

Abstract
The recent advances in the field of immuno-oncology have dramatically changed the therapeutic strategy against advanced malignancies. Bispecific antibody-based immunotherapies have gained momentum in preclinical and clinical investigations following the regulatory approval of the T cell–redirecting antibody blinatumomab. In this review, we focus on emerging and novel mechanisms of action of bispecific antibodies interacting with immune cells with at least one of their arms to regulate the activity of the immune system by redirecting and/or reactivating effector cells toward tumor cells. These molecules, here referred to as bispecific immunomodulatory antibodies, have the potential to improve clinical efficacy and safety profile and are envisioned as a second wave of cancer immunotherapies. Currently, there are more than 50 bispecific antibodies under clinical development for a range of indications, with promising signs of therapeutic activity. We also discuss two approaches for in vivo secretion, direct gene delivery, and infusion of ex vivo gene-modified cells, which may become instrumental for the clinical application of next-generation bispecific immunomodulatory antibodies.
Introduction
The past decade has witnessed a number of cancer immunotherapy breakthroughs, all of which involve the modulation of T cell–mediated immunity. Three major categories of cancer immunotherapies are currently approved for clinical use: (i) immune-checkpoint blockers (ICB), (ii) genetically engineered T cells expressing chimeric antigen receptors (CAR), and (iii) bispecific antibodies (bsAb). Naturally occurring antibodies are bivalent monospecific molecules. The exception are IgG4 molecules which, due to a single amino acid change in the hinge region, are able to exchange a heavy chain–light chain unit (half-antibody exchange) to form new IgG4 hybrid molecules with specificity towards two different antigens (1–3). The seminal design of a man-made bsAb was proposed 60 years ago when antigen-binding fragments (Fabs) from two different polyclonal antibodies raised in rabbits were re-associated into bispecific F(ab')2 molecules (4). This review does not address the expansive topic of antibody engineering, which has produced over 100 bsAb formats described to date; this has recently been covered by several excellent reviews (5, 6). Three building blocks are required for the construction of an effective bsAb: (i) the antigen-binding domains; (ii) the multimerization core that forms a homo- or hetero-multimer; and (iii) the linkers connecting the other blocks (Fig. 1A). The antigen-binding domain can be an antibody fragment, such as a Fab, single-chain fragment variable (scFv), or single-domain antibody (sdAb), or alternatively, an antibody mimetic. Another approach is the use of extracellular domains of natural receptors or ligands for the design of bsAbs (Fig. 1A). Several methods have been used for the multimerization of antibodies, such as linear gene fusions, domain-swapping strategies, and self-associating peptides and protein domains (Fig. 1B). Multiple technology platforms are available for the design of bsAbs, allowing fine-tuning of binding valence, stoichiometry, size, flexibility, and pharmacokinetic properties according to the intended use (Fig. 1A and B; ref. 5).
Figure 1.

Schematic overview of the basic building blocks of a bispecific molecule and some of the leading strategies used for its generation. Three different components are required for the generation of a bsAb: the binding domains, the “multimerization core” that allows homo- or hetero-multimer formation, and the linkers connecting the other blocks (A). The binding domains can be derived from antibodies, such as Fabs, scFvs, or sdAbs, or from natural receptors or ligands. There are multiple technology platforms allowing fine-tuning of binding valence, size, and stoichiometry (A and B). In the simplest setting a bsAb contains one binding site for each antigen (1 + 1), but other formats with 1 + 2, 1 + 3, 2 + 2, or 3 +3 stoichiometries have been generated (A and B). Symmetric IgG-based bsAbs contain unmodified heavy chain (HC) constant regions, whereas asymmetric IgG-based bsAbs contain modified HC to force heterodimerization, such as the knobs-into-holes (kih) strategy. Two antigen-binding domains can be directly fused, resulting in small-sized variable (V) domain-only molecules. Alternatively, two binding domains can be fused to a non-immunoglobulin protein, such as human serum albumin (HSA), or human collagen homotrimerization domains, among others. ATTACK, asymmetric tandem trimerbody for T-cell activation and cancer killing; DART, dual affinity retargeting; DNL, dock-and-Lock; DVD-Ig, dual variable domain immunoglobulin; LiTE, bispecific light T-cell engager; tandAb, tandem diabody.
The multitargeting concept that bsAbs make possible is particularly appealing from a therapeutic point of view because many diseases are multifactorial, involving multiple receptors, ligands, and signaling cascades. Consequently, blockade or modulation of several different pathologic factors and pathways may result in improved therapeutic efficacy. An interesting feature of bsAbs is their potential to display obligate activities, which are emergent functionalities that require two binding specificities to be connected in the same molecule. This can be exploited for innovative therapeutic concepts, for instance, bridging two cell types (trans co-engagement) or engaging two molecules on the same cell membrane (cis co-engagement). In this review, we describe the emerging applications of “bispecific immunomodulatory antibodies,” defined as those obligate bsAbs interacting with at least one of their arms, with innate immune cells and/or adaptive immune cells, to promote recruitment, redirection, derepression and/or reinvigoration of exhausted immune cells. We also review important issues related to the delivery of therapeutic bsAbs in oncology, which may ultimately determine their clinical fate.
BsAbs Redirecting Immune Effector Cells
Strategies for redirecting cells of the adaptative immune system
Polyclonal T-cell redirection: pan-T-cell engagers
The first application of bsAbs in cancer immunotherapy focused on the redirection of T cells towards tumor cells. T-cell engaging bsAbs (TCE) are designed to simultaneously bind to a selected tumor-associated antigen (TAA) on the tumor cell surface and one of the extracellular CD3 subunits (most commonly CD3ϵ) on the T-cell surface (Fig. 2A). TCEs mediate a major histocompatibility complex (MHC)-unrestricted tumor killing by recognizing a predefined TAA, and are applicable to all patients regardless of their MHC haplotype. This also circumvents some of the evasion strategies used by tumors to escape recognition by the immune system, such as loss of MHC expression or defects in antigen-presenting machinery (7). Proof of concept was published in the mid-1980s (8).
Figure 2.
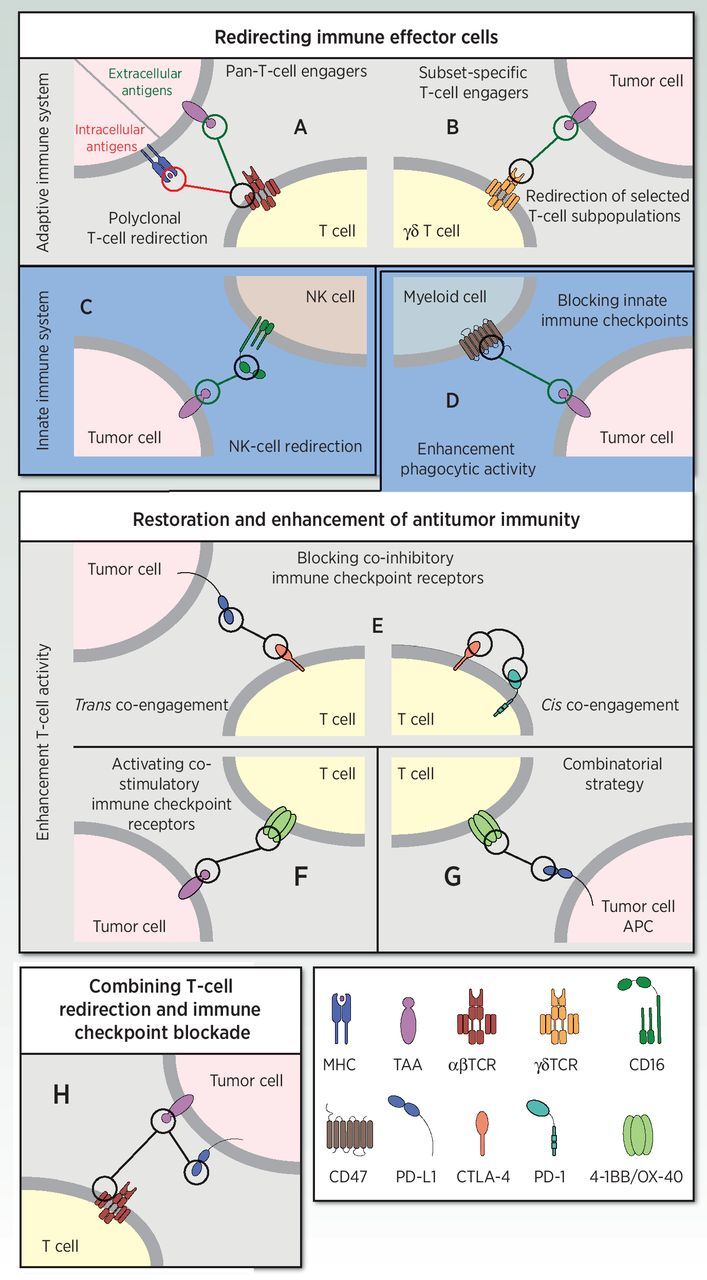
Schematic diagram illustrating the mechanism of action of bispecific immunomodulatory antibodies in cancer immunotherapy strategies. BsAbs redirecting immune effector cells (top box). Redirection of T cells towards extracellular TAA or intracellular neoantigens via pan-T-cell engagers (A) or subset-specific T-cell engagers (B). Redirection of NK cells via bispecific killer-cell engagers (C). BsAbs for the restoration and enhancement of antitumor activity (middle box). Enhancement of phagocytic activity by blocking innate immune checkpoints (“don't eat me”) via anti-TAA x anti-CD47 bsAbs (D). Enhancement of T-cell activity by blocking co-inhibitory immune checkpoint receptors in-trans or in-cis binding (E), activating costimulatory immune checkpoint receptors by TAA-targeted 4-1BB–agonistic bsAbs (F), or a combinational approach using anti–PD-L1 (x anti–4-1BB or anti–OX-40) bsAbs (G). BsAbs combining T-cell redirection and immune checkpoint blockade (bottom box, H).
The first TCE approved for clinical use was catumaxomab in 2009, a hybrid mouse–rat bispecific IgG targeting the epithelial cell adhesion molecule (EpCAM) and CD3ϵ, for the intraperitoneal treatment of malignant ascites (9, 10). However, intravenous administration of catumaxomab was associated with severe adverse events that were attributed to off-target binding of the active Fc domain to other immune cells expressing FcγRs, resulting in cytokine release syndrome (CRS) and T cell–mediated liver toxicity (11, 12). In 2014, blinatumomab, an anti-CD19 × anti-CD3 “bispecific T-cell engager” (BiTE), was approved for the treatment of acute lymphoblastic leukemia (ALL). The BiTE format, also known as a tandem scFv or (scFv)2, is a small-sized Fc-free molecule composed of two scFvs connected by a flexible linker on a single polypeptide (Fig. 1B; ref. 13). Fc-free formats lack the half-life extension of mAbs that is conveyed by the neonatal Fc receptor (FcRn), and as result, blinatumomab has a short serum half-life compared with IgG antibodies, necessitating continuous intravenous infusion to achieve therapeutic serum levels (14). However, the small and flexible BiTE structure, with its closely positioned binding sites, allows the formation of a compact immunologic synapse, similar to that which occurs under physiologic conditions through T-cell receptor (TCR)/MHC interactions (15). The clinical success of blinatumomab has driven the generation of an evolving array of bsAbs, such as the “dual-affinity retargeting” (DART) format (16), and several other TCEs with multivalent TAA binding and monovalent CD3 binding, that is, 2 + 1 (17) and 3 + 1 (18) binding stoichiometries (Fig. 1B). A monovalent interaction with CD3 is preferred, since bivalent CD3 binding (i.e., 2+2 designs) may crosslink the TCR/CD3 complex even without simultaneous binding to TAA-expressing cells (Fig. 1B), leading to a systemic T-cell activation and CRS (19). These multivalent antibodies may enable new strategies to restrict their activity towards tumors, such as the use of low-affinity binding domains to require multivalent engagement of overexpressed TAAs for stable binding (17), or the combined targeting of two different TAAs which are not co-expressed outside the tumor.
The majority of clinical-stage anti-CD3–based pan-T-cell engagers are being developed for the treatment of hematologic malignancies, targeting CD20 for non–Hodgkin lymphoma, B-cell maturation antigen for multiple myeloma, and CD33, CD123 and C-type lectin-like molecule-1 for acute myeloid leukemia (AML; ref. 20). Those targets are also commonly expressed on normal blood cells, but depletion of these cells can be tolerated without inducing severe adverse events. In solid tumors, clinical studies have been initiated using TCEs targeting carcinoembryonic antigen (CEA; ref. 20) and gpA33 (21) for colorectal cancer; EGFR variant III for gliomas (22); prostate-specific membrane antigen for prostate cancer (23); and MUC-1 (20), glypican-3 (24), P-cadherin (25), and B7-H3 (26) for multiple indications (20). For a deeper analysis of TCEs in clinical development, the reader is referred to several recent reviews (20, 27, 28). However, most of these TAAs are also expressed at low levels in normal cells, and their targeting may therefore induce on-target/off-tumor toxicities, complicating the development of TCEs for solid tumors. Hence, it is imperative to develop new strategies to modulate the reactivity, affinity, and/or increasing tumor specificity of bsAbs (29, 30).
TCEs may also target intracellular antigens (Fig. 2A). Neoantigen peptides presented by MHC molecules on cancer cells can be targeted by antibodies known as TCR mimics (TCRm; ref. 31). TCRms recognizing oncoprotein WT1 and PRAME epitopes in the context of HLA-A*0201, fused with an anti-CD3 scFv, were able to selectively activate human T cells to kill cancer cells in vitro and in vivo in ALL, AML, and mesothelioma mouse models (31, 32). Another way to target MHC-presented peptides from intracellular antigens is through the use of engineered TCR peptides fused to an anti-CD3 scFv, which is called the immune-mobilizing monoclonal TCRs against cancer (ImmTAC) format (Fig. 1B; ref. 33). ImmTAC molecules have been reported to effectively redirect T cells to kill cancer cells expressing extremely low surface epitope densities, and to significantly delay the in vivo growth of human multiple myeloma cells (33). An ImmTAC targeting gp100 was well tolerated and showed signs of efficacy in a phase I/II clinical trial in melanoma (34).
Redirection of selected T-cell subpopulations: subset-specific T-cell engagers
An important disadvantage of anti-CD3 TCEs is that a large proportion of the T-cell pool is activated. Most mature T cells use a CD3-associated αβTCR as their antigen recognition structure. Conventional αβ T cells can be subdivided into three major subtypes: CD4+ helper T cells, CD8+ cytotoxic T lymphocytes (CTL), and regulatory T (Treg) cells. It has recently been reported that human primary Tregs activated by blinatumomab inhibit both the proliferation and cytolytic activity of CD8+ CTLs in vitro (35). Therefore, the design of TCEs that can selectively engage individual T-cell subsets would provide an advantage over current anti-CD3 pan-T-cell engagers. Because expression of the co-receptor CD8 is the distinctive feature of CTLs, the selective redirection of primary human CD8+ αβ T cells against human cell lines by anti-CD8 TCEs has recently been explored, but with only limited success (36).
T cells expressing the alternative γδTCR are a minor subset (1–5%) of the T-cell population in peripheral blood, but constitute a major population among intestinal intraepithelial lymphocytes. The Vγ9Vδ2 subtype is the predominant type of circulating γδ T cells in healthy individuals. The successful redirection of the cytotoxic activity of γδ T cells was initially demonstrated in vitro with a bsAb targeting the γδTCR and a human ovarian cancer antigen (Fig. 2B; ref. 37). More recently, a bsAb targeting the Vγ9 chain of the Vγ9Vδ2 TCR and HER-2 in a xenograft tumor model has been reported (38). A VHH-based bispecific construct (Fig. 1B) targeting both Vγ9Vδ2 T cells and EGFR induced potent γδ T-cell activation and tumor cell lysis both in vitro and in a mouse xenograft model (39). An anti-Vγ9 × anti-CD123 bsAb selectively activated Vγ9+ γδ T cells and induced γδ T cell–mediated cytotoxicity against AML cells in vitro and in vivo in a xenograft mouse model (40).
Strategies for redirecting cells of the innate immune system
Natural killer (NK)-cell redirection
NK cells are cytotoxic lymphocytes of the innate immune system that play an important role in tumor immunosurveillance (41). One strategy to redirect NK cells to cancer cells is based on tandem scFvs, called “bispecific killer cell engager” (BiKE) or “trispecific killer cell engager” (TriKE), which have one or two TAA-specific scFv fused to an anti-CD16A (FcγRIIIA) scFv (Fig. 2C; refs. 42–44). Alternatively, the two scFvs can be connected by a modified human IL15 crosslinker, producing a “TRiKE”-like construct that promotes in vivo persistence and activation of NK and T cells in a xenograft mouse tumor model (45). An anti-CD30 × anti-CD16A TandAb (46) has reached phase II clinical development for treatment of Hodgkin's lymphoma and cutaneous T-cell lymphoma (47), and a TriKE consisting of anti-CD33 × anti-CD16A and a modified IL15 linker is currently being studied in phase I for CD33-expressing hematologic tumors (45).
BsAbs for the Restoration and Enhancement of Antitumor Immunity
Strategies for enhancement phagocytic activity
Blocking innate immune checkpoints
It is well established that tumors can escape macrophage-mediated phagocytosis through expression of antiphagocytic (“don't eat me”) signals. CD47 binds to the signal regulatory protein-α (SIRPα) on macrophages to negatively regulate phagocytosis of healthy cells (48). On tumor cells, CD47 is upregulated and functions as an immune checkpoint that modulates macrophage phagocytosis (49). Therefore, strategies aiming to disrupt the CD47–SIRPα axis are being actively investigated for the treatment of several cancer types (50). BsAbs cotargeting CD47 and a TAA such as CD20, CD19, or mesothelin have demonstrated enhanced phagocytosis induction and antitumor activity in vitro and in vivo in murine xenograft models of human non–Hodgkin lymphoma (51) and leukemia (Fig. 2D; ref. 52).
Strategies for enhancement of T-cell activity
Blocking co-inhibitory immune checkpoint receptors
ICBs are mAbs blocking negative regulators of T-cell activation, such as cytotoxic T lymphocyte antigen 4 (CTLA-4), programmed cell death 1 (PD-1), or programmed cell death 1 ligand 1 (PD-L1). The therapeutic efficacy of ICBs depends on preexisting TCR-dependent responses to tumor-specific neoantigens presented by MHC molecules on cancer cells (53). The blockade of inhibitory checkpoints can restore effector functions to exhausted tumor-infiltrating T cells and have shown remarkable efficacy in a wide range of cancers, but their efficacy is limited to 10% to 30% of patients (54).
The success of conventional ICBs, and the improved clinical benefit observed in combination therapies with ICBs (55) has prompted the development of innovative dual therapeutic concepts blocking two inhibitory checkpoints by bridging two cell types or binding two molecules on the surface of the same cell (Fig. 2E). Most of these bsAbs block the PD-1/PD-L1 axis with one arm, and CTLA-4, lymphocyte activation gene 3 (LAG3), or T-cell immunoglobulin mucin 3 (TIM3) with the other arm (56). Currently, the safety and early efficacy of anti–PD-1 × (anti–CTLA-4, anti-LAG3, or anti-TIM3) and anti–PD-L1 × anti–CTLA-4 bsAbs are being evaluated in early clinical trials, whereas many others are in preclinical development (56).
Activating costimulatory immune checkpoint receptors
Costimulatory receptors of the TNF receptor superfamily (TNFRSF), such as CD27, OX40 (CD134), 4‐1BB (CD137), and glucocorticoid-induced TNFR-related protein (GITR) are particularly interesting targets, as they are transiently expressed by T cells following their activation through the TCR (57). The most clinically advanced agonist antibodies against costimulatory checkpoint molecules have reached phase II trials (58). However, off-tumor toxicity has been a major impediment to the clinical development of anti–4-1BB mAbs, and studies in murine models suggest that toxicity involves engagement of antibody Fc with FcγRs (59). Fc-free (60, 61) or Fc-silenced (62, 63) bsAbs, which cross-link 4-1BB in a manner that is dependent on simultaneous TAA engagement, may alleviate off-tumor toxicities that are associated with Fc engagement by FcγRs, as shown in mouse (60, 62, 63) and nonhuman primate models (Fig. 2F; ref. 63). These TAA-targeted 4-1BB–agonistic bsAbs exhibited robust antitumor activity without toxicity, and some of them are in clinical development.
Targeting costimulatory and co-inhibitory immune checkpoint receptors
A combinational approach using both immune checkpoint blockade and T-cell costimulation strategies can be enacted using anti–PD-L1 (× anti–4-1BB or anti-OX40) bsAbs (Fig. 2G). Conditional T-cell costimulation in parallel with PD-L1 checkpoint inhibition elicited potent antitumor immune responses in multiple in vivo tumor models (64), and showed a favorable safety profile in nonhuman primates (65).
BsAbs combining T-cell redirection and immune checkpoint blockade
The upregulation of inhibitory checkpoint receptors represents a resistance mechanism to antitumor immunity that is well established in different T cell–redirecting strategies (66, 67). Sustained inhibitory signaling correlates with a dysfunctional or exhausted state, characterized by a reduced T-cell effector function, proliferative potential, and cytotoxicity (68). Several studies have demonstrated that combination of ICBs and TCEs maximized T-cell killing of tumor cells in vitro (69) and enhanced antitumor activity in a patient-derived xenograft model of leukemia (70). Such evidence has fostered the development of bsAbs aiming to provide locally restricted ICB to T cells redirected by tumor-specific pan-T-cell engagers (Fig. 2H). Therefore, the efficacy of the T-cell engagement is increased while avoiding the immune-related adverse events (irAE) associated with systemic exposure to ICBs. An anti-CD33 × anti-CD3 BiTE fused to the extracellular domain of PD-1, termed “checkpoint inhibitory T-cell engager” (CiTE), resulted in enhanced T cell–mediated cytotoxicity against CD33+PD-L1+ cell lines and strong antitumor effect in preclinical models of AML (Fig. 2H; ref. 71).
Delivery Strategies
Clinical-grade bsAbs
In oncology, approved bsAbs are administered as recombinant proteins (Fig. 3). However, clinical-grade antibodies are extremely expensive and suffer from manufacturing challenges, including poor stability during long-term storage, and a tendency to aggregate over time (72). Purified antibodies requiring higher doses need to be administered through slow intravenous infusions to limit infusion reactions. Furthermore, systemic administration entails some limitations, especially in the case of small-sized bsAb formats, such as BiTEs, where an infusion pump is required for continuous delivery. BiTEs and DARTs have been fused to silenced Fc domains in an attempt to extend their circulatory half-life (73).
Figure 3.

Delivery strategies for bsAb-based therapies. Intravenous administration of recombinant purified bsAb (left) by repeated high-dose bolus injections or continuous infusion. The in vivo secretion of bsAbs (right) can be achieved following direct injection of genetic material using viral vectors of synthetic nucleic acids (RNA or DNA), or by infusion or implantation of autologous or allogenic genetically modified bsAb-secreting cells.
In vivo secretion of bsAbs
The in vivo production of bsAbs can compensate for the rapid renal clearance of Fc-free formats, resulting in a sustained effective antibody concentration and circumventing concerns regarding the formulation and long-term storage of recombinant proteins (74). The two main genetic strategies are based on either direct gene delivery or inoculation of ex vivo genetically modified cells (Fig. 3). The in vivo transfer of bsAb-encoding genetic information might be performed using viral and nonviral vectors. Direct in vivo delivery of synthetic nucleic acid-encoded bsAbs employing messenger RNA and plasmid DNA represents new approaches for the in vivo secretion of bsAbs. Systemic administration of engineered mRNAs encoding Fc-free tumor-specific pan-T-cell engagers resulted in sustained levels of therapeutic TCEs and strong antitumor effect in different mouse models (75). Similarly, the systemic administration of a nonviral DNA vector minicircle encoding an anti-CD3 × anti-CD20 bsAb resulted in the expression of therapeutic levels and an effective antitumor activity in a B-cell lymphoma xenograft mouse model (76). In another study, a single intramuscular injection of synthetic plasmid DNA induced secretion of Fc-free TCEs for 4 months and delayed cancer progression in mice (77). Synthetic nucleic acid platforms have advantages such as rapid product development and simpler manufacturing processes. Several types of oncolytic viruses have been armed with expression cassettes encoding TCEs, demonstrating that the combination of both, direct oncolysis and T cell–mediated killing, enhanced antitumor efficacy compared with the parental counterpart in syngenic and xenograft tumor models (78–80).
Alternatively, genetic material can be delivered into patient-derived cells ex vivo, at which point the bsAb-secreting cells are introduced into the patient. This has some potential advantages over direct gene delivery strategies. For instance, the tumor homing of infused cells and subsequent intratumoral secretion might circumvent issues of tumor penetration and systemic on-target/off-tumor toxicity (81). Indeed, strategies based on the endogenous secretion of TCEs by engineered cells (STAb cells) are currently emerging (81). T cells represent ideal vehicles for STAb therapy due to their capacity to migrate to tumor sites and their ability to act simultaneously as antibody factories and effectors, but other cell types, such as mesenchymal stem cells and endothelial cells, are suitable candidates to be engineered for STAb therapies (81). The potent anti-tumor activity of different types of STAb cells has been demonstrated in multiple in vitro and in vivo models (82).
Conclusions
BsAbs have come to prominence over the past decade, following the FDA's approval of the Fc-free TCE blinatumomab in ALL. The bsAb field has been continually expanding since then, with the constant emergence of novel formats and the development of innovative multitargeting concepts aiming to improve therapeutic efficacy and safety. In this review, we focus on bispecific immunomodulatory antibodies able to regulate the response of the immune system through the specific interaction or interactions with cell surface receptors expressed on immune cells and cancer cells. A multitude of bispecific immunomodulatory antibodies are in clinical trials with promising therapeutic results. We anticipate that through continued innovation and optimization of antibody formats, targets, and delivery systems, treatment strategies using bispecific immunomodulatory antibodies may soon form the leading edge of cancer immunotherapy.
Authors' Disclosures
B. Blanco reports grants from Carlos III Health Institute during the conduct of the study. L. Alvarez-Vallina reports grants from Spanish Ministry of Science and Innovation, CRIS Cancer Foundation, and Spanish Association Against Cancer during the conduct of the study, as well as has a patent for WO/2019/234187 licensed to Leadartis SL. No disclosures were reported by the other author.
Acknowledgments
We thank S.L. Harwood for critically reading the manuscript. Research was supported by grants from the Carlos III Health Institute (PI20/01030), cofounded by the Plan Nacional de Investigación and the European Union to B. Blanco; the Spanish Ministry of Science and Innovation (SAF2017–89437-P, RTC-2017–5944–1), partially supported by the European Regional Development Fund; the CRIS Cancer Foundation (FCRIS-IFI-2018); and the Spanish Association Against Cancer (AECC, 19084) to L. Alvarez-Vallina. C. Domínguez-Alonso was supported by a fellowship from the Spanish Ministry of Science and Innovation (PRE2018–083445).
References
- 1. van der Neut KM, Schuurman J, Losen M, Bleeker WK, Martinez-Martinez P, Vermeulen E, et al. Anti-inflammatory activity of human IgG4 antibodies by dynamic Fab arm exchange. Science 2007;317:1554–7. [DOI] [PubMed] [Google Scholar]
- 2. Labrijn AF, Buijsse AO, van den Bremer ET, Verwilligen AY, Bleeker WK, Thorpe SJ, et al. Therapeutic IgG4 antibodies engage in Fab-arm exchange with endogenous human IgG4 in vivo. Nat Biotechnol 2009;27:767–71. [DOI] [PubMed] [Google Scholar]
- 3. Yang X, Ambrogelly A. Enlarging the repertoire of therapeutic monoclonal antibodies platforms: domesticating half molecule exchange to produce stable IgG4 and IgG1 bispecific antibodies. Curr Opin Biotechnol 2014;30:225–9. [DOI] [PubMed] [Google Scholar]
- 4. Nisonoff A, Rivers MM. Recombination of a mixture of univalent antibody fragments of different specificity. Arch Biochem Biophys 1961;93:460–2. [DOI] [PubMed] [Google Scholar]
- 5. Nunez-Prado N, Compte M, Harwood S, Alvarez-Mendez A, Lykkemark S, Sanz L, et al. The coming of age of engineered multivalent antibodies. Drug Discov Today 2015;20:588–94. [DOI] [PubMed] [Google Scholar]
- 6. Brinkmann U, Kontermann RE. The making of bispecific antibodies. MAbs 2017;9:182–212. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Garrido F. HLA class-I expression and cancer immunotherapy. Adv Exp Med Biol 2019;1151:79–90. [DOI] [PubMed] [Google Scholar]
- 8. Staerz UD, Kanagawa O, Bevan MJ. Hybrid antibodies can target sites for attack by T cells. Nature 1985;314:628–31. [DOI] [PubMed] [Google Scholar]
- 9. Zeidler R, Reisbach G, Wollenberg B, Lang S, Chaubal S, Schmitt B, et al. Simultaneous activation of T cells and accessory cells by a new class of intact bispecific antibody results in efficient tumor cell killing. J Immunol 1999;163:1246–52. [PubMed] [Google Scholar]
- 10. Sebastian M, Kiewe P, Schuette W, Brust D, Peschel C, Schneller F, et al. Treatment of malignant pleural effusion with the trifunctional antibody catumaxomab (Removab) (anti-EpCAM x Anti-CD3): results of a phase 1/2 study. J Immunother 2009;32:195–202. [DOI] [PubMed] [Google Scholar]
- 11. Linke R, Klein A, Seimetz D. Catumaxomab: clinical development and future directions. MAbs 2010;2:129–36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Borlak J, Langer F, Spanel R, Schondorfer G, Dittrich C. Immune-mediated liver injury of the cancer therapeutic antibody catumaxomab targeting EpCAM, CD3 and Fcgamma receptors. Oncotarget 2016;7:28059–74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Mack M, Riethmuller G, Kufer P. A small bispecific antibody construct expressed as a functional single-chain molecule with high tumor cell cytotoxicity. Proc Natl Acad Sci U S A 1995;92:7021–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Goebeler ME, Bargou R. Blinatumomab: a CD19/CD3 bispecific T cell engager (BiTE) with unique anti-tumor efficacy. Leuk Lymphoma 2016;57:1021–32. [DOI] [PubMed] [Google Scholar]
- 15. Roda-Navarro P, Alvarez-Vallina L. Understanding the spatial topology of artificial immunological synapses assembled in T cell-redirecting strategies: a major issue in cancer immunotherapy. Front Cell Dev Biol 2019;7:370. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Moore PA, Zhang W, Rainey GJ, Burke S, Li H, Huang L, et al. Application of dual affinity retargeting molecules to achieve optimal redirected T-cell killing of B-cell lymphoma. Blood 2011;117:4542–51. [DOI] [PubMed] [Google Scholar]
- 17. Bacac M, Fauti T, Sam J, Colombetti S, Weinzierl T, Ouaret D, et al. A novel carcinoembryonic antigen T-cell bispecific antibody (CEA TCB) for the treatment of solid tumors. Clin Cancer Res 2016;22:3286–97. [DOI] [PubMed] [Google Scholar]
- 18. Harwood SL, Alvarez-Cienfuegos A, Nunez-Prado N, Compte M, Hernandez-Perez S, Merino N, et al. ATTACK, a novel bispecific T cell-recruiting antibody with trivalent EGFR binding and monovalent CD3 binding for cancer immunotherapy. Oncoimmunology 2017;7:e1377874. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Chatenoud L, Ferran C, Reuter A, Legendre C, Gevaert Y, Kreis H, et al. Systemic reaction to the anti-T-cell monoclonal antibody OKT3 in relation to serum levels of tumor necrosis factor and interferon-gamma [corrected]. N Engl J Med 1989;320:1420–1. [DOI] [PubMed] [Google Scholar]
- 20. Clynes RA, Desjarlais JR. Redirected T cell cytotoxicity in cancer therapy. Annu Rev Med 2019;70:437–50. [DOI] [PubMed] [Google Scholar]
- 21. Hurwitz H, Crocenzi T, Lohr J, Bonvini E, Johnson S, Moore P, et al. A phase I, first-in-human, open label, dose escalation study of MGD007, a humanized gpA33 × CD3 dual-affinity re-targeting (DART®) protein in patients with relapsed/refractory metastatic colorectal carcinoma. J Immunother Cancer 2014;2:86. [Google Scholar]
- 22. Schaller TH, Snyder DJ, Spasojevic I, Gedeon PC, Sanchez-Perez L, Sampson JH. First in human dose calculation of a single-chain bispecific antibody targeting glioma using the MABEL approach. J Immunother Cancer 2020;8:e000213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Hummel HD, Kufer P, Grullich C, Seggewiss-Bernhardt R, Deschler-Baier B, Chatterjee M, et al. Pasotuxizumab, a BiTE((R)) immune therapy for castration-resistant prostate cancer: Phase I, dose-escalation study findings. Immunotherapy 2021;13:125–41. [DOI] [PubMed] [Google Scholar]
- 24. Ishiguro T, Sano Y, Komatsu SI, Kamata-Sakurai M, Kaneko A, Kinoshita Y, et al. An anti-glypican 3/CD3 bispecific T cell-redirecting antibody for treatment of solid tumors. Sci Transl Med 2017;9:eaal4291. [DOI] [PubMed] [Google Scholar]
- 25. Root AR, Cao W, Li B, LaPan P, Meade C, Sanford J, et al. Development of PF-06671008, a highly potent anti-P-cadherin/anti-CD3 bispecific DART molecule with extended half-life for the treatment of cancer. Antibodies (Basel) 2016;5:6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Tolcher AW, Alley EW, Chichili G, Baughman JE, Moore PA, Bonvini E, et al. Phase 1, first-in-human, open label, dose escalation ctudy of MGD009, a humanized B7-H3 x CD3 dual-affinity re-targeting (DART) protein in patients with B7-H3-expressing neoplasms or B7-H3 expressing tumor vasculature. J Clin Oncol 34:15s, 2016 (suppl; abstr. TPS3105). [Google Scholar]
- 27. Krishnamurthy A, Jimeno A. Bispecific antibodies for cancer therapy: A review. Pharmacol Ther 2018;185:122–34. [DOI] [PubMed] [Google Scholar]
- 28. Suurs FV, Lub-de Hooge MN, de Vries EGE, de Groot DJA. A review of bispecific antibodies and antibody constructs in oncology and clinical challenges. Pharmacol Ther 2019;201:103–19. [DOI] [PubMed] [Google Scholar]
- 29. Mazor Y, Sachsenmeier KF, Yang C, Hansen A, Filderman J, Mulgrew K, et al. Enhanced tumor-targeting selectivity by modulating bispecific antibody binding affinity and format valence. Sci Rep 2017;7:40098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Banaszek A, Bumm TGP, Nowotny B, Geis M, Jacob K, Wolfl M, et al. On-target restoration of a split T cell-engaging antibody for precision immunotherapy. Nat Commun 2019;10:5387. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Dao T, Pankov D, Scott A, Korontsvit T, Zakhaleva V, Xu Y, et al. Therapeutic bispecific T-cell engager antibody targeting the intracellular oncoprotein WT1. Nat Biotechnol 2015;33:1079–86. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Chang AY, Dao T, Gejman RS, Jarvis CA, Scott A, Dubrovsky L, et al. A therapeutic T cell receptor mimic antibody targets tumor-associated PRAME peptide/HLA-I antigens. J Clin Invest 2017;127:2705–18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Liddy N, Bossi G, Adams KJ, Lissina A, Mahon TM, Hassan NJ, et al. Monoclonal TCR-redirected tumor cell killing. Nat Med 2012;18:980–7. [DOI] [PubMed] [Google Scholar]
- 34. Middleton MR, McAlpine C, Woodcock VK, Corrie P, Infante JR, Steven NM, et al. Tebentafusp, a TCR/anti-CD3 bispecific fusion protein targeting gp100, potently activated antitumor immune responses in patients with metastatic melanoma. Clin Cancer Res 2020;26:5869–78. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Duell J, Dittrich M, Bedke T, Mueller T, Eisele F, Rosenwald A, et al. Frequency of regulatory T cells determines the outcome of the T-cell-engaging antibody blinatumomab in patients with B-precursor ALL. Leukemia 2017;31:2181–90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Michalk I, Feldmann A, Koristka S, Arndt C, Cartellieri M, Ehninger A, et al. Characterization of a novel single-chain bispecific antibody for retargeting of T cells to tumor cells via the TCR co-receptor CD8. PLoS One 2014;9:e95517. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Ferrini S, Prigione I, Mammoliti S, Colnaghi MI, Menard S, Moretta A, et al. Re-targeting of human lymphocytes expressing the T-cell receptor gamma/delta to ovarian carcinoma cells by the use of bispecific monoclonal antibodies. Int J Cancer 1989;44:245–50. [DOI] [PubMed] [Google Scholar]
- 38. Oberg HH, Peipp M, Kellner C, Sebens S, Krause S, Petrick D, et al. Novel bispecific antibodies increase gammadelta T-cell cytotoxicity against pancreatic cancer cells. Cancer Res 2014;74:1349–60. [DOI] [PubMed] [Google Scholar]
- 39. de Bruin RCG, Veluchamy JP, Lougheed SM, Schneiders FL, Lopez-Lastra S, Lameris R, et al. A bispecific nanobody approach to leverage the potent and widely applicable tumor cytolytic capacity of Vgamma9Vdelta2-T cells. Oncoimmunology 2017;7:e1375641. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Ganesan R, Chennupati V, Ramachandran B, Hansen MR, Singh S, Grewal IS. Selective recruitment of gammadelta T cells by a bispecific antibody for the treatment of acute myeloid leukemia. Leukemia 2021. Feb 1 [Epub ahead of print]. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Shimasaki N, Jain A, Campana D. NK cells for cancer immunotherapy. Nat Rev Drug Discov 2020;19:200–18. [DOI] [PubMed] [Google Scholar]
- 42. Singer H, Kellner C, Lanig H, Aigner M, Stockmeyer B, Oduncu F, et al. Effective elimination of acute myeloid leukemic cells by recombinant bispecific antibody derivatives directed against CD33 and CD16. J Immunother 2010;33:599–608. [DOI] [PubMed] [Google Scholar]
- 43. Gleason MK, Verneris MR, Todhunter DA, Zhang B, McCullar V, Zhou SX, et al. Bispecific and trispecific killer cell engagers directly activate human NK cells through CD16 signaling and induce cytotoxicity and cytokine production. Mol Cancer Ther 2012;11:2674–84. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Felices M, Lenvik TR, Davis ZB, Miller JS, Vallera DA. Generation of BiKEs and TriKEs to improve NK cell-mediated targeting of tumor cells. Methods Mol Biol 2016;1441:333–46. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Vallera DA, Felices M, McElmurry R, McCullar V, Zhou X, Schmohl JU, et al. IL15 trispecific killer engagers (TriKE) make natural killer cells specific to CD33+ targets while also inducing persistence, in vivo expansion, and enhanced function. Clin Cancer Res 2016;22:3440–50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Wu J, Fu J, Zhang M, Liu D. AFM13: a first-in-class tetravalent bispecific anti-CD30/CD16A antibody for NK cell-mediated immunotherapy. J Hematol Oncol 2015;8:96. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Rothe A, Sasse S, Topp MS, Eichenauer DA, Hummel H, Reiners KS, et al. A phase 1 study of the bispecific anti-CD30/CD16A antibody construct AFM13 in patients with relapsed or refractory Hodgkin lymphoma. Blood 2015;125:4024–31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Brown EJ, Frazier WA. Integrin-associated protein (CD47) and its ligands. Trends Cell Biol 2001;11:130–5. [DOI] [PubMed] [Google Scholar]
- 49. Jaiswal S, Jamieson CH, Pang WW, Park CY, Chao MP, Majeti R, et al. CD47 is upregulated on circulating hematopoietic stem cells and leukemia cells to avoid phagocytosis. Cell 2009;138:271–85. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Feng M, Jiang W, Kim BYS, Zhang CC, Fu YX, Weissman IL. Phagocytosis checkpoints as new targets for cancer immunotherapy. Nat Rev Cancer 2019;19:568–86. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Piccione EC, Juarez S, Liu J, Tseng S, Ryan CE, Narayanan C, et al. A bispecific antibody targeting CD47 and CD20 selectively binds and eliminates dual antigen expressing lymphoma cells. MAbs 2015;7:946–56. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Dheilly E, Moine V, Broyer L, Salgado-Pires S, Johnson Z, Papaioannou A, et al. Selective blockade of the ubiquitous checkpoint receptor CD47 is enabled by dual-targeting bispecific antibodies. Mol Ther 2017;25:523–33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Chamoto K, Hatae R, Honjo T. Current issues and perspectives in PD-1 blockade cancer immunotherapy. Int J Clin Oncol 2020;25:790–800. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Wei SC, Duffy CR, Allison JP. Fundamental mechanisms of immune checkpoint blockade therapy. Cancer Discov 2018;8:1069–86. [DOI] [PubMed] [Google Scholar]
- 55. Wolchok JD, Chiarion-Sileni V, Gonzalez R, Rutkowski P, Grob JJ, Cowey CL, et al. Overall survival with combined nivolumab and ipilimumab in advanced melanoma. N Engl J Med 2017;377:1345–56. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Labrijn AF, Janmaat ML, Reichert JM, Parren PWHI. Bispecific antibodies: a mechanistic review of the pipeline. Nat Rev Drug Discov 2019;18:585–608. [DOI] [PubMed] [Google Scholar]
- 57. Chester C, Sanmamed MF, Wang J, Melero I. Immunotherapy targeting 4–1BB: mechanistic rationale, clinical results, and future strategies. Blood 2018;131:49–57. [DOI] [PubMed] [Google Scholar]
- 58. Lee DH. Update of early phase clinical trials in cancer immunotherapy. BMB Rep 2021;54:70–88. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Li F, Ravetch JV. Antitumor activities of agonistic anti-TNFR antibodies require differential FcgammaRIIB coengagement in vivo. Proc Natl Acad Sci U S A 2013;110:19501–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Compte M, Harwood SL, Munoz IG, Navarro R, Zonca M, Perez-Chacon G, et al. A tumor-targeted trimeric 4–1BB-agonistic antibody induces potent anti-tumor immunity without systemic toxicity. Nat Commun 2018;9:4809. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Compte M, Harwood SL, Erce-Llamazares A, Tapia-Galisteo A, Romero E, Ferrer I, et al. An Fc-free EGFR-specific 4–1BB-agonistic trimerbody displays broad antitumor activity in humanized murine cancer models without toxicity. Clin Cancer Res 2021;27:3167–77. [DOI] [PubMed] [Google Scholar]
- 62. Hinner MJ, Aiba RSB, Jaquin TJ, Berger S, Durr MC, Schlosser C, et al. Tumor-localized costimulatory T-cell engagement by the 4–1BB/HER2 bispecific antibody-anticalin fusion PRS-343. Clin Cancer Res 2019;25:5878–89. [DOI] [PubMed] [Google Scholar]
- 63. Claus C, Ferrara C, Xu W, Sam J, Lang S, Uhlenbrock F, et al. Tumor-targeted 4–1BB agonists for combination with T cell bispecific antibodies as off-the-shelf therapy. Sci Transl Med 2019;11:eaav5989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Kuang Z, Pu P, Wu M, Wu Z, Wang L, Li Y, et al. A novel bispecific antibody with PD-L1-assisted OX40 activation for cancer treatment. Mol Cancer Ther 2020;19:2564–74. [DOI] [PubMed] [Google Scholar]
- 65. Lakins MA, Koers A, Giambalvo R, Munoz-Olaya J, Hughes R, Goodman E, et al. FS222, a CD137/PD-L1 tetravalent bispecific antibody, exhibits low toxicity and antitumor activity in colorectal cancer models. Clin Cancer Res 2020;26:4154–67. [DOI] [PubMed] [Google Scholar]
- 66. Kohnke T, Krupka C, Tischer J, Knosel T, Subklewe M. Increase of PD-L1 expressing B-precursor ALL cells in a patient resistant to the CD19/CD3-bispecific T cell engager antibody blinatumomab. J Hematol Oncol 2015;8:111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67. de Miguel M, Umana P, Gomes de Morais AL, Moreno V, Calvo E. T-cell-engaging therapy for solid tumors. Clin Cancer Res 2021;27:1595–603. [DOI] [PubMed] [Google Scholar]
- 68. Wherry EJ, Kurachi M. Molecular and cellular insights into T cell exhaustion. Nat Rev Immunol 2015;15:486–99. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69. Osada T, Patel SP, Hammond SA, Osada K, Morse MA, Lyerly HK. CEA/CD3-bispecific T cell-engaging (BiTE) antibody-mediated T lymphocyte cytotoxicity maximized by inhibition of both PD1 and PD-L1. Cancer Immunol Immunother 2015;64:677–88. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70. Krupka C, Kufer P, Kischel R, Zugmaier G, Lichtenegger FS, Kohnke T, et al. Blockade of the PD-1/PD-L1 axis augments lysis of AML cells by the CD33/CD3 BiTE antibody construct AMG 330: reversing a T-cell-induced immune escape mechanism. Leukemia 2016;30:484–91. [DOI] [PubMed] [Google Scholar]
- 71. Herrmann M, Krupka C, Deiser K, Brauchle B, Marcinek A, Ogrinc WA, et al. Bifunctional PD-1 x alphaCD3 x alphaCD33 fusion protein reverses adaptive immune escape in acute myeloid leukemia. Blood 2018;132:2484–94. [DOI] [PubMed] [Google Scholar]
- 72. Sanchez-Martin D, Sanz L, Alvarez-Vallina L. Engineering human cells for in vivo secretion of antibody and non-antibody therapeutic proteins. Curr Opin Biotechnol 2011;22:924–30. [DOI] [PubMed] [Google Scholar]
- 73. Arvedson TL, Balazs M, Bogner P, Black K, Graham K, Henn A, et al. Abstract 55: Generation of half-life extended anti-CD33 BiTE® antibody constructs compatible with once-weekly dosing. Cancer Res 2017;77:55. [Google Scholar]
- 74. Sanz L, Blanco B, Alvarez-Vallina L. Antibodies and gene therapy: teaching old ‘magic bullets’ new tricks. Trends Immunol 2004;25:85–91. [DOI] [PubMed] [Google Scholar]
- 75. Stadler CR, Bahr-Mahmud H, Celik L, Hebich B, Roth AS, Roth RP, et al. Elimination of large tumors in mice by mRNA-encoded bispecific antibodies. Nat Med 2017;23:815–7. [DOI] [PubMed] [Google Scholar]
- 76. Pang X, Ma F, Zhang P, Zhong Y, Zhang J, Wang T, et al. Treatment of human B-cell lymphomas using minicircle DNA vector expressing Anti-CD3/CD20 in a mouse model. Hum Gene Ther 2017;28:216–25. [DOI] [PubMed] [Google Scholar]
- 77. Perales-Puchalt A, Duperret EK, Yang X, Hernandez P, Wojtak K, Zhu X, et al. DNA-encoded bispecific T cell engagers and antibodies present long-term antitumor activity. JCI Insight 2019;4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78. Fajardo CA, Guedan S, Rojas LA, Moreno R, Arias-Badia M, de Sostoa J, et al. Oncolytic adenoviral delivery of an egfr-targeting T-cell engager improves antitumor efficacy. Cancer Res 2017;77:2052. [DOI] [PubMed] [Google Scholar]
- 79. Speck T, Heidbuechel JPW, Veinalde R, Jaeger D, von Kalle C, Ball CR, et al. Targeted BiTE expression by an oncolytic vector augments therapeutic efficacy against solid tumors. Clin Cancer Res 2018;24:2128. [DOI] [PubMed] [Google Scholar]
- 80. de SJ, Fajardo CA, Moreno R, Ramos MD, Farrera-Sal M, Alemany R. Targeting the tumor stroma with an oncolytic adenovirus secreting a fibroblast activation protein-targeted bispecific T-cell engager. J Immunother Cancer 2019;7:19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81. Blanco B, Compte M, Lykkemark S, Sanz L, Alvarez-Vallina LT. Cell-redirecting strategies to ‘STAb’ tumors: beyond CARs and bispecific antibodies. Trends Immunol 2019;40:243–57. [DOI] [PubMed] [Google Scholar]
- 82. Blanco B, Ramirez-Fernandez A, Alvarez-Vallina L. Engineering immune cells for in vivo secretion of tumor-specific T cell-redirecting bispecific antibodies. Front Immunol 2020;11:1792. [DOI] [PMC free article] [PubMed] [Google Scholar]