

Abstract
As one of the highest induced genes in activated macrophages, immune-responsive gene 1 (IRG1) encodes a mitochondrial metabolic enzyme catalysing the production of itaconic acid (ITA). While ITA has an anti-inflammatory property, the underlying mechanisms are not fully understood. Here, we show that ITA is a potent inhibitor of the TET family DNA dioxygenases. ITA binds to the same site in TET2 where the co-substrate α-ketoglutarate (α-KG) binds, inhibiting TET2 catalytic activity. Lipopolysaccharides (LPS) treatment, which induces Irg1 expression and ITA accumulation, inhibits Tet activity in macrophages. Transcriptome analysis reveals TET2 is a major target of ITA in suppressing LPS-induced genes, including those regulated by NF-κB and STAT signaling pathways. In vivo, ITA decreases 5hmC level, reduces LPS-induced acute pulmonary edema, lung and liver injury, and protects mice against lethal endotoxaemia depending on the catalytic activity of Tet2. Our study thus identifies ITA as an immune modulatory metabolite that selectively inhibits TET enzymes to dampen the inflammatory responses.
IRG1 plays a key role in anti-pathogen defense, as deletion of Irg1 in mice causes severe defects in response to bacterial and viral infection, and decreased survival1, 2. IRG1 transcription is rapidly induced by pathogen infection and inflammatory conditions primarily in cells of myeloid lineage3. IRG1 encodes a mitochondrial metabolic enzyme, aconitate decarboxylase 1 (ACOD1), that catalyzes the decarboxylation of cis-aconitate to produce the anti-inflammatory metabolite itaconic acid (ITA)4. Several molecular processes are affected by ITA, including succinate dehydrogenase (SDH) inhibition5, resulting in succinate accumulation and metabolic reprogramming6, 7, and alkylation of protein cysteine residues, inducing the electrophilic stress response mediated by NRF2 and IκBζ8, 9 and impairing aerobic glycolysis10. However, the mechanism by which ITA exerts its profound anti-inflammatory effect on regulating gene transcription has not yet been fully elucidated.
Results
Itaconate binds directly to TET2 in a manner similar to α-KG
Deletion of the Irg1 gene or treatment with cell permeable ITA alters the transcriptional signature in response to LPS2. We speculated that ITA may impact epigenetics to influence gene expression, and therefore, we determined the effect of Irg1 expression and ITA accumulation on global histone and DNA de/methylation in transfected HEK293T cells (Fig. 1a). We found that ectopic expression of either wild-type or catalytic inactive mutant Irg1 had little effect on mono-, di-, and trimethylation of all five histone H3 lysine residues (Fig. 1b, 1c). In contrast, expression of wild-type Irg1, but not the catalytic inactive mutant, dramatically reduced Tet2-mediated global 5hmC in cells (Fig. 1d–f). Like α-KG, which is a crucial co-substrate for the activity of TET2, ITA is also a dicarboxylic acid containing a 4- or 5-carboxylate that, in the case of α-KG, forms hydrogen and ionic bonds with H1416, R1896, and S1898 in TET211. Of note, α-KG binds to Fe(II) in a bidentate manner via its C-1 carboxylate and C-2 keto groups, which are lacking in ITA. This raises the possibility that ITA may act as an α-KG antagonist to competitively inhibit TET and other α-KG/Fe(II)-dependent dioxygenases, similar to previously identified oncometabolites, D-2-HG, succinate, and fumarate. Molecular modeling suggests that ITA, like the other α-KG antagonists, can interact with amino acid residues within the active site of TET2 (Extended Data Fig. 1a).
Figure 1. Irg1 expression reduces Tet2-catalyzed 5hmC production in cells.
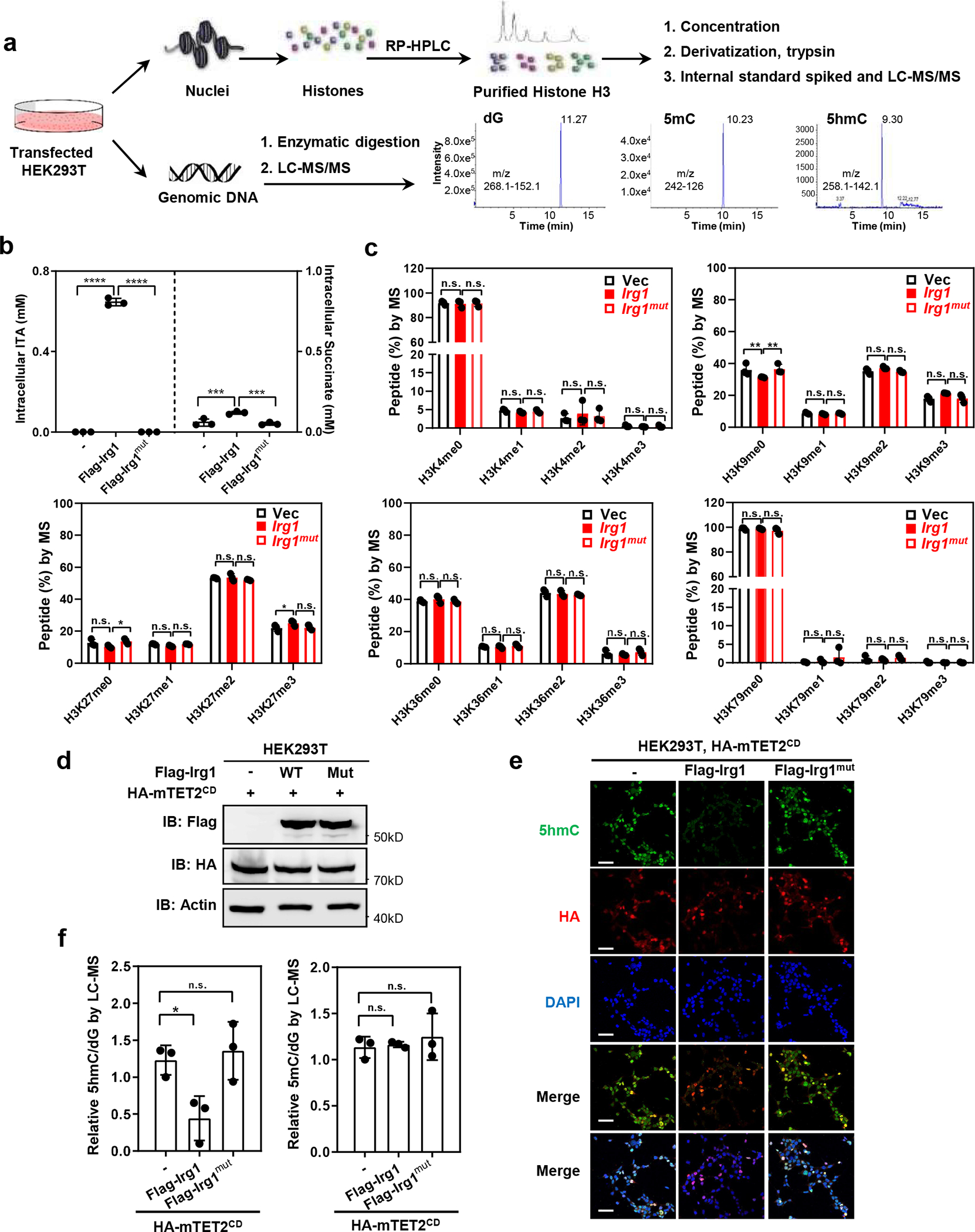
a, Schematic diagram of experimental procedures to determine the effect of Irg1 expression on global histone and DNA de/methylation.
b, Overexpression of Irg1, but not catalytic-inactive mutant, results in elevated intracellular levels of ITA and succinate. Flag-tagged WT or H103A/K207E/K272E mutant Irg1 (referred to as Irg1mut) was transiently overexpressed in HEK293T cells, and the intracellular levels of metabolites were determined by LC-MS/MS.
c, Overexpression of Irg1 does not significantly affect global histone demethylation markers. Transfected HEK293T cells in (b) were subjected to histone methylation quantification by MRM-based LC-MS/MS.
d, Overexpression of Irg1, but not Irg1mut, inhibits Tet2 activity. Flag-tagged wild-type Irg1 or catalytic defect mutant was co-expressed with HA-mTET2CD in HEK293T cells. The ectopically expressed proteins of Irg1 and Tet2 proteins were verified by western-blot using indicated antibodies.
e, f, Overexpression of Irg1, but not Irg1mut, inhibits Tet2-catalyzed 5hmC production. Flag-tagged wild-type Irg1 or catalytic defect mutant was co-expressed with HA-mTET2CD in HEK293T cells and was then subjected to detect 5hmC by immunofluorescence staining using indicated antibodies(e) Data shown represent 3 independent experiments’ in e. Scale bar, 50μM.Genomic DNA from the transfected cells in (d) were also subjected to 5hmC and 5mC quantification by LC-MS/MS (f).
Data shown in b, c, and f are average values with S.D. of n = three independent experiments. Asterisks denote statistical significance with one-way (f) or two-way ANOVA (b) for multiple comparisons. *denotes p < 0.05, ***denotes p < 0.001, ****denotes p < 0.0001 for the indicated comparison. n.s. = not significant.
Using in vitro enzymatic assay based on immobilized substrate DNA and fluorescence detection of 5hmC (Fig. 2a), we found ITA inhibited the catalytic activity of mTET2CD with a half maximal inhibitory concentration (IC50) of 171 μM (Fig. 2b), similar to that of L-2-HG (115 μM) and considerably more potent than D-2-HG (1,057 μM), succinate (> 5mM) and fumarate (> 5 mM). Similar results were obtained when using a commercial TET activity assay kit (Extended Data Fig. 1b). The inhibition of mTET2CD activity by ITA or L-2-HG was overcome dose-dependently by excess α-KG (Fig. 2c). Furthermore, saturation transfer difference (STD) NMR spectroscopy demonstrated that ITA, but not glutamine as a negative control metabolite, bound directly to recombinant human TET2CD and that ITA-TET2CD binding was blocked by α-KG (Fig. 2d). Mutation of either R1896 or S1898 in TET2 reduced its 5hmC-producing activity. These mutations, associated with weakened binding of α-KG in TET2 (Extended Data Fig. 1c, 1d), were found to also abolish the binding of ITA (Fig. 2e). Together, these results demonstrate that ITA binds directly to TET2 in a manner similar to α-KG and potently inhibits TET activity.
Figure 2. Itaconate binds directly to TET2 in a manner similar to α-KG.
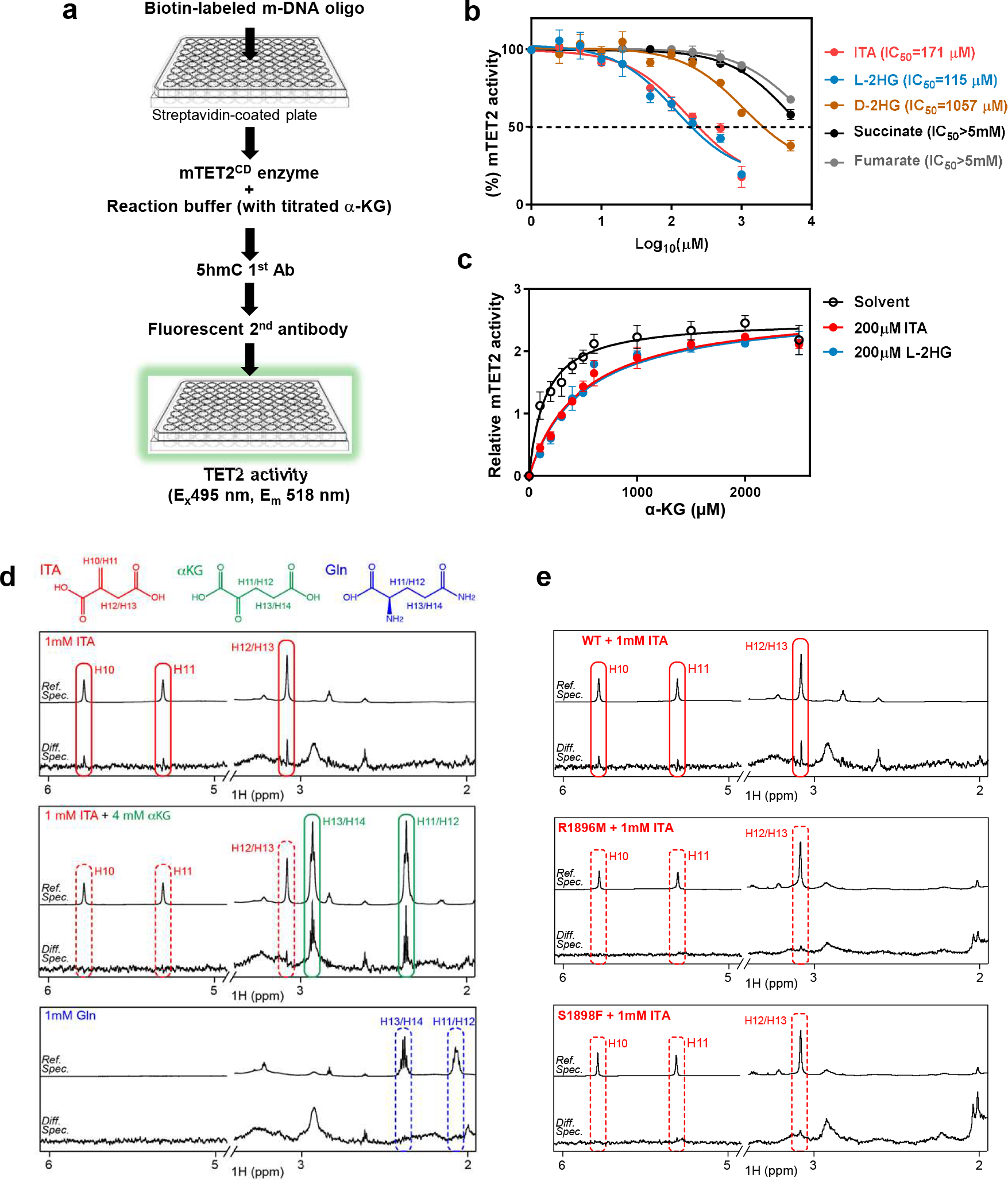
a, Schematic diagram of the workflow for assaying Tet enzyme activity using immobilized substrate DNA and fluorescence detection of 5hmC.
b, IC50 values of indicated metabolites toward mTET2CD were measured by a protocol modified from the method shown in Fig. 2b. Enzyme activity was analyzed using GraphPad software and IC50 values were calculated using the equation of log (inhibitor) vs. response (three parameters). Data shown are average values with S.D. of 3 experiments.
c, α-KG overcomes ITA and L-2HG inhibition of Tet2. The relative Tet2 enzyme activity was measured as in Fig. 1c in the presence of fixed concentration of ITA or L-2-HG and variable concentration of α-KG. Data was analyzed using GraphPad software following Michaelis-Menten equation. Data shown are average values of 2 or 3 experiments.
d, ITA binds to TET2 and is competed off by α-KG. Saturation transfer difference (STD) NMR independent spectroscopy was used to determine the binding of metabolites with recombinant human TET2CD.
e, ITA is unable to bind with TET2 mutants. STD NMR spectroscopy was used to determine the binding of ITA with recombinant proteins of TET2CD mutant, i.e. R1896M or S1898F. Data shown represent 2 independent experiments in d and e.
P values are calculated using two-tailed Student’s t-test for paired comparisons (a). **denotes p < 0.01, ***denotes p < 0.001 for the indicated comparison.
Irg1 produces Itaconate to selectively inhibit TET enzymes in vivo
To determine whether ITA inhibits the TET family enzymes in cells, we treated RAW264.7 mouse macrophages with octyl-ITA (OI) or unmodified ITA and confirmed rapid (< 30 seconds) intracellular accumulation of ITA in a dose-dependent manner and inhibition of SDH by ITA, as seen by the accumulation of succinate (Extended Data Fig. 2a, 2b). OI, like cell-permeable Octyl-L-2HG, accumulated in HEK293T cells overexpressing mTET2CD and dose-dependently reduced 5hmC (Extended Data Fig. 2c, 3a). This reduction was reversed by the addition of increasing amount of cell-permeable dimethyl-α-KG (Extended Data Fig. 3b). Likewise, in HEK293T cells overexpressing TET3, exogenously added OI or ITA led to intracellular accumulation of ITA and decreased global 5hmC (Extended Data Fig. 3c, 3d, 3e and 3f). Unlike L-2-HG, which also inhibits JmjC-domain histone lysine demethylases (KDMs)12, 13, and in accord with Irg1 overexpression (Fig. 1c), addition of OI had no detectable effect on global histone methylation (Extended Data Fig. 3g), supporting a selective inhibition of ITA towards TET enzymes over KDMs.
Given that the anti-inflammatory metabolite ITA is accumulated primarily in cells of myeloid lineage3, we treated Tet2+/+ and Tet2−/− bone marrow-derived macrophages (BMDMs) with either OI or ITA (fig. 3a). In these cells, intracellular accumulation of ITA was observed-in an ITA-dose-dependent manner in Tet2+/+ BMDMs (Extended Data Fig. 3h). Global mapping of 5hmC via DNA immunoprecipitation coupled with high-throughput sequencing (hMeDIP-seq) showed that OI treatment decreased 5hmC at both promoters and intragenic regions in Tet2+/+ BMDMs (Extended Data Fig. 3i). Quantitative LC–MS/MS analysis further confirmed that exogenously added OI or ITA reduced global 5hmC by as much as 95% in Tet2+/+ BMDMs but had little effect in Tet2−/− cells, in which basal levels of 5hmC were already reduced by ~92% (Fig. 3a). These findings thus suggest that Tet2 is the key Tet enzyme controlling 5hmC production in macrophages and can be nearly completely inhibited by exogenous ITA.
Figure 3. Irg1 produces Itaconate to inhibit Tet activity in vivo.
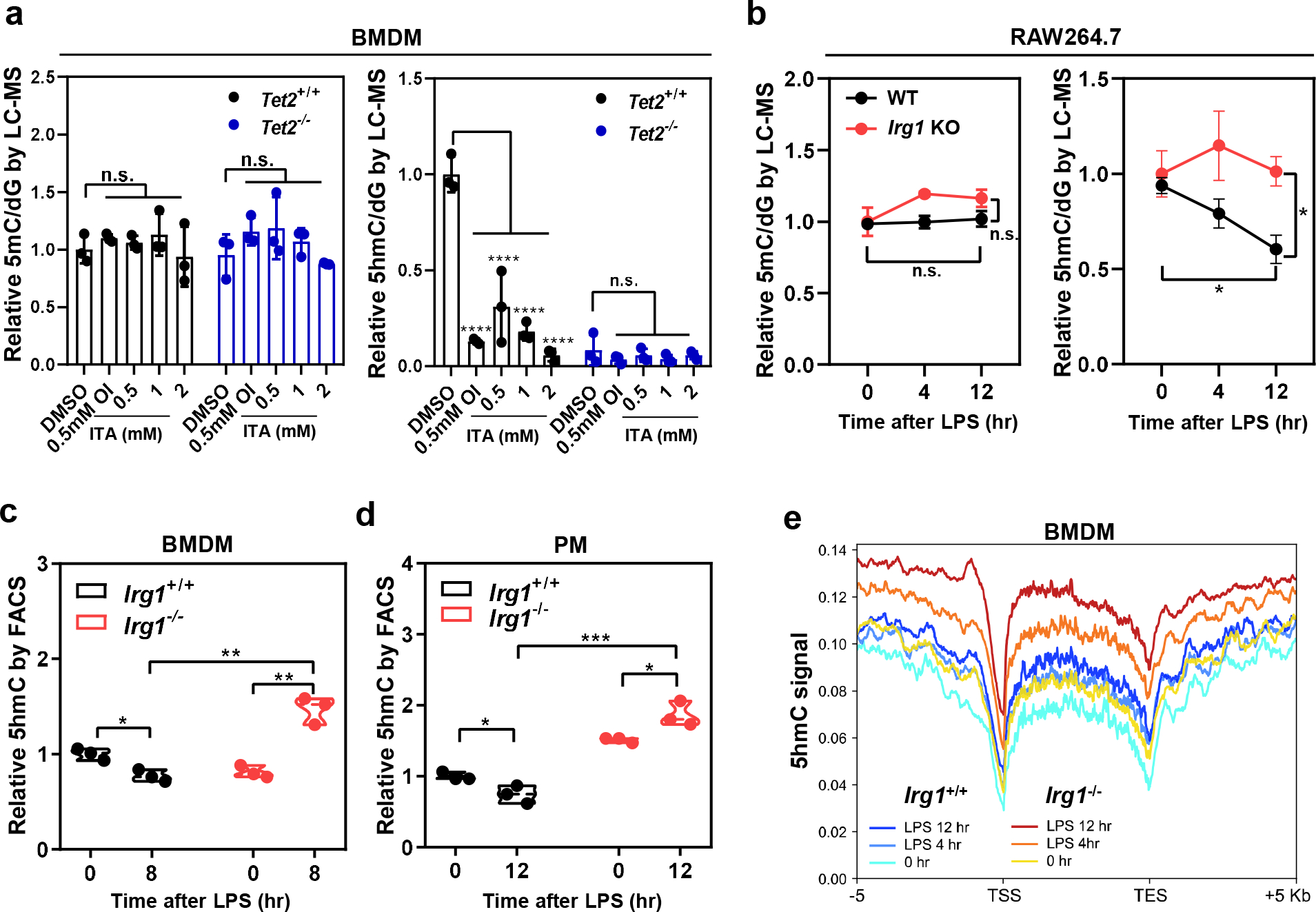
a, Treatment with OI or unmodified ITA reduces genome-wide 5hmC in Tet2+/+ BMDMs, but not Tet2−/− cells. Mouse BMDMs were treated with 0.5 mM OI or increased concentrations of unmodified ITA as indicated for 8 hours. Global levels of 5mC and 5hmC were quantified by LC-MS/MS.
b, LPS treatment reduces genome-wide 5hmC in Irg1-WT but not Irg1-KO RAW264.7 cells. Macrophages were treated with LPS for indicated time, and global levels of 5mC and 5hmC were determined by LC-MS/MS.
c, d, LPS treatment reduces genome-wide 5hmC in Irg1+/+ but not Irg1−/− primary macrophages. Mouse BMDMs (c) and peritoneal macrophage (d) were isolated and then treated with LPS for indicated time. Genomic 5hmC was determined by FACS.
e, Deletion of Irg1 leads to increased 5hmC at promoters and intragenic regions. Irg1+/+ and Irg1−/− BMDMs were treated with LPS for indicated time, following 5hmC mapping by hMeDIP-seq. The normalized density profile for 5hmC across gene body ± 5kb flanking regions is shown. The experiments were performed once.
Shown in a-d are average values with S.D. (a, c) or SEM (b). of three independent experiments. P values are calculated using two-tailed Student’s t-test for paired comparisons (b, c, d) or two-way ANOVA for multiple comparisons (a).*denotes p < 0.05, **denotes p < 0.01, ***denotes p < 0.001, and ****denotes p < 0.0001 for the indicated comparison. n.s. = not significant.
Consistent with previous studies3, 4, LPS treatment rapidly induced Irg1 mRNA within 30 min in RAW264.7 macrophages, followed by protein accumulation within 2 hours (Extended Data Fig. 4a, 4b). As a result, endogenous ITA rapidly accumulated to millimolar levels between 2 to 4 hours after LPS stimulation, and reached as high as 3.75 mM before starting to decline (Extended Data Fig. 4d). This accumulation was completely abolished by deletion of the Irg1 gene (Extended Data Fig. 4c, 4d). LPS treatment also induced the accumulation of succinate but had no significant effect on fumarate or 2-HG levels (Extended Data Fig. 4e). As previously noted14, 15, α-KG levels were also affected, decreasing from ~0.4 mM in untreated macrophages to ~0.2 mM at 4 hours after LPS stimulation (Extended Data Fig. 4e).
In macrophages examined, Tet1 mRNA expression was extremely low, while the mRNA expression of Tet2 and Tet3 was up-regulated and down-regulated by LPS treatment, respectively (Extended Data Fig. 5a). Interestingly, global 5hmC was reduced by LPS stimulation in Irg1-WT RAW264.7 cells but stayed relatively higher in Irg1-KO cells, as determined by LC-MS/MS (Fig. 3b), immunofluorescence staining (Extended Data Fig. 5b), and FACS analysis (Extended Data Fig. 5c). Treatment of LPS-stimulated Irg1-KO RAW264.7 cells with 3-nitropropionic acid (3-NPA), a specific SDH inhibitor, led to intracellular accumulation of succinate to as high as 1.5 mM, but had little effect on global 5hmC (Extended Data Fig. 5d). This result argues against the model that succinate, which is accumulated by ITA-mediated SDH inhibition and itself an inhibitor of Tet enzymes16, is responsible for the LPS-induced TET inhibition. Moreover, the addition of dimethyl-α-KG restored 5hmC in LPS-treated RAW264.7 cells (Extended Data Fig. 5e), further supporting that α-KG and ITA competitively interact with Tet enzymes. In line with cell treatment with exogenous ITA (Extended Data Fig. 3g), LPS induction of ITA had no effect on global histone methylation in RAW264.7 cells (Extended Data Fig. 5f).
In accord with Tet2 transcription, its protein expression was induced by LPS in BMDMs and peritoneal macrophages (PMs) (Extended Data Fig. 5g, 5h). Deletion of Irg1 did not affect Tet2 induction by LPS, but increased 5hmC by 1.9-folds compared to LPS-treated Irg1+/+ BMDMs (Fig. 3c). In PMs, deletion of Irg1 increased both basal and post-LPS levels of 5hmC, with the latter being 2.5-fold higher in Irg1−/− than Irg1+/+ PMs (Fig. 3d). Furthermore, 5hmC mapping demonstrated that deletion of Irg1 increased 5hmC at promoter and intragenic regions in LPS-stimulated BMDMs (Fig. 3e). These results thus suggest that ITA, which is produced by IRG1 during the inflammatory response, selectively inhibits TET enzymes.
Tet2 is a major target of Itaconate to suppress LPS-induced genes in macrophages
Several mechanisms have been proposed for the anti-inflammatory function of ITA17. For instance, ITA may directly inhibit SDH5, resulting in succinate accumulation and metabolic reprogramming6, 7; how succinate accumulation contributes to the anti-inflammatory function of ITA remains unclear. ITA is also known to act as an electrophile in Michael reactions to alkylate cysteine residues in proteins, including KEAP which targets NRF2, and to induce electrophilic stress through activating NRF2- or IκBζ-mediated pathways8, 9. Nonetheless, ITA can also influence the expression of genes, such as IκBζ, IL-6, TNF and ATF3, independently of NRF29, and the precise mechanism by which ITA affects gene expression remains largely unknown. To investigate the role of Tet enzymes as effectors of ITA, we compared the transcriptome changes caused by either OI treatment, which caused inhibition of Tet2 catalytic activity, or Tet2 deletion (Tet2-KO) in LPS-stimulated RAW264.7 cells (Extended Data Fig. 6a). At least 2-fold up- or down-regulation was observed in 3,734 genes when treating Tet2-WT cells with OI, and in 3,717 genes upon Tet2 deletion (Fig. 4a). Considering that the catalytic function of Tet enzymes is typically associated with gene activation, we thus focused on the genes which were down-regulated by OI upon LPS stimulation. While OI treatment led to 1,846 down-regulated genes in Tet2-WT cells, it down-regulated only 807 genes in Tet2-KO cells. Only 493 (27%) out the 1,846 genes down-regulated by OI treatment in Tet2-WT cells were also down-regulated by OI in Tet2-KO cells (Extended Data Fig. 6b), indicating that in absence of Tet2, a large proportion (73%) of genes downregulated by ITA are no longer affected. We further focused on LPS-inducible genes in macrophages. Of 1,846 genes that were down-regulated by OI treatment in Tet2-WT cells, 712 were induced by LPS by at least 2-folds (Extended Data Fig. 6c). Of these 712 genes, 509 (71.5% of the total) were also down-regulated by Tet2 deletion in LPS-stimulated cells (Fig. 4b), whereas only 135 (19.0% of the total) were down-regulated by OI treatment in Tet2-KO cells (Extended Data Fig. 6d). These results thus indicate that a significant fraction of LPS-induced genes is commonly regulated by ITA and Tet2. Many of these genes are involved in the innate immune and inflammatory responses, including signaling pathways mediated by cytokines, such as TNF, Toll-like receptor, and NF-κB (Fig. 4b). K-mean clustering of the 712 LPS-induced and OI-inhibited genes showed similar expression patterns in macrophages between OI treatment and Tet2-KO upon LPS stimulation (Fig. 4c). Together, these data support the notion that ITA acts largely through Tet2 to inhibit the expression of LPS inducible genes in macrophages.
Figure 4. Tet2 is a major target of Itaconate to suppress LPS-induced genes in macrophages.
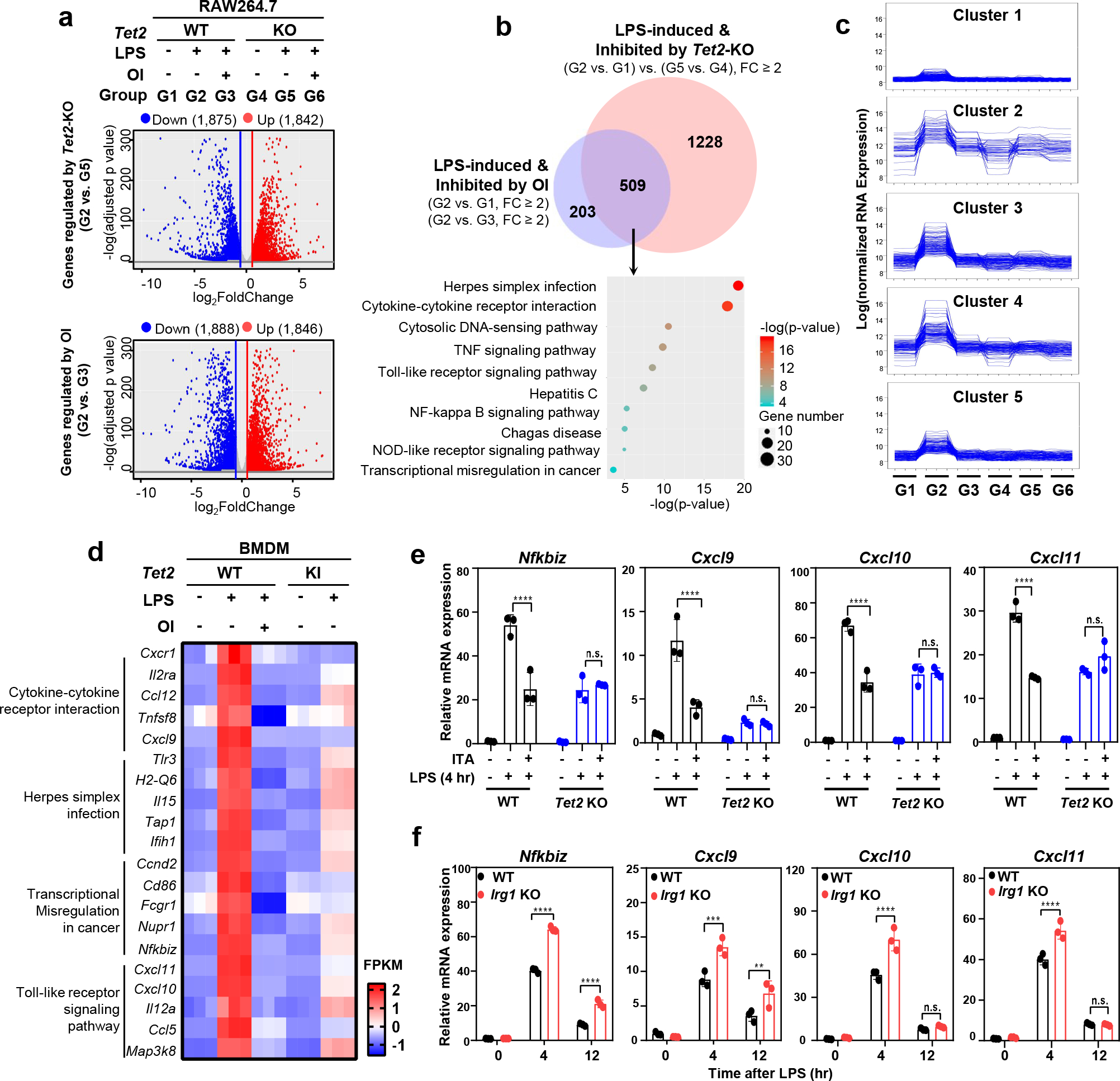
a, Volcano representation of RNA-seq analyses of the effect of OI treatment and Tet2 deletion on regulating gene expression in LPS-treated RAW264.7 cells. All genes differentially expressed with p-value ≤ 0.05 and fold change (FC) ≥2 are highlighted in red (up-regulation) and blue (down-regulation). RNA-seq was performed using three biological replicates. The data shown are average values of three biological replicates.
b, Overlap between LPS-induced genes down-regulated by OI and by Tet2 deletion in RAW264.7 cells is displayed by Venn diagram (p-value < 10−10). Top 10 pathways enriched among the overlapping genes were identified by KEGG pathway analysis.
c, K-mean cluster analysis of 712 which are induced by LPS and down-regulated by OI in RAW264.7 cells, see also Extended Data Fig. 6c.
d, Representative LPS-induced genes inhibited by OI and Tet2 catalytic inactivation in BMDMs. Top LPS-induced genes most significantly affected by OI in BMDMs are selected from 4 signaling pathways that are also affected by OI and Tet2-KO in RAW264.7 macrophages.
e, Unmodified ITA inhibits LPS-induced genes in a Tet2-dependent manner. Tet2-WT and Tet2-KO RAW264.7 cells were pre-treated with unmodified ITA (3 mM) and then stimulated with LPS, following detection of the indicated gene mRNA expression by qRT-PCR.
f, Deletion of Irg1 leads to up-regulation of genes which are induced by LPS but inhibited by either ITA or Tet2 catalytic inactivation. Irg1-WT and Irg1-KO RAW264.7 cells were treated with LPS for indicated time, following determination of indicated gene mRNA expression by qRT-PCR.
Shown in e and f are average values with S.D. of three independent experiments. P values are calculated using two-way ANOVA for multiple comparisons. **denotes p < 0.01, ***denotes p < 0.001, and ****denotes p < 0.0001 for the indicated comparison. n.s. = not significant.
TET2 has both catalytic dependent and independent functions18. To gain more insight into ITA influencing gene expression through inhibiting the catalytic activity of TET2, we generated Tet2H1795R knock-in (KI) mutant mice. H1795 in mouse Tet2 is equivalent to human TET2 H1881, which is recurrently mutated in human acute myeloid leukemia and is essential for TET2 catalytic activity19. We confirmed the catalytic-defective mutation H1795R by genotyping and 5hmC dot-blot assay (Extended Data Fig. 6e, 6f). We then isolated Tet2+/+ and Tet2H1795R (Tet2-KI) BMDMs and treated them with LPS either alone or together with OI, followed by gene expression profiling analysis. Of 1,463 genes that were induced by LPS and down-regulated by OI treatment, 607 (41.5%) were down-regulated by Tet2-KI (Extended Data Fig. 6g). KEGG signaling pathways analysis of these 607 genes revealed that, similarly to our findings in Tet2-KO cells, they were also enriched for genes involved in innate immune and inflammatory responses (Extended Data Fig. 6g), including four pathways dually impaired by both OI treatment and Tet2 deletion in RAW264.7 cells.
Many genes from the four above-mentioned pathways encode chemokines, interleukins, and their receptors (Fig. 4d). Among these genes, Nfkbiz encodes IκBζ, an atypical IκB member and a transcriptional regulator of selective NF-κB target genes. IκBζ regulates IL-6 production in macrophages and other cell types20, as well as IFN-γ production in NK cells21, and it is involved in Th17 development22, psoriasis23, and cellular senescence24. Three (TH1)-type chemokines, Cxcl9, Cxcl10, and Cxcl11, are transcriptionally activated by STAT1-recruited TET2 following interferon treatment25 and attract effector T cells during the inflammatory response26. To provide further evidence supporting TET2 as the major target of ITA, we examined the regulation of selected genes by Irg1/ITA and Tet2 in detail. Our data demonstrated that the effect of exogenous ITA on suppressing LPS-induced chemokine and cytokine genes was completely abolished by Tet2 deletion (Fig. 4e and Extended Data Fig. 7a). To eliminate interference factors from endogenous ITA, we found that shortly after LPS stimulation before endogenous ITA started accumulating, addition of OI still hindered LPS-induced mRNA expression of Nfkbiz, Il-6, and Cxcl9/10/11, in Tet2-WT macrophages, mimicking the effect of Tet2 deletion (Extended Data Fig. 7b). In contrast, LPS-induced mRNA expression of chemokines and cytokines was further increased by Irg1-deletion in macrophages (Fig. 4f).
Members of the NF-κB transcription factor family play a critical role in innate immunity in response to pathogen infections, tissue injury, and tumor inflammation27. Among 16 members of NF-κB, IκB and IKK families, Nfkbiz was the only one whose induction by LPS was hindered by Tet2 inactivation (by either OI treatment or Tet2-KI) or Tet2 deletion (Extended Data Fig. 8a, 8b, 8c). OI treatment failed to affect Nfkbiz mRNA expression in LPS-stimulated Tet2-KO cells (Extended Data Fig. 8d), and treatment with dimethyl-α-KG was able to further increase Nfkbiz mRNA in LPS-stimulated Tet2-WT but not Tet2-KO cells (Extended Data Fig. 8e, 8f). In accord, western blotting analysis confirmed the effects of ITA and α-KG on inhibiting and stimulating LPS-induced IκBζ protein, respectively, in a Tet2-dependent manner (Extended Data Fig. 8g, 8h). Together, these results support TET2 as the major target of ITA function in suppressing LPS-induced genes, including those regulated by both NF-κB and STAT1 immune signaling pathways.
RelA interacts with and recruits Tet2 to Nfkbiz promoter in LPS-activated macrophages
TET2 is known to be recruited to DNA by multiple sequence-specific transcription factors (TFs), including WT119, 28, STAT329, CXXC530, and STAT131. In searching for TFs that might recruit TET2 to regulate LPS-induction of Nfkbiz, we found that LPS stimulated Tet2 binding with NF-κB p65/RelA in RAW264.7 cells (Fig. 5a, 5b). The proximal promoter region of mouse Nfkbiz gene contains three putative canonical κB sites and two of them, κB1 and κB2, are conserved in the human Nfkbiz promoter32 (Fig. 5c). Systematic investigation of TF binding in resting or activated macrophages has previously identified RelA bindings at the Nfkbiz promoter33. We confirmed the binding of RelA to the Nfkbiz promoter and found that it was stimulated by LPS but abolished by RelA knockdown (Fig. 5d). Tet2 bound to the Nfkbiz promoter (Fig. 5e), and this binding was stimulated by LPS in a manner dependent on RelA (Fig. 5f). In accord with Tet2 occupancy, 5hmC in the Nfkbiz promoter was low in unstimulated macrophages, while was increased by 4.3-folds after LPS stimulation, and this increase was abolished by RelA knockdown (Fig. 5g). Along with lower levels of 5hmC at the Nfkbiz promoter, RelA knockdown led to decreased Nfkbiz mRNA levels in RAW264.7 cells during the LPS response, as observed upon TET2 loss (Fig. 5h, 5j). In contrast, Nfkbiz mRNA was up-regulated by Irg1-KO in LPS-stimulated macrophages (Fig. 5h), which was associated with higher 5hmC in the Nfkbiz proximal promoter region (Fig. 5i). Similar to Nfkbiz, the expression of multiple Tet2-dependent cytokine and chemokine genes was also inhibited by RelA knockdown in LPS-stimulated macrophages (Fig. 5j). Taken together, these results suggest that, in response to LPS stimulation, p65 NF-κB/RelA recruits Tet2 to induce DNA demethylation and gene expression.
Figure 5. RelA interacts with and recruits Tet2 to Nfkbiz promoter in LPS-activated macrophages.
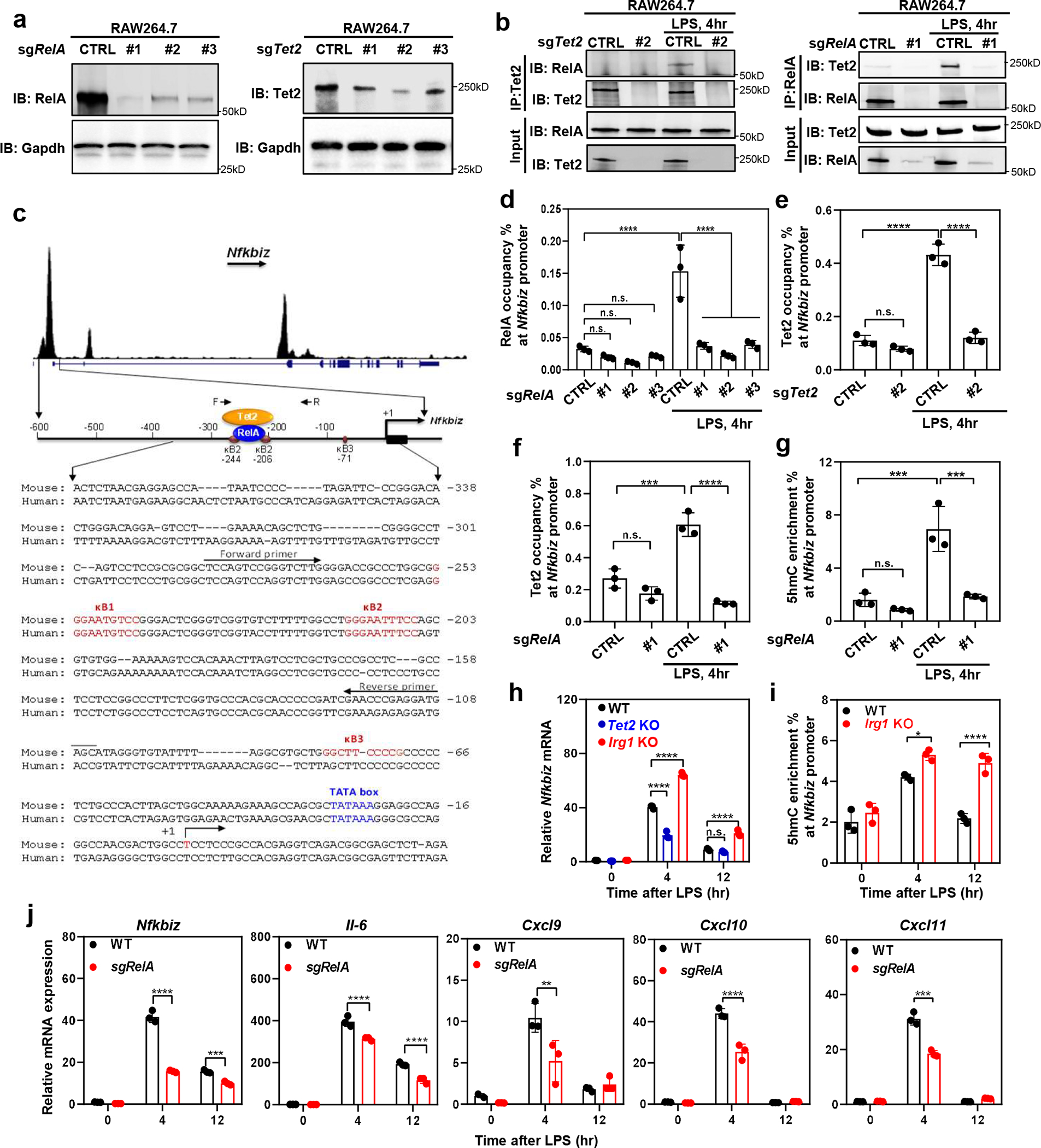
a, Knockdown of RelA or Tet2 in RAW264.7 cells by sgRNA, confirmed by western blot.
b, LPS stimulates RelA-Tet2 binding. RAW264.7 cells were treated with sgRNA against Tet2 (left) or RelA (right) and then challenged without or with LPS for 4 hours, followed by co-immunoprecipitation assay to detect the protein interaction between RelA and Tet2 using indicated antibodies. A representative blot of two independent experiments is shown in (a, b).
c, Top panel shows RelA binding to the promoter of Nfkbiz promoter identified by the systematic ChIP-seq analyses of transcription factor binding in resting and activated macrophages33. Middle panel: Schematic illustration of the proximal promoter region of mouse Nfkbiz gene, cis-regulatory elements and binding of RelA and Tet2 as determined by ChIP assay in Fig. 5. Bottom panel: Nucleotide sequence of the proximal promoter region of mouse and human Nfkbiz genes. Three putative canonical κB sites, transcription start sites were previously identified32.
d, RelA binds to Nfkbiz promoter. RelA occupancy at the promoter region of Nfkbiz was determined by ChIP-qPCR in control and RelA knockdown RAW264.7 cells.
e, Tet2 binds to Nfkbiz promoter. Tet2 occupancy at the promoter region of Nfkbiz was determined by ChIP-qPCR in control and Tet2 knockdown RAW264.7 cells.
f, g, RelA recruits Tet2 to bind and catalyze 5mC hydroxylation of Nfkbiz gene promoter. Tet2 occupancy (f) and 5hmC enrichment (g) were determined by ChIP-qPCR assays in unstimulated and LPS-treated RAW264.7 cells using relevant antibodies.
h, Deletion of Tet2 and Irg1 leads to down-regulation and up-regulation of Nfkbiz mRNA expression, respectively, as determined by qRT-PCR assays.
i, Irg1-WT and Irg1-KO RAW264.7 cells were treated with LPS as indicated. The Nfkbiz promoter enrichment of 5hmC was determined by hMeDIP-qPCR.
j, RAW264.7 with or without RelA knockdown were treated with LPS for indicated time. The mRNA expression was determined by qRT-PCR assays.
Shown in d-j are average values with S.D. of three independent experiments. P values are calculated using one way (d-g) or two-way ANOVA (h, j) for multiple comparisons. *denotes p < 0.05, **denotes p < 0.01, ***denotes p < 0.001, and ****denotes p < 0.0001 for the indicated comparison. n.s. = not significant.
Itaconate reduces LPS-induced mouse mortality in a manner dependent on Tet2
To further investigate the role of ITA-mediated TET2 inhibition in vivo, intraperitoneal injection of LPS was conducted to induce inflammation in mice. We found that at four hours post LPS challenge, ITA in Irg1+/+ peritoneal leukocytes was increased by 13.5-folds (from ~23.5 μM to 316.7 μM) (Extended Data Fig. 9a, 9b). Basal 5hmC was similar between Irg1−/− and Irg1+/+ leukocytes, while it was significantly higher in LPS-challenged Irg1−/− leukocytes than Irg1+/+ cells (Fig. 6a). Quantitative LC-MS/MS analysis confirmed that genomic 5hmC was 2.5-fold higher in Irg1−/− leukocytes than Irg1+/+ cells at four hours after LPS injection (Fig. 6b). ChIP-qPCR analysis demonstrated that 5hmC at the Nfkbiz promoter was stimulated by LPS in WT, but not Tet2−/− leukocytes and, importantly, deletion of Irg1 further increased 5hmC at Nfkbiz promoter by 1.8-fold as compared to WT cells after LPS challenge (Fig. 6c). In LPS-stimulated leukocytes of different genotypes, 5hmC levels at the Nfkbiz promoter closely correlated to its mRNA levels, which were the highest in LPS-challenged Irg1−/− leukocytes followed by WT and then Tet2−/− cells (Fig. 6d). In addition, we also extended this finding to other target genes of NF-κB-TET2 and STAT1-TET2 axes, including Cxcl9, Cxcl10, Cxcl11, Il6, Il-12a, and Il-15 (Fig. 6d).
Figure 6. Itaconate reduces LPS-induced mouse mortality in a manner dependent on Tet2.
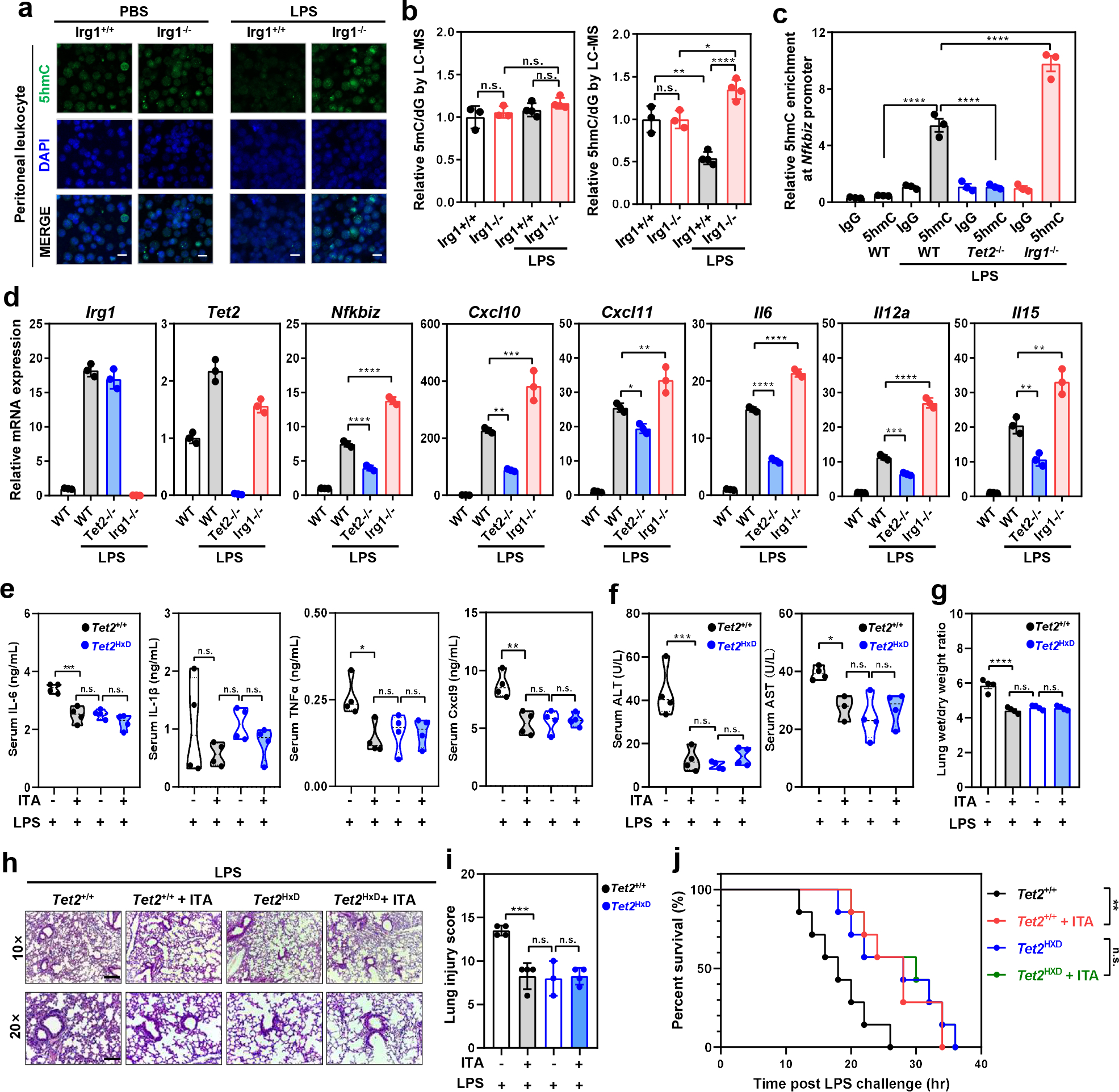
a,b, Peritoneal leukocytes were freshly isolated from indicated mice at 4 hours post i.p. injection of PBS or LPS (20 mg/kg). Genomic 5hmC was detected by immunofluorescence staining (IF, n=3 mice per group) (a) and LC-MS/MS (n=3–4 mice per group) (b). Representative IF result is shown. Scale bar, 20 μm.
c,. As described above in (a-b), the Nfkbiz promoter enrichment of 5hmC was determined by hMeDIP-qPCR in peritoneal leukocytes.
d, As described above in (a-b), mouse peritoneal leukocytes were harvested and analyzed for indicated gene expression by qRT-PCR.
e, Transplanted mice were i.p. injected with ITA prior to LPS challenge (25 mg/kg; n=4 mice per group). At 4 hours post LPS, serum levels of indicated cytokines /chemokines were determined as described in Methods.
f, As described above in (e), serum samples were collected for measurement of ALT and AST activities as described in Methods.
g,h,i, As described above in (e), lung tissues of transplanted mice(n=3–4 mice) were harvested and then subjected to the wet/dry weight ratio calculation (g) as well as HE staining (h). The injury score was determined as described in Methods (i). Representative photomicrographs are shown. Scale bar, 200 μm (upper panels)& 100 μm (lower panels).
j, Transplanted mice were i.p. injected with ITA prior to LPS challenge (25 mg/kg; n=7 mice per group), and the animal survival was monitored and Kaplan-Meier survival curves were determined as described in Methods.
Shown in c, d are average values with S.D. (b, d-f) or SEM. (c,g) of three independent experiments (if not pointed out). P- values are calculated using one-way ANOVA (b-f); As for the percent survival, P values were determined using log-rank (Mantel-Cox) test comparing each 2 groups; *denotes p < 0.05, **denotes p < 0.01, ***denotes p < 0.001, and ****denotes p < 0.0001 for the indicated comparison. n.s. = not significant.
Next, we employed a mouse model of LPS-induced sepsis and found that in line with our findings in isolated leukocytes (Fig. 6d), serum Il-6 was higher in LPS-challenged Irg1−/− mice than Irg1+/+ controls (Extended Data Fig. 9c). Irg1−/− mice were more susceptible to LPS-induced acute lung injury and had higher mortality (Extended Data Fig. 9d, 9e, 9f), confirming the role of Irg1/ITA in dampening LPS-induced inflammatory response. Conversely, intraperitoneal injection of ITA led to reduced 5hmC by as much as 36% in peritoneal leukocytes, while 5mC was unaffected (Extended Data Fig. 10a). ITA treatment was able to attenuate LPS-induced pulmonary edema (wet-to-dry ratio, W/D; Extended Data Fig. 10b) as well as acute lung injury (Extended Data Fig. 10c, 10d), and protected mice against lethal endotoxaemia (Extended Data Fig. 10e). These anti-inflammatory and protective effects of ITA were not observed in Tet2−/− mice (Extended Data Fig. 10b, 10c, 10d, 10f).
To further strengthen the functional significance of the Irg1/ITA/Tet2 axis, chimeric mice were developed by transplanting bone marrow cells from catalytically inactive Tet2HxD knock-in (KI) mutant or WT mice (Extended Data Fig. 10g). The Tet2HxD KI mutant mice contain H1295Y and D1297A double substitution mutations in mouse Tet2, which are equivalent to human TET2 H1382 and H1384, respectively34, and disrupt the binding with the essential cofactor Fe2+. Reconstitution of hemapotoiesis with Tet2HxD bone marrow led to ~50% reduction in global 5hmC in transplanted animals (Extended Data Fig. 10h, 10i). Compared with WT chimeric mice, Tet2HxD chimeric mice had dampened LPS-induced inflammatory responses, including lower serum levels of pro-inflammatory cytokines and chemokines such as Il-6, Tnf-α, Cxcl9 (Fig. 6e), reduced serum activities of ALT and AST (liver damage markers; Fig. 6f), less severe pulmonary edema (Fig. 6g) and lung injury (Fig. 6h, 6i), and prolonged survival (Fig. 6j). Importantly, these molecular, tissue and animal changes caused by the catalytic inactivation of Tet2 were all recapitulated in ITA-pretreated WT chimeric mice upon LPS challenge (Fig. 6e–6j). These results thus provide direct in vivo evidence to support that TET2 is a major functional target of ITA and that the Irg1/ITA/Tet2 axis plays a key physiological role in immune modulation.
Discussion
Four metabolites, D-2HG, succinate, fumarate, and L-2HG, have previously been reported to inhibit TET and other α-KG/Fe(II)-dependent dioxygenases such as histone demethylase KDMs. Among these four metabolites, D-2HG accumulates in cancer cells with mutated isocitrate dehydrogenases (IDH1/2), while L-2HG is produced by the promiscuous activity of either lactate dehydrogenase (LDH) or malate dehydrogenase (MDH) in cells under acidic or hypoxic conditions35. Succinate and fumarate can accumulate as a result of loss-of-function mutations in cancer cells affecting SDH and fumarate dehydrogenase (FH), respectively, as well as under certain conditions such as glutamine replenishment of the TCA cycle36, 37. In contrast to these metabolites, ITA is the primary reaction product of the metabolic enzyme IRG1, whose level remains virtually undetectable in the absence of stimuli and is boosted in cells of myeloid lineage during the inflammatory response. Unlike above-mentioned four metabolites which inhibit both TET and KDM enzymes involved in epigenetic regulation, ITA is a physiological metabolite that selectively inhibits TET enzymes.
Our results presented here connect the function of Irg1/ITA and Tet2 during inflammatory response. Mutations in TET2 lead to clonal hematopoiesis, altered response to immunotherapy, atherosclerosis, and hematopoietic malignancy38–40. Separately, genetic studies in mice have demonstrated a critical function of Irg1 in inflammatory response to wide range of pathogens, such as M. tuberculosis 2, Zika virus 1, Salmonella41 and LPS (Fig. 6). Not only does our study demonstrate that TET2 is inhibited by ITA, but also that TET2 is an important functional target of ITA. Furthermore, this study uncovers three prevoisly undescribed regulatory mechanisms linked to the inflammatory response, that is, the inhibition of TET2 by the anti-inflammatory metabolite ITA, the functional interaction between TET2 and a key inflammatory regulator NF-κB, and the transcriptional activation of a key driver in inflammation, IκBζ, by TET2-mediated DNA demethylation and subsequent downregulation by ITA inhibition of TET2. Together, these findings establish a prevoisly undescribed Irg1/ITA-Tet2-NF-κB-IκBζ axis that mediates inflammatory response.
TET2 is recruited by WT119, 28, STAT329, CXXC530, STAT131, and NF-κB (Fig. 5) to regulate immune cell differentiation, homeostasis and function. STAT and NF-κB mediate two principal signaling pathways during the inflammatory response in different types of immune cells, including macrophages. In this study, we show that Irg1/ITA inhibits Tet2 to dampen LPS-induced expression of both the NF-κB-TET2 and STAT1-TET2 target genes in macrophages, while deletion of Irg1 leads to transcriptional activation of these genes (Fig. 4 and Fig. 6). The inhibition of NF-κB-Tet2 and STAT1-Tet2 by ITA provides a molecular basis of how ITA can exert an inhibitory effect on many proinflammatory genes in both STAT and NF-κB signaling pathways (Fig. 7). Of note, Tet2 can also recruit HDAC to repress the expression of inflammatory genes such as Il-6 18. These findings lead us to speculate the biphasic model for how macrophages use TET2 to regulate gene transcription during inflammatory response: a catalytic-dependent function to transactivate inflammatory genes during early response after being recruited by transcription factors to their target genes, and then switch to enzymatic inhibition by ITA and catalytic-independent repression of gene expression to resolve inflammation.
The broad function of ITA is consistent with the induction of IRG1 by diverse pathogens such as Mycobacterium2, 42, Salmonella 41, influenza A virus 43, West Nile virus 44, Zika virus 1, Marek disease virus 45 and T.gondii 46, as well as induction in human PBMCs of sepsis patients 47. In addition to viral and bacterial infection, IRG1 is also induced in Zebrafish epidermal cells during cutaneous inflammation48, by particulate matter air pollution 49, during peritoneal tumorigenesis50, and embryonic implantation51. The results presented here support the biological relevance of TET inhibition by ITA in broad physiological and pathological processes and provide previously unapprecaited insights into the regulation of these processes. In addition to Tet inhibition, ITA may exert its anti-inflammatory effect through additional mechanisms, such as SDH inhibition to cause succinate accumulation and metabolic reprogramming, protein alkylation to induce electrophilic stress and activation of the KEAP1-NRF2 signaling pathway, and impairment of aerobic glycolysis. It will be important for future studies to parse the contribution of these different mechanisms to the physiological functions of IRG1 in response to different pathogens and endogenous inflammatory conditions.
Methods
Animals and study approval
Tet2−/− mice (JAX stock #023359) and Irg1−/− mice (JAX stock #029340) were purchased from the Jackson Laboratory. Animals were backcrossed for more than 7 generations onto the C57BL/6J and C57BL/6N background, respectively. Animals were maintained at Shanghai Research Center for Model Organisms. Animals were given unrestricted access to a standard diet and tap water. Animal experiments were performed at Fudan Animal Center in accordance with animal welfare guidelines, and the procedures were approved by the Ethics Committee of the Institutes of Biomedical Sciences (IBS), Fudan University. Analysis was performed on 6–10 week-old female mice obtained from this breeding.
Tet2H1795R KI mutant mice were generated using the CRISPR/Cas9 system, by the UNC Animal Models Core (AMC), with direct pronuclear injection of mRNA and sgRNAs targeting Tet2 KI in C57BL/6J embryos. Successful point mutant insertion was confirmed by PCR and DNA sequencing, and a single founder was mated to C57BL/6J females. The animal experiments were approved by the IACUC at UNC–Chapel Hill. Analysis was performed on 6–10 week-old male mice obtained from this breeding.
Tet2HxD KI mutant mice on C57BL/6J background were constructed by using CRISPR/Cas9 system and were kindly provided by Dr. Jiayu Chen and Dr. Shaorong Gao (School of Life Sciences and Technology, Tongji University, Shanghai). Tet2HxD KI and sex-matched wild-type mice were used as donor for bone marrow transplantation (BMT): In brief, C57BL/6 J mice (male, 8–10 weeks old) underwent total body irradiation with a total of 7.5 Gy from a cesium source, and the lethally irradiated recipients were transplanted with 2×106 whole bone marrow cells of donors by tail vein injection. The transplanted animals were maintained on antibiotic water in pathogen-free facilities at Shanghai Children’s Medical Center. The rate of chimerism in each transplanted animal was verified by PCR sequencing upon sacrifice. At 6 weeks after BMT, antibiotic water was withdrawed for 3 days, and the transplanted animals were subjected to a model of LPS-induced sepsis as decribed below. The animal experiments were approved and supervised by the Animal Research Committee (ARC) of Shanghai Jiao Tong University of Medicine with proper ethical regulations. Analysis was performed on 6–10 week-old female mice obtained from this breeding.
This study complies with all relevant ethical regulations approved by the Ethics Committee of the Institutes of Biomedical Sciences (IBS), Fudan University, protocol number is DSF-2020–066 and UNC Institutional Animal Care and Use Committee (IACUC), protocol number is 19–115.
Cell culture
RAW264.7 cells (ATCC® TIB-71™, passage 25) and HEK293T cells (ATCC® CRL-3216™) were acquired from UNC Lineberger Tissue Culture Facility and were maintained in DMEM medium containing 10% FBS, 1% Penicillin/Streptomycin antibiotics. Sf9 insect cells were purchased from ATCC(CRL-1711™) and were maintained in Cytiva SFX-Insect™ Cell Culture Media (Cytiva, SH30278.02).
For isolation of bone marrow-derived macrophages (BMDMs), mice were euthanized in a CO2 chamber and death was confirmed by cervical dislocation. Bone marrow cells were harvested from the femur and the tibia and differentiated in DMEM (containing 10% fetal calf serum, 1% penicillin/streptomycin and 20% and 20 ng/mL M-CSF) for 6 days. 1 × 106 BMDMs per milliliter were used for LPS and/or OI treatments. Unless otherwise stated, the cells were pre-treated with OI (250 μM) for 12 hours and then stimulated with LPS (100 ng/ml). Moreover, the unmodified itaconic acid was adjusted to pH 7.0 with NaOH. Cells were pretreated with unmodified ITA for 12 hs using indicated dose.
For isolation of peritoneal macrophages, peritoneal exudate cells were elicited by intraperitoneal (i.p.) injection of 1 ml of Brewer’s thioglycolate (Sigma) as previously described52. At 24 hours after i.p. injection, cells were harvested by peritoneal lavage using 5 ml of ice-cold, sterile PBS. Total cells were plated on a 90-mm dish and allowed to adhere for 24 hours. Cells were then washed 3 times with cold PBS to remove non-adherent cells, re-plated and treated with LPS (100 ng/ml) for indicated time.
Cell lines were authenticated by monitoring morphology and growth characteristics. No contamination tests were performed. No commonly misidentified cells were used in this study.
Transfection and immunoprecipitation
Transfection of RAW264.7 cells was performed using Amaxa Cell Line Nucleofector Kit V according to the manufacturer’s protocol (Lonza VCA-1003). For the other cell transfection, we used Lipofectamine 2000 (Invitrogen) or polyJet (SignaGen) following the manufacturer’s instruction.
For immunoprecipitation, cells were washed with cold phosphate buffered saline (PBS) and lysed in NP-40 buffer, containing 50 mM Tris-HCl, 300 mM NaCl, 0.3% NP-40, pH 7.4 and protease inhibitor cocktail (Roche) with rotation at 4°C for 40 min. After centrifugation at 13000×g for 15 min at 4°C, the supernatant was incubated with indicated antibodies for 2 hrs, followed by incubation with Protein-A/G for another 1 hr at 4°C.
Histone methylation profiling by LC-MS/MS
Histone methylation was quantified by MRM-based LC-MS/MS method, as described by previous methods 53, 54. In short, the core histones were acid extracted from tissue with 0.4 M H2SO4 and precipitated with trichloroacetic acid (TCA), followed by washing with ice-cold acetone twice. Then the histone protein was treated with N-Hydroxysuccinimide-propionate and digested with trypsin. Finally, the digested peptides were concentrated to dryness. Prior to the LC-MRM-MS, a mixture of isotope labeled histone peptides was added as internal standards to the samples, the histone peptides concentration in the samples was calculated by the peak area ratio of histone peptides to internal standard peptides.
Global DNA de/methylation quantification by LC-MS/MS
Genomic DNA from cultured cells was digested with DNA Degradase Plus (Zymo Research) at 37°C for 3 hrs. Deoxyguanosine (dG) was used as an internal control. The digested samples were then subjected to LC-MS/MS analysis using a ShimazuLC (LC-20AB pump) system coupled with 4000qtrap triple quadrupole mass spectrometer (AB sciex). A C18 column (250mm×2.1mm I.D., 3 μm particle size, ULTIMATE) was used. The mass spectrometer was optimized and set up in selected reaction monitoring (SRM) scan mode for monitoring the [M+H+] of 5mC (m/z 242.1→126.1), 5hmC (m/z 258.1142.1) and dG (m/z 268.1→ 152.1). The Analyst Software was used for analysis (Analyst 1.6).
TET2 in vitro activity assay by dot-blot
In vitro assay of TET2 catalytic activity was performed using methylated dsDNA oligonucleotides as substrate. In brief, Flag-tagged mTET2CD was ectopically expressed in Sf9 insect cells, immunoprecipitated, and eluted with Flag peptide. The reactions were carried out as described before55, denatured and then neutralized. The reaction mixtures were spotted on nitrocellulose membrane, cross-linked and incubated overnight with antibodies recognizing 5mC (Active Motif, 2793764) or 5hmC (Active Motif, 10013602). Immunoblots were developed using secondary horseradish peroxidase (HRP)-coupled antibodies and a chemiluminescence kit.
TET2 in vitro activity assay by a commercial kit
The Epigenase™ 5mC-Hydroxylase TET Activity/Inhibition Assay Kit (P-3086–96) was used to determine TET2 enzyme activity, according to the manufacturer’s protocol. In brief, 1 μL containing 120 ng of Flag-tagged mTET2CD purified from Sf9 cells was added to a total reaction system of 50 μL, with or without metabolites. After a 30-min incubation at 37°C, absorbance at 450 nm was read.
TET2 in vitro activity assay using immobilized substrate DNA
TET2 in vitro activity assay was developed based on immobilized substrate DNA and fluorescence detection of 5hmC. Briefly, two complementary single-stranded and biotin-labelled oligonucleotides were synthesized at Sigma (5’GTATGCCTCATGCmCGGACTTAACTGCAGTG 3’, 3’ CATACGGAGTACGGmCCTGAATTGACGTCAC5’-[Btn]. The superscript ‘m’ indicates the methylation of following cytosine, whereas ‘Btn’ means biotin label). 2 nmol of single-stranded oligonucleotides were mixed in annealing buffer (10 mM Tris, pH 8.0, 50 mM NaCl, 1 mM EDTA), boiled at 99 °C for 5 min and subsequently transferred to 90 °C-preheated water, which was gradually cooled down to room temperature. 50 pmol of annealed double-stranded DNA oligonucleotides were added to Pierce™ Streptavidin Coated High Capacity 96-wells plates (Catalog number: 15525) and immobilized at room temperature for 30 min. After substrate immobilization, reaction buffer (50 mM HEPES buffer (pH 8.0), 50 μM ammonium iron (II) sulfate, 0.1 mM α-KG, 2 mM ascorbic acid, 2.5 mM DTT, 100 mM NaCl, and 1.2 mM ATP55) with or without metabolites was added into each well, followed by addition of 120 ng of purified mTET2CD per assay. The reactions were incubated at 37°C for 30 min with constant shaking on an Eppendorf ThermoMixer (700 r.p.m). 5hmC was detected by a first incubation with anti-5hmC antibody for 3 hrs at room temperature, followed by 1-hr incubation at room temperature with a Fluorescent-dye conjugated secondary antibody or HRP conjugated secondary antibody. The amount of 5hmC was quantified by using a microreader with excitation at 495 nm and emission at 518 nm.
Nuclear magnetic resonance (NMR)
All samples were prepared in a buffer containing 20 mM BisTris (pH 6.5), 140 mM NaCl, 50 μM FeSO4, 0.6 mM BME, and 80% D2O, where ITA and α-KG stocks were first prepared at pH 6.5 with NaOH. Standard Bruker STD NMR measurements were carried out at 25°C on a Bruker Avance III 700 MHz spectrometer equipped with a 5 mm triple-resonance cryogenic probe. With 1H carrier being on water, a 3-second saturation was performed using a 50-millisecond EBurp2 pulse with a bandwidth of 0.75 ppm centered at 0.8 ppm or −40 ppm for difference and reference experiments, respectively. NMR data were processed using Bruker Topspin 3.5.7.
Gene deletion by CRISPR/Cas9 system
Tet2-KO and Irg1-KO RAW264.7 cells were generated through the CRISPR (clustered regularly interspaced short palindromic repeats)/Cas9 system56. Cells were transiently transfected with Cas9 and single-guide RNA (sgRNA) plasmid with EGFP expression (PX458; Addgene plasmid #48138). The gRNA sequences used for targeting Tet2 and Irg1 were AAAGTGCCAACAGATATCCA31 and AGCTTTGCTGGTATGATTCA, respectively. Two days after transfection, single cells expressing EGFP were sorted into 96-well plates by using fluorescence-activated cell sorting (BD, FACSAria™ III). KO clones were validated by DNA sequencing and western-blot analysis.
Intracellular metabolite quantification by LC-MS/MS
Cell number and cell diameter were measured using and automated cell counter (Countstar). C13-labelled ITA was added as internal standard. Cells were fixed by immediate addition of 1 ml 80% (v/v) chilled (−80°C) methanol. Cell extracts were analyzed by ultrahigh performance liquid chromatograph (Acquity UPLC I-Class, Waters) coupled to a Triple Quadrupole Mass Spectrometer (Xevo TQ-XS, Waters). Cells were assumed to have a spherical shape, and intracellular metabolite concentration was calculated using external standard curves and taking into account cellular diameter (d, micrometers) and cell number using the following equation: [metabolite] = metabolite quantity (moles)/((4/3000)π(d/2)3). The data were analyzed using MassLynx V4.2.
Immunofluorescence (IF)
Cells were washed with cold PBS, fixed with 4% PMSF (Sangon) for 15 min, and permeabilized with 0.3% Triton X-100 for 15 min at room temperature. For 5hmC staining, DNA was denatured with 2 N HCl for 30 min and then neutralized with 100 mM Tris-HCl (PH 8.5) for 10 min. After washing 3 times with PBS, samples were incubated with blocking buffer (3% BSA in PBS) for 1 hr, followed by incubation at 4°C overnight with anti-5hmC primary antibody. After washing 3 times with PBS, cells were incubated with Alexa Fluor 594 (Green) conjugated secondary antibody (Invitrogen) at room temperature for 1 hr. Cell nucleus was stained with DAPI (Invitrogen). Images were captured using Leica fluorescence optical microscope.
Global 5hmC quantification by FACS
The FACS assay was performed as previously described57. Cells were re-suspended in FACS buffer (PBS with 1% BSA, 2 mM EDTA) and incubated with Fc blocker for 10 min on ice. After washing with FACS buffer, cells were fixed and permeabilized using the Cell Fixation/permeabilization kit (BD Biosciences). DNA was denatured by adding 2N HCl for 20 min and then neutralized with 10 mM Tris-HCl (pH 8.0) for 20 min. Anti-5hmC antibody (Active Motif, 1:200) and FITC conjugated goat anti-rabbit secondary antibody (1:200; Thermo Fisher Scientific) were used for 5hmC staining. Attune NxT Acoustic Focusing Cytometer( AFC2, Invitrogen) was used to collect data and data were analyzed using the FlowJo (10.6.2) software.
Global 5mC and 5hmC quantification by dot-blot
DNA dot-blot assay was performed as described previously with some modifications19, 31, 58. Briefly, genomic DNA was spotted on a nitrocellulose membrane (Whatman). The membrane was placed under an ultraviolet lamp for 20 min to crosslink DNA, and then blocked with 5% milk in TBS-Tween20 for 1 hr, followed by incubation with the anti-5mC or anti-5hmC antibody at 4°C overnight. After incubation with an HRP-conjugated secondary antibody (GeneScript) for 1 hr at RT, the membrane was washed with TBS-Tween 20 for three times and then scanned by a Typhoon scanner (GE Healthcare), and the quantification of 5mC or 5hmC was done by Image-Quanta software (GE Healthcare).
Hydroxymethylcytosine DNA immunoprecipitation sequencing (hMeDIP-seq)
5hmC mapping was conducted as described by previous methods59. Briefly, mouse BMDMs were pre-treated with or without 0.5 mM OI for 8 hrs, and then stimulated with 100 ng/ml LPS for 4 hrs. From these cells, 10 μg of sonicated and adaptor ligated genomic DNA was used as input, and 5 μL of 5hmC antibody (Active Motif) was used to immunoprecipitate modified DNA. Immunoprecipitated DNA was amplified for Illumina sequencing. Detailed sequencing information of hMeDIP-seq data used in this study were summarized in Supplementary Table 2. The DIP-seq datasets have been deposited in Gene Expression Omnibus (GEO) under the accession number GSE158580.
RNA purification and real-time quantitative PCR (qRT-PCR)
Total RNA was extracted from cells using TRIzol (Invitrogen) as recommended by the manufacturer’s protocol. RNA was reversely transcribed using oligo-dT primers. Diluted cDNA was then used in qRT-PCR reactions containing SYBR Premix ExTaq (TaKaRa) and gene-specific primers. The reactions were performed in a QuantStudio™ 6 Flex Real-Time PCR System (Applied Biosystems). β-Actin was used as a housekeeping control. Primers were listed in Supplementary Table 1.
RNA sequencing (RNA-seq)
RAW264.7 or BMDM cells were treated with or without LPS for 4 hrs and pre-treated with 250 μM OI for 12 hrs and then were subjected to RNA extraction. Each condition was prepared in triplicate for each RNA-seq experiment. Library preparation was done using the Illumina TrueSeq mRNA sample preparation kit and sequencing on Illumina HiSeq4000 machine was performed by High Throughput Genomic Sequencing in Novagene. Paired-end FASTQ sequences were aligned to the mouse genome (mm10). Differential gene expression analysis was performed using the DESeq2 package and selecting raw gene counts output from Rsubread60. Genes with a fold change of >=1.2 or 2.0 (as labelled in the figure captions) and FDR < 0.05 were considered to be significantly differentially expressed. This information was also added in the method of RNA sequencing (RNA-se Datasets have been deposited in Gene Expression Omnibus (GEO) under the accession number GSE148143, GSE148145 and GSE148147.
Chromatin-immunoprecipitation with quantitative PCR (ChIP-qPCR)
ChIP assay was performed as described previously61. Briefly, cells were cross-linked with 1% paraformaldehyde and sonicated at 4°C for 20 min (Bioruptor, low mode). Chromatin was immunoprecipitated at 4°C for 3 hrs with indicated antibodies. Antibody-chromatin complexes were pulled-down using protein A-Sepharose (RepliGen), washed and eluted. After cross-link reversal and proteinase K (TaKaRa) treatment, immunoprecipitated DNA was extracted using a PCR Purification Kit (QIAGEN). The DNA fragments were further analyzed by real-time quantitative PCR using the primers as listed in Supplementary Table 1.
Hydroxymethylated DNA immunoprecipitation with quantitative PCR, hMeDIP-qPCR
The hMeDIP-qPCR assay was performed as previously described62. Briefly, 2 μg genomic DNA was extracted from cultured cells by the phenol-chloroform method and then denatured and immunoprecipitated with anti-5hmC (Active Motif) or rabbit IgG antibody (Millipore) and protein G-sepharose (Invitrogen). Beads were washed for three times and treated with proteinase K for 4 hrs. DNA was extracted using a PCR Purification Kit (QIAGEN) and analyzed by real-time quantitative PCR using the primers as listed in Supplementary Table 1.
Mouse models of LPS-induced sepsis
Mice (8–10 weeks of age) were intraperitoneally injected with 50 mg/kg ITA (pH=7.0) for 12 hrs, prior to LPS challenge whenever indicated. Blood and tissue samples were harvested at indicated times. Whole-blood samples were allowed to coagulate for 15 minutes at room temperature and centrifuged at 3,220×rcf(g) for 5 min to collect serum. Serum levels of Il-6 (Cat. No. 70-EK206/3–96), Il-1β (Cat. No. 70-EK201B/3–96) and Cxcl9 (Cat. No. 70-EK2143/2–96) were determined by using ELISA Kits (Multi Sciences). Serum Tnf-α (Cat. No. E-EL-M0049c) was determined by using a ELISA kit from Elabscience.
To quantify the magnitude of liver injury induced by LPS, serum activities of alanine transaminase (ALT) and aspartate transaminase (AST) were measured using commercial kits (Nanjing Jiancheng). To quantify the magnitude of pulmonary edema induced by LPS, whole-lung tissue of each individual mouse was collected, rinsed to remove surface blood, and patted dry, and the immediate weight was recorded as the wet weight. The tissue was air dried for 1 day and the weight was recorded as the dry weight for calculating the lung wet/dry weight ratio. Furthermore, lung histopathological injury was assessed by hematoxylin and eosin (HE) staining, and acute lung injury was scored as follows: a) alveolar congestion, b) hemorrhage, c) infiltration or aggregation of neutrophils in airspace or vessel wall, and d) thickness of alveolar wall/hyaline membrane formation. Each item was scored on a 5-point scale as follows: 0 = minimal damage, 1+ = mild damage, 2+ = moderate damage, 3+ = severe damage, and 4+ = maximal damage63. For the survival assay, animals were carefully monitored as previously reported8. Humane end point for the experiment was based on body condition, physical condition and unprovoked behavior.
Statistics and reproducibility
Statistical analyses were performed using GraphPad Prism (8 .0) or MS Excel(OFFICE 2016). Summarized views on data that underlie the statistical tests as well as the exact P values areavailable in the source data. The statistical details for each experiment are also provided in the figure legends. In general, statistical analyses were performed with two-tailed Student’s t-test for paired comparisons or one/two-way ANOVA for multiple comparisons. Data shown (if not pointed out) represents the results obtained from triplicate independent experiments with mean +/− Standard Deviation (S.D.) or standard error of the mean (SEM). The values of p< 0.05 were considered statistically significant. No statistical method was used to predetermine sample size. No data were excluded from analyses. RNA-seq data were performed using three biological replicates and hMeDIP-seq was performed once. The RT-qPCR data, matabolites and 5hmC quantification by LC-MS and were performed using three biological replicates unless specified in the legends. The western blot data were performed twice with similar results and a representative blot of two independent experiments is shown in this study. For RT-qPCR data and LC-MS, quantification and statistics were derived from n = 3 independent experiments unless specified in the legends
Data availability
The RNA-seq data that support the findings of this study have been deposited in the Gene Expression Omnibus (GEO) under the following accession codes: GSE148143, GSE148145 and GSE148147. The DIP-seq datasets have been deposited in Gene Expression Omnibus (GEO) under the accession number GSE158580. Source data are provided with this study. All other data supporting the findings of this study are available from the corresponding author on reasonable request.
Extended Data
Extended Data Fig. 1. Itaconate inhibits the catalytic activity of Tet2 in vitro.

a, Computational modeling of TET2-metabolites binding. Current TET2 crystal reveals the binding of indicated metabolites with TET2. The high resolution 2.03 Å structure of TET2 in complex with N-oxalylglycine (NOG) in PDB entry 4NM6 was used as the basis for modeling α-KG and the related oncometabolites. While keeping the Fe(II), protein and solvent heavy atoms fixed, polar hydrogens in the binding site and the ligands were optimized using the MMFF94 energy potential with a reaction field solvation model as implemented in MOE.2018.
b, IC50 of indicated metabolites toward purified mTET2CD measured by using a commercial kit (EpiGentek). Enzyme activity was analyzed using GraphPad software and IC50 values were calculated using the equation of log (inhibitor) vs. response (three parameters). Data represent three independent experiments with similar results.
c, R1896M or S1898F mutation reduces TET2 enzyme activity. As shown, both R1896 and S1898 are α-KG binding sites in the catalytic domain (CD) of human TET2 (upper panel). In vitro TET2 activity was determined by 5hmC dot-blot assays using human TET2CD purified from bacteria, as well as methylated dsDNA oligonucleotides as substrates (lower panel). Data represent two independent experiments with similar results.
d, R1896M mutation, but not S1898F mutation, weakens the binding of TET2 with α-KG. STD NMR spectroscopy was used to determine the binding of α-KG with recombinant proteins of TET2CD mutant. Data represent two independent experiments with similar results.
Extended Data Fig. 2. Octyl-ITA (OI) can enter the cells and be hydrolyzed to itaconate.

a, OI can enter and accumulate in the cell. RAW264.7 cells were treated with different concentrations of OI for different lengths of time, followed by LC-MS/MS analysis of intracellular ITA. Data shown represent average values with S.D. of three independent experiments.
b, Unmodified ITA can enter and accumulate in the cell. RAW264.7 cells were treated with increased concentrations of ITA as indicated, and the intracellular concentration of ITA and succinate was measured by LC-MS/MS. Data shown represent average values with S.D. of four independent experiments.
c, Intracellular accumulation of cell-permeable metabolites. HEK293T cells were treated with cell-permeable octyl-ITA and octyl-L-2-HG for 12 hours as indicated, and the intracellular levels of L-2HG and ITA were quantified by LC-MS/MS. Shown are average values of 2 independent experiments. The intracellular concentrations of indicated metabolites are shown.
Extended Data Fig. 3. Itaconate inhibits Tet activity in cells.
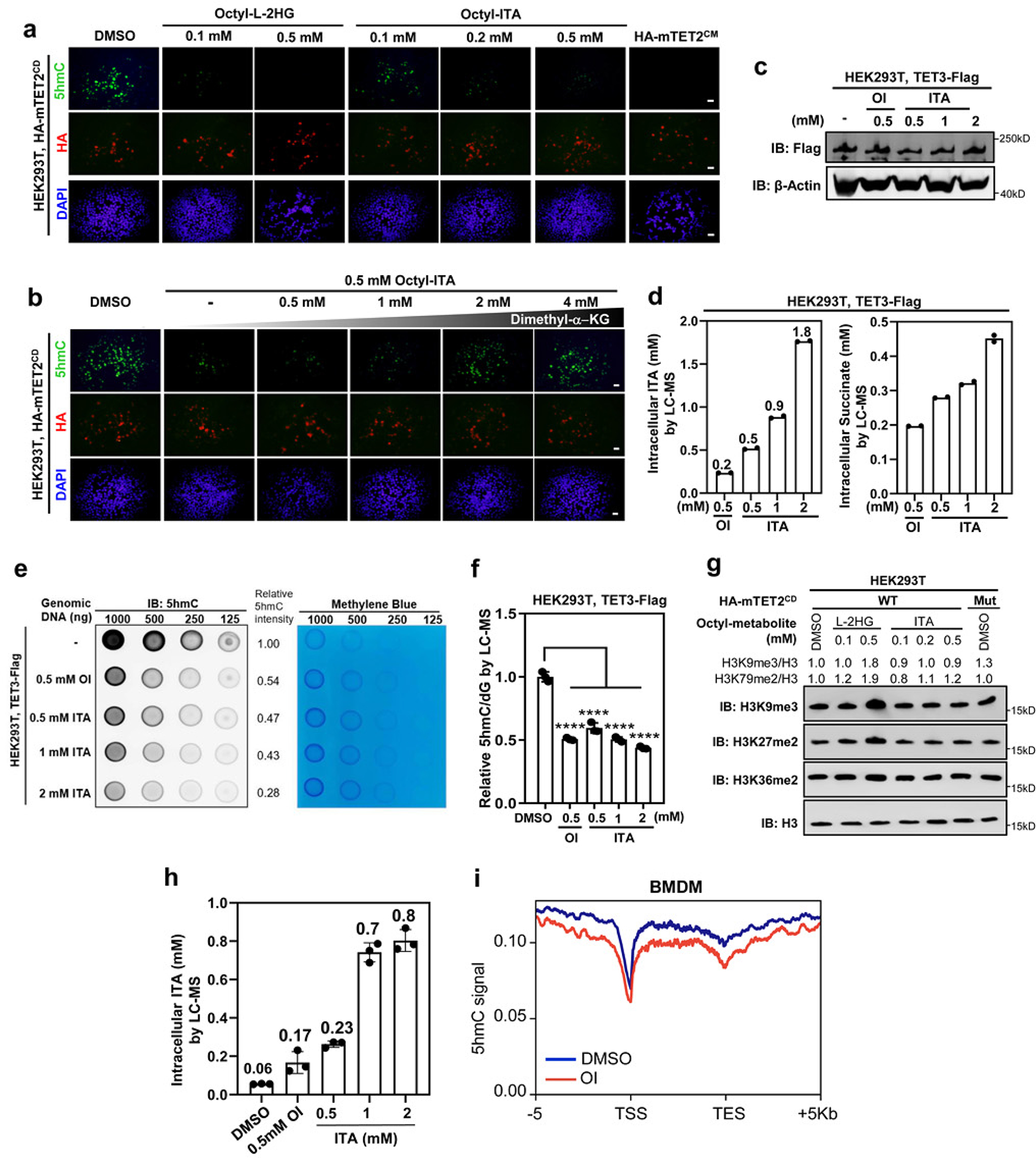
a, HEK293T cells were transiently overexpressed with HA-tagged mTET2CD or catalytic inactive mutant (referred to as mTET2CM), and then treated with indicated cell-permeable metabolites. Global 5hmC was detected by immunofluorescence. Scale bar, 20 μΜ.
b, TET2-overexpressing HEK293T cells in (a) were treated with indicated OI either alone or together with increasing amount of Dimethyl-α-KG. Global 5hmC was detected by immunofluorescence. Scale bar, 20 μM. Data in (a-b) represent two independent experiments with similar results
c, HEK293T cells were transiently overexpressed with Flag-tagged human full-length TET3. The expression of TET3 was determined by western blot with indicated antibodies. A representative blot of two independent experiments is shown.
d, TET3-overexpressing HEK293T cells in (c) were treated with indicated concentrations of OI or ITA. The intracellular levels of ITA and succinate were measured by LC-MS/MS. Data shown are average values of 2 independent experiments.
e, f, TET3-overexpressing HEK293T cells in (c) were treated with indicated concentrations of OI or unmodified ITA, and then global 5hmC was detected by dot-blot (e) and LC-MS/MS (f). Data shown in (f) are average values with S.D. of 3 independent experiments.
g, TET2-overexpressing HEK293T cells in (a) were treated with different amounts of cell-permeable ITA or L-2-HG as indicated, and histone methylation markers were determined by western blot with indicated antibodies. A representative blot of two independent experiments is shown.
h, Tet2+/+ and Tet2−/− BMDMs were treated with 0.5 mM OI or increased concentrations of unmodified ITA for 8 hours, followed by LC-MS/MS analysis to determine intracellular ITA. Data shown are average values with S.D. of 3 independent experiments.
i, Wildtype BMDMs were treated with DMSO or 0.5 mM OI for 8 hours, followed by 5hmC mapping by hMeDIP-seq. The normalized density profile for 5hmC across gene body + 5kb flanking regions is shown. The experiment was performed once.
P values are calculated using one-way ANOVA for multiple comparisons (f). ****denotes p < 0.0001 for the indicated comparison.
Extended Data Fig. 4. LPS induces Itaconate and metabolic reprogramming in macrophages.
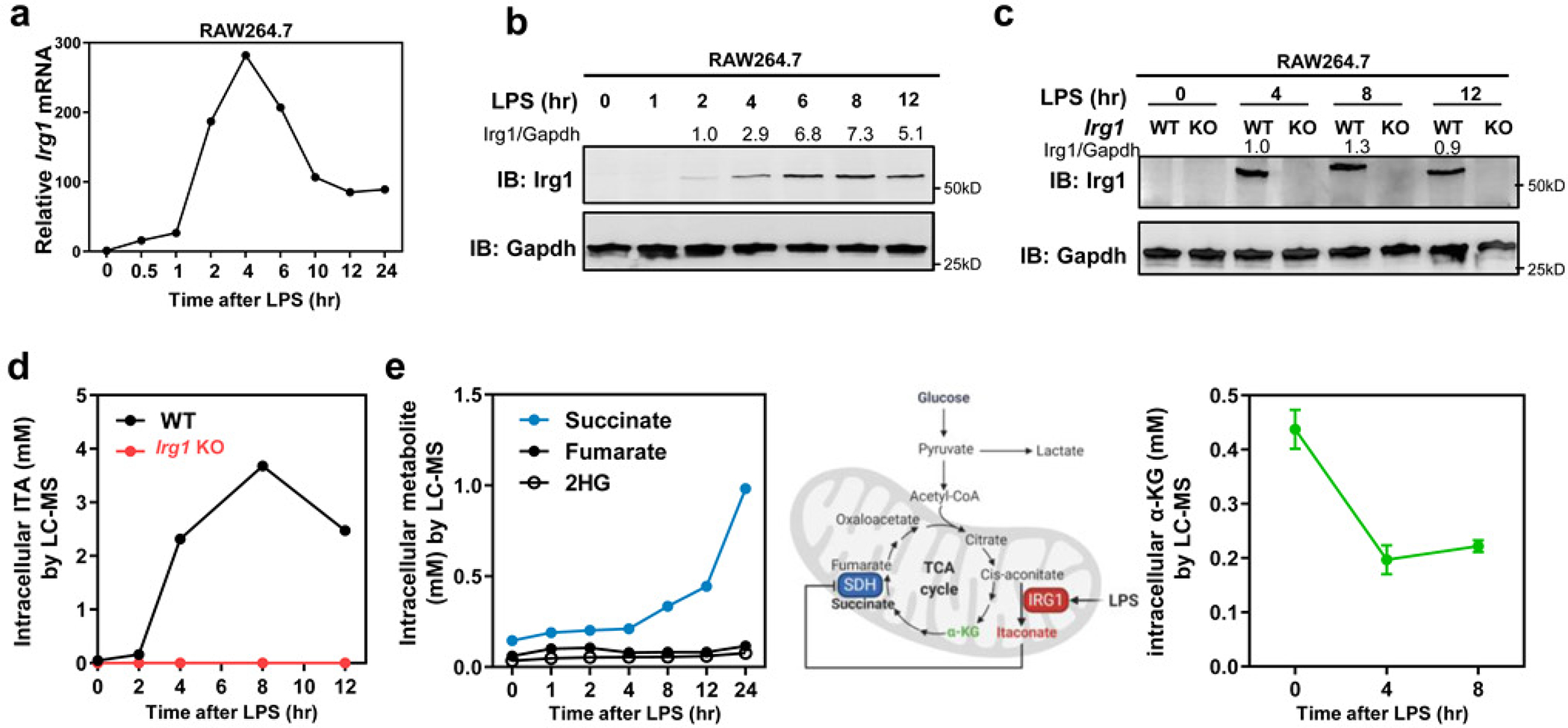
a, b, Irg1 mRNA and protein expression in RAW264.7 cells after LPS treatment, as determined by qRT-PCR and western blot, respectively. Data shown in (a) are average values of 2 (time points: 1–12 hours) or 3 (time points: 0/0.5/24 hours) independent experiments
c, d, LPS-induced ITA accumulation in Irg1-WT but not Irg1-KO RAW264.7 cells. Irg1 protein was detected by western blot (c), and the intracellular concentration of ITA was measured by LC-MS/MS (d). Data shown in (d) are average values of 2 independent experiments. A representative blot of two independent experiments is shown in (b) and (c).
e, Succinate but not fumarate and 2-HG is accumulated, while α-KG is decreased upon LPS treatment in RAW264.7 cells. The intracellular concentrations of indicated metabolites were measured by LC-MS/MS. Data shown are average values of 2 independent experiments (left) or average values with S.D. of 3 independent experiments (right).
Extended Data Fig. 5. LPS-induced Itaconate inhibits Tet activity in macrophages.
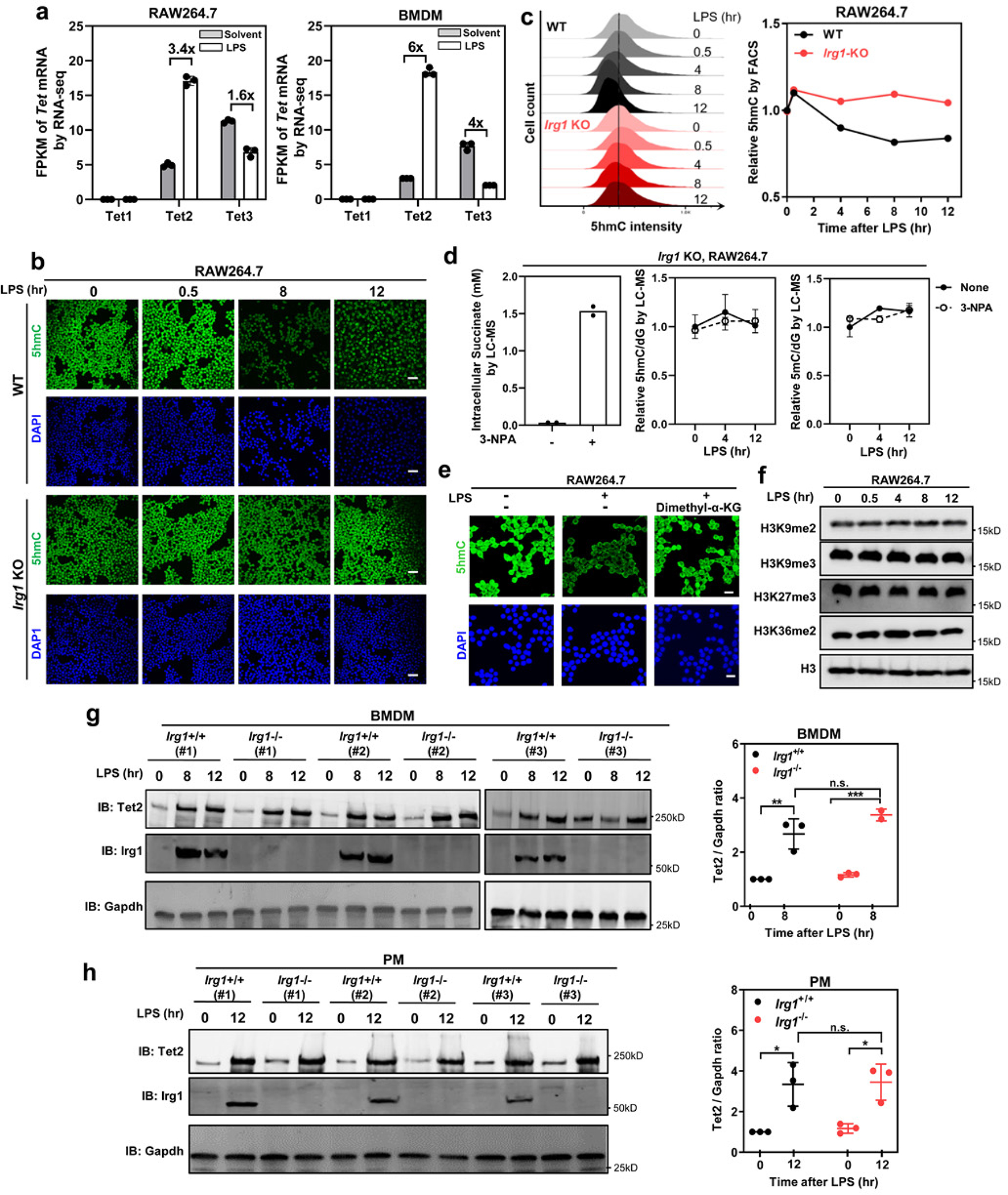
a, In RAW264.7 and mouse BMDMs, relative mRNA expression of Tet1, Tet2 and Tet3 was determined by RNA-seq and plotted according to the fragments per kilobase of transcript per million mapped reads (FPKM) values. Fold changes in Tet gene expression for the indicated comparison are shown. Data shown are average values with S.D. of 3 independent experiments.
b, c, LPS treatment reduces genome-wide 5hmC in Irg1-WT but not Irg1-KO RAW264.7 cells. 5hmC was determined by IF staining (b) and FACS (c) in cells after LPS treatment for indicated time. Scale bar, 50 μm. Data shown in (c, right) are average values of 2 independent experiments.
d, LPS-triggered 5hmC decline is not due to succinate accumulation in macrophages. Irg1-KO RAW264.7 cells were treated with 3-NPA (1 mM), which specifically inhibits SDH and causes intracellular accumulation of succinate (left panel, data shown are average values with 2 independent experiments). Genomic levels of 5mC and 5hmC were determined by LC-MS/MS in these cells after LPS treatment for indicated time (middle and right panels, data shown are average values with S.D. of 3 independent experiments).
e, α-KG restores LPS-induced 5hmC decrease in RAW264.7 cells. Macrophages were challenged with LPS for 12 hours and then treated dimethyl-α-KG (1 mM) for 12 hours. Genomic 5hmC was detected by IF staining. Scale bar, 25 μm.
f, RAW264.7 cells were treated with LPS for indicated time, and then histone methylation markers were determined by western blot using indicated antibodies.
g, h, Mouse BMDMs (g) and thioglycolate elicited peritoneal macrophages (h) were from Irg1+/+ or Irg1−/− mice (n=3 mice per group), and then were either unstimulated or treated with LPS for indicated time. The levels of Tet2 and Irg1 proteins were determined by western blot using indicated antibodies.
P values are calculated using one-way ANOVA (g, h) for multiple comparisons. *denotes p < 0.05, **denotes p < 0.01, and ***denotes p < 0.001 for the indicated comparison. n.s. = not significant. Data represent two independent experiments with similar results in (b, e, f)
Extended Data Fig. 6. Tet2 is the major target of Itaconate in macrophages during LPS response.

a, Principal component analysis (PCA) on the effect of OI treatment and Tet2 deletion on gene expression in LPS-treated RAW264.7 cells. 3D representation of different color-coded RNA-seq data sets corresponding to different Tet2 genotype, LPS and OI treatments. RNA-seq was performed using three biological replicates.
b, 1,846 genes which are down-regulated by OI in Tet2-WT RAW264.7 cells are compared with 807 gene which are down-regulated by OI in Tet2-KO cells (FC ≥ 2). As shown, only 493 (of 1,846) genes were commonly down-regulated by OI in both Tet2-WT and Tet2-KO cells.
c, Identification of LPS-induced genes that are inhibited by OI. The overlap of LPS-induced genes (FC ≥ 2) and OI-inhibited genes (FC ≥ 2) was displayed by Venn diagram (P-value < 10–10). 712 genes were identified and then used for KEGG pathway analysis by online DAVID analysis. Top 10 pathways are listed.
d, 712 genes which are induced by LPS and down-regulated by OI in Tet2-WT RAW264.7 cells are compared with 807 gene which are down-regulated by OI in Tet2-KO cells (FC ≥ 2). As shown, only 135 (of 712) genes were commonly down-regulated by OI in both Tet2-WT and Tet2-KO cells.
e, f, Verification of the Tet2 H1795R KI mice. The KI mutation was confirmed by DNA sequencing (e). Catalytic inactivation of Tet2 was confirmed by 5hmC dot-blot using genomic DNA from hepatocytes (f). The experiments in e–f were performed once.
g, Overlap between LPS-induced genes down-regulated by OI and by H1795R catalytical inactivation in Tet2 in BMDMs is displayed by Venn diagram (p-value < 10–10). Top 10 pathways enriched among the overlapping genes were identified by KEGG pathway analysis.
Extended Data Fig. 7. Itaconate inhibits Tet2-mediated LPS induction of Nfkbiz and other inflammatory genes.
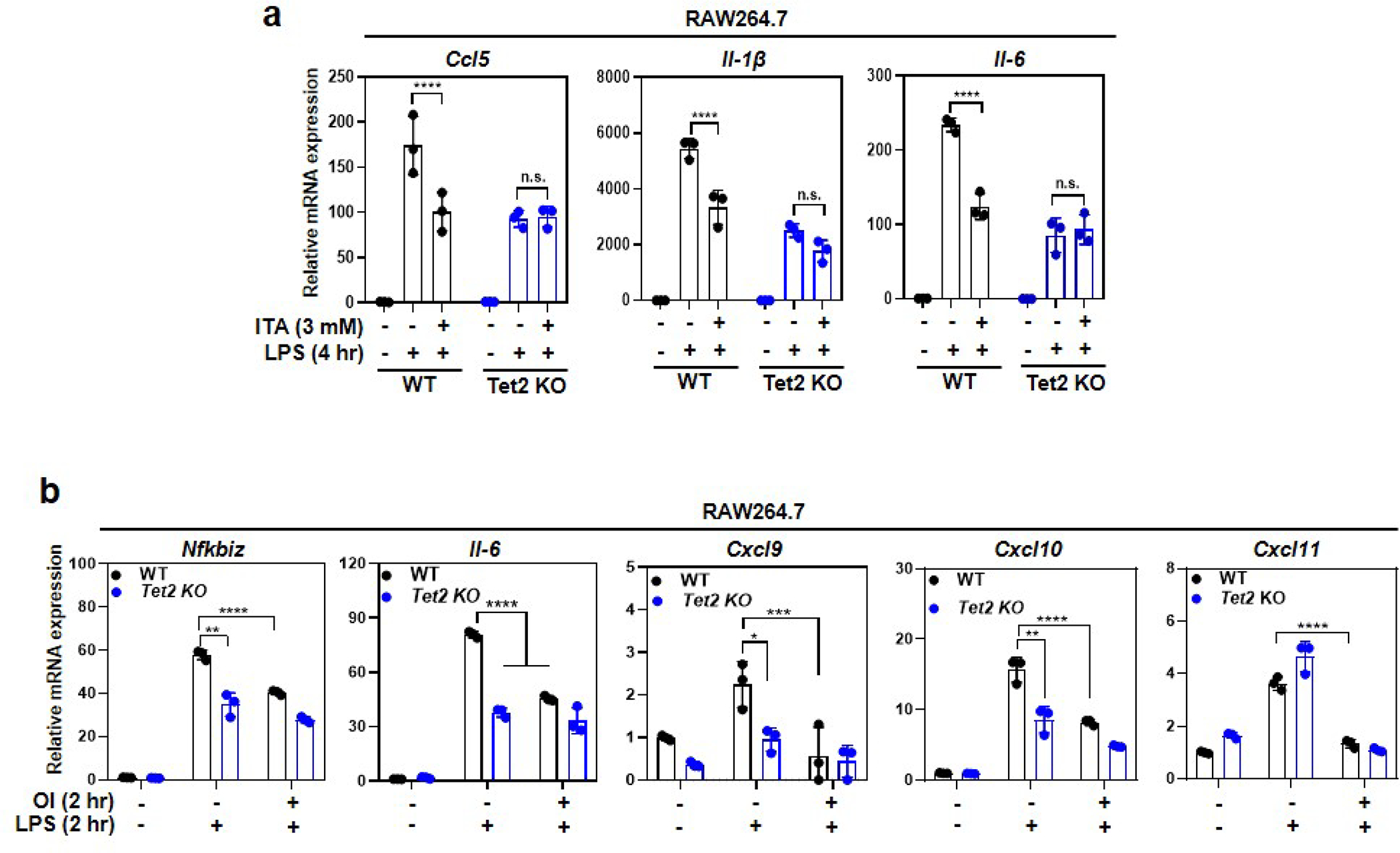
a, Unmodified ITA inhibits LPS-induced genes in a Tet2-dependent manner. Tet2-WT and Tet2-KO RAW264.7 cells were pre-treated with unmodified ITA (3 mM) and then stimulated with LPS for 4 hours, following detection of the indicated gene mRNA expression by qRT-PCR.
b, Addition of OI in a limited time window inhibits LPS-induced genes, mimicking the effect of Tet2 deletion. RAW264.7 cells were stimulated with LPS for 2 hours before endogenous ITA starts to accumulate, and then treated with OI for another 2 hours, following determination of indicated gene mRNA expression by qRT-PCR.
Shown are average values with S.D. of three independent experiments. P values are calculated using two-way ANOVA for multiple comparisons. *denotes p < 0.05, **denotes p < 0.01, ***denotes p < 0.001, and ****denotes p < 0.0001 for the indicated comparison. n.s. = not significant.
Extended Data Fig. 8. Itaconate inhibits Tet2-mediated LPS induction of ΙκΒζ.

a, b, Gene expression of all detectable NfκB family members in RAW164.7 cells (a) and BMDMs (b) with indicated treatments was determined by RNA-seq and represented by heatmaps.
c, Tet2 deletion inhibits LPS-induction of Nfkbiz gene expression in RAW264.7 cells. Tet2-WT and Tet2-KO macrophages were treated with LPS for indicated time. Nfkbiz mRNA was determined by qRT-PCR. Data shown are average values with S.D. of 3 independent experiments.
d, Both ITA and Tet2 deletion inhibits LPS-induction of Nfkbiz. Tet2-WT and Tet2-KO RAW264.7 cells were treated with either LPS alone or with 01 and the level of Nfkbiz mRNA was determined by qRT-PCR. Data shown are average values with S.D. of 3 independent experiments.
e, α-KG activates Nfkbiz mRNA expression in RAW264.7 cells. Macrophages were treated with increased concentrations (0.5,1, 2 mM) of Dimethyl-α-KG together with LPS as indicated. Nfkbiz mRNA was determined by qRT-PCR. Data shown are average values with S.D. of 3 independent experiments.
f, α-KG elevates Nfkbiz mRNA level in Tet2-dependent manner. Tet2-WT and Tet2-K0 RAW264.7 cells were treated with either LPS alone or with cell-permeable α-KG, and the level of Nfkbiz mRNA was determined by qRT-PCR. Data shown are average values with S.D. of 3 independent experiments.
g, h, α-KG restores ΙκΒζ protein blocked by ITA in a Tet2-dependnet manner. Tet2-WT (g) but not Tet2-K0 (h) RAW264.7 cells were treated with LPS and cell-permeable ITA and α-KG as indicated. The levels of ΙκΒζ protein were measured by western blot and normalized against Gapdh. A representative blot of two independent experiments is shown in (g, h).
P values are calculated using two-tailed Student’s t-test for paired comparisons (c), one-way ANOVA (e, d) and two-way ANOVA (f) for multiple comparisons. **denotes p < 0.01, ***denotes p < 0.001, and ****denotes p < 0.0001 for the indicated comparison, n.s. = not significant.
Extended Data Fig. 9. Irgl-deficient mice are more susceptible to LPS-induced acute lung injury and mortality.

a, Flowchart for establishing LPS-induced sepsis mouse model. Briefly, age and sex-matched Irg1−/− or Irg1+/+ mice were i.p. injected with PBS or LPS (20 mg/kg). At 4- or 24-hours post LPS injection, peritoneal leukocytes, serum, and lung were harvested for further analysis.
b, LPS induces ITA accumulation in Irg1+/+ peritoneal leukocytes, but not in those from Irg1−/− mice. Peritoneal leukocytes were freshly isolated as described above in (a), and the intracellular concentration of ITA was measured by LC-MS/MS (n=2–3 mice per group).
c, LPS-challenged Irg1−/− mice exhibit higher serum Il-6 levels than Irg1+/+ controls, as determined by ELISA (n=3–7 mice per group).
d, e, LPS-challenged Irg1−/− mice exhibit more severe lung injury than Irg1+/+ controls. As described above in (a), mouse lungs were harvested and then subjected to histopathological analysis (n=3–6 mice per group). Representative photomicrographs of HE staining are shown (d). Scale bar, 200 μm (upper panels) & 100 μm (lower panels).
f, Irg1−/− mice are more susceptible to LPS-induced mortality. Kaplan-Meier survival curves were determined as described in Methods (n=10 mice per group).
Shown in b, c and e are average values with S.D. P values are calculated using one-way ANOVA for multiple comparisons (c, e). As for the percent survival, P values were determined using log-rank (Mantel-Cox) test comparing each 2 groups (f); *denotes p < 0.05, **denotes p < 0.01 and ****denotes p < 0.0001 for the indicated comparison.
Extended Data Fig. 10. Itaconate reduces LPS-induced mouse mortality in a manner dependent on Tet2.

a, Mice were i.p. injected with ITA prior to LPS challenge (25 mg/kg, n=3 mice per group; left panel). At 2 hours post LPS, peritoneal leukocytes and lung were freshly isolated and then subjected to determination of genomic 5hmC and 5mC by LC-MS/MS (right panels).
b,c,d As described above in (a), mice were i.p. injected with ITA prior to LPS challenge (n=6–10 mice per group). At 2 hours post LPS, lung tissues were harvested and then subjected to the wet/dry weight ratio calculation (b). Meanwhile, lung histopathological injury was assessed by HE staining and the injury score was determined as described in method (c, d). Representative photomicrographs are shown. Scale bar, 200 μm (upper panels) & 100 μm (lower panels).
e,f, As described above in (a), mice were i.p. injected with ITA prior to LPS challenge (Tet2+/+, n=13 mice; Tet2+/+, n=14 mice; Tet2−/−, n=7 mice per group). The animal survival was carefully monitored and Kaplan-Meier survival curves were determined as described in Methods.
g, Flowchart for developing chimeric mice by bone marrow transplantation (BMT).
h, i, The Kl mutations were confirmed by DNA sequencing in bone marrow samples of transplanted animals (h). Catalytic inactivation of Tet2 was also confirmed by 5hmC LC-MS/MS using genomic DNA from bone marrow samples of transplanted animals
j, A schematic model for Itaconate inhibits TET2 to dampen inflammatory response.
Shown are average values with S.D. (a, i) or SEM (b, d). P- values are calculated using two-tailed Student’s t-test for paired comparisons (i) or one-way ANOVA for multiple comparisons (a, b, d); As for the percent survival, P values were determined using log-rank (Mantel-Cox) test comparing each 2 groups; *denotes p < 0.05, **denotes p < 0.01, ***denotes p < 0.001, and ****denotes p < 0.0001 for the indicated comparison, n.s. = not significant.
Supplementary Material
Acknowledgements
We thank Joshua Boyer, Stu Parnham and UNC Biomolecular NMR Spectroscopy Core Facility for the NMR analysis, Matt Lee of Molecular Cornerstones for molecular modeling, Yanhui Xu (Fudan University) for providing human TET2CD expression vector, Jiemin Wong (East China Normal University) for providing human TET3 expression vector, and Nick Brown (UNC-Chapel Hill) for advising TET2 purification, and Jiayu Chen and Shaorong Gao (School of Life Sciences and Technology, Tongji University, Shanghai) for providing Tet2HxD KI mutant mice. We also thank the staff members working at IBS, Fudan University for their help on mass spectrometry analyses. This work was supported by National Key R&D Program of China (2020YFA0803202 and 2016YFA0501800 to D. Y.; 2016YFC1305102 to Y.Z.), the NSFC grants (No. 31871431, No. 31821002 to D. Y.; No. 82103366 to L-L. C.; No. 81671561, No. 81974248 to Y. Z.), the Innovative research team of high-level local universities in Shanghai (SSMU-ZLCX20180501 to D. Y.), and Program for Outstanding Medical Academic Leader (2019LJ19 to Y.Z.). This study was also supported by Samuel Waxman Research Foundation Investigator Award and NIH grant CA163834 (A. S. B. and Y. X).
Footnotes
Competing interests
The authors declare no competing interests.
References
- 1.Daniels BP et al. The Nucleotide Sensor ZBP1 and Kinase RIPK3 Induce the Enzyme IRG1 to Promote an Antiviral Metabolic State in Neurons. Immunity 50, 64–76 e64 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Nair S et al. Irg1 expression in myeloid cells prevents immunopathology during M. tuberculosis infection. J Exp Med 215, 1035–1045 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Lee CG, Jenkins NA, Gilbert DJ, Copeland NG & O’Brien WE Cloning and analysis of gene regulation of a novel LPS-inducible cDNA. Immunogenetics 41, 263–270 (1995). [DOI] [PubMed] [Google Scholar]
- 4.Michelucci A et al. Immune-responsive gene 1 protein links metabolism to immunity by catalyzing itaconic acid production. Proc Natl Acad Sci U S A 110, 7820–7825 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Ackermann WW & Potter VR Enzyme inhibition in relation to chemotherapy. Proc Soc Exp Biol Med 72, 1–9 (1949). [DOI] [PubMed] [Google Scholar]
- 6.Lampropoulou V et al. Itaconate Links Inhibition of Succinate Dehydrogenase with Macrophage Metabolic Remodeling and Regulation of Inflammation. Cell Metab 24, 158–166 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Cordes T et al. Immunoresponsive Gene 1 and Itaconate Inhibit Succinate Dehydrogenase to Modulate Intracellular Succinate Levels. J Biol Chem 291, 14274–14284 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Mills EL et al. Itaconate is an anti-inflammatory metabolite that activates Nrf2 via alkylation of KEAP1. Nature 556, 113–117 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Bambouskova M et al. Electrophilic properties of itaconate and derivatives regulate the IkappaBzeta-ATF3 inflammatory axis. Nature 556, 501–504 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Qin W et al. S-glycosylation-based cysteine profiling reveals regulation of glycolysis by itaconate. Nature chemical biology 15, 983–991 (2019). [DOI] [PubMed] [Google Scholar]
- 11.Hu L et al. Crystal structure of TET2-DNA complex: insight into TET-mediated 5mC oxidation. Cell 155, 1545–1555 (2013). [DOI] [PubMed] [Google Scholar]
- 12.Chowdhury R et al. The oncometabolite 2-hydroxyglutarate inhibits histone lysine demethylases. EMBO Rep 12, 463–469 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Xu W et al. Oncometabolite 2-hydroxyglutarate is a competitive inhibitor of alpha-ketoglutarate-dependent dioxygenases. Cancer cell 19, 17–30 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Seim GL et al. Two-stage metabolic remodelling in macrophages in response to lipopolysaccharide and interferon-gamma stimulation. Nature metabolism 1, 731–742 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Liu PS et al. alpha-ketoglutarate orchestrates macrophage activation through metabolic and epigenetic reprogramming. Nature immunology 18, 985–994 (2017). [DOI] [PubMed] [Google Scholar]
- 16.Xiao M et al. Inhibition of alpha-KG-dependent histone and DNA demethylases by fumarate and succinate that are accumulated in mutations of FH and SDH tumor suppressors. Genes Dev 26, 1326–1338 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.O’Neill LAJ & Artyomov MN Itaconate: the poster child of metabolic reprogramming in macrophage function. Nat Rev Immunol 19, 273–281 (2019). [DOI] [PubMed] [Google Scholar]
- 18.Zhang Q et al. Tet2 is required to resolve inflammation by recruiting Hdac2 to specifically repress IL-6. Nature 525, 389–393 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Wang Y et al. WT1 recruits TET2 to regulate its target gene expression and suppress leukemia cell proliferation. Molecular cell 57, 662–673 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Yamamoto M et al. Regulation of Toll/IL-1-receptor-mediated gene expression by the inducible nuclear protein IkappaBzeta. Nature 430, 218–222 (2004). [DOI] [PubMed] [Google Scholar]
- 21.Miyake T et al. IkappaBzeta is essential for natural killer cell activation in response to IL-12 and IL-18. Proc Natl Acad Sci U S A 107, 17680–17685 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Okamoto K et al. IkappaBzeta regulates T(H)17 development by cooperating with ROR nuclear receptors. Nature 464, 1381–1385 (2010). [DOI] [PubMed] [Google Scholar]
- 23.Johansen C et al. IkappaBzeta is a key driver in the development of psoriasis. Proc Natl Acad Sci U S A 112, E5825–5833 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Alexander E et al. IkappaBzeta is a regulator of the senescence-associated secretory phenotype in DNA damage- and oncogene-induced senescence. J Cell Sci 126, 3738–3745 (2013). [DOI] [PubMed] [Google Scholar]
- 25.Xu H et al. Chronic inflammation in fat plays a crucial role in the development of obesity-related insulin resistance. J Clin Invest 112, 1821–1830 (2003). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Nagarsheth N, Wicha MS & Zou W Chemokines in the cancer microenvironment and their relevance in cancer immunotherapy. Nat Rev Immunol 17, 559–572 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Liu T, Zhang L, Joo D & Sun SC NF-kappaB signaling in inflammation. Signal transduction and targeted therapy 2 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Rampal R et al. DNA hydroxymethylation profiling reveals that WT1 mutations result in loss of TET2 function in acute myeloid leukemia. Cell Rep 9, 1841–1855 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Ichiyama K et al. The methylcytosine dioxygenase Tet2 promotes DNA demethylation and activation of cytokine gene expression in T cells. Immunity 42, 613–626 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Ma S et al. Epigenetic regulator CXXC5 recruits DNA demethylase Tet2 to regulate TLR7/9-elicited IFN response in pDCs. J Exp Med 214, 1471–1491 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Xu YP et al. Tumor suppressor TET2 promotes cancer immunity and immunotherapy efficacy. J Clin Invest 130, 4316–4331 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Yamazaki S, Muta T, Matsuo S & Takeshige K Stimulus-specific induction of a novel nuclear factor-kappaB regulator, IkappaB-zeta, via Toll/Interleukin-1 receptor is mediated by mRNA stabilization. J Biol Chem 280, 1678–1687 (2005). [DOI] [PubMed] [Google Scholar]
- 33.Link VM et al. Analysis of Genetically Diverse Macrophages Reveals Local and Domain-wide Mechanisms that Control Transcription Factor Binding and Function. Cell 173, 1796–1809 e1717 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Zhao Z et al. The catalytic activity of TET2 is essential for its myeloid malignancy-suppressive function in hematopoietic stem/progenitor cells. Leukemia 30, 1784–1788 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Ye D, Guan KL & Xiong Y Metabolism, Activity, and Targeting of D- and L-2-Hydroxyglutarates. Trends in cancer 4, 151–165 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Ryan DG et al. Coupling Krebs cycle metabolites to signalling in immunity and cancer. Nature metabolism 1, 16–33 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Arts RJ et al. Glutaminolysis and Fumarate Accumulation Integrate Immunometabolic and Epigenetic Programs in Trained Immunity. Cell Metab 24, 807–819 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Lio CJ, Yuita H & Rao A Dysregulation of the TET family of epigenetic regulators in lymphoid and myeloid malignancies. Blood 134, 1487–1497 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Jaiswal S & Libby P Clonal haematopoiesis: connecting ageing and inflammation in cardiovascular disease. Nat Rev Cardiol 17, 137–144 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Cook EK, Luo M & Rauh MJ Clonal hematopoiesis and inflammation: Partners in leukemogenesis and comorbidity. Exp Hematol 83, 85–94 (2020). [DOI] [PubMed] [Google Scholar]
- 41.Chen M et al. Itaconate is an effector of a Rab GTPase cell-autonomous host defense pathway against Salmonella. Science 369, 450–455 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Basler T, Jeckstadt S, Valentin-Weigand P & Goethe R Mycobacterium paratuberculosis, Mycobacterium smegmatis, and lipopolysaccharide induce different transcriptional and post-transcriptional regulation of the IRG1 gene in murine macrophages. J Leukoc Biol 79, 628–638 (2006). [DOI] [PubMed] [Google Scholar]
- 43.Preusse M, Tantawy MA, Klawonn F, Schughart K & Pessler F Infection- and procedure-dependent effects on pulmonary gene expression in the early phase of influenza A virus infection in mice. BMC Microbiol 13, 293 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Cho H et al. Differential innate immune response programs in neuronal subtypes determine susceptibility to infection in the brain by positive-stranded RNA viruses. Nat Med 19, 458–464 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Smith J et al. Systems analysis of immune responses in Marek’s disease virus-infected chickens identifies a gene involved in susceptibility and highlights a possible novel pathogenicity mechanism. J Virol 85, 11146–11158 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Safronova A et al. Alarmin S100A11 initiates a chemokine response to the human pathogen Toxoplasma gondii. Nature immunology 20, 64–72 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Li Y et al. Immune responsive gene 1 (IRG1) promotes endotoxin tolerance by increasing A20 expression in macrophages through reactive oxygen species. J Biol Chem 288, 16225–16234 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Hall CJ et al. Epidermal cells help coordinate leukocyte migration during inflammation through fatty acid-fuelled matrix metalloproteinase production. Nat Commun 5, 3880 (2014). [DOI] [PubMed] [Google Scholar]
- 49.Sun KA et al. Endogenous itaconate is not required for particulate matter-induced NRF2 expression or inflammatory response. Elife 9 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Weiss JM et al. Itaconic acid mediates crosstalk between macrophage metabolism and peritoneal tumors. J Clin Invest 128, 3794–3805 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Cheon YP, Xu X, Bagchi MK & Bagchi IC Immune-responsive gene 1 is a novel target of progesterone receptor and plays a critical role during implantation in the mouse. Endocrinology 144, 5623–5630 (2003). [DOI] [PubMed] [Google Scholar]
- 52.Zhang X, Goncalves R & Mosser DM The isolation and characterization of murine macrophages. Current protocols in immunology Chapter 14, Unit 14 11 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Gao J et al. Absolute quantification of histone PTM marks by MRM-based LC-MS/MS. Anal Chem 86, 9679–9686 (2014). [DOI] [PubMed] [Google Scholar]
- 54.Yu Y et al. Quantitative Profiling of Combinational K27/K36 Modifications on Histone H3 Variants in Mouse Organs. J Proteome Res 15, 1070–1079 (2016). [DOI] [PubMed] [Google Scholar]
- 55.Nakagawa T et al. CRL4(VprBP) E3 ligase promotes monoubiquitylation and chromatin binding of TET dioxygenases. Molecular cell 57, 247–260 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Ran FA et al. Genome engineering using the CRISPR-Cas9 system. Nat Protoc 8, 2281–2308 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Lee M, Zhou Y & Huang Y An Engineered Split-TET2 Enzyme for Chemical-inducible DNA Hydroxymethylation and Epigenetic Remodeling. J Vis Exp (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Lv L et al. Vpr Targets TET2 for Degradation by CRL4(VprBP) E3 Ligase to Sustain IL-6 Expression and Enhance HIV-1 Replication. Molecular cell 70, 961–970 e965 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Shen L et al. Genome-wide analysis reveals TET- and TDG-dependent 5-methylcytosine oxidation dynamics. Cell 153, 692–706 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Liao Y, Smyth GK & Shi W The R package Rsubread is easier, faster, cheaper and better for alignment and quantification of RNA sequencing reads. Nucleic acids research 47, e47 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Chen LL et al. SNIP1 Recruits TET2 to Regulate c-MYC Target Genes and Cellular DNA Damage Response. Cell Rep 25, 1485–1500 e1484 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Lan F et al. Recognition of unmethylated histone H3 lysine 4 links BHC80 to LSD1-mediated gene repression. Nature 448, 718–722 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Mikawa K, Nishina K, Takao Y & Obara H ONO-1714, a nitric oxide synthase inhibitor, attenuates endotoxin-induced acute lung injury in rabbits. Anesthesia and analgesia 97, 1751–1755 (2003). [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The RNA-seq data that support the findings of this study have been deposited in the Gene Expression Omnibus (GEO) under the following accession codes: GSE148143, GSE148145 and GSE148147. The DIP-seq datasets have been deposited in Gene Expression Omnibus (GEO) under the accession number GSE158580. Source data are provided with this study. All other data supporting the findings of this study are available from the corresponding author on reasonable request.