

Abstract
Myeloproliferative neoplasms (MPN) are chronic stem cell disorders characterized by enhanced proliferation of myeloid cells, immune deregulation, and drug resistance. JAK2 somatic mutations drive the disease in 50–60% and CALR mutations in 25–30% of cases. Published data suggest that JAK2‐V617F‐mutated MPN cells express the resistance‐related checkpoint PD‐L1. By applying RNA‐sequencing on granulocytes of 113 MPN patients, we demonstrate that PD‐L1 expression is highest among polycythemia vera patients and that PD‐L1 expression correlates with JAK2‐V617F mutational burden (R = 0.52; p < .0001). Single nucleotide polymorphism (SNP) arrays showed that chromosome 9p uniparental disomy (UPD) covers both PD‐L1 and JAK2 in all MPN patients examined. MPN cells in JAK2‐V617F‐positive patients expressed higher levels of PD‐L1 if 9p UPD was present compared to when it was absent (p < .0001). Moreover, haplotype‐based association analyses provided evidence for germline genetic factors at PD‐L1 locus contributing to MPN susceptibility independently of the previously described GGCC risk haplotype. We also found that PD‐L1 is highly expressed on putative CD34+CD38− disease‐initiating neoplastic stem cells (NSC) in both JAK2 and CALR‐mutated MPN. PD‐L1 overexpression decreased upon exposure to JAK2 blockers and BRD4‐targeting agents, suggesting a role for JAK2‐STAT5‐signaling and BRD4 in PD‐L1 expression. Whether targeting of PD‐L1 can overcome NSC resistance in MPN remains to be elucidated in forthcoming studies.
1. INTRODUCTION
Myeloproliferative neoplasms (MPN) are chronic bone marrow (BM) disorders characterized by clonal hematopoiesis, overproduction of myeloid cells, inflammation, and immune deregulation. 1 , 2 The three classical BCR‐ABL1‐negative MPN are essential thrombocythemia (ET), polycythemia vera (PV), and primary myelofibrosis (PMF). The JAK2‐V617F mutation drives the disease in ~95% of PV patients and 50%–60% of all cases with ET and PMF. 3 , 4 , 5 , 6 Somatic mutations affecting CALR are disease drivers in 30%–40% of ET and PMF patients, 7 while MPL mutations account for 5%–10% of all MPN cases. 8 , 9 MPN patients with unknown disease drivers (no driver mutation detected) are referred to as ´triple negative´ MPN. Progression to secondary acute myeloid leukemia (sAML) may occur in all three MPN entities, but is more frequently documented in patients with PMF compared to those with ET and PV. 10 Most of the current therapeutic approaches in MPN are not curative, and disease management focuses on amelioration of symptoms. 11 , 12 The only exception is allogeneic hematopoietic stem cell transplantation, a procedure that is associated with a relatively high mortality risk, and is, therefore, performed only in younger patients with advanced disease or sAML. 11 Lack of curative potential and commonly occurring resistance to drug therapies applied in MPN patients highlight the need for the development of novel therapeutic concepts. One strategy may be to establish drug‐based therapies or antibody‐based immunotherapies capable of targeting and eradicating the disease‐initiating neoplastic stem cells (NSC) in MPN. 13
While immune dysregulation in MPN has been well documented, the mechanisms by which the MPN (stem) cells escape the antitumor immune response remain unclear. 1 Published studies have shown that MPN cells display certain immune checkpoint molecules, including PD‐L1, which may contribute to resistance, and that constitutive activation of JAK2 by the V617F mutation causes PD‐L1 upregulation in MPN cells. 14 , 15
PD‐L1 and PD‐L2 expression by neoplastic cells and binding of these molecules to the PD‐1 receptor on T cells inhibits the antigen‐specific Tcell‐mediated antitumor immune response, and represents a major mechanism of immune escape. 16 , 17 PD‐L1 is not only expressed on cells of many tumors, but also cells within the tumor microenvironment in inflammatory conditions, thus reducing antitumor immunity. 18 , 19 Upregulation of PD‐L1 in cancer cells can be a consequence of PD‐L1 gene amplification, activation of oncogenic signaling pathways, or epigenetic regulation, but it can also be induced by certain pro‐inflammatory cytokines. 20 Blocking of the interaction between the PD‐1 receptor and its two ligands potentiates antitumor T‐cell responses and in line with this notion, checkpoint inhibitors targeting the PD‐1/PD‐L1 interaction have emerged as highly promising anticancer drugs, leading to durable responses in various solid tumors. 21 , 22 , 23 , 24 The use of PD‐1/PD‐L1 interaction‐targeting agents in MPN is currently under investigation, however, it is unclear, which MPN subtypes are most suitable for testing in clinical trials.
The genes encoding the PD‐L1 (CD274, alias PD‐L1), PD‐L2 (CD273, alias PD‐L2), and JAK2 proteins are located in close proximity on the short arm of chromosome 9, which is often affected by copy number neutral loss of heterozygosity through the mechanism of acquired uniparental disomy (UPD) in MPN patients, usually leading to an increase in the JAK2‐V617F mutational burden. 25 UPD of chromosome 9p is the most common chromosomal aberration found in MPN. It affects up to 80% of PV patients, close to half of PMF and sAML patients, and 6%–18% of cases with ET. 26 However, it is unknown whether chromosome 9p UPD affects PD‐L1/2 genes, which have a more centromeric position than JAK2. In particular, it is not known whether chromosome 9p UPD affects PD‐L1/2 expression levels in MPN cells.
In the current study, we analyzed PD‐L1 expression in neoplastic cells in a well‐characterized cohort of MPN patients, for which in‐depth genomic data were available. We combined whole‐transcriptome data with chromosomal aberration profiles based on SNP arrays and mutational profiles from targeted sequencing using a myeloid gene panel. We show that PD‐L1 overexpression in MPN is associated with chromosome 9p UPD and that it correlates with JAK2‐V617F mutational burden in granulocytes. We also found that germline genetic factors at the PD‐L1 gene locus contribute to MPN susceptibility, and we demonstrate that PD‐L1 is upregulated on the cell surface of phenotypically defined MPN NSC.
2. METHODS
2.1. Patients and primary cell isolation
Samples from 181 MPN patients and 14 healthy donors were collected at the Medical University of Vienna and Hanusch Hospital in Vienna, Austria; Elisabethinen Hospital in Linz, Austria; and Fondazione IRCCS Policlinico San Matteo in Pavia, Italy. All patients and healthy donors provided informed consent in accordance with the Declaration of Helsinki. The study was approved by local ethics committees. Patients´ characteristics are shown in Table S1. Diagnostic criteria were applied as described previously. 5 Peripheral blood (PB) samples were collected from healthy donors and MPN patients and BM samples from MPN patients during routine investigations. Granulocytes and mononuclear cells (MNC) were isolated from PB and BM samples according to standard procedures using density gradient centrifugation.
2.2. DNA and RNA isolation and driver mutation analysis
DNA and RNA were isolated from granulocyte fractions of PB samples of MPN patients or healthy donors using standard procedures. 5 JAK2, CALR, and MPL mutational analyses and evaluations of mutational burden (variant allele frequency) were performed as previously described. 5
2.3. RNA‐sequencing and data analysis
The evaluation of the expression of PD‐L1/2 and other selected genes is based on a large RNA‐sequencing data set published in Schischlik et al. 27 Further details are available in Appendix S1.
2.4. Targeted DNA‐sequencing and microarray analysis
Methods used for targeted DNA sequencing with the TruSight Myeloid Sequencing Panel (Illumina, San Diego, CA) and microarray analysis using Genome‐Wide Human SNP 6.0 arrays (Affymetrix, San Diego, CA, USA) are described in the Appendix S1.
2.5. Cell lines and in vitro studies
In vitro studies were performed using HEL, SET‐2, and UT‐7 cell lines. UT‐7 cells were engineered to express various CALR mutants using CRISPR/Cas9 technology as described recently. 28 Methods related to in vitro studies as well as 3H‐thymidine incorporation assay are described in the Appendix S1.
2.6. Evaluation of expression of PD‐L1/2 and PD‐1 on primary MPN cells and cell lines
Heparinized BM or PB samples or cell lines were incubated with various combinations of monoclonal antibodies (Table S2) at room temperature in the dark for 15 min. As normal control, we used commercially available BM cells from healthy donors (Lonza, Basel, CH) as previously described. 29 After incubation, erythrocytes were lysed using fluorescence‐activated cell sorting (FACS)‐Lysing Solution (BD Biosciences, San José, CA, USA). Cells were washed and analyzed by flow cytometry using FACSCanto II (BD Biosciences, San José, CA, USA) essentially as decribed. 30 FlowJo software (version 8.8.7, TreeStar, Ashland, OR, USA) was used for data analysis. Antibody‐staining results were controlled by applying isotype‐matched control antibodies, and were expressed as either percentage of positive cells or as staining index, which represents the ratio of median fluorescence intensities obtained with the specific monoclonal antibody and the isotype‐matched control antibody. Phenotypic NSC were defined as CD34+CD45dimCD38− while progenitors were defined as CD34+CD45dimCD38+ cells. 31 , 32 For detection of T cells, we used monoclonal antibodies against CD45, CD3, CD4, and CD8, while B cells were analyzed using CD19 antibody and NK cells by using a CD56 antibody. Gating strategies were applied as described previously. 30
To assess the effects of drugs on PD‐L1 expression, primary MPN MNC from 6 JAK2‐V617F‐positive patients were incubated in medium or medium containing interferon‐gamma (IFN‐γ) (200 U/mL) in the absence or presence of the BET (Bromodomain and Extra‐Terminal proteins) inhibitor JQ1 (2500 nM), the BET degrader dBET6 (100 nM), ruxolitinib (2500 nM) or dimethyl sulfoxide (DMSO) (control) at 37°C for 24 h. JQ1 and ruxolitinib were obtained from Selleckchem (Houston, TX, USA) and dBET6 from Aobious (Gloucester, MA, USA). Then, cells were harvested and CD34+CD45dimCD38− MPN stem cells were examined for expression of PD‐L1 by multicolor flow cytometry as described above. To assess drug effects on PD‐L1 expression in cell lines, cells were incubated in IFN‐γ (200 U/mL) in the absence or presence of DMSO, JQ1 (100–250 nM), dBET6 (50–500 nM), or ruxolitinib (50–250 nM) at 37°C for 24 h. Then, cells were examined for expression of PD‐L1 by flow cytometry.
2.7. Linkage analysis, association analysis, and haplotype‐based expression analysis
Linkage analysis for the JAK2 and PD‐L1 loci was performed using the LDpair and LDmatrix functions as implemented in the LDlink web‐based toolset (https://ldlink.nci.nih.gov/). Haplotype‐based association analyses were performed using previously published SNP‐array data from 272 MPN patients from Vienna, Austria 33 in conjunction with 1620 Bavarian controls from the German Cooperative Health Research in the Region of Augsburg cohort (KORA). 34 Cohort analyses of genetic data, including haplotype determination were performed using PLINK. 35 Association analyses based on haplotype frequency distributions were performed using Fisher's exact test as implemented in R. 36 Haplotype‐based comparative expression analysis was performed for MPN patients with both SNP‐array and RNA‐Seq data available. Statistical evaluation was performed using unpaired t‐tests as implemented in Prism (version 8.0.0, GraphPad Software, La Jolla, CA, USA).
2.8. Statistical analyses
Statistical analyses were performed using Prism software (Graphpad Software). For comparison of marker expression in various subgroups of patients, analysis of variance was applied. Significance levels in differences in expression of markers and targets on stem‐ and progenitor cells in various groups of patients were analyzed by Student's t‐test with or without Welch's correction. p < .05 was considered statistically significant.
3. RESULTS
3.1. PD‐L1 is overexpressed in myeloid cells in MPN and is highest in PV
To determine the expression levels of PD‐L1 and PD‐L2 in MPN cells, we analyzed the whole‐transcriptome RNA‐sequencing data set of a well‐characterized cohort of 104 patients with chronic phase MPN, 9 patients with post‐MPN sAML, and 14 healthy controls (Figures 1A,B and S1,2). 27 The MPN cohort included 55 patients with PMF, 32 with ET, and 17 with PV. The samples were selected based on the highest possible burden of the disease‐driving mutations in granulocytes in order to ensure that the vast majority of the cells examined would represent the MPN clone. In addition, we chose a balanced number of JAK2‐V617F and CALR‐mutated cases among ET and PMF patients. We observed a significantly higher expression level of PD‐L1 mRNA in patients with PV compared to healthy controls (p < .0001; Figure 1A). To assess the effect of the disease‐driving mutations on PD‐L1 expression in granulocytes, we divided the patient cohorts into subsets based on disease‐driving mutations and diagnosis (Figure 1B). Among both ET and PMF patients, JAK2‐V617F positive cases showed higher PD‐L1 mRNA expression in granulocytes compared to CALR‐mutated MPN patients (p < .005 and p < .01, respectively; Figure 1B). Patients with PV who were all carrying the JAK2‐V617F mutation, displayed the highest expression levels of PD‐L1 mRNA in neoplastic cells.
FIGURE 1.
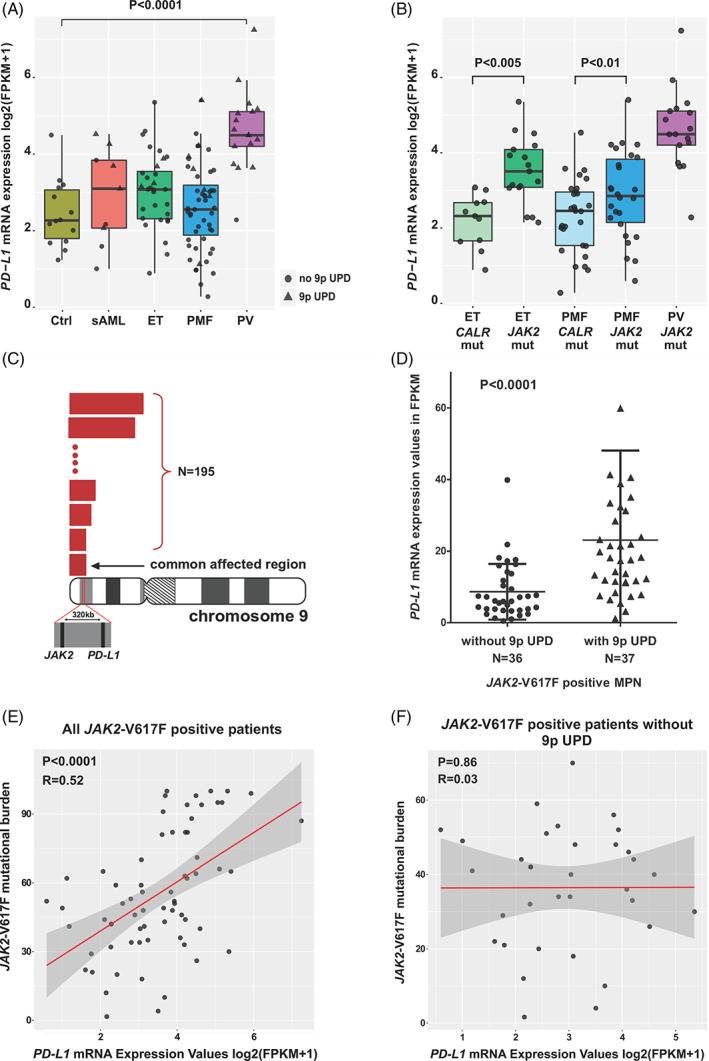
RNA‐sequencing reveals upregulation of PD‐L1 in myeloid cells of patients with polycythemia vera and the role of chromosome 9p UPD in PD‐L1 upregulation in MPN. (A) The box plots show the median (bold horizontal line), interquartile range (box), and total range (whiskers) of PD‐L1 mRNA expression levels detected by RNA‐sequencing of granulocyte samples from MPN and secondary AML patients and healthy donors. Patients with PV displayed significant upregulation of PD‐L1 expression compared to healthy donors and other MPN phenotypes; (B) RNA‐sequencing of granulocyte samples from MPN patients with ET, PMF, and PV revealed higher PD‐L1 expression in JAK2‐V617F mutant patients than in CALR mutation‐driven ET and PMF. The box plots show the median (bold horizontal line), interquartile range (box), and total range (whiskers) of PD‐L1 mRNA expression; (C) Chromosome 9p UPD, detected in 195 MPN patient samples using Human Whole‐genome Affymetrix 6.0 SNP arrays, always targets both JAK2 and PD‐L1 genes, despite a more centromeric position of PD‐L1. (D) PD‐L1 mRNA expression in granulocytes, evaluated by RNA‐sequencing, is significantly higher in JAK2‐V617F‐positive patients who in addition carry a chromosome 9p UPD than in JAK2‐V617F positive patients without this aberration (p < .0001). The horizontal line represents the mean ± standard deviation. (E) PD‐L1 mRNA expression measured by RNA‐sequencing is significantly correlated with the granulocyte JAK2‐V617F mutational burden (R = 0.52; p < .0001). (F) When excluding MPN cases with chromosome 9p UPD from this analysis, the correlation between PD‐L1 mRNA expression and JAK2‐V617F mutational burden in granulocytes of MPN patients is lost (R = 0.03; p = .9). Ctrl, control; ET, essential thrombocythemia; mut, mutant; PMF, primary myelofibrosis; PV, polycythemia vera; sAML, secondary acute myeloid leukemia; UPD, uniparental disomy [Color figure can be viewed at wileyonlinelibrary.com]
Targeted sequencing of a gene panel relevant for myeloid malignancies was performed for 77 patients of the cohort used for whole‐transcriptome sequencing. The most common mutations besides the disease drivers were mutations in TET2 in 14.3%, DNMT3A in 28.6%, and SF3B1 in 10.4% of the patients. However, presence of these additional mutations did not show an effect on PD‐L1 expression in MPN cells (data not shown).
PD‐L2, encoding the other ligand of PD‐1, was expressed at very low levels in MPN cells in all samples analyzed, with PV patients displaying slightly increased expression levels compared to the other patient groups (Figure S2).
3.2. PD‐L1 expression correlates with the JAK2‐V617F mutational burden and is higher in patients with UPD of chromosome 9p
As PD‐L1 and JAK2 are both located on the 9p24.1 locus (320 kb apart; Figure 1C), we hypothesized that the presence of chromosome 9p UPD has an effect on the observed PD‐L1 expression in MPN cells, which would explain why MPN cells in patients with PV have higher PD‐L1 expression levels compared to other disease entities. To test this hypothesis, we analyzed a previously published cohort of MPN patients for whom we had available data from Genome‐wide Human Affymetrix 6.0 SNP arrays. 33 We detected chromosome 9p UPD in 195 MPN patients, while 15 patients carried three copies of chromosome 9p (9p gains), out of 408 patients analyzed. In all of the identified MPN cases with chromosome 9p aberrations, we observed that the commonly affected region invariably covers both, the JAK2 and PD‐L1 genes (Figure 1C). As PD‐L1 has a more centromeric position, it is possible that this gene represents the second target of chromosome 9p UPD in MPN.
To determine the presence of 9p UPD in our cohort of patients characterized by RNA‐sequencing, we analyzed the SNP‐array data that was available for 73 JAK2‐V617F positive patients, and found that 37 patients carried the 9p UPD, while in 36 patients this aberration was absent. To assess the effect of the 9p UPD on PD‐L1 expression in MPN cells, we compared the PD‐L1 mRNA expression between JAK2‐V617F‐positive patients with and without 9p UPD, and observed a significantly higher expression of PD‐L1 in MPN cells in patients with 9p UPD (p < .0001; Figure 1D). In addition, we found that the presence of the 9p UPD leads to higher PD‐L1 expression in various MPN subsets (Figure S3). Next, we performed correlation studies, which revealed that PD‐L1 levels correlate with the JAK2‐V617F mutational burden in granulocytes of MPN patients (p < .0001, R = 0.52; Figure 1E). The correlation of PD‐L1 expression with the JAK2‐V617F mutational burden was lost when cases with 9p UPD were excluded from these analyses (p = .9, R = 0.03; Figure 1F).
3.3. Germline genetic variation at the PD‐L1 gene locus affects MPN risk possibly through acting on PD‐L1 expression
Previous data have shown that a common haplotype referred to as GGCC (also: 46/1) haplotype preferentially acquires JAK2‐V617F and thus confers susceptibility for MPN. 37 Our data suggest that the presence of chromosome 9p UPD might be relevant for PD‐L1 upregulation also at the more distal PD‐L1 locus. Therefore, we evaluated a possible role of germline genetic variation at the PD‐L1 locus and its interplay with the GGCC haplotype at the distal JAK2 locus. We performed linkage analysis, including the GGCC haplotype and an SNP within the 3’UTR of PD‐L1 (rs4143815), as there were no suitable coding SNPs available. We did not observe a strong linkage disequilibrium (LD) between the JAK2 and PD‐L1 loci (D′ = 0.1171, R 2 = .0112), however, the presence of a haplotype block (genomic region with low recombination rate and low haplotype diversity) spanning both loci at weak LD could not be excluded (Figure 2A). Next, we determined the frequencies of the possible JAK2/PD‐L1 haplotypes in a large SNP‐array‐typed MPN cohort (n = 272) 27 and a population‐matched non‐MPN control cohort (n = 1620; Table S3). Haplotype‐based association analyses on the observed counts suggested the PD‐L1 rs4143815 minor allele as risk factor for MPN susceptibility independent of the JAK2 GGCC haplotype tagged by rs10974944 (Table 1). This was observed on both GGCC risk (rs10974944 minor allele; p = .04) and GGCC protective (rs10974944 major allele; p = .02) backgrounds (Table 1). These observations are supported by a GGCC‐independent effect of the PD‐L1 haplotype on expression at the larger locus, including both JAK2 and PD‐L1, although a modest number of informative samples was available for this analysis (Figure 2B,C). Specifically, the presence of the PD‐L1 rs4143815 minor allele results in increased JAK2 expression on both JAK2 GGCC protective (major) and risk (minor) background, albeit lacking formal statistical significance (Figure 2B). A similar overall effect can be observed for PD‐L1 expression, where the presence of the PD‐L1 rs4143815 minor allele on GGCC risk background results in increased PD‐L1 expression (p = .03; Figure 2C).
FIGURE 2.
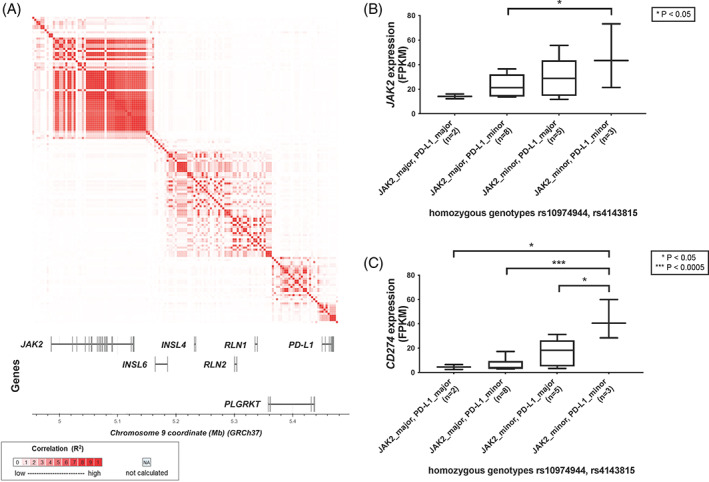
Evaluation of germline genetic variation at the PD‐L1 locus. (A) Linkage analyses for the JAK2 and PD‐L1 loci suggest the lack of strong LD between the loci (pairwise LD metrics for rs10974944‐rs4143815: D′ = 0.1171, R2 = 0.0112), while low correlations are observed all across the region. (B,C) Expression of JAK2 (B) and PD‐L1 (C) in MPN patients (n = 18) homozygous for both rs10974944 (JAK2 locus) and rs4143815 (PD‐L1 locus). Significance levels as determined by unpaired t‐tests are shown if p < .05 [Color figure can be viewed at wileyonlinelibrary.com]
TABLE 1.
Association analysis for an MPN patient cohort (n = 272) versus a non‐MPN control cohort (n = 1620) for haplotypes, including both JAK2 (rs10974944) and PD‐L1 (rs4143815); results from homozygous calls at both loci are shown
| Reference haplotype | Test haplotype | OR | 95% CI lower | 95% CI upper | p‐value | Effect tested |
|---|---|---|---|---|---|---|
| JAK2_major, PD‐L1_major | JAK2_major, PD‐L1_minor | 2.15 | 0.94 | 4.61 | .04 | PD‐L1 minor allele alone |
| JAK2_major, PD‐L1_major | JAK2_minor, PD‐L1_major | 7.24 | 3.85 | 13.62 | 2.74E−10 | JAK2 minor allele alone |
| JAK2_major, PD‐L1_major | JAK2_minor, PD‐L1_minor | 18.83 | 8.4 | 43.66 | 4.23E−14 | JAK2 and PD‐L1 minor alleles combined |
| JAK2_minor, PD‐L1_minor | JAK2_minor, PD‐L1_major | 0.39 | 0.15 | 0.94 | .02 | PD‐L1 major allele alone |
| JAK2_minor, PD‐L1_minor | JAK2_major, PD‐L1_minor | 0.11 | 0.04 | 0.31 | 1.78E−06 | JAK2 major allele alone |
| JAK2_minor, PD‐L1_minor | JAK2_major, PD‐L1_major | 0.05 | 0.02 | 0.12 | 4.23E−14 | JAK2 and PD‐L1 major alleles combined |
3.4. PD‐L1 is expressed on the cell surface of NSC in MPN patients
As we demonstrated the PD‐L1 mRNA upregulation in myeloid cells of MPN patients, we wanted to confirm these findings at the protein level. To investigate if the phenotypically defined (CD34+CD45dimCD38−) NSC in MPN patients express PD‐L1 on their cell surface, we analyzed cells isolated from fresh BM samples of 49 MPN patients and 7 healthy controls by flow cytometry (Figures 3A,B and S4). PD‐L1 surface expression was assessed in both, CD34+CD45dimCD38− and CD34+CD45dimCD38+ MPN cell populations. Stem cells and progenitor cells in MPN patients showed a significantly higher surface expression of PD‐L1 compared to stem and progenitor cells in healthy controls (p < .0001) (Figures 3A and S5A). Both JAK2‐mutated and CALR‐mutated MPN patients showed significantly higher surface expression of PD‐L1 on NSC and progenitor cells compared to normal stem and progenitor cells (Figures 3B and S5B). By contrast, PD‐L1 expression on CD34+cell subsets in the three triple‐negative MPN patients tested, did not significantly differ from PD‐L1 levels detected on CD34+ cells in healthy controls (Figures 3B and S5B). Among the MPN patients analyzed by flow cytometry, 7 were diagnosed with PV, 28 with ET, 6 with PMF, and 5 with post‐MPN AML. Surprisingly, no significant difference in the surface expression of PD‐L1 on NSC measured by flow cytometry was observed among the four different phenotypes (Figures S4 and S6). This may be explained by low JAK2‐V617F burden in the seven PV patients we analyzed by FACS. NSC of MPN patients did not express PD‐L2 on the cell surface as measured by flow cytometry (Figure S7A,C) and the same was observed for progenitor cells (Figure S7B).
FIGURE 3.
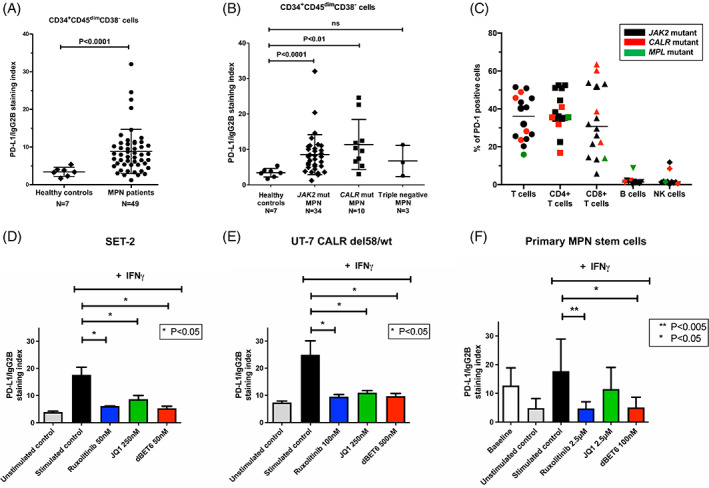
PD‐L1 is upregulated on the surface of stem cells isolated from bone marrow samples of MPN patients and can be downregulated by ruxolitinib and dBET6. (A,B) PD‐L1 expression was assessed by multicolor flow cytometry on CD34+CD45dimCD38− putative neoplastic stem cells isolated from the bone marrow of MPN patients (N = 49) or healthy donors (N = 7). The horizontal line represents the mean ± standard deviation. (B) Both JAK2 and CALR mutant MPN patients showed an upregulation of PD‐L1 on stem cells when compared to healthy donors (p < .001 and p < .01, respectively). The horizontal line represents the mean ± standard deviation. (C) Expression of PD‐1 on T cells, B cells, and NK cells from fresh bone marrow samples of MPN patients was assessed using multicolor flow cytometry. The results are shown as percentage of PD‐1 positive cells and each datapoint represents one patient, while the horizontal line represents the median value. PD‐1 was expressed on both CD4+ and CD8+ T cells, but was not found to be expressed on B and NK cells of the majority of the samples analyzed. (D) SET‐2 and UT‐7 CALR del58/wt (E) cells were incubated with medium or medium containing 200 U/mL of IFN‐γ with or without indicated concentrations of ruxolitinib, JQ1, or dBET6. Drug concentrations were selected at IC20‐IC30 for each cell line. Expression of PD‐L1 was evaluated using flow cytometry upon 24 h of incubation. The expression of PD‐L1 is shown as the staining index, which represents the ratio of median fluorescence intensity of PD‐L1 and matched isotype control. The experiments were performed in triplicate and graphs represent mean ± standard deviation. (F) Primary MNC from six independent JAK2‐V617F positive MPN patients were incubated with medium or medium containing 200 U/mL of IFN‐γ with or without the indicated concentrations of ruxolitinib, JQ1, or dBET6 for 24 h. Upon incubation, PD‐L1 expression on CD34+CD45dimCD38− cells was evaluated using multicolor flow cytometry. The expression of PD‐L1 is shown as the staining index, which represents the ratio of median fluorescence intensity of PD‐L1 antibody and the isotype‐matched control antibody. The graph represents mean ± standard deviation of six independent experiments [Color figure can be viewed at wileyonlinelibrary.com]
Finally, we found that both CD4+ and CD8+ T cells isolated from fresh BM samples of MPN patients (N = 16) express the PD‐L1 receptor PD‐1, and that this expression is not influenced by the presence of the MPN disease‐driving mutations JAK2, CALR, or MPL (Figures 3C and S8). In almost all MPN patients tested (N = 12), B cells and NK cells did not express PD‐1 on their cell surface (Figures 3C and S8).
3.5. PD‐L1 expression in MPN (stem) cells can be modulated by ruxolitinib and BRD4‐targeting drugs and both agents suppress proliferation of MPN cells
We evaluated the effects of ruxolitinib and of the BRD4‐targeting drugs JQ1 and dBET6 on IFN‐γ‐induced expression of PD‐L1 in the JAK2‐V617F‐positive cell lines HEL and SET‐2, and UT‐7 cells carrying CALR mutations. In all tested JAK2‐V617F‐ and CALR‐mutated cell lines, ruxolitinib, JQ1, and dBET6 were found to downregulate IFN‐γ‐induced PD‐L1 expression after 24 h as measured by flow cytometry, while no significant difference was observed in UT‐7 wild type cells upon treatment (Figures 3D,E and S9). We also examined drug effects on primary MPN stem cells obtained from six patients with JAK2‐V617F‐positive MPN. We found that ruxolitinib and dBET6 completely suppress IFN‐γ‐induced expression of PD‐L1 in phenotypically defined (CD34+CD45dimCD38−) MPN NSC, while JQ1 did not show a significant effect (Figure 3F). Interestingly, ex vivo culturing of primary MPN stem cells in Roswell Park Memorial Institute (RPMI) medium for 24 h (in the absence of IFN‐γ) led to reduced PD‐L1 levels compared to baseline levels measured in stem cells in fresh BM samples. Finally, we were able to show that JQ1 and dBET6 dose‐dependently suppress the growth of primary MPN cells (Figure S10) and the same effect was observed with JQ1 and dBET6 in the MPN‐related cell lines HEL, SET‐2, and UT‐7 cells carrying CALR mutations (Figure S11).
4. DISCUSSION
Previously published data have demonstrated that JAK2‐V617F‐positive MPN cells display PD‐L1, a major immune checkpoint antigen mediating immunological resistance in neoplastic cells in diverse hematologic malignancies. 14 So far, little is known about the regulation and function of expression of PD‐L1 in MPN cells. We here show that PD‐L1 expression in MPN cells is highest in patients with PV, correlates with the JAK2‐V617F burden, and is associated with chromosome 9p UPD. Moreover, we show that PD‐L1 is not only expressed on mature myeloid cells in MPN, but also on the disease‐initiating NSC in all MPN patients tested. Finally, our data show that JAK2‐targeting and BRD4/MYC‐targeting drugs counteract the IFN‐γ‐induced expression of PD‐L1 on MPN cells and MPN NSC.
Although responses to PD‐L1/PD‐1‐targeting therapy have been documented even in cancers where PD‐L1 expression was low, high PD‐L1 expression is often used as a biomarker and predictor of response to PD‐L1/PD‐1‐targeting therapy in several types of cancer, including Hodgkin lymphoma. 21 , 23 , 38 , 39 , 40 Patients with Hodgkin lymphoma often carry amplifications of the chromosome 9p24.1 locus containing PD‐L1, PD‐L2, and JAK2 genes, leading to high PD‐L1 and PD‐L2 protein expression. 41 , 42 , 43 In MPN, chromosome 9p is affected by UPD most commonly in PV, but also in patients with PMF and ET. 26 The 9p UPD is considered to be a late event in the clonal evolution of MPN, occurring after the acquisition of JAK2‐V617F and leading to amplification of oncogenic mutation to homozygosity and an increase in mutational burden. 25 However, several studies have provided evidence that 9p UPD can occur prior to JAK2‐V617F in up to 10% of MPN patients, indicating that there might be a second target of this chromosomal aberration. 44 , 45 We here show that PD‐L1 is always affected by chromosome 9p aberrations in MPN. The fact that PD‐L1 expression in MPN cells correlates with the JAK2‐V617F mutational burden and that among JAK2‐V617F positive patients, those with homozygous JAK2‐V617F mutation display higher PD‐L1 expression, indicate that the mutant JAK2 dosage and subsequent activation of the JAK–STAT pathway, lead to PD‐L1 upregulation, which is in line with the results published by Prestipino et al. 14
Acquisition of chromosome 9p UPD and subsequent upregulation of PD‐L1 expression on affected cells may contribute to a positive selection of JAK2‐V617F homozygous stem cells. As we are not capable of modeling chromosome 9p UPD without the presence of JAK2‐V617F, we attempted to evaluate the influence of germline genetic factors on expression of PD‐L1. While we did not observe a strong LD between the GGCC haplotype and the 3’UTR of PD‐L1, we could show that carriers of the minor allele of the PD‐L1 variant rs4143815 are enriched in an MPN cohort. Of note, this enrichment was observed independent of the GGCC haplotype, establishing a novel risk factor for MPN susceptibility, albeit at a modest effect size as compared to the adjacent JAK2 GGCC risk locus. While the GGCC haplotype reflects strongly increased PD‐L1 expression levels, either directly through germline regulatory mechanisms or through increased acquisition of JAK2‐V617F, our results demonstrate an additive, but independent effect of the “PD‐L1 risk haplotype” tagged by the minor allele of rs4143815 on PD‐L1 expression. As our analyses were performed in an MPN cohort, it remains to be determined whether similar effects can be observed also in cancer cells without JAK2‐V617F mutation. Overall, these data provide first evidence for a role of germline factors at the PD‐L1 locus in MPN pathogenesis independent of the JAK2 GGCC risk haplotype. However, potential presence of a haplotype block at weak LD prevents a full dissection of the two loci by genetic means, thus requiring functional studies.
We demonstrated that stem and progenitor cells from MPN patients express high levels of PD‐L1, regardless of the JAK2 and CALR mutational status. This finding might stem from the external factors present in the BM microenvironment, which can affect PD‐L1 expression. The involvement of the IFN‐γ signaling through JAK1/2‐STAT axis in expression of PD‐L1 has been well documented. 46 , 47 Altered expression of cytokines is considered to be a hallmark of MPN and measurable levels of IFN‐γ in the plasma or sera of patients with MPN have been reported in several studies, albeit with conflicting results. 48 , 49 , 50 We could demonstrate that the PD‐L1 expression in CD34+CD45dimCD38− stem cells of JAK2‐V617F positive MPN patients spontaneously decreases when the cells are incubated ex vivo in RPMI medium for 24 h, and that the stimulation with IFN‐γ restores PD‐L1 expression to normal baseline levels. Therefore, we cannot rule out that the difference in PD‐L1 expression among the MPN phenotypes, or between the stem and progenitor cell populations and granulocytes in the case of CALR positive patients could at least in part be explained by extrinsic factors, such as different IFN‐γ levels. An alternative explanation would be that PD‐L1 levels are differentially regulated by cytokines or oncogenic machineries in different types of cells. PD‐L1 upregulation on progenitor cells and stem cells of CALR positive MPN patients is in line with the findings of Bozkus et al. who demonstrated that these patients develop T cells specific for the mutant CALR and that targeting the PD‐L1/PD‐1 interaction can lead to restoration of this T‐cell response against mutant CALR in MPN patients. 51
The ability of BET inhibitors, such as JQ1, to suppress constitutive or IFN‐γ induced PD‐L1 expression in certain cancer cell lines as well as stem cells of chronic myeloid leukemia patients were previously described. 52 , 53 In line with these results, we show that treatment with the JAK2 inhibitor ruxolitinib or the BET‐degrader dBET6, but not BET inhibitor JQ1, leads to downregulation of PD‐L1 expression in CD34+CD45dimCD38− stem cells in JAK2‐V617F positive MPN patients, while all three drugs exerted inhibitory effects in JAK2 and CALR‐mutated cell lines. It remains unknown whether these drug effects on PD‐L1 expression play a role in vivo in patients with MPN and may support or facilitate the cytoreductive capabilities of these agents against MPN (stem) cells. In this regard, it is worth noting that ruxolitinib exerts modest effects on depleting MPN cells and reducing the JAK2‐V617F mutant allele burden in MPN patients (only 10%–20% reduction) and also showed little effects on disease progression to post‐MPN sAML. 54 , 55 It is also of interest to note that BET protein bromodomain targeting agents were previously shown to be highly active against post‐MPN sAML cells, and to exert a synergistic effect with ruxolitinib. 56 , 57 As previous reports also demonstrated synergistic effects of anti‐PD‐1 antibodies with BET inhibitor JQ1, 52 our data indicates that such applications should be assessed in future studies in the context of MPN.
In conclusion, our data show that PD‐L1 is expressed abundantly in MPN cells, including MPN‐initiating phenotypically defined CD34+CD45dimCD38− NSC. In addition, we demonstrate that PD‐L1 levels are highest in neoplastic cells in patients with PV, correlate with the JAK2‐V617F burden and with chromosome 9p UPD, and are promoted by IFN‐γ exposure through a BRD4/MYC‐dependent pathway. We also provide first evidence that germline genetic factors at the PD‐L1 locus contribute to MPN susceptibility independently of the GGCC (46/1) risk haplotype. Since PD‐L1 is a major resistance‐mediating immune checkpoint, these data may have clinical implications and may pave the way for the development and application of new PD‐L1‐blocking therapies in MPN.
CONFLICTS OF INTEREST
R.K. has received honoraria from and served on the advisory board of AOP Orphan Pharmaceuticals AG, has received honoraria from Pharma Essentia, and has equity ownership in MyeloPro Diagnostics and Research GmbH. H.G. has been a consultant to and received honoraria and research funding from AOP Orphan Pharmaceuticals AG; has received honoraria from Novartis, Celgene, and Janssen‐Cilag; has been a consultant to Roche, MyeloPro Diagnostics, and Research GmbH; and has received personal fees from PharmaEssentia. P.V. received honoraria from Novartis, Incyte and BMS/Celgene. The remaining authors declare no competing financial interests.
Supporting information
Appendix S1 Supporting Information
ACKNOWLEDGMENTS
This work was supported by the Austrian Science Fund (FWF) grant F4704‐B20 to P.V. The investigations conducted at Fondazione IRCCS Policlinico San Matteo, Pavia, Italy, were supported by the Associazione Italiana per la Ricerca sul Cancro (AIRC), Milan, Italy (AIRC 5 × 1000 project #21267 and International Accelerator project #22796). In addition, E.R. was supported by a grant from the Italian Ministry of Health for young researchers (GR‐2016‐02361272). We thank the team of the Biomedical Sequencing Facility at CeMM for assistance with next‐generation sequencing and data analysis.
Milosevic Feenstra JD, Jäger R, Schischlik F, et al. PD‐L1 overexpression correlates with JAK2‐V617F mutational burden and is associated with 9p uniparental disomy in myeloproliferative neoplasms. Am J Hematol. 2022;97(4):390-400. doi: 10.1002/ajh.26461
Funding information This work was supported by the Austrian Science Fund (FWF) grant F4704‐B20 to P.V. The investigations conducted at Fondazione IRCCS Policlinico San Matteo, Pavia, Italy, were supported by the Associazione Italiana per la Ricerca sul Cancro (AIRC), Milan, Italy (AIRC 5 × 1000 project #21267 and International Accelerator project #22796). In addition, ER was supported by a grant from the Italian Ministry of Health for young researchers (GR‐2016‐02361272).
DATA AVAILABILITY STATEMENT
The RNA‐sequencing and microarray data have been deposited in the following repositories: European Genome‐phenome Archive; Accession ID: EGAS00001003486 and Array express; Accession ID: E‐MTAB‐1845.
REFERENCES
- 1. Barosi G. An immune dysregulation in MPN. Curr Hematol Malig Rep. 2014;9:331‐339. [DOI] [PubMed] [Google Scholar]
- 2. Vainchenker W, Kralovics R. Genetic basis and molecular pathophysiology of classical myeloproliferative neoplasms. Blood. 2017;129:667‐679. [DOI] [PubMed] [Google Scholar]
- 3. Baxter EJ, Scott LM, Campbell PJ, et al. Acquired mutation of the tyrosine kinase JAK2 in human myeloproliferative disorders. Lancet. 2005;365:1054‐1061. doi: 10.1016/S0140-6736(05)71142-9 [DOI] [PubMed] [Google Scholar]
- 4. James C, Ugo V, Le Couedic JP, et al. A unique clonal JAK2 mutation leading to constitutive signalling causes polycythaemia vera. Nature. 2005;434:1144‐1148. doi: 10.1038/nature03546 [DOI] [PubMed] [Google Scholar]
- 5. Kralovics R, Passamonti F, Buser AS, et al. A gain‐of‐function mutation of JAK2 in myeloproliferative disorders. N Engl J Med. 2005;352:1779‐1790. doi: 10.1056/NEJMoa051113 [DOI] [PubMed] [Google Scholar]
- 6. Levine RL, Wadleigh M, Cools J, et al. Activating mutation in the tyrosine kinase JAK2 in polycythemia vera, essential thrombocythemia, and myeloid metaplasia with myelofibrosis. Cancer Cell. 2005;7:387‐397. doi: 10.1016/j.ccr.2005.03.023 [DOI] [PubMed] [Google Scholar]
- 7. Klampfl T, Gisslinger H, Harutyunyan AS, et al. Somatic mutations of calreticulin in myeloproliferative neoplasms. N Engl J Med. 2013;369:2379‐2390. doi: 10.1056/NEJMoa1311347 [DOI] [PubMed] [Google Scholar]
- 8. Pardanani AD, Levine RL, Lasho T, et al. MPL515 mutations in myeloproliferative and other myeloid disorders: a study of 1182 patients. Blood. 2006;108:3472‐3476. doi: 10.1182/blood-2006-04-018879 [DOI] [PubMed] [Google Scholar]
- 9. Pikman Y, Lee BH, Mercher T, et al. MPLW515L is a novel somatic activating mutation in myelofibrosis with myeloid metaplasia. PLoS Med. 2006;3:e270. doi: 10.1371/journal.pmed.0030270 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Abdulkarim K, Girodon F, Johansson P, et al. AML transformation in 56 patients with Ph‐ MPD in two well defined populations. Eur J Haematol. 2009;82:106‐111. doi: 10.1111/j.1600-0609.2008.01163.x [DOI] [PubMed] [Google Scholar]
- 11. Asher S, McLornan DP, Harrison CN. Current and future therapies for myelofibrosis. Blood Rev. 2020;42:100715. doi: 10.1016/j.blre.2020.100715 [DOI] [PubMed] [Google Scholar]
- 12. Guglielmelli P, Vannucchi AM. Current management strategies for polycythemia vera and essential thrombocythemia. Blood Rev. 2020;42:100714. doi: 10.1016/j.blre.2020.100714 [DOI] [PubMed] [Google Scholar]
- 13. Braun LM, Zeiser R. Immunotherapy in myeloproliferative diseases. Cell. 2020;9(6):1559. doi: 10.3390/cells9061559 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Prestipino A, Emhardt AJ, Aumann K, et al. Oncogenic JAK2(V617F) causes PD‐L1 expression, mediating immune escape in myeloproliferative neoplasms. Sci Transl Med. 2018;10:eaam7729. doi: 10.1126/scitranslmed.aam7729 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Wang JC, Chen C, Kundra A, et al. Programmed cell death receptor (PD‐1) ligand (PD‐L1) expression in Philadelphia chromosome‐negative myeloproliferative neoplasms. Leuk Res. 2019;79:52‐59. doi: 10.1016/j.leukres.2019.02.010 [DOI] [PubMed] [Google Scholar]
- 16. Freeman GJ, Long AJ, Iwai Y, et al. Engagement of the PD‐1 immunoinhibitory receptor by a novel B7 family member leads to negative regulation of lymphocyte activation. J Exp Med. 2000;192:1027‐1034. doi: 10.1084/jem.192.7.1027 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Latchman Y, Wood CR, Chernova T, et al. PD‐L2 is a second ligand for PD‐1 and inhibits T cell activation. Nat Immunol. 2001;2:261‐268. doi: 10.1038/85330 [DOI] [PubMed] [Google Scholar]
- 18. Curiel TJ, Wei S, Dong H, et al. Blockade of B7‐H1 improves myeloid dendritic cell‐mediated antitumor immunity. Nat Med. 2003;9:562‐567. doi: 10.1038/nm863 [DOI] [PubMed] [Google Scholar]
- 19. Zou W, Wolchok JD, Chen L. PD‐L1 (B7‐H1) and PD‐1 pathway blockade for cancer therapy: mechanisms, response biomarkers, and combinations. Sci Transl Med. 2016;8(328):328rv4. doi: 10.1126/scitranslmed.aad7118 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Cha JH, Chan LC, Li CW, Hsu JL, Hung MC. Mechanisms controlling PD‐L1 expression in cancer. Mol Cell. 2019;76(3):359‐370. doi: 10.1016/j.molcel.2019.09.030 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Garon EB, Rizvi NA, Hui R, et al. Pembrolizumab for the treatment of non‐small‐cell lung cancer. N Engl J Med. 2015;372:2018‐2028. doi: 10.1056/NEJMoa1501824 [DOI] [PubMed] [Google Scholar]
- 22. Wolchok JD, Kluger H, Callahan MK, et al. Nivolumab plus ipilimumab in advanced melanoma. N Engl J Med. 2013;369:122‐133. doi: 10.1056/NEJMoa1302369 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Topalian SL, Hodi FS, Brahmer JR, et al. Safety, activity, and immune correlates of anti‐PD‐1 antibody in cancer. N Engl J Med. 2012;366:2443‐2454. doi: 10.1056/NEJMoa1200690 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Brahmer JR, Tykodi SS, Chow LQ, et al. Safety and activity of anti‐PD‐L1 antibody in patients with advanced cancer. N Engl J Med. 2012;366:2455‐2465. doi: 10.1056/NEJMoa1200694 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Kralovics R, Guan Y, Prchal JT. Acquired uniparental disomy of chromosome 9p is a frequent stem cell defect in polycythemia vera. Exp Hematol. 2002;30:229‐236. doi: 10.1016/s0301-472x(01)00789-5 [DOI] [PubMed] [Google Scholar]
- 26. Wang L, Wheeler DA, Prchal JT. Acquired uniparental disomy of chromosome 9p in hematologic malignancies. Exp Hematol. 2016;44:644‐652. doi: 10.1016/j.exphem.2015.11.005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Schischlik F, Jager R, Rosebrock F, et al. Mutational landscape of the transcriptome offers putative targets for immunotherapy of myeloproliferative neoplasms. Blood. 2019;134:199‐210. doi: 10.1182/blood.2019000519 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Jia R, Balligand T, Atamanyuk V, et al. Hematoxylin binds to mutant calreticulin and disrupts its abnormal interaction with thrombopoietin receptor. Blood. 2020;137:1920‐1931. doi: 10.1182/blood.2020006264 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Herrmann H, Sadovnik I, Eisenwort G, et al. Delineation of target expression profiles in CD34+/CD38− and CD34+/CD38+ stem and progenitor cells in AML and CML. Blood Adv. 2020;4:5118‐5132. doi: 10.1182/bloodadvances.2020001742 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Eisenwort G, Sadovnik I, Schwaab J, et al. Identification of a leukemia‐initiating stem cell in human mast cell leukemia. Leukemia. 2019;33:2673‐2684. doi: 10.1038/s41375-019-0460-6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Ivanov D, Milosevic Feenstra JD, Eisenwort G, et al. Phenotyping of disease‐initiating CD34+/CD38─ stem cells in BCR‐ABL1─ MPN reveals expression of multiple cytokine receptors and resistance‐related antigens. Blood. 2020;136(Supplement 1):53. doi: 10.1182/blood-2020-140477 [DOI] [Google Scholar]
- 32. Lysenko V, Wildner‐Verhey van Wijk N, Zimmermann K, et al. Enhanced engraftment of human myelofibrosis stem and progenitor cells in MISTRG mice. Blood Adv. 2020;4(11):2477‐2488. doi: 10.1182/bloodadvances.2019001364 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Klampfl T, Harutyunyan A, Berg T, et al. Genome integrity of myeloproliferative neoplasms in chronic phase and during disease progression. Blood. 2011;118:167‐176. doi: 10.1182/blood-2011-01-331678 [DOI] [PubMed] [Google Scholar]
- 34. Holle R, Happich M, Lowel H, Wichmann HE, Group MKS . KORA‐‐a research platform for population based health research. Gesundheitswesen. 2005;67(Suppl 1):S19‐S25. doi: 10.1055/s-2005-858235 [DOI] [PubMed] [Google Scholar]
- 35. Purcell S, Neale B, Todd‐Brown K, et al. PLINK: a tool set for whole‐genome association and population‐based linkage analyses. Am J Hum Genet. 2007;81:559‐575. doi: 10.1086/519795 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. R Core Team . R: A Language and Environment for Statistical Computing. Foundation for Statistical Computing; 2013. [Google Scholar]
- 37. Olcaydu D, Harutyunyan A, Jager R, et al. A common JAK2 haplotype confers susceptibility to myeloproliferative neoplasms. Nat Genet. 2009;41:450‐454. doi: 10.1038/ng.341 [DOI] [PubMed] [Google Scholar]
- 38. Herbst RS, Soria JC, Kowanetz M, et al. Predictive correlates of response to the anti‐PD‐L1 antibody MPDL3280A in cancer patients. Nature. 2014;515:563‐567. doi: 10.1038/nature14011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Maleki Vareki S, Garrigos C, Duran I. Biomarkers of response to PD‐1/PD‐L1 inhibition. Crit Rev Oncol Hematol. 2017;116:116‐124. doi: 10.1016/j.critrevonc.2017.06.001 [DOI] [PubMed] [Google Scholar]
- 40. Ansell SM, Lesokhin AM, Borrello I, et al. PD‐1 blockade with nivolumab in relapsed or refractory Hodgkin's lymphoma. N Engl J Med. 2015;372:311‐319. doi: 10.1056/NEJMoa1411087 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Roemer MG, Advani RH, Ligon AH, et al. PD‐L1 and PD‐L2 genetic alterations define classical Hodgkin lymphoma and predict outcome. J Clin Oncol. 2016;34:2690‐2697. doi: 10.1200/JCO.2016.66.4482 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Roemer MGM, Redd RA, Cader FZ, et al. Major histocompatibility complex class II and programmed death ligand 1 expression predict outcome after programmed death 1 blockade in classic hodgkin lymphoma. J Clin Oncol. 2018;36:942‐950. doi: 10.1200/JCO.2017.77.3994 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Green MR, Monti S, Rodig SJ, et al. Integrative analysis reveals selective 9p24.1 amplification, increased PD‐1 ligand expression, and further induction via JAK2 in nodular sclerosing Hodgkin lymphoma and primary mediastinal large B‐cell lymphoma. Blood. 2010;116:3268‐3277. doi: 10.1182/blood-2010-05-282780 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Vilaine M, Olcaydu D, Harutyunyan A, et al. Homologous recombination of wild‐type JAK2, a novel early step in the development of myeloproliferative neoplasm. Blood. 2011;118:6468‐6470. doi: 10.1182/blood-2011-08-372813 [DOI] [PubMed] [Google Scholar]
- 45. Wang L, Swierczek SI, Lanikova L, et al. The relationship of JAK2(V617F) and acquired UPD at chromosome 9p in polycythemia vera. Leukemia. 2014;28:938‐941. doi: 10.1038/leu.2014.20 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Dong H, Strome SE, Salomao DR, et al. Tumor‐associated B7‐H1 promotes T‐cell apoptosis: a potential mechanism of immune evasion. Nat Med. 2002;8:793‐800. doi: 10.1038/nm730 [DOI] [PubMed] [Google Scholar]
- 47. Garcia‐Diaz A, Shin DS, Moreno BH, et al. Interferon receptor signaling pathways regulating PD‐L1 and PD‐L2 expression. Cell Rep. 2017;19:1189‐1201. doi: 10.1016/j.celrep.2017.04.031 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Vaidya R, Gangat N, Jimma T, et al. Plasma cytokines in polycythemia vera: phenotypic correlates, prognostic relevance, and comparison with myelofibrosis. Am J Hematol. 2012;87:1003‐1005. doi: 10.1002/ajh.23295 [DOI] [PubMed] [Google Scholar]
- 49. Obro NF, Grinfeld J, Belmonte M, et al. Longitudinal cytokine profiling identifies GRO‐alpha and EGF as potential biomarkers of disease progression in essential Thrombocythemia. Hemasphere. 2020;4:e371. doi: 10.1097/HS9.0000000000000371 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Tabarroki A, Rogers HJ, Visconte V, et al. The molecular and cytokine profile of triple‐negative (JAK2 V617F, JAK2 exon 12, MPL negative) myelofibrosis, a myeloproliferative neoplasm with distinct Clinico‐pathologic characteristics. Blood. 2012;120:3805. doi: 10.1182/blood.V120.21.3805.3805 [DOI] [Google Scholar]
- 51. Bozkus CC, Roudko V, Finnigan JP, et al. Shared Neoantigen‐induced T‐cell immunity directed against mutated Calreticulin in myeloproliferative neoplasms. Cancer Discov. 2019;9:1192‐1207. doi: 10.1158/2159-8290.CD-18-1356 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Hogg SJ, Vervoort SJ, Deswal S, et al. BET‐bromodomain inhibitors engage the host immune system and regulate expression of the immune checkpoint ligand PD‐L1. Cell Rep. 2017;18:2162‐2174. doi: 10.1016/j.celrep.2017.02.011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Zhu H, Bengsch F, Svoronos N, et al. BET bromodomain inhibition promotes anti‐tumor immunity by suppressing PD‐L1 expression. Cell Rep. 2016;16:2829‐2837. doi: 10.1016/j.celrep.2016.08.032 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Vannucchi AM, Kiladjian JJ, Griesshammer M, et al. Ruxolitinib versus standard therapy for the treatment of polycythemia vera. N Engl J Med. 2015;372(5):426‐435. doi: 10.1056/NEJMoa1409002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Verstovsek S, Mesa RA, Gotlib J, et al. A double‐blind placebo‐controlled trial of ruxolitinib for myelofibrosis. N Engl J Med. 2012;366(9):799‐807. doi: 10.1056/NEJMoa1110557 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Saenz DT, Fiskus W, Manshouri T, et al. BET protein bromodomain inhibitor‐based combinations are highly active against post‐myeloproliferative neoplasm secondary AML cells. Leukemia. 2017;31:678‐687. doi: 10.1038/leu.2016.260 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Saenz DT, Fiskus W, Qian Y, et al. Novel BET protein proteolysis‐targeting chimera exerts superior lethal activity than bromodomain inhibitor (BETi) against post‐myeloproliferative neoplasm secondary (s) AML cells. Leukemia. 2017;31:1951‐1961. doi: 10.1038/leu.2016.393 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Appendix S1 Supporting Information
Data Availability Statement
The RNA‐sequencing and microarray data have been deposited in the following repositories: European Genome‐phenome Archive; Accession ID: EGAS00001003486 and Array express; Accession ID: E‐MTAB‐1845.