

Abstract
Background
Atrial fibrillation (AF) can lead to the loss of microvascular integrity thereby enhancing AF progression. Mechanistically, the pro‐coagulant state that drives the risk of stroke in patients with AF may also play a causal role in microvascular loss. Direct oral anticoagulants (DOACs), the preferred anticoagulants for AF, can target factors upstream (factor Xa [FXa]) or downstream (thrombin) in the coagulation cascade and mediate differential vascular effects through interaction with protease‐activated receptors (PARs).
Objective
To investigate the potential effect of different DOACs on vascular integrity.
Methods
To model the impact of DOACs on vascular integrity, we utilized platelet‐free plasma in thrombin generation assays and endothelial barrier assays under identical experimental conditions. These multifactorial systems provide all coagulation factors and their respective natural inhibitors in physiological ratios in combination with the pro‐coagulant endothelial surface on which coagulation is initiated. Furthermore, the system provides pro‐ and anti‐barrier factors and monitoring both assays simultaneously permits coupling of thrombin kinetics to endothelial barrier dynamics.
Results
We provide evidence that the anti‐FXa DOAC rivaroxaban and the anti‐thrombin DOAC dabigatran are efficient in blocking their target proteases. However, while rivaroxaban could preserve endothelial barrier function, dabigatran failed to protect endothelial integrity over time, which could be prevented in the presence of a custom‐made peptide that blocks thrombin’s exosite‐I.
Conclusions
Proteolytically inactive thrombin in complex with dabigatran evokes loss of barrier function that can be prevented by a protease‐activated receptor‐1 mimicking peptide blocking thrombin’s exosite‐I.
Keywords: anticoagulant drugs, atrial fibrillation, coagulation, direct thrombin inhibitors, endothelium
Essentials.
The effect of different direct oral anticoagulants on the endothelial component in atrial fibrillation is unknown.
Endothelial integrity was monitored in a plasma milieu permitting linkage of thrombin kinetics and barrier dynamics.
Dabigatran, in contrast to rivaroxaban, exerts an adverse non‐hemostatic effect on vascular integrity.
Exosite‐I, still accessible in dabigatran‐bound thrombin, may destabilize the endothelial barrier.
1. INTRODUCTION
Atrial fibrillation (AF) produces a hypercoagulable state, which may provoke pro‐fibrotic, pro‐hypertrophic, and pro‐inflammatory responses. 1 Currently, direct oral anticoagulants (DOACs) are the preferred choice of treatment for stroke prevention in AF. 2 , 3 Based on the preference of the practitioner, 4 DOACs either directed against factor Xa (FXa) or thrombin are prescribed. Anti‐FXa DOACs such as rivaroxaban, apixaban, and edoxaban inhibit the coagulation system upstream in the coagulation cascade, thereby preventing the formation of thrombin. The direct thrombin inhibitor (DTI) dabigatran allows for the formation of FXa and thrombin, but inhibits thrombin activity as soon as it is formed. 5 Both types of DOACs interact with the catalytically active site of their specific target protease in a reversible fashion, 6 while retaining all other coagulation proteases. Next to their effect in hemostasis, activated coagulation factors exert non‐hemostatic functions through interaction with protease‐activated receptors (PARs) expressed by endothelial cells (ECs) and many other cell types. 7
All PARs share a conserved mechanism of activation, in which protease cleavage events result in a neo‐exposed amino acid sequence that acts as tethered ligand and reassociates with the receptor to trigger a structural rearrangement leading to receptor activation. With regard to thrombin, its catalytic site recognizes the protease‐activated receptor‐1 (PAR‐1) sequence LDPR41‐S42 and a second interaction occurs between thrombin’s anion‐binding exosite‐I and an N‐terminal region of PAR‐1, which contains an acidic, hirudin‐like domain. This secondary interaction facilitates efficient cleavage of PAR‐1 at the R41‐S42 peptide bond, exposing the tethered ligand domain SFLLRN that binds intramolecularly to the receptor and promotes transmembrane signaling. 8
Vascular PAR signaling plays a key role in modulating diverse cellular activities. For instance, thrombin‐induced PAR‐1 signaling exerts barrier‐disruptive responses, while activated protein C (APC)‐induced PAR‐1 signaling and FXa‐induced PAR‐2 signaling can drive barrier protective responses on endothelial cells. 7
These observations imply that targeting individual proteases with different DOACs may have pronounced, differential effects on non‐hemostatic functions, despite comparable anticoagulant efficacies.
In the current study we set out to simultaneously study the coagulant potential and barrier integrity of ECs in vitro, an early step in endothelial destabilization and microvascular rarefaction. Tumor necrosis factor alpha (TNF‐α) was used to stimulate the ECs, because upregulation of plasma–TNF‐α is linked to AF and AF progression. 9 This experimental setup was combined with platelet‐free plasma (PFP), which provides the system with both pro‐ and anti‐barrier and pro‐ and anti‐coagulant factors at physiological concentrations, permitting linkage of endothelial barrier dynamics with thrombin kinetics.
We demonstrate that the DOACs rivaroxaban and dabigatran fulfilled their anti‐coagulant profile. However, while rivaroxaban could support barrier function, dabigatran could not maintain endothelial integrity. Dabigatran‐bound thrombin was catalytically inactive, but was still able to interact with PAR‐1 through exosite‐I, leading to destabilization of the endothelial barrier.
These results indicate that cellular function should be considered when evaluating safety and efficacy of anticoagulants. When translated to the clinical arena, DOACs specifically directed against FXa activity may well be preferred to the DTI dabigatran as they may spare microvascular integrity, prevent atrial remodelling, and dampen the vicious circle of coagulation induced AF–substrate formation.
2. MATERIALS AND METHODS
2.1. Cell culture
Human umbilical vein endothelial cells (HUVECs) were isolated and cultured in T75‐flask (precoated with 1% gelatin) and maintained in EGM2 medium (Promocell). The cells were incubated at 37°C in a humidified incubator under 5% CO2. The medium was changed every 2 or 3 days until the cells reached 80% confluence. Cells were then trypsinized and re‐seeded for further experiments. Only cells that underwent two to three passages were used for the experiments.
2.2. Reagents
Recombinant TNF‐α was purchased from Life Technologies. Rivaroxaban and dabigatran (Alsachim) were weighed and dissolved in DMSO; aliquots were stored at −20°C. Recombinant hirudin was dissolved in H2O and stored at −20°C (LabNed). PPACK was obtained from Calbiochem (Merck Biosciences). Plasma‐derived α‐thrombin and corn trypsin inhibitor (CTI; final concentration 70 µg/ml) were obtained from Haematologic Technologies.
2.3. Plasma preparation
Venous blood from healthy consenting volunteers not taking any medication was obtained from the biobank of the Leiden University Medical Center. All healthy volunteers signed a written informed consent. The Medical Ethics Committee of the Leiden University Medical Center approved the study protocol.
Blood was collected into Vacutainer® tubes containing ACD (Becton Dickinson). Whole blood was centrifuged at 200 g for 20 min at room temperature to prepare platelet‐rich plasma (PRP). PFP was prepared by centrifuging PRP twice at 1200 g for 15 min at room temperature and the depletion of platelets was confirmed by automated cell counter (Sysmex). All functional assays were performed in freshly prepared PRP and PFP.
Additional methods are available in the supporting information Appendix S1.
3. RESULTS
3.1. Activated endothelial cells as source of active tissue factor
Inflammatory biomarkers such as TNF‐α are found to be significantly increased in patients with AF, supporting a direct association between inflammation and AF. 10 To mimic this inflammatory condition, HUVECs were incubated with increasing concentrations of TNF‐α and the expression of full‐length tissue factor (TF) was measured. Under basal conditions, TF mRNA or surface expression of TF protein (antibody 10H10) was undetectable, while HUVECs exposed to TNF‐α (1–10 ng/ml, 5 h) induced TF mRNA (Figure 1A) and protein expression in a dose‐dependent fashion as measured by flow cytometry (Figure 1D). TF protein expression and localization was confirmed by immunohistochemical staining of permeabilized HUVECs (Figure 1C). Interestingly, those ECs maximally responsive to TNF‐α expressed large amounts of TF‐protein, which coincided with the release of von Willebrand factor (VWF) from their Weibel Palade bodies, a known additional prothrombotic effect of TNF‐α stimulation. 11
FIGURE 1.
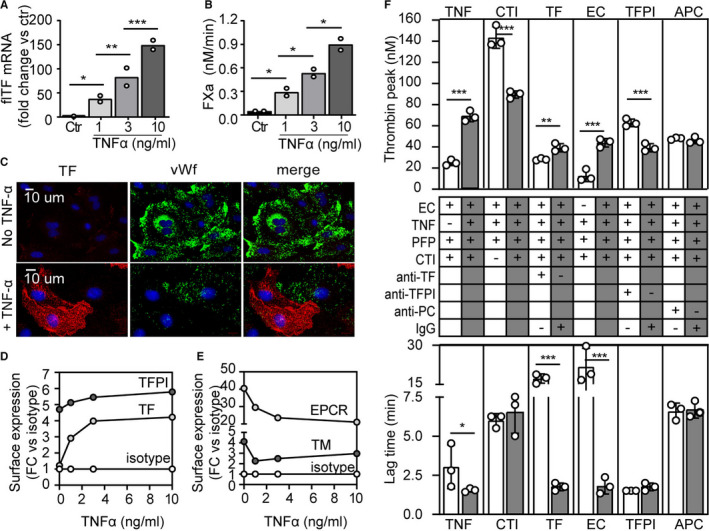
Tissue factor (TF)‐mediated thrombin generation in PFP incubated on ECs. A, HUVECs stimulated with TNF‐α (0–10 ng/ml, 5 h) show enhanced TF‐mRNA expression. (B, HUVECs stimulated with TNF‐α (0–10 ng/ml, 5 h) generate FXa due to expression of active TF on their surface. Data represent two independent experiments; means were compared using one‐way analysis of variance with Tukey’s multiple comparisons test. *P < .05, **P < .01; ***P < .001. C, Immunofluorescence images of permeabilized HUVECs stimulated with TNF‐α (5 ng/ml for 5 h) show TF‐protein expression stained with 10H10 and VWF–protein secretion. D‐E, HUVECs stimulated with TNF‐α (0–10 ng/mL, 5 hrs) show an enhanced surface expression of (D) TF and TFPI and a decreased expression of (E) EPCR and TM as measured by flow cytometry. F, Thrombin generation assays were performed on monolayers of confluent HUVECs, expressed as thrombin peak (nM) or lag time (min) measured in separate experiments: ± TNF‐α (1 µg/ml, 5 h); ± CTI to inhibit the intrinsic pathway component FXIIa; ± function‐blocking antibody directed against TF (5G9, 50 μg/ml) or an IgG control (50 μg/mL); ± ECs as source of TF and phospholipids; ± function‐blocking antibody against TFPI (ADG4903, 5 µg/ml) or an IgG‐control (5 µg/ml); ± antibody HPC4 (100 μg/ml) to inhibit activation of PC or an IgG control (100 μg/ml). Individual experiments were performed with different HUVEC batches and PFP donors in which the reference condition EC + TNF‐α + PFP is depicted with gray bars and the varied condition with white bars. Mean ± SD per conditions (at least three independent experiments) was compared using a 2‐tailed Student’s t‐test. *P < .05, **P < .01, ***P < .001. CTI, corn trypsin inhibitor; ECs, endothelial cells; EPCR, endothelial protein C receptor; FACS, fluorescence‐activated cell sorting; HUVECs, human umbilical vein endothelial cells; PC, protein C; PFP, platelet free plasma; TFPI, tissue factor pathway inhibitor; TM, thrombomodulin; TNF‐α; tumor necrosis factor alpha; VWF, von Willebrand factor
The functional activity of TF was studied by incubating HUVEC monolayers with purified FX and factor VIIa (FVIIa) and FXa generation was monitored. Incubation of the HUVECs with TNF‐α resulted in a dose‐dependent activation of factor X (FX; Figure 1B). These results confirm that under these pro‐inflammatory conditions, the vascular endothelium is a source of procoagulant TF.
To investigate other phenotypic changes upon TNF‐α stimulation, surface expression of tissue factor pathway inhibitor (TFPI), thrombomodulin (TM), and endothelial protein C receptor (EPCR) was measured by fluorescence‐activated cell sorting (FACS) on dissociated ECs and expressed as fold change of an isotype control (IT; Figure 1D,E and Figure S1 in supporting information for histographs). Data of one HUVEC donor are shown, but other EC isolations gave similar results. TNF‐α stimulation resulted in a decrease of the protein C (PC)‐related receptors TM and EPCR and a slight increase in TFPI.
3.2. Thrombin generation on endothelial cells
Next, to assess the procoagulant state of the TNF‐α–conditioned ECs, thrombin generation in PFP was measured on a monolayer of HUVECs. PFP rather than isolated coagulation factors was used to administer all coagulation factors and their respective inhibitors in normal ratios. In this way, the interplay of endogenous pro‐ and anti‐coagulation mediators such as antithrombin and components of the PC pathway is maximally preserved. Thus, this experimental setup ensures thrombin formation in a physiologically relevant environment. PFP incubated on TNF‐α–activated ECs showed a significantly increased thrombin peak and shortened lag time over non‐activated ECs (Figure 1F, TNF).
To examine the contribution of contact activation under these conditions the FXIIa inhibitor CTI (70 µg/ml) was added to the system, which resulted in a significantly reduced thrombin peak compared to the untreated condition (Figure 1F, CTI). These results show that addition of CTI to PFP to inhibit the intrinsic pathway of coagulation is essential to examine the TF‐dependent activation of coagulation (extrinsic pathway) and therefore CTI was added in all following experiments.
To confirm the principal role of TF under these circumstances, a function‐neutralizing monoclonal antibody (Mab) of TF (5G9) was added to PFP. Addition of 5G9 markedly prolonged the lag time of thrombin generation on TNF‐α–activated ECs and a significantly decreased thrombin peak compared to the IgG control (Figure 1F, TF). Activation of PFP in the absence of ECs showed a very low thrombin activity and substantial prolongation of the lag time, confirming the contribution of ECs as a source of TF (Figure 1F, EC).
Next, we studied the potential contribution of TFPI. TFPI is expressed by the endothelium and secreted following thrombin formation, 12 upon which it serves as a FXa‐dependent inhibitor of the TF‐FVIIa complex on the EC surface. 13 The function blocking antibody ADG4903 was used, which neutralizes TFPI by interfering with the binding of FXa. Blocking TFPI significantly increased thrombin activity but did not affect the lag time (Figure 1F, TFPI). To investigate the contribution of APC in thrombin generation, antibody HPC4 was added to the plasma, which blocks PC activation mediated by the thrombin–TM complex. 14 , 15 This antibody did not affect thrombin peak nor lag time (Figure 1F, APC).
3.3. Rivaroxaban and dabigatran effectively block thrombin generation on endothelial cells
To determine the concentrations required to completely block FXa activity and thrombin generation in PFP, increasing concentrations of the DOACs rivaroxaban and dabigatran were added. Rivaroxaban significantly reduced the thrombin peak by 88% at a concentration of 1 μM and completely blocked thrombin generation at 10 μM (Figure S2A‐B in supporting information). In addition, a dose‐dependent and significant prolongation in lag time was observed for reactions with increasing concentrations of rivaroxaban, with the lag time exceeding 120 min at 10 μM (Figure S2C). Dabigatran significantly reduced the thrombin peak by 95% at a concentration of 3 μM and completely blocked thrombin activity at 10 μM (Figure S2D‐E). Furthermore, increasing concentrations of dabigatran significantly prolonged the lag time to more than 120 min at 10 μM (Figure S2F).
Estimation of the IC50 revealed that this value for dabigatran was approximately 4‐fold higher (IC50 2 µM) relative to rivaroxaban (IC50 0.5 μM). These results imply that the coagulation cascade is more efficiently inhibited at the level of FXa by rivaroxaban, thereby preventing the formation of thrombin, than by direct thrombin inhibition by dabigatran. In following experiments, 10 μM DOACs were used to ensure the absence of proteolytically active thrombin in the system.
3.4. Rivaroxaban maintains endothelial cell integrity, while dabigatran does not
An electric cell‐substrate impedance sensor (ECIS) assay was used to determine the effect of PFP on EC barrier function (Figure 2A). The experimental conditions for the ECIS assay were identical to those of the thrombin generation assay, to ensure that EC integrity measurements overlap with those of the thrombin generation. In short, ECs activated with TNF‐α (1 ng/ml) for TF expression were washed and incubated with PFP and EC resistance was measured. In general, the EC washing procedure temporarily led to a loss in barrier function, but over time, the resistance curve showed a recovery (Figure 2A). This barrier‐recovery capacity is a marker of EC integrity and is expressed as percentage of maximal recovery measured before the washing procedure (barrier‐reference).
FIGURE 2.
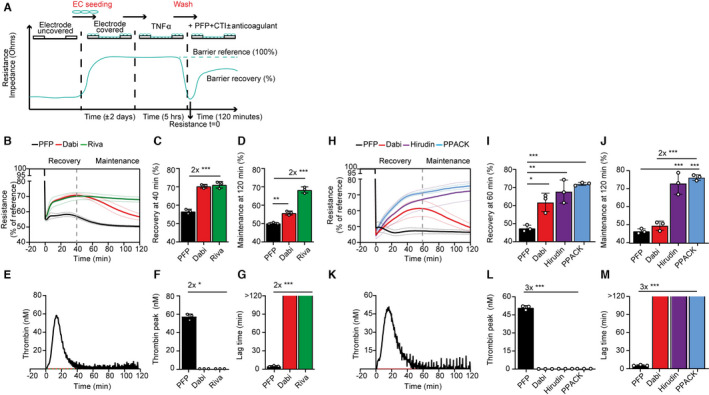
Rivaroxaban maintains EC integrity, while dabigatran does not. A, Schematic of ECIS experimental setup. The barrier‐recovery capacity was expressed as percentage of maximal recovery measured before the washing procedure (barrier reference). B and H, Examples of ECIS barrier recovery curves over time on TNF‐α–stimulated HUVECs from one PFP donor and an EC donor (mean ± SD values of triplicate wells in dotted lines) measured from −20 to 120 min of PFP, PFP + dabigatran (Dabi), PFP + rivaroxaban (Riva), PFP + hirudin (Hirudin), and PFP + PPACK (PPACK). Recovery phase = 0–40/60 min; maintenance phase = 40/60–120 min, varying per experiment. C and I, Barrier resistance values of the recovery phase (three wells per condition at 40/60 min) and (D and J) maintenance phase (at 120 min). E and K, Examples of thrombin generation curves over time from the same plasma‐ and EC donors used in (B) and (H), respectively. Quantification of thrombin generation expressed as (F and L) thrombin peak (mean ± SD in nM) and (G and M) lag time (mean ± SD in min). Means were compared using one‐way analysis of variance with Tukey’s multiple comparisons test. *P < .05, **P < .01, ***P < .001. ECs, endothelial cells; ECIS, electric cell‐substrate impedance sensor; HUVECs, human umbilical vein endothelial cells; PFP, platelet‐free plasma; SD, standard deviation; TNF‐α; tumor necrosis factor alpha
Barrier function was assessed with PFP either with or not supplemented with 10 μM rivaroxaban or dabigatran. PFP without coagulation inhibitors did not rescue barrier function (Figure 2B), which coincided with the generation of high thrombin levels (Figure 2E‐F). Strikingly, assessment of EC barrier function by PFP supplemented with 10 μM rivaroxaban revealed a biphasic rescue (Figure 2B). The period from 0–40 min was defined as recovery phase and from 40–120 min as maintenance phase and these periods were quantified by calculating average values at 40 min (recovery, Figure 2C) and 120 min (maintenance, Figure 2D). Interestingly, dabigatran‐anticoagulated plasma did support the recovery phase to the same extent as rivaroxaban (Figure 2C), but barrier function in the maintenance phase declined significantly over time (Figure 2D), while thrombin was maximally inhibited (Figure 2, 3, 4, 5; mean ± standard deviation [SD] of triplicate wells). These results indicate that prevention of thrombin generation by rivaroxaban protects the EC barrier more efficiently than dabigatran‐inhibited thrombin.
FIGURE 3.
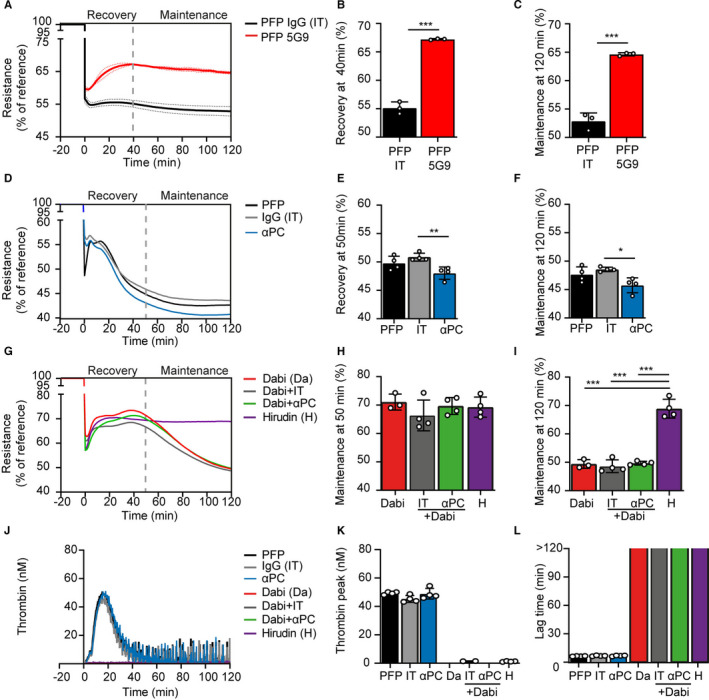
Role of TF and APC on EC barrier function. A, Representation of ECIS barrier recovery curves over time on TNF‐α–stimulated HUVECs with PFP in the absence or presence of function‐blocking TF antibody (5G9, 50 µg/ml) or an isotype IgG (IT 50 µg/ml). B, Quantification of the barrier resistance at the recovery phase (at 40 min) and C, maintenance phase (at 120 min) shows that blockage of TF supports barrier recovery significantly (two‐tailed Student’s t‐test, P < .001). D, Barrier recovery curves with the quantification for the (E) recovery phase at 50 min and the (F) maintenance phase at 120 min showed that inhibition of PC activation with antibody HPC4 (αPC, 100 μg/ml) reduced the barrier in the recovery and maintenance phase in the absence of dabigatran compared to isotype IgG (IT, 100 μg/ml). In the presence of dabigatran, HPC4 had no effect on the (E) recovery phase nor the (F) maintenance phase compared to IT. G, Thrombin generation curves with quantification expressed as (H) thrombin peak (nM) and (I) lag time (min) showed no effect of HPC4 versus IT in PFP and no effect of HPC4 versus IT in PFP + dabigatran. Means were compared using one‐way analysis of variance with Tukey’s multiple comparisons test. *P < .05, **P < .01, ***P < .001. αPC = HPC4 antibody (100 μg/ml); APC, activated protein C; Da, dabigatran; EC, endothelial cell; ECIS, electric cell‐substrate impedance sensor; H, hirudin; HUVECs, human umbilical vein endothelial cells; IT, IgG isotype control (100 μg/ml); PC, protein C; PFP, platelet‐free plasma; SD, standard deviation; TF, tissue factor; TNF‐α; tumor necrosis factor alpha
FIGURE 4.
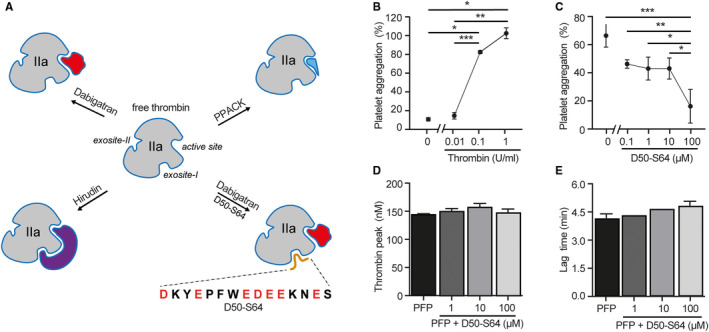
Thrombin binding capacity of peptide D50‐S64. A, Schematic illustration of free thrombin with its active site and exosite‐I and ‐II and in complex with different direct thrombin inhibitors. Dabigatran is a non‐peptidic, reversible inhibitor interacting with thrombin’s active site only, leaving exosite‐I and ‐II available. Hirudin binding is irreversible and will interact with both the active site and exosite‐I. PPACK is a peptide derivative, which forms an irreversible interaction with the active site, leaving exosite‐I available. Peptide D50‐S64 was designed, which mimics the PAR‐1 sequence that will interact with exosite‐I of thrombin. Incubation of thrombin with both peptide and dabigatran will reflect the situation of hirudin‐blocked thrombin. B, Light transmission aggregometry was performed with purified platelets (200.106/ml) incubated with thrombin (0–1 U/ml). C, Thrombin‐induced (0.1 U/ml) platelet aggregation was dose‐dependently inhibited with peptide D50‐S64 (0–100 µM). Individual points of three separate experiments with three different platelet donors are shown. A standardized CAT assay was performed using pooled PFP, to which purified TF and phospholipids were added. A representative thrombin generation curve of PFP ± a dose response of peptide D50‐S64 (0–100 μM) is shown. The peptide did not affect (D) peak height (nM) or (E) lag time (min) of thrombin generation. Mean ± standard deviation were compared using one‐way analysis of variance with Tukey’s multiple comparisons test. *P < .05, **P < .01, ***P < .001. CAT, calibrated automated thrombogram; PAR‐1, protease‐activated receptor‐1 ; PFP, platelet‐free plasma; TF, tissue factor
FIGURE 5.
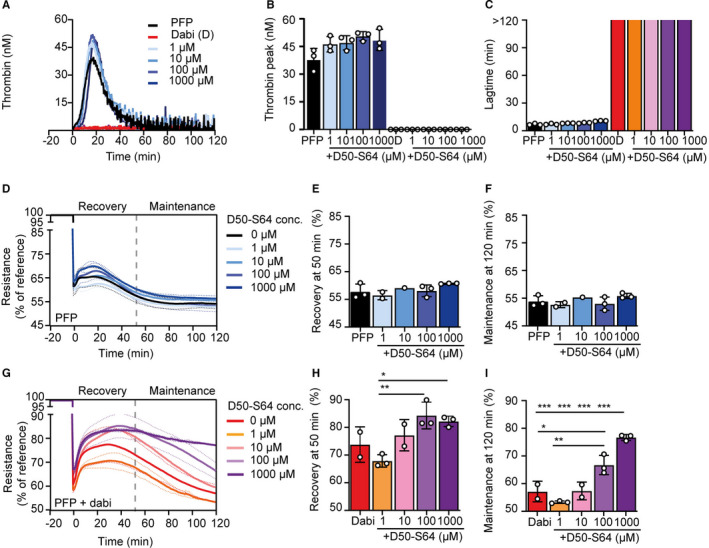
The effect of peptide D50‐S64 on thrombin generation and barrier function. A, Representation from one plasma‐donor and an endothelial cell donor of thrombin generation (nM) over time in platelet‐free plasma (PFP), PFP supplemented with dabigatran (D or dabi), peptide D50‐S64 (1–1000 µM), or dabigatran plus peptide D50‐S64 (1–1000 µM) incubated on tumor necrosis factor‐‐stimulated human umbilical vein endothelial cells as source of tissue factor and phospholipids. B, Quantification of thrombin peak values (mean ± standard deviation [SD] in nM) and (C) lag time (min) showed no effect of peptide D50‐S64 compared to PFP alone and no extra effect in combination with dabigatran. D, An example of electric cell‐substrate impedance sensor (ECIS) recovery curves over time (mean values of triplicate wells with SD in dotted lines) in the absence and (G) presence of dabigatran (PFP + dabi) in combination with increasing dose of peptide D50‐S64 (1–1000 µM). E, Quantification for the recovery phase (at 50 min) and (F) the maintenance phase (at 120 min) showed no effect of peptide D50‐S64 on barrier recovery in the absence of dabigatran. H, In the presence of dabigatran, peptide D50‐S64 had no effect on the recovery phase of the barrier, but (I) in the maintenance phase addition of peptide D50‐S64 showed a dose‐dependent rescue of the barrier function. Data represent mean ± SD from triplicate wells; means were compared using one‐way analysis of variance. *P < .05, **P < .01, ***P < .001
Figure 2B is an example of a technical replicate obtained from one donor in triplicate wells per condition and shows average values ± SD in dotted lines, illustrating the robustness of the ECIS measurements within one experiment. However, between‐donor (n = 3) differences were observed in barrier function (Figure S3A‐C in supporting information) and thrombin generation (Figure S3D‐F), showing biological variations in PFP donors in combination with different EC isolations. Thus, in each experiment a dabigatran‐associated significant decline in barrier function could be observed in the maintenance phase, but these experiments could not be combined to generate a biological replicate.
Despite these biological variations, we observed corresponding dynamics between endothelial barrier function and thrombin kinetics as illustrated by the shaded areas in Supplementary (Figure S3A‐F, reviewed in detail in the discussion) indicating that these processes are interrelated.
To confirm the role of TF‐mediated coagulation on barrier function, TF function‐blocking Mab 5G9 was added to PFP and compared to PFP supplemented with an IT. The ECs showed barrier recovery and a stable maintenance phase when TF activity was blocked (Figure 3A‐C). To evaluate the contribution of the APC pathway in barrier function, PC activation was inhibited with the blocking antibody HPC4 (αPC) and compared to an IT. Interestingly, while again thrombin generation and lag time were not affected in PFP (Figure 3J‐L), a slight but significant decrease in barrier function was observed both in the recovery‐ and the maintenance phase (Figure 3D‐F). In the presence of dabigatran, however, blocking activation of PC had no effect on barrier function (Figure 3G‐I).
3.5. The thrombin inhibitors hirudin and PPACK are barrier protective
To gain a mechanistic understanding on the pharmacodynamic effects of thrombin inhibition on TNF‐α–stimulated EC barrier function, we next assessed the effect of the thrombin inhibitors hirudin and PPACK.
While dabigatran is a reversible, univalent, non‐peptidic DTI that interacts with thrombin’s active site, hirudin is an irreversible bivalent DTI that interferes with both the active site and exosite‐I and PPACK is an irreversible, peptide‐derived DTI that only binds to the active site (Figure 4A). Hirudin (1 μM) and PPACK (100 μM) effectively blocked thrombin activity to the same extent as dabigatran (Figure 2K), resulting in similar thrombin peak values (Figure 2L, mean ± SD of triplicate wells) and lag times (Figure 2M). Hirudin‐ and PPACK‐anticoagulated PFP supported the barrier function in the recovery phase in a similar manner to dabigatran (Figure 2H‐I). In addition, in the presence of hirudin and PPACK, EC barrier function was sustained in the maintenance phase, while again with dabigatran the barrier function deteriorated significantly over time (Figure 2H,J). Please note that Figure 2H‐M are examples obtained from one donor, but these results were confirmed with a second donor (Figure S4 in supporting information). These results show a difference in effect on barrier function of the non‐peptidic, reversible DTI dabigatran compared to the peptidic, irreversible DTI PPACK, while both maintain the accessibility of thrombin’s exosite‐I and show complete inhibition of catalytic activity of thrombin.
To investigate a possible contribution of exosite‐1 exposure of the thrombin–dabigatran complex in the loss of barrier function, a PAR‐1–mimicking peptide was synthesized.
3.6. PAR‐1 peptide D50‐S64 inhibits thrombin‐dependent platelet aggregation
Thrombin is an important mediator of platelet aggregation and acts initially through binding of exosite‐I to PAR‐1 expressed by the platelets and subsequently by cleaving PAR‐1 through its proteolytic active site. 16 To specifically study the role of thrombin exosite‐I in PAR‐1 activation, a synthetic peptide mimicking the PAR‐1 sequence that interacts with exosite‐I (D50‐S64) 8 was designed (Figure 4A) and synthesized (Figure S5 in supporting information).
To assess the functional impact of the PAR‐1–mimicking peptide D50‐S64 on thrombin activity, we measured thrombin‐dependent aggregation of washed platelets. A non‐saturating concentration of thrombin (0.1 U/ml) was used that resulted in 80% of the maximal platelet aggregation response (Figure 4B). Next, thrombin‐induced platelet aggregation in the presence of increasing concentrations of the peptide D50‐S64 (0.1–100 µM) resulted in a dose‐dependent inhibition of platelet aggregation to a residual 10% at the highest dose (Figure 4C). We concluded that the inhibitory effect of peptide D50‐S64 on thrombin‐mediated platelet aggregation strongly supports binding of the peptide to the exosite‐I of purified thrombin, thereby preventing thrombin‐induced platelet activation by PAR‐1.
3.7. The PAR‐1 peptide D50‐S64 does not affect thrombin generation in the absence or presence of ECs
To assess whether the PAR‐1 peptide also affects the generation/activity of thrombin, the standardized calibrated automated thrombogram (CAT) assay was used, 17 in which purified phospholipids and TF are added to plasma. The activity of the thrombin formed in solution is measured in real time using a fluorogenic substrate. In this assay, addition of peptide D50‐S64 (1–100 µM) to the plasma did not affect the thrombin peak values (Figure 4D; duplicate donors) nor the lag time (Figure 4E) calculated from the thrombin generation assay with pooled plasma. These results indicate that blockage of exosite‐I by peptide D50‐S64 does not affect TF‐induced thrombin generation in the absence of PAR‐1–expressing ECs.
Next, we established the effect of peptide D50‐S64 in thrombin generation on ECs stimulated with TNF‐α (Figure 5A; example of one donor). Increasing concentrations of peptide D50‐S64 (1–1000 µM) added to PFP in the absence or presence of 10 µM dabigatran had no effect on thrombin activity as measured by thrombin peak (Figure 5B; mean ± SD of triplicate wells) or lag time (Figure 5C). Thus, also in the presence of TF‐ and PAR‐1–expressing ECs, PAR‐1 peptide D50‐S64 did not affect thrombin generation/activity.
3.8. PAR‐1 peptide D50‐S64 in combination with dabigatran rescues EC barrier function
To assess the effect of the peptide D50‐S64 on EC barrier function, increasing concentration (1–1000 µM) was added to PFP and an ECIS assay was performed. Addition of peptide D50‐S64 had no effect on the recovery nor on the maintenance phase (Figure 5D‐F; example of 1 donor in triplicate wells).
Interestingly, when adding peptide D50‐S64 (1–1000 µM) in combination with dabigatran (10 µM) the recovery phase was not affected, but the loss of barrier function in the maintenance phase was prevented (Figure 5G‐I). To be able to repeat the experiment with three other PFP donors with the available amount of peptide, the condition with peptide concentration 1000 µM was omitted and hirudin was added (Figure S6 in supporting information). At the concentration of 100 µM peptide D50‐S64 in combination with dabigatran, barrier function was rescued to the same level as hirudin in all three donors (Figure S6J,K,L), although resistance profiles varied between different donors as observed previously. These data demonstrate that exosite‐I exposed in dabigatran‐bound thrombin is involved in the PAR‐1–mediated deterioration of barrier function, while thrombin’s catalytic activity was completely blocked.
4. DISCUSSION
Atrial fibrillation‐induced I/R episodes promote loss of microvascular integrity, leading to microvascular leakage, which is a key component in the development of fibrosis, 18 , 19 the substrate for AF progression. 20 To date DOACs are the preferred choice of anticoagulant treatment for stroke prevention in AF. However, DOACs can exhibit non‐hemostatic vascular effects via PARs and thereby may affect the vascular component of AF progression in addition to their anti‐thrombotic action. 21 , 22 , 23 To investigate the non‐hemostatic effects of DOACs, an in vitro barrier function assay (ECIS) was used as proof‐of‐concept for the role of DOACs on vascular integrity.
Simultaneous performance of ECIS and thrombin generation using ECs and PFP, providing both pro‐ and anti‐coagulant and pro‐ and anti‐barrier factors, permitted linking thrombin kinetics with endothelial barrier dynamics.
The pro‐inflammatory nature present in AF was mimicked by TNF‐α stimulation of ECs, as an upregulation of plasma TNF‐α is linked to AF and AF progression. 9 , 10 Characterization of the determinants of thrombin generation showed that the TNF‐α–stimulated ECs provided TF by de novo synthesis (Figure 1), albeit at a low concentration. This was concluded from the fact that CTI, an inhibitor of the intrinsic pathway of coagulation, lowered the formation of thrombin, which is reported to only occur at low TF concentration (<1 pM). 17 Therefore, CTI was added in all experiments to ensure that our data were only dependent on the TF‐mediated extrinsic pathway. The contribution of ECs as source of TF has been debated, but its expression has been confirmed in the cardiac microvasculature of patients suffering from chronic ischemic heart disease such as AF. 24 The ECs also provided TFPI (Figure 1D), the endogenous inhibitor of the FVIIa–TF complex and one of the strongest thrombin‐generating determinants at low TF, 25 as blocking TFPI increased thrombin generation by approximately 50% (Figure 1F, TFPI).
Another determinant present in our system is the APC pathway, as the endothelium is a potential source of TM and EPCR, which in concert with PC and protein S will provide a negative feedback loop by inactivating FVa and FVIIIa. However, TNF‐α stimulation attenuated surface expression of TM and EPCR already at 1 ng/ml TNF‐α (Figure 1E), which is consistent with the literature, 26 thus potentially suppressing this pathway, which has been confirmed in patients with AF. 27 When assessing the contribution of APC to thrombin generation, inhibition of activation of PC using antibody HPC4 showed no detectable effect.
Collectively, the actual TF‐mediated thrombin activity measured in the system is the net result of the interplay between pro‐ and counteracting pathways present under physiological circumstances.
Interestingly, comparing thrombin generation and ECIS, corresponding dynamics were visible over time reflecting the different stages in thrombin generation (Figure S3D‐F, Figures S4C‐D and S6D‐F) on endothelial barrier recovery (Figure S3A‐C, Figures S4A‐B and S6A‐C).
During the initiation phase of thrombin generation (defined as the phase in which 0‐1/6th of the total amount of thrombin is formed 25 ), FXa and low amounts of thrombin (approximately 50 pM) activate PAR‐2 28 and PAR‐1, 29 respectively, circumstances which both have been shown to support barrier function. This can explain the initial recovery of the barrier (phase 2 in Figure S3). During the propagation phase of thrombin activation (phase 5), FXa activates FV 30 and thrombin activates FV and FVIII thereby leading to an explosive burst of thrombin, which subsequently disappears in the termination phase (phase 6) when thrombin is mainly inhibited by antithrombin. 25 The increasing amounts of proteolytically active thrombin generated during phases 5 and 6 will interact with PAR‐1 on the EC surface leading to cleavage of the receptor with subsequent intracellular signaling resulting in a barrier plateau in phase 3, probably due to competitive action between pro‐ and anti‐barrier effects.
In our system, inhibition of the PC pathway had no detectable effect on the thrombin activity, but showed a slightly decreased barrier function in both the recovery and maintenance phase, indicating that APC‐mediated barrier protection was active (Figure 3, 4, 5).
In phase 7 all active thrombin is catalytically inhibited, while thrombin generation is halted due to exhaustion of the system. In this phase the barrier function deteriorates and the balance may shift toward leakage (phase 4), which may indicate that it takes some time for the ECs to react to the thrombin‐induced cleavage of PAR‐1.
In the presence of rivaroxaban no thrombin was formed, which coincided with fast recovery of barrier function and a fully sustained maintenance phase (Figure 2B and Figure S3). In the presence of dabigatran (10 μM), FXa will facilitate a thrombin propagation phase through activation of FV, 30 yielding cumulative formation of thrombin–dabigatran complexes over time. The EC barrier started to leak in the maintenance phase. Furthermore, under these circumstances, blocking PC activation with HPC4 had no effect on barrier function (Figure 3, 4, 5), indicating that APC was not involved in the deterioration of the barrier in the presence of dabigatran.
However, this phenomenon could be prevented by blocking exosite‐I of dabigatran‐inhibited thrombin with the PAR‐1–derived peptide D50‐S64 and was comparable to the action of hirudin, which blocks thrombin’s active site and exosite‐I. For complete recovery of barrier function to the level of hirudin, a rather high concentration (100 µM, Figure S6J‐L) of peptide D50‐S64 was required, which may be explained by the fact that the peptide may also be absorbed by prothrombin as has been shown for a very similar peptide (N49‐N62). 31
Interestingly, when thrombin was inhibited with PPACK, which is a peptidic, irreversible inhibitor of thrombin that also keeps exosite‐I available (Figure 4A), barrier function was consolidated in the maintenance phase to the level of hirudin (Figure 2J and Figure S4G‐H). Explanation for these differences between PPACK and dabigatran must be based on either the (ir)reversibility or the intrinsic structure of these two monovalent DTIs. We consider reversibility of dabigatran inhibition unlikely as explanation, because experiments were performed with saturating concentrations (10 μM, Figure S2). Furthermore, if the loss of barrier function in the maintenance phase were mediated by thrombin released from the thrombin–dabigatran complex, this proteolytically active thrombin would also be able to cleave the fluorogenic substrate and this was not the case (Figure 2, 3, 4, 5).
From a structural point of view, PPACK and the non‐peptide inhibitor N‐alpha‐(2‐naphthylsulfonylglycyl)‐4‐amidinophenylalanine piperidide (NAPAP), from which dabigatran is developed, show large differences in docking geometry; 32 also, PPACK ultimately forms a covalent bond with the active site Ser of thrombin. This difference in interaction may evoke different allosteric changes in exosite‐I, which has also been shown when comparing two non‐peptidic inhibitors such as dabigatran and argatroban. These DTIs have different synthetic structures, yet engage the same subsites in the active site of thrombin, but evoke distinct responses in exosite‐I–dependent interaction with the substrate γAγA‐fibrin, showing a decreased binding of the thrombin–dabigatran complex, and an increased binding of the thrombin–argatroban complex. 33 These data show that different active site–directed thrombin inhibitors can elicit unique responses mediated by allosteric changes in exosite‐I. Hence, we propose that the effects of dabigatran‐bound thrombin on the loss of endothelial barrier function may be due to the specific structural conformation of the dabigatran molecule leading to specific allosteric modulation of exosite‐I.
Interestingly, Chen et al. 34 reported that dabigatran‐bound, catalytically inactive thrombin incubated on ECs resulted in enhanced surface expression of PAR‐1, which was then susceptible to cleavage by thrombin or to activation by the thrombin‐receptor activating peptide (TRAP) TFLLRNPNDK.
Furthermore, a similar finding has been reported for platelets: AF patients taking a clinically defined dose of dabigatran (median plasma concentration 0.17/IQR 0.21 µM) showed enhanced TRAP‐induced platelet aggregation, 35 which coincided with enhanced PAR‐1 receptor density on the platelets. 35 , 36 This dabigatran‐mediated effect was not observed with other platelet agonists such as collagen, ADP, or arachidonic acid. 36 Moreover, this phenomenon was specific for dabigatran, as patients on rivaroxaban (mean plasma concentration 0.48 ± 0.25 µM) did not exhibit enhanced platelet aggregation. 35
Our results indicate that the potential enhanced expression of PAR‐1 on the endothelial surface mediated by dabigatran–thrombin complex occurred in PFP conditions with natural (anti)‐coagulation factors present and within 1 h, but without extra addition of a thrombin‐like PAR‐1 agonist.
Our multifactorial experimental setup undoubtedly not only activates the extrinsic pathway of coagulation but may also trigger fibrinolysis and/or complement, thus potentially generating other agonists that may cleave the putative surface‐stabilized PAR‐1 receptor in our system. Vascular ECs are a source of tissue plasminogen activator (tPA) and maintain a high plasminogen activation potential on their surface. 37 , 38 Although plasmin can target PAR‐1 at several cleavage sites, when plasmin is present in excess of active thrombin (which is the case when dabigatran is present), it can cleave at Arg41‐Ser42 and activate PAR‐1 on platelets. 39 Also, the complement system, potentially active in our system, may produce C4a, which has been shown to activate PAR‐1. 40 However, deciphering candidate agonists that potentially activate surface‐stabilized PAR‐1 by dabigatran‐complexed thrombin is beyond the scope of this article. We realize that we are using supra‐therapeutic concentrations of dabigatran and rivaroxaban in our assays (10 µm), which is about 10 times higher as measured in the circulation of patients. 35 However, these concentrations were dictated by the necessity to completely inhibit the target proteases. We speculate that the prolonged exposure in vivo with normo‐therapeutic concentrations of DOACs versus the short exposure (<1 h) with supra‐therapeutic concentrations as in our experiments may yield comparable effects.
Collectively, our experimental setup is uniquely suited to monitor both thrombin generation and endothelial barrier function simultaneously and our results imply that cellular function should be taken into account when evaluating safety and efficacy of anticoagulants.
Our results indicate that while thrombin is catalytically fully inhibited by dabigatran, it may still be able to interact with PAR‐1, potentially leading to enhanced expression of the receptor on the endothelial surface, where it may be subject to activation by other proteases then thrombin, leading to subsequent loss of barrier function. Because rivaroxaban prevents the formation of thrombin and can maintain EC integrity, treatment with rivaroxaban and not dabigatran may be the preferred anticoagulant therapy of choice for AF patients.
CONFLICTS OF INTEREST
The authors declare no competing financial interests.
AUTHOR CONTRIBUTIONS
S.C.D. designed experiments, analyzed and interpreted data, generated figures, and wrote the manuscript; S.M.A. synthesized and purified the peptide; H.M.H.S., T.M.H., M.H.A.B., and H.H.V. provided advice and materials; A.J.v.Z. generated figures and edited the manuscript; H.C.d.B. designed experiments, analyzed and interpreted data, generated figures, and wrote the manuscript.
Supporting information
Appendix S1
ACKNOWLEDGMENTS
The authors wish to thank René van Oerle (CARIM Lab, Maastricht University Medical Center) and Annemarie van Oeveren‐Rietdijk (Department of Nephrology, Leiden University Medical Center) for performing thrombin generation analysis and technical support.
Dólleman SC, Agten SM, Spronk HMH, et al. Thrombin in complex with dabigatran can still interact with PAR‐1 via exosite‐I and instigate loss of vascular integrity. J Thromb Haemost. 2022;20:996–1007. doi: 10.1111/jth.15642
Anton Jan van Zonneveld and Hetty C. de Boer share senior authorship.
Manuscript handled by: Roger Preston
Final decision: Roger Preston, 10 January 2022
Funding information
This work was supported by the Netherlands Cardiovascular Research Initiative: an initiative with support of the Dutch Heart Foundation CVON program Reappraisal of Atrial fibrillation: interaction between hyperCoagulability, Electrical remodelling, and Vascular destabilization in the progression of AF (RACE V, NHS2010B233).
REFERENCES
- 1. Spronk HM, De Jong AM, Verheule S, et al. Hypercoagulability causes atrial fibrosis and promotes atrial fibrillation. Eur Heart J. 2017;38(1):38‐50. [DOI] [PubMed] [Google Scholar]
- 2. Steffel J, Verhamme P, Potpara TS, et al. The 2018 European Heart Rhythm Association Practical Guide on the use of non‐vitamin K antagonist oral anticoagulants in patients with atrial fibrillation: executive summary. Europace. 2018;20(8):1231‐1242. [DOI] [PubMed] [Google Scholar]
- 3. Lip GY, Fauchier L, Freedman SB, et al. Atrial fibrillation. Nat Rev Dis Primers. 2016;2:16016. [DOI] [PubMed] [Google Scholar]
- 4. Tritschler T, Castellucci LA. It's time for head‐to‐head trials with direct oral anticoagulants. Thromb Res. 2019;180:64‐69. [DOI] [PubMed] [Google Scholar]
- 5. Gulpen AJ, Ten Cate‐Hoek AJ, Ten Cate H. Upstream versus downstream thrombin inhibition. Expert Rev Cardiovasc Ther. 2016;14(11):1273‐1282. [DOI] [PubMed] [Google Scholar]
- 6. Gong IY, Kim RB. Importance of pharmacokinetic profile and variability as determinants of dose and response to dabigatran, rivaroxaban, and apixaban. Can J Cardiol. 2013;29(7 Suppl):S24‐S33. [DOI] [PubMed] [Google Scholar]
- 7. Rezaie AR. Protease‐activated receptor signalling by coagulation proteases in endothelial cells. Thromb Haemost. 2014;112(5):876‐882. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Vu TK, Wheaton VI, Hung DT, Charo I, Coughlin SR. Domains specifying thrombin‐receptor interaction. Nature. 1991;353(6345):674‐677. [DOI] [PubMed] [Google Scholar]
- 9. Guo Y, Lip GY, Apostolakis S. Inflammation in atrial fibrillation. J Am Coll Cardiol. 2012;60(22):2263‐2270. [DOI] [PubMed] [Google Scholar]
- 10. Li J, Solus J, Chen Q, et al. Role of inflammation and oxidative stress in atrial fibrillation. Heart Rhythm. 2010;7(4):438‐444. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Tannenbaum SH, Gralnick HR. Gamma‐interferon modulates von Willebrand factor release by cultured human endothelial cells. Blood. 1990;75(11):2177‐2184. [PubMed] [Google Scholar]
- 12. Lupu C, Lupu F, Dennehy U, Kakkar VV, Scully MF. Thrombin induces the redistribution and acute release of tissue factor pathway inhibitor from specific granules within human endothelial cells in culture. Arterioscler Thromb Vasc Biol. 1995;15(11):2055‐2062. [DOI] [PubMed] [Google Scholar]
- 13. Wood JP, Ellery PE, Maroney SA, Mast AE. Biology of tissue factor pathway inhibitor. Blood. 2014;123(19):2934‐2943. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Stearns DJ, Kurosawa S, Sims PJ, Esmon NL, Esmon CT. The interaction of a Ca2+‐dependent monoclonal antibody with the protein C activation peptide region. Evidence for obligatory Ca2+ binding to both antigen and antibody. J Biol Chem. 1988;263(2):826‐832. [PubMed] [Google Scholar]
- 15. Taylor FB Jr, Chang A, Esmon CT, D'Angelo A, Vigano‐D'Angelo S, Blick KE. Protein C prevents the coagulopathic and lethal effects of Escherichia coli infusion in the baboon. J Clin Invest. 1987;79(3):918‐925. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Coughlin SR. Protease‐activated receptors and platelet function. Thromb Haemost. 1999;82(2):353‐356. [PubMed] [Google Scholar]
- 17. Hemker HC, Giesen P, AlDieri R, et al. The calibrated automated thrombogram (CAT): a universal routine test for hyper‐ and hypocoagulability. Pathophysiol Haemost Thromb. 2002;32(5–6):249‐253. [DOI] [PubMed] [Google Scholar]
- 18. Skalidis EI, Hamilos MI, Karalis IK, Chlouverakis G, Kochiadakis GE, Vardas PE. Isolated atrial microvascular dysfunction in patients with lone recurrent atrial fibrillation. J Am Coll Cardiol. 2008;51(21):2053‐2057. [DOI] [PubMed] [Google Scholar]
- 19. Xu Z, Castellino FJ, Ploplis VA. Plasminogen activator inhibitor‐1 (PAI‐1) is cardioprotective in mice by maintaining microvascular integrity and cardiac architecture. Blood. 2010;115(10):2038‐2047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Wyse DG, Van Gelder IC, Ellinor PT, et al. Lone atrial fibrillation: does it exist? J Am Coll Cardiol. 2014;63(17):1715‐1723. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Coughlin SR. Thrombin signalling and protease‐activated receptors. Nature. 2000;407(6801):258‐264. [DOI] [PubMed] [Google Scholar]
- 22. Borensztajn K, Peppelenbosch MP, Spek CA. Coagulation factor Xa signaling: the link between coagulation and inflammatory bowel disease? Trends Pharmacol Sci. 2009;30(1):8‐16. [DOI] [PubMed] [Google Scholar]
- 23. Troyanovsky B, Alvarez DF, King JA, Schaphorst KL. Thrombin enhances the barrier function of rat microvascular endothelium in a PAR‐1‐dependent manner. Am J Physiol Lung Cell Mol Physiol. 2008;294(2):L266‐L275. [DOI] [PubMed] [Google Scholar]
- 24. Reichman‐Warmusz E, Domal‐Kwiatkowska D, Matysiak N, et al. Tissue factor is unregulated in microvascular endothelial cells of patients with heart failure. J Clin Pathol. 2016;69(3):221‐225. [DOI] [PubMed] [Google Scholar]
- 25. Castoldi E, Rosing J. Thrombin generation tests. Thromb Res. 2011;127(Suppl 3):S21‐S25. [DOI] [PubMed] [Google Scholar]
- 26. Cohen CT, Turner NA, Moake JL. Production and control of coagulation proteins for factor X activation in human endothelial cells and fibroblasts. Sci Rep. 2020;10(1):2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Negreva M, Georgiev S, Vitlianova K. Decreased activity of the protein C anticoagulant pathway in the early hours of paroxysmal atrial fibrillation. Clin Appl Thromb Hemost. 2017;23(7):793‐799. [DOI] [PubMed] [Google Scholar]
- 28. Feistritzer C, Lenta R, Riewald M. Protease‐activated receptors‐1 and ‐2 can mediate endothelial barrier protection: role in factor Xa signaling. J Thromb Haemost. 2005;3(12):2798‐2805. [DOI] [PubMed] [Google Scholar]
- 29. Bae JS, Yang L, Manithody C, Rezaie AR. The ligand occupancy of endothelial protein C receptor switches the protease‐activated receptor 1‐dependent signaling specificity of thrombin from a permeability‐enhancing to a barrier‐protective response in endothelial cells. Blood. 2007;110(12):3909‐3916. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Schuijt TJ, Bakhtiari K, Daffre S, et al. Factor Xa activation of factor V is of paramount importance in initiating the coagulation system: lessons from a tick salivary protein. Circulation. 2013;128(3):254‐266. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Billur R, Sabo TM, Maurer MC. Thrombin exosite maturation and ligand binding at ABE II help stabilize PAR‐binding competent conformation at ABE I. Biochemistry. 2019;58(8):1048‐1060. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Brandstetter H, Turk D, Hoeffken HW, et al. Refined 2.3 A X‐ray crystal structure of bovine thrombin complexes formed with the benzamidine and arginine‐based thrombin inhibitors NAPAP, 4‐TAPAP and MQPA. A starting point for improving antithrombotics. J Mol Biol. 1992;226(4):1085‐1099. [DOI] [PubMed] [Google Scholar]
- 33. Yeh CH, Stafford AR, Leslie BA, Fredenburgh JC, Weitz JI. Dabigatran and argatroban diametrically modulate thrombin exosite function. PLoS One. 2016;11(6):e0157471. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Chen B, Soto AG, Coronel LJ, Goss A, van Ryn J, Trejo J. Characterization of thrombin‐bound dabigatran effects on protease‐activated receptor‐1 expression and signaling in vitro. Mol Pharmacol. 2015;88(1):95‐105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Olivier CB, Weik P, Meyer M, et al. TRAP‐induced platelet aggregation is enhanced in cardiovascular patients receiving dabigatran. Thromb Res. 2016;138:63‐68. [DOI] [PubMed] [Google Scholar]
- 36. Achilles A, Mohring A, Dannenberg L, et al. Dabigatran enhances platelet reactivity and platelet thrombin receptor expression in patients with atrial fibrillation. J Thromb Haemost. 2017;15(3):473‐476. [DOI] [PubMed] [Google Scholar]
- 37. Urano T, Castellino FJ, Suzuki Y. Regulation of plasminogen activation on cell surfaces and fibrin. J Thromb Haemost. 2018;16(8):1487‐1497. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Suzuki Y, Yasui H, Brzoska T, Mogami H, Urano T. Surface‐retained tPA is essential for effective fibrinolysis on vascular endothelial cells. Blood. 2011;118(11):3182‐3185. [DOI] [PubMed] [Google Scholar]
- 39. Kuliopulos A, Covic L, Seeley SK, Sheridan PJ, Helin J, Costello CE. Plasmin desensitization of the PAR1 thrombin receptor: kinetics, sites of truncation, and implications for thrombolytic therapy. Biochemistry. 1999;38(14):4572‐4585. [DOI] [PubMed] [Google Scholar]
- 40. Wang H, Ricklin D, Lambris JD. Complement‐activation fragment C4a mediates effector functions by binding as untethered agonist to protease‐activated receptors 1 and 4. Proc Natl Acad Sci USA. 2017;114(41):10948‐10953. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Appendix S1