

Abstract
Pemigatinib is a fibroblast growth factor receptor 1–3 inhibitor used to treat cholangiocarcinoma. A compartmental population pharmacokinetics model was developed using data from 318 patients with cancer enrolled in a phase 1 dose‐escalation/dose‐expansion study, a phase 1 Japanese PK bridging study, and a phase 2 cholangiocarcinoma study. The final model for pemigatinib was a 2‐compartment disposition model with first‐order absorption and linear elimination. All fixed‐ and random‐effect parameters were estimated with good precision, and no apparent biases in the overall model fit were observed. For females, the estimated typical pemigatinib absorption rate constant (ka) and oral clearance (CL/F) were estimated (1.49 L/h and 10.3 L/h, respectively). For males, the typical apparent clearance and ka are 19.0% higher and 56.5% lower, respectively, compared with females. Typical apparent volume of distribution of the central compartment (Vc/F) and peripheral compartment for a 73.3‐kg patient was estimated to be 122.0 L and 80.1 L, respectively; both increased with body weight. Phosphate binder coadministration decreases typical pemigatinib CL/F by 14.1%. Proton pump inhibitor coadministration increases typical pemigatinib apparent Vc/F by 24.4%. Phosphate binders and sex contribute a <20% change to CL/F. The impact of the investigated covariates on pemigatinib pharmacokinetics are not clinically significant.
Keywords: advanced malignancy, FGFR inhibitor, INCB054828, pemigatinib, population pharmacokinetics
Introduction
Dysregulated fibroblast growth factor receptor (FGFR) signaling as a result of genomic alterations in FGFR are key oncogenic drivers in a range of human cancers, including cholangiocarcinoma. 1 , 2 , 3 , 4 As such, selective FGFR inhibitors have been investigated for the treatment of various FGFR‐driven malignancies. 5 , 6 , 7 , 8 , 9 , 10 Pemigatinib (INCB054828) is a selective and potent inhibitor of the FGFR family (FGFR1, FGFR2, and FGFR3) of tyrosine kinase receptors. 4 The pivotal phase 2, open‐label, multicenter FIGHT‐202 study (NCT02924376) evaluated the safety of pemigatinib in previously treated patients with metastatic cholangiocarcinoma harboring FGFR2 fusions or rearrangements. 7 Findings from this study led to pemigatinib being approved for the treatment of adults with previously treated, unresectable, locally advanced, or metastatic cholangiocarcinoma with FGFR2 fusions or rearrangements by the US Food and Drug Administration, European Medicines Agency, and Japan Pharmaceuticals and Medical Devices Agency, making pemigatinib the first targeted therapy approved in this setting. 11 , 12 , 13 , 14 A global phase 3 study assessing the efficacy and safety of pemigatinib compared with gemcitabine plus cisplatin in the first‐line setting in patients with advanced cholangiocarcinoma harboring FGFR2 fusions or rearrangement is ongoing. 9
Pemigatinib is a Biopharmaceutics Classification System Class II compound with pH‐dependent solubility but high permeability. 15 Following multiple‐dose administration in the fasted state in patients with advanced malignancies, pemigatinib exhibited near‐complete and rapid absorption achieving a maximum plasma drug concentration (Cmax) at ≈1 to 2 hours postdose with linear pharmacokinetics (PK) over a dose range of 1 to 20 mg. 16 Pemigatinib exhibits almost complete absorption (1.3% of the administered radioactive dose was recovered as unchanged pemigatinib in feces) (data on file) and low renal clearance (1.19% of total clearance). The metabolism and elimination of pemigatinib have been extensively evaluated in a human absorption, distribution, metabolism, and elimination (ADME) study as well as a battery of in vitro studies (data on file). In the human ADME study, known metabolites accounted for 72% of total radioactivity, of which M2 (O‐desmethyl pemigatinib) and its secondary metabolites (M7, M8, and M9) accounted for 76.9% of the metabolite burden in urine and feces. In vitro metabolism studies indicate that cytochrome P450 (CYP) 3A is solely responsible for M2 formation from pemigatinib. The terminal elimination half‐life of pemigatinib is ≈15 hours and accumulation ratio was ≈1.6 for area under the plasma concentration–time curve from time 0 to 24 hours (AUC0‐24). 16
A CYP3A4‐mediated drug‐drug interaction (DDI) study showed that coadministration of a strong CYP3A4 inhibitor (itraconazole) or a strong CYP3A4 inducer (rifampin) with pemigatinib resulted in a clinically significant change in the exposure of pemigatinib; the pemigatinib AUC increased by ≈90%, and decreased by 85%, when coadministered with itraconazole and rifampin, respectively. 17 Clinical DDI data–validated physiologically based pharmacokinetic (PBPK) modeling predicted a >50% increase and >50% decrease in AUC when coadministered with a strong or moderate CYP3A4 inhibitor or inducer, respectively (data on file). 15 Taken together, these data suggest that the dose of pemigatinib should be reduced when a strong CYP3A4 inhibitor is coadministered, and coadministration of a strong or moderate CYP3A4 inducer should be avoided.
A hepatic impairment study showed that there was evidence of a clinically significant pemigatinib AUC increase (74%) for patients with severe hepatic impairment, and pemigatinib AUC increase (46%) for patients with moderate hepatic impairment is covered by safety margin (therapeutic dose of 13.5 mg once daily and highest safe dose of 20 mg once daily). 18 A renal impairment study showed that there was evidence of a clinically significant pemigatinib AUC increase (59%) for patients with severe renal impairment. 18 The exposure difference between patients with end‐stage renal disease before or after hemodialysis and patients with normal renal function (geometric mean ratio of 76.8% and 91.3%, respectively) was not clinically meaningful. 18
The effect of food on pemigatinib was modest; in a fed state, the median time to Cmax was delayed by 4.02 hours postdose, which was statistically significant compared with the fed state (P = 0.0013). However, this observed effect of high‐fat and high‐calorie diet on pemigatinib steady state was not clinically significant (data on file). The effects of proton pump inhibitors (PPIs) and histamine‐2 (H2)‐receptor antagonists on pemigatinib exposure were modest (AUC decreased by 8% with esomeprazole and increased by 3% with ranitidine) and not clinically meaningful based on results of an acid‐reducing agent–mediated DDI study. 17
The purpose of this analysis was to develop a population PK model for pemigatinib to describe the PK of pemigatinib in patients with all tumor types. A covariate analysis was performed to evaluate pemigatinib PK in a subpopulation.
Methods
Data
Three clinical studies were conducted in accordance with the International Council for Harmonisation Guideline for Good Clinical Practice, including the archiving of essential documents, the principles of the Declaration of Helsinki, and other applicable local ethical and legal requirements. The protocol and all amendments were reviewed and approved by qualified institutional review board/independent ethics committee before enrollment of patients at each site. All patients were provided an institutional review board/independent ethics committee–approved informed consent form before study entry.
Data from 3 clinical studies were used in the population PK of pemigatinib: study 1 was a phase 1/2 dose‐escalation and dose‐expansion study (monotherapy and combination therapy) in patients with advanced malignancies (FIGHT‐101 [NCT02393248]; N = 157); study 2 was a phase 1 monotherapy study in Japanese patients with advanced malignancies (FIGHT‐102 [NCT03235570]; N = 25); study 3 was a phase 2 monotherapy study in patients with advanced/metastatic or surgically unresectable cholangiocarcinoma (FIGHT‐302 [NCT02924376]; N = 136). 7 A complete summary of the study designs, treatments administered, and PK sampling is provided in Table 1. Patients received once‐daily doses of pemigatinib on a 2‐weeks‐on therapy and 1‐week‐off therapy schedule or a continuous administration regimen in the dose‐escalation and dose‐expansion study. In a Japanese PK bridging study, Japanese patients were treated with pemigatinib as intermittent dosing at 9 mg and 13.5 mg. In a phase 2 cholangiocarcinoma study, patients received a once‐daily dose of pemigatinib at 13.5 mg on a 2‐weeks‐on therapy and 1‐week‐off therapy schedule. Sparse sampling approach was used in the phase 2 study, and rich sampling was collected in the 2 phase 1 studies.
Table 1.
Studies Included in the Population PK Analysis
| Study | Phase | Population | Study Site Location | Number of Patients | Dosing | Dosing Duration | PK Sampling Times |
|---|---|---|---|---|---|---|---|
| 1 |
Phase 1/2, open‐label, dose‐escalation study FIGHT‐101 (NCT02393248) |
Advanced malignancies |
United States Denmark |
157 (monotherapy and combination therapy) |
Part 1 (dose escalation): 1 mg, 2 mg, 4 mg, 6 mg, 9 mg, 13.5 mg, 18 mg, 20 mg Part 2 (dose expansion): 9 mg, 13.5 mg |
Once daily oral dose on a 2‐weeks‐on/1‐week‐off therapy or continuous schedule (1 cycle = 21 days) |
|
| 2 |
Phase 1, open‐label, dose‐escalation (3 + 3 design), dose‐expansion study FIGHT‐102 |
Japanese patients with advanced malignancies | Japan | 25 |
Part 1 (dose escalation): 9 mg, 13.5 mg Part 2 (dose expansion): 13.5 mg |
Once daily oral dose on a 2‐weeks‐on/1‐week‐off therapy (1 cycle = 21 days) |
|
| 3 |
Phase 2, open‐label, single‐arm, multicenter study FIGHT‐202 (NCT02924376) 7 |
Patients with advanced/metastatic or surgically unresectable cholangiocarcinoma who have failed at least 1 previous treatment |
United States European Union a United Kingdom Asia b |
136 | 13.5 mg | Once‐daily oral dose on a 2‐weeks‐on/1‐week‐off therapy schedule |
|
C, cycle; D, day; PK, pharmacokinetics
Belgium, France, Germany, Israel, Italy, Spain.
Japan, Republic of Korea, Taiwan, Thailand.
Analytical Methods
Analysis of plasma samples was performed using a validated, liquid chromatography–tandem mass spectrometry method previously described in Ji T, et al. 18 The assay had a linear range of 1 nM (0.488 ng/mL) to 1000 nM (488 ng/mL), with a limit of quantitation of 1 nM (0.488 ng/mL). Pemigatinib concentrations in plasma were expressed in mass concentration units. The validation of the methods was performed regarding accuracy, precision, selectivity, lower limit of quantification, calibration curve, matrix effect, carryover, and stability.
Covariates
Time‐independent predictors of PK variability explored included disease‐related parameters (cancer type, FGFR2 alteration, renal impairment, and hepatic impairment), demographics (sex, race, age, weight, and body mass index), treatment (monotherapy and combination therapy), and clinical laboratory assessments (alanine aminotransferase [ALT], aspartate aminotransferase [AST], alkaline phosphatase, total bilirubin, albumin, and creatinine clearance). Time‐dependent variables explored included concomitant medications (phosphate binders, diuretics, PPIs, H2‐receptor antagonists, and CYP3A4 inhibitors and inducers; Supplementary Table S1). To overcome the limitations of a small sample size, a specific drug interaction analysis was performed only if ≥5% of the patient population were on the concomitant medication involved.
Data Handling
The concentration record was excluded from the analysis when a scheduled PK sample was not collected. The relevant concentration was deleted from the analysis dataset when date or time for a PK sample was missing. Missing baseline clinical laboratory values and vital signs were imputed by the values at screening, or at the visit closest to the baseline visit. The population median value was used to replace all other missing continuous covariates. The number 99 was assigned, and the group was examined as a separate category in the covariate testing for all missing categorical covariates. The covariate was not used in the analysis when a significant number (>15%) of this covariate value was missing. Plasma concentrations <1 nM (0.488 ng/mL; the assay's lower limit of quantitation) were assigned a value of zero and not included in the data analysis.
Population PK Analysis Method
Pemigatinib PK parameters were evaluated by testing 1‐ and 2‐compartment disposition models with first‐order absorption and linear elimination. The magnitude of interindividual variability (IIV) was initially estimated for absorption rate constant (ka), apparent oral clearance (CL/F), apparent volume of distribution for the central compartment (Vc/F), apparent intercompartmental clearance (Q/F), and apparent volume of distribution for the tissue (peripheral) compartment (Vp/F) parameters. An exponential error model was used to characterize IIV. Both proportional error and additive error models were used to evaluate the magnitude of residual variability, using a first‐order conditional estimation method with interaction.
Post hoc Bayesian estimation, defined as maximum posteriori estimate of η, was used in this study to assess population PK model–estimated PK parameters. After a final base model was identified, covariate effects were evaluated for the model parameters ka, CL/F, Vc/F, and Vp/F. Covariates to be formally tested for statistical significance in non‐mixed effects modeling (NONMEM) were initially identified using a generalized additive model (GAM) analysis screening tool. Typical values of structural PK parameters were made a function of the covariate, to allow incorporation of candidate covariates into the PK model as fixed‐effect parameters. Covariate parameters were added in a stepwise univariate fashion during the model forward selection process and subtracted stepwise in the reduction (backward elimination) process for NONMEM regression analysis. The significance of incorporating parameters into or removing parameters from the population model was examined using the likelihood ratio test. Covariates considered significant in the forward selection process contributed at least a 3.84 unit reduction in the objective function (α = 0.05, 1 degree of freedom [df]), and covariates considered significant when removed from the model contributed at least a 7.88 unit increase in the objective function value (α = 0.005, 1 df). The error models for IIV and residual variability in the full multivariable model were evaluated following completion of backward selection. Finally, potential simplifications of covariate equations in the model were evaluated, such as redefining significant discrete group covariates to fewer groups or reducing power functions to linear functions (power term ≈1.0). Bootstrap analysis (a statistical procedure that resamples a single dataset to create many simulated samples) consisting of repeated simulation of concentrations by 500 times was used to calculate 95% confidence intervals (CIs) of population PK model–estimated parameters. The accuracy and robustness of the final population PK model were investigated using a visual predictive check (VPC) method, consisting of repeated simulation of concentrations by 500 times to generate quartile bands of the simulated data.
SAS version 9.4 (SAS Institute, Inc., Cary, North Carolina, USA) was used to prepare data and perform exploratory data analyses. NONMEM® (Version 7.4, Icon Development Solutions, Ellicott City, Maryland, USA) was used to perform PK analyses, and the GFortran Compiler 4.6 NONMEM runs were executed using PDX‐Pop® for NONMEM® (Version 5.2.2, Icon Development Solutions). Diagnostic graphs and GAM were performed using xpose4.6.1 (Uppsala, Sweden) in R 3.5.1. Bootstrapping and VPC were performed using PsN 5.0.0 (Uppsala, Sweden).
Results
Data Description
In total, 2304 plasma pemigatinib concentration records from 157 patients with advanced malignancies enrolled in a phase 1 dose‐escalation and dose‐expansion study, 272 plasma pemigatinib concentration records from 25 Japanese patients with advanced malignancies enrolled in a Japan PK bridging study, and 392 plasma pemigatinib concentration records from 136 patients with cholangiocarcinoma enrolled in a phase 2 study were available for population PK modeling.
The patient demographic characteristics and laboratory values, and co‐comedication information are shown in Tables 2 and 3. The population was ≈45% male, ranged in age from 21 to 79 years (median, 59 years), and although primarily White (68.2%), included 28 Japanese patients (8.8%) from the Japanese PK bridging study (N = 25) and the phase 2 cholangiocarcinoma study (N = 3). The median body weight and body mass index were 73.3 kg (range, 39.8‐156.0 kg) and 25.8 kg/m2 (range, 14.3‐58.2 kg/m2), respectively. The median values of Modified Diet in Renal Disease (MDRD) glomerular filtration rate, AST, and total bilirubin were 85 mL/min/1.73 m2, 29 U/L, and 8.55 µM/L, respectively. In terms of renal function, 146 (45.9%), 134 (42.1%), and 38 (11.9%) patients were classified as normal renal function, mild renal impairment, and moderate renal impairment, respectively, by MDRD glomerular filtration rate criterion, 19 whereas in terms of hepatic function, 213 (67%), 94 (29.4%), and 11 (3.5%) patients were classified as normal hepatic function, and mild and moderate hepatic dysfunction, respectively, based on the National Cancer Institute's hepatic impairment classification. No patient with severe renal and no patient with hepatic impairment was included in the population PK dataset. Seventy‐four patients (23.3%) and 13 patients (4.1%) in the analysis dataset reported taking weak and moderate CYP3A4 inhibitors, respectively, for the duration of the study period at the time of PK sample collection. Twenty‐nine patients (9.1%) reported taking at least 1 weak CYP3A4 inducer at the time of PK sample collection. For acid‐reducing agents, 87 patients (26.9%) and 36 patients (11.1%) in the analysis dataset reported taking PPI and H2‐receptor antagonists, respectively, for the duration of the study period at the time of PK sample collection. Hyperphosphatemia, an expected on‐target pharmacologic effect of FGFR inhibition, was managed with diet modifications, phosphate binders, and diuretics. A total of 117 patients (36.1%) and 24 patients (7.4%) in the analysis dataset reported taking phosphate binders and diuretics, respectively, for the duration of the study period at the time of PK sample collection.
Table 2.
Patient Characteristics, Clinical Lab Tests, and Prognostic Factors at Baseline
| Patient Characteristics (N = 318) | Mean (SD) | Median (Range) |
|---|---|---|
| Age, y | 57.6 (12.4) | 59.0 (21.0‐79.0) |
| Weight, kg | 76.4 (21.1) | 73.3 (39.8‐156.0) |
| Body mass index, kg/m2 | 27.0 (6.6) | 25.8 (14.3‐58.2) |
| MDRD, mL/min/1.73 m2 | 90.2 (29.0) | 85.0 (33.0‐229.0) |
| Total bilirubin, mg/100 mL | 0.626 (0.450) | 0.503 (0‐3.30) |
| Albumin, g/L | 38.2 (5.2) | 39.0 (21.0‐68.0) |
| Alkaline phosphatase, U/L | 195.0 (259.0) | 118.0 (8.91‐3580.0) |
| Alanine aminotransferase, U/L | 28.3 (21.6) | 22.5 (1.8‐142.0) |
| Aspartate aminotransferase, U/L | 38.8 (30.1) | 29.0 (1.71‐206.0) |
| Baseline serum phosphate concentration, mg/dL | 3.5 (0.6) | 3.6 (1.6‐5.1) |
| Baseline serum creatinine concentration, mg/100 mL | 0.833 (0.245) | 0.789 (0.390‐1.79) |
| Sex, n (%) | ||
| Female | 175 (55.0) | |
| Male | 143 (45.0) | |
| Race/ethnicity, n (%) | ||
| Asian | 23 (7.2) | |
| Black or African American | 20 (6.3) | |
| Hispanic | 19 (6.0) | |
| Japanese | 28 (8.8) | |
| Other | 11 (3.5) | |
| White | 217 (68.2) | |
| Dosing regimen, n (%) | ||
| Continuous | 51 (16.0) | |
| Intermittent | 267 (84.0) | |
| Hyperphosphatemia, n (%) | ||
| No | 103 (32.4) | |
| Yes | 215 (67.6) | |
| NCI hepatic impairment classification, n (%) | ||
| Mild | 94 (29.6) | |
| Moderate | 11 (3.5) | |
| Normal | 213 (67.0) | |
| Renal impairment category, n (%) | ||
| Mild | 134 (42.1) | |
| Moderate | 38 (11.9) | |
| Normal | 146 (45.9) | |
| FGFR2 alteration, n (%) | ||
| No | 215 (67.6) | |
| Yes | 103 (32.4) |
MDRD, Modified Diet in Renal Disease; NCI, National Cancer Institute; SD, standard deviation.
Table 3.
Concomitant Medication
| Medication | Type | Pooled (N = 318), n (%) |
|---|---|---|
| CYP3A4 inhibitor | Weak | 74 (23.3) |
| Moderate | 13 (4.1) | |
| CYP3A4 inducer | Weak | 29 (9.1) |
| Moderate | 1 (0.3) | |
| Acid‐reducing agent | Proton pump inhibitors | 87 (26.9) |
| Histamine‐2‐receptor antagonists | 36 (11.1) | |
| Phosphate management agent | Phosphate binder | 117 (36.1) |
| Diuretics | 24 (7.4) |
CYP, cytochrome P450.
Note: Refer to Supplementary Table S1 for the list of concomitant medications evaluated.
Base Structure Model
Log‐transformed concentration data were used for modeling. The base structural PK model was a 2‐compartment disposition model with first‐order absorption and linear elimination. All fixed‐effect parameters were estimated with very good precision (percent relative standard error [%RSE] <10%). The first‐order ka was estimated to be 0.997 h−1. CL/F was estimated to be 10.7 L/h, with estimates for Vc/F and Vp/F of 134 L and 87.0 L, respectively. Q/F was 14.5 L/h. The magnitude of estimated IIV for Vc/F and Vp/F was 44.6% coefficient of variation (CV) and 37.3% CV, respectively. The poorest precision was found for the estimate of IIV in Vp/F (48.9 %RSE). The estimate of residual variability (standard deviation [SD]) was 0.401. A relatively large IIV was observed for ka (133% CV) in the base model.
Covariate Analysis
Forward Selection of Covariates
The covariates identified in the GAM analysis were added to the model during the first phase of forward selection process including the effect of sex, phosphate binders, hyperphosphatemia, CYP3A4 inducers, and albumin on CL/F; the effect of body weight, PPI, sex, and cancer type on Vc/F; the effect of tumor type, treatment, age, body weight, and MDRD on Vp/F; and the effect of tumor type, sex, renal function, PPI, H2‐receptor antagonist, and albumin on ka. During the evaluation of CYP3A4 inducers on CL/F, the patient who took a moderate CYP3A4 inducer was grouped with the patients who took weak CYP3A4 inducers because only 1 patient took a moderate CYP3A4 inducer. The effects of renal impairment and hepatic impairment on clearance were also evaluated, although they were not identified as significant covariates by GAM analysis. After 7 steps of the covariate model–building process, weak CYP3A4 inducers, sex, and phosphate binders on CL/F, body weight, and PPI on Vc/F, body weight on Vp/F, and sex on ka were identified as predictors for PK parameters. Inclusion of these 7 covariates reduced objective function value by −12.1, −9.86, −9.42, −82.2, −15.3, −15.1, and −15.1, respectively, and reduced IIV of CL/F, Vc/F, Vp/F, and ka by 2.5%, 12.8%, 13.5%, and 7.0%, respectively.
Backward Selection of Covariates
Removing any of the covariates identified in the forward selection process did not generate statistically significant change of value of the objective function. Therefore, no covariate was removed in the backward elimination process.
Model Refinement
The omega matrix (the estimated variance‐covariance matrix of interindividual random effects [η]) for CL/F and Vc/F was added in the model after backward elimination process due to the observation of correction between CL/F and Vc/F covariance. The effect of weak CYP3A4 inducers on CL/F was statistically significant. However, the 95% CI of the effects of CYP3A4 inducer on clearance included zero. Therefore, the effect of weak CYP3A4 inducers on CL/F was removed. The value of IIV on the Vp/F estimate was close to zero and the 95% CI IIV estimate included zero. In addition, removing IIV on Vp/F was not statistically significant for the change of the value of the objective function. Thus, IIV on Vp/F was removed to generate the final population PK model.
Final Model
The final PK model was parameterized in terms of ka, CL/F, Vc/F, Q/F, and Vp/F, and all the estimated fixed‐effect and random‐effect parameters were estimated with good precision (%RSE <29%; Table 4). The magnitude of the IIV was moderate for CL/F (43.4 %CV) and Vc/F (35.1 %CV) but high for ka (127 %CV). Residual variability was moderate (0.401 SD). The equations to predict the typical CL/F, Vc/F, Vp/F, and ka of pemigatinib are provided below in Equations I, II, III, and IV.
| (I) |
| (II) |
| (III) |
| (IV) |
Table 4.
Parameter Estimates and Standard Errors From the Pemigatinib Final Population PK Model
| Final Parameter Estimate | Magnitude of Interindividual Variability (%CV) | |||||
|---|---|---|---|---|---|---|
| Parameter | Population Mean | %RSE | 95% CI | Final Estimate | %RSE | 95% CI |
| ka, h−1 | 1.49 | 12.3 | 1.11‐1.91 | 127 | 12.3 | 112, 143 |
| CL/F, L/h | 9.00 | 3.86 | 8.38‐9.77 | 43.4 | 10.0 | 38.9, 47.7 |
| Vc/F, L | 161 | 7.08 | 140‐188 | 35.1 | 25.0 | 24.9, 42.8 |
| Vp/F, L | 80.1 | 7.07 | 66.0‐91.2 | NE | NE | NE |
| Q/F, L/h | 16.0 | 13.0 | 11.1‐20.1 | NE | NE | NE |
| Phosphate binder on CL | 0.141 | 26.4 | 0.0697‐0.223 | NA | NA | NA |
| Sex (male vs female) on CL | 0.190 | 26.4 | 0.0812‐0.300 | NA | NA | NA |
| Body weight (median = 73.3 kg) on Vc/F | 0.738 | 17.9 | 0.512‐1.01 | NA | NA | NA |
| Proton pump inhibitor on Vc/F | –0.244 | 23.0 | –0.344‐0.130 | NA | NA | NA |
| Body weight on Vp/F | 1.22 | 28.2 | 0.338‐1.74 | NA | NA | NA |
| Omega matrix for CL/F and Vc/F | 0.122 | 15.9 | 0.0853‐0.162 | NA | NA | NA |
| RV, SD | 0.401 | 6.83 | 0.374‐0.429 | NA | NA | NA |
| Minimum value of the objective function, −1157.266 | ||||||
CI, confidence interval; CL/F, apparent oral clearance; CV, coefficient of variation; ka, first‐order absorption rate constant; NA, not available; NE, not estimated; PK, pharmacokinetics; Q/F, apparent intercompartmental clearance; RSE, relative standard error; RV, residual variability; SD, standard deviation; Vc/F, apparent central volume of distribution; Vp/F, apparent peripheral volume of distribution.
95%CI of population PK model–estimated parameters were calculated by bootstrap analysis consisting of repeated simulation of concentrations by 500 times.
Where:
CL/Fji is the predicted typical value of apparent clearance (L/h) for the jth subject on ith visit,
Vc/Fji is the predicted typical value of apparent central volume of distribution (L) for the jth subject on ith visit,
Vp/Fj is the predicted typical value of apparent peripheral volume of distribution (L) for the jth subject,
kaj is the predicted typical value of rate of absorption constant for the jth subject,
SEXNj is the flag variable for sex in the jth subject, where 1 is for male and 2 is for female,
BWTj is the body weight (kg) of the jth subject, and
BINDERji is the flag variable for phosphate binders in the jth subject on ith visit, where 1 is for used and zero is for not used.
The goodness‐of‐fit plots for the final PK model (Figure 1) showed that an under‐prediction bias at higher predicted concentrations is not severe at either the population (dependent variable vs population predicted concentration [PRED]) or the individual level (dependent variable vs individual predicted concentration [IPRED]). The horizontal cone‐shaped pattern exhibited in the plot of residuals versus predicted concentrations supports the use of the additive residual variability model. The plot of individual weighted residuals (|IWRES|) vs IPRED shows no trend from low to high individual‐predicted concentrations. The plot of conditional WRES vs PRED exhibits an equivalent scattering of data above and below the zero line, which is homogeneous across the entire range of model‐predicted concentrations. This is indicative of a relatively unbiased fit. A similar trend was observed on conditional WRES vs time since first dose, indicating a relatively unbiased fit independent of time. The shrinkage, defined as the reduction in the effects of sampling variation and calculated as 1‐SD(η)/ω where η are the between individual variation terms and ω is the population model estimate of the SD in η of ηCL, ηVc, ηka, and ε are 7.64%, 21.9%, 25.8%, and 9.04%, respectively, supporting that the diagnostic plots are informative.
Figure 1.
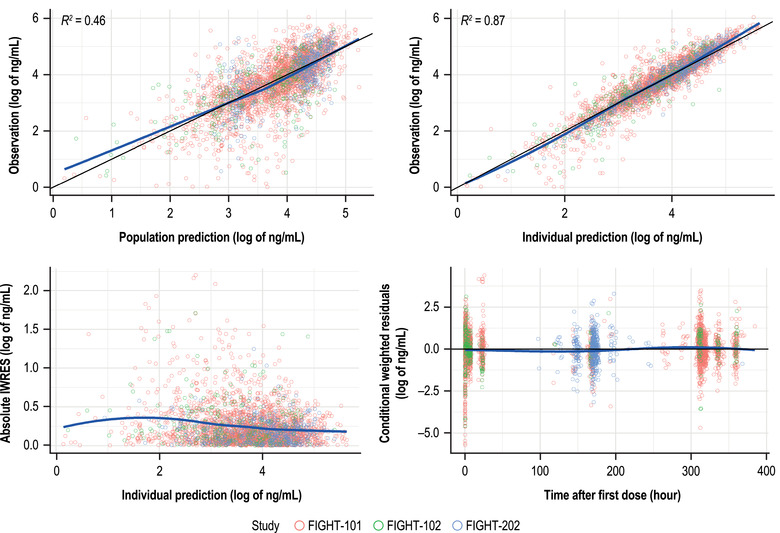
Goodness‐of‐fit plots for the final population PK model. The solid blue line represents the locally weighted scatterplot smoothing; the black line represents the linear regression. IWRES, individual weighted residuals; PK, pharmacokinetics.
Based on the evaluable population, the estimated mean (SD) half‐life of pemigatinib was 16.0 (5.76) hours. The typical pemigatinib ka and CL/F were estimated at 1.49 h−1 and 10.3 L/h, respectively, for female patients. For male patients, the typical CL/F is 19.0% higher and the typical ka value is 56.5% lower compared with female patients. The typical Vc/F and Vp/F for a patient with a body weight of 73.3 kg were estimated to be 122.0 L and 80.1 L, respectively. The Vc/F and Vp/F are increased with increasing body weight. Coadministration with phosphate binders decreases typical pemigatinib CL/F by 14.1%. Coadministration with PPI increases typical pemigatinib Vc/F by 24.4%. The contribution of phosphate binders and sex to the change of CL/F are <30%, and they are not clinically meaningful.
The effects of various covariates on the CL/F of pemigatinib are summarized in Figure 2. The clearances in patients with mild or moderate renal impairment or hepatic impairment are not statistically significantly different from the patients with normal renal or hepatic function. The geometric mean ratio (90%CI) for mild hepatic impairment vs normal hepatic function, moderate hepatic impairment vs normal hepatic function, mild renal impairment vs normal renal function, and moderate renal impairment vs normal renal function is 0.999 (0.918‐1.09), 1.04 (0.842‐1.29), 0.992 (0.914‐1.08), and 0.967 (0.853‐1.10), respectively. A similar CL/F was observed between cholangiocarcinoma and noncholangiocarcinoma cancer patients (geometric mean ratio of CL/F, 0.974) and patients with FGFR2 rearrangement vs patients who are negative for FGFR2 rearrangement (geometric mean ratio of CL/F, 1.07), indicating patients with FGFR2‐rearranged cholangiocarcinoma have no impact on the exposure of pemigatinib. Japanese patients had similar CL/F compared with Western patients (geometric mean ratio of CL/F, 0.998). The CL/F of Hispanic patients was higher than non‐Hispanic White patients (geometric mean ratio of CL/F, 1.19), but the difference was not statistically significant.
Figure 2.
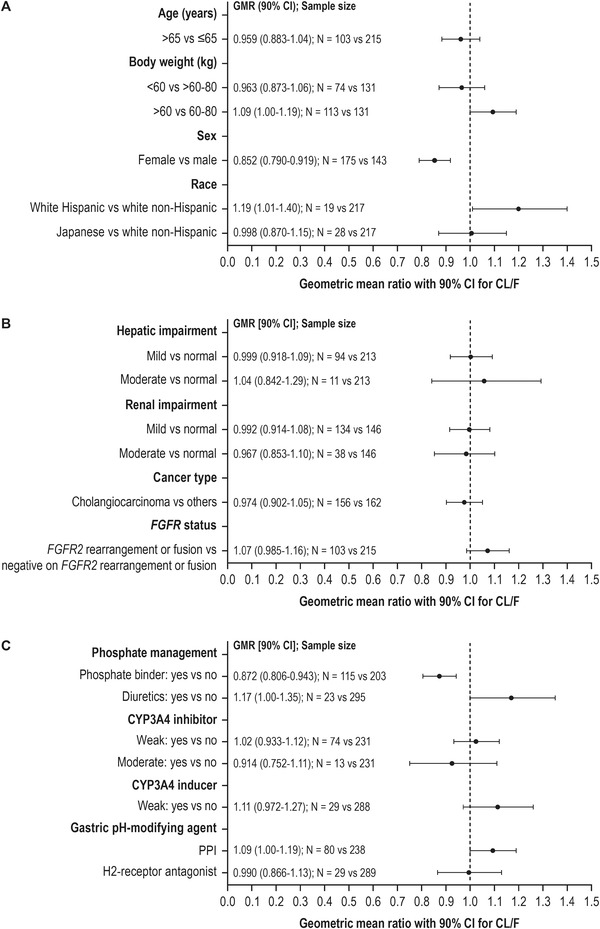
Covariate evaluation of pemigatinib oral clearance using final population PK model for (A) demographic variables, (B) drug‐related variables, and (C) concomitant medications. Data represent GMR ± 90%CI for CL/F. CI, confidence interval; CL/F, apparent oral clearance; CYP, cytochrome P450; FGFR2, fibroblast growth factor receptor 2; GMR, geometric mean ratio; PK, pharmacokinetics.
Model Evaluation
The VPC on the final PK model showed that the majority of the observed data fell within the 90% prediction interval and the median of the prediction interval tracks the middle of the observed data. Figure 3 illustrates the 90% prediction intervals, derived from the simulated datasets, overlaid on the observed pemigatinib concentration vs time since last dose data. The slight deviations from the center of the colored bands represent the plasma samples included in the analysis that were collected outside the window of the prespecified PK collection schedule. The sparse samples within these time intervals contribute to this slight deviation from the center of the color bands. Overall, the VPC indicates there are no apparent biases in the overall model fit for the data.
Figure 3.
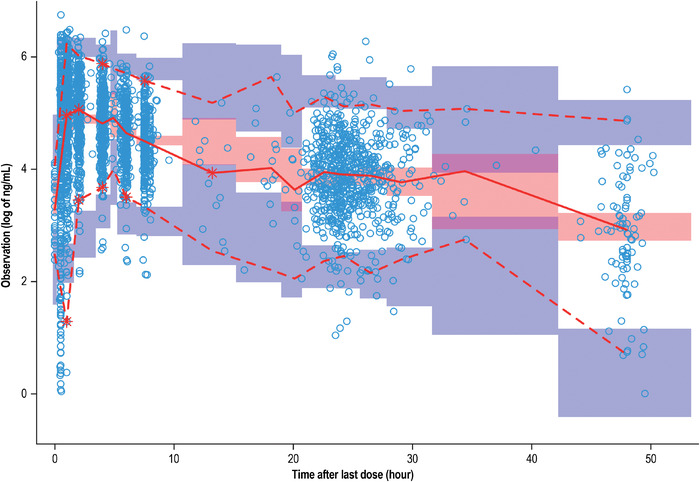
Visual predictive check of the final population PK model. Red solid line: predicted median; red dashed line: predicted 5% and 95%; blue circle: observed; purple band: 95%CI for predicted 5% and 95%; red band: 95% CI for predicted median; CI, confidence interval; PK, pharmacokinetics.
Discussion
This population PK analysis included data from 318 patients with cancer from 3 clinical studies. A 2‐compartment PK model with first‐order absorption and first‐order elimination sufficiently described the data included in this analysis.
A covariate analysis was performed to identify factors predictive of variability in pemigatinib PK. In addition to the sex effect on CL/F and the influence of body weight on Vc/F and Vp/F, concomitant use of phosphate binders and PPI was found to be a statistically significant predictor of CL/F and Vc/F, respectively. Sex was identified as the only statistically significant predictor of ka. None of the other intrinsic and extrinsic factors such as concomitant medications and none of the laboratory indices of kidney and liver function were found to be significant descriptors of IIV in PK parameters.
The typical ka value is 56.5% higher for female patients compared with male patients, and it is difficult to explain this difference. The contribution of sex to the change of oral clearance is <20% and is not clinically meaningful. The Cmax could be largely affected by ka. However, compared with population PK post hoc–estimated Cmax steady state (156 nM [76.05 ng/mL]) for male patients in the analysis dataset, female patients are predicted to have a Cmax steady state (Cmax,ss) of 198 nM (96.53 ng/mL), a 27% increase in Cmax,ss, indicating the impact is not clinically meaningful because the exposure‐response curve between Cmax and efficacy 7 or safety (data on file) was not steep. The typical CL/F value of female patients is 19% lower than male patients, which could be due to the higher body weight for male patients. However, body weight was not identified as a significant predictor for CL/F. The effect of sex on the pemigatinib clearance is not clinically meaningful. Sex was determined as a predictor for CL/F; females had lower clearance than men, indirectly implying the body weight effect on CL/F since females had lighter body weight than men. Both effects of weight and sex were tested in model building and sex was determined as a better predictor for CL/F. The plot of eta (the addition of a random variable drawn from a normal distribution on a parameter) on CL/F vs weight showed even distribution around zero, further suggesting that sex was a better predictor for clearance than weight after inclusion of sex effect.
Hyperphosphatemia is an expected on‐target pharmacologic effect of FGFR inhibition. Nearly 68% of the patients included in this analysis developed hyperphosphatemia after treatment with pemigatinib, which is managed with a low phosphate diet and introduction of phosphate binders. The decrease in pemigatinib CL/F with coadministration of phosphate binders is unknown. The contribution of phosphate binders to the change in pemigatinib CL/F is not clinically meaningful.
Coadministration with a weak CYP3A4 inducer was a significant predictor of CL/F and increased typical pemigatinib CL/F by 24.2% in the full multivariate PK model. However, the 95%CI of the effects of a CYP3A4 inducer on clearance included zero. In addition, coadministration with a weak CYP3A4 inducer resulted in a <30% reduction of CL/F and was not clinically meaningful. Therefore, the weak CYP3A4 inducer was removed from the final model as a covariate for CL/F. There was only 1 patient who received a concomitant moderate CYP3A4 inducer in this analysis. The post hoc–estimated CL/F value for this patient was 22.8 L/h, which is ≈100% higher than that of pemigatinib alone (11.7 L/h). This result is consistent with the PBPK modeling result, which showed a >50% reduction of AUC with coadministration of moderate CYP3A4 inducers. 15 PBPK modeling showed an ≈50% increase in pemigatinib AUC when coadministered with a moderate CYP3A4 inhibitor and no DDI between pemigatinib and a weak CYP3A4 inhibitor. The moderate CYP3A4 inhibitor was not identified as a predictor of pemigatinib clearance, which could be due to the small number of evaluable patients using moderate CYP3A4 inhibitors. The acid‐reducing agent–mediated DDI study showed that that there is only a modest effect on the overall exposure of pemigatinib following coadministration with esomeprazole (a PPI) or ranitidine (an H2‐receptor antagonist). 20 Therefore, it is expected that a PPI and an H2‐receptor antagonist are not predictors of pemigatinib PK.
Body weight was identified as a significant predictor for Vc/F and Vp/F. The Vc/F and Vp/F are increased with increasing body weight. In addition, the concomitant use of PPIs increased Vc/F by 24.4%, which is consistent with the observation of increase in terminal‐phase volume of distribution by coadministration with esomeprazole. 20
Similarities in PK parameters were observed among the 3 studies in this population PK analysis. The geometric mean CL/F of 10.9 L/h in this analysis is comparable to those calculated by the noncompartmental analysis approach (geometric mean CL/F ranged from 9.88 to 11.7 L/h) in the phase 1 dose‐escalation and dose‐expansion study. 16
As the renal clearance of pemigatinib is low (1% of total clearance), 18 a clinically significant effect of mild and moderate renal impairment on the pemigatinib PK was not expected. In the current study, mild and moderate renal impairment were not found to be statistically significant predictors for CL/F. The comparisons of post hoc Bayesian‐estimated CL/Fs between renal dysfunction and normal renal function showed that the geometric mean ratio and 90%CI for mild renal impairment vs normal renal function and moderate renal impairment vs normal renal function is 0.992 (0.914‐1.08) and 0.967 (0.853‐1.10), respectively. Therefore, no dose adjustment is recommended for the patients with mild and moderate renal impairment. In vitro and in vivo data showed that liver metabolism is the predominant clearance pathway of pemigatinib in humans. In the current study, mild and moderate hepatic impairment were not found to be statistically significant predictors for CL/F. The comparisons of post hoc Bayesian‐estimated CL/Fs between hepatic dysfunction and normal function showed that the geometric mean ratio and 90%CI for mild hepatic impairment vs normal hepatic function and moderate hepatic impairment vs normal hepatic function is 0.999 (0.918‐1.09) and 1.04 (0.842‐1.29), respectively. Therefore, no dose adjustment is recommended for the patients with mild and moderate hepatic impairment.
Currently, pemigatinib is approved for treatment of advanced/metastatic or surgically unresectable cholangiocarcinoma with a FGFR2 fusion or other rearrangement in patients who have progressed after at least 1 prior line of systemic therapy. It is critical to evaluate the exposure difference for cholangiocarcinoma versus noncholangiocarcinoma cancer patients as well as patients with FGFR2 rearrangement or fusion vs patients who are negative for FGFR2 rearrangement or fusion. The cancer type was not identified as a predictor for PK parameters. Comparisons of post hoc Bayesian‐estimated CL/Fs showed that a similar CL/F was observed between cholangiocarcinoma and noncholangiocarcinoma cancer patients (geometric mean ratio of CL/F, 0.974) and patients with FGFR2 rearrangement or fusion vs patients who are negative for FGFR2 rearrangement or fusion (geometric mean ratio of CL/F, 1.07), indicating patients with cholangiocarcinoma and patients with FGFR2 rearrangement or fusion have no impact on the exposure of pemigatinib.
Race was not a predictor for CL/F. In particular, comparisons of post hoc Bayesian‐estimated CL/Fs showed that Japanese patients had similar CL/F compared with Western patients (geometric mean ratio of CL/F, 1.0). Though a slightly higher CL/F in Hispanic patients was estimated compared with non‐Hispanic White patients (geometric mean ratio of CL/F, 1.2), there were only 19 Hispanic patients and the difference was not statistically significant.
Overall, the impact of age, body weight, race/ethnicity (Hispanic, Japanese), creatinine clearance, clinical laboratory values (albumin, total bilirubin, AST, ALT, and alkaline phosphatase), concomitant medication (weak/moderate CYP3A4 inhibitors, diuretics, PPI, and H2‐receptor antagonist), renal and hepatic impairment (mild, moderate), tumor type (cholangiocarcinoma), and FGFR2 rearrangement or fusion on pemigatinib clearance were not statistically significant.
Conclusions
The population PK model adequately described the PK of pemigatinib in patients with cancer. The covariate analysis results suggest that the impact of sex and the use of phosphate binder as co‐medication on PK are not clinically meaningful and no dose adjustment is recommended for patients with mild or moderate renal impairment and patients with hepatic impairment. These results support the label language used for pemigatinib.
Conflicts of Interest
All authors are employees and shareholders of Incyte Corporation. The study was funded by Incyte Corporation, Wilmington, Delaware.
Editorial assistance was provided by Envision Pharma Group, Inc. (Philadelphia, Pennsylvania), and funded by Incyte Corporation.
Data‐Sharing Statement
The datasets generated and/or analyzed during the current study are available upon reasonable request (email: yeleswaram@incyte.com).
Supporting information
Supporting information, Additional supplemental information can be found by clicking the Supplements link in the PDF toolbar or the Supplemental Information section at the end of the web‐based version of this article.
Ji Tao, Chen Xuejun, Liu Xiang, Yeleswaram Swamy. Population Pharmacokinetics Analysis of Pemigatinib in Patients With Advanced Malignancies. Clinical Pharmacology in Drug Development. 2022;00:1‐12. 10.1002/cpdd.1038
References
- 1. Greulich H, Pollock PM. Targeting mutant fibroblast growth factor receptors in cancer. Trends Mol Med. 2011;17(5):283‐292. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Gallo LH, Nelson KN, Meyer AN, Donoghue DJ. Functions of fibroblast growth factor receptors in cancer defined by novel translocations and mutations. Cytokine Growth Factor Rev. 2015;26(4):425‐449. [DOI] [PubMed] [Google Scholar]
- 3. Arai Y, Totoki Y, Hosoda F, et al. Fibroblast growth factor receptor 2 tyrosine kinase fusions define a unique molecular subtype of cholangiocarcinoma. Hepatology. 2014;59(4):1427‐1434. [DOI] [PubMed] [Google Scholar]
- 4. Liu PCC, Koblish H, Wu L, et al. INCB054828 (pemigatinib), a potent and selective inhibitor of fibroblast growth factor receptors 1, 2, and 3, displays activity against genetically defined tumor models. PLoS One. 2020;15(4):e0231877. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Javle M, Lowery M, Shroff RT, et al. Phase II study of BGJ398 in patients with FGFR‐altered advanced cholangiocarcinoma. J Clin Oncol. 2018;36(3):276‐282. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Mazzaferro V, El‐Rayes BF, M Droz Dit Busset, et al. Derazantinib (ARQ 087) in advanced or inoperable FGFR2 gene fusion‐positive intrahepatic cholangiocarcinoma. Br J Cancer. 2019;120(2):165‐171. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Abou‐Alfa GK, Sahai V, Hollebecque A, et al. Pemigatinib for previously treated, locally advanced or metastatic cholangiocarcinoma: a multicentre, open‐label, phase 2 study. Lancet Oncol. 2020;21(5):671‐684. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Loriot Y, Necchi A, Park SH, et al. Erdafitinib in locally advanced or metastatic urothelial carcinoma. N Engl J Med. 2019;381(4):338‐348. [DOI] [PubMed] [Google Scholar]
- 9. Bekaii‐Saab TS, Valle JW, Van Cutsem E, et al. FIGHT‐302: first‐line pemigatinib vs gemcitabine plus cisplatin for advanced cholangiocarcinoma with FGFR2 rearrangements. Future Oncol. 2020;16(30):2385‐2399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Merz V, Zecchetto C, Simionato F, et al. A phase II trial of the FGFR inhibitor pemigatinib in patients with metastatic esophageal‐gastric junction/gastric cancer trastuzumab resistant: the FiGhTeR trial. Ther Adv Med Oncol. 2020;12:1758835920937889. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Hoy SM. Pemigatinib: first approval. Drugs. 2020;80(9):923‐929. [DOI] [PubMed] [Google Scholar]
- 12. Merz V, Zecchetto C, Melisi D. Pemigatinib, a potent inhibitor of FGFRs for the treatment of cholangiocarcinoma. Future Oncol. 2021;17(4):389‐402. [DOI] [PubMed] [Google Scholar]
- 13. Rizzo A, Ricci AD, Brandi G. Pemigatinib: hot topics behind the first approval of a targeted therapy in cholangiocarcinoma. Cancer Treat Res Commun. 2021;27:100337. [DOI] [PubMed] [Google Scholar]
- 14. Roskoski R Jr. Properties of FDA‐approved small molecule protein kinase inhibitors: a 2021 update. Pharmacol Res. 2021;165:105463. [DOI] [PubMed] [Google Scholar]
- 15. Ji T, Zhang Y, Overholt H, Chen X, Yeleswaram S. Evaluation of CYP3A4‐mediated and transporter‐mediated drug‐drug interaction potential for pemigatinib using physiologically‐based pharmacokinetic modeling [abstract]. Clin Pharmacol Ther. 2020;107(Suppl S1):PII‐123. [Google Scholar]
- 16. Ji T, Lihou C, Asatiani E, et al. Pharmacokinetics and pharmacodynamics of pemigatinib, a potent and selective inhibitor of FGFR 1, 2, and 3, in patients with advanced malignancies. Mol Cancer Ther. 2019;18(12 Suppl):C071. [Google Scholar]
- 17. Ji T, Rockich K, Epstein N, et al. Evaluation of drug‐drug interactions of pemigatinib in healthy participants. Eur J Clin Pharmacol. 2021;77:1887‐1897. [DOI] [PubMed] [Google Scholar]
- 18. Ji T, Rockich K, Epstein N, et al. Evaluation of the pharmacokinetics of pemigatinib in patients with impaired hepatic or renal function [published online ahead of print June 24, 2021]. Br J Clin Pharmacol. 10.1111/bcp.14954. [DOI] [PubMed] [Google Scholar]
- 19. Levey AS, Stevens LA, Schmid CH, et al. A new equation to estimate glomerular filtration rate. Ann Intern Med. 2009;150(9):604‐612. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Ji T, Rockich K, Epstein N, Chen X, Punwani N, Yeleswaram S. Evaluation of drug‐drug interactions of INCB054828 in healthy volunteers. Clin Pharmacol Ther. 2019;105(Suppl S83):PIII‐008. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supporting information, Additional supplemental information can be found by clicking the Supplements link in the PDF toolbar or the Supplemental Information section at the end of the web‐based version of this article.