R. , Eric Larson, C. J. , … SciPy 1.0 Contributors . (2020). SciPy 1.0: Fundamental algorithms for scientific computing in Python. Nature Methods, 17(3), 261–272. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Webster, B. L. , Webster, J. P. , Gouvras, A. N. , Garba, A. , Lamine, M. S. , Diaw, O. T. , Seye, M. M. , Tchuem Tchuenté, L.‐A. , Simoonga, C. , Mubila, L. , Mwanga, J. R. , Lwambo, N. J. S. , Kabatereine, N. B. , Lange, C. N. , Kariuki, C. , Mkoji, G. M. , Rollinson, D. , & Stothard, J. R. (2013). DNA ‘barcoding’of Schistosoma mansoni across sub‐Saharan Africa supports substantial within locality diversity and geographical separation of genotypes. Acta Tropica, 128(2), 250–260. 10.1016/j.actatropica.2012.08.009 [DOI] [PubMed] [Google Scholar]
- Webster, J. P. , Davies, C. M. , Hoffman, J. I. , Ndamba, J. , Noble, L. R. , & Woolhouse, M. E. (2001). Population genetics of the schistosome intermediate host Biomphalaria pfeifferi in the Zimbabwean highveld: Implications for co‐evolutionary theory. Annals of Tropical Medicine and Parasitology, 95(2), 203–214. 10.1080/00034980120041062 [DOI] [PubMed] [Google Scholar]
- Webster, J. P. , Gower, C. M. , & Blair, L. (2004). Do hosts and parasites coevolve? Empirical support from the Schistosoma system. American Naturalist, 164(Suppl. 5), S33–51. 10.1086/424607 [DOI] [PubMed] [Google Scholar]
- Webster, J. P. , & Woolhouse, M. E. J. (1998). Selection and strain specificity of compatibility between snail intermediate hosts and their parasitic schistosomes. Evolution, 52(6), 1627–1634. 10.1111/j.1558-5646.1998.tb02243.x [DOI] [PubMed] [Google Scholar]
- Weir, B. S. , & Cockerham, C. C. (1984). Estimating F‐statistics for the analysis of population structure. Evolution, 38(6), 1358–1370. 10.1111/j.1558-5646.1984.tb05657.x [DOI] [PubMed] [Google Scholar]
- Woolhouse, M. E. , & Webster, J. P. (2000). In search of the red queen. Parasitol Today, 16(12), 506–508. 10.1016/s0169-4758(00)01820-2 [DOI] [PubMed] [Google Scholar]
- Yalcindag, E. , Elguero, E. , Arnathau, C. , Durand, P. , Akiana, J. , Anderson, T. J. , Aubouy, A. , Balloux, F. , Besnard, P. , Bogreau, H. , Carnevale, P. , D'Alessandro, U. , Fontenille, D. , Gamboa, D. , Jombart, T. , Le Mire, J. , Leroy, E. , Maestre, A. , Mayxay, M. , … Prugnolle, F. (2012). Multiple independent introductions of Plasmodium falciparum in South America. Proceedings of the National Academy of Sciences of the United States of America, 109(2), 511–516. 10.1073/pnas.1119058109 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Fig S1
Table S1
Data Availability Statement
Data used in this paper were previously published (Berriman et al., 2009; Chevalier et al., 2019; Crellen et al., 2016; International Helminth Genomes Consortium, 2019; Le Clec'h et al., 2021) under multiple NCBI BioProject (PRJNA439266, PRJNA560070, PRJEB522, PRJEB526, PRJNA743359 and PRJNA773498) and NCBI Short Read Archive (ERR046038, ERR103049, ERR103050, ERR119614, ERR119615, ERX284221, ERR310938, ERR539846, ERR539847, ERR539848, and ERR9974) accessions.
Code availability: Scripts, notebooks, and environmental yaml files are available at https://github.com/nealplatt/sch_man_nwinvasion/releases/tag/v0.2 (last accessed October 21, 2021) or https://doi.org/10.5281/zenodo.5590460 (last accessed October 21, 2021).