

Abstract
Summary
Use of PacBio sequencing for characterizing barcoded libraries of genetic variants is on the rise. However, current approaches in resolving PacBio sequencing artifacts can result in a high number of incorrectly identified or unusable reads. Here, we developed a PacBio Read Alignment Tool (PacRAT) that improves the accuracy of barcode-variant mapping through several steps of read alignment and consensus calling. To quantify the performance of our approach, we simulated PacBio reads from eight variant libraries of various lengths and showed that PacRAT improves the accuracy in pairing barcodes and variants across these libraries. Analysis of real (non-simulated) libraries also showed an increase in the number of reads that can be used for downstream analyses when using PacRAT.
Availability and implementation
PacRAT is written in Python and is freely available (https://github.com/dunhamlab/PacRAT).
Supplementary information
Supplemental data are available at Bioinformatics online.
1 Introduction
Improvements in sequencing technology have greatly expanded our capacity not just for detecting genetic variants but also for assessing their function. Approaches such as deep mutational scanning (DMS) use a library of variants to characterize the impact of genetic variation on protein structure, gene expression and gene function across many fields (Kinney and McCandlish, 2019). Such variant libraries are commonly ‘barcoded’ with short random DNA tags, which allows for short-read sequencing of multiple time points and removes the step of directly sequencing entire variants at every time point. Barcodes are associated with library variants, which is a simple process if the variable region and the linked barcode can be sequenced together with short-read methods. However, with longer variable regions and/or distance to the barcode this becomes increasingly costly, challenging and time-consuming (Hiatt et al., 2010). Many studies are turning to PacBio sequencing to generate high-quality long reads that span the entirety of the barcode and variable region (Amorosi et al., 2021; Matreyek et al., 2018; Ollodart et al., 2021; Yeh et al., 2021).
Compared to Illumina short-read sequencing, PacBio generates much longer continuous reads, though with the disadvantage of an increased error rate, which can be as high as 15% (Wenger et al., 2019). To reduce errors, highly accurate reads are generated from circular consensus sequencing (CCS), which circularizes molecules by ligating single-stranded SMRTbellTM adaptors and allowing a single DNA molecule to be sequenced multiple times. Each pass through the molecule produces a subread, and these subreads are collapsed to form a more accurate consensus sequence. Although PacBio chemistry has improved, errors are still pervasive in CCS reads. Compared to Illumina NovaSeq, the PacBio mismatch rate is 17× lower, but indel rates are 181× higher (Wenger et al., 2019). Such indel errors are more prominent in CCS reads with lower numbers of subreads, often those from longer target sequences, as fewer complete passes are contained within the average read length. Therefore, additional error correction steps are required to improve reliability and maximize information gained from PacBio consensus reads.
In this article, we present PacBio Read Alignment Tool (‘PacRAT’) which maximizes the number of usable reads for barcode-variant mapping while reducing sequencing errors found in CCS reads.
2 Materials and methods
PacRAT is a reference-free, highly reliable approach for linking barcodes to variants and improves upon the output of a previously used method, Assembly By PacBio (ABP) (Matreyek et al., 2018). ABP assigns barcodes to variants by taking either the highest-frequency sequence associated with each barcode or the highest average quality sequence associated with each barcode. Crucially, this assigned variant often retains sequencing errors (most commonly 1 bp indels), depending on the coverage and quality of reads associated with each barcode.
To fix this issue, our program assigns barcodes to variants by aligning CCS reads with the same barcode using multiple sequence alignment (MSA) (Edgar, 2004). A consensus sequence is then generated from these alignments. If no ambiguous nucleotides are present, this consensus sequence is used as the final, error-corrected sequence. However, if ambiguities in these alignments persist, a second pairwise alignment is performed with EMBOSS Needle (Rice et al., 2000) between the highest average quality CCS read and the derived consensus sequence. Sites with ambiguities are resolved by taking the nucleotide sequence from the highest quality read. Thresholds for determining consensus sequences can be specified by the user, with the default requiring that the majority of sequences share the same base at each position.
3 Results and conclusions
To assess the accuracy of PacRAT, we computationally generated eight DMS libraries with a range of sizes (759–4086 bp). Using SimLoRD, a PacBio SMRT read simulator (Stöcker et al., 2016), we simulated CCS reads in triplicate from these DMS libraries with 10×, 5× and 3× read coverage (Fig. 1 and Supplementary Fig. S1). A one-way ANOVA test and post hoc Tukey test showed that barcode-variant mapping significantly improves with PacRAT in nearly all libraries (Fig. 1, Supplementary Fig. S1 and Supplementary Table S1). For the largest simulated library (4086 bp), PacRAT correctly identified the variant for 97% of barcodes, whereas ABP only identified 14% (Fig. 1). Efficiency decrease is amplified as variable region lengths increase since MSA is able to correct many of the errors present in longer libraries. 5× and 3× downsamples of our libraries correctly matched 72% and 41% of barcodes from the longest simulated library, respectively. In a smaller 2.1 kb library, we observed that 99.7%, 91.5% and 72.7% of barcodes were correctly identified with PacRAT at 10×, 5× and 3× coverage, respectively. These results indicate that PacRAT can accommodate reduced sequencing read requirements while maintaining the accuracy of data, which could reduce library sequencing costs and improve throughput.
Fig. 1.
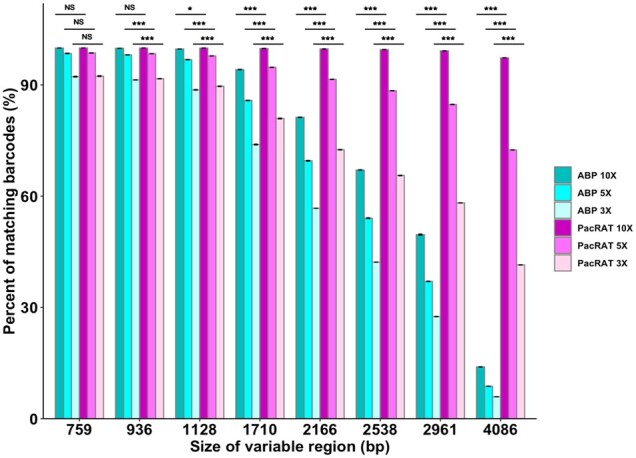
Comparison of correct variant identification between ABP and PacRAT in simulated libraries with different lengths of variable regions. Variant libraries of eight different variable region lengths were generated using SimLoRD (Stöcker et al., 2016) with parameters −xn 8e−03 2.54 5500 −xs 2.78e−03 −0.48 825 48303.073 1.469 chosen to better simulate longer variable regions (Supplementary Table S1) at 10×, 5× and 3× read coverage. Significance values are derived from a one-way ANOVA test and Tukey post hoc comparisons (see Supplementary Table S1): NS, not significant. P < 0.05 (*), P < 0.0005 (***)
To verify that our program improves barcode-variant mapping with real PacBio data, we re-analyzed two published variant libraries created for genes MSH2 (Ollodart et al., 2021) and CYP2C9 (Amorosi et al., 2021). Additional information on the generation and characteristics of these libraries can be found in Supplementary Tables S2 and S3. The MSH2 library was generated at 22X coverage via DNA synthesis, where specific single amino acid variants were requested from Twist Biosciences. PacRAT decreased the number of unexpected variants from 16.8% of barcodes analyzed with ABP to 10.5% with PacRAT (Supplementary Table S3). Downsamples of this library showed consistent results, and we are unable to distinguish if these errors are sequencing or synthesis errors.
For the ∼118 000-barcode CYP2C9 library, which was generated by NNK mutagenesis at each codon via inverse PCR, PacRAT increased the number of barcodes associated with on-target variants from 36 108 barcodes to 39 847 barcodes, while reducing the number of barcodes associated with indels from 66 761 barcodes to 61 551 barcodes. Barcodes associated with indels are expected to have low functional scores, but with ABP only, we observed 3043 barcodes associated with indels that had scores similar to wild-type variants, likely indicating a variant misclassification. PacRAT was able to reduce the number of these misclassifications by 48.4%.
PacRAT improves upon existing PacBio library barcode-variant mapping pipelines by implementing MSAs to resolve the variant sequence associated with each barcode. Our approach improves the data quality and reduces the costs of sequencing by utilizing reads that were assigned incorrect sequences or otherwise discarded in previous methods.
Supplementary Material
Acknowledgements
We thank to Phil Green for his original suggestion of this approach. We thank Jochen Weile, Atina Cote and Fritz Roth for the helpful suggestions to improve PacRAT. We also thank to Nick Popp for beta testing PacRAT.
Funding
Research reported in this publication was supported by the National Institute of General Medical Sciences of the National Institutes of Health (NIH) under award numbers [R01 GM132162 and R01 GM101091]; the National Human Genome Research Institute of the NIH [T32 HG00035 to C.J.A.]; National Science Foundation Graduate Research Fellowships Program [DGE-1762114 to C.C.Y.]; the Bonita and David Brewer Fellowship to S.S. The research of M.J.D. was supported in part by a Faculty Scholar grant from the Howard Hughes Medical Institute.
Conflict of Interest: none declared.
Data Availability
Scripts for simulations are available on the Github link provided below the abstract. Other data are reanalyzed datasets that are publicly available and can be searched by going to those referenced publications.
Contributor Information
Chiann-Ling C Yeh, Department of Genome Sciences, University of Washington, Seattle, WA 98195, USA.
Clara J Amorosi, Department of Genome Sciences, University of Washington, Seattle, WA 98195, USA.
Soyeon Showman, Department of Genome Sciences, University of Washington, Seattle, WA 98195, USA; Molecular and Cellular Biology Program, University of Washington, Seattle, WA 98195, USA.
Maitreya J Dunham, Department of Genome Sciences, University of Washington, Seattle, WA 98195, USA.
References
- Amorosi C.J. et al. (2021) Massively parallel characterization of CYP2C9 variant enzyme activity and abundance. Am. J. Hum. Genet., 108, 1735–1751. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Edgar R.C. (2004) MUSCLE: multiple sequence alignment with high accuracy and high throughput. Nucleic Acids Res., 32, 1792–1797. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hiatt J.B. et al. (2010) Parallel, tag-directed assembly of locally derived short sequence reads. Nat. Methods, 7, 119–122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kinney J.B., McCandlish D.M. (2019) Massively parallel assays and quantitative sequence–function relationships. Annu. Rev. Genomics Hum. Genet., 20, 99–127. [DOI] [PubMed] [Google Scholar]
- Matreyek K.A. et al. (2018) Multiplex assessment of protein variant abundance by massively parallel sequencing. Nat. Genet., 50, 874–882. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ollodart A.R. et al. (2021) Multiplexing mutation rate assessment: determining pathogenicity of Msh2 variants in Saccharomyces cerevisiae. Genetics, 218, iyab058. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rice P. et al. (2000) EMBOSS: the European Molecular Biology Open Software Suite. Trends Genet., 16, 276–277. [DOI] [PubMed] [Google Scholar]
- Stöcker B.K. et al. (2016) SimLoRD: simulation of long read data. Bioinformatics, 32, 2704–2706. [DOI] [PubMed] [Google Scholar]
- Wenger A.M. et al. (2019) Accurate circular consensus long-read sequencing improves variant detection and assembly of a human genome. Nat. Biotechnol., 37, 1155–1162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yeh C.-L.C. et al. (2021) High-throughput functional analysis of natural variants in yeast. bioRxiv. https://doi.org/10.1101/2021.02.26.433108.
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
Scripts for simulations are available on the Github link provided below the abstract. Other data are reanalyzed datasets that are publicly available and can be searched by going to those referenced publications.