

Abstract
Thousands of new metabolic and regulatory enzymes have evolved by gene duplication and divergence since the dawn of life. New enzyme activities often originate from promiscuous secondary activities that have become important for fitness due to a change in the environment or a mutation. Mutations that make a promiscuous activity physiologically relevant can occur in the gene encoding the promiscuous enzyme itself, but can also occur elsewhere, resulting in increased expression of the enzyme or decreased competition between the native and novel substrates for the active site. If a newly useful activity is inefficient, gene duplication/amplification will set the stage for divergence of a new enzyme. Even a few mutations can increase the efficiency of a new activity by orders of magnitude. As efficiency increases, amplified gene arrays will shrink to provide two alleles, one encoding the original enzyme and one encoding the new enzyme. Ultimately, genomic rearrangements eliminate co-amplified genes and move newly evolved paralogs to a distant region of the genome.
Keywords: enzyme evolution, gene duplication, promiscuity, directed evolution, innovation-amplification-divergence model
Introduction
The genome of the last universal common ancestor (LUCA) contained on the order of 500 genes [1]. The genomes of extant free-living organisms are much larger. E. coli K12 MG1655 has 4566 genes. The Chardonnay grape has 38,020 [2]. The diversification of life since the LUCA has clearly been fueled by the evolution of new genes. New genes can evolve de novo from noncoding DNA, by gene fusions that combine previously existing domains, and by gene duplication and divergence. This review will focus on evolution of new enzymes by gene duplication and divergence.
Susumu Ohno proposed in 1970 that new functions arise after gene duplication by mutations in one allele while the other maintains its original function [3] (Figure 1). Neofunctionalization via the Ohno model is unlikely because loss of one copy by deletion, drift, counter-selection due to the cost of the duplicated region (see further below), or an inactivating mutation is orders of magnitude more likely than acquisition of a mutation that confers a new function (Figure 2) [4]. Consequently, Bergthorrson et al. proposed the Innovation-Amplification-Divergence (IAD) model [4] (Figure 1), which suggests the more likely scenario that a promiscuous side-function first becomes important for fitness, providing selective pressure to maintain two or more gene copies while mutations improve the new function. Once a sufficiently good new gene emerges, extraneous copies can be lost, leaving behind two paralogs. The IAD model is similar to the classic subfunctionalization model, in which the ancestral functions of a multifunctional protein are divided among paralogs, but differs in that gene duplication is immediately advantageous. This is an important distinction because gene duplication is costly, and deletion of a duplicated gene that does not confer a selective advantage will likely occur before mutations that lead to subfunctionalization.
Figure 1.
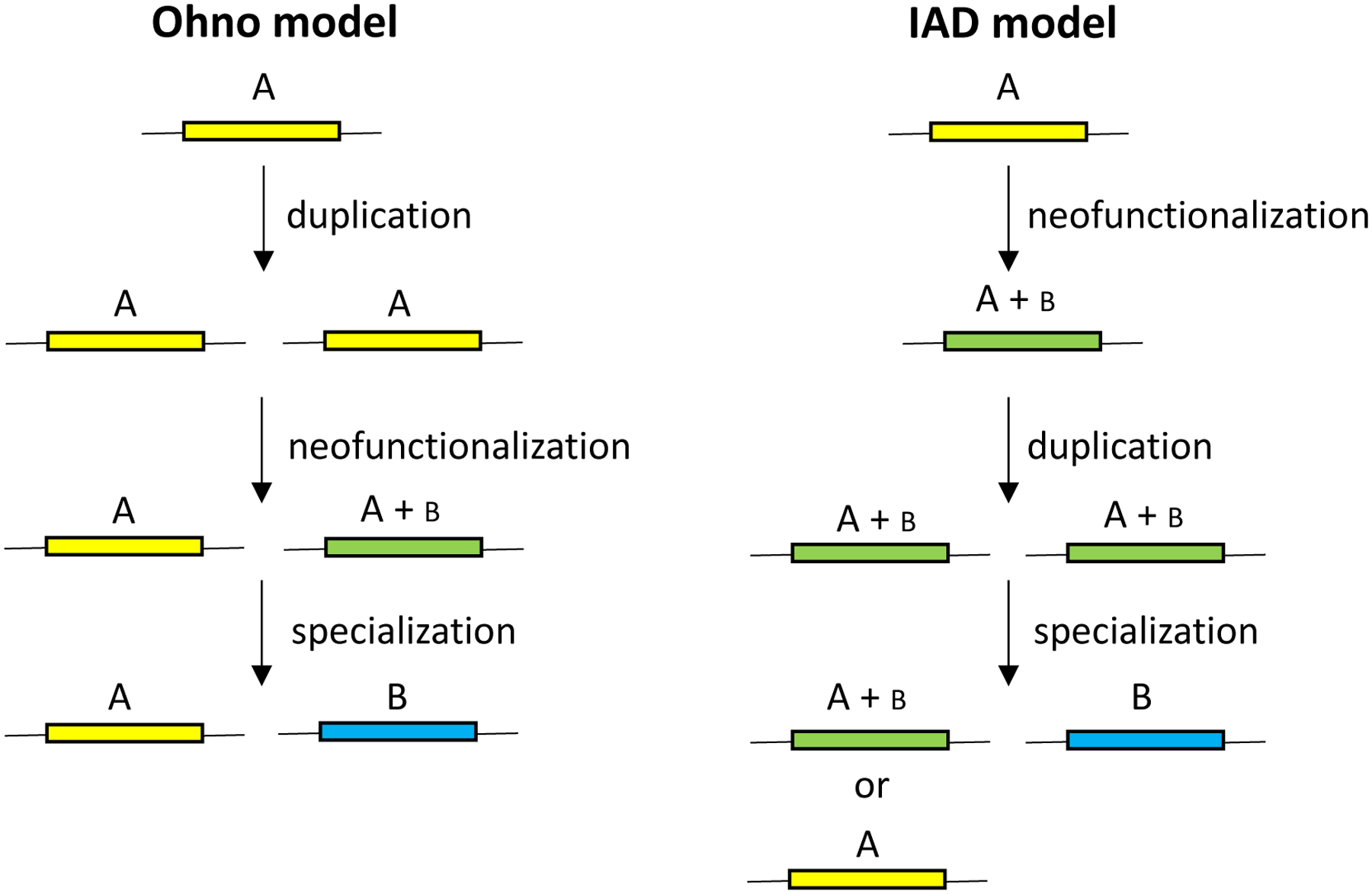
Two models for the evolution of new genes. In the Ohno model, duplication occurs before neofunctionalization. In the IAD model, neofunctionalization occurs before gene duplication. After neofunctionalization in either model, selection for increased gene dosage can lead to further amplification. Only duplications are shown for simplicity.
Figure 2.
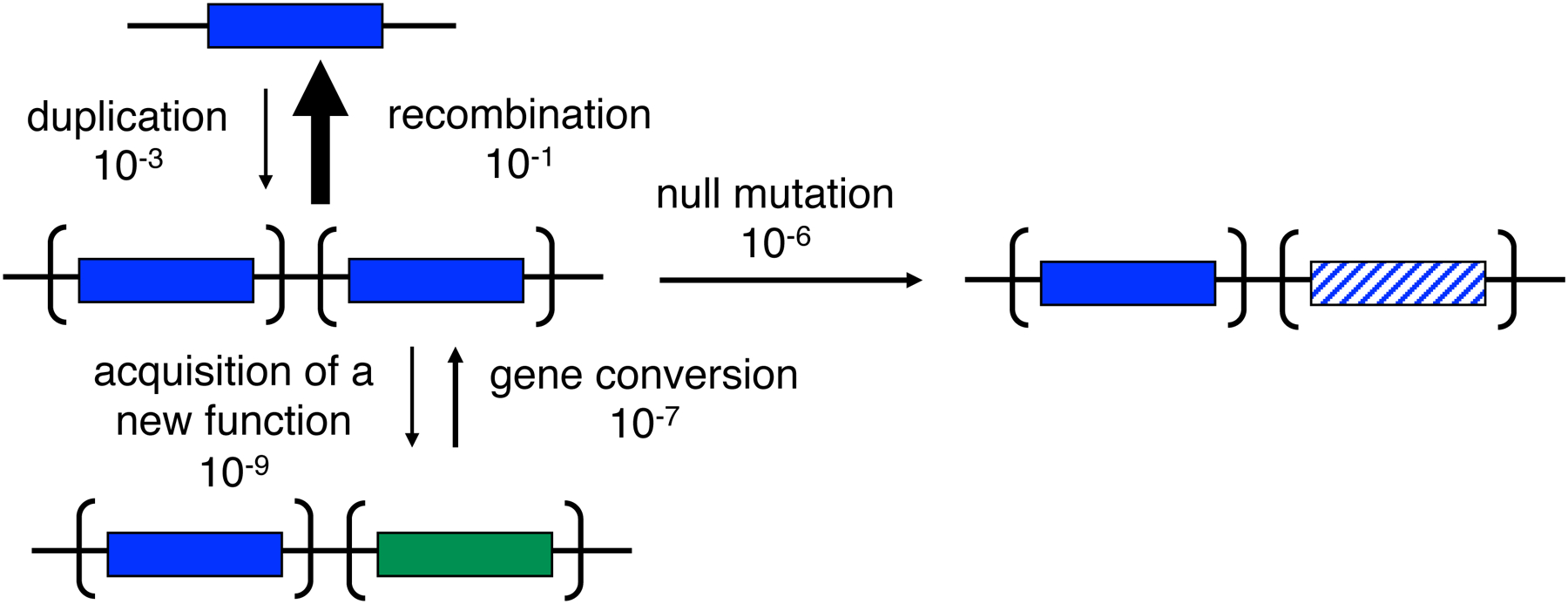
Estimated rates of processes affecting a redundant gene copy. Acquisition of a new function is orders of magnitude less likely than loss of function via deletion, drift, mutations or gene conversion. Reprinted with permission from Proc Natl Acad Sci USA 104(43):17004–9. Copyright 2007 National Academy of Sciences.
Gene duplication can occur by whole-genome duplication (WGD), chromosomal duplication, or smaller scale segmental duplication that usually encompasses several genes. WGDs have played a major role in the evolution of eukaryotes. One or more rounds of WGD occurred in Saccharomyces cerevisiae [5, 6] and Paramecium species [7, 8]. WGD and even whole-genome triplication have been especially common in plants [9]. Two rounds of WGD occurred in vertebrates before the divergence of fish and tetrapods at least 450 million years ago [10]. Additional WGDs occurred in some fish lineages, the most recent about 8.2 million years ago in the common carp [11].
Although WGDs are rare over evolutionary time, they have contributed substantially to the expansion of eukaryotic genomes. An average of 25% of vertebrate genes belong to families derived from two rounds of ancient WGD [12]. Paralogs arising from WGD are called ohnologs in honor of Susumu Ohno. Although most gene copies formed by WGD are lost or pseudogenized [6, 9, 13], duplicates produced by WGD persist longer than those produced by segmental duplication [7, 14], most likely because WGD does not perturb gene balance in ways that hamper fitness. The extended lifetime of gene duplicates in the context of WGD may provide some opportunity for neofunctionalization. However, even if neofunctionalization occurs occasionally after WGD, ohnologs account for a minority of duplicated genes in most plants [9], and the same is likely true for most other organisms. Thus, most evolutionary innovation has likely occurred via the IAD mechanism rather than the Ohno mechanism.
Interestingly, ohnologs are enriched in different functional classes than those derived from small-scale duplications. Ohnologs in vertebrates are enriched in proteins involved in developmental processes, cell differentiation and intracellular signaling [15]. In contrast, paralogs in bacteria, which arise from segmental duplications, are enriched in proteins involved in amino acid transport and metabolism, transcription, inorganic ion metabolism, carbohydrate metabolism, defense mechanisms, and energy production and conversion [16]. These patterns suggest that WGD may provide opportunities for divergence of genes for which local segmental duplication would disrupt gene balance and lead to selection for loss of the duplicated region.
The extent of gene duplication and divergence
The extent of innovation by gene duplication and divergence has been enormous. Gene duplication and divergence began even before the LUCA [17–20]. By the emergence of the last common ancestors of eukaryotes (LECA), bacteria (LBCA) and archaea (LACA), about 20% of genes already had paralogs. The LECA is predicted to have had 4137 genes that fall into 2150 clusters with an average cluster size of 1.9. The 995 genes in the LBCA fall into 798 clusters, and the 1028 genes in the LACA into 861 clusters. The paralogs in these lineages likely arose by both gene duplication and divergence and acquisition of genes by horizontal gene transfer. Among the 2150 LECA clusters, 171 show evidence of such pseudoparalogs [17].
During the billions of years since the LECA, LBCA and LACA, evolution of paralogs has continued on a massive scale. In plants, at least 50% of genes have arisen due to segmental duplication, WGD, or even whole-genome triplication [21]. In bacteria, paralogous proteins account for between 7 and 41% of the genome [16]. In E. coli, 68% of enzymes, 82% of transporters, and 79% of regulatory proteins belong to paralogous groups [22]. The number of paralogous enzymes and transcription factors in bacteria and archaea increases with genome size, but there is considerable variation among genomes of the same size (Figure 3) [23, 24]. On average, 28% of enzymes and 40% of transcription factors are paralogous.
Figure 3.
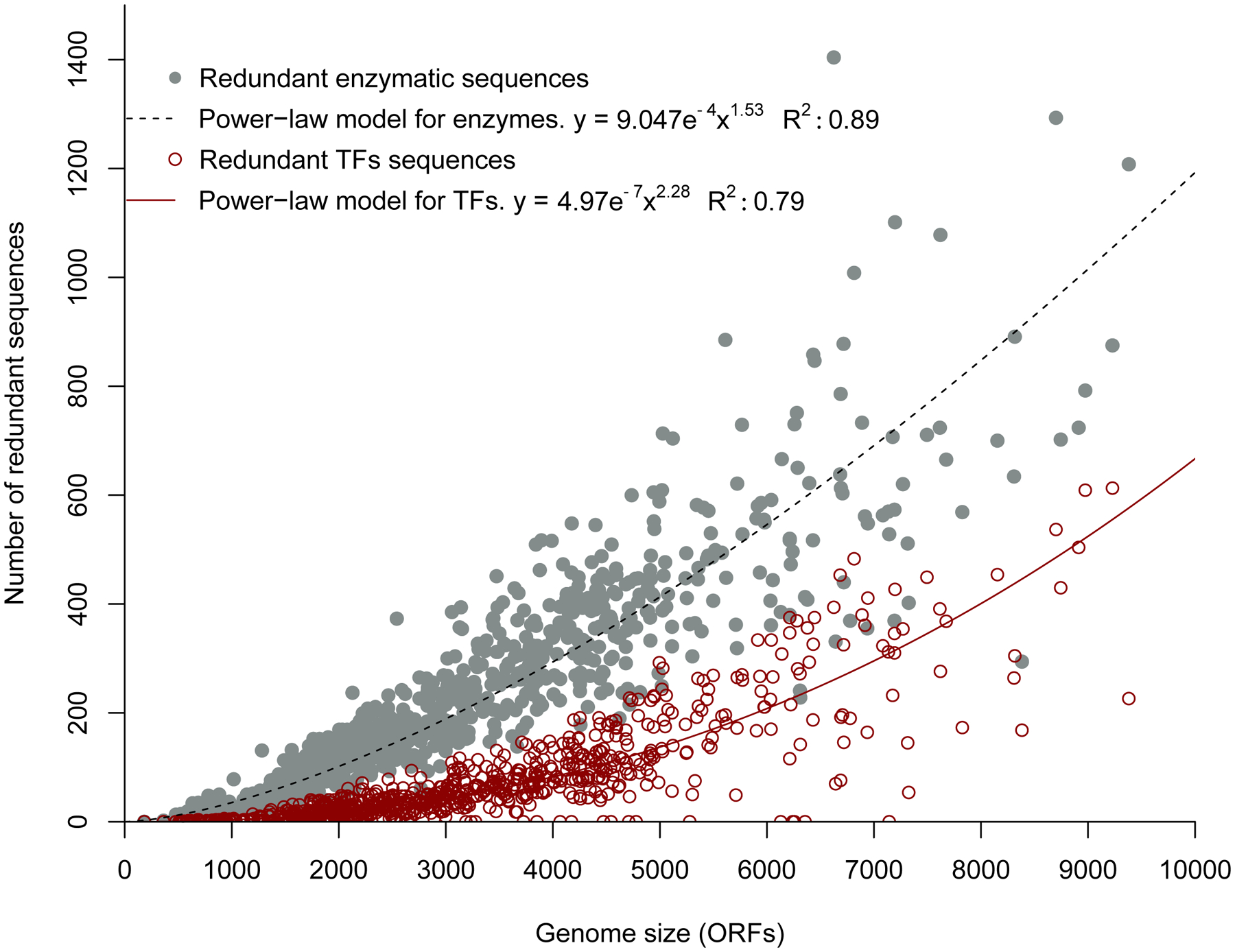
Number of redundant enzyme (grey) and transcription factor (red) sequences (i.e. paralogs) in 794 bacterial and archaeal genomes. Paralogs are defined as sequences with ≥ 30% sequence identity over ≥ 60% of the sequence, with an E-value of < 10e−5. Reprinted from PLoS One 8(7):e69707, 2013.
Not all paralogs in a genome have arisen by gene duplication and divergence within that organism or one of its progenitors. Homologous genes can be acquired by horizontal gene transfer. Eukaryotes also acquired paralogous genes via the symbiotic events that led to mitochondria and chloroplasts. The mitochondrial NAD+-dependent isocitrate dehydrogenase is clearly derived from the alpha-proteobacterial progenitor of the mitochondria, and is only distantly related to the NADP+-dependent isozymes found in the cytoplasm and the peroxisome [25].
What can be achieved by duplication and divergence of genes encoding enzymes
Divergence between duplicated genes can change the regulation, cellular localization, and/or function of enzymes. For example, E. coli has three isozymes of 3-deoxy-7-phosphoheptulonate synthase that catalyze the first step in the pathway for synthesis of aromatic amino acids. Divergence after gene duplication has led to differential feedback inhibition by aromatic amino acids; each isozyme is inhibited by only one of the three aromatic amino acids, allowing flux into the pathway to be controlled in response to levels of each of the end products.
Gene duplication and divergence has led to differential cellular localization of two isozymes of NADP-dependent isocitrate dehydrogenase, IDH1 and IDH2, in eukaryotes. IDH1 is found in both the cytoplasm and the peroxisome [26]. Cytoplasmic IDH1 produces NADPH for fatty acid synthesis and reduction of glutathione disulfide to glutathione, a major cellular defense against oxidative damage. Peroxisomal IDH1 provides NADPH for cholesterol synthesis. IDH2 is localized to the mitochondria. IDH2 produces NADPH for reduction of glutathione disulfide to glutathione and protects mitochondria against oxidative damage [27].
Duplication and divergence of genes encoding enzymes often alters substrate specificity. For example, the S1 family of serine proteases utilizes a conserved His-Asp-Ser catalytic triad to cleave peptide bonds, but divergence of the substrate binding pocket has led to trypsin-like enzymes that cleave after Lys or Arg, chymotrypsin-like enzymes that cleave after a hydrophobic amino acid, and elastase-like enzymes that cleave after Ala [28]. Similarly, duplication and divergence of kinase genes has generated 518 human kinases [29]. Most (478) belong to a single superfamily, the eukaryotic protein kinase superfamily (Figure 4). Five additional atypical protein kinase families have 2–6 members each. This vast expansion of ancestral kinases allows cells to modulate the behavior of proteins involved in signaling, gene expression, cell proliferation, differentiation, apoptosis, cytoskeletal rearrangement, motility, metabolism and vesicle transport.
Figure 4.
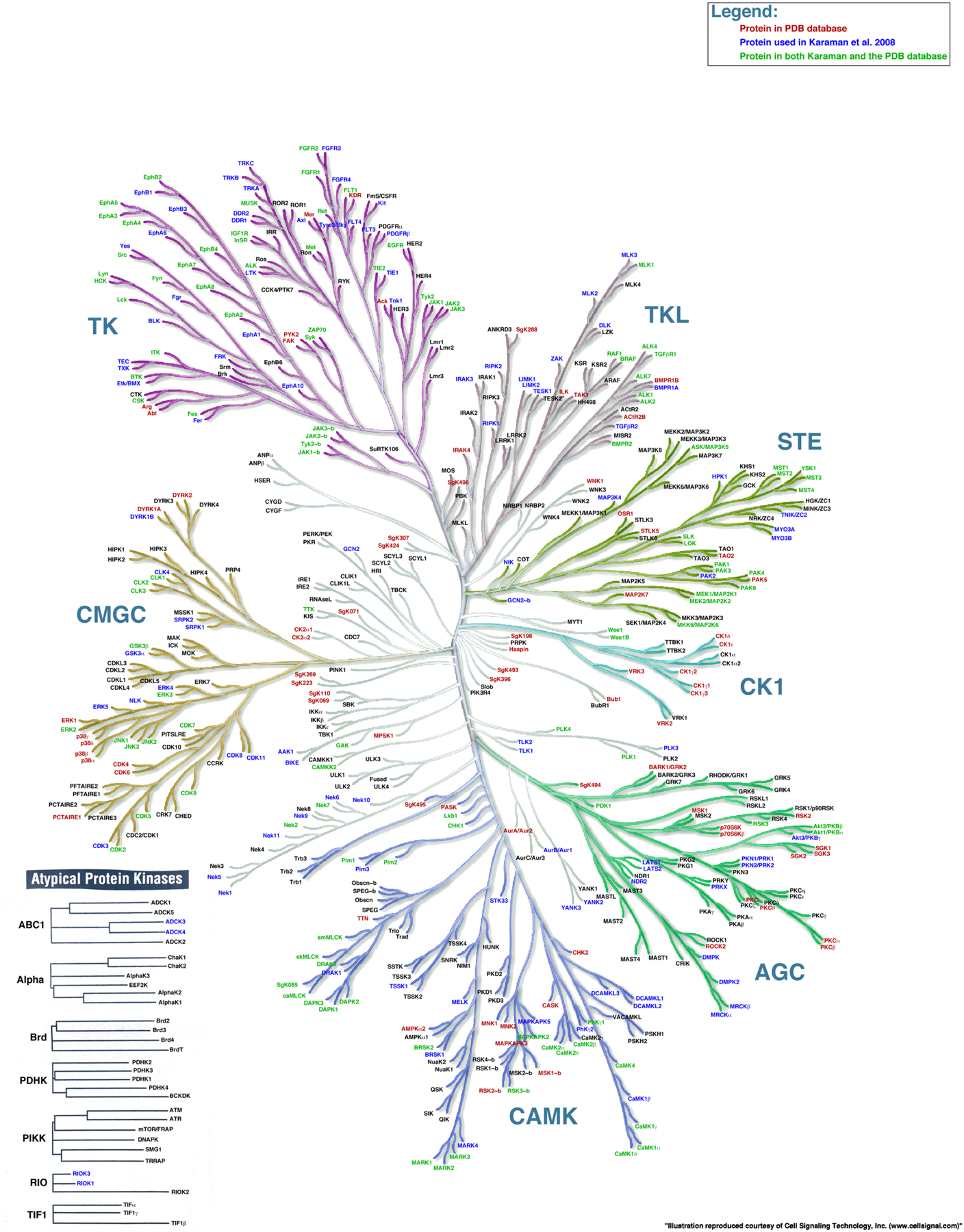
The human protein kinome. AGC, contains PKA, PKG, and PKC families; CAMK, calcium/calmodulin-dependent protein kinase; CK1, casein kinase 1; CMGC, contains CDK, MAPK, GSK3, and CLK families; STE, homologs of yeast Sterile 7, Sterile 11, Sterile 20 kinases; TK, tyrosine kinases; TKL, tyrosine-kinase-like. Reprinted from [91].
Gene duplication and divergence can also lead to evolution of new catalytic mechanisms. Mechanistically diverse enzyme superfamilies exploit a common ancestral capability such as stabilization of an enolate (enolase superfamily), deprotonation of water for attack on an electrophilic substrate (amidohydrolase superfamily), or attack of an active site Asp on a substrate to form a covalent intermediate (haloacid dehalogenase superfamily) [30]. In each case, mutations have both altered substrate binding pockets and introduced new catalytic groups that enable catalysis of diverse reactions. For example, enzymes in the enolase superfamily catalyze elimination of H2O and NH3, racemization, epimerization and lactonization reactions [31].
IAD Step 1: neofunctionalization
A strict interpretation of the term neofunctionalization would be emergence of a new function that was not previously present. However, in the context of discussions about gene duplication and divergence, neofunctionalization refers to emergence of a physiologically relevant function, which can occur either by a mutation that does indeed generate a new function, or a change in circumstances that makes a previously existing promiscuous function important for fitness.
Mutations can lead to a novel enzymatic activity by removing steric hindrance in the vicinity of the active site, thereby allowing binding of molecules that are too big to fit in the ancestral active site. Alternatively, mutations can alter charged residues in the active site, either relieving unfavorable charge-charge interactions with novel substrates or providing new attractive interactions. However, it is important to recognize that neofunctionalization does not necessarily require a mutation. An inefficient promiscuous activity of a pre-existing enzyme may become important for fitness due to change in the environment. For example, a promiscuous enzyme might be recruited to destroy a newly encountered toxin such as an antibiotic or anthropogenic pollutant, or to convert a new compound in the environment into a common metabolite to enable an organism to exploit a new source of carbon and energy. A newly encountered competitor or predator might be deterred by a novel secondary product produced by a promiscuous enzyme.
The hundreds of enzymes in any proteome provide a vast reservoir of potential activities that can serve as the starting point for evolution of new enzymes by gene duplication and divergence. However, many potentially useful promiscuous activities are not available at a level that can impact fitness, either because they are expressed at low levels or because they are simply too inefficient. Further, the productivity of a promiscuous reaction can be compromised by competition from the enzyme’s native substrate.
Eq. 1 expresses the rate of a promiscuous reaction in the presence of a native substrate (Nat) for the simple case of a unimolecular reaction or a bimolecular reaction in which the enzyme is saturated with the other substrate. (Equations for the rates of more complex reactions are more involved [32], but in every case the native substrate acts as a competitive inhibitor of the promiscuous reaction.) This equation suggests that flux through a promiscuous reaction can be improved in multiple ways, including increasing the concentration of the enzyme, increasing the concentration of the promiscuous substrate (Prom), decreasing the concentration of the native substrate (Nat), altering the active site to increase kcat,Prom or decrease KM,Prom, and increasing KM,Nat for the native substrate.
| Eq. 1 |
The recruitment of a mutant of E. coli ProA (γ-glutamyl phosphate reductase) to replace ArgC (N-acetylglutamyl phosphate reductase) provides an illustration. ProA and ArgC catalyze the reduction of an acyl phosphate to an aldehyde in the pathways for proline and arginine synthesis, respectively (Figure 5). ProA has an inefficient ability to catalyze reduction of N-acetylglutamyl phosphate, but wild-type ProA is too inefficient to substitute for ArgC in a ΔargC strain. However, a mutation that changes Glu383 to Ala in the active site enables ProA to take over the function of ArgC and support growth on glucose as a sole carbon source. The mutation increases kcat/KM for the promiscuous activity by 156-fold and decreases kcat/KM for the native activity by 1700-fold [33]. The net effect increases flux by both increasing kcat/KM for the new substrate and decreasing the ability of the native substrate to inhibit the newly important reaction.
Figure 5.
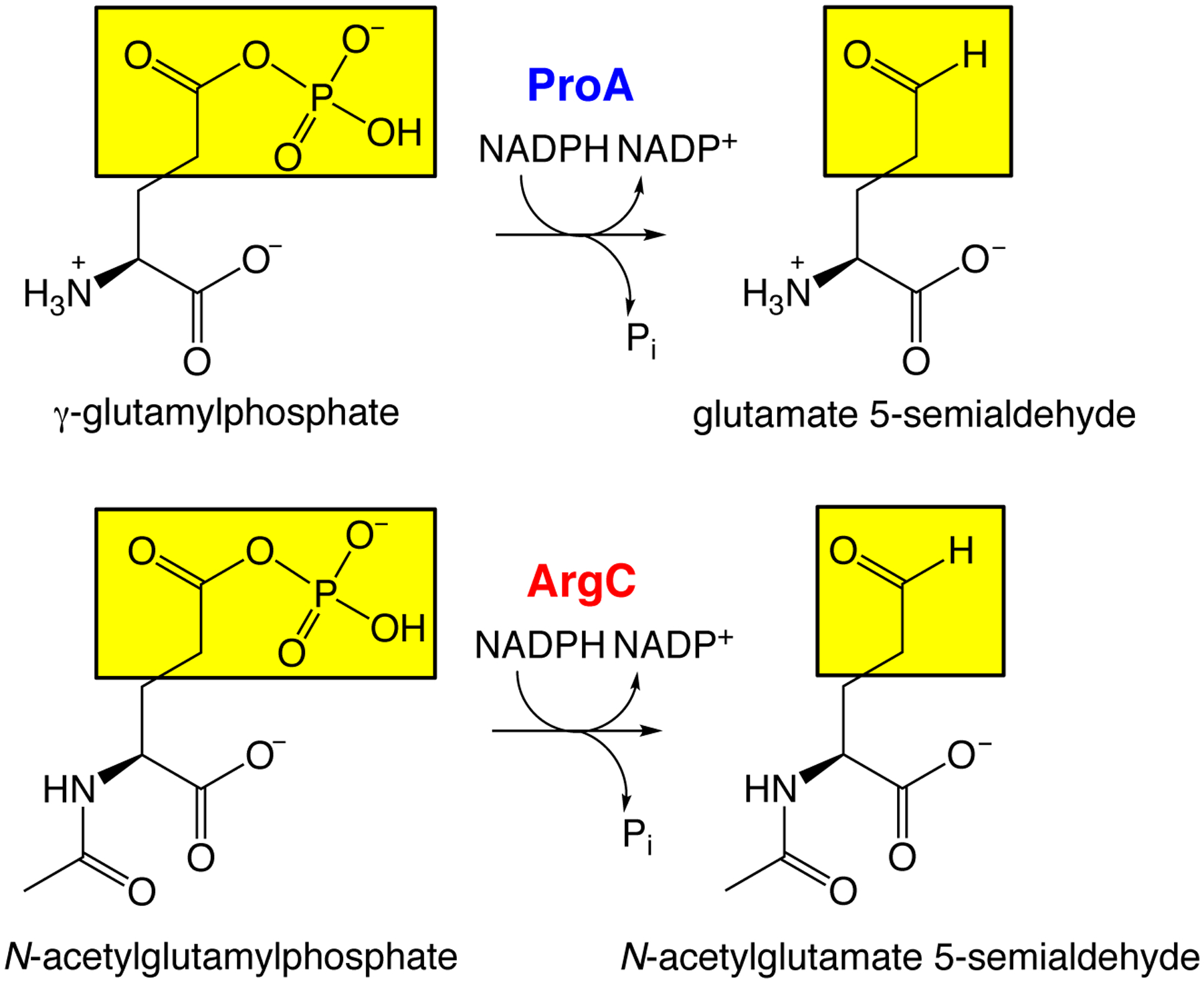
ProA and ArgC catalyze reduction of an acyl phosphate to an aldehyde in the pathways for synthesis of proline and arginine, respectively. ProA has an inefficient promiscuous activity with N-acetylglutamyl phosphate.
Importantly, mutations that allow a promiscuous activity to become physiologically relevant need not occur in the gene encoding the promiscuous enzyme. Expression of a potentially useful promiscuous enzyme can be elevated by mutations in genes encoding regulatory proteins, either relieving repression or activating transcription under circumstances in which the enzyme would not normally be expressed. For example, p-nitrophenol degradation enzymes in Pseudomonas sp. Strain WBC-3 can degrade 2-chloro-4-nitrophenol, but PnpR, the transcriptional regulator that activates their expression, does not respond to 2-chloro-4-nitrophenol. A point mutation enables PnpR to respond to 2-chloro-4-nitrophenol and thereby allows the bacterium to express the promiscuous enzymes and degrade a new carbon source [34].
Mutations that affect the expression or activities of other enzymes in the metabolic network can improve the rate of a promiscuous reaction by altering concentrations of the native and/or promiscuous substrates. An example is the recruitment of a promiscuous activity of 3-phosphoglycerate dehydrogenase (SerA) to catalyze dehydrogenation of erythronate in a novel pathway assembled from promiscuous enzymes that restores PLP synthesis in a laboratory-evolved strain of E. coli lacking PdxB (4-phospherythronate dehydrogenase) (Figure 6) [35]. The level of 3-phosphoglycerate, the native substrate for SerA, is diminished in this strain due to a deletion in pgl that results in diversion of glyceraldehyde 3-phosphate from glycolysis toward the pentose phosphate pathway and a point mutation in gapA that decreases the activity of glyceraldehyde 3-phosphate dehydrogenase in the glycolytic pathway by 5-fold. The combined effect of these mutations diminishes levels of downstream glycolytic intermediates, including 3-phosphophglycerate. According to Eq. 1, the decreased level of 3-phosphoglycerate should relieve competitive inhibition of the newly important erythronate dehydrogenase activity of SerA. An additional factor may have been the decreased serine level, which is formed by a three-step pathway beginning with 3-phosphoglycerate. Since SerA is subject to feedback inhibition by serine, a decrease in the serine level will ensure that the enzyme is available for PLP synthesis even if serine levels are adequate.
Figure 6.
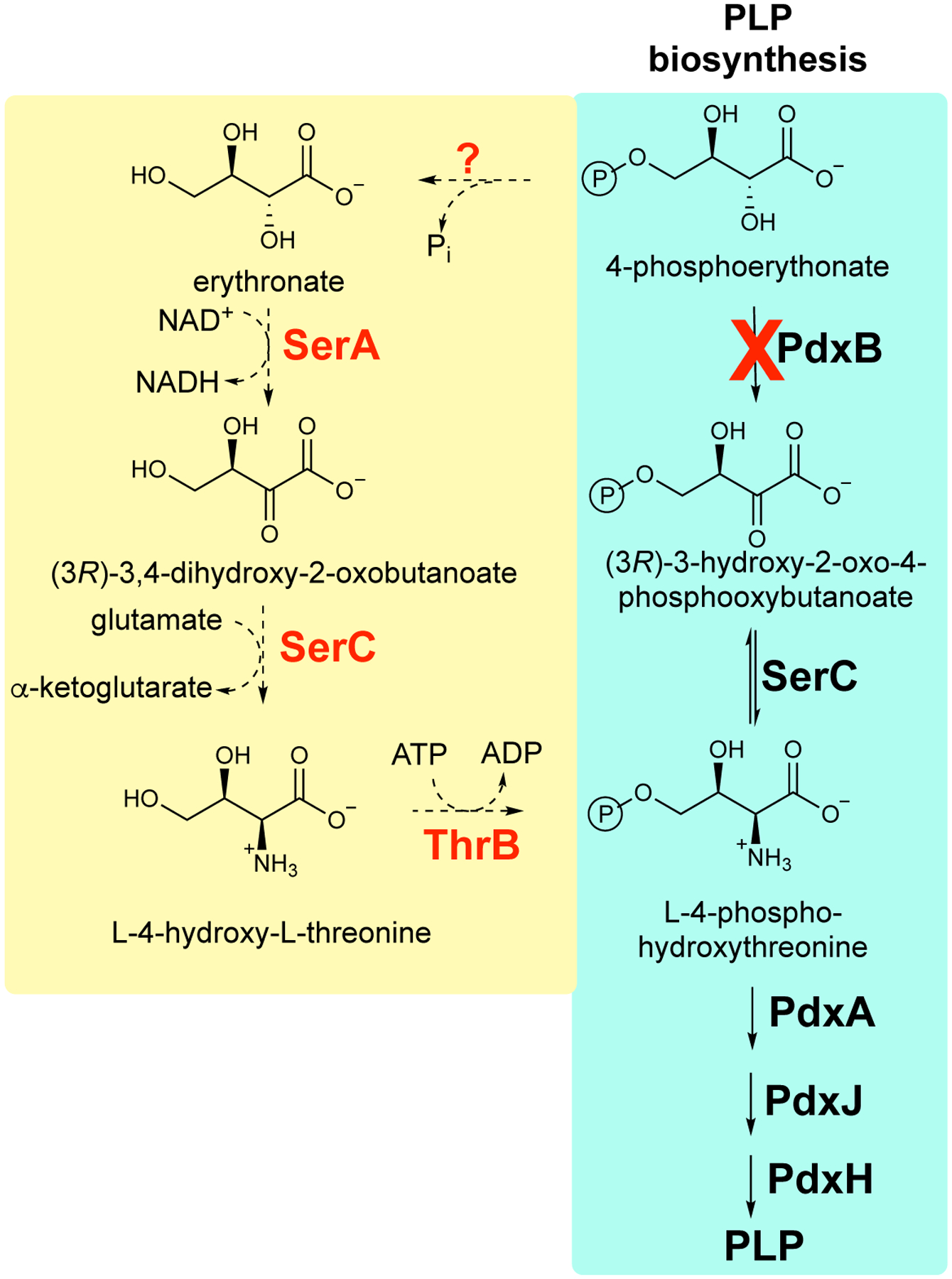
A novel pathway patched together from promiscuous enzyme activities restores synthesis of PLP in ΔpdxB E. coli by bypassing the block in the pathway. Promiscuous enzymes that normally serve other functions are highlighted in red. SerA, 3-phosphoglycerate dehydrogenase; SerC, phosphoserine/phosphohydroxythreonine aminotransferase; ThrB, homoserine kinase.
The examples described above demonstrate that neofunctionalization can occur by several mechanisms, some of which do not require a mutation in the gene encoding a promiscuous enzyme. In the case of an environmental change, neofunctionalization may not require a mutation at all. Subsequent events depend upon whether a bifunctional or generalist enzyme is sufficient, or whether evolution of a new specialist enzyme is required to improve fitness. A generalist enzyme may be ideal, for example, for catalyzing detoxification reactions. A broad-specificity enzyme that can detoxify multiple toxins may be preferable to a large suite of specialized enzymes that require greater investment of resources and may be too specific to handle previously unencountered toxins. On the other hand, duplication and divergence is preferable for when there is an adaptive conflict between two functions, such as inhibition of flux through one pathway due to a competing substrate, or to a need to independently regulate levels of two activities in response to environmental conditions.
IAD Step 2: Duplication
Gene duplication is the pre-requisite for evolution of a new specialized enzyme by the IAD mechanism. Duplications occur at a rate of 3.4 × 10−6 per gene/generation in S. cerevisiae [36], 10−7 per gene/generation in C. elegans [37] and 1.3 × 10−7 per gene/generation in Drosophila melanogaster [36–38]. Importantly, these duplication rates are orders of magnitude higher than the rates of point mutations [36–38], so are often the most facile way to increase the cellular activity of an inefficient enzyme. The steady-state frequency of duplication (which is determined by the ratio of the rate of duplication and the rate at which duplicates are lost) depends strongly on genomic location. In S. enterica, steady-state frequencies of gene duplicates vary between 5.8 × 10−5 and 3.2 × 10−2 per cell at different loci. Thus, about 10% of the population will harbor a duplication someplace in the genome [39].
The most common duplication events [40] are tandem duplications formed by unequal crossing over between daughter chromosomes prior to cell division. This process does not require long stretches of sequence identity, as shown by the examples of junction sequences formed by unequal crossing in Acinetobacter baylyi ADP1 (Figure 7) [41]. Duplications can also form as a consequence of template switching at stalled replication forks, which results in inverted tandem duplications [42]. Finally, duplicates can be deposited in remote locations when a mRNA is reverse-transcribed and the cDNA is integrated at a random site in the genome. Because these genes lack promoters, they are usually non-functional and decay into pseudogenes. However, some retrogenes have acquired new functions due to altered gene expression, localization, or fusion with other genes at the site of integration [43].
Figure 7.
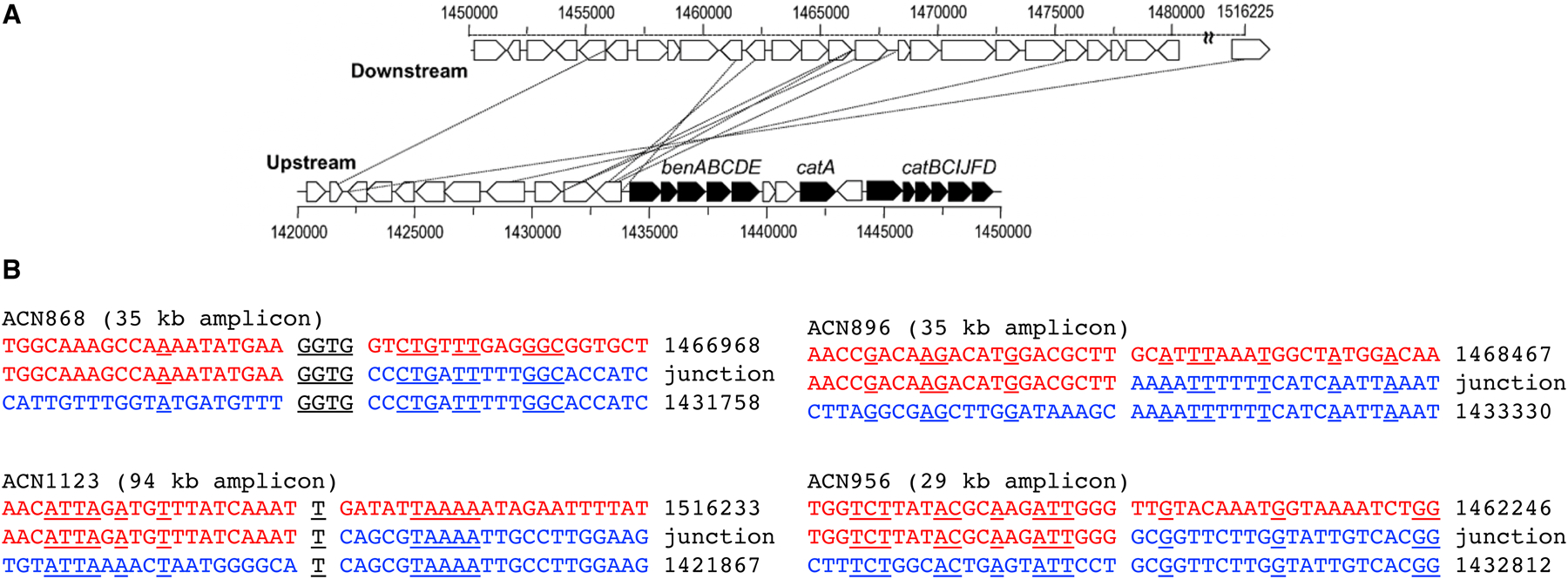
DNA sequences involved in recombination during duplication of regions surrounding the ben and cat genes in A. baylyi ADP1 observed after selection for growth on benzoate (Ben+) or anthraniliate (Ant+). The top line in B is the parental downstream sequence, and the bottom line is the parental upstream sequence. The middle line is the sequence formed at the junction. Identical nucleotides are underlined. Part A reprinted with permission from Mol Microbiol. 83(3):520–35, 2012.
With the exception of duplication via retrotransposition, the events that lead to gene duplication do not respect gene boundaries. Duplicated regions can include partial genes and many genes. Duplicated regions in C. elegans populations grown under selection spanned 1–121 protein-coding genes [44]. A duplicated 92 kb region encompassing 89 genes was identified in a natural isolate of Streptococcus agalactiae [45].
Most segmental duplications are detrimental due to the cost of maintaining extra DNA, transcribing genes, and producing proteins from the duplicated region. Indeed, a comparison of the rate of duplication formation in D. melanogaster with polymorphisms in populations suggests that >99% of duplications are deleterious and are rapidly purged from populations [38]. Protein synthesis is the most substantial cost associated with duplication, accounting for approximately 94 and 98% of the total cost of a gene in E. coli and S. cerevisiae, respectively [46]. Thus, duplication of regions with highly expressed proteins are especially costly. Even small duplications impose significant fitness costs. Growth of E. coli in the presence of the β-lactam antibiotic meropenem resulted in 20–80-fold amplification of 8–16 kb regions surrounding a plasmid-encoded bla gene. When meropenem was omitted from the medium, the adapted strains grew 42–58% more slowly than the parental strain. Each extra kb of DNA reduced fitness by 0.15% [47].
The costs associated with segmental duplication also depend on the functions of the duplicated genes. Overexpression of regulatory kinases can perturb a host of downstream processes; overexpression of DYRK1A (dual-specificity tyrosine phosphorylation-regulated kinase 1A) caused by trisomy 21 in humans perturbs expression of 239 genes and activates pathways involved in neurofibrillary degeneration and β-amyloidosis [48]. Significant costs can also arise from perturbations of gene balance. For example, excessive activity of thymidylate synthase, which produces dihydrofolate, relative to that of the downstream enzyme dihydrofolate reductase, results in accumulation of dihydrofolate, which is toxic [49]. Overexpression of proteins that are part of complexes can lead to imbalances in stoichiometry and wasteful destruction of excess subunits.
The long stretches of homologous DNA in direct tandem duplications facilitate further unequal crossing over, leading to generation of daughter cells with either more or fewer copies of the duplicated segment (Figure 8). In the absence of selection, amplification will be even less tolerated than duplication. However, if increasing the dosage of a gene under selection in a duplicated region is beneficial, daughter cells that acquire additional copies will be more fit, up to a point. Consequently, populations rapidly move toward a copy number that balances the benefit of multiple copies of the gene under selection with the cost of maintaining the segmental amplification. Amplification of a β-lactamase gene under selection during growth of E. coli in the presence of meropenem resulted in 20–50 copies within 42 generations [50].
Figure 8.
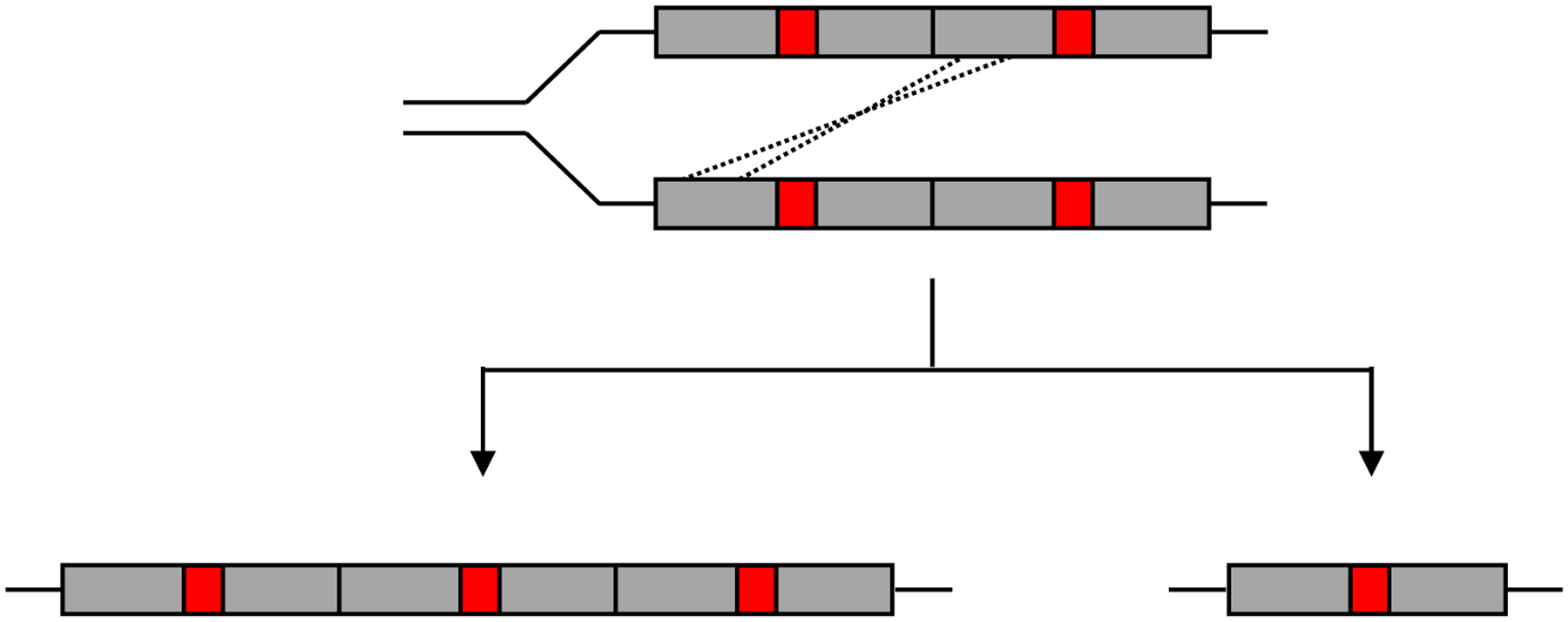
Unequal crossing-over between homologous regions of duplicated segments during genome replication gives rise to daughter cells with either more or fewer copies of the duplicated segment.
When selection is very strong, segmental amplifications can increase the size of the genome substantially. More than 50 copies of a 22-kb segment accumulated in a strain of E. coli in which the inefficiency of a newly recruited promiscuous enzyme limited fitness [51]. The amplified region increased the size of the genome by 24%. Growth of a strain of A. baylyi that lacks transcriptional activators for benzoate degradation on benzoate as a sole carbon source resulted in amplification of the weakly expressed genes for degradation of benzoate and catechol (which is formed from benzoate by the first two steps in the pathway). One evolved strain accumulated 105 copies of a 27 kb segment of DNA, a total of 2.8 Mb of DNA added to a 3.6 Mb genome [41].
The number of copies at steady-state will depend upon the benefit of increasing dosage of the gene under selection and the cost of co-amplifying neighboring genes. This cost might be ameliorated by mechanisms that decrease gene expression or minimize the perturbation of metabolic or regulatory networks caused by gene imbalances. For example, transcription of the Trp operon in bacteria and archaea is inhibited by a repressor that binds to the operator in the presence of tryptophan [52]. A second, more diverse, layer of regulation also monitors the level of tryptophan. In E. coli, translation of a Trp-containing leader peptide in the first cistron of the trp operon results in formation of a transcription terminator when charged tRNATrp is available, thus preventing unnecessary transcription of the downstream Trp synthesis genes. Other mechanisms accomplish the same thing in B. subtilis [53] and Euryarchaea [54]. Thus, the trp operon should be repressed in the presence of adequate tryptophan, even if it is present in multiple copies.
The immediate impact of gene duplication/amplification on mRNA and protein levels is unknown. Previous studies have focused upon yeast strains, cancer cells, and other organisms in which copy number variation has existed for long periods of time, during which compensatory mutations may have occurred. Further, most previous studies have not compared cells that were isogenic except for a copy number variation. Nevertheless, these studies provide some hints about the impact of copy number variation on gene expression. mRNA levels generally increase in proportion to copy number, although with considerable variability. Transcription of 11–36% of amplified genes is lower than expected based on gene copy number in pairs of wild yeast strains that differ in the number of copies of chromosomes 8 or 16 but are otherwise isogenic [55], suggesting the possibility of feedback mechanisms that normalize expression of some genes when copy number is perturbed. On the other hand, increased gene dosage may outstrip the capacity of the normal regulatory processes, leading to excessive transcription. Mutation accumulation lines of C. elegans that were bottlenecked at one individual at each passage show an average of a 3-fold increase in transcription from duplicated genes (Figure 9) [56]. However, the range of values was wide, ranging from 0.7–5, indicating that the story may be different for every gene.
Figure 9.
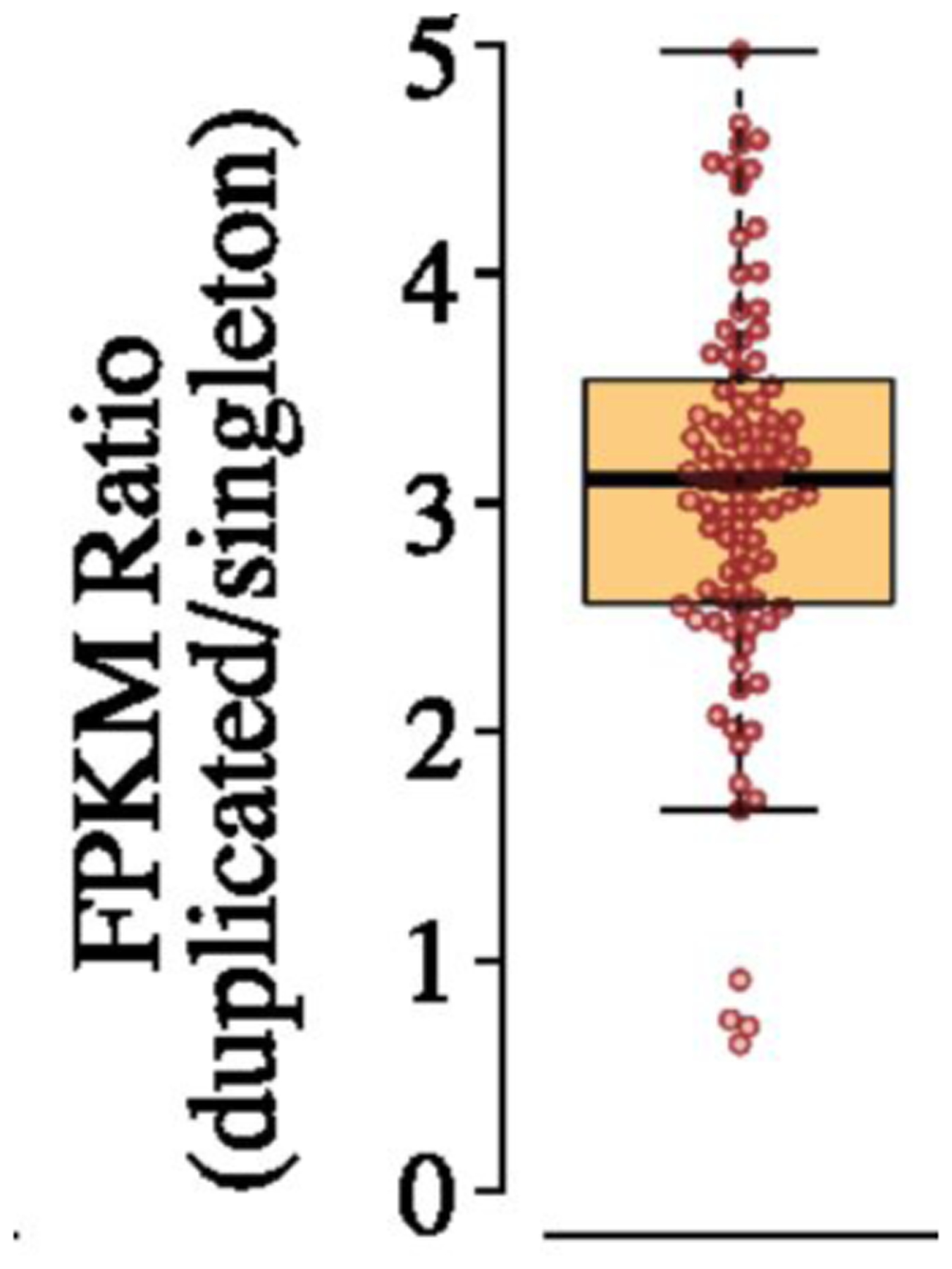
Fold increase in transcription for duplicated genes in 17 mutation-accumulation lines of C. elegans relative to lines in which the genes were not duplicated. FPKM, fragments per kilobase of exon model per million mapped reads. Reprinted with permission from Proc Natl Acad Sci USA 115(28):7386–91, 2018.
Since producing protein is more costly than producing mRNA [46], protein overexpression from an amplified gene is the most critical contribution to the cost of gene duplication/amplification. An analysis of 52 cancer-related proteins in 251 breast cancer specimens examined the relationships between copy number, gene expression and protein expression [57]. Only 8 proteins (Group A) showed correlations between copy number and both gene and protein expression (Figure 10). None of the other 34 proteins showed a correlation between copy number and protein expression, even though some showed a correlation between copy number and gene expression. The relatively constant protein levels could be due to either homeostatic mechanisms or to additional mutations in the cancer cells, which are obviously far from isogenic.
Figure 10.

Correlations between mRNA and protein levels and gene copy number for 52 proteins in 251 breast cancer cell lines. Proteins are divided into groups based on patterns of correlations. Only Group A proteins show significant correlations between copy number and protein levels. Red arrows indicate proteins for which correlations are improved after reducing the effect of samples with little variability in copy number. CN, copy number; GX, gene expression; PX, protein expression. Reprinted from Mol Oncol. 7(3):704–18, 2013. Influence of DNA copy number and mRNA levels on the expression of breast cancer related proteins. Myhre S, Lingjaerde OC, Hennessy BT, Aure MR, Carey MS, Alsner J, et al.. Published under a Creative Commons Attribution (CC BY) License.
It is difficult to extrapolate the effects of long-standing duplications on mRNA and protein expression to the immediate impacts of duplication because selection against amplification of highly expressed regions and possibly mutations that ameliorate overexpression of proteins from amplified segments obscure the picture. Thus, the extent to which the impacts of co-amplification of genes in the neighborhood of a gene under selection can be buffered by homeostatic transcriptional and post-transcriptional mechanisms is unclear. Does it matter? It might. In an extreme case, a problematic neighboring gene might prevent duplication from happening in the first place. Within a population, clones that amplify small regions should have a lesser burden due to co-amplified genes and should therefore attain higher copy numbers and have the greatest chance to acquire beneficial mutations. Clones that acquire a mutation that dampens expression of co-amplified genes may be able to acquire additional copies of the gene under selection and therefore evolve a new enzyme more quickly, but may ultimately have to acquire a reversion or compensatory mutation to restore proper expression. In a microbial community, genes encoding a newly useful activity in different species may occur in different genomic contexts. Species that can amplify a gene under selection while incurring the smallest costs due to co-amplification of neighboring genes may succeed in evolving a new enzyme first and then outcompete other species.
IAD Step 3: Improvement of a newly important activity
Once neofunctionalization and gene duplication have occurred, the stage is set for divergence of a new enzyme. The details of this process - the initial level of the new activity, the mutations that improved it, and the order in which they occurred - have largely been obscured by time. Many paralogs are highly diverged; 80% of paralogs in E. coli are <50% identical [24]. At such low levels of sequence identity, is difficult to distinguish between substitutions that enhanced a new function and those accumulated by neutral drift. Bioinformatic analyses can often pinpoint active site changes that contributed to a new function, but beneficial changes further from the active site may not be obvious. However, experimental investigations of recently diverged enzymes and reconstructed ancestral proteins as well as efforts to evolve new enzymes in the laboratory have provided important insights into the divergence process.
The level of a newly useful promiscuous activity at the time a new enzyme began to evolve can be assessed if the ancestral enzyme is available. Ancestral enzymes are rarely identifiable in nature, but a fortunate pair of discoveries revealed a striking relationship between atrazine chlorohydrolase and melamine deaminase (Figure 11). Atrazine chlorohydrolase (AtzA), first identified in a Pseudomonad isolated from atrazine-contaminated soil, is 98% identical to melamine deaminase (TriA) from a Pseudomonad isolated from effluent from a melamine manufacturing plant. Since melamine production began 40 years prior to use of atrazine began in 1958, it is more likely that AtzA evolved from TriA than the other way around, although this cannot be proven. TriA is an efficient melamine deaminase, with a kcat/KM of 20,810 M−1s−1. Its promiscuous activity with atrazine has a kcat/KM of 60 M−1s−1.
Figure 11.
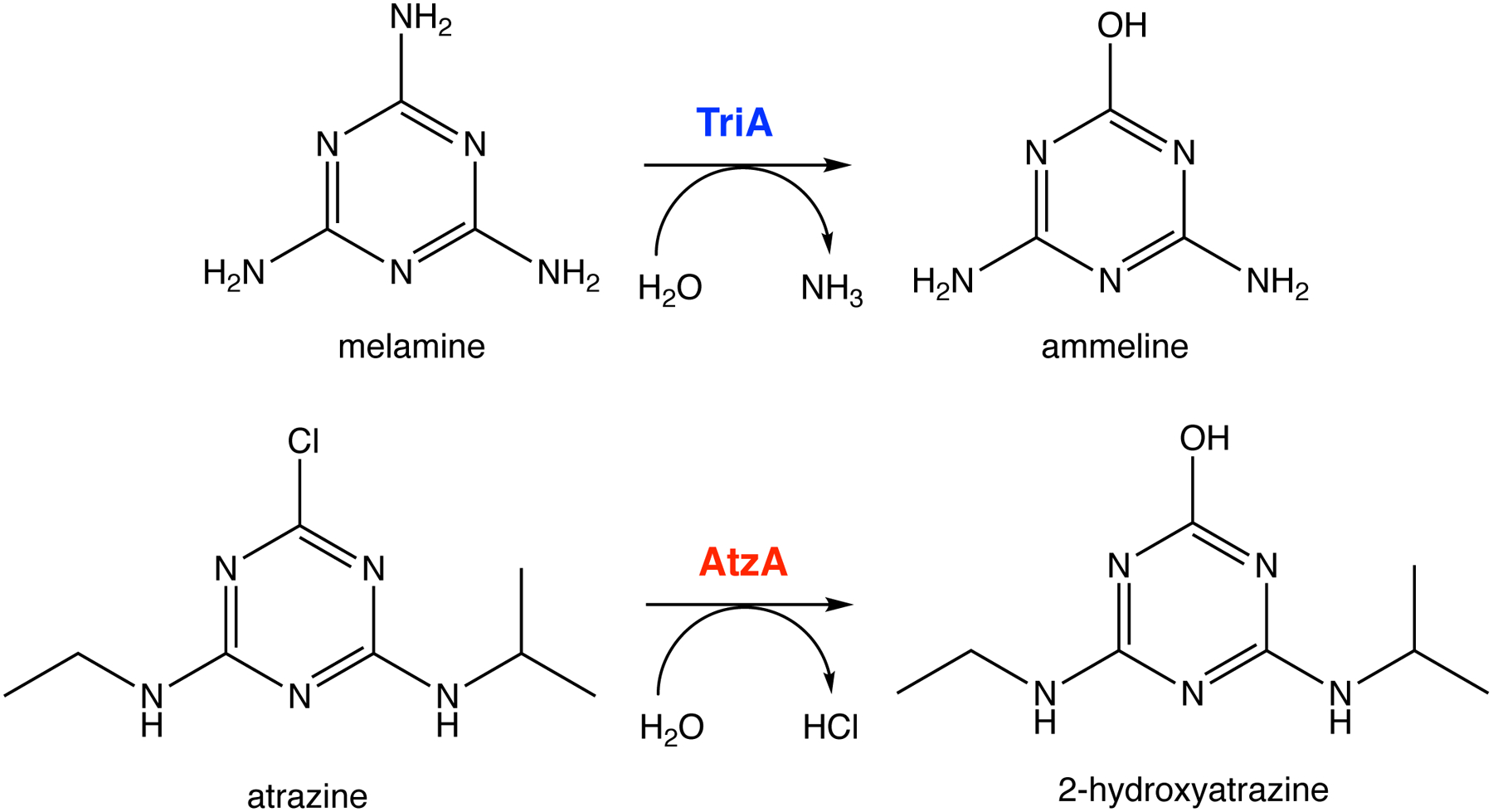
The reactions catalyzed by melamine deaminase (TriA) and atrazine chlorohydrolase (AtzA). AtzA may have evolved from TriA, which has a weak promiscuous activity with atrazine.
Another recently evolved enzyme, methyl parathion hydrolase, allows bacteria in soil contaminated with the insecticide methyl parathion to access a novel source of carbon and phosphate. Phylogenetic analysis suggests that methyl parathion hydrolase evolved from an ancestral dihydrocoumarin hydrolase after the introduction of methyl parathion in the 1950s (Figure 12). The reconstructed ancestor of methyl parathion hydrolase and dihydrocoumarin hydrolase proved to be an efficient dihydrocoumarin hydrolase (kcat/KM = 2.1 × 106 M−1s−1), with an inefficient promiscuous ability to hydrolyze methyl parathion (kcat/KM = 27 M−1s−1) [58]. These examples show that promiscuous activities that are orders of magnitude less efficient than typical metabolic enzymes can serve as the starting point for evolution of new enzymes in nature. In the laboratory, even less efficient promiscuous activities suffice; a promiscuous phenylphosphonatase activity of an arylsulfatase with a kcat/KM of only 0.015 M−1s−1 was improved 105-fold by successive rounds of directed evolution [59].
Figure 12.
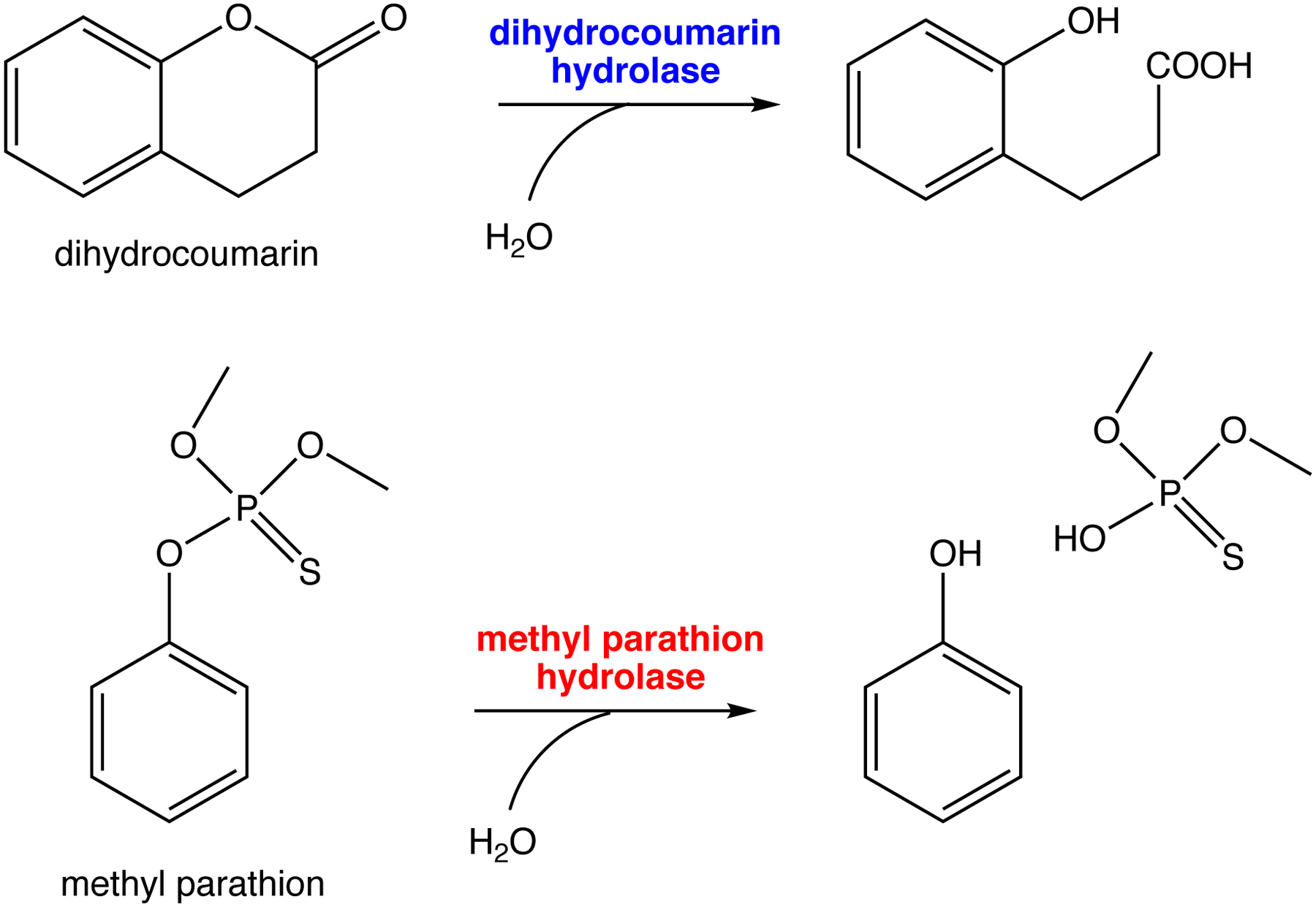
Reactions catalyzed by dihydrocoumarin hydrolase and methyl parathion hydrolase. The reconstructed ancestor of extant dihydrocoumarin hydrolases and methyl parathion hydrolases is an efficient dihydrocoumarin hydrolase with an inefficient promiscuous methylparathion hydrolase activity.
Although most promiscuous activities are inefficient, some are remarkably efficient. For example, a bifunctional alanine racemase/glutamate racemase from Thermotoga maritima has a highly efficient promiscuous cystathionine β-lyase activity with a kcat/KM that is 11- and 1200-fold higher than those for its physiological glutamate racemase and alanine racemase activities, respectively [60]. A reconstructed ancestor of mammalian paraoxonases, detoxification enzymes that are named for their promiscuous paraoxonase activity, had substantial activity with methyl parathion, an anthropogenic pesticide that would not appear for many millions of years [61]. Such efficient promiscuous activities most likely occur with substrates that are not typically encountered by the enzyme, so that there has been no selective pressure to sculpt the active site to exclude a molecule that can compete with the native substrate.
Just a few mutations can improve an initially inefficient activity by an impressive amount. A single point mutation in the active site of muconate lactonizing enzyme II from Pseudomonas sp. P54 improves kcat/KM for a promiscuous o-succinylbenzoate synthase activity by at least 6 orders of magnitude to 1.9 × 103 M−1s−1 [62]. Three amino acid changes in TriA (C331S, D328N and L84F) increase the kcat/KM for atrazine hydrolysis to 12,300 M−1s−1, 84% of that of AtzA [63].
As described above, improving the in vivo efficiency of a newly needed reaction is not just a matter of increasing kcat or decreasing KM for the new substrate. If the native substrate is present, natural selection should result in a strong tradeoff between the native and new activities, as mutations that increase KM for the native substrate will increase the velocity of the new reaction (Eq. 1).
Although substantial improvements can be achieved with a few mutations, optimization of a new activity is often more difficult. Atrazine chlorohydrolase has a kcat/KM of 1.5 × 104 M−1s−1 [63]. For comparison, the median kcat/KM for 1942 enzymes is 1.3 × 105 M−1s−1 [64]. The KM for atrazine is high, exceeding the solubility limit of atrazine, and molecular docking suggests that binding of atrazine forces amino acid side chains in the active site into strained conformers, and that non-productive binding modes are common. Four mutations identified by site-saturation mutagenesis of active site residues improved kcat/KM by 20-fold [65], so there is room for improvement [65]. The efficiency of atrazine chlorohydrolase may be modest because there has been insufficient time for a highly specific and efficient enzyme to evolve. Alternatively, selective pressure for improvement may have ceased because its catalytic performance is good enough that further improvement would not provide any additional fitness benefit to the bacterium.
The finding that optimization is difficult is not limited to natural evolution of new enzymes. Efforts to evolve new enzymes by directed evolution commonly encounter a pattern of diminishing returns in which initial mutations have large effects on efficiency, but later mutations make progressively smaller contributions [59, 66, 67]. However, multiple rounds of directed evolution sometimes succeed in evolving enzymes with catalytic efficiencies comparable to well-evolved enzymes. For example, six rounds of directed evolution including both error-prone PCR and DNA shuffling produced a paraoxonase from a promiscuous N-acylhomoserine lactonase with a kcat/KM of 1.1 × 105 M−1s−1 [67].
Beneficial mutations often remodel active sites so that new substrates are better positioned to interact with the active site machinery. This can be achieved by altering the substrate binding pocket, the positions of active site residues, or both. For example, a promiscuous phosphotriesterase activity of Bacillus thuringiensis N-acylhomoserine lactonase AiiA was improved 1000-fold by directed evolution [67]. Replacement of Val60 and Phe64 at the periphery of the active site with smaller residues allowed Phe68 to swing down into the active site (Figure 13). Molecular dynamics simulations suggest that Phe68 stacks upon the p-nitrophenyl group of the paraoxon, improving its orientation relative to the attacking water. Similarly, a 100,000-fold improvement of a promiscuous phenylphosphonate hydrolase activity of an aryl sulfatase was achieved by directed evolution, primarily due to two mutations that enlarged the active site while leaving the positions of five catalytic residues unchanged [59]. Molecular dynamics simulations suggest that the substrate in the evolved enzyme binds closer to the active site nucleophile and a Lys that stabilizes the developing negative charge on the leaving group, and is better positioned for attack by the nucleophile.
Figure 13.
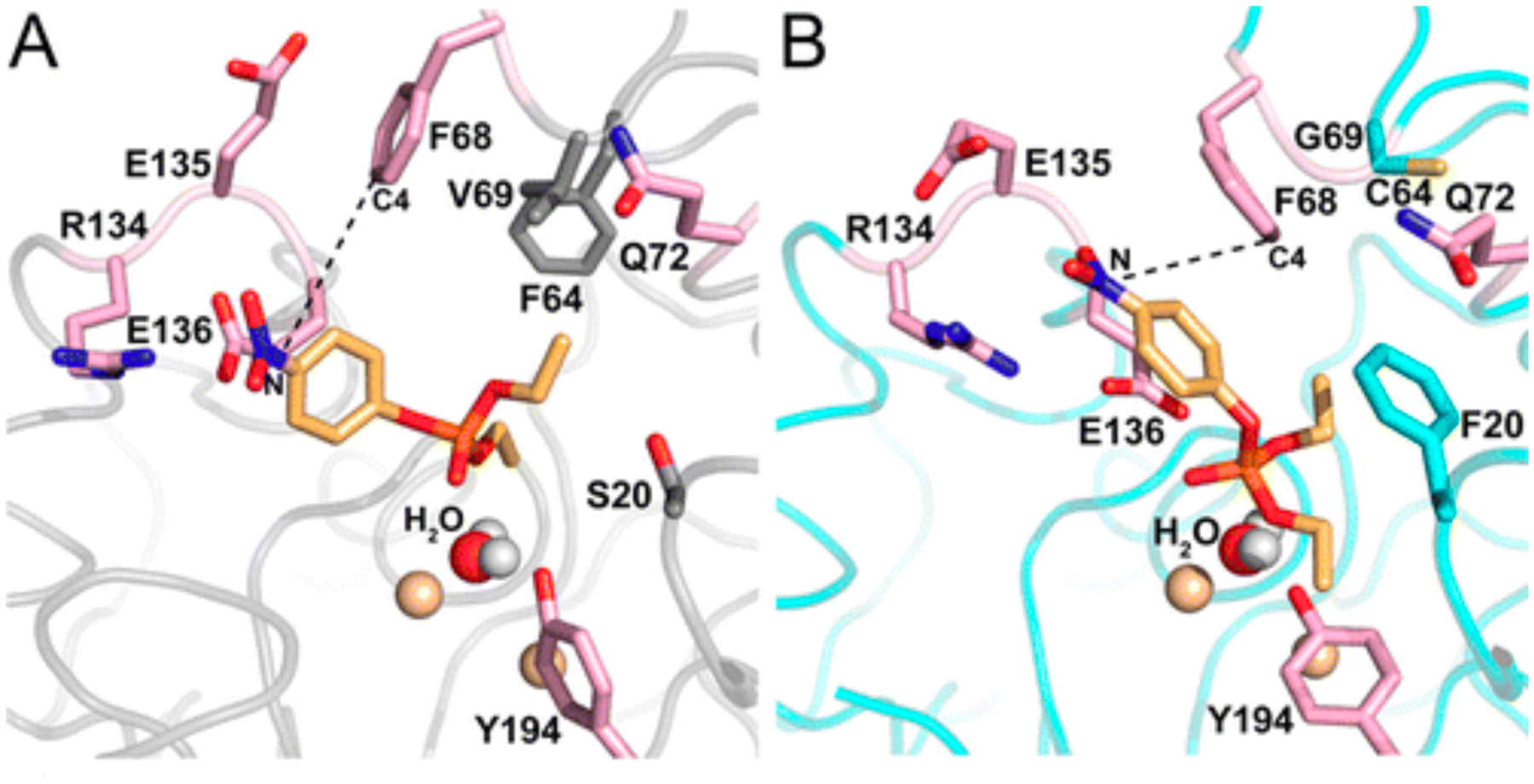
Paraoxon docked into the active site of wild-type AiiA (left) and a mutant enzyme with six mutations, three of which (S20F, V69G and F64C) reshape the active site. The position of the nucleophilic water and the two metal ions to which it is coordinated (gold spheres) are unchanged. In the wild-type enzyme, the substrate is not positioned correctly for in-line attack or water. Repositioning of Phe68 orients the substrate more appropriately. Reprinted with permission from Biochemistry 55(32):4583–93, 2106. Copyright 2016 American Chemical Society.
Beneficial mutations can also introduce new catalytic groups. In mechanistically diverse superfamilies, some of the catalytic machinery of the ancestor has been retained through cycles of gene duplication and divergence, but mechanisms have been diversified by changes in catalytic residues in the active site. For example, enzymes in the enolase superfamily share an ancestral ability to stabilize an enolate using an active site Mg++. Three highly divergent and functionally diverse families within the superfamily (Figure 14A) have retained that ancestral catalytic capability, but the fate of the enolate intermediate is determined by the nature of residues that protrude into the active site from the loops surrounding the active site (Figure 14B) [68].
Figure 14.
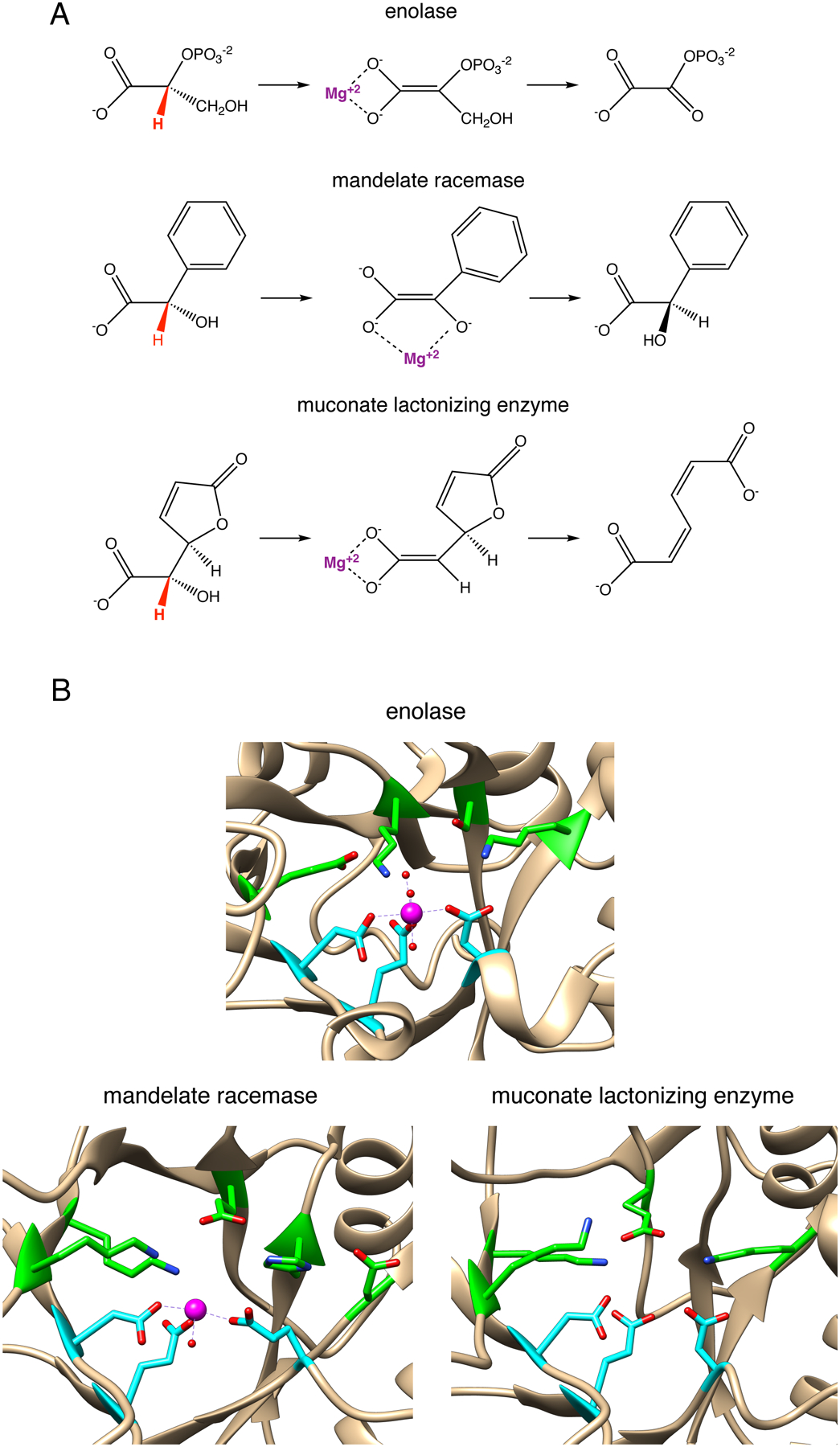
A) Reactions catalyzed by members of the enolase superfamily. B) Active sites of enzymes in the enolase superfamily. The three residues that coordinate the active site Mg++ are highlighted in cyan. Catalytic residues are highlighted in green. Molecular graphics analysis was performed with UCSF Chimera v. 1.13, developed by the Resource for Biocomputing, Visualization, and Informatics at the University of California, San Francisco, with support from NIH P41-GM103311 [92].
Mutations can also improve a promiscuous activity by altering protein dynamics and skewing the ensemble of active site conformations toward those that most effectively catalyze the new reaction. The experimental evolution of TrpF activity in HisA illustrates the importance of optimizing loop motions. HisA and TrpF catalyze Amadori rearrangements in the pathways for synthesis of histidine and tryptophan, respectively (Figure 15). HisA is a typical (βα)8 barrel in which loops at the catalytic face contribute to substrate binding and catalysis. Trp145 in loop 5 stacks with the carboxamide aminoimidazole moiety of the substrate for HisA, N′-[(5′-phosphoribosyl(formimino]-5-aminoimidazole-4-carboxamide ribonucleotide (ProFAR) (Figure 15B) [69]. An enzyme capable of inefficiently catalyzing both reactions was generated from HisA by duplicating codons 13–15 and changing Asp10 to Gly [70]. Duplication of codons 13–15 introduces an additional Val-Val-Arg into loop 1. The duplicated Arg (Arg15[c]) extends into the active site and is predicted to stack with the aromatic ring of the TrpF substrate, phosphoribosyl anthranilate (PRA) (Figure 15C). The extended loop 1 prevents loop 5 from adopting the conformation needed to bind ProFAR, so the HisA(dup13–15) enzyme lacks HisA activity. The D10G mutation restores some HisA activity by increasing the flexibility of loop 1, allowing the enzyme to adopt two conformations, one in which loop 5 interacts with the HisA substrate and one in which loop 1 interacts with the TrpF substrate [71].
Figure 15.
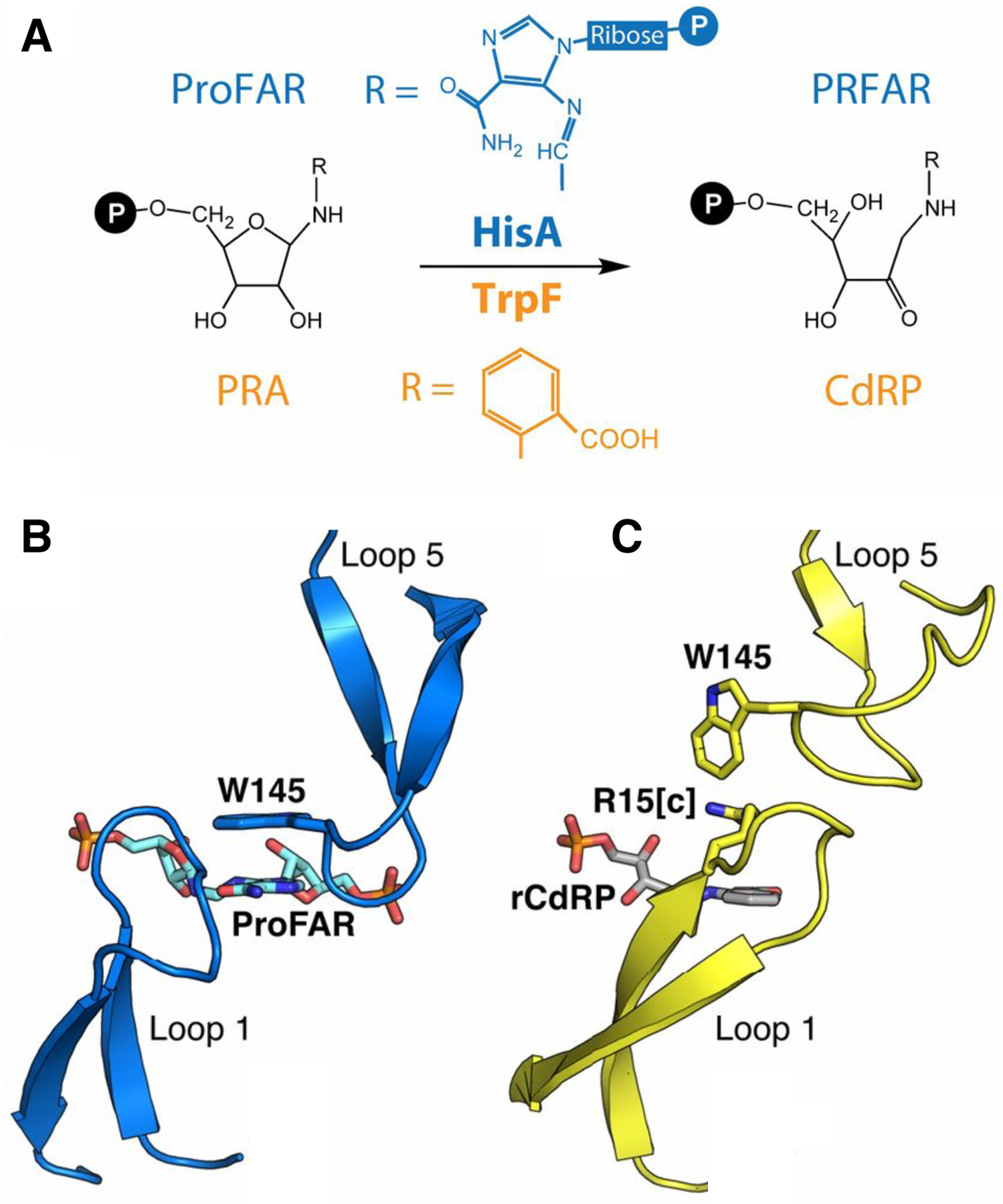
A) HisA and TrpF catalyze Amadori rearrangements of structurally different substrates. B) The ProFAR substrate in the active site of the catalytically inactive D7N D176A HisA (PDB 5A5W). C) A TrpF product analog, rCdRP (reduced 1′-(2′-carboxyphenylamino)-1′-deoxyribulose 5′-phosphate), positioned in the active site of HisA(D7N/dup13–15/D10G) based upon its position in the active site of the ortholog PriA (PDB 2Y85). Reprinted with permission from Proc Natl Acad Sci USA 114(18):4727–32, 2017. Structural and functional innovations in the real-time evolution of new (betaalpha)8 barrel enzymes. Newton MS, Guo X, Soderholm A, Nasvall J, Lundstrom P, Andersson DI, et al.
Näsvall et al. evolved several lineages of Salmonella enterica in which the inefficient bifunctional HisA (dup13–15/D10G) supported synthesis of both histidine and tryptophan for 3000 generations [70]. The gene encoding the enzyme amplified and then either diverged to encode two specialist enzymes or evolved toward a more efficient generalist enzyme. The best TrpF specialist acquired three additional mutations and reached a kcat/KM of 1.8 × 103 M−1s−1. (For comparison, kcat/KM for wild-type E. coli TrpF is 6.8 × 106 M−1s−1 [72].) Two mutations – Q24L and V15bM – appear to promote TrpF activity by stabilizing loop 1 in a conformation favoring TrpF activity.
Beneficial mutations need not occur in the active site. Mutations in the second or third shell around the active site can subtly alter the shape of the active site or influence side chain or loop motions to favor a new reaction. Beneficial mutations distant from the active site are commonly found during both natural evolution and directed evolution [73]. Three of the nine amino acids that confer atrazine chlorohydrolase activity on the melamine deaminase TriA are in the second or third shell around the substrate access channel and active site [74]. Even more strikingly, only one of 17 amino acid changes accumulated during directed evolution of E. coli aspartate aminotransferase toward improved activity with branched chain amino acids involves a residue in direct contact with the substrate [75]. A global look at the impact of mutations outside the active site is provided by the effects of 6500 non-synonymous mutations in amiE, which encodes an aliphatic amidase, on growth of E. coli when an aliphatic amide provides the only nitrogen source. AmiE prefers small amide substrates and turns over the bulky isobutyramide poorly. Wrenbeck et al. found 395 mutations (many involving different substitutions at the same position) that improved specificity for isobutyramide, but did not affect activity with acetamide and propionamide [76]. A change of Trp138 in the active site to either Ala or Gly created room for the larger substrate. Most of the other beneficial substitutions, however, were ≥ 9Å away from the active site, and distributed throughout the protein (Figure 16).
Figure 16.
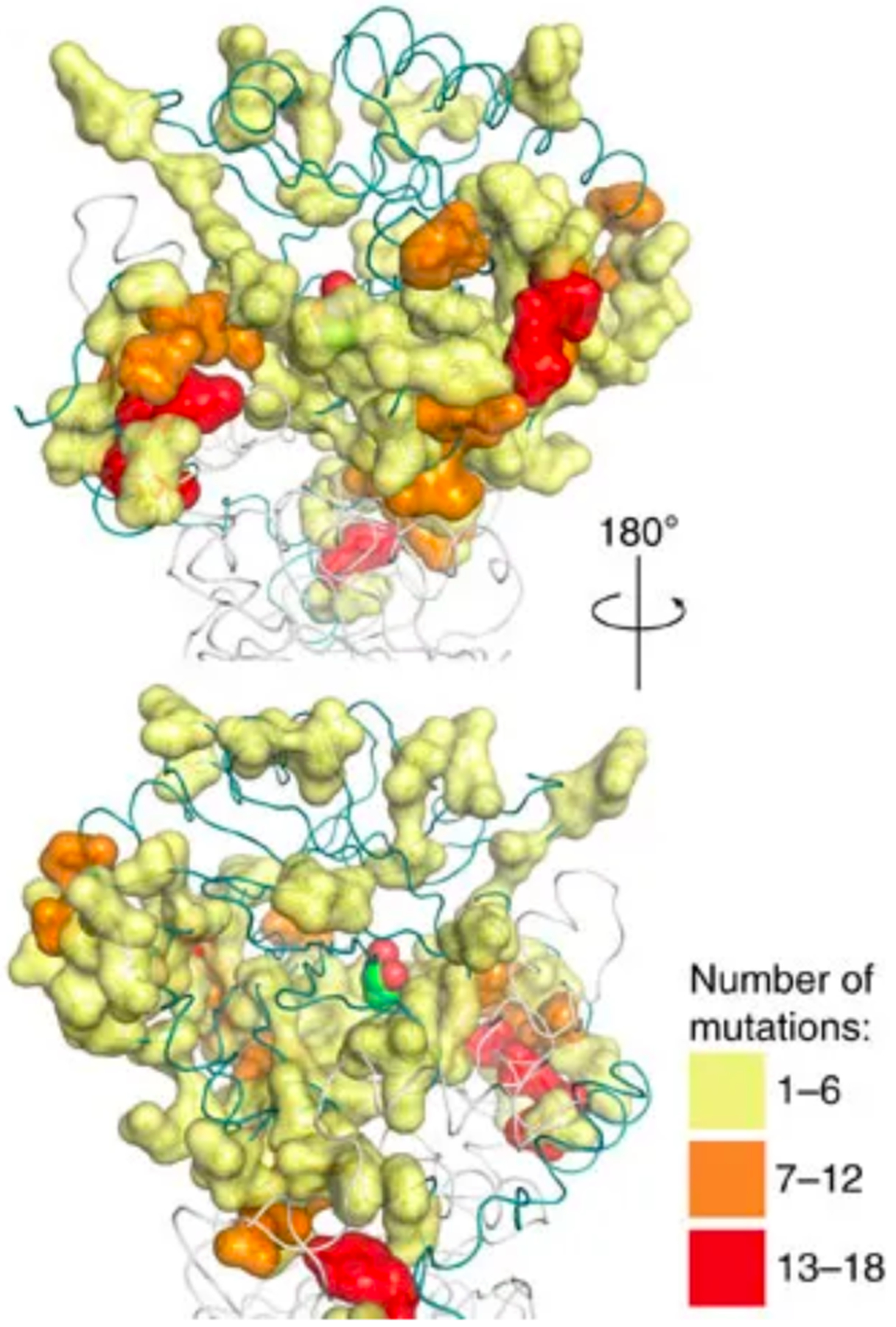
Sites in the P. aeruginosa aliphatic amidase AmiE at which mutations improved growth of E. coli in the presence of isobutyramide as a sole nitrogen source. The active site is marked by a space-filled ligand covalently attached to the active site Cys166. Colors indicate the number of substitutions that were found to be favorable at each position in the protein. Reproduced from Nat Commun. 8:15695, 2017 under a Creative Commons license (http://creativecommons.org/licenses/by/4.0/).
While the order in which mutations improved a new activity during past episodes of divergence after gene duplication/amplification cannot be discerned, several laboratory studies have recapitulated possible trajectories from ancestral to derived enzymes. Weinreich et al. generated all possible intermediates between an ancestral TEM β-lactamase that is specific for penicillin and a naturally evolved enzyme that improves resistance to cefotaxime, a third-generation cephalosporin, by 100,000-fold [77]. The gene encoding the evolved enzyme had four missense mutations and a non-coding mutation upstream of the coding sequence. Of the 120 possible trajectories toward the evolved enzyme, only 18 avoided a step in which resistance decreased due to pervasive sign epistasis; four of the five mutations were neutral or detrimental in some allelic backgrounds.
A similar finding emerged from an examination of trajectories for the evolution of Pseudomonas sp. WBC-3 methyl parathion hydrolase from a dihydrocoumarin hydrolase described above. Many of the 32 differences between the derived and ancestral enzymes likely arose by neutral drift. However, five changes in the active site are clearly important, as their reversion to the predicted ancestral state diminished methyl parathion hydrolase activity by 900-fold and restored most of the dihydrocoumarin hydrolase activity. Yang et al. constructed the 32 possible intermediates between methyl parathion hydrolase and a progenitor in which the ancestral residues had been restored at the five critical positions. Figure 17 shows that only 19 of the possible 120 trajectories avoid a step in which activity decreases [58].
Figure 17.
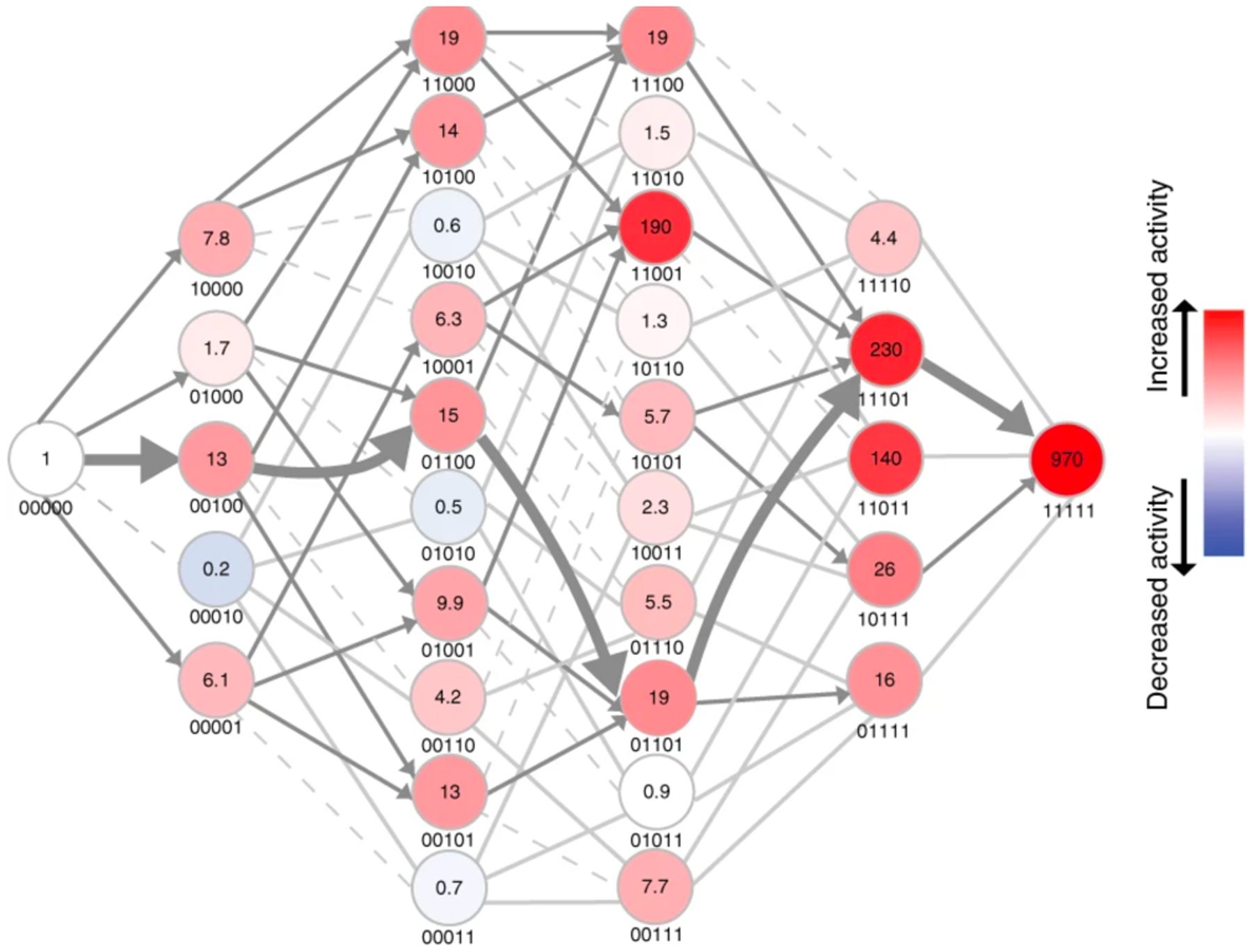
The adaptive landscape between methyl parathion hydrolase (right) and an ancestor in which five key residues had been reverted to the ancestral state (left). The numbers under each node indicate the absence (0) or presence (1) of the derived residue at positions 73, 193, 258, 271 and 273. Colors indicate the mean methyl parathion hydrolase activity in lysates for three biological replicates. Dashed light grey lines indicate paths that are evolutionarily inaccessible because they go through an intermediate with decreased activity. Grey arrows indicate steps in which activity is increased. Reprinted by permission from Springer Nature: Nat Chem Biol. 15(11):1120–8, 2019. Higher-order epistasis shapes the fitness landscape of a xenobiotic-degrading enzyme, Yang G, Anderson DW, Baier F, Dohmen E, Hong N, Carr PD, et al. Copyright 2019.
Not surprisingly, evolution of a new enzyme often requires tradeoffs between the ancestral and new activities as the active site changes to accommodate a new substrate. For example, three mutations that enhance TrpF activity in the inefficient bifunctional HisA/TrpF (HisA(dup13–15/D10G)) enzyme described above by 35-fold diminish the HisA activity to an undetectable level (Figure 18A) [71]. The strong trade-off between HisA and TrpF activities minimizes inhibition of the evolving TrpF activity by the native substrate for HisA in cells in which both substrates are present. On the other hand, promiscuous activities can sometimes be substantially improved without significantly impairing the native activity. Mutations that improved the promiscuous paraoxonase activity of an N-acylhomoserine lactonase by 1000-fold decreased the native activity by only 3-fold (Figure 18B) [67]. In this case, the native substrate was not present, and improvement of the promiscuous activity did not require mutations that minimize binding of the native substrate. Weak trade-offs have also been observed in other directed evolution experiments in which native substrates were absent [78].
Figure 18.
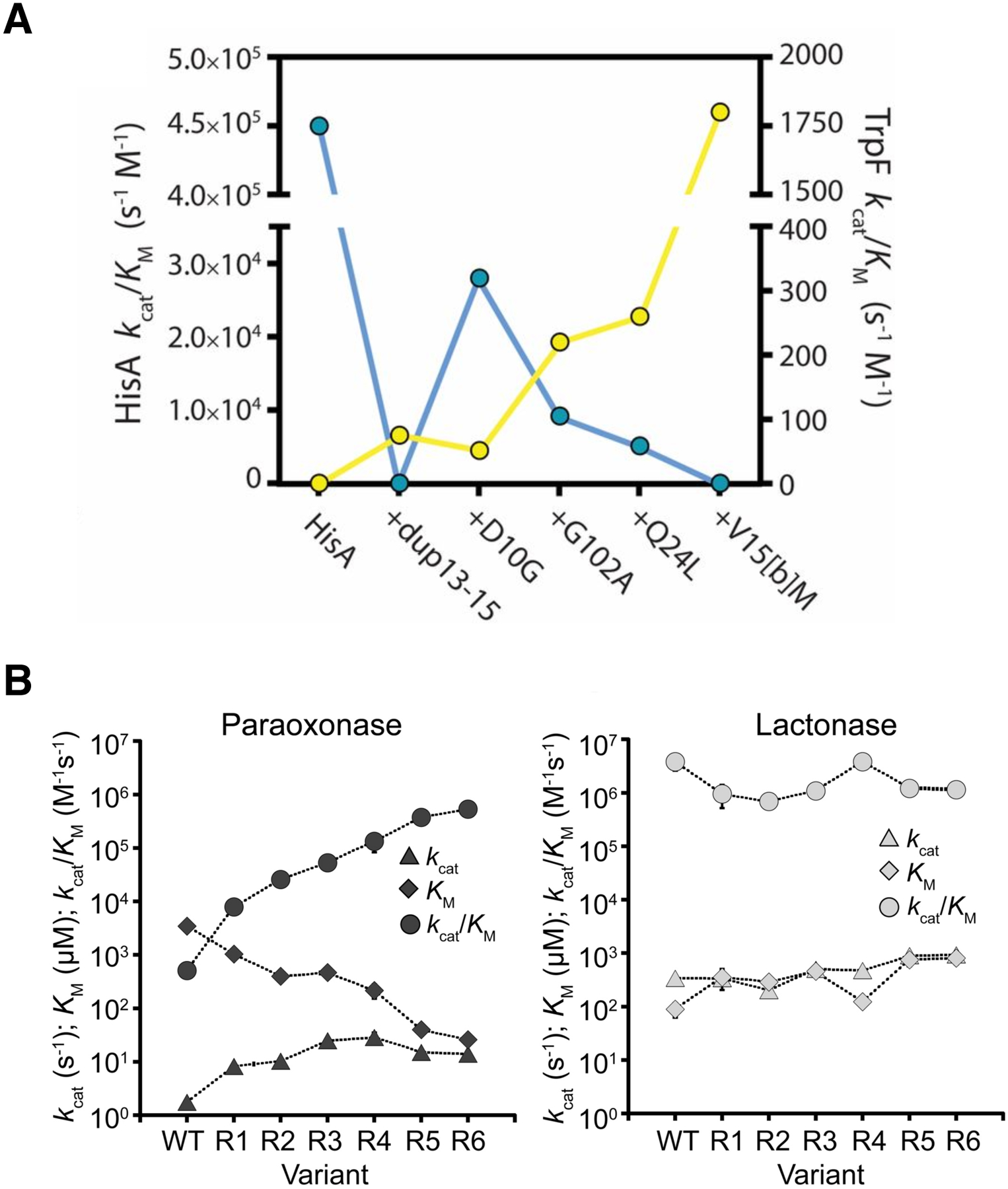
Tradeoffs between the original and new activities of an evolving enzyme. A) A strong tradeoff between TrpF (yellow) and HisA (blue) activities in HisA. An allele encoding HisA(dup13–15/D10G) evolved into a specialist TrpF in S. enterica in which this bifunctional enzyme supported synthesis of both histidine and tryptophan, but poorly. Reprinted with permission from Proc Natl Acad Sci USA 114(18):4727–32, 2017. Newton MS, Guo X, Soderholm A, Nasvall J, Lundstrom P, Andersson DI, et al. Structural and functional innovations in the real-time evolution of new (betaalpha)8 barrel enzymes. B) A weak tradeoff between the original homoserine lactonase activity and the promiscuous paraoxonase activity of AiiA through six rounds of directed evolution. Reprinted with permission from Biochemistry 55:4583–4593, 2016. Yang G, Hong N, Baier F, Jackson CJ, Tokuriki N. Conformational tinkering drives evolution of a promiscuous activity through indirect mutational effects. Copyright 2016 American Chemical Society.
Finally, it is important to note that evolution of a new enzyme is not only about improving catalytic function. Evolution of new transcriptional regulation is often required, as well, as the transcriptional regulation of the ancestral enzyme will often be inappropriate for the diverged enzyme. Indeed, transcription of paralogs is generally substantially different. For example, regulation of the paralogs maltodextrin phosphorylase and glycogen phosphorylase in E. coli is completely different, as expected given their roles in degradation of different polysaccharides (Figure 19). New regulatory mechanisms can arise by mutations in a promoter that bring a gene under the control of an existing transcriptional regulator, mutations that alter the binding specificity of an existing transcriptional regulator, and/or mutations that change the ligand specificity of a transcriptional regulator. Such mutations can occur before gene duplication, and indeed may be required to allow sufficient enzyme to be expressed and start the process of evolution of a specialized enzyme. Alternatively, they can occur during or after gene duplication and divergence.
Figure 19.
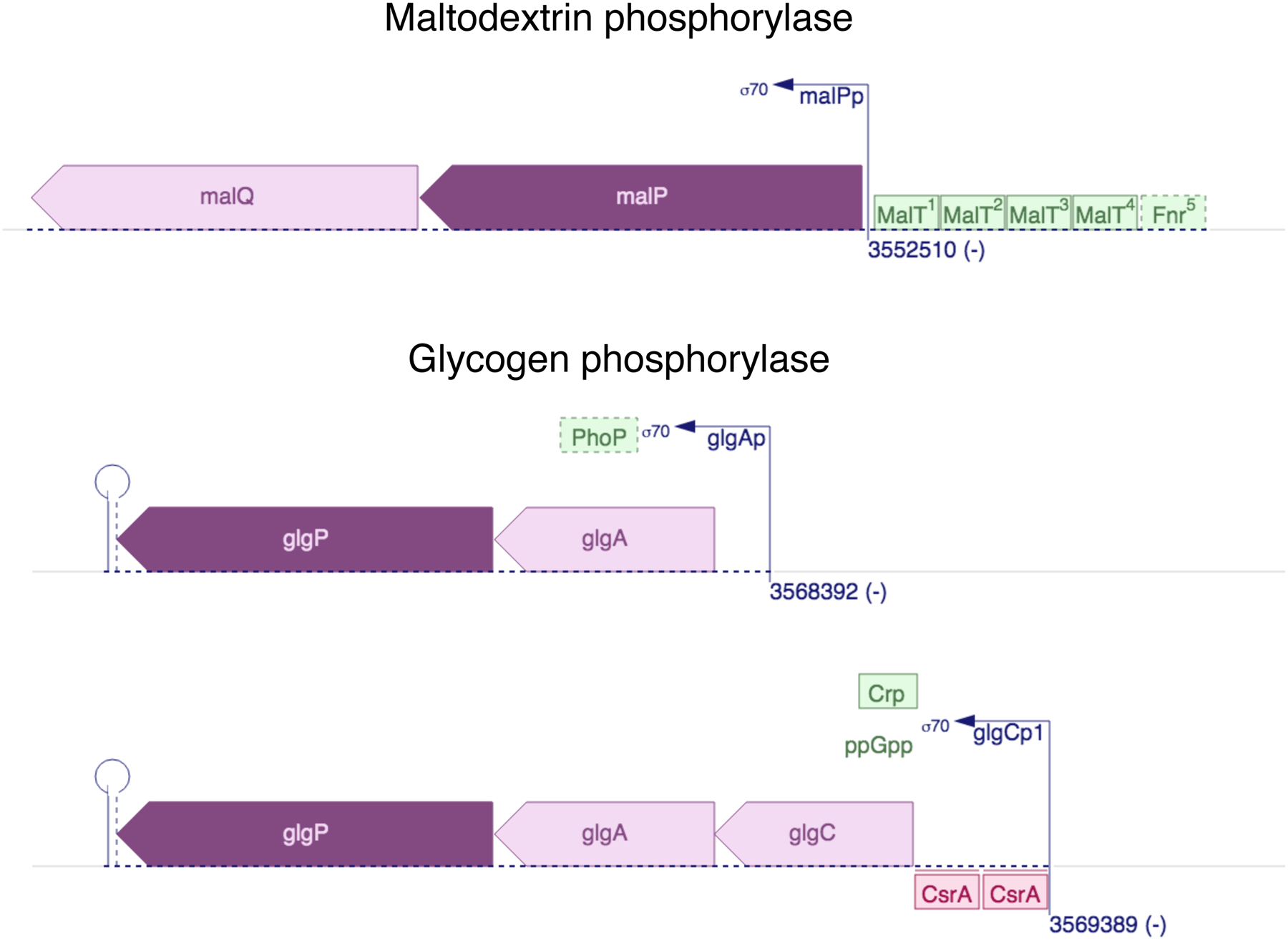
Divergent regulation of genes encoding E. coli maltodextrin phosphorylase (malP) and glycogen phosphorylase (glpG), paralogs with 48% sequence identity. glpG is transcribed from two different promoters. Green and red boxes indicate proteins that activate and repress transcription, respectively. The locations of the binding sites for the glycogen phosphorylase promoters are not known. Diagrams from Ecocyc Version 23.5 [93].
IAD Step 4: Deamplification and genome remodeling
As the function of an evolving enzyme improves, fewer gene copies are needed to maintain fitness, and deamplification can improve fitness by decreasing the metabolic burden associated with co-amplified genes. Deamplification can occur in stages. For example, the gene encoding E383A ProA (ProA*), which has a weak N-acetylglutamyl phosphate reductase activity (Figure 5), amplified rapidly during evolution of E. coli lacking N-acetylglutamyl phosphate reductase (ArgC) on glucose + proline [79]. (Proline was added so that ProA* was free to evolve toward a neo-ArgC in the absence of a requirement for its original function.) Figure 20 shows amplification and later deamplification of proA* in one evolved lineage. The initial increase in growth rate was due to a mutation that corrects the known pyrimidine synthesis deficiency in K12 strains of E. coli [79–81]. Following this mutation, a 41 kb region surrounding proA* amplified to six copies. Soon thereafter, a mutation that changed Phe372 to Leu near the active site improved the neo-ArgC activity of the enzyme by 4-fold. The copy number of proA** dropped as a consequence, but only to three. Presumably additional mutations are required to create an enzyme that is good enough to allow deamplification to a single copy.
Figure 20.
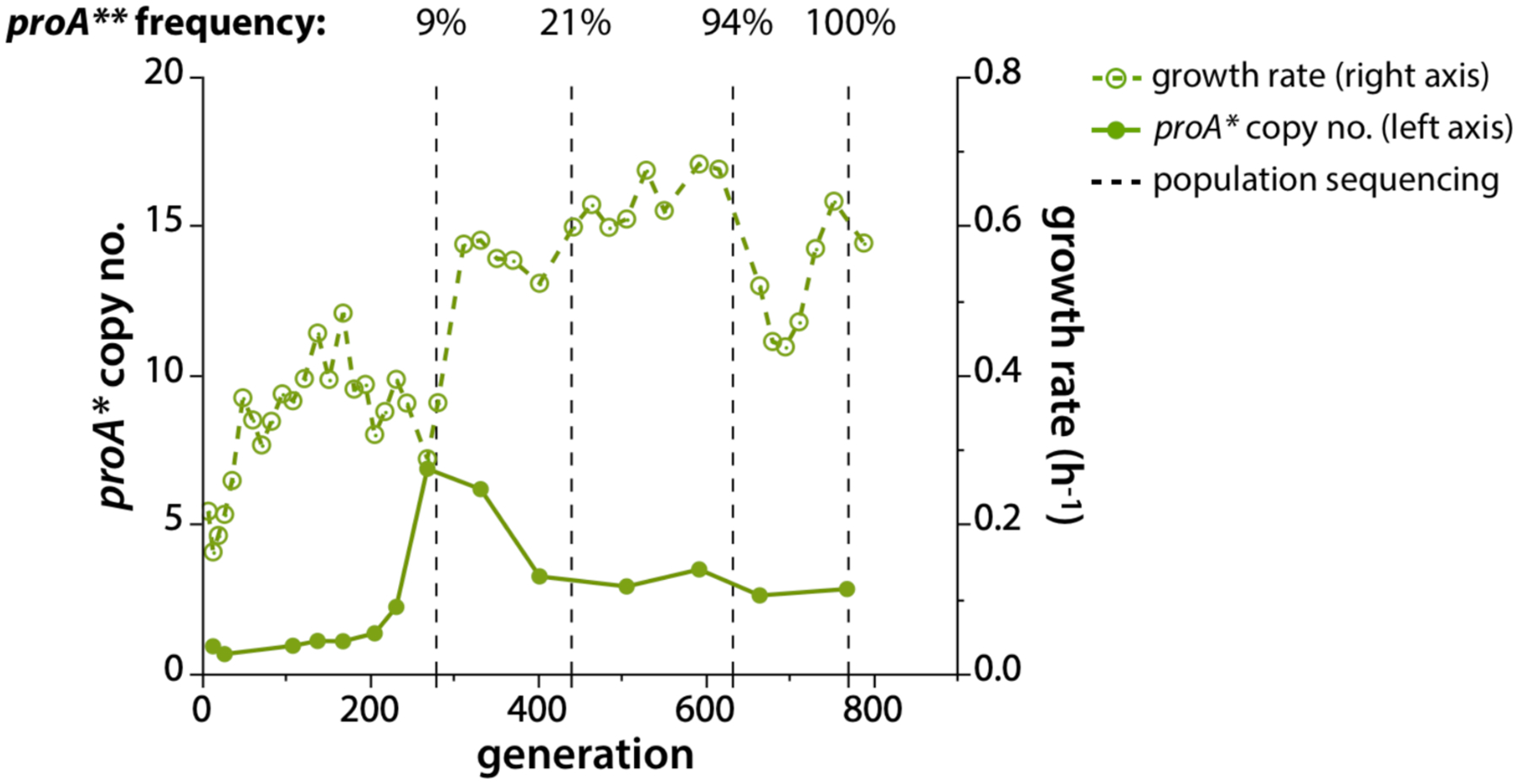
A gene encoding ProA* (E383A ProA), which has weak ArgC activity, amplifies to 6 copies in ΔargC proA* E. coli prior to a mutation that changes Phe372 to Leu. Subsequently, proA**, which encodes E383A F372L ProA, deamplifies to three copies. Reprinted from eLife 8:e53535, 2019: Mutations that improve efficiency of a weak-link enzyme are rare compared to adaptive mutations elsewhere in the genome. Morgenthaler AB, Kinney WR, Ebmeier CC, Walsh CM, Snyder DJ, Cooper VS, et al.
When two specialist enzymes evolve by amplification and divergence, an amplified array can shrink down to two copies. In this situation, the extra copies of co-amplified genes, which impose an unnecessary metabolic burden, need to be eliminated. A glimpse into how this can happen is provided by remodeling of duplicated regions in S. enterica carrying a plasmid encoding a fused lacI-lacZ gene with a leaky +1 frameshift mutation [82]. Growth of these cells on lactose as a sole carbon source selects for amplification of the lac region because leaky expression from multiple copies is required to produce sufficient β-galactosidase. In most colonies, duplication occurred between two identical copies of an IS3 insertion element, resulting in 134 kb repeats. In some of these colonies, subsequent recombination between short repetitive sequences resulted in deletions that spared the lac gene, but eliminated unnecessary DNA, resulting in shorter repeat units. Cells with the shorter repeats were able to amplify the region under selection to a higher degree than cells with the longer repeats and thus had a higher probability of acquiring a reversion mutation that restored the proper reading frame of lacZ. In this case, remodeling occurred prior to mutations that solved the problem of inadequate β-galactosidase expression, but such remodeling could also occur during or after divergence of two paralogs. Figure 21 illustrates how remodeling can begin to eliminate extraneous DNA when only one gene in a duplicated gene array is under selection.
Figure 21.
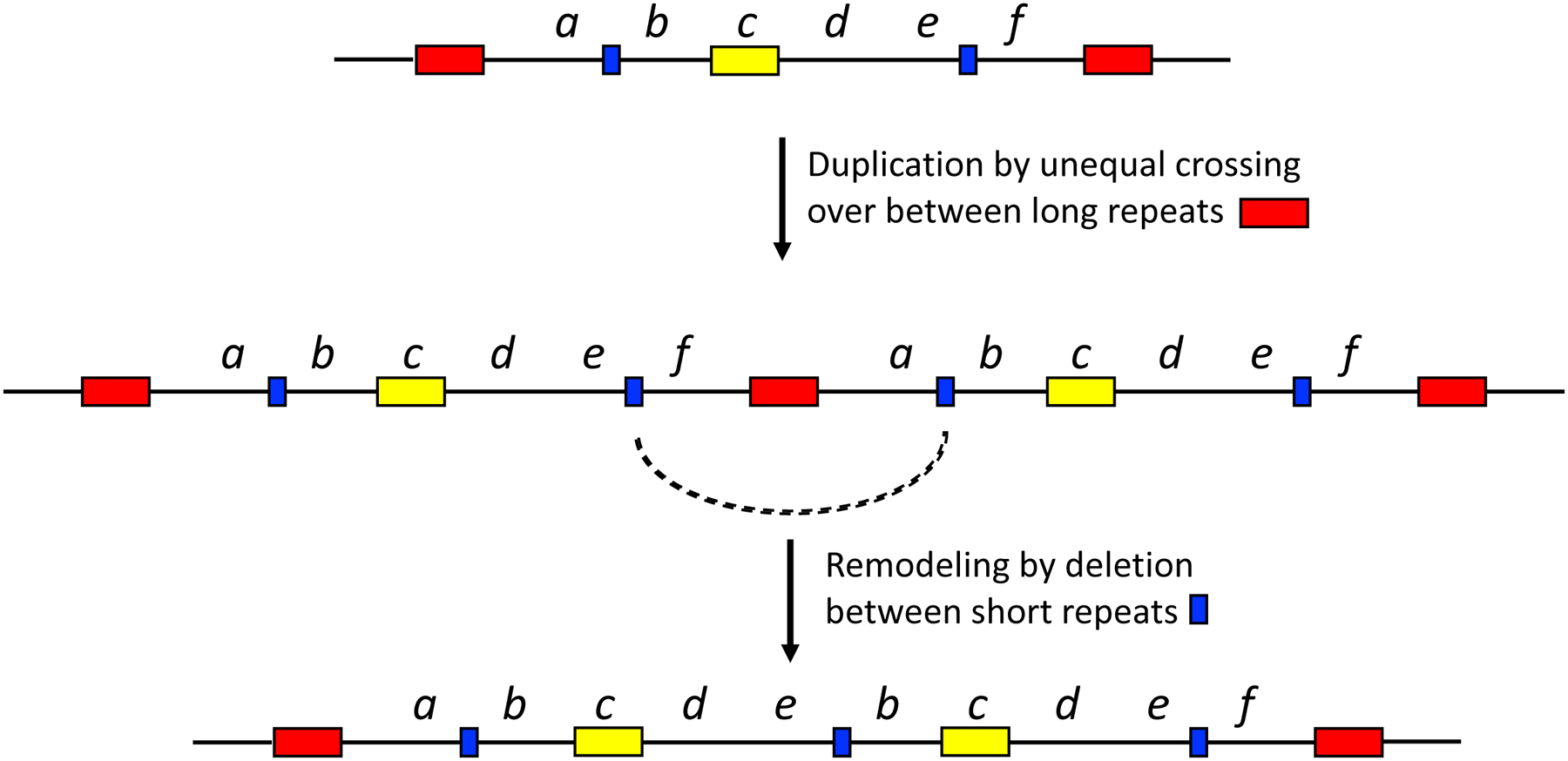
Remodeling within a segmental duplication removes some extraneous DNA. The yellow box indicates the gene under selection.
The type of remodeling described in the previous paragraph is only part of the picture of genome remodeling after paralog divergence. Larger scale genomic rearrangements tend to move paralogous genes apart over time. Paralogous genes in E. coli are nearly always separated (https://www.genome.wisc.edu/pub/expression/paratab.txt). The same is true in eukaryotes. Within chromosomes, duplicate pairs with high levels of sequence identity, which are presumed to be the result of recent duplication events, are closer together than older duplicates in several eukaryotes [40], suggesting a gradual process that moves paralogs apart.
Paralogous genes can also be relocated to other chromosomes. Eleven % of duplicate pairs in C. elegans with KS = 0, indicating very recent duplication, are already found on different chromosomes [83]. (Ks is the number of substitutions per synonymous site. Ks increases with time as synonymous mutations, most of which are silent, accumulate.) Strikingly, 64% of older duplicate pairs (0 < KS < 0.1) are found on different chromosomes. Similarly, in humans, the most recently duplicated pairs in small gene families are found on the same chromosome, but 44% of older duplicate pairs (0.025 < KS < 0.1) are found on different chromosomes [84]. Although these studies did not address whether both genes in duplicated pairs encode functional products, and whether one has acquired a new function, they clearly indicate a tendency for gene duplicates to be separated soon after their formation. The paucity of spatially close paralogous genes suggests that there is selective pressure to separate them, possibly because extensive regions of sequence similarity between recently diverged paralogs would permit recombination during genome replication that could result in gene loss and/or generation of chimeric and probably dysfunctional proteins. The mechanisms that scatter paralogs throughout the genome are unknown.
A curious feature of the human genome is that duplicated genes tend to be found in blocks containing genes derived by duplication of loci in disparate regions of the genome [85]. Over 400 duplication blocks contain 9–541 duplicated segments ranging in size from 5 kb to 4.3 Mb that have ≥90% identity to an ancestral locus elsewhere in the genome. Figure 22 shows, for example, a duplication block on chromosome 2 that contains segments duplicated from elsewhere on chromosome 2 as well as nine other chromosomes. Why and how duplicated genes are collected in these large blocks is not clear.
Figure 22.
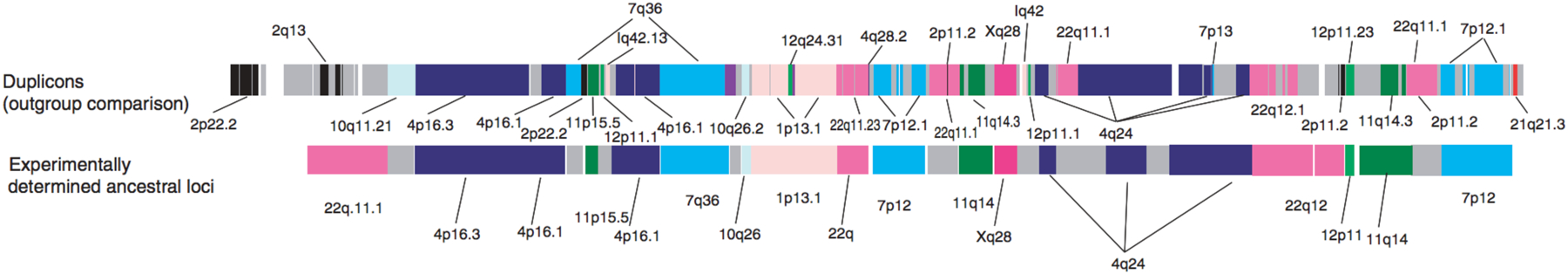
A duplication block in the 2p11 region of human chromosome 2. Each segment is labelled according the location of the presumed ancestral copy. The upper diagram shows computationally predicted ancestral loci of segments in the duplication block, and the lower diagram shows segments whose ancestral loci have been determined experimentally. (Some of the shortest segments have not been experimentally addressed.) Most of the duplicated segments come from other chromosomes. Reprinted by permission from Springer Nature: Nature Genetics, 39(11):1361–8. Ancestral reconstruction of segmental duplications reveals punctuated cores of human genome evolution, Z. Jiang et al. Copyright 2007.
Conclusion
Gene duplication and divergence has occurred throughout the history of life on earth, beginning even before the last universal common ancestor of bacteria, archaea and eukaryotes approximately 3.8 billion years ago. Many episodes of new enzyme evolution happened in the remote past. We can recognize homologous enzymes by sequence and structural similarity, but in most cases bioinformatics can reveal only patterns of active site residues that are required for catalysis of a particular reaction. The order of mutations that led to emergence of a new enzyme is obscured by the extensive sequence divergence that has occurred since the initial gene duplication event.
Great progress toward understanding the evolution of new enzymes by gene duplication and divergence has been made in the past 20 years. High-throughput substrate profiling experiments have opened our eyes to the astonishing extent of promiscuity available in extant enzymes [86–88]. Multiple mechanisms for gene duplication are now recognized [42]. Technological advances in high-throughput sequencing, directed evolution, ancestral sequence reconstruction and structural biology have provided a wealth of information about the impact of mutations on evolving enzyme activities, as well as insights into the accessibility of mutational trajectories between initial promiscuous activities and more efficient evolved enzymes. However, gaps in our understanding remain. There is a dawning recognition that protein dynamics contribute to promiscuity [89] and that mutations can improve new activities by altering protein dynamics, but there are few examples at this point [71, 90]. We know that transcriptional regulation changes as new enzymes evolve, but the interplay of mutations that alter regulation and those that alter enzymatic activity has not been explored experimentally. Finally, we know that extraneous DNA is removed after duplication and divergence and paralogs move apart in the genome, but the mechanisms responsible for this critical stage are not known. There is more to learn.
Acknowledgements
This work was supported by NIH/NIGMS R01GM134044 and NASA Astrobiology Institute NNA15BB04A.
Abbreviations:
- ArgC
N-acetylglutamyl phosphate reductase
- AtzA
atrazine chlorohydrolase
- IAD
innovation-amplification-divergence
- IDH
isocitrate dehydrogenase
- LACA
last archaeal common ancestor
- LBCA
last bacterial common ancestor
- LECA
last eukaryotic common ancestor
- LUCA
last universal common ancestor
- ProA
γ-glutamyl phosphate reductase
- SerA
3-phosphoglycerate dehydrogenase
- TriA
melamine deaminase
- WGD
whole-genome duplication
Footnotes
Conflicts of interest
None.
References
- 1.Kannan L, Li H, Rubinstein B & Mushegian A (2013) Models of gene gain and gene loss for probabilistic reconstruction of gene content in the last universal common ancestor of life, Biol Direct. 8, 32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Zhou Y, Minio A, Massonnet M, Solares E, Lv Y, Beridze T, Cantu D & Gaut BS (2019) The population genetics of structural variants in grapevine domestication, Nat Plants. 5, 965–979. [DOI] [PubMed] [Google Scholar]
- 3.Ohno S (1970) Evolution by gene duplication., Springer-Verlag, New York. [Google Scholar]
- 4.Bergthorsson U, Andersson DI & Roth JR (2007) Ohno’s dilemma: evolution of new genes under continuous selection, Proc Natl Acad Sci U S A. 104, 17004–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Wolfe KH & Shields DC (1997) Molecular evidence for an ancient duplication of the entire yeast genome, Nature. 387, 708–13. [DOI] [PubMed] [Google Scholar]
- 6.Kellis M, Birren BW & Lander ES (2004) Proof and evolutionary analysis of ancient genome duplication in the yeast Saccharomyces cerevisiae, Nature. 428, 617–24. [DOI] [PubMed] [Google Scholar]
- 7.Aury JM, Jaillon O, Duret L, Noel B, Jubin C, Porcel BM, Segurens B, Daubin V, Anthouard V, Aiach N, Arnaiz O, Billaut A, Beisson J, Blanc I, Bouhouche K, Camara F, Duharcourt S, Guigo R, Gogendeau D, Katinka M, Keller AM, Kissmehl R, Klotz C, Koll F, Le Mouel A, Lepere G, Malinsky S, Nowacki M, Nowak JK, Plattner H, Poulain J, Ruiz F, Serrano V, Zagulski M, Dessen P, Betermier M, Weissenbach J, Scarpelli C, Schachter V, Sperling L, Meyer E, Cohen J & Wincker P (2006) Global trends of whole-genome duplications revealed by the ciliate Paramecium tetraurelia, Nature. 444, 171–8. [DOI] [PubMed] [Google Scholar]
- 8.McGrath CL, Gout JF, Doak TG, Yanagi A & Lynch M (2014) Insights into three whole-genome duplications gleaned from the Paramecium caudatum genome sequence, Genetics. 197, 1417–28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Qiao X, Li Q, Yin H, Qi K, Li L, Wang R, Zhang S & Paterson AH (2019) Gene duplication and evolution in recurring polyploidization-diploidization cycles in plants, Genome Biol. 20, 38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Dehal P & Boore JL (2005) Two rounds of whole genome duplication in the ancestral vertebrate, PLoS Biol. 3, e314. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Li JT, Hou GY, Kong XF, Li CY, Zeng JM, Li HD, Xiao GB, Li XM & Sun XW (2015) The fate of recent duplicated genes following a fourth-round whole genome duplication in a tetraploid fish, common carp (Cyprinus carpio), Sci Rep. 5, 8199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Singh PP & Isambert H (2020) OHNOLOGS v2: a comprehensive resource for the genes retained from whole genome duplication in vertebrates, Nucleic Acids Res. 48, D724–D730. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Brunet FG, Roest Crollius H, Paris M, Aury JM, Gibert P, Jaillon O, Laudet V & Robinson-Rechavi M (2006) Gene loss and evolutionary rates following whole-genome duplication in teleost fishes, Mol Biol Evol. 23, 1808–16. [DOI] [PubMed] [Google Scholar]
- 14.Lynch M (2007) The origins of genome architecture, Sinauer Associates, Inc., Sunderland, Massachusetts. [Google Scholar]
- 15.Singh PP, Arora J & Isambert H (2015) Identification of ohnolog genes originating from whole genome duplication in early vertebrates, based on synteny comparison across multiple genomes, PLoS Comput Biol. 11, e1004394. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Gevers D, Vandepoele K, Simillon C & Van de Peer Y (2004) Gene duplication and biased functional retention of paralogs in bacterial genomes, Trends Microbiol. 12, 148–54. [DOI] [PubMed] [Google Scholar]
- 17.Makarova KS, Wolf YI, Mekhedov SL, Mirkin BG & Koonin EV (2005) Ancestral paralogs and pseudoparalogs and their role in the emergence of the eukaryotic cell, Nucleic Acids Res. 33, 4626–38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Gogarten JP, Kibak H, Dittrich P, Taiz L, Bowman EJ, Bowman BJ, Manolson MF, Poole RJ, Date T, Oshima T, Konishi J, Denda K & Yoshida M (1989) Evolution of the vacuolar H+-ATPase: implications for the origin of eukaryotes, Proc Natl Acad Sci U S A. 86, 6661–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Brown JR & Doolittle WF (1995) Root of the universal tree of life based on ancient aminoacyl-tRNA synthetase gene duplications, Proc Natl Acad Sci U S A. 92, 2441–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Gribaldo S & Cammarano P (1998) The root of the universal tree of life inferred from anciently duplicated genes encoding components of the protein-targeting machinery, J Mol Evol. 47, 508–16. [DOI] [PubMed] [Google Scholar]
- 21.Panchy N, Lehti-Shiu M & Shiu SH (2016) Evolution of gene duplication in plants, Plant Physiol. 171, 2294–316. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Liang P, Labedan B & Riley M (2002) Physiological genomics of Escherichia coli protein families, Physiol Genomics. 9, 15–26. [DOI] [PubMed] [Google Scholar]
- 23.Martinez-Nunez MA, Poot-Hernandez AC, Rodriguez-Vazquez K & Perez-Rueda E (2013) Increments and duplication events of enzymes and transcription factors influence metabolic and regulatory diversity in prokaryotes, PLoS One. 8, e69707. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Pushker R, Mira A & Rodriguez-Valera F (2004) Comparative genomics of gene-family size in closely related bacteria, Genome Biol. 5, R27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Wang P, Lv C & Zhu G (2015) Novel type II and monomeric NAD+ specific isocitrate dehydrogenases: phylogenetic affinity, enzymatic characterization, and evolutionary implication, Sci Rep. 5, 9150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Geisbrecht BV & Gould SJ (1999) The human PICD gene encodes a cytoplasmic and peroxisomal NADP(+)-dependent isocitrate dehydrogenase, J Biol Chem. 274, 30527–33. [DOI] [PubMed] [Google Scholar]
- 27.Jo SH, Son MK, Koh HJ, Lee SM, Song IH, Kim YO, Lee YS, Jeong KS, Kim WB, Park JW, Song BJ & Huh TL (2001) Control of mitochondrial redox balance and cellular defense against oxidative damage by mitochondrial NADP+-dependent isocitrate dehydrogenase, J Biol Chem. 276, 16168–76. [DOI] [PubMed] [Google Scholar]
- 28.Rawlings ND, Barrett AJ, Thomas PD, Huang X, Bateman A & Finn RD (2018) The MEROPS database of proteolytic enzymes, their substrates and inhibitors in 2017 and a comparison with peptidases in the PANTHER database, Nucleic Acids Res. 46, D624–D632. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Manning G, Whyte DB, Martinez R, Hunter T & Sudarsanam S (2002) The protein kinase complement of the human genome, Science. 298, 1912–34. [DOI] [PubMed] [Google Scholar]
- 30.Brown SD, Gerlt JA, Seffernick JL & Babbitt PC (2006) A gold standard set of mechanistically diverse enzyme superfamilies, Genome Biol. 7, R8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Gerlt JA, Babbitt PC, Jacobson MP & Almo SC (2012) Divergent evolution in enolase superfamily: strategies for assigning functions, J Biol Chem. 287, 29–34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Segel IH (1975) Enzyme kinetics: behavior and analysis of rapid equilibrium and steady-state enzyme systems, John Wiley & Sons, Inc., New York. [Google Scholar]
- 33.Khanal A, Yu McLoughlin S, Kershner JP & Copley SD (2015) Differential effects of a mutation on the normal and promiscuous activities of orthologs: implications for natural and directed evolution, Mol Biol Evol. 32, 100–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Deng S-K, Zhang W-M, Wang J-P, Gao Y-Z, Xu Y & Zhou N-Y (2019) Single point mutation in the transcriptional regulator PnpR renders Pseudomonas sp. strain WBC-3 capable of utilizing 2-chloro-4-nitrophenol, Intl Biodeterioration & Biodegradation. 143, 104732. [Google Scholar]
- 35.Kim J, Flood JJ, Kristofich MR, Gidfar C, Morgenthaler AB, Fuhrer T, Sauer U, Snyder D, Cooper VS, Ebmeier CC, Old WM & Copley SD (2019) Hidden resources in the Escherichia coli genome restore PLP synthesis and robust growth after deletion of the essential gene pdxB, Proc Natl Acad Sci U S A. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Lynch M, Sung W, Morris K, Coffey N, Landry CR, Dopman EB, Dickinson WJ, Okamoto K, Kulkarni S, Hartl DL & Thomas WK (2008) A genome-wide view of the spectrum of spontaneous mutations in yeast, Proc Natl Acad Sci U S A. 105, 9272–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Lipinski KJ, Farslow JC, Fitzpatrick KA, Lynch M, Katju V & Bergthorsson U (2011) High spontaneous rate of gene duplication in Caenorhabditis elegans, Curr Biol. 21, 306–10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Schrider DR, Houle D, Lynch M & Hahn MW (2013) Rates and genomic consequences of spontaneous mutational events in Drosophila melanogaster, Genetics. 194, 937–54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Anderson P & Roth J (1981) Spontaneous tandem genetic duplications in Salmonella typhimurium arise by unequal recombination between rRNA (rrn) cistrons, Proc Natl Acad Sci U S A. 78, 3113–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Achaz G, Netter P & Coissac E (2001) Study of intrachromosomal duplications among the eukaryote genomes, Mol Biol Evol. 18, 2280–8. [DOI] [PubMed] [Google Scholar]
- 41.Seaton SC, Elliott KT, Cuff LE, Laniohan NS, Patel PR & Neidle EL (2012) Genome-wide selection for increased copy number in Acinetobacter baylyi ADP1: locus and context-dependent variation in gene amplification, Mol Microbiol. 83, 520–35. [DOI] [PubMed] [Google Scholar]
- 42.Reams AB & Roth JR (2015) Mechanisms of gene duplication and amplification, Cold Spring Harb Perspect Biol. 7, a016592. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Kaessmann H, Vinckenbosch N & Long M (2009) RNA-based gene duplication: mechanistic and evolutionary insights, Nat Rev Genet. 10, 19–31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Farslow JC, Lipinski KJ, Packard LB, Edgley ML, Taylor J, Flibotte S, Moerman DG, Katju V & Bergthorsson U (2015) Rapid increase in frequency of gene copy-number variants during experimental evolution in Caenorhabditis elegans, BMC Genomics. 16, 1044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Brochet M, Couve E, Zouine M, Poyart C & Glaser P (2008) A naturally occurring gene amplification leading to sulfonamide and trimethoprim resistance in Streptococcus agalactiae, J Bacteriol. 190, 672–80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Lynch M & Marinov GK (2015) The bioenergetic costs of a gene, Proc Natl Acad Sci U S A. 112, 15690–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Adler M, Anjum M, Berg OG, Andersson DI & Sandegren L (2014) High fitness costs and instability of gene duplications reduce rates of evolution of new genes by duplication-divergence mechanisms, Mol Biol Evol. 31, 1526–35. [DOI] [PubMed] [Google Scholar]
- 48.Lepagnol-Bestel AM, Zvara A, Maussion G, Quignon F, Ngimbous B, Ramoz N, Imbeaud S, Loe-Mie Y, Benihoud K, Agier N, Salin PA, Cardona A, Khung-Savatovsky S, Kallunki P, Delabar JM, Puskas LG, Delacroix H, Aggerbeck L, Delezoide AL, Delattre O, Gorwood P, Moalic JM & Simonneau M (2009) DYRK1A interacts with the REST/NRSF-SWI/SNF chromatin remodelling complex to deregulate gene clusters involved in the neuronal phenotypic traits of Down syndrome, Hum Mol Genet. 18, 1405–14. [DOI] [PubMed] [Google Scholar]
- 49.Schober AF, Mathis AD, Ingle C, Park JO, Chen L, Rabinowitz JD, Junier I, Rivoire O & Reynolds KA (2019) A two-enzyme adaptive unit within bacterial folate metabolism, Cell Rep. 27, 3359–3370 e7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Lind PA & Andersson DI (2013) Fitness costs of synonymous mutations in the rpsT gene can be compensated by restoring mRNA base pairing, PLoS One. 8, e63373. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Kershner JP, Yu McLoughlin S, Kim J, Morgenthaler A, Ebmeier CC, Old WM & Copley SD (2016) A synonymous mutation upstream of the gene encoding a weak-link enzyme causes an ultrasensitive response in growth rate, J Bacteriol. 198, 2853–63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Otwinowski Z, Schevitz RW, Zhang RG, Lawson CL, Joachimiak A, Marmorstein RQ, Luisi BF & Sigler PB (1988) Crystal structure of trp repressor/operator complex at atomic resolution, Nature. 335, 321–9. [DOI] [PubMed] [Google Scholar]
- 53.Yanofsky C (2004) The different roles of tryptophan transfer RNA in regulating trp operon expression in E. coli versus B. subtilis, Trends Genet. 20, 367–74. [DOI] [PubMed] [Google Scholar]
- 54.Xie Y & Reeve JN (2005) Regulation of tryptophan operon expression in the archaeon Methanothermobacter thermautotrophicus, J Bacteriol. 187, 6419–29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Hose J, Yong CM, Sardi M, Wang Z, Newton MA & Gasch AP (2015) Dosage compensation can buffer copy-number variation in wild yeast, Elife. 4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Konrad A, Flibotte S, Taylor J, Waterston RH, Moerman DG, Bergthorsson U & Katju V (2018) Mutational and transcriptional landscape of spontaneous gene duplications and deletions in Caenorhabditis elegans, Proc Natl Acad Sci U S A. 115, 7386–7391. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Myhre S, Lingjaerde OC, Hennessy BT, Aure MR, Carey MS, Alsner J, Tramm T, Overgaard J, Mills GB, Borresen-Dale AL & Sorlie T (2013) Influence of DNA copy number and mRNA levels on the expression of breast cancer related proteins, Mol Oncol. 7, 704–18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Yang G, Anderson DW, Baier F, Dohmen E, Hong N, Carr PD, Kamerlin SCL, Jackson CJ, Bornberg-Bauer E & Tokuriki N (2019) Higher-order epistasis shapes the fitness landscape of a xenobiotic-degrading enzyme, Nat Chem Biol. 15, 1120–1128. [DOI] [PubMed] [Google Scholar]
- 59.Miton CM, Jonas S, Fischer G, Duarte F, Mohamed MF, van Loo B, Kintses B, Kamerlin SCL, Tokuriki N, Hyvonen M & Hollfelder F (2018) Evolutionary repurposing of a sulfatase: A new Michaelis complex leads to efficient transition state charge offset, Proc Natl Acad Sci U S A. 115, E7293–E7302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Ferla MP, Brewster JL, Hall KR, Evans GB & Patrick WM (2017) Primordial-like enzymes from bacteria with reduced genomes, Mol Microbiol. 105, 508–524. [DOI] [PubMed] [Google Scholar]
- 61.Bar-Rogovsky H, Hugenmatter A & Tawfik DS (2013) The evolutionary origins of detoxifying enzymes: the mammalian serum paraoxonases (PONs) relate to bacterial homoserine lactonases, J Biol Chem. 288, 23914–27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Schmidt DM, Mundorff EC, Dojka M, Bermudez E, Ness JE, Govindarajan S, Babbitt PC, Minshull J & Gerlt JA (2003) Evolutionary potential of (beta/alpha)8-barrels: functional promiscuity produced by single substitutions in the enolase superfamily., Biochemistry. 42, 8387–8393. [DOI] [PubMed] [Google Scholar]
- 63.Noor S, Taylor MC, Russell RJ, Jermiin LS, Jackson CJ, Oakeshott JG & Scott C (2012) Intramolecular epistasis and the evolution of a new enzymatic function, PLoS One. 7, e39822. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Bar-Even A, Noor E, Savir Y, Liebermeister W, Davidi D, Tawfik DS & Milo R (2011) The moderately efficient enzyme: evolutionary and physicochemical trends shaping enzyme parameters, Biochemistry. 50, 4402–10. [DOI] [PubMed] [Google Scholar]
- 65.Scott C, Jackson CJ, Coppin CW, Mourant RG, Hilton ME, Sutherland TD, Russell RJ & Oakeshott JG (2009) Catalytic improvement and evolution of atrazine chlorohydrolase, Appl Environ Microbiol. 75, 2184–91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Tokuriki N, Jackson CJ, Afriat-Jurnou L, Wyganowski KT, Tang R & Tawfik DS (2012) Diminishing returns and tradeoffs constrain the laboratory optimization of an enzyme, Nat Commun. 3, 1257. [DOI] [PubMed] [Google Scholar]
- 67.Yang G, Hong N, Baier F, Jackson CJ & Tokuriki N (2016) Conformational tinkering drives evolution of a promiscuous activity through indirect mutational effects, Biochemistry. 55, 4583–93. [DOI] [PubMed] [Google Scholar]
- 68.Babbitt PC, Hasson M, Wedekind JE, Palmer DJ, Lies MA, Reed GH, Rayment I, Ringe D, Kenyon GL & Gerlt JA (1996) The enolase superfamily: a general strategy for enzyme-catalyzed abstraction of the a-protons of carboxylic acids., Biochemistry. 35, 16489–16501. [DOI] [PubMed] [Google Scholar]
- 69.Soderholm A, Guo X, Newton MS, Evans GB, Nasvall J, Patrick WM & Selmer M (2015) Two-step ligand binding in a (betaalpha)8 barrel enzyme: Substrate-bound structures shed new light on the catalytic cycle of HisA, J Biol Chem. 290, 24657–68. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Nasvall J, Sun L, Roth JR & Andersson DI (2012) Real-time evolution of new genes by innovation, amplification, and divergence, Science. 338, 384–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Newton MS, Guo X, Soderholm A, Nasvall J, Lundstrom P, Andersson DI, Selmer M & Patrick WM (2017) Structural and functional innovations in the real-time evolution of new (betaalpha)8 barrel enzymes, Proc Natl Acad Sci U S A. 114, 4727–4732. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Hommel U, Eberhard M & Kirschner K (1995) Phosphoribosyl anthranilate isomerase catalyzes a reversible Amadori reaction, Biochemistry. 34, 5429–39. [DOI] [PubMed] [Google Scholar]
- 73.Wilding M, Hong N, Spence M, Buckle AM & Jackson CJ (2019) Protein engineering: the potential of remote mutations, Biochem Soc Trans. 47, 701–711. [DOI] [PubMed] [Google Scholar]
- 74.Peat TS, Newman J, Balotra S, Lucent D, Warden AC & Scott C (2015) The structure of the hexameric atrazine chlorohydrolase AtzA, Acta Crystallogr D Biol Crystallogr. 71, 710–20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Oue S, Okamoto A, Yano T & Kagamiyama H (1999) Redesigning the substrate specificity of an enzyme by cumulative effects of the mutations of non-active site residues, J Biol Chem. 274, 2344–9. [DOI] [PubMed] [Google Scholar]
- 76.Wrenbeck EE, Azouz LR & Whitehead TA (2017) Single-mutation fitness landscapes for an enzyme on multiple substrates reveal specificity is globally encoded, Nat Commun. 8, 15695. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Weinreich DM, Delaney NF, DePristo MA & Hartl DL (2006) Darwinian evolution can follow only very few mutational paths to fitter proteins, Science. 312, 111–114. [DOI] [PubMed] [Google Scholar]
- 78.Khersonsky O & Tawfik DS (2010) Enzyme promiscuity: a mechanistic and evolutionary perspective, Annu Rev Biochem. 79, 471–505. [DOI] [PubMed] [Google Scholar]
- 79.Morgenthaler AB, Kinney WR, Ebmeier CC, Walsh CM, Snyder DJ, Cooper VS, Old WM & Copley SD (2019) Mutations that improve efficiency of a weak-link enzyme are rare compared to adaptive mutations elsewhere in the genome, Elife. 8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Jensen KF (1993) The Escherichia coli K-12 “wild types” W3110 and MG1655 have an rph frameshift mutation that leads to pyrimidine starvation due to low pyrE expression levels, J Bacteriol. 175, 3401–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Conrad TM, Joyce AR, Applebee MK, Barrett CL, Xie B, Gao Y & Palsson BO (2009) Whole-genome resequencing of Escherichia coli K-12 MG1655 undergoing short-term laboratory evolution in lactate minimal media reveals flexible selection of adaptive mutations, Genome Biol. 10, R118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Kugelberg E, Kofoid E, Reams AB, Andersson DI & Roth JR (2006) Multiple pathways of selected gene amplification during adaptive mutation, Proc Natl Acad Sci U S A. 103, 17319–24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Katju V & Lynch M (2003) The structure and early evolution of recently arisen gene duplicates in the Caenorhabditis elegans genome, Genetics. 165, 1793–803. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Bu L & Katju V (2015) Early evolutionary history and genomic features of gene duplicates in the human genome, BMC Genomics. 16, 621. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Jiang Z, Tang H, Ventura M, Cardone MF, Marques-Bonet T, She X, Pevzner PA & Eichler EE (2007) Ancestral reconstruction of segmental duplications reveals punctuated cores of human genome evolution, Nat Genet. 39, 1361–8. [DOI] [PubMed] [Google Scholar]
- 86.Huang H, Pandya C, Liu C, Al-Obaidi NF, Wang M, Zheng L, Toews Keating S, Aono M, Love JD, Evans B, Seidel RD, Hillerich BS, Garforth SJ, Almo SC, Mariano PS, Dunaway-Mariano D, Allen KN & Farelli JD (2015) Panoramic view of a superfamily of phosphatases through substrate profiling, Proc Natl Acad Sci U S A. 112, E1974–83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Martinez-Martinez M, Coscolin C, Santiago G, Chow J, Stogios PJ, Bargiela R, Gertler C, Navarro-Fernandez J, Bollinger A, Thies S, Mendez-Garcia C, Popovic A, Brown G, Chernikova TN, Garcia-Moyano A, Bjerga GEK, Perez-Garcia P, Hai T, Del Pozo MV, Stokke R, Steen IH, Cui H, Xu X, Nocek BP, Alcaide M, Distaso M, Mesa V, Pelaez AI, Sanchez J, Buchholz PCF, Pleiss J, Fernandez-Guerra A, Glockner FO, Golyshina OV, Yakimov MM, Savchenko A, Jaeger KE, Yakunin AF, Streit WR, Golyshin PN, Guallar V, Ferrer M & The Inmare C (2018) Determinants and prediction of esterase substrate promiscuity patterns, ACS Chem Biol. 13, 225–234. [DOI] [PubMed] [Google Scholar]
- 88.Andorfer MC, Grob JE, Hajdin CE, Chael JR, Siuti P, Lilly J, Tan KL & Lewis JC (2017) Understanding flavin-dependent halogenase reactivity via substrate activity profiling, ACS Catal. 7, 1897–1904. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.James LC & Tawfik DS (2003) Conformational diversity and protein evolution--a 60-year-old hypothesis revisited, Trends Biochem Sci. 28, 361–8. [DOI] [PubMed] [Google Scholar]
- 90.Campbell E, Kaltenbach M, Correy GJ, Carr PD, Porebski BT, Livingstone EK, Afriat-Jurnou L, Buckle AM, Weik M, Hollfelder F, Tokuriki N & Jackson CJ (2016) The role of protein dynamics in the evolution of new enzyme function, Nat Chem Biol. 12, 944–950. [DOI] [PubMed] [Google Scholar]
- 91.Chartier M, Chenard T, Barker J & Najmanovich R (2013) Kinome Render: a stand-alone and web-accessible tool to annotate the human protein kinome tree, PeerJ. 1, e126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Huang CC, Couch GS, Pettersen EF & Ferrin TE (1996) Chimera: An extensible molecular modeling application constructed using standard components., Pac Symp Biocomputing. 1, 724. [Google Scholar]
- 93.Keseler IM, Mackie A, Santos-Zavaleta A, Billington R, Bonavides-Martinez C, Caspi R, Fulcher C, Gama-Castro S, Kothari A, Krummenacker M, Latendresse M, Muniz-Rascado L, Ong Q, Paley S, Peralta-Gil M, Subhraveti P, Velazquez-Ramirez DA, Weaver D, Collado-Vides J, Paulsen I & Karp PD (2017) The EcoCyc database: reflecting new knowledge about Escherichia coli K-12, Nucleic Acids Res. 45, D543–D550. [DOI] [PMC free article] [PubMed] [Google Scholar]