

Abstract
The treatment of advanced and metastatic kidney cancer has entered a golden era with the addition of more therapeutic options, improved survival and new targeted therapies. Tyrosine kinase inhibitors, mammalian target of rapamycin (mTOR) inhibitors and immune checkpoint blockade have all been shown to be promising strategies in the treatment of renal cell carcinoma (RCC). However, little is known about the best therapeutic approach for individual patients with RCC and how to combat therapeutic resistance. Cancers, including RCC, rely on sustained replicative potential. The cyclin-dependent kinases CDK4 and CDK6 are involved in cell-cycle regulation with additional roles in metabolism, immunogenicity and antitumour immune response. Inhibitors of CDK4 and CDK6 are now commonly used as approved and investigative treatments in breast cancer, as well as several other tumours. Furthermore, CDK4/6 inhibitors have been shown to work synergistically with other kinase inhibitors, including mTOR inhibitors, as well as with immune checkpoint inhibitors in preclinical cancer models. The effect of CDK4/6 inhibitors in kidney cancer is relatively understudied compared with other cancers, but the preclinical studies available are promising. Collectively, growing evidence suggests that targeting CDK4 and CDK6 in kidney cancer, alone and in combination with current therapeutics including mTOR and immune checkpoint inhibitors, might have therapeutic benefit and should be further explored.
The current era of targeted therapy and precision medicine for cancer treatment, including renal cell carcinoma (RCC), was unimaginable only a few decades ago. Patients with early-stage RCC have a good prognosis and are often cured with surgery alone, but advanced and metastatic disease have a much worse prognosis, with low 5-year survival (~13%)1. Traditional chemotherapeutics and radiotherapy have never shown considerable efficacy in metastatic RCC2, and the first real therapeutic advance came in the mid-1990s with the success of high-dose IL-2, which suggested that harnessing the immune system might be effective in RCC treatment3. Unfortunately, IL-2 treatment in metastatic RCC caused extreme, limiting adverse events including hepatic and renal dysfunction3. In the early 2000s, the success of tyrosine kinase inhibitors (TKIs), such as sunitinib, and mechansitic target of rapamycin (mTOR) inhibitors, such as everolimus, started a new era of targeted therapy for the treatment of metastatic RCC and these agents quickly became the gold standard in RCC therapy4,5. Many of the genes implicated in RCC development and progression are involved in tyrosine kinase signalling pathways, including MET, vascular endothelial growth factor receptor (VEGFR), and fibroblast growth factor receptor (FGFR) pathways6 and the mTOR pathway7, but limited success in predicting responsiveness to TKI and mTOR inhibitors has been observed, owing to a lack of effective biomarkers8. Another advance in the treatment of metastatic RCC was made with the testing and FDA approval of novel immune checkpoint inhibitors (ICIs) including those targeting programmed cell death 1 (PD1) or programmed cell death 1 ligand 1 (PDL1) and cytotoxic T lymphocyte associated protein 4 (CTLA4) in the 2010s8–10. Checkpoint inhibitors ultimately induce T cell activation in order to promote immune-mediated killing of cancer cells11. Checkpoint inhibitors in combination with TKI treatment increased disease stability and improved both overall survival and progression-free survival (PFS) in patients with RCC8–10; however, intrinsic and acquired resistance to these agents, as well as the paucity of biomarkers for RCC8 make it difficult to predict patients’ response to treatment and identify the best sequence of therapies for individual patients.
In the mid-2010s, when checkpoint inhibitors were gaining FDA approval for the treatment of RCC, therapeutics specifically targeting cyclin-dependent kinase 4 (CDK4) and CDK6 were approved by the FDA for the treatment of breast cancer12–14. CDK4 and CDK6 are involved in cell-cycle entry at the G1/S checkpoint, and they require chaperoning by the CDC37–heat shock protein 90 (HSP90) chaperone system for stabilization and activation15,16. The holoenzyme complexes containing D-type cyclins and CDK4 and CDK6 canonically phosphorylate the tumour suppressor retinoblastoma (Rb), leading to the release of E2F transcriptional repression needed for G1/S transition15.
Non-specific CDK inhibitors were tested in clinical trials in haematological and solid malignancies but induced high levels of toxic effects17,18; thus, the development of inhibitors specifically targeting both CDK4 and CDK6 provided a new potential clinical therapeutic window in cancer17,18. FDA approval has been given to three specific CDK4 and CDK6 inhibitors (referred to here as CDK4/6 inhibitors) — palbociclib (ibrance), ribociclib (KISQALI) and abemaciclib (verzenio) — for the treatment of hormone receptor (HR)-positive and human epidermal growth factor receptor 2 (HER2)-negative (HR+ and HER2−) breast cancer and are currently under clinical investigation in many other cancers19,20. Many of the regimens approved in breast cancer are CDK4/6 inhibitors administered in combination with anti-oestrogen therapies12–14,21,22, and results from preclinical and clinical studies demonstrated that CDK4/6 inhibitors work remarkably well in combination with hormone therapy in breast cancer, or with kinase inhibitors (such as TKIs and mTOR inhibitors) in a variety of cancers, owing to cross-regulation between CDK and mTOR signalling pathways23. Additional work highlighted a role for CDK4 and CDK6 inhibition in augmenting tumour immunogenicity and antitumour immune response23. Several ongoing clinical trials use CDK4/6 inhibitors in combination with immune checkpoint blockade in prostate, breast, head and neck cancer and liposarcoma24–28. The success of CDK4/6 inhibitors in combination with mTOR29–35 and ICIs24–28,36–40 in preclinical and clinical models suggests that these inhibitors could be ideal therapeutics in RCC, as, in this setting, they might also work synergistically with therapeutics currently used in RCC treatment4,5.
In this Perspective, we discuss the relevant preclinical and clinical knowledge on CDK4 and CDK6 function and inhibition to support the idea that further exploration of the role of these therapeutics in kidney cancer is warranted.
CDK4 and CDK6 structure and role in the cell cycle
CDKs are necessary for the ordered progression of the cell cycle and function in the integration of cellular signals to control cell-cycle checkpoints41. As the name suggests, the function of CDKs requires the formation of holoenzyme complexes with cyclin proteins42. The concentration of cyclins varies throughout the cell cycle and their binding to CDKs promotes CDK kinase activation42. CDK4 and the closely related CDK6 both depend on D-type cyclins, the expression of which increases in early G1 in response to various mitogenic stimuli43. CDK4 and CDK6 are largely functionally homologous and mice lacking either gene individually are viable43–46. Notable differences in CDK4 and CDK6 function also exist: CDK4 is involved in hypothalamic–pituitary axis signalling, fertility and insulin production, whereas CDK6-null animals have mild haematopoietic defects44–46. In general, most studies focus on one protein or the other, with the bulk of the literature concentrating on CDK4; studies usually do not differentiate between CDK4 and CDK6 when non-specific inhibitors are used. CDK4, in complex with D-type cyclins, phosphorylates Rb47 (FIG. 1). Rb directly binds to the activation domain of the transcription factor E2F and inactivates it, thereby preventing cells from entering S phase41; however, phosphorylation of Rb disrupts its interaction with E2F1, enabling E2F-mediated transcription of its target genes and consequent exit from the G1 phase of the cell cycle47. The cyclin D–CDK4 complex recognizes and binds two distinct sites in the C terminus of Rb, leading to Rb phosphorylation48. CDK4-mediated phosphorylation of Rb prevents its cleavage by caspase 3 (as part of the apoptotic cascade) antagonizing the induction of apoptosis49. CDK4 and CDK6 activity also promotes the assembly of the pre-replication complex (needed to coordinate DNA replication in S phase) through the regulation of Rb–E2F-mediated transcription of target genes50. Additionally, CDK4 and CDK6 delay senescence, thereby extending cellular lifespan in a kinase-activity-dependent manner through an as-yet unidentified mechanism51,52.
Fig. 1 |. CDK4 stabilization and canonical function in the cell cycle.
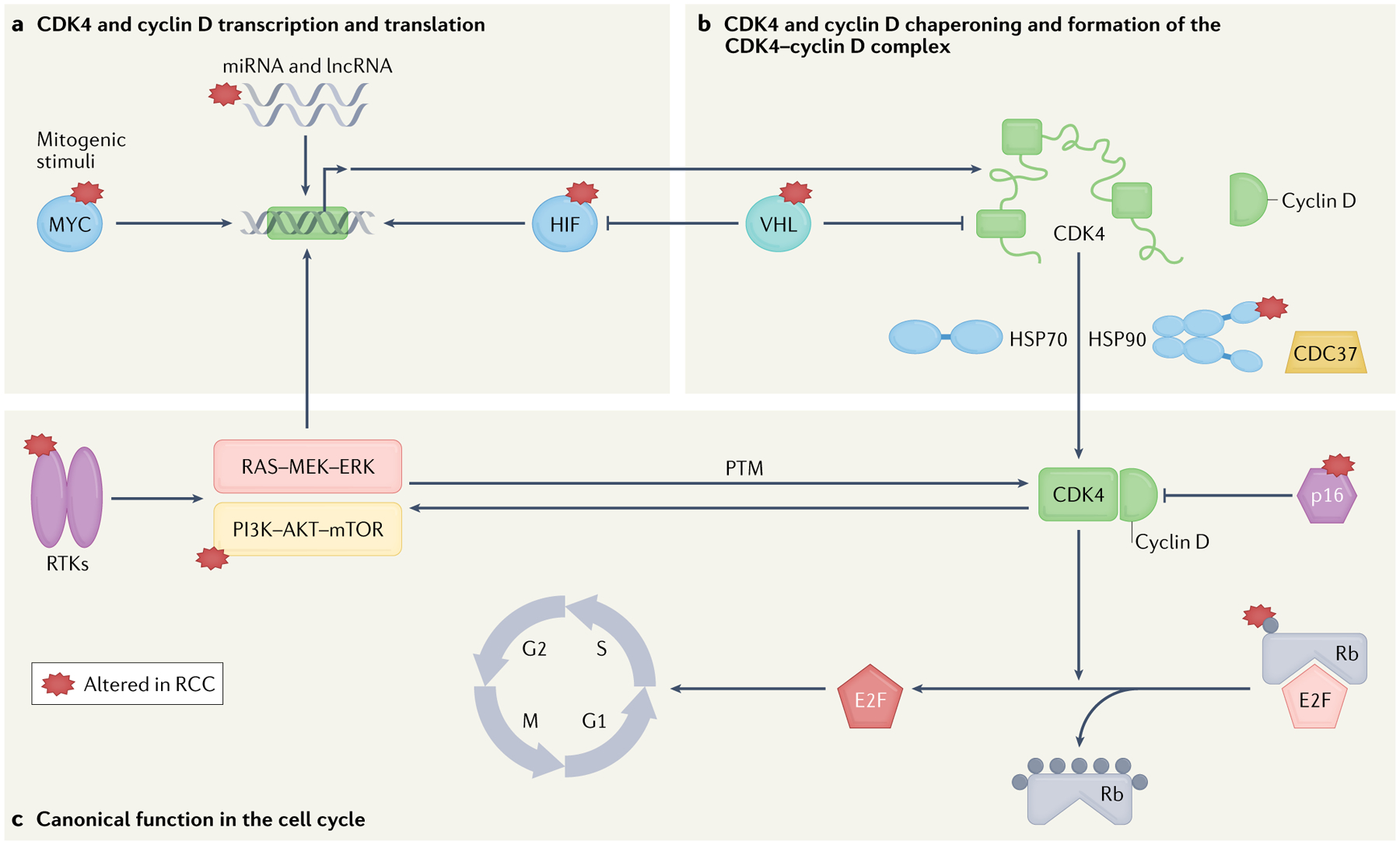
a | Cyclin D and cyclin-dependent kinase 4 (CDK4) transcription and translation can be activated by mitogenic stimuli43 and long non-coding RNAs (lncRNAs)227,228, or inhibited by the action of several microRNAs (miRNAs)225,226. b | CDK4–cyclin D complex assembly and stabilization require components of the chaperone machinery such as heat shock protein 90 (HSP90) in complex with the co-chaperone CDC37 (HSP90–CDC37) and HSP70, which assist folding and stabilization of cyclin D and CDK4 (REFS71,72,124–128). c | Canonically, CDK4 phosphorylates the tumour suppressor retinoblastoma (Rb; grey dots), enabling the transcription factor E2F to enter the nucleus and transcribe its target genes, promoting progression through the S phase of the cell cycle15. The tumour suppressor p16 is an endogenous inhibitor of CDK4 function65. Several signalling cascades have a role in the transcriptional regulation of CDK4–cyclin D kinase complex including MYC55, RAS–mitogen-activated protein kinase kinase (MEK)–extracellular signal-regulated kinase (ERK)117 and hypoxia-inducible factor (HIF)60,234. Von Hippel–Lindau (VHL) regulates CDK4 by controlling the expression of CDK4 and HIF59,60. Reciprocal activating mechanisms exist between CDK4 and the phosphoinositide 3-kinase (PI3K)–AKT–mechanistic target of rapamycin (mTOR) pathway (mTOR regulates CDK4–T172 phosphorylation, whereas CDK4 phosphorylates the endogenous mTOR inhibitor tuberous sclerosis complex 2 (TSC2))63,116,212,213. Many genes and proteins involved in CDK4 function and regulation are known to be altered in renal cell carcinoma (RCC, starred proteins). PTM, post-translational modification; RTK, receptor tyrosine kinase.
Sustained replicative potential and loss of cell-cycle control is a hallmark of cancer53 and a mutual requirement for CDK4 and the transcription factor MYC exists in malignant transformation54 (FIG. 1). CDK4 is a direct transcriptional target of MYC and is necessary for MYC-driven cell-cycle entry55, which is in part dependent on the regulation of cyclin-dependent kinase inhibitor 1B (p27), an endogenous cell cycle inhibitor56,57. Elevated p27 levels in Myc−/− cells were shown to make it impossible for cyclin D–CDK4 complexes to be activated, probably owing to alterations in the stoichiometry and relative abundance of cyclin D and p27 (REF.56). The function of CDK4 in regulating S phase entry is partially mediated by the control of p27 activity: CDK4 sequesters p27, preventing p27-mediated inhibition of the cyclin E–CDK2 complex and enabling G1/S transition57. Cdk4−/−p27−/− double knockout mouse embryonic fibroblasts (MEFs) displayed partially restored kinetics of the G0/S transition57. Cdk4−/− MEFs displayed similar growth to their wild-type counterpart under continuous growth conditions but their ability to re-enter S phase after a period of serum starvation was considerably impaired57. However, investigations carried out in Drosophila, which only has single CDK4 and cyclin D genes, demonstrated that cell-cycle progression is not impaired in the absence of Cdk4, but cell and organismal growth were compromised58. Hypoxia-inducible factor-prolyl hydroxylase (Hph) is an oxygen-dependent enzyme that adds hydroxyl groups to proline residues on target proteins (such as hypoxia-inducible factor (HIF)), which allows them to be recognized by the E3-ubiquitin ligase Von Hippel–Lindau (VHL) and subsequently ubiquitinated and targeted for degradation59. Loss-of-function mutations in Hph specifically suppressed the cell-growth function of CDK4–cyclin D complexes in Drosophila60, showing that Hph is an important mediator of cyclin D–CDK4 signalling and a regulator of cell growth60, although the effectors of Hph in this context have not been identified. This study provided an early link between the oxygen-sensing mechanism regulating HIF levels (deregulation of which is central to RCC development)6,7 and CDK4 (REF.60) (FIG. 1). In conclusion, CDK4 and CDK6 control cell-cycle progression through several distinct mechanisms.
Non-canonical role of CDK4 and CDK6 in metabolism
CDK4 and CDK6 are also involved in the control of metabolism, including insulin signalling and glucose metabolism46,61–64 (FIG. 2). The role of CDK4 and CDK6 in metabolism might be crucial in RCC, which is often considered to be a metabolic disease7. Distinct subtypes of hereditary and sporadic RCC tumorigenesis arise from mutations in tumour suppressors such as folliculin, hamartin, tuberin and fumarate hydratase, which are also involved in cellular metabolism and nutrient sensing pathways such as mTOR signalling7. The first indication that CDK4 might have a role in metabolism came from Cdk4-deficient mice that are viable, but small, and develop insulin-deficient diabetes as a result of decreased pancreatic β-islet cells46. The expression of a CDK4 mutant that blocks interaction with the protein p16 (encoded by CDKN2A), which binds to and inhibits CDK4 kinase activity65, in this mouse model resulted in increased CDK4 activity and subsequently led to abnormal β-cell proliferation and pancreatic hyperplasia46. These effects can potentially be explained by defective insulin signalling, as mice with disrupted insulin signalling are phenotypically similar to CDK4-deficient mice46. CDK4 has also been shown to regulate insulin secretion from β-cells in a mouse model through E2F1-regulated expression (mediated by CDK4) of Kir6.2, a component of the KATP channel involved in insulin secretion66. The role of CDK4 in glucose metabolism has been shown to be independent of its role in cell-cycle regulation in a mouse model of diabetes, both in mouse hepatocytes and in whole animals61. In this mouse model, in response to insulin, cyclin D–CDK4 was shown to phosphorylate and activate the acetyltransferase general control non-repressed protein 5 (GCN5)61. GCN5 acetylates and suppresses hepatic glucose production via inhibition of the transcriptional co-activator peroxisome proliferator-activated receptor-γ coactivator-1α (PGC1α)61, which, therefore, links insulin signalling to the expression of glucose and lipid metabolic genes61. Furthermore, cyclin D3–CDK6 was found to phosphorylate and inhibit 6-phosphofructokinase and pyruvate kinase M2, two important regulatory glycolytic enzymes, to redirect glycolytic intermediates to the pentose phosphate and serine pathways in a cellular model of T cell acute lymphoblastic leukaemia62. Treatment with CDK4/6 inhibitors in this model reduced the flux through the pentose phosphate and serine pathways, leading to depletion of NADPH and glutathione and a subsequent increase in reactive oxygen species and apoptosis of tumour cells62. Taken together, several independent lines of evidence support a role for CDK4 in the regulation of cellular metabolic pathways (FIG. 2).
Fig. 2 |. Cellular functions and regulation of CDK4.
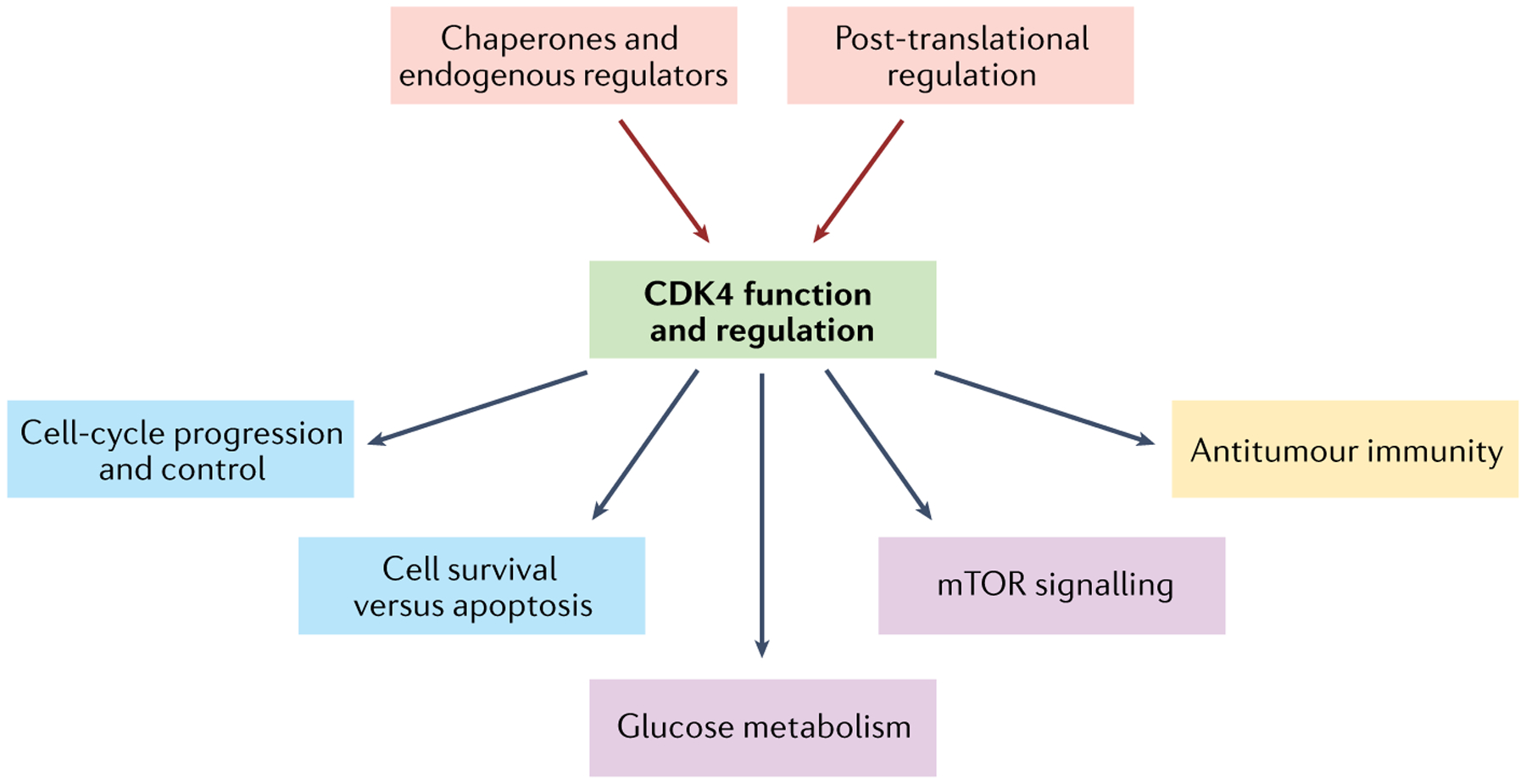
Cyclin-dependent kinase 4 (CDK4) activity is subject to multiple levels of regulation (red), including endogenous regulators93,94,97,109,110 and molecular chaperones (necessary for CDK4 folding and stabilization)71,72,124–128 as well as post-translational modifications (involved in CDK4 activation)112,113,116,117. Once activated, CDK4 exerts multiple functions in the cell: in addition to its canonical role in regulating cell cycle progression and cell survival (blue)15,57, CDK4 also promotes glucose metabolism and metabolic and survival signalling pathways through the mTOR axis (purple)62–64. Last, CDK4 has a role in immune modulation and antitumour immunity (yellow)23,36–40,219. mTOR, mammalian target of rapamycin.
CDK4 has also been shown to be involved in the regulation of lysosomal function, autophagic flux and fatty acid oxidation (FAO) through the regulation of the mTOR pathway67. The nutrient-sensing kinase 5′ adenosine monophosphate-activated protein kinase (AMPK) inhibits mTOR signalling68. Treatment of several human cell lines with CDK4/6 inhibitors in combination with an AMPK activator induced autophagy and impaired lysosomal function, leading to cell death and tumour regression owing to the lack of metabolic intermediates needed to synthetize macromolecules to survive67. CDK4 has also been shown to repress FAO by directly phosphorylating AMPKα2, which controls FAO through phosphorylation of acetyl-CoA-carboxylase69. Pharmacological inhibition or knockout of CDK4 in a mouse model caused an increase in oxidative metabolism and exercise capacity69; this effect is dependent on AMPK, as CDK4 inhibition failed to increase FAO in AMPK-knockout cells69. The involvement of CDK4 and CDK6 in the regulation of metabolic processes further supports the idea of inhibiting these kinases in RCC, in which both proliferation and metabolic signalling are dysregulated7, particularly through alterations in oxygen and nutrient sensing signalling pathways.
Regulation of CDK4 and CDK6 function
CDK4 and CDK6 function is regulated by multiple factors, including endogenous interacting partners, post-translational modifications (PTMs) and the molecular chaperone HSP9015,70–72 (FIGS 1,2).
Inhibition of CDK4 and CDK6 by endogenous proteins.
Many levels of regulation of CDK4 and CDK6 function, including endogenous interacting partners and PTMs, have been reported15,70 (FIG. 2). CDK4/6–cyclin D complex activation and inhibition are controlled by two distinct classes of regulatory subunits: the CDK interacting protein/kinase inhibitory protein (Cip/Kip) family, including p21Cip1 (CDKN1A)73–76, p27Kip1 (CDKN1B)77–79 and p57Kip2 (CDKN1C)80,81; and the INK4 family, including p15INK4b (CDKN2B)82, p16INK4a (CDKN2A)65, p18INK4c (CDKN2C)83,84 and p19INK4d (CDKN2D)84,85. Cip/Kip family members can act on a broad spectrum of CDK–cyclin complexes, inhibiting CDK1, CDK2, CDK4 and CDK6, whereas INK4 proteins exclusively inactivate CDK4–cyclin D and CDK6–cyclin D complexes65,86–89. INK4 proteins function as tumour suppressors, and loss-of-function mutations in these proteins cause carcinogenesis90. For example, CDKN2A maps to a region on chromosome 9p21 (REF.91) that was found to be deleted in an array of various cancer types, including RCC, suggesting a tumour suppressive function for CDKN2A92. Structural work using circular dichroism and nuclear magnetic resonance (NMR) spectroscopy showed that purified p16 has low structural stability and a highly dynamic structure93. Interaction of p16 with CDK4 occurs between ankyrin-like repeats in p16 (structural domains frequently implicated in mediating protein–protein interaction)93, and the N-terminal portion of CDK4, which overlaps with the binding site for cyclin D94. Binding of p16 to CDK4 inhibits CDK4 kinase activity65. A point mutation in CDK4 (CDK4–R24C) is sufficient to abrogate its binding with p16 (REF.95), suggesting that the R24 site of CD4K is involved in binding with p16. Additional CDK4 sites important for p16 binding were identified using scanning mutagenesis that changed charged residues to neutral alanines and included residues 22, 25, 97 and 281 of CDK4 (REF.95). Mice homozygous for the CDK4–R24C mutation develop multiple tumours and are at increased susceptibility to chemical carcinogens such as 7,12-dimethylbenz[a]anthracene (DMBA)96,97. MEFs from homozygous CDK4R24C mice are susceptible to oncogene-induced transformation96,97, have an increased proliferation rate and escape both replicative senescence and contact-mediated growth inhibition, demonstrating the importance of p16-mediated inhibition of CDK4 in cell growth97.
Specific alterations in the cyclin D–CDK4–Rb axis are frequent events in many cancers98,99, including RCC99. One study analysing 42 glioblastoma tissue samples found deletions, mutations or loss of heterozygosity in either RB1 (encoding Rb) or CDKN2A in 86% of samples, and changes in these two genes were generally mutually exclusive, implying that the CDK4–Rb axis is a crucial regulatory pathway in glioblastoma100. Mutation and deletion of CDKN2A, including germline mutations, are also frequent events in melanoma101 and were shown to correlate with sensitivity to the CDK4/6 inhibitor palbociclib in a panel of 47 melanoma cell lines, whereas RB1 loss predicted resistance to palbociclib101, again indicating the mutual exclusivity of these pathways. Interestingly, an increased risk of melanoma has been observed in patients diagnosed with RCC and vice versa102. Kidney cancer is often diagnosed after melanoma and is generally of a lower stage at diagnosis in patients with a history of melanoma than in those without102. In a study including 42 patients with both RCC and melanoma, the renal tumour was diagnosed concomitantly or after melanoma in 83% of patients102, but germline CDKN2A mutations were only identified in patients with a known familial melanoma syndrome (2 of 42 total patients), suggesting that a potential genetic predisposition underlying melanoma and RCC coexistence is partially independent from CDKN2A102. However, somatic CDKN2A mutation, deletion or hypermethylation was found in 15.8% of renal tumours overall in The Cancer Genome Atlas (TCGA) database (n = 894 total samples) and was altered in all examined histological subtypes103. CDKN2A alterations were also associated with worse overall survival across different types of RCC on Kaplan–Meier analysis (P < 0.0001)103. Collectively, these results suggest that the regulation of CDK4 activity by the tumour suppressor p16 is important in RCC, and CDK4 activity modulation could be harnessed in RCC treatment.
Several germline mutations causing inherited genetic syndromes can predispose patients to developing RCC7, including Von Hippel–Lindau syndrome (VHL), Birt–Hogg–Dubé syndrome (FLCN), hereditary papillary RCC (MET) and hereditary leiomyomatosis (FH)7,104–108. Germline missense mutations in CDKN2B (encoding the endogenous CDK4 inhibitor protein p15INK4B) were identified in one family with features of hereditary RCC in which none of the gene mutations known to predispose to RCC was found109. Functional evaluation in colony formation and cell-growth assays demonstrated that these mutations impaired the tumour suppressive function of CDKN2B109.
p16 expression has been demonstrated to increase under hypoxic conditions in a monkey kidney cell line (CV-1), where p16 binds CDK4 under hypoxia, but not under aerobic conditions in non-transformed cells110. Regulation of p16 expression under hypoxia is of particular interest for RCC, as the most frequent RCC subtype (clear cell RCC (ccRCC)) is characterized by a dysregulation of oxygen sensing regulatory pathways7. Furthermore, p16 is regulated by phosphorylation, and phosphorylated p16 preferentially interacts with CDK4 (REF.111). Identified phosphorylation sites on p16 include serines 7, 8, 140 and 152, which are located outside the conserved CDK4 interaction domain, and are instead positioned within the regions mutated in sporadic and inherited cancers111. All deletions and mutations affecting p16 expression or function ultimately abolish p16-mediated inhibition of CDK4, generating uncontrolled, pro-tumorigenic CDK4 activity99. Control of CDK4 activity by the tumour suppressor p16 is the most widely studied endogenous CDK4 inhibition strategy, and further work is necessary to understand the functional spectrum of CDK4-inhibitory proteins.
Post-translational regulation of CDK4 and CDK6.
CDK4 function is in part regulated by tyrosine phosphorylation (FIG. 2), which is important for the control of DNA damage checkpoints112. CDK4 was found to be tyrosine phosphorylated in quiescent, arrested rat fibroblasts and then dephosphorylated upon cell cycle entry112. Furthermore, CDK4 catalytic activity requires phosphorylation on T172, which is independent of assembly with cyclin D113. This phosphorylation is mediated by CDK7 (REF.114) and can also be mediated by JUN N-terminal kinases (JNKs)115. CDK4-T172 phosphorylation increases in response to mitogenic stimuli in part through the mTORC1 signalling116,117, which links CDK4 and CDK6 activity with nutrient sensing and cell metabolism.
HSP90-mediated chaperoning of CDK4 and CDK6.
Many proteins require assistance by chaperone proteins for proper folding and activation118. HSP90 is an abundant molecular chaperone involved in the conformational maturation, folding and activation of its target proteins, including many oncogenic kinases such as CDK4 (REFS16,119,120) (FIGS 1,2). A large cohort of co-chaperones facilitates protein interaction with HSP90 and regulates HSP90 ATPase activity, thereby promoting target protein folding and activation121–123. The kinase-specific co-chaperone CDC37 is highly specialized for promoting conformational development of kinase target proteins including CDKs, such as CDK4 (REFS124,125). CDC37 decelerates the ATPase activity of HSP90 to enable protein kinases to interact with the chaperone122. CDK4 and CDC37 directly interact and CDC37–HSP90 chaperoning mediates stabilization of CDK4, preferentially associating with the apo-fraction of CDK4 not bound to cyclin D71,72. However, CDC37 also helps mediate the assembly of cyclin D–CDK4 complexes126,127. Furthermore, cyclin D1 stability is regulated by the molecular chaperone HSP70 (REF.128). Cyclin D1 binding to HSP70 is increased by CDK-dependent phosphorylation of HSP70 in response to changes in cellular conditions such as DNA damage, leading to cyclin D1 degradation128 (FIG. 1).
Structural analysis of CDK4 in complex with CDC37 and HSP90 has provided a great deal of insight into the chaperoning and activation of this and other kinase clients125,129–133. The first structural view of HSP90 interacting with a client protein (CDK4) was solved using single-particle electron microscopy and suggested that HSP90 induces a conformational change of the bound kinase through its ATPase activity131. The 3.9 Å cryogenic electron microscopy (cryoEM) structure of full-length HSP90, CDK4 and CDC37 enabled visualization of this multi-protein complex assembly and provided a detailed picture of the interactions within the complex129. The cryoEM structure of the HSP90–CDC37–CDK4 complex revealed that CDK4, partially unfolded, is stabilized and protected by CDC37 and HSP90 (REF.129); the authors of the study proposed that ATP hydrolysis and subsequent opening of the HSP90 structure gives CDK4 the opportunity to properly fold and, in the case of failure, another chaperone cycle can commence129. Using this information, molecular modelling studies have demonstrated reciprocal changes in kinase protein and chaperone stability and suggest that the CDC37–HSP90 machinery protects proteins from degradation by exploiting areas of inherent instability in the kinase structure124.
CDC37–HSP90 chaperoning of kinases, including CDK4, is also regulated by post-translational modifications134. Casein kinase 2-mediated phosphorylation of CDC37 on S13 (CDC37-S13) is a prerequisite for CDC37 association with CDK4, as well as other kinases135; phosphorylated CDC37 can then recruit CDK4 to HSP90 for chaperoning, leading to CDK4 stability and kinase activity135,136. Notably, in the cryoEM structure, CDC37 is phosphorylated on S13 and, therefore, is stabilized in a kinase-interacting conformation125,129; subsequent dephosphorylation of CDC37-S13 by the phosphatase co-chaperone protein phosphatase 5 (PP5) is then required for full kinase activation and release132,137. A series of tyrosine phosphorylation events on CDC37 is also important for CDK4 chaperoning138: tyrosine phosphorylations of CDC37 (CDC37-Y4 and CDC37-Y298) were shown to be involved in the dissociation of CDK4 from CDC37 after recruitment to HSP90, followed by phosphorylation of HSP90 on Y197, which released CDC37 from the kinase–HSP90 complex, as demonstrated in immunoprecipitation assays138,139. Finally, phosphorylation of HSP90 on Y627 led to the dissociation of CDK4 and remaining co-chaperones from HSP90, as shown in transfected COS7 cells138.
Overall, CDC37 acts as a protein quality-control checkpoint and the chaperone machinery helps to stabilize and regulate the activity of CDK4 and other kinases125,129,140. Thus, CDC37 and HSP90 both support oncogenic processes and, in fact, CDC37 has been shown to promote colorectal cancer growth specifically through CDK4 activation in human colon cancer cell lines and xenografts141. The HSP90–CDC37 chaperone machinery, therefore, provides an additional target for the allosteric inhibition of CDK4 in cancer therapy141. Disruption of the HSP90–CD37–kinase complex with an HSP90 inhibitor or CDC37-knockdown leads to cell-cycle arrest, apoptosis and decreased proliferation in various tumour cell lines140,142,143. Furthermore, short peptides specifically targeting the CDC37–HSP90 interaction have been developed using a computational approach144 and have been shown to efficiently disrupt CDC37–HSP90 interaction and compromise the stability of CDK4 in HEK293 cells, although they exhibited limited cytotoxicity144. Using a similar strategy, peptides mimicking CDK4 specifically disrupted its interaction with HSP90 and led to the induction of apoptosis in ccRCC 786-O cells145.
CDK4/6 inhibition in murine and multiple myeloma cell lines was also effectively induced by promoting their E3 ubiquitin ligase-mediated degradation, through molecules known as proteolysis-targeting chimaeras (PROTACs)146. Taken together, these studies highlight the importance of molecular chaperones in mediating stability and function of CDK4 and how targeting this machinery could provide an adjunctive mechanism for CDK4 inhibition. However, the most successful and well-studied strategy for CDK4 inhibition to date is the competition with ATP binding at the catalytic site147.
Development of CDK4/6 inhibitors
The initially developed non-specific CDK inhibitors had high levels of toxic effects, in large part owing to inhibition of CDK2 and lack of specificity17,18. Some of these agents were used in phase I and phase II clinical trials but had high levels of dose-limiting toxic effects including deep venous thrombosis, severe diarrhoea and electrolyte abnormalities17,18. Considering that the CDK4–cyclin D–Rb axis is frequently mutated across tumour subtypes15, specific inhibitors for these kinases were sought. One of the first specific small-molecule inhibitors of CDK4 tested preclinically was CINK4 (a chemical inhibitor of CDK4)148. CINK4 was able to specifically inhibit CDK4 in in vitro kinase assays, cause growth arrest and decrease Rb phosphorylation in osteosarcoma and colon cancer cells, and slow the growth of colon cancer xenografts148. Further attempts were made to refine CDK4-specific inhibitors, and using CDK4 structural information as guidance led to success149. Obtaining a crystal structure of CDK4 to aid inhibitor design was initially challenging, as purifying and crystallizing the protein was difficult149. However, the crystal structure of the related family member CDK2 had been solved150,151; thus, a CDK4 mimic was created by swapping the CDK4 ATP-binding pocket into CDK2, which revealed that CDK4 has a larger binding pocket than CDK2, enabling the design of slightly larger compounds that were able to specifically bind and inhibit CDK4, but did not fit the binding pocket of CDK2 (REF.152). Computational methods further helped define inhibitor selectivity for CDK4 over CDK2 and other related kinases153,154. Different molecular modelling strategies were used to understand the differences between CDK4 and CDK2 that impart selectivity for CDK4 inhibitors (including CINK and PD-0183812, another small-molecule CDK4/6-specific inhibitor154); upon binding of either CINK or PD-0183812 to CDK4, the motion of a disordered loop within the protein was reduced, whereas loop flexibility was largely unaffected in CDK2, suggesting a stronger structural effect of these inhibitors on CDK4 than CDK2 (REF.154). In this study, the selectivity of these compounds for CDK4 was also the result of reduced solvent accessibility at the active site of CDK4 compared with CDK2, in addition to the stability of the hydrogen bonds formed between the inhibitor and a lysine residue within the active site binding pocket of CDK4 (REF.154). With the knowledge obtained by studies on CDK4 inhibitor specificity, several classes of compounds have been tested as specific CDK4 inhibitors155.
Among CDK4 inhibitors that progressed to FDA approval are palbociclib and ribociclib (approved in 2015 and 2017, respectively), both of which are pyrimidine derivatives156,157. Structure-based modification led to the refinement of palbociclib (PD-0332991), which showed remarkable selectivity for CDK4/6 compared with 36 other protein kinases, including the closely related CDK2, and demonstrated promising pharmacokinetics, with efficacy at daily dosing and >50% oral bioavailability156,157. Initial studies demonstrated that palbociclib treatment led to G1 arrest, decrease in Rb phosphorylation and regression of colon cancer tumour xenografts156. Ribociclib (LEE011) also showed good selectivity for CDK4/6 over other related kinases, as well as efficacy in both enzymatic and cell-based assays158. The third CDK4/6 inhibitor to receive FDA approval, abemaciclib (LY2835219, approved in 2017), has been shown to inhibit CDK4/6 with low nanomolar potency and lead to G1 arrest specifically in Rb-proficient cells, suggesting that Rb status could be used to help predict responsiveness to this inhibitor159; moreover, abemaciclib was also effective and well tolerated with oral administration in mouse xenograft studies159–162. Unlike palbociclib and ribociclib, abemaciclib is more selective for CDK4 than CDK6 and also possesses inhibitory selectivity towards CDK9, although limited work delineating unique features specifically based on this selectivity is available23,163. Differences in the selectivity of these inhibitors towards CDK4/6 compared with other kinases could underlie subtle differences in their intended and off-target effects, as well as predict the effectiveness of these drugs in combinations. In general, literature rarely draws a distinction between CDK4 and CDK6 in discussing the function of these proteins or small-molecule inhibitors targeting CDK4 or CDK6. Many of the features predicted to impart selectivity of small-molecule inhibitors for CDK4 or CDK6, such as the non-conserved aspects of the ATP-binding pocket, were seen in co-crystal structures of these compounds bound to monomeric CDK6163 (FIG. 3). Differences in structure and potency exist between palbociclib and abemaciclib, but common gene expression features create a composite signature of response to CDK4/6 inhibition164. Overall, the structure-guided development of CDK4/6 specific inhibitors (palbociclib, ribociclib and abemaciclib) enabled the progression of these inhibitors to clinical approval, as they were better tolerated than the previously developed non-specific CDK inhibitors.
Fig. 3 |. Structure of CDK6 in complex with CDK4/6 inhibitors.
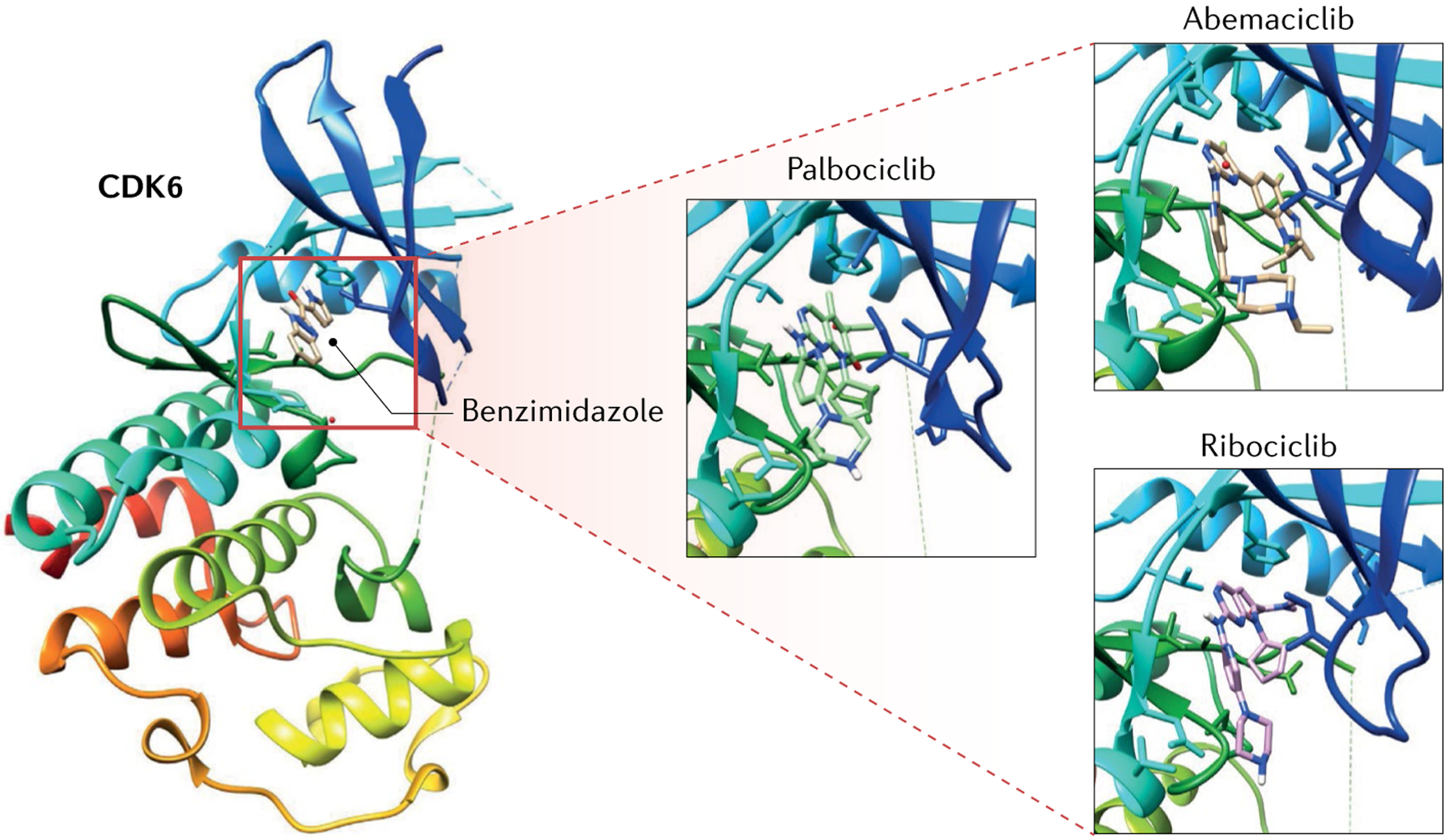
Crystal structure of cyclin-dependent kinase 6 (CDK6) in complex with its ligand benzimidazole bound to the catalytic cleft of CDK6 (Protein Data Bank (PDB) code: 4AUA, red square in the crystal structure on the left)265. The catalytic cleft of CDK6 contains the ATP-binding pocket and lies between the N-domain (red, orange, yellow and green) and the C-domain (cyan and blue). Close up of CDK6 catalytic cleft (squares on the right) in complex with the ATP-competitive CDK4 and CDK6 inhibitors palbociclib (PDB code: 5L2I), abemaciclib (PDB code: 5L2S) and ribociclib (PDB code: 5L2T)163.
CDK4/6 inhibition in breast cancer — road to FDA approval
CDK4/6 inhibitors were first used clinically in the treatment of HR+ and HER2− advanced breast cancer21. This cancer is primarily treated with anti-oestrogens, similar to the antiandrogen therapies used in prostate cancer165,166, and CDK4 inhibitors were found to work best in combination with these therapies167. Preclinical studies found that CDK4/6 inhibitors preferentially killed oestrogen-receptor positive (ER+) breast cancer cell lines and worked synergistically with anti-oestrogens167. Several factors can mediate this synergism, including evidence that ER+ breast cancers generally retain an Rb expression signature168 and that cyclin D1 transcription is oestrogen responsive169. The phase II and III clinical trials PALOMA12,21, MONALEESA13,22 and MONARCH14 led to the approval of palbociclib, ribo ciclib and abemaciclib, respectively, in combination with anti-oestrogen therapy, either aromatase inhibitors or fulvestrant, between 2015 and 2018 (REFS12–14,21,22) (FIG. 4). Abemaciclib is the only inhibitor approved as a single agent and administered continuously, as both palbociclib and ribociclib demonstrated dose-limiting neutropenia19,170 and are, therefore, administered continuously for 3 weeks, followed by 1 week off19. Meta-analyses of these pivotal trials demonstrated increased PFS (HR 0.55, P < 0.00001, n = 4,580 patients171) as well as overall survival (HR 0.79, P = 0.004, n = 4,580 patients171) in patients treated with CDK4/6 inhibitors in combination with anti-oestrogen therapy compared with anti-oestrogen therapy alone171–174. Results from the pivotal trial PALOMA, testing the efficacy of palbociclib plus letrozole (an aromatase inhibitor), showed a better PFS in patients treated with the combination therapy (24.8 months (95% CI 22.1 to not estimable)) than in patients treated with letrozole alone (14.5 months (95% CI 12.9–17.1 months))21. Pooled analysis of results from phase III studies highlighted that CDK4/6 inhibitors have a positive effect on PFS in patients with breast cancer if used as first-line combination therapy (HR 0.55 (95% CI 0.49–0.62)), or as a second-line and beyond combination therapy (HR 0.56 (95% CI 0.49–0.64))175. Furthermore, several trials, including NATALEE176 and MonarchE177, examining the role of CDK4/6 inhibition in the adjuvant setting, are ongoing178. Preliminary results from several clinical trials indicate that CDK4/6 inhibition could also have efficacy in HER2+ and triple-negative breast cancer (TNBC)19,179. The success of CDK4/6 inhibitors in ER+HER2− cancer led to FDA approval of these small molecules for breast cancer treatment in combination with anti-oestrogen therapy, but the efficacy of CDK4/6 inhibitors has also been consistently explored in several other cancers180–185.
Fig. 4 |. Timeline of CDK4/6 inhibitor development and approval of systemic therapies for renal cell carcinoma.
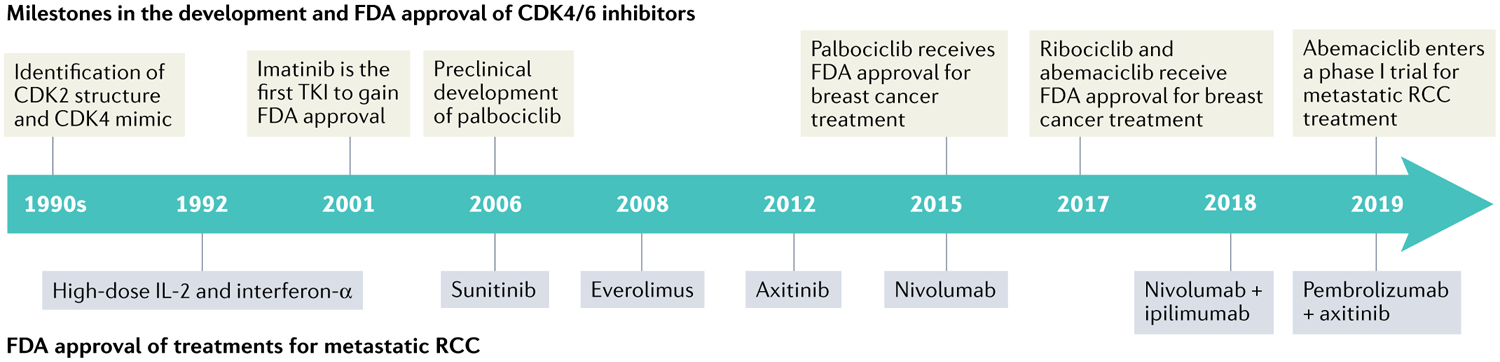
Timeline showing milestones in the development and FDA approval of cyclin-dependent kinase 4 (CDK4) and CDK6 inhibitors (above)12–14,21,22. Also, FDA approval of various systemic and targeted therapies for the treatment of advanced and metastatic renal cell carcinoma (RCC, below) is shown3–5,8–10. TKI, tyrosine kinase inhibitor.
CDK4/6 inhibition in other cancers
Palbociclib, ribociclib and abemaciclib have been tested in preclinical models of several cancers, many of which have causative alterations in the CDK4/CDK6 pathway, and clinical investigations are currently ongoing180–185. CDK4/6 inhibitors have not found notable success as single agents, but the relationship between alterations in CDK4/6 signalling and response to therapy provides an interesting framework for examination of these agents in RCC, as has been done in other cancers180–185. Germline alterations in the CDK4/6 pathway, including nonsense mutations, missense mutations and splice variants, have been documented in familial melanoma186–188, and palbociclib was found to induce cytostasis and delay tumour growth in a xenograft model of melanoma189. Mantle cell lymphoma frequently results from a translocation involving CCND1 (encoding cyclin D1) and has shown sensitivity to palbociclib in one phase I trial, with PFS >1 year in 5 of 17 patients190–192. CDK4 is also frequently mutated in liposarcoma, with over 90% of liposarcomas harbouring CDK4 amplifications193. A phase II study of palbociclib in patients with progressive liposarcoma (n = 29 evaluable for the primary end point) exceeded the primary end point and reported 66% PFS (90% CI 51–100%) at 12 weeks, suggesting that CDK4/6 inhibition is promising in this setting194. Additionally, preclinical efficacy of CDK4/6 inhibitors, particularly palbociclib, was observed with a dose-dependent decrease in the viability of adrenocortical carcinoma cell lines195. Taken together, these studies exemplify the broad applicability of CDK4/6 inhibitors in cancer treatment.
Owing to its tumour suppressive role and its frequent loss in cancer, p16 has been examined as a stratifying marker to predict responsiveness to CDK4/6 inhibitors and has been used as a selection criterion in some preclinical and clinical studies30,196–198. A preclinical study using cellular and xenograft models of gastric cancer demonstrated that palbociclib selectively inhibited proliferation and migration in CDKN2A-mutated cells but not in cells with wild-type CDKN2A196, indicating that CDK2NA status might predict sensitivity to CDK4/6 inhibition. Additionally, in a phase II trial of palbociclib in previously treated patients with advanced, progressive non-small-cell lung cancer with p16-null disease (assessed using immunohistochemistry), half of the patients achieved stable disease for 4–10.5 months30. CDK4/6 inhibitors have shown only modest benefit in unselected patients with serous ovarian cancer197, but results from a case report demonstrated ongoing response to palbociclib and letrozole treatment in a heavily pre-treated patient with homozygous CDKN2A deletion, with ongoing decrease of lesion size (46% decrease from baseline) on CT scan after 1 year of therapy197. Homozygous CDKN2A deletion is a rare genetic event in ovarian cancer, but these observations suggest that patient selection could be of paramount importance in predicting response to treatment197.
Results of a small phase II study of palbociclib in patients with metastatic, platinum-refractory urothelial carcinoma were disappointing198; patients were selected for p16 loss and intact Rb expression using immunohistochemistry and only 2 of 12 patients met the primary end point of PFS at 4 months, whereas the overall survival in the study cohort was 6.3 months198. The results of this trial suggest that robust preclinical studies to identify tumour characteristics, such as genetic alterations, are needed in urothelial as well as other cancers for optimal patient selection and rational choice of combination therapies. Examination of CDK4/6 inhibitors in various cancers demonstrates that the identification of mechanisms or expression signatures that predict responsiveness in preclinical settings must be taken with caution into the clinical arena owing to the complexity of intended and possible off-target effects. These studies also highlight the need to understand the mechanisms of resistance to single-agent therapy and how it can be overcome with combination therapies.
Resistance to CDK4/6 inhibitors
Intrinsic and acquired resistance to CDK4/6 inhibitors has been documented and examined, similar to other targeted cancer therapeutics, but this work is still largely preclinical18. An intact Rb pathway is thought to be required for CDK4/6 inhibitor efficacy, and Rb-null status is predictive of intrinsic resistance, as observed in a mouse model of mammary carcinoma23,36. One systematic screening of hundreds of different cancer cell lines found that genomic alterations leading to elevation of D-type cyclin levels were associated with enhanced sensitivity to abemaciclib, whereas CDKN2A alterations were less predictive of abemaciclib sensitivity199. Palbociclib does not bind to CDK4 in cells insensitive to the drug, in part because CDK4–p16 interaction precludes palbociclib binding200. Results from a retrospective analysis of cultured breast cancer cell lines167 showed that high p16 levels were predictive of resistance to CDK4/6 inhibition, but low p16 levels did not necessarily predict sensitivity to these inhibitors, suggesting that additional determinants of CDK4/6 inhibitor sensitivity might exist200. Results of another preclinical study in breast cancer cell lines showed that CDK6 overexpression and low CDK4 expression were associated with ribociclib resistance and that increased p21 levels positively correlated with ribociclib sensitivity, indicating a role for p21 in the development of ribociclib resistance201. Accordingly, stabilization of p53 via co-treatment with a murine double minute 2 (MDM2) antagonist resulted in p21 accumulation and increased sensitivity to CDK4/6 inhibitors in both resistant patient-derived xenografts and a mouse melanoma model202. Interestingly, upregulation of cyclin D1 secondary to CDK4/6 inhibitor treatment was shown to sequester p21 and reduce the ability of p21 to inhibit CDK2 (REF.202), which is necessary for proliferation in CDK4/6 inhibitor-treated cells, indicating that CDK2 is the effector of p21 function in mediating sensitivity to CDK4/6 inhibitors202. Cyclin E overexpression has also been observed concomitantly with CDK4/6 inhibitor resistance in breast cancer cell lines and patient samples, and cyclin E expression likely contributes to this resistance203. Cyclin E activates CDK2; therefore, resistance to CDK4/6 inhibitors can be overcome by CDK2 blockade203, although no CDK2-specific inhibitors are currently approved. Furthermore, PTMs of CDK4, including the activating T172 phosphorylation (which is a central rate-limiting step for cell-cycle initiation)204, was also found to be predictive of palbociclib sensitivity in breast cancer cell lines and primary tumour samples205.
Crosstalk between the CDK4–cyclin D–Rb axis and other signalling pathways also has an important role in mediating sensitivity and resistance to CDK4/6 inhibition206. A rapid adaptive resistance mechanism to CDK4/6 inhibition has been seen preclinically in a KRAS-driven pancreatic cancer model117. This response was the result of KRAS-dependent post-transcriptional upregulation of cyclin D1 and cyclin E1, which also occurs in an mTOR and mitogenic signalling-dependent manner117 and could be attenuated by mTOR inhibition across a variety of pancreatic cancer patient-derived xenograft lines117. In this study, CDK2 activation and the formation of the cyclin D–CDK4 complex, even in the presence of CDK4/6 inhibition, were hypothesized to be implicated in resistance to CDK4/6 inhibitors117. Alterations and rewiring of various kinase signalling pathways are important contributors to CDK4/6 inhibitor resistance and are new potential therapeutic targets to exploit207. In a CRISPR–dCas9 screen aimed at examining the effect of various gene deletions in the T24 bladder cancer cell line, several signalling pathways were identified as potential mediators of palbociclib resistance208, including receptor tyrosine kinases (RTKs), phosphoinositide 3-kinase (PI3K)–AKT–mTOR, Ras–mitogen-activated protein kinase (MAPK), Janus kinase (JAK)–signal transducer and activator of transcription (STAT), and Wnt208. Importantly, the combination of palbociclib and inhibitors targeting these kinases demonstrated a reduction in cell growth in cell culture and xenograft models of bladder cancer, highlighting the potential of combination therapies in preclinical studies208.
Several CDK4/6 inhibitor resistance mechanisms have been identified199–203, but the pharmacological development of inhibitors targeting these mechanisms is still lacking. The identification of target proteins responsible for CDK4/6 inhibitor resistance might be essential to the clinical success of these therapeutics; thus, further elucidation of biomarkers of cellular resistance to CDK4/6 inhibitors is necessary.
Synergism with mTOR inhibitors
Activation of the mTOR pathway and the upstream RTK and PI3K–AKT pathways have been implicated in resistance to CDK4/6 inhibition as well as in the regulation of these kinases206. Agents specifically targeting mTOR and other signalling kinases are approved for the treatment of various cancers209 and have been a mainstay in the treatment of advanced and metastatic RCC210,211. Combination of CDK4/6 inhibitors with mTOR inhibition has, therefore, been an area of great research interest in cancer treatment (FIG. 5).
Fig. 5 |. CDK4 at the interface between mTOR signalling and immune checkpoint regulation.
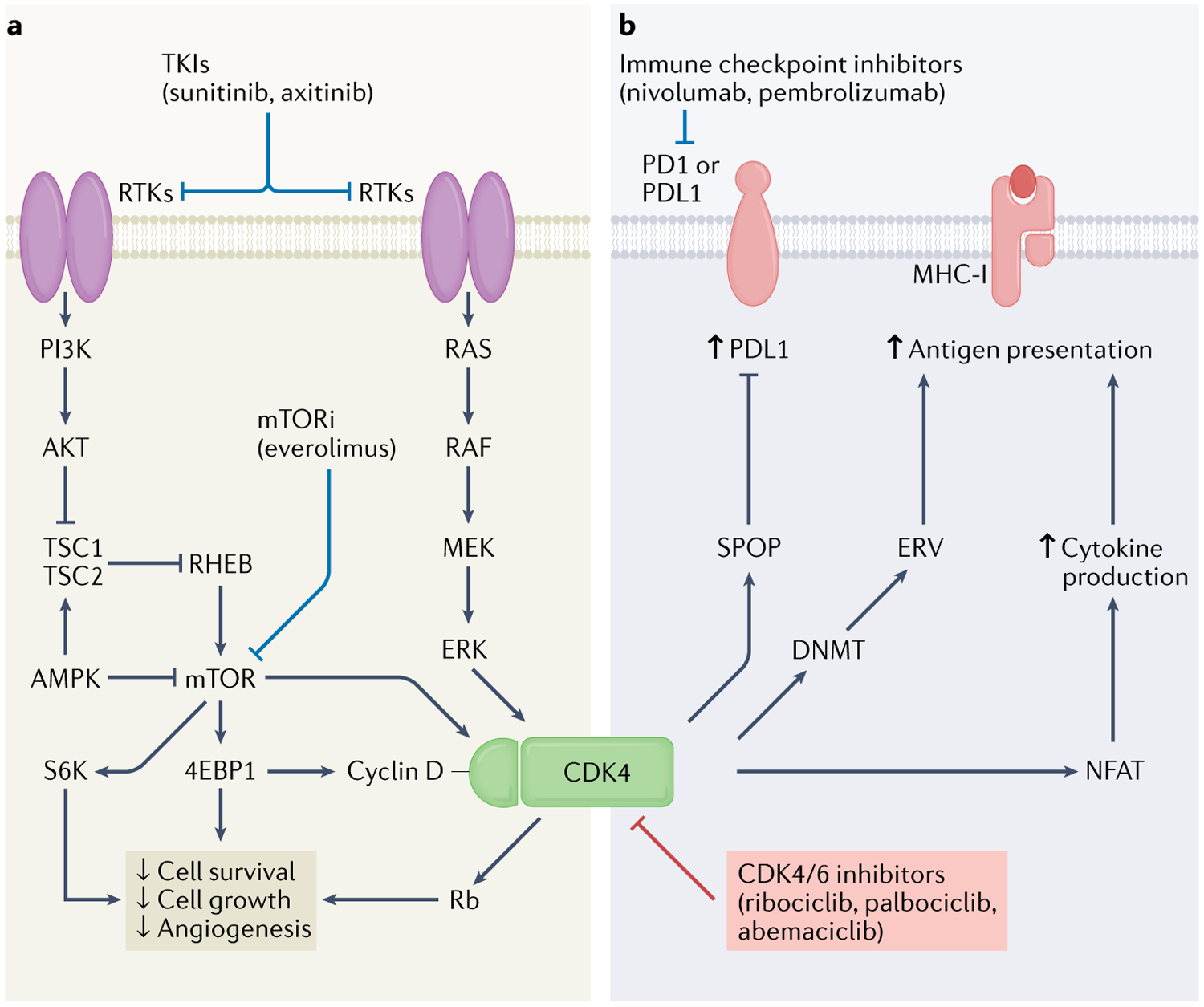
Cyclin-dependent kinase 4 (CDK4)–cyclin D complex function is in part regulated through receptor tyrosine kinase (RTK) signalling axes such as RAS–mitogen-activated protein kinase kinase (MEK)–extracellular signal-regulated kinase (ERK)117 and phosphoinositide 3-kinase (PI3K)–AKT–mechanistic target of rapamycin (mTOR)7 and their downstream effectors. Collectively, these pathways work to affect cell and tumour growth and survival. a | Tyrosine kinase inhibitors (TKIs, such as sunitinib and axitinib) and mTOR inhibitors (such as everolimus) can work synergistically with CDK4/6 inhibitors (ribociclib, palbociclib and abemaciclib) to reduce cell survival, cell growth and angiogenesis63,208. b | CDK4 also alters tumour immune response36–40,219. CDK4 inhibitors can work synergistically with immune checkpoint inhibitors (such as nivolumab and pembrolizumab) to stimulate antitumour immune response through several mechanisms, including functional modulation of the upstream regulator of programmed cell death 1 ligand 1 (PDL1) speckle-type POZ protein (SPOP) (leading to upregulation of PDL1)40, as well as DNA methyltransferase (DNMT)36 (leading to increased antigen presentation) and nuclear factor of activated T cells (NFAT)37 (leading to increased cytokine production and antigen presentation). CDK6 has not been included in the figure, owing to the discrepancy in the availability of high-quality data. 4EBP1, 4E-binding protein1; AMPK, AMP-activated protein kinase; ERV, endogenous retrovirus; MHC-I, major histocompatibility complex class I; PD1, programmed cell death 1; Rb, retinoblastoma; S6K, S6 kinase; TSC, tuberous sclerosis complex.
CDK4 activity is regulated by mTOR, through mTOR-dependent phosphorylation of CDK4-T172 (REFS116,212). Insights into the mechanism behind the synergy of mTOR with CDK4/6 inhibition came from studies in cell culture and mouse models using abemaciclib, and suggest that CDK4 and CDK6 can activate mTOR by phosphorylating the tumour suppressor tuberous sclerosis complex 2 (TSC2) on S1217 and S1452 (REF.63). TSC2 normally exerts inhibitory control on mTORC1; thus, CDK4 or CDK6 phosphorylation of TSC2 releases mTOR from TSC2-mediated endogenous inhibition, much like Rb phosphorylation by CDK4 or CDK6 releases E2F63. Cyclin D was also found to be a binding partner of TSC2 (REF.213). Co-overexpression of cyclin D and CDK4 or CDK6 in human embryonic kidney fibroblasts increased TSC2 phosphorylation and decreased TSC2 levels213, compromising TSC2 function213; following TSC2 inhibition, mTOR signalling was initiated, leading to increased phosphorylation of the downstream targets of mTOR, 4E-binding protein 1 (4EBP1) and S6 kinase (S6K), and promoting cell growth213.
Taken together, these results indicate that the effects of CDK4/6 inhibitors on the suppression of cell proliferation and metabolism partially depend on the inhibition of mTOR signalling. Interestingly, in TNBC cells, palbociclib alone led to upregulation of AKT and mTOR signalling29; moreover, synergistic effects of sustained combination treatment with palbociclib and PI3K–mTOR inhibitors in these cells showed increased cell death29. In the same study, downregulation of glucose metabolism was observed in TNBC cells treated with palbociclib and this effect was enhanced by the addition of the PI3K inhibitor BYL719 under both normoxic and hypoxic conditions. These data indicate that inhibition of CDK4/6 also negatively affects the function of HIF1 (REF.29). The mTOR pathway is crucial for nutrient sensing and metabolic signalling214; thus, the synergism and crosstalk of the mTOR and CDK4/6 pathways highlights the importance of integrating nutrient status and cellular metabolism with cell-cycle control, which is of particular interest considering the biology of RCC.
Results of a growth inhibition screen in p16-null non-small-cell lung cancer cell lines in which several targeted and cytotoxic agents were combined with palbociclib showed that only mTOR inhibitors had a synergistic effect with palbociclib30. Preclinical work in ER+ breast cancer cell lines and xenografts demonstrated the most robust growth arrest and delay in resistance development when mTOR inhibition was combined with CDK4/6 inhibition and hormonal therapy31. In these models, the addition of the mTOR inhibitor AZD2014 enhanced the effects of CDK4/6 inhibition on E2F-mediated transcription31 (FIG. 5); furthermore, CDK4/6 inhibitor-resistant MCF7 cell lines reactivated the CDK–Rb–E2F pathway but retained sensitivity to mTOR inhibition31. These data suggest that mTOR inhibition is a therapeutic option in the case of resistance to CDK4/6 inhibitors. Synergism between palbociclib and the second-generation pan-mTOR inhibitor MLN0128 has also been shown in ER− breast cancer and TNBC cell lines32. In a cell-culture model of T cell acute lymphoblastic leukaemia, treatment with mTOR inhibitors and the glucocorticoid dexamethasone acted in synergy with ribociclib, whereas many drugs traditionally used in relapsed T cell acute lymphoblastic leukaemia, such as methotrexate and doxorubicin, which act on rapidly proliferating cells, behaved as ribociclib antagonists215. These data indicate that the drug mechanism of action is essential to identify efficacious combination therapies. Synergism between palbociclib and the mTOR inhibitor MLN0128 was also shown in a mouse model of intrahepatic cholangiocarcinoma, in which either agent alone reduced tumour growth and the combination therapy led to tumour regression33. In this context, mTOR inhibition counteracted the upregulation of cyclin D seen with palbociclib alone33. Similarly, palbociclib in combination with the mTOR inhibitor everolimus showed enhanced ability to disrupt metabolism and cell growth in preclinical models of glioblastoma compared with the single agents34. Additionally, everolimus treatment enhanced the blood–brain barrier permeability of palbociclib in mice, which is promising for the treatment of intracranial metastases in several malignancies, including RCC34. Combination of the mTOR inhibitor temsirolimus and palbociclib also showed a synergistic effect in cell-line models of diffuse intrinsic pontine glioma, a particularly lethal paediatric brain tumour35.
In summary, inhibition of CDK4/6 in combination with mTOR inhibitors shows improved efficacy when compared with the respective monotherapies. These findings are of particular relevance to RCC given the prominent role of mTOR hyperactivation in the pathogenesis of this disease and the use of mTOR inhibitors in RCC treatment7,9.
Role of CDK4 and CDK6 in immune modulation and checkpoint inhibitor response
CDK4 is well known to have a role in T cell proliferation and cytokine responsiveness216,217, and several studies have explored the role of CDK4 and CDK6 in antitumour immunity as well as the synergism of CDK4/6 inhibition with ICIs (FIGS 2,5). Results of one study using a variety of preclinical models of solid tumours including cell lines and xenografts showed that, in addition to tumour cell cycle arrest, treatment with CDK4/6 inhibitors triggered antitumour immunity primarily via two mechanisms, both of which are associated with decreased DNA methyltransferase 1 (DNMT1) levels, a downstream target of the CDK–Rb–E2F transcriptional axis36. First, activation of endogenous retroviral elements resulting from decreased DNA methylation enhances intracellular levels of double-stranded RNA and antigen presentation through a phenomenon known as ‘viral mimicry’36, and, second, CDK4/6 inhibition suppresses the proliferation of regulatory T cells36. Together, these effects promote tumour cell clearance through T cell response, which is enhanced by checkpoint-blockade therapy36. Furthermore, transcriptomic analysis of serial biopsy samples obtained from patients (n = 23) with breast cancer treated with CDK4/6 inhibitors showed the activation of antitumour immunity36.
CDK4/6 inhibitors have also been found to enhance the activation of effector T cells and infiltration of T cells into tumours, thereby augmenting the response to anti-PD1 therapy37–39. This effect was, at least in part, mediated by CDK6-dependent regulation of the activity of the transcription factor nuclear factor of activated T cells (NFAT), which induces T cell activation218; the combination of CDK4/6 inhibitors with anti-PD1 blockade was also associated with high production of the T cell-activating cytokine IL-2, although the mechanism remains to be elucidated37. In another study, abemaciclib alone delayed tumour growth in a mouse syngeneic colon carcinoma tumour model and was associated with a T cell inflammatory signature38, whereas combination with anti-PDL1 therapy led to persistent complete tumour regression in 2 of 10 animals38. In mouse models of colon and mammary carcinoma, responses to combined therapy were more effective and robust with phased administration of abemaciclib and anti-PDL1, than sequential administration of abemaciclib and anti-PDL1 (REF.38), suggesting that the immunomodulatory effect of CDK4/6 inhibition does not persist after treatment discontinuation. Combination treatment with CDK4/6 inhibitors and anti-PD1 therapy in melanoma xenograft models demonstrated enhanced growth inhibition compared with monotherapy with either agent and was associated with increased tumour immune cell infiltration39. Additionally, in a cohort of patients with advanced melanoma, copy number gain of CDK4 was associated with innate resistance to checkpoint inhibitor therapy, although the mechanisms were not elucidated39.
Experiments in cell models of cervical, breast, colon and prostate cancer have demonstrated that PDL1 expression is affected by CDK4/6 inhibitor treatment40,219. PDL1 undergoes ubiquitination and degradation by the E3-ligase cullin 3–speckle-type POZ protein (SPOP)40. In preclinical mouse models of colon carcinoma CDK4/6 inhibition decreased the phosphorylation of SPOP, promoting its degradation and ultimately increasing the stability of PDL1 (REF.40). In this study, tumour regression and overall survival were improved in mice treated with ICIs in combination with CDK4/6 inhibitors40. At the end of the study, 8 of 12 mice treated with combination therapy were alive with minimalto-no tumour burden, whereas the same response was observed only in 3 of 14 mice in the ICIs-alone group, and no survivors were reported in the untreated group40. This effect could, in part, result from the increase in PDL1 expression caused by CDK4/6 inhibitors, which often correlates with tumour responsiveness to ICIs220,221. Other mechanisms have been proposed to contribute to the increase in PDL1 expression upon CDK4/6 inhibitor treatment219. PDL1 is a transcriptional target of NF-κB that is selectively upregulated upon RB1 knockdown or CDK4/6 inhibition219 through CDK4/6-mediated phosphorylation of Rb (Rb-S249/T252), which promotes its interaction with NF-κB219. In a cell model of prostate adenocarcinoma, the binding of phosphorylated Rb-S249/T252 to NF-κB has been shown to block the transcription of PDL1 (REF.219); treatment of prostate adenocarcinoma cells with a phosphomimetic Rb peptide suppressed the upregulation of PDL1 induced by radiotherapy, which promotes immunostimulation via induction of immunogenic cell death219,222.
Overall, several mechanisms contribute to the good response to combination therapy with CDK4/6 inhibitors and immune checkpoint blockade, and much more remains to be discovered, including whether these mechanisms are cell-cycle dependent or independent. Several clinical trials examining the efficacy of combination therapy with CDK4/6 inhibitors and ICIs are ongoing in various cancers, including prostate, breast, and head and neck cancer24–28. Additionally, as checkpoint inhibitors have seen clinical success in RCC8–10, combination of CDK4/6 inhibition with checkpoint blockade could prove very effective in RCC.
Potential of CDK4/6 as targets in RCC
No dedicated trials to test CDK4/6 inhibitors in kidney cancer have been completed so far, but one is ongoing223 and a great deal of evidence suggests the potential utility of these drugs in RCC. Standard therapeutics for systemic therapy in RCC include TKIs (such as sunitinib)5, mTOR inhibitors (such as everolimus)4 and ICIs (such as nivolumab, ipilimumab and pembrolizumab)8–10 (FIG. 5). Unfortunately, an understanding of how to predict response to therapy and how to rationally combat intrinsic and acquired resistance is still limited. CDK4/6 inhibitors have been successful in a variety of cancers and particularly promising in preclinical studies in combination with mTOR and other kinase inhibitors, as well as with checkpoint inhibitors31,33,34,36–40,219, suggesting an opportunity to optimize the use of these therapeutics in RCC.
The potential importance of CDK4/6 inhibitors in RCC is supported by work on microRNAs (miRNAs), which are short, non-coding RNAs that regulate target mRNAs by modulating their translation or stability224. Some miRNAs have a tumour-suppressive function, in part mediated by the targeting and inhibition of CDK4/6 and the downstream effects on the cell cycle, and are downregulated in RCC225 (FIG. 1). The tumour suppressor miRNA miR-1 was found to be downregulated in a majority (36 of 41) of ccRCC tissue samples compared with normal tissue samples, and low miR-1 correlated with advanced cancer stage (P = 0.013) and poor overall survival (P = 0.012)225. Overexpression of miR-1 in cell culture and xenograft models of RCC inhibited proliferation and metastasis, whereas miR-1 silencing promoted these processes225. Importantly, miR-1 targets the expression of CDK4 and CDK6, as well as the proteins Caprin1 and Slug, which are involved in epithelial-to-mesenchymal transition, a process important for metastasis225. Another miRNA, miR-206, showed tumour-suppressive properties in RCC, and was found to be significantly downregulated in 5 ccRCC cell lines as well as 39 of 42 ccRCC tissue samples compared with paired normal tissue (P < 0.01)226. miR-206 was found to induce cell-cycle arrest directly targeting the transcripts of CDK4, CDK9 and CCND1 (REF.226) and low miR-206 levels correlated with increased protein expression of CDK4, CDK9 and cyclin D1 in tumour tissue samples from patients with ccRCC compared with matched normal tissue226. Similarly, long non-coding RNAs, which regulate translation of mRNAs and are involved in diverse cellular functions, have a role in the regulation of RCC growth through CDK4/6 signalling227,228. The long non-coding RNA DUXAP10 was shown to be upregulated in RCC (n = 72 paired RCC and adjacent normal tissues) and correlated with poor clinical prognosis in patients with RCC (n = 88 total patients, P = 0.0037)228. Knockdown of DUXAP10 in ccRCC cell lines correlated with decreased cell growth and downregulation of cyclin D and CDK4 (REF.228), indicating a role for DUXAP10-mediated regulation of RCC growth via modulation of CDK4 expression.
Mutation or loss of CDKN2A and CDKN2B are frequent events in RCC and associated with worse overall survival across various histological subtypes, including clear cell, papillary type 1 and type 2, and chromophobe RCC103. Mutation, hypermethylation, or deletion of CDKN2A are examples of molecular alterations that correlated with decreased survival across the entire cohort of RCC subtypes in one major comprehensive molecular analysis of RCC tissue samples103. Partial deletion of chromosome 9p, which contains CDKN2A, is a frequent event specifically in ccRCC (21% of 110 tumour specimens)103; results from an integrated proteogenomic analysis by the Clinical Proteomic Tumour Analysis Consortium showed that loss of chromosome 9p is associated with an upregulation of mTOR signalling effectors in tumour tissues from treatment-naive patients with RCC compared with adjacent normal tissue229. Evaluation of a panel of 25 RCC cell lines showed a wide range of sensitivity to palbociclib with IC50 values ranging from 25 nM to 700 nM (REF.230). In this study, following palbociclib treatment, RCC cells underwent G0/G1 cell cycle arrest and late apoptosis, and loss of CDKN2A, CDKN2B and E2F1 was significantly associated with palbociclib sensitivity (P = 0.021, P = 0.047 and P = 0.033, respectively)230. Varying degrees of sensitivity to ribociclib have also been shown in different RCC cell lines (76–280 nM)228. The mechanisms through which alterations in the CDK4 signalling axis, including CDKN2A loss, affect CDK4/6 inhibitor sensitivity in RCC are not completely understood and warrant further investigation.
The most common histological subtype of kidney cancer is ccRCC2,7. The majority of ccRCCs harbour loss-of-function mutations in VHL, encoding the recognition subunit of an E3 ubiquitin ligase complex2,7. Under normoxic conditions HIF becomes prolyl-hydroxylated, which enables recognition by VHL and subsequent proteasomal degradation of HIF59. Under hypoxia, HIF is no longer targeted for degradation, accumulates and induces transcription of target genes needed for angiogenesis and survival231. In ccRCC, VHL is non-functional2,7 and HIF accumulates even in normoxia, transcribing its downstream targets, such as VEGF, and contributing to ccRCC survival6. HIF and its downstream pathway components are therapeutic targets approved or under investigation for the treatment of ccRCC7,232. Analysis of primary ccRCC tumour samples and matched non-malignant kidney tissue showed an upregulation of cyclin D1 and CDK4 concomitant to HIF overexpression as a result of VHL mutation or hypermethylation233. Additional work in ccRCC cells has shown that upregulation of the HIF isoform HIF2 as a consequence of VHL loss promotes cyclin D1 expression, suggesting a direct role for HIF in CDK4-mediated cell cycle regulation234. Similarly, loss of chromosome 3p, which harbours the VHL locus, or specific mutations in VHL correlate with the upregulation of the G1/S transition229. Additionally, inducible MYC activation in a Vhl and Cdkn2A double-knockout mouse model led to bona fide ccRCC development, whereas Vhl loss or MYC activation alone led to only modest clear cell changes in the developed tumours235. These studies highlight the importance of alterations in the CDK4/6–CDKN2A axis in ccRCC development.
Enhanced lethality in VHL-null ccRCC cells compared with isogenic cells with restored VHL was tested in a screen using short hairpin RNA vectors against a total of 88 kinases236. Treatment with short hairpin RNA targeting CDK6 displayed preference for VHL-null cells, demonstrating a selective therapeutic effect236. CDK6 selectivity for VHL-null cells was independent of HIF, and a similar enhanced lethality in VHL-null cells compared with VHL-restored cells was observed with a derivative of SB-210878 (REF.237), a small-molecule CDK4/6 inhibitor236. These data suggest that VHL status can provide a predictive indication of response to CDK4/6 inhibitors. Synthetic lethality between VHL loss and CDK4/6 inhibition was confirmed in cultured ccRCC cell lines, as well as in an RNAi screen in Drosophila cells, suggesting a fundamental physiological dependency on both genes and supporting the evaluation of CDK4/6 inhibitors for ccRCC treatment238. The synthetic lethality between VHL and CDK4/6 was found to be HIF independent238 despite transcriptional induction of cyclin D1 by HIF2α238. Combination of palbociclib with the HIF2α inhibitor PT2399 led to a synergistic anti-proliferative response in cells with constitutively upregulated HIF transcriptional activity and improved survival in a mouse xenograft model of ccRCC238. Mice treated with the combination of palbociclib and PT2399 had a median survival >100 days compared with the ~50 days median survival for vehicle-treated mice (P < 0.0001), and 3 of 11 mice in the combination arm remained tumour free and alive 175 days after initial treatment238. Additionally, palbociclib showed improved overall survival (P = 0.0012) and antitumour activity against xenografts that are not dependent on HIF transcriptional activity, although no enhanced efficacy was observed in combination with PT2399, owing to the fact that the tumour does not depend on HIF signalling238. Taken together, these studies clearly demonstrate a link between VHL loss, HIF activation and modulation of cell-cycle regulation via the CDK4/6–cyclin D axis. Therapeutic targeting of CDK4/6 in combination with HIF inhibition (which is also approved for treatment of patients with VHL syndrome232) provides additional benefit in a context of hyperactive HIF-dependent transcription, such as VHL loss238.
The combination of TKIs and CDK4/6 inhibitors has also been explored in ccRCC239. Curcumin, a component of the spice turmeric was demonstrated to have anticancer activity against various cancer cells240 and is known to exert some of its functions by acting as an inhibitor of CDK4 (REF.241). Moreover, addition of curcumin potentiated the growth inhibitory effect of sunitinib in 786-O ccRCC cells242. A similar effect was observed with the Scutellaria baicalensis Georgi-derived natural compound wogonin, which was found to inhibit the CDK4–Rb pathway in 786-O cells243. Treatment with wogonin or palbociclib was able to overcome sunitinib resistance in 786-O cells, highlighting the potential for therapeutic targeting of CDK4 in TKI-resistant RCC243. In another study, abemaciclib alone and in combination with the TKI sunitinib239 decreased cellular viability and increased apoptosis in ccRCC cells239. Additionally, abemaciclib as a single agent or in combination with sunitinib led to regression of 786-O xenografts, and tumour regression was also observed with addition of abemaciclib following pre-treatment with sunitinib239. The combination of abemaciclib and sunitinib is currently under clinical investigation in a phase I trial in metastatic RCC (NCT03905889)223. To date, NCT03905889 is the only trial testing CDK4/6 inhibitors specifically in RCC, but other active clinical trials are ongoing for various pan-cancer indications for which patients with RCC might be eligible (TABLE 1). Many of these trials are investigating the combination of CDK4/6 inhibitors with other therapeutics, such as TKIs, and conventional chemotherapeutics223,244–246 (TABLE 1); inclusion criteria also take into account predictive markers such as CDK4/6 amplification, and p16 or Rb expression180–183,244,247–256 (TABLE 1). Overall, evidence suggests that use of CDK4/6 inhibitors in RCC is promising and more preclinical and clinical examination is warranted.
Table 1 |.
Current CDK4/6 inhibitor trials in rCC and pan-cancer settings
| CDK4/6 inhibitor | Companion drug | Companion drug target | Cancer type | Phase, study status | Altered CDK signalling genesa | NCT identifier | Refs |
|---|---|---|---|---|---|---|---|
| Abemaciclib | Sunitinib | VEGF | Metastatic RCC | Phase I recruiting | NA | NCT03905889 | 26 |
| None | NA | Solid tumours | Phase II recruiting | CCND1, CCND2 CCND3, CDK4, CDK6 | NCT03310879 | 247 | |
| None | NA | Advanced and/or metastatic cancers | Phase I completed | NA | NCT02919696 | 185,257 | |
| Fulvestrant | ER | Advanced cancer | Phase I active not recruiting | NA | NCT01394016 | 184,258 | |
| Paclitaxel | None | Advanced or metastatic solid tumours | Phase Ib/II not yet recruiting | CCND1, CCND2 CCND3, CDK4, CDK6 | NCT04594005 | 244 | |
| Abemaciclib or palbociclib | None | NA | Advanced solid tumours, non-Hodgkin lymphoma, multiple myeloma | Phase II recruiting | CDK4 or CDK6 amplification, CDKN2A deletion or mutation | NCT02693535 | 180–182,248 |
| Palbociclib | None | NA | Advanced malignant solid neoplasm | Phase II recruiting | CCND1, CCND2 or CCND3 amplification or CDK4 or 6 amplification and RB1-positive | NCT02465060 | 183,249 |
| None | NA | Advanced solid tumours, non-Hodgkin lymphoma, multiple myeloma | Phase II recruiting | CDKN2A, CDK4, CCND1, SMARCA4 | NCT03297606 | 250 | |
| None | NA | Rare tumours, refractory tumours | Phase II recruiting | CDK4 or CDK6 mutation or amplification | NCT03239015 | 251 | |
| Telaglenastat | Glutaminase | Solid tumours NSCLC, CRC | Phase I and II recruiting | KRAS mutation | NCT03965845 | 259 | |
| Gedatolisib | PI3K–mTOR | Advanced SCLC, pancreatic cancer, head and neck cancer, solid tumours | Phase I recruiting | CDK4 or CDK6, PI3K activity | NCT03065062 | 252 | |
| Trametinib | MEK | Solid tumours | Phase I completed | NA | NCT02065063 | 245 | |
| PD-0325901 | MEK | Metastatic or unresectable NSCLC and solid tumours | Phase I/II active not recruiting | KRAS mutation | NCT02022982 | 246 | |
| 5-FU, oxaliplatin | Antimetabolite, alkylating agent | Advanced solid tumours | Phase I active not recruiting | RB1-positive | NCT01522989 | 253 | |
| None | NA | Advanced malignant solid tumours, advanced and refractory lymphomas | Phase II active not recruiting | CCND1, CCND2, CCND3 amplification | NCT04439201 | 260 | |
| Taselisib, pictilisib | PI3K | Advanced solid tumours, breast cancer | Phase I active, not recruiting | Hyperactive PI3K–AKT pathway | NCT02389842 | 261 | |
| Ribociclib | Gemcitabine | Antimetabolite | Metastatic solid tumours | Phase I active not recruiting | Amplification of CDK4, CDK6, CCND1 or CCND3; expression of RB1, CDKN2A | NCT03237390 | 254 |
| HDM201 | p53–MDM2 | Malignant solid tumour | Phase II recruiting | CDK4, CDK6 CCND1 or CCND3 amplification, CDKN2A deletion, RB1-positive, TP53 wild type | NCT04116541 | 255 | |
| CS3002 | None | NA | Advanced solid tumours | Phase I recruiting | NA | NCT04162301 | 262 |
| HS-10342 | None | NA | Advanced solid tumours | Phase I recruiting | NA | NCT04060511 | 263 |
| PF-07220060 | Letrozole, fulvestrant | Aromatase, ER | Liposarcoma, colorectal, prostate, breast, lung adenocarcinoma, solid tumours | Phase I not yet recruiting | CDK4 or CCND1 amplification for solid tumours | NCT04557449 | 256 |
| TQB3303 | None | NA | Advanced malignant solid tumour | Phase I not yet recruiting | NA | NCT04275050 | 264 |
CDK, cyclin-dependent kinase; CRC, colorectal cancer; ER, oestrogen receptor; MEK, mitogen-activated protein kinase kinase; MDM2, murine double minute 2; mTOR, mammalian target of rapamycin; NA, not applicable; NCT, national clinical trial; NSCLC, non-small-cell lung cancer; PI3K, phosphoinositide 3-kinase; Rb, retinoblastoma; RCC, renal cell carcinoma; SCLC, small cell lung cancer; VEGF, vascular endothelial growth factor.
Altered CDK signalling genes column indicates genes in which abnormalities are required for inclusion criteria; specific types of alteration are indicated when appropriate.
Conclusions
Remarkable progress in the treatment of advanced and metastatic kidney cancer has been made over the past two decades. Despite the success of TKIs and mTOR inhibitors such as sunitinib and everolimus, as well as that of immune checkpoint blockade with agents such as nivolumab, ipilimumab and pembrolizumab, intrinsic and acquired resistance to these therapeutics is a problem. Inhibition of CDK4/6 with palbociclib, ribociclib or abemaciclib has become a standard therapy for HR+ and HER2− breast cancer and shows great clinical promise in a wide variety of other cancers. Additionally, emerging evidence suggests that CDK4/6 inhibitors can work synergistically with TKIs, mTOR inhibitors and ICIs. The success of combination therapies, coupled with limited but promising evidence of a role for CDK4 and CDK6 in RCC, suggests that further preclinical and clinical exploration of CDK4/6 inhibition in this cancer is warranted. RCC is traditionally considered a metabolic disease, and alterations of various metabolic and nutrient sensing signalling pathways in this cancer are crucial for pathogenesis and survival. CDK4 and CDK6 are involved in the integration of metabolic and nutrient sensing signalling pathways to control cell-cycle progression. An intimate relationship between CDK4/6 and master regulators of cell growth pathways, such as mTOR and HIF, exists; thus, preclinical studies to understand the mechanisms through which CDK4/6 pathways lead to RCC progression are required. Upfront identification of regulatory mechanisms of CDK4/6 activity and markers for sensitivity and resistance to CDK4/6 inhibitors will help in the rational selection of patients and combination therapies for future clinical trials examining the efficacy of CDK4/6 inhibitors in RCC.
Acknowledgements
The authors are grateful to W. Marston Linehan (Urologic Oncology Branch, NCI) for his scientific contribution. This work was partly supported by the National Institute of General Medical Sciences with the NIH (grants R35GM139584, R01GM124256 and DoD KC190038 (M.M.), and R01GM139932 (D.B.)). The content is solely the responsibility of the authors and does not necessarily represent the official views of the NIH. This work was also supported by funds from SUNY Upstate Medical University and the Upstate Foundation (D.B. and M.M.).
Footnotes
Competing interests
The authors declare no competing interests.
References
- 1.Siegel RL, Miller KD & Jemal A Cancer statistics, 2020. CA Cancer J. Clin 70, 7–30 (2020). [DOI] [PubMed] [Google Scholar]
- 2.Hsieh FS et al. Palbociclib induces activation of AMPK and inhibits hepatocellular carcinoma in a CDK4/6-independent manner. Mol. Oncol 11, 1035–1049 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.McDermott DF et al. Randomized phase III trial of high-dose interleukin-2 versus subcutaneous interleukin-2 and interferon in patients with metastatic renal cell carcinoma. J. Clin. Oncol 23, 133–141 (2005). [DOI] [PubMed] [Google Scholar]
- 4.Motzer RJ et al. Efficacy of everolimus in advanced renal cell carcinoma: a double-blind, randomised, placebo-controlled phase III trial. Lancet 372, 449–456 (2008). [DOI] [PubMed] [Google Scholar]
- 5.Motzer RJ et al. Sunitinib versus interferon alfa in metastatic renal-cell carcinoma. N. Engl. J. Med 356, 115–124 (2007). [DOI] [PubMed] [Google Scholar]
- 6.Shen C & Kaelin WG Jr The VHL/HIF axis in clear cell renal carcinoma. Semin. Cancer Biol 23, 18–25 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Linehan WM et al. The metabolic basis of kidney cancer. Cancer Discov. 9, 1006–1021 (2019). [DOI] [PubMed] [Google Scholar]
- 8.Rini BI et al. Pembrolizumab plus axitinib versus sunitinib for advanced renal-cell carcinoma. N. Engl. J. Med 380, 1116–1127 (2019). [DOI] [PubMed] [Google Scholar]
- 9.Motzer RJ et al. Nivolumab versus everolimus in advanced renal-cell carcinoma. N. Engl. J. Med 373, 1803–1813 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Motzer RJ et al. Nivolumab plus ipilimumab versus sunitinib in advanced renal-cell carcinoma. N. Engl. J. Med 378, 1277–1290 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Topalian SL, Drake CG & Pardoll DM Immune checkpoint blockade: a common denominator approach to cancer therapy. Cancer Cell 27, 450–461 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Turner NC et al. Palbociclib in hormone-receptor-positive advanced breast cancer. N. Engl. J. Med 373, 209–219 (2015). [DOI] [PubMed] [Google Scholar]
- 13.Hortobagyi GN et al. Ribociclib as first-line therapy for HR-positive, advanced breast cancer. N. Engl. J. Med 375, 1738–1748 (2016). [DOI] [PubMed] [Google Scholar]
- 14.Sledge GW Jr et al. MONARCH 2: abemaciclib in combination with fulvestrant in women with HR+/HER2− advanced breast cancer who had progressed while receiving endocrine therapy. J. Clin. Oncol 35, 2875–2884 (2017). [DOI] [PubMed] [Google Scholar]
- 15.Baker SJ & Reddy EP CDK4: a key player in the cell cycle, development, and cancer. Genes Cancer 3, 658–669 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Schopf FH, Biebl MM & Buchner J The HSP90 chaperone machinery. Nat. Rev. Mol. Cell Biol 18, 345–360 (2017). [DOI] [PubMed] [Google Scholar]
- 17.Bai J, Li Y & Zhang G Cell cycle regulation and anticancer drug discovery. Cancer Biol. Med 14, 348–362 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Knudsen ES & Witkiewicz AK The strange case of CDK4/6 inhibitors: mechanisms, resistance, and combination strategies. Trends Cancer 3, 39–55 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Pernas S, Tolaney SM, Winer EP & Goel S CDK4/6 inhibition in breast cancer: current practice and future directions. Ther. Adv. Med. Oncol 10, 1758835918786451 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.O’Leary B, Finn RS & Turner NC Treating cancer with selective CDK4/6 inhibitors. Nat. Rev. Clin. Oncol 13, 417–430 (2016). [DOI] [PubMed] [Google Scholar]
- 21.Finn RS et al. Palbociclib and letrozole in advanced breast cancer. N. Engl. J. Med 375, 1925–1936 (2016). [DOI] [PubMed] [Google Scholar]
- 22.Yardley DA MONALEESA clinical program: a review of ribociclib use in different clinical settings. Future Oncol. 15, 2673–2686 (2019). [DOI] [PubMed] [Google Scholar]
- 23.Klein ME, Kovatcheva M, Davis LE, Tap WD & Koff A CDK4/6 inhibitors: the mechanism of action may not be as simple as once thought. Cancer Cell 34, 9–20 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.US National Library of Medicine. ClinicalTrials.gov https://ClinicalTrials.gov/show/NCT04751929 (2021). [DOI] [PubMed]
- 25.US National Library of Medicine. ClinicalTrials.gov https://ClinicalTrials.gov/show/NCT04438824 (2021). [DOI] [PubMed]
- 26.US National Library of Medicine. ClinicalTrials.gov https://ClinicalTrials.gov/show/NCT04360941 (2021). [DOI] [PubMed]
- 27.US National Library of Medicine. ClinicalTrials.gov https://ClinicalTrials.gov/show/NCT04213404 (2021). [DOI] [PubMed]
- 28.US National Library of Medicine. ClinicalTrials.gov https://ClinicalTrials.gov/show/NCT03294694 (2020). [DOI] [PubMed]
- 29.Cretella D et al. The anti-tumor efficacy of CDK4/6 inhibition is enhanced by the combination with PI3K/AKT/mTOR inhibitors through impairment of glucose metabolism in TNBC cells. J. Exp. Clin. Cancer Res 37, 72 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Gopalan PK et al. CDK4/6 inhibition stabilizes disease in patients with p16-null non-small cell lung cancer and is synergistic with mTOR inhibition. Oncotarget 9, 37352–37366 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Michaloglou C et al. Combined inhibition of mTOR and CDK4/6 is required for optimal blockade of E2F function and long-term growth inhibition in estrogen receptor-positive breast cancer. Mol. Cancer Ther 17, 908–920 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Yamamoto T, Kanaya N, Somlo G & Chen S Synergistic anti-cancer activity of CDK4/6 inhibitor palbociclib and dual mTOR kinase inhibitor MLN0128 in pRb-expressing ER-negative breast cancer. Breast Cancer Res. Treat 174, 615–625 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Song X et al. Combined CDK4/6 and Pan-mTOR inhibition is synergistic against intrahepatic cholangiocarcinoma. Clin. Cancer Res 25, 403–413 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Olmez I et al. Combined CDK4/6 and mTOR inhibition is synergistic against glioblastoma via multiple mechanisms. Clin. Cancer Res 23, 6958–6968 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Asby DJ et al. Combined use of CDK4/6 and mTOR inhibitors induce synergistic growth arrest of diffuse intrinsic pontine glioma cells via mutual downregulation of mTORC1 activity. Cancer Manag. Res 10, 3483–3500 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Goel S et al. CDK4/6 inhibition triggers anti-tumour immunity. Nature 548, 471–475 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Deng J et al. CDK4/6 inhibition augments antitumor immunity by enhancing T-cell activation. Cancer Discov. 8, 216–233 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Schaer DA et al. The CDK4/6 inhibitor abemaciclib induces a T cell inflamed tumor microenvironment and enhances the efficacy of PD-L1 checkpoint blockade. Cell Rep. 22, 2978–2994 (2018). [DOI] [PubMed] [Google Scholar]
- 39.Yu J et al. Genetic aberrations in the CDK4 pathway are associated with innate resistance to PD-1 blockade in Chinese patients with non-cutaneous melanoma. Clin. Cancer Res 25, 6511–6523 (2019). [DOI] [PubMed] [Google Scholar]
- 40.Zhang J et al. Cyclin D-CDK4 kinase destabilizes PD-L1 via cullin 3-SPOP to control cancer immune surveillance. Nature 553, 91–95 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Malumbres M & Barbacid M Cell cycle, CDKs and cancer: a changing paradigm. Nat. Rev. Cancer 9, 153–166 (2009). [DOI] [PubMed] [Google Scholar]
- 42.Satyanarayana A & Kaldis P Mammalian cell-cycle regulation: several Cdks, numerous cyclins and diverse compensatory mechanisms. Oncogene 28, 2925–2939 (2009). [DOI] [PubMed] [Google Scholar]
- 43.Sherr CJ D-type cyclins. Trends Biochem. Sci 20, 187–190 (1995). [DOI] [PubMed] [Google Scholar]
- 44.Sherr CJ & Roberts JM Living with or without cyclins and cyclin-dependent kinases. Genes Dev. 18, 2699–2711 (2004). [DOI] [PubMed] [Google Scholar]
- 45.Malumbres M et al. Mammalian cells cycle without the D-type cyclin-dependent kinases CDK4 and CDK6. Cell 118, 493–504 (2004). [DOI] [PubMed] [Google Scholar]
- 46.Rane SG et al. Loss of CDK4 expression causes insulin-deficient diabetes and CDK4 activation results in beta-islet cell hyperplasia. Nat. Genet 22, 44–52 (1999). [DOI] [PubMed] [Google Scholar]
- 47.Kato J, Matsushime H, Hiebert SW, Ewen ME & Sherr CJ Direct binding of cyclin D to the retinoblastoma gene product (pRb) and pRb phosphorylation by the cyclin D-dependent kinase CDK4. Genes Dev. 7, 331–342 (1993). [DOI] [PubMed] [Google Scholar]
- 48.Pan W, Cox S, Hoess RH & Grafstrom RH A cyclin D1/cyclin-dependent kinase 4 binding site within the C domain of the retinoblastoma protein. Cancer Res. 61, 2885–2891 (2001). [PubMed] [Google Scholar]
- 49.Wallace M & Ball KL Docking-dependent regulation of the Rb tumor suppressor protein by Cdk4. Mol. Cell Biol 24, 5606–5619 (2004). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Braden WA, McClendon AK & Knudsen ES Cyclin-dependent kinase 4/6 activity is a critical determinant of pre-replication complex assembly. Oncogene 27, 7083–7093 (2008). [DOI] [PubMed] [Google Scholar]
- 51.Ruas M et al. CDK4 and CDK6 delay senescence by kinase-dependent and p16INK4a-independent mechanisms. Mol. Cell Biol 27, 4273–4282 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Brookes S et al. Evidence for a CDK4-dependent checkpoint in a conditional model of cellular senescence. Cell Cycle 14, 1164–1173 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Matthews HK, Bertoli C & de Bruin RAM Cell cycle control in cancer. Nat. Rev. Mol. Cell Biol 23, 74–88 (2021). [DOI] [PubMed] [Google Scholar]
- 54.Haas K et al. Mutual requirement of CDK4 and Myc in malignant transformation: evidence for cyclin D1/CDK4 and p16INK4A as upstream regulators of Myc. Oncogene 15, 179–192 (1997). [DOI] [PubMed] [Google Scholar]
- 55.Hermeking H et al. Identification of CDK4 as a target of c-MYC. Proc. Natl Acad. Sci. USA 97, 2229–2234 (2000). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Obaya AJ, Kotenko I, Cole MD & Sedivy JM The proto-oncogene c-myc acts through the cyclin-dependent kinase (Cdk) inhibitor p27Kip1 to facilitate the activation of CDK4 and CDK6 and early G1 phase progression. J. Biol. Chem 277, 31263–31269 (2002). [DOI] [PubMed] [Google Scholar]
- 57.Tsutsui T et al. Targeted disruption of CDK4 delays cell cycle entry with enhanced p27(Kip1) activity. Mol. Cell Biol 19, 7011–7019 (1999). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Meyer CA et al. Drosophila CDK4 is required for normal growth and is dispensable for cell cycle progression. EMBO J. 19, 4533–4542 (2000). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Jaakkola P et al. Targeting of HIF-alpha to the von Hippel-Lindau ubiquitylation complex by O2-regulated prolyl hydroxylation. Science 292, 468–472 (2001). [DOI] [PubMed] [Google Scholar]
- 60.Frei C & Edgar BA Drosophila cyclin D/Cdk4 requires Hif-1 prolyl hydroxylase to drive cell growth. Dev. Cell 6, 241–251 (2004). [DOI] [PubMed] [Google Scholar]
- 61.Lee Y et al. Cyclin D1-Cdk4 controls glucose metabolism independently of cell cycle progression. Nature 510, 547–551 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Wang H et al. The metabolic function of cyclin D3-CDK6 kinase in cancer cell survival. Nature 546, 426–430 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Romero-Pozuelo J, Figlia G, Kaya O, Martin-Villalba A & Teleman AA CDK4 and CDK6 couple the cell-cycle machinery to cell growth via mTORC1. Cell Rep. 31, 107504 (2020). [DOI] [PubMed] [Google Scholar]
- 64.Krall AS & Christofk HR Cell cycle: division enzyme regulates metabolism. Nature 546, 357–358 (2017). [DOI] [PubMed] [Google Scholar]
- 65.Serrano M, Hannon GJ & Beach D A new regulatory motif in cell-cycle control causing specific inhibition of cyclin D/CDK4. Nature 366, 704–707 (1993). [DOI] [PubMed] [Google Scholar]
- 66.Annicotte JS et al. The CDK4-pRB-E2F1 pathway controls insulin secretion. Nat. Cell Biol 11, 1017–1023 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Martinez-Carreres L et al. CDK4 regulates lysosomal function and mTORC1 activation to promote cancer cell survival. Cancer Res. 79, 5245–5259 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Hardie DG, Ross FA & Hawley SA AMPK: a nutrient and energy sensor that maintains energy homeostasis. Nat. Rev. Mol. Cell Biol 13, 251–262 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Lopez-Mejia IC et al. CDK4 phosphorylates AMPKalpha2 to inhibit its activity and repress fatty acid oxidation. Mol. Cell 68, 336–349.e6 (2017). [DOI] [PubMed] [Google Scholar]
- 70.Nebenfuehr S, Kollmann K & Sexl V The role of CDK6 in cancer. Int. J. Cancer 147, 2988–2995 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Dai K, Kobayashi R & Beach D Physical interaction of mammalian CDC37 with CDK4. J. Biol. Chem 271, 22030–22034 (1996). [DOI] [PubMed] [Google Scholar]
- 72.Stepanova L, Leng X, Parker SB & Harper JW Mammalian p50Cdc37 is a protein kinase-targeting subunit of Hsp90 that binds and stabilizes Cdk4. Genes Dev. 10, 1491–1502 (1996). [DOI] [PubMed] [Google Scholar]
- 73.Gu Y, Turck CW & Morgan DO Inhibition of CDK2 activity in vivo by an associated 20K regulatory subunit. Nature 366, 707–710 (1993). [DOI] [PubMed] [Google Scholar]
- 74.Harper JW, Adami GR, Wei N, Keyomarsi K & Elledge SJ The p21 Cdk-interacting protein Cip1 is a potent inhibitor of G1 cyclin-dependent kinases. Cell 75, 805–816 (1993). [DOI] [PubMed] [Google Scholar]
- 75.Xiong Y et al. p21 is a universal inhibitor of cyclin kinases. Nature 366, 701–704 (1993). [DOI] [PubMed] [Google Scholar]
- 76.Noda A, Ning Y, Venable SF, Pereira-Smith OM & Smith JR Cloning of senescent cell-derived inhibitors of DNA synthesis using an expression screen. Exp. Cell Res 211, 90–98 (1994). [DOI] [PubMed] [Google Scholar]
- 77.Polyak K et al. p27Kip1, a cyclin-Cdk inhibitor, links transforming growth factor-beta and contact inhibition to cell cycle arrest. Genes Dev. 8, 9–22 (1994). [DOI] [PubMed] [Google Scholar]
- 78.Polyak K et al. Cloning of p27Kip1, a cyclin-dependent kinase inhibitor and a potential mediator of extracellular antimitogenic signals. Cell 78, 59–66 (1994). [DOI] [PubMed] [Google Scholar]
- 79.Toyoshima H & Hunter T p27, a novel inhibitor of G1 cyclin-Cdk protein kinase activity, is related to p21. Cell 78, 67–74 (1994). [DOI] [PubMed] [Google Scholar]
- 80.Lee MH, Reynisdottir I & Massague J Cloning of p57KIP2, a cyclin-dependent kinase inhibitor with unique domain structure and tissue distribution. Genes Dev. 9, 639–649 (1995). [DOI] [PubMed] [Google Scholar]
- 81.Matsuoka S et al. p57KIP2, a structurally distinct member of the p21CIP1 Cdk inhibitor family, is a candidate tumor suppressor gene. Genes Dev. 9, 650–662 (1995). [DOI] [PubMed] [Google Scholar]
- 82.Hannon GJ & Beach D p15INK4B is a potential effector of TGF-beta-induced cell cycle arrest. Nature 371, 257–261 (1994). [DOI] [PubMed] [Google Scholar]
- 83.Guan KL et al. Growth suppression by p18, a p16INK4/MTS1- and p14INK4B/MTS2-related CDK6 inhibitor, correlates with wild-type pRb function. Genes Dev. 8, 2939–2952 (1994). [DOI] [PubMed] [Google Scholar]
- 84.Hirai H, Roussel MF, Kato JY, Ashmun RA & Sherr CJ Novel INK4 proteins, p19 and p18, are specific inhibitors of the cyclin D-dependent kinases CDK4 and CDK6. Mol. Cell Biol 15, 2672–2681 (1995). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Chan FK, Zhang J, Cheng L, Shapiro DN & Winoto A Identification of human and mouse p19, a novel CDK4 and CDK6 inhibitor with homology to p16ink4. Mol. Cell Biol 15, 2682–2688 (1995). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.LaBaer J et al. New functional activities for the p21 family of CDK inhibitors. Genes Dev. 11, 847–862 (1997). [DOI] [PubMed] [Google Scholar]
- 87.James MK, Ray A, Leznova D & Blain SW Differential modification of p27Kip1 controls its cyclin D-cdk4 inhibitory activity. Mol. Cell Biol 28, 498–510 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Blain SW, Montalvo E & Massague J Differential interaction of the cyclin-dependent kinase (Cdk) inhibitor p27Kip1 with cyclin A-Cdk2 and cyclin D2-Cdk4. J. Biol. Chem 272, 25863–25872 (1997). [DOI] [PubMed] [Google Scholar]
- 89.Russo AA, Tong L, Lee JO, Jeffrey PD & Pavletich NP Structural basis for inhibition of the cyclin-dependent kinase Cdk6 by the tumour suppressor p16INK4a. Nature 395, 237–243 (1998). [DOI] [PubMed] [Google Scholar]
- 90.Roussel MF The INK4 family of cell cycle inhibitors in cancer. Oncogene 18, 5311–5317 (1999). [DOI] [PubMed] [Google Scholar]
- 91.Takita J et al. Deletion map of chromosome 9 and p16 (CDKN2A) gene alterations in neuroblastoma. Cancer Res. 57, 907–912 (1997). [PubMed] [Google Scholar]
- 92.Nobori T et al. Deletions of the cyclin-dependent kinase-4 inhibitor gene in multiple human cancers. Nature 368, 753–756 (1994). [DOI] [PubMed] [Google Scholar]
- 93.Boice JA & Fairman R Structural characterization of the tumor suppressor p16, an ankyrin-like repeat protein. Protein Sci. 5, 1776–1784 (1996). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Coleman KG et al. Identification of CDK4 sequences involved in cyclin D1 and p16 binding. J. Biol. Chem 272, 18869–18874 (1997). [DOI] [PubMed] [Google Scholar]
- 95.Ceha HM, Nasser I, Medema RH & Slebos RJ Several noncontiguous domains of CDK4 are involved in binding to the P16 tumor suppressor protein. Biochem. Biophys. Res. Commun 249, 550–555 (1998). [DOI] [PubMed] [Google Scholar]
- 96.Sotillo R et al. Wide spectrum of tumors in knock-in mice carrying a Cdk4 protein insensitive to INK4 inhibitors. EMBO J. 20, 6637–6647 (2001). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Rane SG, Cosenza SC, Mettus RV & Reddy EP Germ line transmission of the Cdk4R24C mutation facilitates tumorigenesis and escape from cellular senescence. Mol. Cell Biol 22, 644–656 (2002). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Foulkes WD, Flanders TY, Pollock PM & Hayward NK The CDKN2A (p16) gene and human cancer. Mol. Med 3, 5–20 (1997). [PMC free article] [PubMed] [Google Scholar]
- 99.Jardim DL et al. Cyclin pathway genomic alterations across 190,247 solid tumors: leveraging large-scale data to inform therapeutic directions. Oncologist 26, e78–e89 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Ueki K et al. CDKN2/p16 or RB alterations occur in the majority of glioblastomas and are inversely correlated. Cancer Res. 56, 150–153 (1996). [PubMed] [Google Scholar]
- 101.Young RJ et al. Loss of CDKN2A expression is a frequent event in primary invasive melanoma and correlates with sensitivity to the CDK4/6 inhibitor PD0332991 in melanoma cell lines. Pigment. Cell Melanoma Res 27, 590–600 (2014). [DOI] [PubMed] [Google Scholar]
- 102.Maubec E et al. Characteristics of the coexistence of melanoma and renal cell carcinoma. Cancer 116, 5716–5724 (2010). [DOI] [PubMed] [Google Scholar]
- 103.Ricketts CJ et al. The cancer genome atlas comprehensive molecular characterization of renal cell carcinoma. Cell Rep. 23, 313–326.e5 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Latif F et al. Identification of the von Hippel-Lindau disease tumor suppressor gene. Science 260, 1317–1320 (1993). [DOI] [PubMed] [Google Scholar]
- 105.Nickerson ML et al. Mutations in a novel gene lead to kidney tumors, lung wall defects, and benign tumors of the hair follicle in patients with the Birt-Hogg-Dube syndrome. Cancer Cell 2, 157–164 (2002). [DOI] [PubMed] [Google Scholar]
- 106.Zbar B et al. Hereditary papillary renal cell carcinoma: clinical studies in 10 families. J. Urol 153, 907–912 (1995). [PubMed] [Google Scholar]
- 107.Grubb RL 3rd et al. Hereditary leiomyomatosis and renal cell cancer: a syndrome associated with an aggressive form of inherited renal cancer. J. Urol 177, 2074–2079; discussion 2079–2080 (2007). [DOI] [PubMed] [Google Scholar]
- 108.Launonen V et al. Inherited susceptibility to uterine leiomyomas and renal cell cancer. Proc. Natl Acad. Sci. USA 98, 3387–3392 (2001). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Jafri M et al. Germline mutations in the CDKN2B tumor suppressor gene predispose to renal cell carcinoma. Cancer Discov. 5, 723–729 (2015). [DOI] [PubMed] [Google Scholar]
- 110.Zygmunt A, Tedesco VC, Udho E & Krucher NA Hypoxia stimulates p16 expression and association with cdk4. Exp. Cell Res 278, 53–60 (2002). [DOI] [PubMed] [Google Scholar]
- 111.Gump J, Stokoe D & McCormick F Phosphorylation of p16INK4A correlates with Cdk4 association. J. Biol. Chem 278, 6619–6622 (2003). [DOI] [PubMed] [Google Scholar]
- 112.Jinno S, Hung SC & Okayama H Cell cycle start from quiescence controlled by tyrosine phosphorylation of Cdk4. Oncogene 18, 565–571 (1999). [DOI] [PubMed] [Google Scholar]
- 113.Kato JY, Matsuoka M, Strom DK & Sherr CJ Regulation of cyclin D-dependent kinase 4 (cdk4) by cdk4-activating kinase. Mol. Cell Biol 14, 2713–2721 (1994). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Bisteau X et al. CDK4 T172 phosphorylation is central in a CDK7-dependent bidirectional CDK4/CDK2 interplay mediated by p21 phosphorylation at the restriction point. PLoS Genet. 9, e1003546 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Colleoni B et al. JNKs function as CDK4-activating kinases by phosphorylating CDK4 and p21. Oncogene 36, 4349–4361 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Blancquaert S et al. cAMP-dependent activation of mammalian target of rapamycin (mTOR) in thyroid cells. Implication in mitogenesis and activation of CDK4. Mol. Endocrinol 24, 1453–1468 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Knudsen ES et al. Cell cycle plasticity driven by MTOR signaling: integral resistance to CDK4/6 inhibition in patient-derived models of pancreatic cancer. Oncogene 38, 3355–3370 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Hartl FU, Bracher A & Hayer-Hartl M Molecular chaperones in protein folding and proteostasis. Nature 475, 324–332 (2011). [DOI] [PubMed] [Google Scholar]
- 119.Walton-Diaz A et al. Contributions of co-chaperones and post-translational modifications towards Hsp90 drug sensitivity. Future Med. Chem 5, 1059–1071 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Woodford MR et al. Targeting Hsp90 in non-cancerous maladies. Curr. Top. Med. Chem 16, 2792–2804 (2016). [DOI] [PubMed] [Google Scholar]
- 121.Sima S & Richter K Regulation of the Hsp90 system. Biochim. Biophys. Acta Mol. Cell Res 1865, 889–897 (2018). [DOI] [PubMed] [Google Scholar]
- 122.Mayer MP & Le Breton L Hsp90: breaking the symmetry. Mol. Cell 58, 8–20 (2015). [DOI] [PubMed] [Google Scholar]
- 123.Rohl A, Rohrberg J & Buchner J The chaperone Hsp90: changing partners for demanding clients. Trends Biochem. Sci 38, 253–262 (2013). [DOI] [PubMed] [Google Scholar]
- 124.Czemeres J, Buse K & Verkhivker GM Atomistic simulations and network-based modeling of the Hsp90-Cdc37 chaperone binding with Cdk4 client protein: a mechanism of chaperoning kinase clients by exploiting weak spots of intrinsically dynamic kinase domains. PLoS One 12, e0190267 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125.Verba KA & Agard DA How Hsp90 and Cdc37 lubricate kinase molecular switches. Trends Biochem. Sci 42, 799–811 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126.Lamphere L et al. Interaction between Cdc37 and Cdk4 in human cells. Oncogene 14, 1999–2004 (1997). [DOI] [PubMed] [Google Scholar]
- 127.Zhao Q, Boschelli F, Caplan AJ & Arndt KT Identification of a conserved sequence motif that promotes Cdc37 and cyclin D1 binding to Cdk4. J. Biol. Chem 279, 12560–12564 (2004). [DOI] [PubMed] [Google Scholar]
- 128.Truman AW et al. CDK-dependent Hsp70 Phosphorylation controls G1 cyclin abundance and cell-cycle progression. Cell 151, 1308–1318 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129.Verba KA et al. Atomic structure of Hsp90-Cdc37-Cdk4 reveals that Hsp90 traps and stabilizes an unfolded kinase. Science 352, 1542–1547 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130.Hallett ST et al. Differential regulation of G1 CDK complexes by the Hsp90-Cdc37 chaperone system. Cell Rep. 21, 1386–1398 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131.Vaughan CK et al. Structure of an Hsp90-Cdc37-Cdk4 complex. Mol. Cell 23, 697–707 (2006). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 132.Vaughan CK et al. Hsp90-dependent activation of protein kinases is regulated by chaperone-targeted dephosphorylation of Cdc37. Mol. Cell 31, 886–895 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133.Mollapour M et al. Swe1Wee1-dependent tyrosine phosphorylation of Hsp90 regulates distinct facets of chaperone function. Mol. Cell 37, 333–343 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134.Backe SJ, Sager RA, Woodford MR, Makedon AM & Mollapour M Post-translational modifications of Hsp90 and translating the chaperone code. J. Biol. Chem 295, 11099–11117 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 135.Miyata Y & Nishida E CK2 binds, phosphorylates, and regulates its pivotal substrate Cdc37, an Hsp90-cochaperone. Mol. Cell Biochem 274, 171–179 (2005). [DOI] [PubMed] [Google Scholar]
- 136.Oberoi J et al. Structural and functional basis of protein phosphatase 5 substrate specificity. Proc. Natl Acad. Sci. USA 113, 9009–9014 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 137.Sager RA, Dushukyan N, Woodford M & Mollapour M Structure and function of the co-chaperone protein phosphatase 5 in cancer. Cell Stress. Chaperones 25, 383–394 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 138.Xu W et al. Dynamic tyrosine phosphorylation modulates cycling of the HSP90-P50(CDC37)-AHA1 chaperone machine. Mol. Cell 47, 434–443 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 139.Bachman AB et al. Phosphorylation induced cochaperone unfolding promotes kinase recruitment and client class-specific Hsp90 phosphorylation. Nat. Commun 9, 265 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 140.Smith JR et al. Restricting direct interaction of CDC37 with HSP90 does not compromise chaperoning of client proteins. Oncogene 34, 15–26 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 141.Lawless N, Blacklock K, Berrigan E & Verkhivker G Structural bioinformatics and protein docking analysis of the molecular chaperone-kinase interactions: towards allosteric inhibition of protein kinases by targeting the hsp90-cdc37 chaperone machinery. Pharmaceuticals 6, 1407–1428 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 142.Zhu J et al. Cdc37 facilitates cell survival of colorectal carcinoma via activating the CDK4 signaling pathway. Cancer Sci. 109, 656–665 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 143.Wang Z, Wei W, Sun CK, Chua MS & So S Suppressing the CDC37 cochaperone in hepatocellular carcinoma cells inhibits cell cycle progression and cell growth. Liver Int. 35, 1403–1415 (2015). [DOI] [PubMed] [Google Scholar]
- 144.D’Annessa I et al. Design of disruptors of the Hsp90-Cdc37 interface. Molecules 25, 360 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 145.Paladino A et al. Chemical perturbation of oncogenic protein folding: from the prediction of locally unstable structures to the design of disruptors of hsp90-client interactions. Chemistry 26, 9459–9465 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 146.Steinebach C et al. Systematic exploration of different E3 ubiquitin ligases: an approach towards potent and selective CDK6 degraders. Chem. Sci 11, 3474–3486 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 147.Sherr CJ, Beach D & Shapiro GI Targeting CDK4 and CDK6: from discovery to therapy. Cancer Discov. 6, 353–367 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 148.Soni R et al. Selective in vivo and in vitro effects of a small molecule inhibitor of cyclin-dependent kinase 4. J. Natl Cancer Inst 93, 436–446 (2001). [DOI] [PubMed] [Google Scholar]
- 149.Martin MP, Endicott JA & Noble MEM Structure-based discovery of cyclin-dependent protein kinase inhibitors. Essays Biochem. 61, 439–452 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 150.De Azevedo WF Jr et al. Structural basis for specificity and potency of a flavonoid inhibitor of human CDK2, a cell cycle kinase. Proc. Natl Acad. Sci. USA 93, 2735–2740 (1996). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 151.Gray NS et al. Exploiting chemical libraries, structure, and genomics in the search for kinase inhibitors. Science 281, 533–538 (1998). [DOI] [PubMed] [Google Scholar]
- 152.Ikuta M et al. Crystallographic approach to identification of cyclin-dependent kinase 4 (CDK4)-specific inhibitors by using CDK4 mimic CDK2 protein. J. Biol. Chem 276, 27548–27554 (2001). [DOI] [PubMed] [Google Scholar]
- 153.McInnes C et al. Structural determinants of CDK4 inhibition and design of selective ATP competitive inhibitors. Chem. Biol 11, 525–534 (2004). [DOI] [PubMed] [Google Scholar]
- 154.Park H, Yeom MS & Lee S Loop flexibility and solvent dynamics as determinants for the selective inhibition of cyclin-dependent kinase 4: comparative molecular dynamics simulation studies of CDK2 and CDK4. Chembiochem 5, 1662–1672 (2004). [DOI] [PubMed] [Google Scholar]
- 155.Guan H, Du Y, Han W, Shen J & Li Q Development of selective cyclin-dependent kinase 4 inhibitors for antineoplastic therapies. Anticancer. Agents Med. Chem 17, 646–657 (2017). [DOI] [PubMed] [Google Scholar]
- 156.Fry DW et al. Specific inhibition of cyclin-dependent kinase 4/6 by PD 0332991 and associated antitumor activity in human tumor xenografts. Mol. Cancer Ther 3, 1427–1438 (2004). [PubMed] [Google Scholar]
- 157.Toogood PL et al. Discovery of a potent and selective inhibitor of cyclin-dependent kinase 4/6. J. Med. Chem 48, 2388–2406 (2005). [DOI] [PubMed] [Google Scholar]
- 158.Cho YS et al. 4-(Pyrazol-4-yl)-pyrimidines as selective inhibitors of cyclin-dependent kinase 4/6. J. Med. Chem 53, 7938–7957 (2010). [DOI] [PubMed] [Google Scholar]
- 159.Gelbert LM et al. Preclinical characterization of the CDK4/6 inhibitor LY2835219: in-vivo cell cycle-dependent/independent anti-tumor activities alone/in combination with gemcitabine. Invest. N. Drugs 32, 825–837 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 160.Wright MD & Abraham MJ Preclinical discovery and development of abemaciclib used to treat breast cancer. Expert Opin. Drug Discov 16, 485–496 (2021). [DOI] [PubMed] [Google Scholar]
- 161.O’Brien N et al. Preclinical activity of abemaciclib alone or in combination with antimitotic and targeted therapies in breast cancer. Mol. Cancer Ther 17, 897–907 (2018). [DOI] [PubMed] [Google Scholar]
- 162.Torres-Guzman R et al. Preclinical characterization of abemaciclib in hormone receptor positive breast cancer. Oncotarget 8, 69493–69507 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 163.Chen P et al. Spectrum and Degree of CDK drug interactions predicts clinical performance. Mol. Cancer Ther 15, 2273–2281 (2016). [DOI] [PubMed] [Google Scholar]
- 164.Knudsen ES, Hutcheson J, Vail P & Witkiewicz AK Biological specificity of CDK4/6 inhibitors: dose response relationship, in vivo signaling, and composite response signature. Oncotarget 8, 43678–43691 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 165.Roberto M et al. CDK4/6 inhibitor treatments in patients with hormone receptor positive, Her2 negative advanced breast cancer: potential molecular mechanisms, clinical implications and future perspectives. Cancers 13, 332 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 166.Singer EA, Golijanin DJ, Miyamoto H & Messing EM Androgen deprivation therapy for prostate cancer. Expert Opin. Pharmacother 9, 211–228 (2008). [DOI] [PubMed] [Google Scholar]
- 167.Finn RS et al. PD 0332991, a selective cyclin D kinase 4/6 inhibitor, preferentially inhibits proliferation of luminal estrogen receptor-positive human breast cancer cell lines in vitro. Breast Cancer Res. 11, R77 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 168.Ertel A et al. RB-pathway disruption in breast cancer: differential association with disease subtypes, disease-specific prognosis and therapeutic response. Cell Cycle 9, 4153–4163 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 169.Altucci L et al. 17beta-Estradiol induces cyclin D1 gene transcription, p36D1-p34cdk4 complex activation and p105Rb phosphorylation during mitogenic stimulation of G1-arrested human breast cancer cells. Oncogene 12, 2315–2324 (1996). [PubMed] [Google Scholar]
- 170.Dickler MN et al. MONARCH 1, a phase II study of abemaciclib, a CDK4 and CDK6 inhibitor, as a single agent, in patients with refractory HR+/HER2− metastatic breast cancer. Clin. Cancer Res 23, 5218–5224 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 171.Li J et al. Cyclin-dependent kinase 4 and 6 inhibitors in hormone receptor-positive, human epidermal growth factor receptor-2 negative advanced breast cancer: a meta-analysis of randomized clinical trials. Breast Cancer Res. Treat 180, 21–32 (2020). [DOI] [PubMed] [Google Scholar]
- 172.Wang L et al. CDK4/6 inhibitors plus endocrine therapy improve overall survival in advanced HR+/HER2− breast cancer: a meta-analysis of randomized controlled trials. Breast J. 26, 1439–1443 (2019). [DOI] [PubMed] [Google Scholar]
- 173.Deng Y et al. CDK4/6 inhibitors in combination with hormone therapy for HR+/HER2− advanced breast cancer: a systematic review and meta-analysis of randomized controlled trials. Clin. Breast Cancer 18, e943–e953 (2018). [DOI] [PubMed] [Google Scholar]
- 174.Messina C et al. CDK4/6 inhibitors in advanced hormone receptor-positive/HER2-negative breast cancer: a systematic review and meta-analysis of randomized trials. Breast Cancer Res. Treat 172, 9–21 (2018). [DOI] [PubMed] [Google Scholar]
- 175.Gao JJ et al. CDK4/6 inhibitor treatment for patients with hormone receptor-positive, HER2-negative, advanced or metastatic breast cancer: a US Food and Drug Administration pooled analysis. Lancet Oncol. 21, 250–260 (2020). [DOI] [PubMed] [Google Scholar]
- 176.US National Library of Medicine. ClinicalTrials.gov https://ClinicalTrials.gov/show/NCT03701334 (2022). [DOI] [PubMed]
- 177.Johnston SRD et al. Abemaciclib combined with endocrine therapy for the adjuvant treatment of HR+, HER2−, node-positive, high-risk, early breast cancer (monarchE). J. Clin. Oncol 38, 3987–3998 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 178.Curigliano G & Loibl S CDK4/6 inhibitors in breast cancer: one more step towards reduced mortality. Lancet Oncol. 21, 191–192 (2020). [DOI] [PubMed] [Google Scholar]
- 179.Tan AR et al. Trilaciclib plus chemotherapy versus chemotherapy alone in patients with metastatic triple-negative breast cancer: a multicentre, randomised, open-label, phase 2 trial. Lancet Oncol. 20, 1587–1601 (2019). [DOI] [PubMed] [Google Scholar]
- 180.Al Baghdadi T et al. Sunitinib in patients with metastatic colorectal cancer (mCRC) with FLT-3 amplification: results from the targeted agent and profiling utilization registry (TAPUR) study. Target. Oncol 15, 743–750 (2020). [DOI] [PubMed] [Google Scholar]
- 181.Alva AS et al. Pembrolizumab in patients with metastatic breast cancer with high tumor mutational burden: results from the targeted agent and profiling utilization registry (TAPUR) study. J. Clin. Oncol 39, 2443–2451 (2021). [DOI] [PubMed] [Google Scholar]
- 182.Fisher JG et al. Cetuximab in patients with breast cancer, non-small cell lung cancer, and ovarian cancer without KRAS, NRAS, or BRAF mutations: results from the targeted agent and profiling utilization registry (TAPUR) study. Target. Oncol 15, 733–741 (2020). [DOI] [PubMed] [Google Scholar]
- 183.Flaherty KT et al. Molecular landscape and actionable alterations in a genomically guided cancer clinical trial: National Cancer Institute Molecular Analysis for Therapy Choice (NCI-MATCH). J. Clin. Oncol 38, 3883–3894 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 184.Tate SC et al. A population pharmacokinetic and pharmacodynamic analysis of abemaciclib in a phase I clinical trial in cancer patients. Clin. Pharmacokinet 57, 335–344 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 185.Zhang J et al. A randomized phase I study of abemaciclib in Chinese patients with advanced and/or metastatic cancers. Target. Oncol 16, 177–187 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 186.Hussussian CJ et al. Germline p16 mutations in familial melanoma. Nat. Genet 8, 15–21 (1994). [DOI] [PubMed] [Google Scholar]
- 187.Kamb A et al. Analysis of the p16 gene (CDKN2) as a candidate for the chromosome 9p melanoma susceptibility locus. Nat. Genet 8, 23–26 (1994). [DOI] [PubMed] [Google Scholar]
- 188.Zuo L et al. Germline mutations in the p16INK4a binding domain of CDK4 in familial melanoma. Nat. Genet 12, 97–99 (1996). [DOI] [PubMed] [Google Scholar]
- 189.Teh JLF et al. Metabolic adaptations to MEK and CDK4/6 cotargeting in uveal melanoma. Mol. Cancer Ther 19, 1719–1726 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 190.Perez-Galan P, Dreyling M & Wiestner A Mantle cell lymphoma: biology, pathogenesis, and the molecular basis of treatment in the genomic era. Blood 117, 26–38 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 191.Leonard JP et al. Selective CDK4/6 inhibition with tumor responses by PD0332991 in patients with mantle cell lymphoma. Blood 119, 4597–4607 (2012). [DOI] [PubMed] [Google Scholar]
- 192.Martin P et al. A phase I trial of palbociclib plus bortezomib in previously treated mantle cell lymphoma. Leuk. Lymphoma 60, 2917–2921 (2019). [DOI] [PubMed] [Google Scholar]
- 193.Binh MB et al. MDM2 and CDK4 immunostainings are useful adjuncts in diagnosing well-differentiated and dedifferentiated liposarcoma subtypes: a comparative analysis of 559 soft tissue neoplasms with genetic data. Am. J. Surg. Pathol 29, 1340–1347 (2005). [DOI] [PubMed] [Google Scholar]
- 194.Dickson MA et al. Phase II trial of the CDK4 inhibitor PD0332991 in patients with advanced CDK4-amplified well-differentiated or dedifferentiated liposarcoma. J. Clin. Oncol 31, 2024–2028 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 195.Hadjadj D et al. A hypothesis-driven approach identifies CDK4 and CDK6 inhibitors as candidate drugs for treatments of adrenocortical carcinomas. Aging 9, 2695–2716 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 196.Huang S et al. CDK4/6 inhibitor suppresses gastric cancer with CDKN2A mutation. Int. J. Clin. Exp. Med 8, 11692–11700 (2015). [PMC free article] [PubMed] [Google Scholar]
- 197.Frisone D et al. Durable response to palbociclib and letrozole in ovarian cancer with CDKN2A loss. Cancer Biol. Ther 21, 197–202 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 198.Rose TL et al. Phase II trial of palbociclib in patients with metastatic urothelial cancer after failure of first-line chemotherapy. Br. J. Cancer 119, 801–807 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 199.Gong X et al. Genomic aberrations that activate D-type cyclins are associated with enhanced sensitivity to the CDK4 and CDK6 inhibitor abemaciclib. Cancer Cell 32, 761–776.e6 (2017). [DOI] [PubMed] [Google Scholar]
- 200.Green JL et al. Direct CDKN2 modulation of CDK4 alters target engagement of CDK4 inhibitor drugs. Mol. Cancer Ther 18, 771–779 (2019). [DOI] [PubMed] [Google Scholar]
- 201.Iida M et al. The p21 levels have the potential to be a monitoring marker for ribociclib in breast cancer. Oncotarget 10, 4907–4918 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 202.Vilgelm AE et al. MDM2 antagonists overcome intrinsic resistance to CDK4/6 inhibition by inducing p21. Sci. Transl. Med 11, eaav7171 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 203.Pandey K et al. Combined CDK2 and CDK4/6 inhibition overcomes palbociclib resistance in breast cancer by enhancing senescence. Cancers 12, 3566 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 204.Bockstaele L et al. Regulated activating Thr172 phosphorylation of cyclin-dependent kinase 4(CDK4): its relationship with cyclins and CDK “inhibitors”. Mol. Cell Biol 26, 5070–5085 (2006). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 205.Raspe E et al. CDK4 phosphorylation status and a linked gene expression profile predict sensitivity to palbociclib. EMBO Mol. Med 9, 1052–1066 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 206.Pandey K et al. Molecular mechanisms of resistance to CDK4/6 inhibitors in breast cancer: a review. Int. J. Cancer 145, 1179–1188 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 207.Pancholi S et al. Tumour kinome re-wiring governs resistance to palbociclib in oestrogen receptor positive breast cancers, highlighting new therapeutic modalities. Oncogene 39, 4781–4797 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 208.Tong Z et al. Functional genomics identifies predictive markers and clinically actionable resistance mechanisms to CDK4/6 inhibition in bladder cancer. J. Exp. Clin. Cancer Res 38, 322 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 209.Hua H et al. Targeting mTOR for cancer therapy. J. Hematol. Oncol 12, 71 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 210.Voss MH, Molina AM & Motzer RJ mTOR inhibitors in advanced renal cell carcinoma. Hematol. Oncol. Clin. North. Am 25, 835–852 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 211.Ghidini M et al. Clinical development of mTor inhibitors for renal cancer. Expert Opin. Investig. Drugs 26, 1229–1237 (2017). [DOI] [PubMed] [Google Scholar]
- 212.Paternot S & Roger PP Combined inhibition of MEK and mammalian target of rapamycin abolishes phosphorylation of cyclin-dependent kinase 4 in glioblastoma cell lines and prevents their proliferation. Cancer Res. 69, 4577–4581 (2009). [DOI] [PubMed] [Google Scholar]
- 213.Zacharek SJ, Xiong Y & Shumway SD Negative regulation of TSC1–TSC2 by mammalian D-type cyclins. Cancer Res. 65, 11354–11360 (2005). [DOI] [PubMed] [Google Scholar]
- 214.Kim J & Guan KL mTOR as a central hub of nutrient signalling and cell growth. Nat. Cell Biol 21, 63–71 (2019). [DOI] [PubMed] [Google Scholar]
- 215.Pikman Y et al. Synergistic drug combinations with a CDK4/6 inhibitor in T-cell acute lymphoblastic leukemia. Clin. Cancer Res 23, 1012–1024 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 216.Modiano JF, Domenico J, Szepesi A, Lucas JJ & Gelfand EW Differential requirements for interleukin-2 distinguish the expression and activity of the cyclin-dependent kinases Cdk4 and Cdk2 in human T cells. J. Biol. Chem 269, 32972–32978 (1994). [PubMed] [Google Scholar]
- 217.Modiano JF, Mayor J, Ball C, Fuentes MK & Linthicum DS CDK4 expression and activity are required for cytokine responsiveness in T cells. J. Immunol 165, 6693–6702 (2000). [DOI] [PubMed] [Google Scholar]
- 218.Macian F NFAT proteins: key regulators of T-cell development and function. Nat. Rev. Immunol 5, 472–484 (2005). [DOI] [PubMed] [Google Scholar]
- 219.Jin X et al. Phosphorylated RB promotes cancer immunity by inhibiting NF-kappaB activation and PD-L1 expression. Mol. Cell 73, 22–35 e26 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 220.Iwai Y et al. Involvement of PD-L1 on tumor cells in the escape from host immune system and tumor immunotherapy by PD-L1 blockade. Proc. Natl Acad. Sci. USA 99, 12293–12297 (2002). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 221.Herbst RS et al. Predictive correlates of response to the anti-PD-L1 antibody MPDL3280A in cancer patients. Nature 515, 563–567 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 222.Sato H, Okonogi N & Nakano T Rationale of combination of anti-PD-1/PD-L1 antibody therapy and radiotherapy for cancer treatment. Int. J. Clin. Oncol 25, 801–809 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 223.US National Library of Medicine. ClinicalTrials.gov https://ClinicalTrials.gov/show/NCT03905889 (2022). [DOI] [PubMed]
- 224.O’Brien J, Hayder H, Zayed Y & Peng C Overview of MicroRNA biogenesis, mechanisms of actions, and circulation. Front. Endocrinol 9, 402 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 225.Xiao H et al. MiR-1 downregulation correlates with poor survival in clear cell renal cell carcinoma where it interferes with cell cycle regulation and metastasis. Oncotarget 6, 13201–13215 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 226.Xiao H et al. miR-206 functions as a novel cell cycle regulator and tumor suppressor in clear-cell renal cell carcinoma. Cancer Lett. 374, 107–116 (2016). [DOI] [PubMed] [Google Scholar]
- 227.Statello L, Guo CJ, Chen LL & Huarte M Gene regulation by long non-coding RNAs and its biological functions. Nat. Rev. Mol. Cell Biol 22, 96–118 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 228.Chen D, Sun X, Zhang X & Cao J Inhibition of the CDK4/6-Cyclin D-Rb pathway by ribociclib augments chemotherapy and immunotherapy in renal cell carcinoma. Biomed. Res. Int 2020, 9525207 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 229.Clark DJ et al. Integrated proteogenomic characterization of clear cell renal cell carcinoma. Cell 179, 964–983.e31 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 230.Logan JE et al. PD-0332991, a potent and selective inhibitor of cyclin-dependent kinase 4/6, demonstrates inhibition of proliferation in renal cell carcinoma at nanomolar concentrations and molecular markers predict for sensitivity. Anticancer. Res 33, 2997–3004 (2013). [PubMed] [Google Scholar]
- 231.Semenza GL Targeting HIF-1 for cancer therapy. Nat. Rev. Cancer 3, 721–732 (2003). [DOI] [PubMed] [Google Scholar]
- 232.Jonasch E et al. Belzutifan for renal cell carcinoma in Von Hippel-Lindau disease. N. Engl. J. Med 385, 2036–2046 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 233.Atkins DJ et al. Concomitant deregulation of HIF1alpha and cell cycle proteins in VHL-mutated renal cell carcinomas. Virchows Arch. 447, 634–642 (2005). [DOI] [PubMed] [Google Scholar]
- 234.Schodel J et al. Common genetic variants at the 11q13.3 renal cancer susceptibility locus influence binding of HIF to an enhancer of cyclin D1 expression. Nat. Genet 44, 420–425 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 235.Bailey ST et al. MYC activation cooperates with Vhl and Ink4a/Arf loss to induce clear cell renal cell carcinoma. Nat. Commun 8, 15770 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 236.Bommi-Reddy A et al. Kinase requirements in human cells. III. Altered kinase requirements in VHL−/− cancer cells detected in a pilot synthetic lethal screen. Proc. Natl Acad. Sci. USA 105, 16484–16489 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 237.Zhu G et al. Synthesis, structure-activity relationship, and biological studies of indolocarbazoles as potent cyclin D1-CDK4 inhibitors. J. Med. Chem 46, 2027–2030 (2003). [DOI] [PubMed] [Google Scholar]
- 238.Nicholson HE et al. HIF-independent synthetic lethality between CDK4/6 inhibition and VHL loss across species. Sci. Signal 12, eaay0482 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 239.Small J, Washburn E, Millington K, Zhu J & Holder SL The addition of abemaciclib to sunitinib induces regression of renal cell carcinoma xenograft tumors. Oncotarget 8, 95116–95134 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 240.Anand P, Sundaram C, Jhurani S, Kunnumakkara AB & Aggarwal BB Curcumin and cancer: an “old-age” disease with an “age-old” solution. Cancer Lett. 267, 133–164 (2008). [DOI] [PubMed] [Google Scholar]
- 241.Mukhopadhyay A et al. Curcumin-induced suppression of cell proliferation correlates with down-regulation of cyclin D1 expression and CDK4-mediated retinoblastoma protein phosphorylation. Oncogene 21, 8852–8861 (2002). [DOI] [PubMed] [Google Scholar]
- 242.Debata PR et al. Curcumin potentiates the ability of sunitinib to eliminate the VHL-lacking renal cancer cells 786-O: rapid inhibition of Rb phosphorylation as a preamble to cyclin D1 inhibition. Anticancer. Agents Med. Chem 13, 1508–1513 (2013). [DOI] [PubMed] [Google Scholar]
- 243.Wang Y et al. Wogonin induces apoptosis and reverses sunitinib resistance of renal cell carcinoma cells via inhibiting CDK4-RB pathway. Front. Pharmacol 11, 1152 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 244.US National Library of Medicine. ClinicalTrials.gov https://ClinicalTrials.gov/show/NCT04594005 (2021). [DOI] [PubMed]
- 245.US National Library of Medicine. ClinicalTrials.gov https://ClinicalTrials.gov/show/NCT02065063 (2018). [DOI] [PubMed]
- 246.US National Library of Medicine. ClinicalTrials.gov https://ClinicalTrials.gov/show/NCT02022982 (2022). [DOI] [PubMed]
- 247.US National Library of Medicine. ClinicalTrials.gov https://ClinicalTrials.gov/show/NCT03310879 (2022). [DOI] [PubMed]
- 248.US National Library of Medicine. ClinicalTrials.gov https://ClinicalTrials.gov/show/NCT02693535 (2022). [DOI] [PubMed]
- 249.US National Library of Medicine. ClinicalTrials.gov https://ClinicalTrials.gov/show/NCT02465060 (2022). [DOI] [PubMed]
- 250.US National Library of Medicine. ClinicalTrials.gov https://ClinicalTrials.gov/show/NCT03297606 (2021). [DOI] [PubMed]
- 251.US National Library of Medicine. ClinicalTrials.gov https://ClinicalTrials.gov/show/NCT03239015 (2020). [DOI] [PubMed]
- 252.US National Library of Medicine. ClinicalTrials.gov https://ClinicalTrials.gov/show/NCT03065062 (2022). [DOI] [PubMed]
- 253.US National Library of Medicine. ClinicalTrials.gov https://ClinicalTrials.gov/show/NCT01522989 (2020). [DOI] [PubMed]
- 254.US National Library of Medicine. ClinicalTrials.gov https://ClinicalTrials.gov/show/NCT03237390 (2021). [DOI] [PubMed]
- 255.US National Library of Medicine. ClinicalTrials.gov https://ClinicalTrials.gov/show/NCT04116541 (2021). [DOI] [PubMed]
- 256.US National Library of Medicine. ClinicalTrials.gov https://ClinicalTrials.gov/show/NCT04557449 (2022). [DOI] [PubMed]
- 257.US National Library of Medicine. ClinicalTrials.gov https://ClinicalTrials.gov/show/NCT02919696 (2020). [DOI] [PubMed]
- 258.US National Library of Medicine. ClinicalTrials.gov https://ClinicalTrials.gov/show/NCT01394016 (2021). [DOI] [PubMed]
- 259.US National Library of Medicine. ClinicalTrials.gov https://ClinicalTrials.gov/show/NCT03965845 (2021). [DOI] [PubMed]
- 260.US National Library of Medicine. ClinicalTrials.gov https://ClinicalTrials.gov/show/NCT04439201 (2022). [DOI] [PubMed]
- 261.US National Library of Medicine. ClinicalTrials.gov https://ClinicalTrials.gov/show/NCT02389842 (2019). [DOI] [PubMed]
- 262.US National Library of Medicine. ClinicalTrials.gov https://ClinicalTrials.gov/show/NCT04162301 (2021). [DOI] [PubMed]
- 263.US National Library of Medicine. ClinicalTrials.gov https://ClinicalTrials.gov/show/NCT04060511 (2019). [DOI] [PubMed]
- 264.US National Library of Medicine. ClinicalTrials.gov https://ClinicalTrials.gov/show/NCT04275050 (2020). [DOI] [PubMed]
- 265.Cho YS et al. Fragment-based discovery of 7-azabenzimidazoles as potent, highly selective, and orally active CDK4/6 inhibitors. ACS Med. Chem. Lett 3, 445–449 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]