

Abstract
Cannabinoid receptor 1 (CB1R), a G protein‐coupled receptor, plays a fundamental role in synaptic plasticity. Abnormal activity and deregulation of CB1R signaling result in a broad spectrum of pathological conditions. CB1R signaling is regulated by receptor desensitization including phosphorylation of residues within the intracellular C terminus by G protein‐coupled receptor kinases (GRKs) that may lead to endocytosis. Furthermore, CB1R signaling is regulated by the protein Src homology 3‐domain growth factor receptor‐bound 2‐like (SGIP1) that hinders receptor internalization, while enhancing CB1R association with β‐arrestin. It has been postulated that phosphorylation of two clusters of serine/threonine residues, 425SMGDS429 and 460TMSVSTDTS468, within the CB1R C‐tail controls dynamics of the association between receptor and its interaction partners involved in desensitization. Several molecular determinants of these events are still not well understood. We hypothesized that the dynamics of these interactions are modulated by SGIP1. Using a panel of CB1Rs mutated in the aforementioned serine and threonine residues, together with an array of Bioluminescence energy transfer‐based (BRET) sensors, we discovered that GRK3 forms complexes with Gβγ subunits of G proteins that largely independent of GRK3’s interaction with CB1R. Furthermore, CB1R interacts only with activated GRK3. Interestingly, phosphorylation of two specific residues on CB1R triggers GRK3 dissociation from the desensitized receptor. SGIP1 increases the association of GRK3 with Gβγ subunits of G proteins, and with CB1R. Altogether, our data suggest that the CB1R signalosome complex is dynamically controlled by sequential phosphorylation of the receptor C‐tail and is also modified by SGIP1.
Keywords: cannabinoid receptor 1, G protein‐coupled receptor kinase, G protein‐coupled receptors, phosphorylation, SGIP1, β‐arrestin
Cannabinoid receptor 1 C‐tail contains two serine/threonine clusters 456SMGDS429 and 460TMSVSTDTS468 that are important for desensitization of the activated receptor. We found that these motifs play distinct roles in the interaction of CB1R with GRK3 and β‐arrestin2. CB1R requires catalytically active GRK3 in order to interact with β‐arrestin2. Furthermore, SGIP1 profoundly modifies the signalosome of CB1R during desensitization. These findings provide new insight into the mechanism of signaling and desensitization of CB1R.
Abbreviations
- BRET
bioluminescence energy transfer
- Cat. no.
catalogue number
- CB1R
cannabinoid receptor 1
- CME
clathrin‐mediated endocytosis
- CNS
central nervous system
- DMEM
Dulbecco's Modified Eagle Medium
- ECS
endocannabinoid system
- FBS
fetal bovine serum
- FCHo
FCH/F‐BAR domain only protein
- GPCR
G protein‐coupled receptor
- GRK
G protein‐coupled receptor kinase
- HEK293
human embryonic kidney 293
- IP1
inositol monophosphate
- mBRET
milliBRET
- PBS
phosphate‐buffered saline
- PBST
phosphate‐buffered saline solution with Tween
- PH
pleckstrin homology
- RH
regulator of G protein signaling homology
- Rluc
Renilla luciferase
- RRID
Research Resource Identifier (see scicrunch.org)
- SCP1
src homology 3‐domain growth factor receptor‐bound 2‐like 1
- SR141716
rimonabant
- WIN
WIN 55,212–2 mesylate ‐ [(3R)‐2,3‐dihydro‐5‐methyl‐3‐(4‐morpholinylmethyl)pyrrolo[1,2,3‐de]‐1,4‐benzoxazin‐6‐yl]‐1‐naphthalenyl‐methanone, monomethanesulfonate
- YFP
yellow fluorescent protein
- βarr
β‐arrestin
Highlights.
• Activation of GRK3 is required for its interaction with CB1R
• CB1R‐GRK3 interaction is regulated by phosphorylation of specific CB1R residues
• Recruitment of GRK3 and β‐arrestin2 to CB1R is dependent on distinct phosphorylation patterns
• SGIP1 profoundly modifies the dynamics of signalosome interactions during CB1R desensitization
1. INTRODUCTION
Cannabinoid receptors, together with their endogenous ligands, endocannabinoids, and the enzymes responsible for their synthesis and degradation, constitute the endocannabinoid system (ECS). Cannabinoid receptor 1 (CB1R), a member of the G protein‐coupled receptor (GPCR) family, is a central molecule of the ECS. In the central nervous system (CNS), CB1R is principally located presynaptically on many GABAergic, glutamatergic, cholinergic, serotonergic, and noradrenergic neurons. CB1R is involved in fine‐tuning of synaptic transmission. Activation of the CB1R suppresses neurotransmitter release in these synapses (Haring, Marsicano, Lutz & Monory, 2007; Kirilly, Hunyady & Bagdy, 2013; Marsicano & Lutz, 1999). It is expressed in circuits important for the processing of anxiety, fear, stress, motor, cognitive and social functions, including the basal ganglia, hippocampus, prefrontal cortex, amygdala and cerebellum (Herkenham et al., 1990; Katona & Freund, 2012). ECS malfunction is linked to numerous pathological states of the nervous system (Araque, Castillo, Manzoni & Tonini, 2017; Pacher & Kunos, 2013).
The regulation of CB1R by the common mechanism leads to tolerance development in vivo. In our previous study, we observed an altered development of tolerance to chronic treatment with THC in SGIP1 knock out mice compared with wild‐type mice in selected tasks (cannabinoid tetrad tests: (catalepsy, anti‐nociception, hypothermia, and suppression of motor coordination). Moreover, withdrawal signs were atypical the in absence of SGIP1 in the knock out animals (Dvorakova et al., 2021). CB1 receptor signaling is tightly regulated. Understanding the mechanisms leading to the development of tolerance is central for rational pharmacological management of disease states in which the endocannabinoid system is involved. Upon stimulation, CB1R follows the desensitization process common for most GPCRs that typically involve phosphorylation of serine/threonine residues within the C terminus by GRKs and possibly other kinases. β‐arrestin is consequently recruited to the receptor leading to G protein uncoupling. β‐arrestin undergoes conformational changes upon interaction with the phosphorylated receptor and serves as a central molecule orchestrating machinery leading to the endocytosis, and subsequent degradation, or recycling of the receptor back to plasma membrane (Cahill, Thomsen & Tarrasch, 2017; Fletcher‐Jones et al., 2020; Leterrier, Bonnard, Carrel, Rossier & Lenkei, 2004).
Intriguingly, different from other GPCRs, CB1R internalization is tightly controlled by interaction with the protein Src homology 3‐domain growth factor receptor‐bound 2‐like endophilin interacting protein 1 (SGIP1) (Dvorakova et al., 2021; Hajkova et al., 2016). SGIP1 together with FCH/F‐BAR domain only protein 1 and 2 (FCHO1/2) belong to muniscin family of proteins involved in GPCR internalization. Contrary to FCHO1/2 that are important initiators of endocytosis (Henne et al., 2007), SGIP1 hinders CB1R internalization (Hajkova et al., 2016). SGIP1 domain organization differs from FCHO1/2 within their N‐termini. FCHO1/2 proteins have their N‐terminal portion folded to form F‐Bar domains that are important for initiation of plasma membrane invagination during early stages of CME pit formation (Henne et al., 2007). The N‐terminus of SGIP1 contains membrane phospholipid‐binding (MP) domain that has no sequence similarity to the F‐Bar motives, but also interacts with the plasma membrane (Uezu, Horiuchi & Kanda, 2007). Most likely, the interaction of the MP domain with the plasma membrane does not impose invagination of the membrane within the nascent pit formation. SGIP1 associates with the CB1R in presynaptic elements of neurons and by blocking endocytosis of the activated CB1Rs causes, among other, consequences of prolonged association between the desensitized CB1R and β‐arrestin2 (Hajkova et al., 2016).
Stimulation of CB1R triggers several signaling pathways, including GRK activation (Jin et al., 1999; Nogueras‐Ortiz & Yudowski, 2016), that mediate phosphorylation of serine/threonine residues of the CB1R C‐termini, leading to β‐arrestins recruitment (Al‐Zoubi, Morales & Reggio, 2019; Garcia, Brown, Hille & Mackie, 1998; Moore, Milano & Benovic, 2007). GRKs are serine/threonine protein kinases with a common modular structure consisting of a catalytic domain within a Regulator of G protein signaling homology (RH) domain that is flanked by N‐terminal α‐helical domain (αN‐helix) and a variable C‐terminal lipid‐binding region. GRKs are soluble proteins that utilize distinct mechanisms to bring them to the close proximity of membrane‐embedded GPCRs. Unlike other families of GRKs that localize to the membrane via palmitoylation (GRK1/7) or prenylation (GRK4/5/6) of C‐termini, GRK2/3 use unique pleckstrin homology (PH) domain to bring the kinases to the vicinity of GPCRs (Gurevich, Tesmer, Mushegian & Gurevich, 2012; Koch, Inglese, Stone & Lefkowitz, 1993). Upon activation of G proteins, GRK2/3 interact with Gβγ via PH domain, recruiting kinases to the membrane and proximity of the activated receptor. This process appears to contribute to allosteric activation of the receptor‐phosphorylation activity of GRK2/3, as disruption of interaction of GRK2 with Gβγ results in inhibition of GRK2‐driven receptor phosphorylation (Carman et al., 2000; Lodowski et al., 2005; Pitcher, Inglese & Higgins, 1992).
Two regions within the CB1R C‐tail contain clusters of serine and threonine residues that are phosphorytalable. One motif is between residues 425 and 429, namely 425SMGDS429 and another is between residues 460 and 468, 460TMSVSTDTS468. Both motives were identified as important for β‐arrestin2 recruitment (Bakshi, Mercier & Pavlopoulos, 2007; Blume, Patten & Eldeeb, 2017; Daigle, Kwok & Mackie, 2008; Hsieh, Brown, Derleth & Mackie, 1999; Jin et al., 1999; Morgan et al., 2014; Singh, Bakshi, Mercier, Makriyannis & Pavlopoulos, 2011; Straiker, Wager‐Miller & Mackie, 2012); however, the precise roles of each of these two regions are still not clear.
SGIP1 interacts with CB1R C‐terminal domain and hinders its internalization. In this study, we investigated the dynamics of the association between GRK3 and CB1R and the role of phosphorylation of residues within both the 425SMGDS429 and 460TMSVSTDTS468 motifs using an alanine scanning approach together with complementary bioluminescence resonance energy transfer‐based (BRET) sensors.
Characterization of interactions involved in CB1R desensitization represents an important step in understanding all phases of cannabinoid signaling. Here we aimed to study if β‐arrestin2 binding to the activated CB1R is dependent on GRK2/3 activity that phosphorylates the CB1R C‐tail in both motifs, and if GRK3 association with the receptor is also controlled by the phosphorylation of these motifs. In the presence of SGIP1, internalization of CB1R is hindered, with profound functional and behavioral consequences (Dvorakova et al., 2021; Hajkova et al., 2016).
2. METHODS
The experiments in this manuscript are not pre‐registered and no blinding procedure was performed in this manuscript. No statistical method is employed to predetermine the sample size of the experiments. No randomization methods were used.
2.1. Chemicals
Reagents for cell culture and transfection were purchased from Thermo Fisher Scientific Inc. (USA). CB1R agonist WIN 55,212‐2 mesylate (WIN) was obtained from Tocris R&D, USA (cat. no. 1038) and GRK2/3 inhibitor cmpd101 was purchased from Hello Bio Ltd., UK (cat. no. HB2840).
2.2. Expression vectors and mutagenesis
Expression vectors for SGIP1‐Flag, β‐arrestin2‐Rluc, GαI1‐Rluc8, Gβ2‐Flag, Gγ2‐YFP (Gβγ in the following text), and empty vector pRK6 used in this study have been described previously (Brule et al., 2014; Charest & Bouvier, 2003; Hajkova et al., 2016). SGIP1‐mCherry was constructed in house. Fragment containing full coding sequence of SGIP1 obtained by PCR amplification from the previously characterized plasmid (Hajkova et al., 2016) was inserted into a prk5_mCherry using BamHI/SalI restriction sites creating SGIP1 fused with N‐terminal mCherry tag. The CB1R mutant variants were produced either by PCR mutagenesis using QuikChange II Site‐directed mutagenesis kit (Agilent, USA, cat. no. 200524) following the manufacturer's instructions using modified primers (Liu & Naismith, 2008) or by replacing C‐tail sequence of full‐length human CB1R by synthetized CB1R C‐tail DNA fragments (GeneCust, France). Plasmid coding full‐length CB1R S425A, S429A that served as a template for mutagenesis was from the laboratory of Ken Mackie from Indiana University, Bloomington, Indiana, USA (Jin et al., 1999). Newly synthesized mutant CB1Rs variants were fused with either YFP (C‐terminal) or SNAP‐Tag (N‐terminal). Synthetic RLuc8 coding sequence (Addgene, France, cat. no. 87121) was cloned in‐frame downstream of the human GRK3 coding sequence and a triple glycine residues linker was included between the tag and GRK3 sequence and subcloned into pcDNA3. All constructs were sequenced prior their use. All plasmids were propagated in E. Coli DH5α (NEB, USA, cat. no. C2987H). The plasmids were purified using Qiagen Midiprep kits (Qiagen, Germany, cat. no. 12123).
2.3. Cell culture and transfections
Human embryonic kidney 293 (HEK293, RRID:CVCL_0063) cells were grown in Dulbecco's Modified Eagle Medium (DMEM) (Thermo Fisher Scientific, cat. no. 21969035) containing 10% fetal bovine serum (FBS) and maintained at 37°C, 95% humidity, and 5% CO2. Twenty‐four hours before the experiment, 150 ng DNA/well was used to transiently transfect 5 × 104 cells/well in 96‐well plates (Merck, Germany, cat. no. M0187‐32EA) coated with poly‐l‐ornithine (Merck, Germany, cat. no. P4957) using Lipofectamine™ 2000 (Thermo Fisher Scientific, cat. no. 11668019) according to the manufacturer's instructions. HEK293 cell line was used up to 30th passage. The cells used in this manuscript are not listed as a commonly misidentified cell line by the International Cell Line Authentication Committee. No authentication has been conducted during the experiments.
2.4. Imaging
Cells were seeded onto culture dishes for microscopy and transfected by correspondent plasmids. Live cells were imaged at 37°C using inverted fluorescent microscope Leica DMI6000 with confocal extension Leica TCS SP5 AOBS TANDEM confocal superfast scanner, objective 63 × 1.4 oil (Leica Microsystems). Samples were excited with argon laser 514 nm and detected with a HyD detector in the range 535–545 nm. Microscopic images were processed in ImageJ.
2.5. Bioluminescence resonance energy transfer assays
To assess the association dynamics between studied molecules, bioluminescence resonance energy transfer (BRET) assay was used. Cells were seeded and transiently transfected as described in cell culture and transfections (Donthamsetti, Quejada, Javitch, Gurevich & Lambert, 2015). Twenty‐four hours after transfection, cells were washed with PBS, and coelenterazine h (NanoLight, cat. no. CAT#301) was added to a final concentration 5 μM. The stimulation of the cells by agonist was performed 5 min later. BRET signal detection was done using Mithras LB 940 microplate reader (Berthold Technologies, Germany) equipped with donor (480 ± 20 nm) and acceptor (540 ± 40 nm) filters. The BRET signal ratio was calculated as the emission of the energy acceptor molecules (540 ± 40 nm) divided by the emission of the energy donor molecules (480 ± 20 nm). The data are presented as the agonist‐promoted milliBRET (mBRET) change that was calculated by subtracting the BRET ratio obtained in the absence of agonist from the one obtained following agonist application and multiplied by 1000 (Hamdan et al., 2007) (Figure S1e).
2.6. Inositol monophosphate accumulation
The extent of inositol monophosphate (IP1) accumulation was measured using a IPOne HTRF kit (Cisbio Bioassays, cat. no. 62IPAPEJ) according to the recommendations of the manufacturer. Briefly, cells were seeded and transiently transfected with CB1R, or the variant, together with chimeric G protein Gqi9 (1:1 ratio). Gqi9 allows Gi/o‐coupled GPCRs to couple to Gq to produce IP1(Brule et al., 2014). Twenty‐four hours after the transfection, cells were incubated in the presence of receptor agonist for 20 min at 37°C, and then cryptate‐labeled anti‐IP1 and D2‐labeled IP1 antibodies were added for 1 h at the 21°C. Native IP1 produced by cells compete with d2‐labeled IP1 (acceptor of energy) for binding of anti‐IP1‐Cryptate (donor of energy). The fluorescence was detected at 665 and 620 nm using a PHERAstar plate reader (BMG Labtechnologies, Germany). The HTRF signal was calculated as the ratio of 665/620 nm emission multiplied by 10,000. The specific measured HTRF signal (energy transfer) is inversely proportional to the concentration of IP1 in the cells. The data were normalized against the minimal and maximal IP1 accumulation in cells driven by specific CB1R variant.
2.7. Immunoblot analysis
Expression levels of CB1R‐YFP mutant variants were assessed using western blot analysis. Briefly, HEK293 cells transfected with a particular CB1R‐YFP variant or empty plasmid pRK6 (mock) were washed with ice‐cold PBS and harvested in PBS complemented with cOmplete™ EDTA‐free Protease Inhibitor Cocktail tablet (Merck, Germany, cat. no. 4693132001) followed by centrifugation 13,000 g for 10 min at 4°C. Supernatants were aspirated and the pellets were resuspended in PBS with protease inhibitor. Afterward, the cells were disrupted by ultrasonication (IKA, Germany) and the total amount of protein in each lysate was determined using Bradford Reagent‐based assay (Sigma‐Aldrich, Czech Republic, cat. no. B6916‐500ML) following the manufacturer's instructions. The samples were then treated in SDS–PAGE treatment buffer (0.25 M Tris–Cl, 8% SDS, 20% glycerol, 0.02% bromophenol blue, 0.04 M DTT, pH 6.8) for 10 min at 85°C.
Lysates were separated by 10% SDS–PAGE. Subsequently, the proteins were transferred to nitrocellulose membrane (Pall Corporation, cat. no. 66485) and the membrane was blocked in 5% blotting‐grade powdered milk (Carl Roth, Germany, cat. no. 68514‐61‐4) in PBST buffer. Afterward, the membrane was cut into two pieces and labeled either with primary antibody mouse anti‐GFP (1:400, Roche, CH, Cat#11814460001) followed by secondary antibody labeling goat anti‐mouse IgG‐HRP antibody (1:10,000, Promega, Cat#W4021) for detection of CB1R‐YFP variants or with primary antibody rabbit anti‐actin (1:500, Sigma‐Aldrich, Czech Republic, Cat#SAB4291137) followed by secondary goat anti‐rabbit IgG‐HRP antibody (1:10,000, Promega, Cat#W4018) for the detection of actin to check the equal loading and protein transfer.
The proteins of interest were visualized by chemiluminescence using the SuperSignal West PICO chemiluminescent substrate (Thermo Fisher Scientific, cat. no. 34579) and detected on the LAS‐300 system (Fujifilm).
2.8. Statistical analysis
Unless stated otherwise, data represent the mean ± SEM of at least three experiments of independent cell preparations performed in triplicates. Sample sizes used for the study were determined based on previous studies of a similar nature (Brule et al., 2014; Hajkova et al., 2016). Post hoc power analysis of sample size showed that the sample sizes in the experiments were sufficient for statistical validity. The analysis was performed using G*Power 3.1.9.4 software with following parameters: power = 0.8, alpha = 0.05, effect size f = 0.25. No exclusion criteria were pre‐determined. The data were not assessed for normality and no test for outliers was conducted. Statistical analysis was performed using two‐way ANOVA followed by Sidak's multiple comparisons test using GraphPad Prism 7 (GraphPad Software, Inc.).
2.9. Ethical statement
Ethical approval was not required for this study.
3. RESULTS
3.1. Blocker of GRK3 catalytic activity cmpd101 obliterates GRK3 interaction with Gβγ and CB1R
During GPCR‐mediated signaling, Gβγ subunits may interact with GRK3 (Carman et al., 2000; Lodowski et al., 2005). In HEK293 cells co‐expressing CB1R, GRK3‐Rluc8, and Gβγ‐YFP (Figure S1a), WIN‐induced activation of CB1R resulted in a rapid GRK3‐Rluc8‐Gβγ‐YFP association, as shown by the increase of BRET signal (Figure 1a). In this experiment, the GRK2/3 inhibitor cmpd101 significantly reduced the interaction between GRK3‐Rluc8 and Gβγ‐YFP, upon WIN stimulation (mBRET values ± SEM in 15 min: CB1R = 161 ± 7.34; CB1R + cmpd101 = 49.1 ± 3.24). The GRK2/3 inhibitor cmpd101 binds to the GRK2/3 active site and renders the kinase catalytically inactive (Thal, Yeow, Schoenau, Huber & Tesmer, 2011; Ikeda S 2007). WIN potency in the presence of cmpd101 was not significantly changed, as observed in WIN‐induced GRK3‐Rluc8‐Gβγ‐YFP association (Figure 1b). Therefore, GRK3 in its active state is required for optimal association with Gβγ.
FIGURE 1.
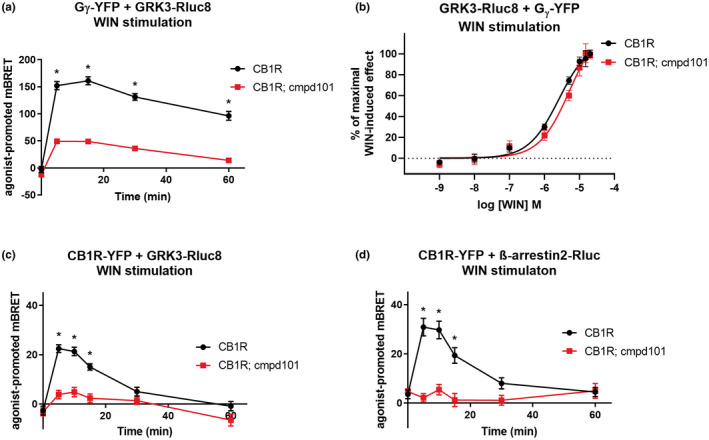
Consequences of GRK3 inactivation on the protein–protein interactions. HEK293 cells were transiently cotransfected with the following plasmid combinations: CB1R‐YFP + GRK3‐RLuc8 + empty plasmid pRK6 (2:1:2 ratio), CB1R‐SNAP + GRK3‐Rluc8 + Gβ‐flag + Gγ‐YFP (2:1:1:2 ratio) or CB1R‐YFP + β‐arrestin2‐Rluc + empty plasmid pRK6 (2:1:2 ratio). After 16 h, cells were pretreated for 30 min with cmpd101 (30 μM) prior to stimulation with the CB1R agonist WIN55212‐2 (WIN, 1 μM) where indicated. (a) Kinetic profiles of GRK3‐RLuc8 and Gγ‐YFP association dynamics in cmpd101 treated and nontreated cells. (b) Dose–response curves of GRK3‐RLuc8 and Gγ‐YFP association dynamics in cmpd101 treated and nontreated cells after CB1R stimulation with increasing concentrations of WIN. Twenty‐four hours after transfection, 5 μM coelenterazine h was added, cells were stimulated with increasing concentrations of WIN and the increase in BRET signal was measured 15 min after WIN application. (c) Kinetic profiles of GRK3‐RLuc8 recruitment by WIN‐activated CB1R‐YFP in HEK293 cells pretreated or not treated with cmpd101. (d) Kinetic profiles of β‐arrestin2‐Rluc recruitment by activated CB1R‐YFP in cmpd101 pretreated and non‐pretreated cells. Data represent the mean ± SEM of three experiments of independent cell preparations performed in three technical replicates. Data of graphs (a), (c), and were normalized against the maximal response of cmpd101 untreated cells. *p ≤ 0.05 (full statistical analysis is disclosed in Table S1)
Next, we studied whether the activity of GRK3 is required for its recruitment to CB1R. In HEK293 cells, we transiently expressed CB1R‐YFP and GRK3‐RLuc8 to monitor their association using BRET signal analysis (Figure S1b). Application of the CB1R agonist WIN induced a rapid increase in BRET efficiency, consistent with the formation of CB1R‐GRK3 complexes (Figure 1c). This was completely suppressed by pretreatment with the CB1R selective inverse agonist rimonabant (SR141716), while WIN application had no effect on the BRET signals in HEK293 cells transfected with the metabotropic glutamate receptor mGluR1a (Figure S2a). GRK2/3 activity blocker cmpd101 almost completely suppressed the agonist‐driven increase in BRET signal between CB1R‐YFP and the GRK3‐RLuc8 (mBRET values ± SEM in 5 min: CB1R = 22.45 ± 1.57; CB1R + cmpd101 = 3.88 ± 1.73) (Figure 1c), suggesting that GRK3 catalytic activity is required for its association with the activated CB1R.
3.2. GRK2/3 catalytic activity inhibitor cmpd101 prevents recruitment OF β‐arrestin2 BY activated CB1R
Phosphorylation of GPCRs results in β‐arrestin recruitment. In cells expressing β‐arrestin2‐Rluc with CB1R‐YFP (Figure S1c), a BRET signal increase was evident upon CB1R activation by WIN. This increase was obliterated upon the pretreatment of the cells with cmpd101 (mBRET values ± SEM in 10 min: CB1R = 29.72 ± 3.57; CB1R + cmpd101 = 5.46 ± 2.08) (Figure 1d). We verified that cmpd101 did not affect the expression of CB1R‐YFP (Figure S3a). Thus, the recruitment of β‐arrestin2 to agonist‐stimulated CB1R is dependent on the catalytic activity of GRK2/3.
3.3. Site‐directed mutagenesis within CB1R C‐tail
To study the role of GRK2/3 in CB1R regulation, we mutated serine and threonine residues within the 425SMGDS429 and 460TMSVSTDTS468 motifs (Figure 2a). We mutated the residues either into alanine residues that do not undergo phosphorylation or into negatively charged aspartic acids residues that partially mimic a phosphorylated state (Figure 2a). CB1R variants containing mutations in 425SMGDS429 motif are referred as CB1R_2X, mutants in 460TMSVSTDTS468 motif as CB1R_6X, and receptors simultaneously mutated in both motifs as CB1R_8X. Based on the mutation type, X is either A (in the case of alanine mutations) or D (in the case of aspartic acid mutations) (Figure 2a). We verified the levels of expression of WT and mutated CB1R forms by immunoblotting (Figure S2d). The multiple bands detected in western blot do not appear in a sample derived from the cells transfected with empty vector (mock), and likely represent distinct CB1R forms such as receptor dimers and /or post‐translationally modified receptors (glycosylation etc.) (de Jesus, Salles, Meana & Callado, 2006; Wager‐Miller, Westenbroek & Mackie, 2002). Proper cellular localization of the proteins was analyzed by confocal fluorescent microscopy (Figure 2b). All mutant receptors were functional, as monitored using the BRET‐based Gαi1‐Gβγ protein activity sensors (Figure S1d, S2b, c). Activation of mutated CB1R by WIN55,212–2 (WIN) (1 µM) in transiently transfected HEK293 cells induced a decrease of the BRET signal resulting from the dissociation/conformational change of Gαi1‐Rluc8–Gγ‐YFP complex. Prior to adding the agonist, the BRET signal remained stable and then declined upon WIN application over 10 min (Figure S2b, c). This establishes that the CB1R C‐tail phosphorylation mutants retain the ability to activate Gαi1 protein signaling pathway.
FIGURE 2.
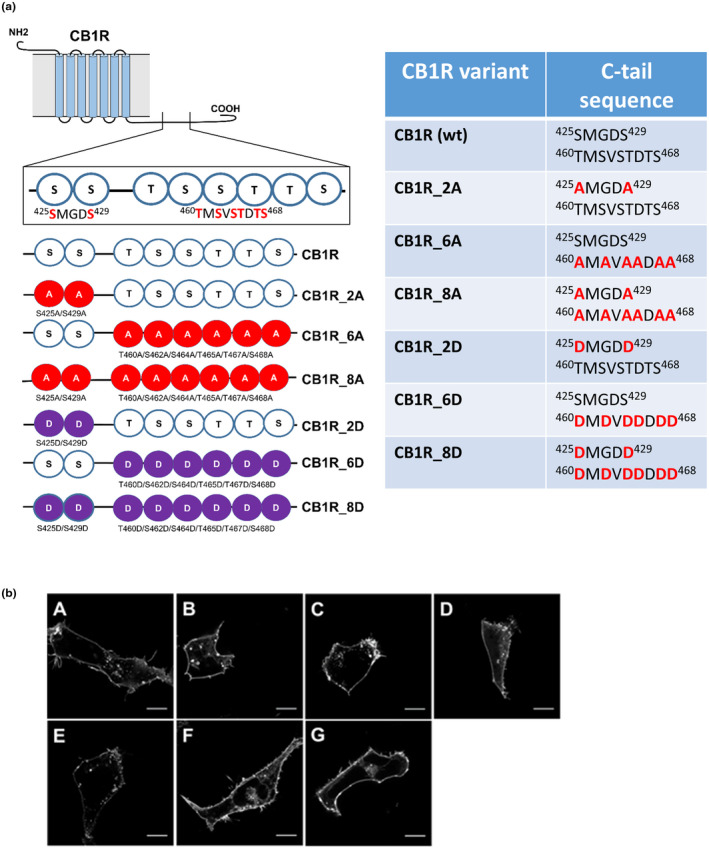
Schematic depiction of CB1R mutants within the C‐tail and characterization of their cellular distribution. (a) List of CB1R mutants and their corresponding sequences. Two regions of CB1R: 425SMGDS429 and 460TMSVSTDTS468 contain serine/threonine residues that are possibly phosphorylated during desensitization of CB1R. CB1R C‐tail phosphorylation mutants were created according to the scheme: A—mutation into alanine, D—mutation into aspartic acid. (b) CB1R and mutant CB1Rs are predominantly localized on the cellular membrane. HEK293 cells were transiently transfected with CB1R‐YFP variant. Twenty‐four hours after transfection, cells were visualized using fluorescent microscope. A single confocal section through the equatorial plane of the cells is shown. Legend: (A) CB1R, (B) CB1R_2A, (C) CB1R_6A, (D) CB1R_8A, (E) CB1R_2D, (F) CB1R_6D, (G) CB1R_8D. Scale bar represents 10 μm
3.4. G protein activation and signaling by CB1R mutants is not altered
Next we tested, wheter the efficiency with which the receptors drive G protein activity is affected by C‐tail mutations. We found that an inability to phosphorylate various residues within CB1R C‐tail regions 425SMGDS429 and 460TMSVSTDTS468 does not affect acute Gαi1 protein activation, as the extent and potency of the activation of Gαi1 proteins was similar in all tested CB1R mutants (logEC50 for Gαi1 activation; CB1R = −7.169; CB1R_2A = −7.128; CB1R_6A = −7.007; CB1R_8A = −7.146) (Figure 3a). The G protein signaling driven by mutant CB1Rs was not altered as well, seeing that the production of IP1 by chimeric G protein Gqi9 was not changed (logEC50 for Gqi9 activation; CB1R = −6.202; CB1R_2A = −6.353; CB1R_6A = −6.311; CB1R_8A = −6.170) (Figure 3b).
FIGURE 3.
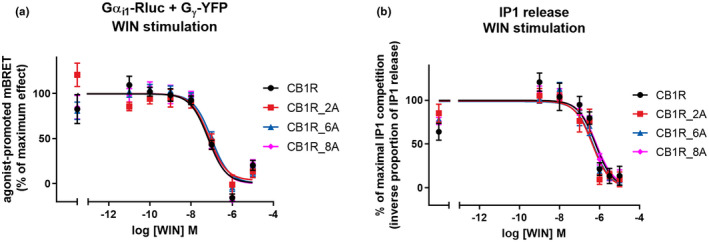
G protein signaling is not changed in CB1R mutants. (a) C‐tail mutations do not significantly change acute CB1R mediated Gαi protein activation. HEK293 cells were transiently cotransfected with CB1R‐SNAP variant, Gαi‐Rluc8, Gβ‐Flag, Gγ‐YFP (2:1:1:1 ratio). Twenty‐four hours after transfection, 5 μM coelenterazine h was added, cells were stimulated with increasing concentrations of WIN and the decrease in BRET signal was measured 15 min after WIN application. (b) CB1R mutants retain the ability to cause IP1 release induced by WIN. HEK293 cells were transiently cotransfected with CB1R‐SNAP variant and chimeric G protein Gqi9. Twenty‐four hours after transfection, cells were stimulated with increasing concentrations of WIN. The extent of IP1 accumulation was measured 20 min after WIN application. Data represent the mean ± SEM of three experiments of independent cell preparations performed in three technical replicates. Data were normalized against the maximal WIN‐ induced response
3.5. Formation of GRK3‐Gβγ complexes is only partially influenced by CB1R phosphorylation state
In cells co‐expressing GRK3‐Rluc8 and Gβ2‐Flag, Gγ2‐YFP (Figure S1a), the application of 1 μM WIN caused a prompt increase in BRET signal (Figure 4a). Subsequently, we used the CB1R mutants to study whether GRK3 association with Gβγ subunits is affected by CB1R C‐tail phosphorylation patterns. WIN stimulation of the CB1R_2A was followed by a rapid increase in BRET signal due to the formation of GRK3‐Gβγ complexes (mBRET values ± SEM at 5 min CB1R = 138.3 ± 1.93; CB1R_2A = 157.5 ± 7.42) (Figure 4b).
FIGURE 4.
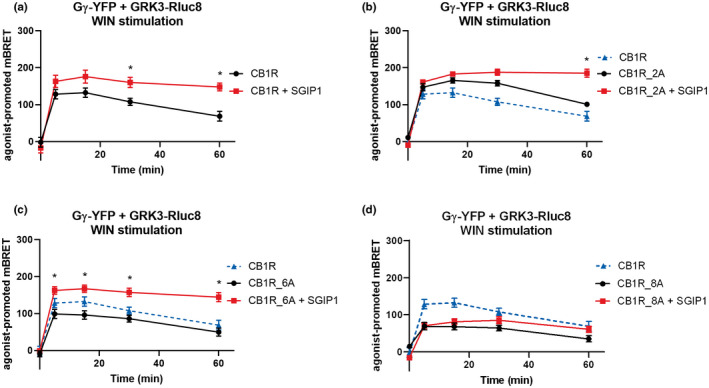
Formation of GRK3‐Gβγ complexes is partially independent of CB1R C‐tail phosphorylation. The association of GRK3‐Gβγ complexes in the mutant receptors that interact with partner proteins is enhanced in SGIP1 presence. HEK293 were transiently co‐transfected with CB1R, GRK3‐Rluc8, Gγ‐YFP, Gβ, and empty vector/SGIP1‐mCherry (1:1:2:1:2 ratio). Twenty‐four hours after transfection, cells were stimulated by 1 μM WIN. (a) Kinetic profile of GRK3 recruitment to Gγ in CB1R in the presence and absence of SGIP1. (b) Kinetic profile of GRK3 recruitment to Gγ in CB1R, CB1R_2A, and CB1R_2A + SGIP1. (c) Kinetic profile of GRK3 recruitment to Gγ driven by CB1R and CB1R_6A in presence/absence of SGIP1. (d) Kinetic profile of GRK3 recruitment to Gγ driven by CB1R and CB1R_8A in presence/absence of SGIP1. Data represent the mean ± SEM of three experiments of independent cell preparations performed in three technical replicates. *p ≤ 0.05 (full statistical analysis is disclosed in Table S2 and Table S3)
The stimulation of the CB1R_6A produced an increase in BRET efficiency, although to a lower extent of the WT CB1R activation‐induced BRET response (mBRET values ± SEM at 5 min: CB1R = 138.3 ± 1.93; CB1R_6A = 91.77 ± 2.90) (Figure 4c). Interestingly, the same result was obtained with the CB1R_8A transfected cells (mBRET values ± SEM at 5 min: CB1R = 138.3 ± 1.93; CB1R_8A = 83.82 ± 10.57) (Figure 4d).
These results suggest that GRK3‐Gβγ interactions are only partially dependent on CB1R phosphorylation and that phosphorylation of the CB1R C‐terminal motif is not required for GRK3‐Gβγ interaction.
3.6. GRK3‐Gβγ association is strengthened and prolonged in the presence of SGIP1
We previously reported that SGIP1 increased interactions between CB1R and β arrestin2 (Hajkova et al., 2016). We now show that the CB1R‐driven GRK3 interaction with Gβγ subunits is also increased and prolonged in the presence of SGIP1. Indeed, in cells co‐expressing GRK3‐Rluc8, Gγ‐YFP, CB1R, WIN stimulation of CB1R was followed by a rapid increase in BRET signal due to the formation of GRK3‐Gβγ complexes that was significantly enhanced and prolonged in the presence of co‐expressed SGIP1 (mBRET values ± SEM in 30 min: CB1R = 107.6 ± 9.73; CB1R + SGIP1 = 160.3 ± 13.91) (Figure 4a).
We also tested whether different CB1R C‐tail phosphorylation patterns affect the ability of SGIP1 to enhance GRK3‐Gβγ association. Activation of CB1R_2A in the presence SGIP1 resulted in significantly prolonged BRET signal in comparison with BRET produced by cells without SGIP1 expression (mBRET values ± SEM in 60 min: CB1R_2A = 101.1 ± 5.92; CB1R_2A + SGIP1 = 185.3 ± 10.76) (Figure 4b). Interestingly, the GRK3‐Gβγ association was significantly augmented immediately after stimulation of CB1R_6A when coexpressed with SGIP1 (mBRET values ± SEM in 5 min: CB1R_6A = 99.04 ± 12.50; CB1R_6A + SGIP1 = 162.6 ± 10.40) (Figure 4c). In cells expressing CB1R_8A, the dynamics of the association between GRK3‐Gβγ after WIN stimulation was reminiscent of that without SGIP1 coexpression (Figure 4d). The expression of Gγ‐YFP or GRK3‐Rluc8 was not modified when coexpressed with SGIP1‐mCherry (Figure S4g–i).
In the presence of SGIP1, the interaction between GRK3 and Gβγ that occurs during CB1R desensitization is enhanced, or prolonged, or both.
3.7. The two C‐terminal motifs differentially control GRK3–CB1R interaction
We next studied the relationship between the phosphorylation pattern of CB1R and the recruitment of GRK3 (Figure S1a). In cells co‐expressing GRK3‐Rluc8 and CB1R‐YFP, the application of 1 μM WIN resulted in a rapid increase of BRET signal (Figure 5a).
FIGURE 5.
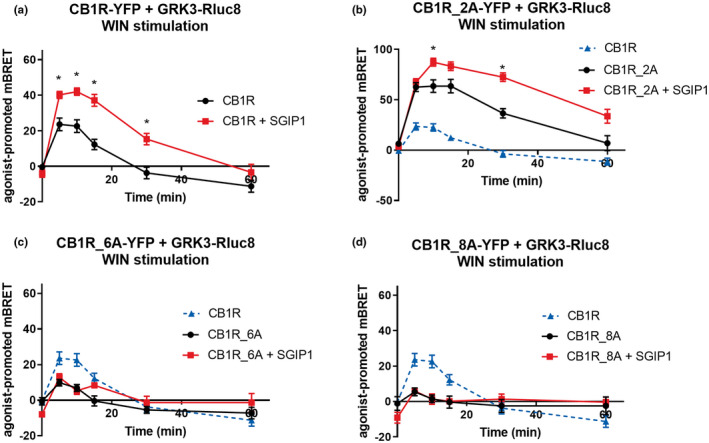
C‐tail multisite phosphorylation is crucial for GRK3 recruitment and dissociation. The association of CB1R‐GRK3 in CB1R mutants that interact with GRK3 is increased by SGIP1. HEK293 cells were transiently co‐transfected with the plasmids coding CB1R‐YFP variant + GRK3‐RLuc8 + empty plasmid pRK6/SGIP1‐mCherry (2:1:2 ratio). Cells were stimulated by WIN55212‐2 (WIN, 1 μM). (a) Kinetic profile of GRK3 recruitment to CB1R in the presence and absence of SGIP1. (b) Kinetic profile of GRK3 recruitment to CB1R, CB1R_2A, and CB1R_2A + SGIP1. (c) Kinetic profile of GRK3 recruitment to CB1R and CB1R_6A in the presence or absence of SGIP1. (d) Kinetic profile of GRK3 recruitment to CB1R, CB1R_8A, and CB1R_8A + SGIP1. Data represent the mean ± SEM of three experiments of independent cell preparations performed in three technical replicates. *p ≤ 0.05 (full statistical analysis is disclosed in Tables S4 and S5)
We observed that preventing phosphorylation of the short motif 425SMGDS429 increased and prolonged the interaction with GRK3 as the agonist stimulation of CB1R_2A‐YFP resulted in a greater BRET signal between the receptor and GRK3‐Rluc8 compared with the CB1R‐YFP/GRK3‐Rluc8 pair (mBRET values ± SEM in 5 min: CB1R = 23.62 ± 3.54; CB1R_2A = 63.56 ± 6.28) (Figure 5b). GRK3 recruitment profile by activated CB1R_2D‐YFP was similar to that obtained with the CB1R‐YFP (CB1R = 23.62 ± 3.54; CB1R_2D = 31.69 ± 3.75) (Figure 7b). The alanine mutation of serine residues in 425SMGDS429 (CB1R_2A) increased the BRET signal between GRK3 and CB1R, while the aspartic acid mutations (CB1R_2D) had no effect relative to WT. Such data are consistent with the possible phosphorylation of these sites decreasing the interaction between GRK3 and CB1R, by either limiting the association, or accelerating dissociation.
FIGURE 7.
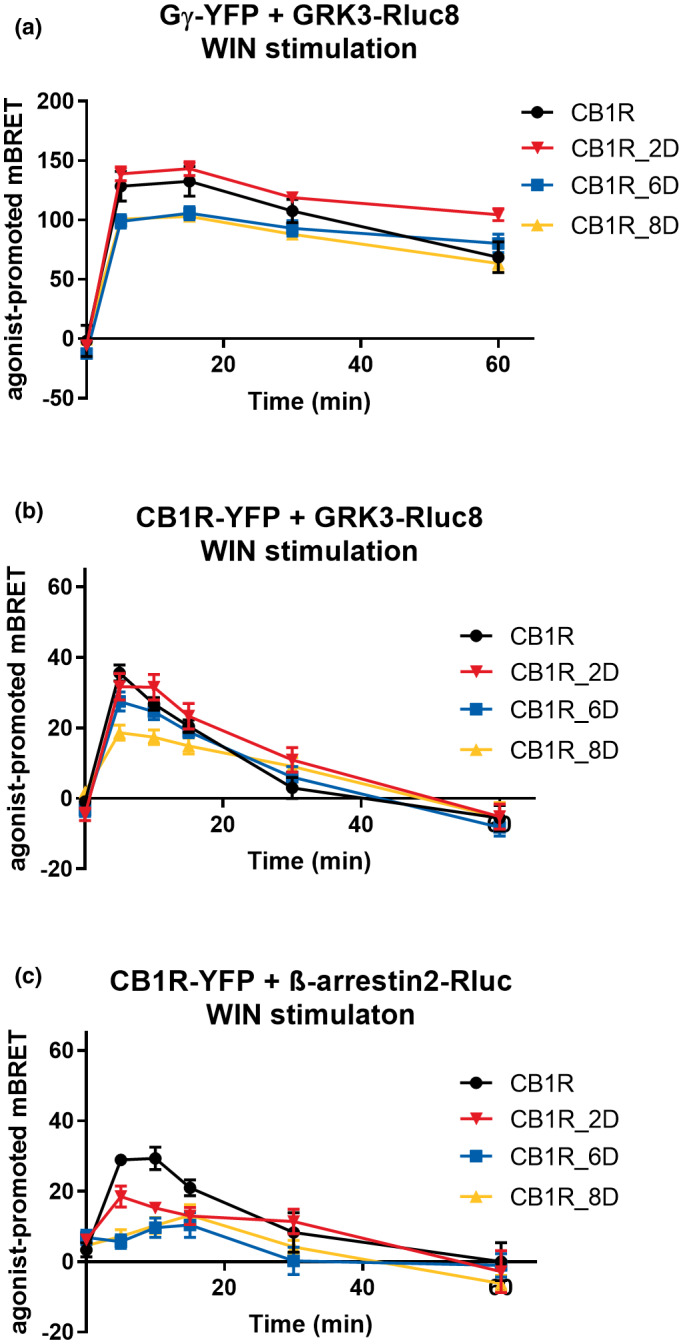
Protein–protein interactions driven by phosphomimetic CB1R mutants. HEK293 were transiently co‐transfected with either CB1R‐YFP variant, GRK3‐Rluc8 and empty plasmid pRK6 or CB1R_YFP variant, β‐arrestin2‐Rluc, and empty plasmid pRK6 or CB1R‐SNAP, GRK3‐Rluc8, Gγ‐YFP, Gβ, and empty vector pRK6 (1:1:2:1:2 ratio). Twenty‐four hours after the transfection, cells were stimulated by 1 μM WIN. (a) Kinetics of GRK3 recruitment to Gγ driven by CB1R, CB1R_2D, CB1R_6D, CB1R_8D. (b) Kinetics of GRK3 recruitment to CB1R, CB1R_2D, CB1R_6D, CB1R_8D. (c) Kinetics of β‐arrestin2 recruitment to CB1R, CB1R_2D, CB1R_6D, CB1R_8D. Data represent the mean ± SEM from three experiments of independent cell preparations performed in triplicate. (full statistical analysis is disclosed in Table S8)
Interestingly, a much lower BRET signal was observed in cells expressing CB1R_6A‐YFP (mutation of the serine/threonine residues of the motif 460TMSVSTDTS468) (mBRET values ± SEM in 5 min: CB1R = 23.62 ± 3.54; CB1R_6A = 9.88 ± 1.709) (Figure 5c). The response was abolished in the presence of CB1R‐8A (Figure 5d). This means that the phosphorylatable amino acids of the long motif of 460TMSVSTDTS468 are required for GRK3–CB1R interaction. Replacing the same residues by aspartate lead to BRET responses similar to that obtained with the WT CB1R (mBRET values ± SEM in 5 min: CB1R = 35.54 ± 2.26; CB1R_6D = 27.50 ± 2.68; CB1R_8D = 18.62 ± 2.16) (Figure 7b).
Expression of the mutated receptors was similar to that of wildtype CB1R and the expression of GRK3‐Rluc8 was not modified by coexpression with any of the mutated receptors (Figure S3b, c).
3.8. CB1R‐GRK3 association is augmented in the presence of SGIP1
Interestingly, SGIP1, known to increase increased CB1R‐β‐arrestin2 interaction (Hajkova et al., 2016), also favored CB1R–GRK3 interaction. In HEK293 cells coexpressing CB1R‐YFP variants, GRK3‐Rluc8 and either SGIP1‐mCherry or an empty vector pRK6, activation of CB1R in the presence of SGIP1 resulted in a significantly stronger and prolonged GRK3 recruitment (mBRET values ± SEM in 5 min: CB1R = 23.62 ± 3.54; CB1R + SGIP1 = 40.16 ± 2.01) (Figure 5a). SGIP1 further increases interaction of CB1R_2A with GRK3‐RLuc8 (mBRET values ± SEM in 10 min: CB1R_2A = 63.56 ± 6.28; CB1R_2A + SGIP1 = 87.26 ± 3.92) (Figure 5b), but could not rescue the interaction between either CB1R_6A or CB1R_8A and GRK3 (Figure 5c, d). The expression of the receptor mutants or GRK3‐Rluc8 was not affected when coexpressed with SGIP1‐mCherry (Figure S4a–c). These results suggest that GRK3 recruitment to CB1R is strengthened and prolonged in the presence of SGIP1.
3.9. Phosphorylation of the CB1R C‐tail regulates β‐arrestin2 interaction
When the second motif cannot be phosphorylated, not only is GRK3 recruitment to CB1R lost, but the recruitment of β‐arrestin2 is also prevented. Upon WIN stimulation, both CB1R_2A‐YFP and CB1R_2D‐YFP mutants were able to recruit β‐arrestin2‐Rluc; however, to a significantly decreased extent in comparison with wildtype CB1R (mBRET values ± SEM in 5 min: CB1R = 29.33 ± 1.93; CB1R_2A = 15.53 ± 3.28; CB1R_2D = 18.48 ± 2.96) (Figures 6b and 7c).
FIGURE 6.
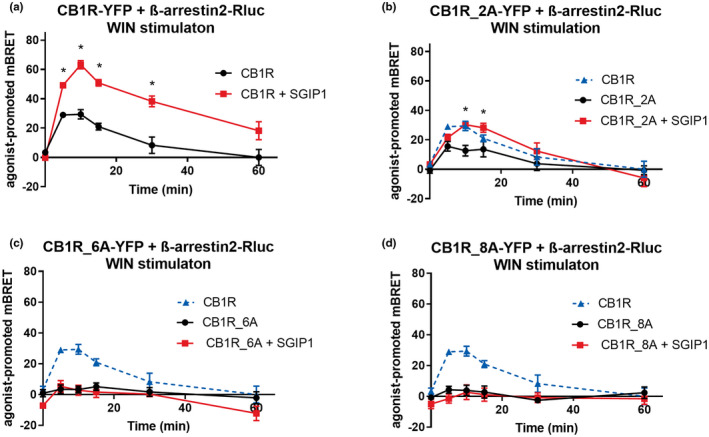
β‐arrestin2 binding is affected by the phosphorylation pattern of the CB1R C‐tail. SGIP1 enhances the association of CB1R‐β‐arrestin2 in cases the mutant receptors interact with β‐arrestin2. HEK293 cells were transiently co‐transfected with the CB1R‐YFP variants, β‐arrestin2‐Rluc, and empty vector/SGIP1‐mCherry (2:1:2 ratio). Cells were stimulated by 1 μM WIN. (a) Kinetic profile of β‐arrestin2 recruitment to CB1R in the presence/absence of SGIP1. (b) Kinetic profile of β‐arrestin2 recruitment to CB1R, CB1R_2A, and CB1R_2A + SGIP1. (c) Kinetic profile of β‐arrestin2 recruitment to CB1R and CB1R_6A in the presence/absence of SGIP1. (d) Kinetic profile of β‐arrestin2 recruitment to CB1R, CB1R_8A, and CB1R_8A + SGIP1. Data represent the mean ± SEM from three experiments of independent cell preparations performed in triplicate. *Represents p ≤ 0.05 (full statistical analysis is disclosed in Tables S6 and S7)
On the other hand, activation of CB1R_6A and CB1R_8A mutants resulted in severely impaired β‐arrestin2 recruitment (mBRET values ± SEM in 5 min: CB1R = 29.33 ± 1.93; CB1R_6A = 3.49 ± 2.77; CB1R_8A = 4.28 ± 2.24) (Figure 6c, d). The expression of β‐arrestin2‐Rluc was not modified by the coexpression of the mutated receptors (Figure S4d, e).
While the 425SMGDS429 motif's participation in βarrestin2 recruitment is clear from the reduced recruitment efficiency of the CB1R_2A mutant, data for the CB1R_6A mutant show that phosphorylatable sites in the 460TMSVSTDTS468 motif are a necessary minimum that the 425SMGDS429 phosphorylation sites cannot compensate for, as the alanine mutation of the 460TMSVSTDTS468 completely abrogated βarrestin2 recruitment.
3.10. SGIP1 strengthens the formation of CB1R‐β–arrestin2 complexes in β‐arrestin2‐interacting receptors
As observed for GRK3–CB1R interaction, SGIP1 could increase the interaction between β‐arrestin2 and CB1R (mBRET values ± SEM in 10 min: CB1R = 26.59 ± 7.08; CB1R + SGIP1 = 60.39 ± 3.53) (Figure 6a) even if the short 425SMGDS429 motif serine/threonine residues are mutated (mBRET values ± SEM in 10 min: CB1R_2A = 12.6 ± 3.60; CB1R_2A + SGIP1 = 30.30 ± 1.651) (Figure 6b). In contrast, SGIP1 did not recover β‐arrestin2 recruitment to CB1R_6A and CB1R_8A that contains the non‐phosphorylatable long motif, 460TMSVSTDTS468 (Figure 6c, d). As a control to the above experiments, we verified the equivalent expression levels of receptors, β‐arrestin2‐Rluc and SGIP1‐mCherry (Figure S4d–f).
While SGIP1 strengthens and prolongs β‐arrestin2 recruitment to the mutated receptors that interact with β‐arrestin2 (CB1R, CB1R_2A), SGIP1 alone is insufficient to rescue this interaction as seen in the case of CB1R_6A and CB1R_8A mutants, which were unable to recruit β‐arrestin2 regardless of SGIP1 presence.
3.11. Protein interactions driven by phosphomimetic CB1R mutants
Next, we examined the protein–protein interactions induced by phosphomimetic CB1R variants containing aspartic acid substitutions for serines/threonines in 425SMGDS429 and 460TMSVSTDTS468 motifs.
First, we tested these mutants for the capability to induce GRK3 recruitment to Gβγ subunits. While the stimulation of CB1R_2D formed GRK3‐Gβγ kinetic pattern similar to the one of CB1R, GRK3 association with Gβγ induced by CB1R_6D and CB1R_8D produced a slightly decreased BRET efficiency (mBRET values ± SEM in 5 min: CB1R = 128.49 ± 12.59; CB1R_2D = 138.81 ± 5.96; CB1R_6D = 98.91 ± 5.60; CB1R_8D = 100.727 ± 2.76) (Figure 7a). However, neither of GRK3‐Gβγ kinetics profiles produced by aspartic acid mutants were significantly different from the wild‐type receptor (Table S8a).
Next, we studied GRK3 interaction with aspartic acid CB1R mutants. We observed that the GRK3 interaction with CB1R_2D and CB1R_6D mutants was akin to the interaction of GRK3 with the wild‐type receptor. Contrary, the BRET signal of CB1R_8D was significantly decreased in comparison with the CB1R (mBRET values ± SEM in 5 min: CB1R = 35.54 ± 2.26; CB1R_2D = 31.69 ± 3.79; CB1R_6D = 27.5 ± 2.68; CB1R_8D = 18.61 ± 2.16) (Figure 7b) (Table S8B).
Lastly, we investigated the recruitment of β‐arrestin2 to aspartic acid CB1R variants. Interestingly, all the aspartic acid CB1R mutants showed impaired ability to recruit β‐arrestin2, as observed by the lower BRET efficiencies in comparison with CB1R (mBRET values ± SEM in 5 min: CB1R = 29.93 ± 1.51; CB1R_2D = 18.48 ± 2.96; CB1R_6D = 5.7 ± 1.97; CB1R_8D = 7.17 ± 1.95) (Figure 7c) (Table S8C).
3.12. GRK3 and β‐arrestin2 interaction with CB1R is regulated by different phosphorylation patterns of its C‐tail
We created additional CB1R‐YFP mutants within both the short and the long motifs in order to more precisely identify the residues involved for GRK3 and arrestin recruitment to CB1R (Figure 8a). The mutant CB1R_425SMGDS429_460AMSVSADAS468 (with all threonines mutated into alanine residues) interacted with GRK3 with a similar profile as the wildtype receptor up to the peak at 5 min, but at later time points the BRET signal for this mutant was prolonged compared with wildtype CB1R (Figure 8b). Thus, CB1R_425SMGDS429_460AMSVSADAS468 produced longer, or a spatially different association with GRK3 than wildtype CB1R (mBRET values ± SEM in 30 min: CB1R = 8.82 ± 4.03; CB1R_425SMGDS429_460AMSVSADAS468 = 27.11 ± 3.71). This effect was further increased by mutation of the serines of the short motif (as observed above) (Figure 8b).
FIGURE 8.
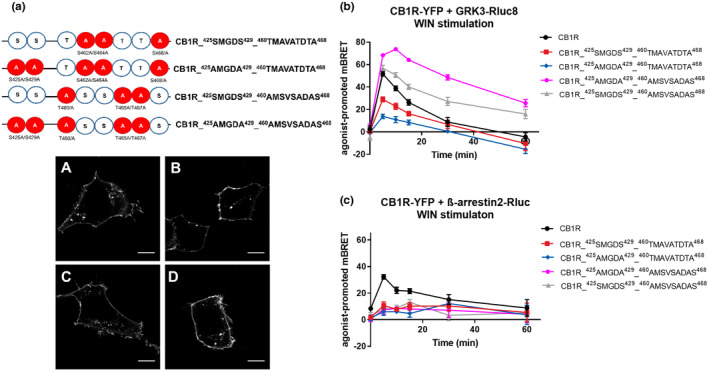
GRK3 and β‐arrestin2 interactions with CB1R depend on unique phosphorylation patterns. (a) Schematic depiction of additional constructed CB1R mutants and their cellular localization. A—mutation into alanine. HEK293 cells were transiently transfected with CB1R‐YFP variant. Twenty‐four hours after transfection, cells were visualized using fluorescent microscope. (A) CB1R_425SMGDS429_460TMAVATDTA468, (B) CB1R_425AMGDA429_460TMAVATDTA468, (C) CB1R_425SMGDS429_460AMSVSADAS468, (D) CB1R_425AMGDA429_460AMSVSADAS468. Scale bar represents 10 μm. (b) Kinetic profile of GRK3 recruitment to CB1R and CB1R mutants. (c) Kinetic profile of β‐arrestin2 recruitment to CB1R and CB1R mutants. Data represent the mean ± SEM from three independent cell preparations experiments performed in triplicate
In the next set of experiments, we used CB1R with mutated serine residues within the 425SMGDS429 and, at the same time, all the threonine residues in the distal C‐terminus were mutated into alanine residues, yielding CB1R_425AMGDA429_460AMSVSADAS468. This mutant receptor had an even more robust and profoundly extended association with GRK3 compared with wildtype receptor (mBRET values ± SEM in 30 min: CB1R = 8.82 ± 4.03; CB1R_425AMGDA429_460AMSVSADAS468 = 48.58 ± 2.34) (Figure 8b). This further confirms that serine residues phosphorylation within the 425SMGDS429 is important for the dissociation of GRK3 from CB1R likely in addition to the role of the threonine residues in the long motif.
In contrast, as expected according to the loss of GRK recruitment by CB1R‐6A or 8A, the CB1R_425SMGDS429_460TMAVATDTA468 mutant had a reduced ability to recruit GRK3 compared with CB1R (mBRET values ± SEM in 5 min: CB1R = 51.69 ± 2.04; CB1R_425SMGDS429_460TMAVATDTA468 = 28.95 ± 2.18). Interestingly, parallel mutations of the serine residues in the case of CB1R_ 425AMGDA429_460TMAVATDTA468 mutant further reduced GRK3 association (mBRET values ± SEM in 5 min: CB1R = 51.69 ± 2.04; CB1R_425AMGDA429_460TMAVATDTA468 = 13.81 ± 2.03) (Figure 8b). The expression of the mutant receptors was similar to that of the CB1R (Figure S3f). This confirms that the serine residues of 460TMSVSTDTS468 motif are crucial for GRK3 recruitment or for its strong interaction with CB1R.
We detected only a marginal interaction of these four CB1R mutants with β‐arrestin2 (Figure 8c). Therefore, precise and extensive phosphorylation of both motifs is required for optimal interaction of CB1R with β‐arrestin2. Phosphorylation of most residues within the second motif is required, as their mutation completely suppressed the receptor–β‐arrestin2 interaction.
4. DISCUSSION
This study describes previously unknown molecular mechanisms and interactions of molecules following activation of the CB1R as it undergoes desensitization. GRK3 interaction with G protein Gβγ subunits, as well as with the receptor, leads to the phosphorylation of several residues within CB1R C‐terminal tail. The kinase activity therefore leads to subsequent events resulting in the recruitment of β‐arrestin2 to the CB1R.
We aimed at dissecting the specificity of the phosphorylation patterns to drive selected interactions within the signalosome that results in desensitization of CB1R. We also show that SGIP1, a recently detected CB1R interacting partner, has profound effects on the extent and kinetics of the signalosome. The interaction between CB1R and GRK3 is enhanced, and dynamics of this interaction is extended, and also interactions GRK3 with G protein βγ dimer is enhanced in the presence of SGIP1.
4.1. GRK2/3 activation is a key step in CB1R desensitization
GRK2/3 execute a significant step in the desensitization of numerous GPCRs (Appleyard et al., 1999; Bawa, Altememi, Eikenburg & Standifer, 2003; Celver, Lowe, Kovoor, Gurevich & Chavkin, 2001; Dautzenberg & Hauger, 2001; Dautzenberg, Wille, Braun & Hauger, 2002; Ishii et al., 1994; Luo, Busillo, Stumm & Benovic, 2017). Activation of G proteins recruits GRK2/3 via interaction with G protein Gβγ dimer and fosters association between GRK2/3 and the receptor, which is consequently phosphorylated within its intracellular C terminus by the kinase (Homan & Tesmer, 2015; Nogues et al., 2017). Desensitization of CB1R expressed in Xenopus oocytes was shown to be dependent on the presence of both GRK3 and β‐arrestin2 (Daigle, Kearn & Mackie, 2008; Daigle, Kwok, et al., 2008; Gyombolai, Boros, Hunyady & Turu, 2013). Our data are in accord with these findings: the kinase activity of GRK2/3 is essential for efficient β‐arrestin2 recruitment to CB1R.
4.2. GRK3–Gβγ interaction
Cmpd101 binds deep in the active site (site responsible for kinase activity) of GRK2/3, but also partially binds to ATP site, thus preventing these kinases to achieve catalytically active state (Ikeda S, 2007; Thal et al., 2011). Following receptor activation, GRK2/3 interacts with the membrane‐associated Gβγ dimers (Boughton et al., 2011; Daaka et al., 1997; Touhara, Inglese, Pitcher, Shaw & Lefkowitz, 1994). In our experiments, cmpd101 inhibited the interaction of GRK3 with G protein βγ dimer, albeit not completely (Figure 1a). The WIN potency in the presence of cmpd101 was not changed (Figure 1b), thus, such a large decrease in BRET is best interpreted as smaller amounts of GRK3–Gβγ complexes formed in the cells. However, one cannot firmly exclude that the lower BRET results from a larger distance, or a different relative orientation between Rluc and YFP, although this is not supported by the structural data (Thal et al., 2011).
Interestingly, to a large extent, GRK3 was able to form complexes with Gβγ independently of CB1R C‐tail phosphorylation patterns, albeit the interaction was slightly modified. As the C‐tail mutation of CB1R did not alter G protein activation (Figure 3a, b), the quantity of released Gβγ available for GRK3 recruitment was not modified. These observations suggest that GRK3 interaction with Gβγ partially depends on the receptor as both receptor and G proteins are always part of the same "receptosome.”
4.3. GRK3–CB1R association
In the presence of cmpd101, recruitment of GRK3 to CB1R is blocked (Figure 1c), demonstrating that GRK3 has to be in an active conformation to interact with the activated CB1R. The residues on the CB1R C‐tail that may undergo GRK2/3 driven phosphorylation are distributed in two clusters, 425SMGDS429 and 460TMSVSTDTS468. GRK3/CB1R recruitment assays with serine/threonine residues mutated within these regions to alanine or aspartate residues identified two major findings. Firstly, stimulation of CB1R_2A (S425A, S429A mutations) resulted in significantly enhanced and prolonged GRK3 association, compared with the wild‐type CB1R. Mutagenesis within the 460TMSVSTDTS468 motif in CB1R_6A resulted only in partial attenuation of GRK3 recruitment to the receptor. Mutation of all serine/threonine residues to alanine within both regions, 425SMGDS429 and 460TMSVSTDTS468 (CB1R_8A), abrogated GRK3 recruitment to the activated receptor (Figure 5d). Secondly, GRK3 recruitment profile of the CB1R_2D mutant, in which aspartate residues mimic the phosphorylated state (S423D, S429D), resembled that of CB1R WT (Figure 7b). CB1R WT with phosphorylatable S423 and S429 have comparable dynamics of interaction between GRK3 and CB1R_2D (with mutations S423D and S429D). This partial recovery of the dynamics might be due to the fact that phosphoserine modification contains double negative charges, while aspartic acid has only a single charge, and perhaps this is insufficient for proper GRK3 binding. Also, phosphorylation of the residues is a dynamic process, and this is not reflected by the use of the constitutively charged aspartic acid in the corresponding CB1R mutants. Our interpretation is that indeed, phosphorylation of these residues (or one of them) is involved in the regulation of this interaction.
Based on these observations, we propose a hypothesis that phosphorylation of residues within the 425SMGDS429 region by GRK3 regulates the dynamics of GRK3–CB1R association. Following their interaction, phosphorylation of residues within 460TMSVSTDTS468 region favors the association, but subsequent phosphorylation of residues from 425SMGDS429, in turn, expedites the dissociation of GRK3 from the receptor. This may avert spatial hindrance and allows β‐arrestin2 association with the receptor.
SGIP1 modifies CB1R association with GRK3, with enhanced impact on their transient interaction in later phases of the desensitization (Figure 5a). This finding further underlines the role of the CB1R‐SGIP1 interaction on the dynamics of the interactions within the signalosome during CB1R desensitization.
4.4. CB1R–β‐arrestin2 interaction
Upon treatment with cmpd101, recruitment of β‐arrestin2 to the activated CB1R was significantly constrained (Figure 1c). We attribute this inhibition of β‐arrestin2 recruitment to the inhibition of GRK2/3 activation impairing CB1R phosphorylation. Previously, using quantitative analysis of fluorescent confocal images, mutant CB1R_2A (mutations of serine residues within the 425SMGDS429 motif named CB1R S425A/S429A in the preceding study) recruited β‐arrestin‐2 to the plasma membrane (Daigle, Kearn, et al., 2008). We revealed an impaired association of β‐arrestin‐2 with this mutant (Figure 6b). Therefore, for both CB1R and the mutated CB1R_2A mobilize β‐arrestin2 from the cytoplasm to the plasmalemma, however, our data show that levels of protein–protein association of β‐arrestin2 are decreased in the case of CB1R_2A. The aforementioned study used quantitative analysis of fluorescent confocal images that reveal recruitment from the cytoplasm toward the membrane, while BRET technology allows a more specific depiction of protein–protein interactions. Moreover, our study may have a different receptor number than the system used previously. A possibility exists that CB1R_2A–β‐arrestin2 complex acquires a different conformational state than with the wildtype receptor, decreasing BRET signal efficiency (Cahill et al., 2017; Nuber et al., 2016). Phosphorylation of residues within the 425SMGDS429 and 460TMSVSTDTS468 regions in CB1R_8A impaired CB1R‐mediated translocation of β‐arrestin2 to the membrane, and their mutation to alanine residues prevented internalization (Daigle, Kwok, et al., 2008; Jin et al., 1999) (Figure 6d). Here, our data are in agreement with the previous report, in which mutation of all eight phosphorylation sites results in loss of the ability to recruit β‐arrestin2 from the cytoplasm.
Our observations support the hypothesis that the 460TMSVSTDTS468 motif serves as an initiation docking site for β‐arrestin2 (Blume et al., 2017; Jin et al., 1999; Morgan et al., 2014). We hypothesize that the decreased β‐arrestin2 interaction with CB1R_2A is a consequence of protracted GRK3‐CB1R_2A interaction. Due to higher affinity between GRK3 and CB1R_2A and GRK3, this kinase sterically hinders β‐arrestin2 interaction with the CB1R_2A. This is further supported by the β‐arrestin2 interaction with CB1R_2D mutant, which has the S425 and S429 mutated into aspartate (Figure 7c), albeit these mutations only partially mimic the phosphorylated state of the corresponding residues. Mutations of the serine/threonine residues within 460TMSVSTDTS468 region into aspartate residues resulted in decrease of the BRET signal (Figure 7c). This suggests, that these residues are phosphorylated selectively, and that a certain pattern, or a “bar code” mode for proper CB1R‐ β‐arrestin‐2 association. This possibility was tested in more detail.
4.5. GRK3 AND β‐arrestin2 interactions with CB1R depend on unique phosphorylation patterns
To further explore the phosphorylation patterns needed for the interaction between CB1R and GRK3, or β‐arrestin2, we constructed additional CB1R mutants with several combinations of alanine replacements within 460TMSVSTDTS468 region. The substitution of only serine residues with alanine residues decreased the receptor's ability to recruit GRK3. In contrast, mutation of all threonine into alanine residues within the same region resulted in a mutant with extended CB1R‐GRK3 association. This was even more significant if S425A and S429A mutations were also included (Figure 8b). These results further illustrate that phosphorylation of 425SMGDS429 may cause dissociation of GRK3 from the receptor. In contrast, phosphorylation of serine residues within the 460TMSVSTDTS468 region is crucial for the formation of CB1R–GRK3 complexes. Conversely, the substitution of the threonine by alanine residues within this region did not prevent the CB1R–GRK3 association, suggesting that phosphorylation of threonine residues is not indispensable for the GRK3 association with CB1R.
All the CB1R variants with different triple alanine mutations in the extreme C‐tail exhibited similarly decreased, but not completely diminished, recruitment of β‐arrestin2 (Figure 8c). Hence, serine and threonine residues within 460TMSVSTDTS468 motif, all contribute to β‐arrestin2 recruitment. This contrasts with the phosphorylation pattern needed for the GRK3–CB1R interaction. Of importance, depending on certain phosphorylation patterns, β‐arrestins can acquire a wide range of conformation states. These different conformations, imposed by distinct phosphorylation patterns of the C termini, may also lead to differential BRET efficiencies (Latorraca et al., 2020; Lee et al., 2016; Nobles, Xiao & Ahn, 2011; Nuber et al., 2016). Our results may also point to sequential and/or cumulative phosphorylation of serine/threonine residues in 460TMSVSTDTS468 of CB1R, as those dynamics may play a role in β‐arrestin2 recruitment.
The μ‐opioid receptor also has a clustering of serine/threonine residues within two regions. Miess and colleagues described that phosphorylation of two serine/threonine clusters within the C‐terminal region of the μ‐opioid receptor controls the dynamics of GRK2 and β‐arrestin2 recruitment (Miess, Gondin & Yousuf, 2018). The extreme C‐tail serine/threonine cluster was involved in GRK2 and β‐arrestin2 recruitment, while the proximal serine/threonine region was involved in the stability of these interactions. Our data harmonize with these findings. GRK3 and β‐arrestin2 interactions with CB1R are regulated by different phosphorylation patterns of the receptor C‐tail. A recent study performed by Møller and colleagues disclosed that in the case of the μ‐opioid receptor, GRK2 and GRK3 have distinct impacts on β‐arrestin2 association (Moller et al., 2020), pointing to the complexity of the process, and likely to unique particulars of the desensitization process among different receptors.
4.6. SGIP1 augments GRK3‐Gβγ, GRK3‐CB1R, and β‐arrestin2–CB1R interactions
Previously, we described that G protein activation by CB1R is not affected by SGIP1 (Hajkova et al., 2016). Subsequent events dependent on CB1R C‐tail phosphorylation that would result in clathrin‐mediated endocytosis (CME) are profoundly influenced by SGIP1. SGIP1 stalls CME, and the phosphorylated receptor remains on the cell surface. We proposed a hypothesis that aims at depicting how the relationship between SGIP1 and CB1R affects events that follow CB1R desensitization; during CB1R desensitization, β‐arrestins interact with the phosphorylated CB1R. The temporary association between phosphorylated CB1R and β‐arrestin terminates as the receptor is internalized. SGIP1 stalls CB1R internalization. Therefore, the β‐arrestin interaction with desensitized CB1R persists longer in the presence of SGIP1. The consequence of stabilizing CB1R at the cell surface by SGIP1 is that dissociation of β‐arrestin2 from CB1R that follows internalization occurs more slowly, as described in our earlier study (Hajkova et al., 2016).
The interaction of GRK3 with Gβγ subunits following activation of the CB1R was also modified by SGIP1 in this study (Figure 4). Interestingly, the most evident effect of SGIP1 on the dynamics of this protein–protein association is at the early phase, but it persists through the observed period. Also, we show that the presence of SGIP1 results in enhancement of GRK3 association with both CB1R and CB1R_2A, but not with CB1R_6A, and CB1R_8A mutants that lose the ability to associate with GRK3 and β‐arrestin2 (Figures 5c, d, 6c, d). These prolonged interactions are likely the result of stalled CB1R internalization produced by SGIP1.
SGIP1 modifies most of the interactions within the CB1R desensitization interactome (Figure 9). The most likely explanation is that this is due to the interference with internalization. We show the rate of CB1R internalization in the presence of SGIP1 as detected in our previous study. Furthermore, our results indicate that the C‐tail phosphorylation patterns of CB1R do not affect SGIP1 association with CB1R. This is in agreement with our previous observations using yeast two hybrid screen with mutated C terminus of CB1R mimicking phosphorylated state of the residues. Using such mutants did not have any effect on the association between CB1R and SGIP1 (Figure 1a in (Hajkova et al., 2016)). Therefore, CB1R association with SGIP1 is independent of the modification. Future studies may discern, whether the effect of SGIP1 has an impact on the phosphorylation patterns of CB1R during desensitization.
FIGURE 9.
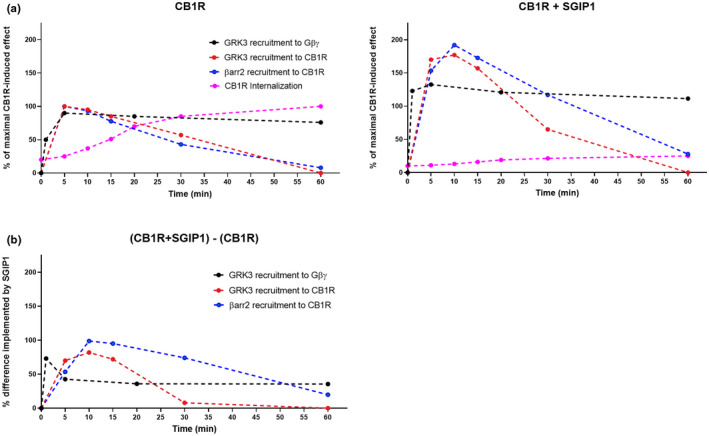
Graphic depiction of events relevant to CB1R desensitization. (a) Kinetics of events during desensitization of CB1R in the absence (left panel) and the presence (right panel) of SGIP1 upon CB1R stimulation with 1 µM WIN in HEK293 cells. Internalization data were taken from the previous study (Hajkova et al., 2016). All the experiments were performed in the same experimental system under similar conditions. (b) The difference in interactions implemented by SGIP1. Values were calculated by subtracting the kinetics values of CB1R + SGIP1 with the values of CB1R. Figure (b) was constructed using calculated data from the experiments shown in (a)
5. CONCLUSION
GRK3 has to reach an active conformation in order to fully interact with Gβγ dimer and CB1R to phosphorylate the CB1R C‐tail. The interaction of GRK3 and Gβγ is allosterically modulated by the CB1R–GRK3 complex and by SGIP1. Phosphorylation of serine residues within the 425SMGDS429 region by GRK3 causes relaxation of the GRK3–CB1R association. The phosphorylation arrangement, called a “bar code” of serine/threonine residues within 460TMSVSTDTS468 region, required for the receptor to interact with GRK3 and β‐arrestin2, has distinct implications on the receptor association with GRK3 and Gβγ. Alike in the case of CB1R interaction with β‐arrestin, SGIP1 enhances the association of GRK3 with both Gβγ subunits of G proteins and CB1R. Therefore, SGIP1 regulates levels of interactions between molecules that are part of the temporal CB1R signalosome that establishes during its desensitization.
CONFLICTS OF INTEREST
All authors report no biomedical financial interests or potential conflicts of interest.
AUTHOR CONTRIBUTIONS
Matej Gazdarica, Laurent Prezeau, and Jaroslav Blahos designed research; Matej Gazdarica and Judith Noda performed research; Ken Mackie contributed new reagents or analytic tools; Matej Gazdarica, Oleh Duridyvka, Vendula Novosadova, Jean‐Philippe Pin, Laurent Prezeau, and Jaroslav Blahos analyzed data; and Matej Gazdarica, Ken Mackie, Jean‐Philippe Pin, Laurent Prezeau, and Jaroslav Blahos wrote the paper.
OPEN RESEARCH BADGES
This article has earned an Open Materials badge for making publicly available the components of the research methodology needed to reproduce the reported procedure and analysis. All materials are available at https://cos.io/our‐services/open‐science‐badges/.
Supporting information
Supplementary Material
ACKNOWLEDGMENTS
This project was supported by Grant Agency of Czech Republic (19‐24172S), LM2015040 (Czech Centre for Phenogenomics) and by RVO: 68378050‐KAV‐NPUI by the Czech Academy of Sciences. Some pharmacology and BRET experiments were performed using the ARPEGE Pharmacology‐Screening‐Interactome platform facility (UMS Biocampus), Montpelier, France. For help with editing the text, we are thankful to Anna Karimova. The Graphical abstract was created using Biorender software.
All experiments were conducted in compliance with the ARRIVE guidelines.
Gazdarica, M. , Noda, J. , Durydivka, O. , Novosadova, V. , Mackie, K. , Pin, J.‐P. , Prezeau, L. , & Blahos, J. (2022). SGIP1 modulates kinetics and interactions of the cannabinoid receptor 1 and G protein‐coupled receptor kinase 3 signalosome. Journal of Neurochemistry, 160, 625–642. 10.1111/jnc.15569
Laurent Prezeau and Jaroslav Blahos contributed equally to this work.
DATA AVAILABILITY STATEMENT
The data that support the findings of this study are available from the corresponding author upon reasonable request.
REFERENCES
- Al‐Zoubi, R. , Morales, P. , & Reggio, P. H. (2019). Structural insights into CB1 receptor biased signaling. International Journal of Molecular Sciences, 20(8), 1837. 10.3390/ijms20081837 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Appleyard, S. M. , Celver, J. , Pineda, V. , Kovoor, A. , Wayman, G. A. , & Chavkin, C. (1999). Agonist‐dependent desensitization of the kappa opioid receptor by G protein receptor kinase and beta‐arrestin. Journal of Biological Chemistry, 274, 23802–23807. [DOI] [PubMed] [Google Scholar]
- Araque, A. , Castillo, P. E. , Manzoni, O. J. , & Tonini, R. (2017). Synaptic functions of endocannabinoid signaling in health and disease. Neuropharmacology, 124, 13–24. 10.1016/j.neuropharm.2017.06.017 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bakshi, K. , Mercier, R. W. , & Pavlopoulos, S. (2007). Interaction of a fragment of the cannabinoid CB1 receptor C‐terminus with arrestin‐2. FEBS Letters, 581, 5009–5016. 10.1016/j.febslet.2007.09.030 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bawa, T. , Altememi, G. F. , Eikenburg, D. C. , & Standifer, K. M. (2003). Desensitization of alpha 2A‐adrenoceptor signalling by modest levels of adrenaline is facilitated by beta 2‐adrenoceptor‐dependent GRK3 up‐regulation. British Journal of Pharmacology, 138, 921–931. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blume, L. C. , Patten, T. , Eldeeb, K. et al (2017). Cannabinoid receptor interacting protein 1a competition with beta‐arrestin for CB1 receptor binding sites. Molecular Pharmacology, 91, 75–86. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boughton, A. P. , Yang, P. , Tesmer, V. M. , Ding, B. , Tesmer, J. J. , & Chen, Z. (2011). Heterotrimeric G protein beta1gamma2 subunits change orientation upon complex formation with G protein‐coupled receptor kinase 2 (GRK2) on a model membrane. Proceedings of the National Academy of Sciences of the United States of America, 108, E667–673. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brule, C. , Perzo, N. , Joubert, J. E. , Sainsily, X. , Leduc, R. , Castel, H. , & Prezeau, L. (2014). Biased signaling regulates the pleiotropic effects of the urotensin II receptor to modulate its cellular behaviors. The FASEB Journal, 28, 5148–5162. 10.1096/fj.14-249771 [DOI] [PubMed] [Google Scholar]
- Cahill, T. J. 3rd , Thomsen, A. R. , Tarrasch, J. T. et al (2017). Distinct conformations of GPCR‐beta‐arrestin complexes mediate desensitization, signaling, and endocytosis. Proceedings of the National Academy of Sciences of the United States of America, 114, 2562–2567. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carman, C. V. , Barak, L. S. , Chen, C. , Liu‐Chen, L. Y. , Onorato, J. J. , Kennedy, S. P. , Caron, M. G. , & Benovic, J. L. (2000). Mutational analysis of Gbetagamma and phospholipid interaction with G protein‐coupled receptor kinase 2. Journal of Biological Chemistry, 275, 10443–10452. [DOI] [PubMed] [Google Scholar]
- Celver, J. P. , Lowe, J. , Kovoor, A. , Gurevich, V. V. , & Chavkin, C. (2001). Threonine 180 is required for G‐protein‐coupled receptor kinase 3‐ and beta‐arrestin 2‐mediated desensitization of the mu‐opioid receptor in Xenopus oocytes. Journal of Biological Chemistry, 276, 4894–4900. [DOI] [PubMed] [Google Scholar]
- Charest, P. G. , & Bouvier, M. (2003). Palmitoylation of the V2 vasopressin receptor carboxyl tail enhances beta‐arrestin recruitment leading to efficient receptor endocytosis and ERK1/2 activation. Journal of Biological Chemistry, 278, 41541–41551. [DOI] [PubMed] [Google Scholar]
- Daaka, Y. , Pitcher, J. A. , Richardson, M. , Stoffel, R. H. , Robishaw, J. D. , & Lefkowitz, R. J. (1997). Receptor and G betagamma isoform‐specific interactions with G protein‐coupled receptor kinases. Proceedings of the National Academy of Sciences of the United States of America, 94, 2180–2185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Daigle, T. L. , Kearn, C. S. , & Mackie, K. (2008). Rapid CB1 cannabinoid receptor desensitization defines the time course of ERK1/2 MAP kinase signaling. Neuropharmacology, 54, 36–44. 10.1016/j.neuropharm.2007.06.005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Daigle, T. L. , Kwok, M. L. , & Mackie, K. (2008). Regulation of CB1 cannabinoid receptor internalization by a promiscuous phosphorylation‐dependent mechanism. Journal of Neurochemistry, 106, 70–82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dautzenberg, F. M. , & Hauger, R. L. (2001). G‐protein‐coupled receptor kinase 3‐ and protein kinase C‐mediated desensitization of the PACAP receptor type 1 in human Y‐79 retinoblastoma cells. Neuropharmacology, 40, 394–407. 10.1016/S0028-3908(00)00167-2 [DOI] [PubMed] [Google Scholar]
- Dautzenberg, F. M. , Wille, S. , Braun, S. , & Hauger, R. L. (2002). GRK3 regulation during CRF‐ and urocortin‐induced CRF1 receptor desensitization. Biochemical and Biophysical Research Communications, 298, 303–308. 10.1016/S0006-291X(02)02463-4 [DOI] [PubMed] [Google Scholar]
- de Jesus, M. L. , Salles, J. , Meana, J. J. , & Callado, L. F. (2006). Characterization of CB1 cannabinoid receptor immunoreactivity in postmortem human brain homogenates. Neuroscience, 140, 635–643. 10.1016/j.neuroscience.2006.02.024 [DOI] [PubMed] [Google Scholar]
- Donthamsetti, P. , Quejada, J. R. , Javitch, J. A. , Gurevich, V. V. , & Lambert, N. A. (2015). Using bioluminescence resonance energy transfer (BRET) to characterize agonist‐induced arrestin recruitment to modified and unmodified G protein‐coupled receptors. Current Protocols in Pharmacology, 70(1), 11–12. 10.1002/0471141755.ph0214s70 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dvorakova, M. , Kubik‐Zahorodna, A. , Straiker, A. , Sedlacek, R. , Hajkova, A. , Mackie, K. , & Blahos, J. (2021). SGIP1 is involved in regulation of emotionality, mood, and nociception and tunes in vivo signaling of Cannabinoid Receptor 1. British Journal of Pharmacology, 178(7), 1588–1604. 10.1111/bph.15383 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fletcher‐Jones, A. , Hildick, K. L. , Evans, A. J. , Nakamura, Y. , Henley, J. M. , & Wilkinson, K. A. (2020). Protein interactors and trafficking pathways that regulate the cannabinoid type 1 receptor (CB1R). Frontiers in Molecular Neuroscience, 13, 108. 10.3389/fnmol.2020.00108 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Garcia, D. E. , Brown, S. , Hille, B. , & Mackie, K. (1998). Protein kinase C disrupts cannabinoid actions by phosphorylation of the CB1 cannabinoid receptor. Journal of Neuroscience, 18, 2834–2841. 10.1523/JNEUROSCI.18-08-02834.1998 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gurevich, E. V. , Tesmer, J. J. , Mushegian, A. , & Gurevich, V. V. (2012). G protein‐coupled receptor kinases: more than just kinases and not only for GPCRs. Pharmacology & Therapeutics, 133, 40–69. 10.1016/j.pharmthera.2011.08.001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gyombolai, P. , Boros, E. , Hunyady, L. , & Turu, G. (2013). Differential beta‐arrestin2 requirements for constitutive and agonist‐induced internalization of the CB1 cannabinoid receptor. Molecular and Cellular Endocrinology, 372, 116–127. [DOI] [PubMed] [Google Scholar]
- Hajkova, A. , Techlovska, S. , Dvorakova, M. , Chambers, J. N. , Kumpost, J. , Hubalkova, P. , Prezeau, L. , & Blahos, J. (2016). SGIP1 alters internalization and modulates signaling of activated cannabinoid receptor 1 in a biased manner. Neuropharmacology, 107, 201–214. 10.1016/j.neuropharm.2016.03.008 [DOI] [PubMed] [Google Scholar]
- Hamdan, F. F. , Rochdi, M. D. , Breton, B. , Fessart, D. , Michaud, D. E. , Charest, P. G. , Laporte, S. A. , & Bouvier, M. (2007). Unraveling G protein‐coupled receptor endocytosis pathways using real‐time monitoring of agonist‐promoted interaction between beta‐arrestins and AP‐2. Journal of Biological Chemistry, 282, 29089–29100. [DOI] [PubMed] [Google Scholar]
- Haring, M. , Marsicano, G. , Lutz, B. , & Monory, K. (2007). Identification of the cannabinoid receptor type 1 in serotonergic cells of raphe nuclei in mice. Neuroscience, 146, 1212–1219. 10.1016/j.neuroscience.2007.02.021 [DOI] [PubMed] [Google Scholar]
- Henne, W. M. , Kent, H. M. , Ford, M. G. J. , Hegde, B. G. , Daumke, O. , Butler, P. J. G. , Mittal, R. , Langen, R. , Evans, P. R. , & McMahon, H. T. (2007). Structure and analysis of FCHo2 F‐BAR domain: a dimerizing and membrane recruitment module that effects membrane curvature. Structure, 15, 839–852. 10.1016/j.str.2007.05.002 [DOI] [PubMed] [Google Scholar]
- Herkenham, M. , Lynn, A. B. , Little, M. D. , Johnson, M. R. , Melvin, L. S. , de Costa, B. R. , & Rice, K. C. (1990). Cannabinoid receptor localization in brain. Proceedings of the National Academy of Sciences, 87, 1932–1936. 10.1073/pnas.87.5.1932 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Homan, K. T. , & Tesmer, J. J. (2015). Molecular basis for small molecule inhibition of G protein‐coupled receptor kinases. ACS Chemical Biology, 10, 246–256. 10.1021/cb5003976 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hsieh, C. , Brown, S. , Derleth, C. , & Mackie, K. (1999). Internalization and recycling of the CB1 cannabinoid receptor. Journal of Neurochemistry, 73, 493–501. 10.1046/j.1471-4159.1999.0730493.x [DOI] [PubMed] [Google Scholar]
- Ikeda, S. , Keneko, M. & Fujiwara, S. (2007) Cardiotonic agent comprising GRK inhibitor. (T. P. C. Ltd ed.). Ikeda S, Keneko M, and Fujiwara S, Japan.
- Ishii, K. , Chen, J. , Ishii, M. , Koch, W. J. , Freedman, N. J. , Lefkowitz, R. J. , & Coughlin, S. R. (1994). Inhibition of thrombin receptor signaling by a G‐protein coupled receptor kinase. Functional specificity among G‐protein coupled receptor kinases. Journal of Biological Chemistry, 269, 1125–1130. 10.1016/S0021-9258(17)42230-7 [DOI] [PubMed] [Google Scholar]
- Jin, W. , Brown, S. , Roche, J. P. , Hsieh, C. , Celver, J. P. , Kovoor, A. , Chavkin, C. , & Mackie, K. (1999). Distinct domains of the CB1 cannabinoid receptor mediate desensitization and internalization. Journal of Neuroscience, 19, 3773–3780. 10.1523/JNEUROSCI.19-10-03773.1999 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Katona, I. , & Freund, T. F. (2012). Multiple functions of endocannabinoid signaling in the brain. Annual Review of Neuroscience, 35, 529–558. 10.1146/annurev-neuro-062111-150420 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kirilly, E. , Hunyady, L. , & Bagdy, G. (2013). Opposing local effects of endocannabinoids on the activity of noradrenergic neurons and release of noradrenaline: relevance for their role in depression and in the actions of CB1 receptor antagonists. Journal of Neural Transmission, 120, 177–186. 10.1007/s00702-012-0900-1 [DOI] [PubMed] [Google Scholar]
- Koch, W. J. , Inglese, J. , Stone, W. C. , & Lefkowitz, R. J. (1993). The binding site for the beta gamma subunits of heterotrimeric G proteins on the beta‐adrenergic receptor kinase. Journal of Biological Chemistry, 268, 8256–8260. 10.1016/S0021-9258(18)53090-8 [DOI] [PubMed] [Google Scholar]
- Latorraca, N. R. , Masureel, M. , Hollingsworth, S. A. , Heydenreich, F. M. , Suomivuori, C.‐M. , Brinton, C. , Townshend, R. J. L. , Bouvier, M. , Kobilka, B. K. , & Dror, R. O. (2020). How GPCR phosphorylation patterns orchestrate arrestin‐mediated signaling. Cell, 183(1813–1825), e1818. 10.1016/j.cell.2020.11.014 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee, M. H. , Appleton, K. M. , Strungs, E. G. , Kwon, J. Y. , Morinelli, T. A. , Peterson, Y. K. , Laporte, S. A. , & Luttrell, L. M. (2016). The conformational signature of beta‐arrestin2 predicts its trafficking and signalling functions. Nature, 531, 665. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leterrier, C. , Bonnard, D. , Carrel, D. , Rossier, J. , & Lenkei, Z. (2004). Constitutive endocytic cycle of the CB1 cannabinoid receptor. Journal of Biological Chemistry, 279, 36013–36021. 10.1074/jbc.M403990200 [DOI] [PubMed] [Google Scholar]
- Liu, H. T. , & Naismith, J. H. (2008). An efficient one‐step site‐directed deletion, insertion, single and multiple‐site plasmid mutagenesis protocol. BMC Biotechnology, 8(1), 91. 10.1186/1472-6750-8-91 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lodowski, D. T. , Barnhill, J. F. , Pyskadlo, R. M. , Ghirlando, R. , Sterne‐Marr, R. , & Tesmer, J. J. (2005). The role of G beta gamma and domain interfaces in the activation of G protein‐coupled receptor kinase 2. Biochemistry, 44, 6958–6970. [DOI] [PubMed] [Google Scholar]
- Luo, J. , Busillo, J. M. , Stumm, R. , & Benovic, J. L. (2017). G protein‐coupled receptor kinase 3 and protein kinase C phosphorylate the distal C‐terminal tail of the chemokine receptor CXCR4 and mediate recruitment of beta‐arrestin. Molecular Pharmacology, 91, 554–566. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marsicano, G. , & Lutz, B. (1999). Expression of the cannabinoid receptor CB1 in distinct neuronal subpopulations in the adult mouse forebrain. European Journal of Neuroscience, 11, 4213–4225. 10.1046/j.1460-9568.1999.00847.x [DOI] [PubMed] [Google Scholar]
- Miess, E. , Gondin, A. B. , Yousuf, A. et al (2018). Multisite phosphorylation is required for sustained interaction with GRKs and arrestins during rapid mu‐opioid receptor desensitization. Science Signalling, 11(539). https://doi/10.1126/scisignal.aas9609 [DOI] [PubMed] [Google Scholar]
- Moller, T. C. , Pedersen, M. F. , van Senten, J. R. , Seiersen, S. D. , Mathiesen, J. M. , Bouvier, M. , & Brauner‐Osborne, H. (2020). Dissecting the roles of GRK2 and GRK3 in mu‐opioid receptor internalization and beta‐arrestin2 recruitment using CRISPR/Cas9‐edited HEK293 cells. Scientific Reports, 10, 17395. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moore, C. A. C. , Milano, S. K. , & Benovic, J. L. (2007). Regulation of receptor trafficking by GRKs and arrestins. Annual Review of Physiology, 69, 451–482. 10.1146/annurev.physiol.69.022405.154712 [DOI] [PubMed] [Google Scholar]
- Morgan, D. J. , Davis, B. J. , Kearn, C. S. , Marcus, D. , Cook, A. J. , Wager‐Miller, J. , Straiker, A. , Myoga, M. H. , Karduck, J. , Leishman, E. , Sim‐Selley, L. J. , Czyzyk, T. A. , Bradshaw, H. B. , Selley, D. E. , & Mackie, K. (2014). Mutation of putative GRK phosphorylation sites in the cannabinoid receptor 1 (CB1R) confers resistance to cannabinoid tolerance and hypersensitivity to cannabinoids in mice. Journal of Neuroscience, 34, 5152–5163. 10.1523/JNEUROSCI.3445-12.2014 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nobles, K. N. , Xiao, K. , Ahn, S. et al (2011). Distinct phosphorylation sites on the beta(2)‐adrenergic receptor establish a barcode that encodes differential functions of beta‐arrestin. Science Signalling, 4, ra51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nogueras‐Ortiz, C. , & Yudowski, G. A. (2016). The multiple waves of cannabinoid 1 receptor signaling. Molecular Pharmacology, 90, 620–626. 10.1124/mol.116.104539 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nogues, L. , Reglero, C. , Rivas, V. , Neves, M. , Penela, P. , & Mayor, F. Jr (2017). G‐protein‐coupled receptor kinase 2 as a potential modulator of the hallmarks of cancer. Molecular Pharmacology, 91, 220–228. 10.1124/mol.116.107185 [DOI] [PubMed] [Google Scholar]
- Nuber, S. , Zabel, U. , Lorenz, K. , Nuber, A. , Milligan, G. , Tobin, A. B. , Lohse, M. J. , & Hoffmann, C. (2016). beta‐Arrestin biosensors reveal a rapid, receptor‐dependent activation/deactivation cycle. Nature, 531, 661–664. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pacher, P. , & Kunos, G. (2013). Modulating the endocannabinoid system in human health and disease successes and failures. FEBS Journal, 280, 1918–1943. 10.1111/febs.12260 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pitcher, J. A. , Inglese, J. , Higgins, J. B. et al (1992). Role of beta gamma subunits of G proteins in targeting the beta‐adrenergic receptor kinase to membrane‐bound receptors. Science, 257, 1264–1267. [DOI] [PubMed] [Google Scholar]
- Singh, S. N. , Bakshi, K. , Mercier, R. W. , Makriyannis, A. , & Pavlopoulos, S. (2011). Binding between a distal C‐terminus fragment of cannabinoid receptor 1 and arrestin‐2. Biochemistry, 50, 2223–2234. 10.1021/bi1018144 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Straiker, A. , Wager‐Miller, J. , & Mackie, K. (2012). The CB1 cannabinoid receptor C‐terminus regulates receptor desensitization in autaptic hippocampal neurones. British Journal of Pharmacology, 165, 2652–2659. 10.1111/j.1476-5381.2011.01743.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thal, D. M. , Yeow, R. Y. , Schoenau, C. , Huber, J. , & Tesmer, J. J. (2011). Molecular mechanism of selectivity among G protein‐coupled receptor kinase 2 inhibitors. Molecular Pharmacology, 80, 294–303. 10.1124/mol.111.071522 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Touhara, K. , Inglese, J. , Pitcher, J. A. , Shaw, G. , & Lefkowitz, R. J. (1994). Binding of G‐protein beta‐gamma‐subunits to pleckstrin homology domains. Journal of Biological Chemistry, 269, 10217–10220. 10.1016/S0021-9258(17)34048-6 [DOI] [PubMed] [Google Scholar]
- Uezu, A. , Horiuchi, A. , Kanda, K. et al (2007). SGIP1 alpha is an endocytic protein that directly interacts with phospholipids and Eps15. Journal of Biological Chemistry, 282(36), 26481–26489. 10.1074/jbc.M703815200 [DOI] [PubMed] [Google Scholar]
- Wager‐Miller, J. , Westenbroek, R. , & Mackie, K. (2002). Dimerization of G protein‐coupled receptors: CB1 cannabinoid receptors as an example. Chemistry and Physics of Lipids, 121, 83–89. 10.1016/S0009-3084(02)00151-2 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary Material
Data Availability Statement
The data that support the findings of this study are available from the corresponding author upon reasonable request.