

Summary
The origin of sweetpotato, a hexaploid species, is poorly understood, partly because the identity of its tetraploid progenitor remains unknown. In this study, we identify, describe and characterize a new species of Ipomoea that is sweetpotato’s closest tetraploid relative known to date and probably a direct descendant of its tetraploid progenitor.
We integrate morphological, phylogenetic, and genomic analyses of herbarium and germplasm accessions of the hexaploid sweetpotato, its closest known diploid relative Ipomoea trifida, and various tetraploid plants closely related to them from across the American continent.
We identify wild autotetraploid plants from Ecuador that are morphologically distinct from Ipomoea batatas and I. trifida, but monophyletic and sister to I. batatas in phylogenetic analysis of nuclear data.
We describe this new species as Ipomoea aequatoriensis T. Wells & P. Muñoz sp. nov., distinguish it from hybrid tetraploid material collected in Mexico; and show that it likely played a direct role in the origin of sweetpotato’s hexaploid genome. This discovery transforms our understanding of sweetpotato’s origin.
Keywords: crop wild relatives, Ecuador, genomics, herbarium specimens, Ipomoea aequatoriensis, new species, tetraploid
Short abstract
See also the Commentary on this article by Särkinen et al., 234: 1107–1108.
Introduction
Sweetpotato, Ipomoea batatas (L.) Lam., is a hexaploid species thought to have originated via allopolyploidy from a diploid and a tetraploid ancestor (Yang et al., 2017). Ipomoea trifida (Kunth) G. Don, a Circum‐Caribbean species, was recently confirmed as sweetpotato’s closest diploid relative and most likely the direct descendant of its diploid progenitor (Muñoz‐Rodríguez et al., 2018). In contrast, the identity of the sweetpotato’s closest tetraploid relative remains unknown. Identifying this entity is key to untangling the evolutionary history of sweetpotato, understanding its contemporary diversity and assembling its large allohexaploid genome.
Whilst preparing a monograph of all American species of Ipomoea L. (Wood et al., 2020), our attention was drawn to herbarium specimens from Ecuador identified as I. batatas but differing in their shorter and blunter sepals (Fig. 1a,b), sepal morphology being an important taxonomic character in Ipomoea (Austin, 1978; Wood et al., 2020). These specimens were restricted to coastal Ecuador (Fig. 1c) and were of wild provenance, in contrast to most populations of I. batatas that are only known from cultivation or as escapes.
Fig. 1.
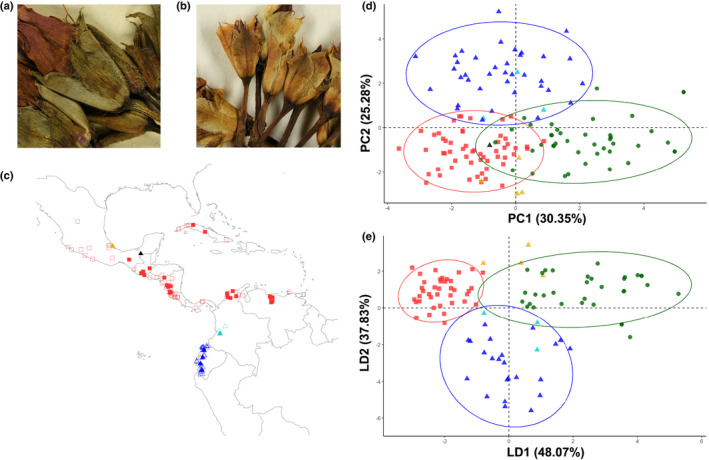
Ipomoea aequatoriensis is morphologically distinct from Ipomoea batatas and Ipomoea trifida. Sepals are (a) oblong/ovate in cultivated I. batatas (Balls 5483) and (b) obovate in I. aequatoriensis (Jativa and Epling. 1191). (c) Map of the Americas showing the distribution of specimens included in the morphological analysis. Closed symbols indicate specimens also included in the genomic analyses. All hexaploid I. batatas specimens in this study are of cultivated origin and are not included in the map. (d) Principal component analysis and (e) linear discriminant analysis of 12 quantitative morphological traits widely used in sweetpotato morphological studies. Ellipses indicate 95% confidence level. In (c–e), I. batatas (green dots), I. aequatoriensis (blue triangles), I. trifida (red squares), hybrids Ipomoea tabascana (black triangle) and I. batatas var. apiculata (orange triangles). The Colombian specimens affinis to I. aequatoriensis are indicated by light blue triangles.
As part of our research, in parallel to studying herbarium specimens, we also grew tetraploid Ipomoea specimens from seeds available in germplasm collections (Supporting Information Table S1). Tetraploid collections (2n = 4x = 60) are of particular interest because their ploidy is intermediate between hexaploid sweetpotato (2n = 6x = 90) and its closest diploid relative, I. trifida (2n = 2x = 30), meaning that they may represent intermediate stages in sweetpotato evolution. The germplasm material studied by us included other tetraploid specimens from the same areas of Ecuador as the distinctive herbarium material we had identified during our studies (Figs S1–S5), as well as material of the Mexican sweetpotato variety I. batatas var. apiculata J.A. McDonald & D.F. Austin and the Mexican hybrid species Ipomoea tabascana J.A. McDonald & D.F. Austin, both of them also tetraploids (Notes S1). Ipomoea tabascana is a modern hybrid between I. batatas and I. trifida known from a single collection (McDonald & Austin, 1990; Srisuwan et al., 2006). Modern tetraploid hybrids such as this may confound data interpretation, hence the importance of including in our study examples of known hybrid origin: it is essential to be able to distinguish between truly autotetraploid entities and other tetraploids of modern hybrid origin.
To place the Ecuadorian specimens in a phylogenetic context, we conducted a preliminary phylogenetic analysis using rpl32‐trnL, a small, noncoding, rapidly‐evolving chloroplast DNA region. Ipomoea batatas contains two different chloroplast lineages (Roullier et al., 2013), the ancestral lineage (chloroplast lineage 1) and a second, more recent lineage that is most likely the result of introgression with chloroplast capture from I. trifida (lineage 2) (Muñoz‐Rodríguez et al., 2018). The preliminary analysis of this small chloroplast DNA region showed that the herbarium specimens and the germplasm material from Ecuador were the same entity, and that they were more closely related to sweetpotato chloroplast lineage 1 than to any other lineage (Methods S1; Fig. S6). A subsequent literature review showed that we were not the first to recognize these tetraploids from Ecuador (Martin & Jones, 1972; Martin et al., 1974; Austin et al., 1993), but previous studies lacked the taxonomic and phylogenetic framework required to accurately infer their relationship with sweetpotato.
Here, we provide the first comprehensive study of these Ecuadorian tetraploids and show that they represent a distinct species that is sweetpotato’s closest wild relative. We describe this new species as Ipomoea aequatoriensis T. Wells & P. Muñoz and show it is most likely the direct descendant of the sweetpotato’s tetraploid progenitor.
Materials and Methods
Herbarium collections and germplasm material
We studied American material from germplasm collections (CIP and USDA) and herbaria (AAU, BM, E, FL, FTG, GUAY, HUEFS, K, LPB, OXF, QAC, QAP, QCA, QCNE, RB, ST, US, USZ, XAL, acronyms according to Thiers (2018)). We included specimens of cultivated hexaploid I. batatas (L.) Lam. and diploid I. trifida (Kunth) G. Don from across their geographical distribution; representatives of the 14 other close wild relatives of sweetpotato (Wood et al., 2020) including the Mexican hybrid tetraploid I. tabascana J.A. McDonald & D.F. Austin, known from a single collection (Notes S1) (McDonald & Austin, 1990; Austin et al., 1991); two specimens of the tetraploid sweetpotato variety I. batatas var. apiculata J.A. McDonald & D.F. Austin, also from Mexico and apparently restricted to the vicinity of the city of Veracruz (Notes S1); the tetraploid material from Ecuador; a tetraploid accession from Colombia; and multiple herbarium collections of wild plants resembling the tetraploid Ecuadorian material and collected in the same geographical area (Dodson & Gentry, 1978; Austin, 1982; Dodson et al., 1985; McDonald & Austin, 1990; Wood et al., 2020). See Tables S2 and S3 for passport data of all specimens and indication of analyses they were included in.
Quantitative morphological analyses
Character selection and measurement
We identified and analysed herbarium specimens and germplasm material of I. trifida (57 specimens), I. batatas (55 specimens), the Ecuadorian tetraploids (44 specimens), I. tabascana (one specimen) and I. batatas var. apiculata (five specimens) (Table S2). We measured 12 morphological characters found to be informative in taxonomic treatments of Ipomoea or commonly used to study sweetpotato germplasm collections (Table S2) (Austin, 1978; Huaman, 1991; Wood et al., 2020). Measurements were taken using digital callipers or, in the case of digitized herbarium specimens, using the biological‐image analysis software Fiji (Schindelin et al., 2012).
Clustering analyses
We first ran a principal component analysis (PCA) to investigate phenotypic clustering between I. batatas, I. trifida and the various tetraploid entities. We used factominer package v.2.4 (Lê et al., 2008) in R and divided the tetraploid material into three groups based on geographical distribution and past determinations: (1) Ecuadorian, (2) I. tabascana, and (3) I. batatas var. apiculata. We then used R package mass v.7.3.54 (Venables et al., 2002) to assess how well individual specimens could be classified into their assigned groups through a linear discriminant analysis (LDA). We plotted the results of both analyses using the ggplot2 package v.3.3.5 (Wickham, 2016), with ellipses depicting 95% confidence level added using the stat_ellipse function.
Analysis of genomic data
We sequenced 13 new specimens using Illumina whole genome sequencing and incorporated them in our previously‐existing dataset of sweetpotato crop wild relatives (CWRs) (Muñoz‐Rodríguez et al., 2018) (Table S2). This material included six Ecuadorian tetraploids (PI 561246, PI 561248, PI 561255, PI 561258, K300/CH71.3, CH81.2), one Colombian tetraploid (K500/CH80.3), diploid I. trifida specimens from Colombia (F. de la Puente 1054) and Mexico (F. de la Puente 2961), three I. batatas var. apiculata (D.F. Austin 7480, PI 518474 and K233) and one I. tabascana (PI 518479).
DNA processing and sequencing
We extracted DNA using the Plant Tissue Mini protocol for Qiagen DNEasy Plant Mini Kit. We created genomic libraries using the NEBNext Ultra DNA Library Prep Kit for Illumina v.3.0 (New England BioLabs, Ipswich, MA, USA). Sequencing was done at Novogene facilities in Cambridge, UK, using Illumina NovoSeq6000. We obtained 150 bp paired end whole genome data, on average 11 Gb per sample. We filtered the sequence files using default parameters in trimmomatic (Bolger et al., 2014) and checked the quality of the reads using FastQC. We used default settings in BBtools’ tadpole (https://sourceforge.net/projects/bbmap/) to correct the reads.
Assembly of single copy nuclear regions for phylogenetic analysis
We assembled 386 putative single copy nuclear DNA regions of all samples using a reference‐guided assembly. A detailed description of how these nuclear regions were identified is provided in Methods S2. We mapped the reads to the reference 386 nuclear probes using BBMap (paired only = t, local = t). We used SAMtools (Danecek et al., 2021) to extract all reads mapped to the reference probes and to remove duplicate reads, and Picard Tools (http://broadinstitute.github.io/picard) to realign the reads mapped around indels. We used BCFtools (Danecek et al., 2021) for variant calling, indel normalization and variant filtering, and VCFtools’ vcf‐sort (Danecek et al., 2011) to sort the VCF files.
Phylogenetic analysis of nuclear DNA regions
We used phylogenetic analysis of nuclear data to confirm the close relationship between the Ecuadorian tetraploids and sweetpotato. We used consensus sequences and included the tetraploids from Ecuador and Colombia, 10 I. batatas specimens, 10 I. trifida specimens, one I. tabascana specimen, two I. batatas var. apiculata specimens, one specimen of each of the other 14 species closely related to sweetpotato and one I. cryptica J.R.I. Wood & Scotland as outgroup (Muñoz‐Rodríguez et al., 2018, 2019). We obtained consensus sequences from VCF variant files using BCFtools consensus (Danecek et al., 2021) and masked all positions in the consensus sequences with read coverage lower than 5×. The use of consensus sequences in phylogenetic analysis can obscure potential subgenome differentiation in polyploids. However, the lack of a reference genome makes it impossible to assign the alleles in the nuclear regions to specific subgenomes. To minimize the potential effects of divergent subgenomes, we only included likely homozygous variant positions in this analysis. Heterozygous sites were therefore masked and not considered in the main phylogenetic analysis but were included in additional phylogenetic analyses (Methods S3).
We used the biopython script sequence_cleaner to remove sequences shorter than 500 bp or with more than 10% ambiguous sites. We excluded three further regions of the analysis (solyc06g073230.2.1_1, solyc08g043170.2.1_1 and solyc11g012820.1.1_1) as none of the sequences in those regions passed the filters, as well as one I. batatas var. apiculata herbarium specimen (D.F. Austin 7480) with almost 80% missing data.
We aligned each of the regions independently using Mafft v.7.310 (Katoh & Standley, 2013) and removed poorly aligned regions using Gblocks (half gaps) (Castresana, 2000; Talavera & Castresana, 2007). We generated summary files of all edited alignments using Amas (Borowiec, 2016) (Table S4). A further 12 alignments that had no variable sites were excluded, so this analysis was done using 371 putative single‐copy nuclear DNA regions. We also used Amas to concatenate the alignments.
We inferred three different phylogenies: (1) partitioned maximum likelihood (ML) analysis of concatenated alignments with automated model selection + merge in Iq‐Tree v.1.6.12 (Nguyen et al., 2015; Kalyaanamoorthy et al., 2017); (2) Approximate ML analysis of unpartitioned concatenated alignments in multi‐threaded double‐precision FastTree v.2.1.1054,71 (GTR + gamma model); and (3) independent gene tree inference using Iq‐Tree v.1.6.12 with automated model selection followed by species tree inference using the coalescent in Astral III (Zhang et al., 2018). We used the GNU parallel tool (Tange, 2011) to parallelize and speed up several steps in the pipeline.
Principal component analysis
We conducted a PCA of I. batatas, I. trifida, the Ecuadorian tetraploids, the Colombian tetraploid specimen K500/CH81.3 and the hybrids I. tabascana and I. batatas var. apiculata. We used a subset of 20 I. batatas and 20 I. trifida samples to try to minimize bias due to uneven population sizes compared to the other entities (Privé et al., 2020). We also conducted additional analyses including all I. batatas and I. trifida samples instead of a subset (Methods S4).
We mapped the nuclear reads to a sweetpotato sample (accession CIP 400435) and called variants using the same procedure as earlier. We filtered out all variants with coverage lower than 5× and ran a linkage disequilibrium pruning step using Plink (‐‐indep‐pairwise 50 10 0.1). We then used Plink (‐‐double‐id ‐‐allow‐extra‐chr ‐‐set‐missing‐var‐ids @:# ‐‐make‐bed ‐‐pca ‐‐geno 0.20 ‐‐snps‐only ‐‐max‐alleles 2) (Chang et al., 2015; Purcell, 2021) for the PCA and plotted the results using tidyverse (Wickham et al., 2019) and ggplot2 (Wickham, 2016) in Rstudio (RStudio Team, 2021). This analysis used 419 single nucleotide polymorphisms (SNPs) from across the 386 Ipomoea nuclear regions, both homozygous and heterozygous.
K‐mer analyses
We used GenomeScope2.0 (Ranallo‐Benavidez et al., 2020) to assess heterozygosity from k‐mer frequencies of raw, unaligned sequencing reads, in a representative Ecuadorian sample (PI 561248) sequenced at high‐coverage. Relative frequency patterns can then be used to infer whether a tetraploid sample is autopolyploid or allopolyploid. We carried out initial k‐mer counting and histogram construction on the filtered but unaligned sequencing reads using Jellyfish (Marçais & Kingsford, 2011). We ran both Jellyfish and GenomeScope2.0 with a maximum coverage of 100 000 and the default k‐mer value of 21. We also ran the same analysis in three Mexican hybrid tetraploids sequenced at lower coverage (Methods S5).
Assembly of whole chloroplast genomes
We used GetOrganelle (‐F embplant_pt; SPAdes options: "‐‐threads 20 ‐‐only‐assembler ‐k 21,33,55,77,93") (Jin et al., 2018) to de novo assemble the chloroplast genomes of the new samples. When GetOrganelle failed to produce a circular genome assembly in the first attempt, we ran a second attempt using ‐‐reduce‐reads‐for‐coverage INF and ‐‐max‐reads INF options. GetOrganelle successfully assembled all samples except one I. trifida sample (F. de la Puente 2961). To assemble the genome of this one sample, we used a reference‐guided assembly using I. trifida (F. de la Puente 1054) as reference.
Phylogenetic network using chloroplast genomes
This analysis includes all I. batatas, I. trifida and I. tabascana specimens from our previous study, together with the 15 newly sequenced samples. We aligned the whole chloroplast genome sequences using Mafft v.7.310 (FFT‐NS‐2) and removed poorly aligned regions using Gblocks (no gaps). We used PopArt (http://popart.otago.ac.nz) to infer a Median Joining Network (reticulation tolerance 0.50 (Bandelt et al., 1999)) with 602 segregating sites, 182 of them parsimony‐informative.
Results
Morphological differentiation
The tetraploid Ecuadorian and Colombian material form a cluster distinct from I. batatas and I. trifida in PCA and LDA of 12 morphological characters (Fig. 1e,f). The PCA shows three clusters corresponding to I. batatas, I. trifida and the Ecuadorian/Colombian material, with some overlap at the margins, predominantly between I. trifida and I. batatas (Fig. 1e). Hybrid specimens from Mexico, i.e. I. tabascana (PI 518479) and I. batatas var. apiculata (PI 518474), fall close to or within the clusters of I. trifida and I. batatas. The three distinct clusters were more pronounced in the LDA trained on 80% of the data (Fig. 1e), which yielded a 90% success rate in accurately identifying the test data and recovered the Mexican hybrids within I. trifida.
Genomic differentiation
The phylogenetic analysis of nuclear regions recovers the six tetraploid specimens from Ecuador and one from Colombia in a clade sister to hexaploid I. batatas (Fig. 2a). This relationship is recovered in all methods of phylogenetic inference with strong support (Figs S7, S8). In addition, the tetraploid Ecuadorian and Colombian specimens also form a distinct group from I. batatas and I. trifida in the different PCA using nuclear SNPs (Figs 2b, S9). The analysis using a subset of I. batatas and I. trifida samples, shown in Fig. 2(b), aimed at preventing bias due to uneven population sizes (Privé et al., 2020). In this analysis, the Ecuadorian and Colombian tetraploids partially overlap with I. trifida in principal component one (PC1) but clearly separate from all entities, including I. trifida, in principal component two (PC2). The single specimen of the Mexican hybrid I. tabascana and the three I. batatas var. apiculata specimens are intermediate between I. trifida and I. batatas in PC1 but cluster with both species in PC2 (Fig. 2b).
Fig. 2.

Molecular analyses identify Ipomoea aequatoriensis as a distinct entity, phylogenetically distinct and isolated in the genetic space. (a) Approximate maximum likelihood analysis of 371 single‐copy nuclear DNA regions. Numbers on the branches indicate Shimodaira–Hasegawa‐like support values; black dots indicate branches with 100% support. (b) Principal component analysis of Ipomoea batatas (green), Ipomoea trifida (red), I. aequatoriensis (blue) and the hybrids Ipomoea tabascana and I. batatas var. apiculata (black and orange respectively). Principal component analysis inferred using 419 single nucleotide polymorphisms (SNPs) from across the 386 nuclear probes. Ellipses indicate multivariate t‐distribution. The Colombian specimen K500/CH81.3 discussed throughout the text is indicated in light blue.
The analysis of nucleotide heterozygosity patterns suggests that the Ecuadorian tetraploids have a genomic structure consistent with an autopolyploid origin, with proportions of aaab consistently higher than aabb (Table S5; Notes S1). This pattern is indicative of two identical or highly similar subgenomes originating from a whole genome duplication (Ranallo‐Benavidez et al., 2020). The same analysis for the hybrid I. tabascana and two specimens of I. batatas var. apiculata, albeit using lower coverage data (Methods S2), shows instead a higher proportion of aabb than aaab (Table S5; Notes S1), which suggests that two distinct subgenomes have been derived from a recent hybridization event.
Analysis of whole chloroplast genomes
The Median Joining phylogenetic network inferred using 602 segregating sites from the alignment of whole chloroplast genomes shows the Ecuadorian plants are associated with the ancestral sweetpotato lineage 1, whereas the single Colombian specimen we sequenced (K500/80.3) is associated with the sweetpotato lineage 2. The hybrid I. tabascana and I. batatas var. apiculata are also associated with sweetpotato lineage 2.
Discussion
Ipomoea aequatoriensis is a distinct species
We have identified a group of plants from Ecuador that are distinct from cultivated sweetpotato and from all sweetpotato CWRs known to date. These tetraploid plants are of wild provenance, morphologically and geographically coherent, most likely autotetraploid, isolated in the genetic space, and form a monophyletic group most closely related to sweetpotato in phylogenetic analysis of nuclear data. Their distinctiveness justifies recognition as a new species I. aequatoriensis T. Wells & P. Muñoz. A formal diagnosis is presented here. Specimen citation, full description and ecological notes are provided in the Notes S2. Specimens from Colombia, although possibly also part of this species, require further study and are not formally included in I. aequatoriensis (see Notes S2).
Ipomoea aequatoriensis T. Wells & P. Muñoz, sp. nov. (Illustration in Fig. S10)
TYPE: ECUADOR. Esmeraldas Province, Quinindé. Austin, D.F. 7803 (holotype FTG, Isotype CIP).
Diagnosis
This species is most closely related to I. batatas (L.) Lam. (Figs 2a, 3) which it resembles in corolla size, dense sub‐umbellate inflorescence and pubescent ovary, but differs in possessing sepals that are consistently shorter (outer: < 7 vs > 7 mm; inner: < 10 mm vs > 12 mm) and stems that are thinner (1–3 mm vs 2–6 mm diameter) with longer internodes (6–16 cm vs 2–10 cm), consistent with a twining (rather than trailing) habit. It also closely resembles I. trifida (Kunth) G. Don, particularly in the twining habit and chartaceous sepals, but differs in having obtuse sepals (80°–160° vs 20°–70°) and laxer, more obviously umbellate inflorescences with a greater number of flowers (5–24 vs 2–12) and mostly entire, larger leaves (4–14 cm vs 2–10 cm long).
Fig. 3.
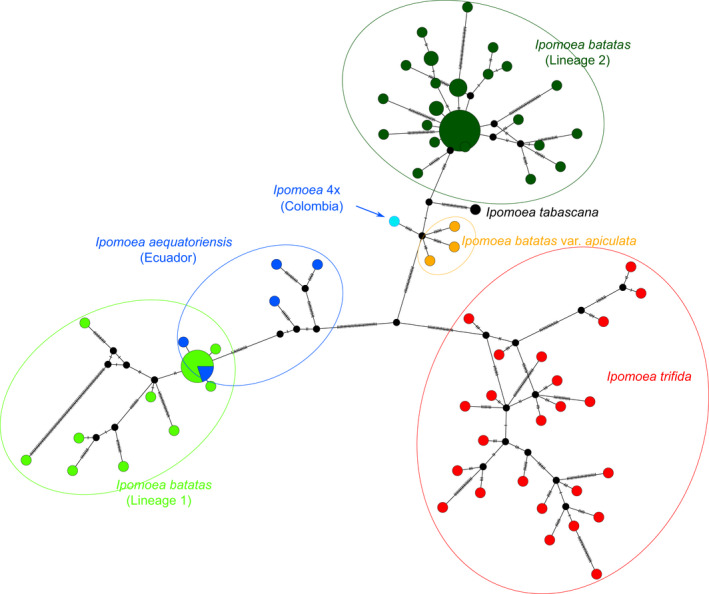
The analysis of chloroplast genomes shows Ipomoea aequatoriensis is associated with the sweetpotato ancestral lineage. Median Joining phylogenetic network inferred using 602 segregating sites (182 parsimony‐informative) and showing the relationships between Ipomoea batatas, Ipomoea trifida, I. aequatoriensis and the hybrid entities, Ipomoea tabascana and I. batatas var. apiculata. The one Colombian specimen sequenced (K500/CH80.3), indicated with an arrow, seems to carry a chloroplast related to sweetpotato lineage 2 chloroplast; we excluded it from our diagnosis of I. aequatoriensis pending further investigation. The size of the circles indicates the number of samples, with samples grouping in larger circles being identical for the sites studied.
Identifying the tetraploid progenitor of sweetpotato
A major barrier to understanding the origin and evolution of sweetpotato remains the difficulty of assembling its large allohexaploid genome (Isobe et al., 2019), which comprises three subgenomes: two identical (BBBB) and one slightly different (AA) in an AABBBB structure (Ting & Kehr, 1953; Ting et al., 1957; Jones, 1965; Magoon et al., 1970; Nishiyama et al., 1975; Shiotani & Kawase, 1987; Srisuwan et al., 2006; Gao et al., 2011; Yang et al., 2017). These subgenomes are most likely derived from a hybridization event between a diploid progenitor that contributed the AA subgenome and a tetraploid progenitor that contributed the BBBB subgenomes (Fig. 4). The AA subgenome is most likely derived from a diploid ancestor shared with I. trifida (Yang et al., 2017; Muñoz‐Rodríguez et al., 2018), but the tetraploid progenitor that contributed the BBBB subgenomes remains unidentified (Yang et al., 2017).
Fig. 4.
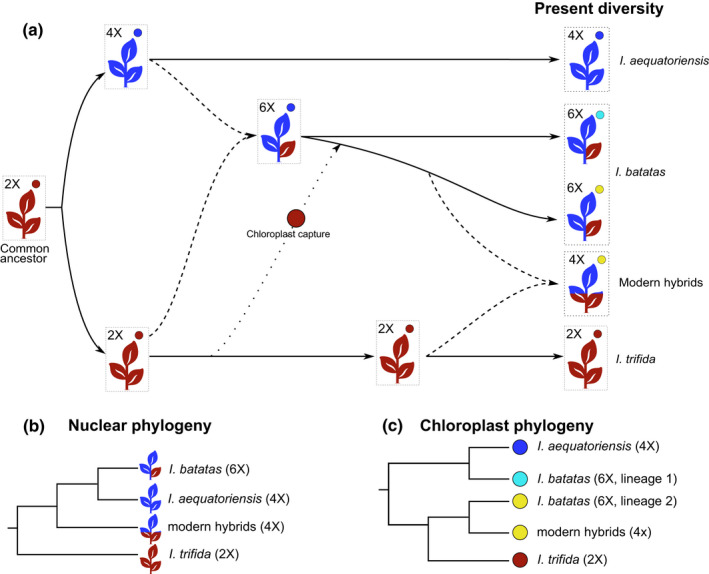
One of several possible scenarios of sweetpotato evolution and origin of current diversity. Tetraploid plants closely related to sweetpotato have two different origins: plants from Ecuador represent direct descendants from the autotetraploid progenitor of hexaploid Ipomoea batatas, whereas plants from Mexico and Central America are the result of a more recent hybridization between hexaploid I. batatas and diploid Ipomoea trifida. (a) One possible scenario, congruent with the data currently available, is presented here. An autotetraploid would have arisen from a whole genome duplication of a diploid common ancestor with I. trifida. This autotetraploid would have hybridized with the diploid ancestor to produce an allohexaploid. Subsequent introgression between the diploid ancestor lineage and the allohexaploid would result in chloroplast capture from I. trifida, explaining the two distinct I. batatas lineages in the chloroplast phylogenies. This separate lineage would keep a hexaploid nuclear genome but a chloroplast most similar to the diploid progenitor, and therefore to modern I. trifida, than to the ancestral sweetpotato lineage. Red and blue colours indicate the proportion of diploid (AA, red) and tetraploid (BBBB, blue) ancestral genomes in the different entities. Small, coloured circles represent the chloroplast. Dashed lines indicate hybridization and dotted line indicates introgression with chloroplast capture. (b) Summary nuclear phylogeny depicting the relationship between modern taxa, with Ipomoea aequatoriensis most closely related to I. batatas. (c) Summary chloroplast phylogeny depicting the relationship between modern taxa, with I. aequatoriensis most closely related to I. batatas lineage 1, the ancestral sweetpotato lineage.
The new autotetraploid species I. aequatoriensis is the closest wild relative of sweetpotato identified to date, and our results strongly suggest it is the direct descendant of sweetpotato’s tetraploid progenitor. A possible scenario for this is presented in Fig. 4, and there are four lines of evidence for this conclusion. First, the wild provenance of the samples we studied, which were not cultivated, feral or derived from breeding programmes (Notes S3). Second, I. aequatoriensis is consistently recovered as monophyletic and sister to I. batatas in nuclear phylogenies, regardless of the method of phylogenetic inference, both in our study (Figs 2a, S7, S8) and in a recent pre‐print (Yan et al., 2021). Third, its genetic structure is indicative of an autopolyploid origin (Table S5; Notes S1), a requirement for the tetraploid progenitor of the sweetpotato because of the AABBBB structure of the sweetpotato genome. Fourth, I. aequatoriensis is most closely related to sweetpotato lineage 1 – the ancestral sweetpotato lineage – in the analyses of chloroplast genomes in our study (Fig. 3) and that by Roullier et al. (2013).
Poor taxonomy and modern hybrids complicate sweetpotato studies
Previous attempts to identify sweetpotato’s tetraploid progenitor have been hampered by taxonomic confusion, the lack of a well‐resolved phylogenetic framework for sweetpotato and its closest relatives or the inclusion of probably feral specimens (Jones, 1967; Nishiyama, 1971; Martin & Jones, 1972; Austin, 1988; Roullier et al., 2013) (Table S1).
In addition, the existence of modern hybrids between I. batatas and its closest diploid relative, I. trifida, further complicates data interpretation. This is because hybridization between I. batatas (3n gametes) and I. trifida (1n gametes) will most likely produce a tetraploid (Orjeda et al., 1991), as in the case of I. tabascana (Austin, 1977; Jarret et al., 1992; Bohac et al., 1993; Srisuwan et al., 2006). Because of their parentage, such tetraploids are closely related to hexaploid I. batatas in nuclear phylogenies (Figs 2a, S7). Therefore, studies that rely purely on phylogenetic analysis of nuclear DNA sequence data are likely to confuse these putative hybrid tetraploids with the autotetraploid progenitor of hexaploid I. batatas (Yan et al., 2021) (Notes S3). However, the incorporation of other lines of evidence confirms the hybrid origin of these tetraploid entities and shows they cannot be the tetraploid progenitor of sweetpotato. First, I. batatas var. apiculata is recovered with the known hybrid I. tabascana in all phylogenies (Figs 2a, S7, S8) and both entities are in an intermediate position between I. trifida and I. batatas in the PCAs using nuclear genomic variants (Fig. 2b), implying a highly similar genetic structure. Second, k‐mer analysis of these samples suggests that they possess two distinct subgenomes (Table S5; Notes S1). The k‐mer analyses require confirmation using higher‐coverage sequence data (Methods S2), but our results are consistent with their apparent hybrid origin (Srisuwan et al., 2006; Muñoz‐Rodríguez et al., 2018). Third, the hybrid entities are most closely related in the chloroplast analysis to the derived sweetpotato chloroplast lineage 2 (Fig. 3), which is the result of introgression with I. trifida and therefore postdates the origin of I. batatas (Muñoz‐Rodríguez et al., 2018). Finally, the PCA and LDA using morphology (Fig. 1d,e) consistently show these specimens cluster with either I. trifida or I. batatas, instead of forming a distinct group as is the case of I. aequatoriensis.
In summary, a broader consideration of collection history, nuclear and chloroplast sequence data, and genomic structure, enables the identification of modern tetraploid hybrids, such as I. tabascana and I. batatas var. apiculata, and rules them out as sweetpotato’s tetraploid progenitor (Fig. 4). Our results also suggest I. batatas var. apiculata should be treated as a distinct entity of hybrid origin akin to I. tabascana rather than a subspecies of I. batatas (Notes S1). Although we have not been able to study all the material listed in earlier studies of tetraploid plants (Table S1; Notes S4), future studies that consider the different criteria presented here should allow the classification of those specimens as either ancient autotetraploids or modern hybrids, and further clarify their relationship to sweetpotato.
Ipomoea aequatoriensis, a key finding for sweetpotato studies
The identification of the closest living relative of the tetraploid progenitor of sweetpotato is key to untangling its genomic history and contemporary diversity. Ipomoea aequatoriensis has all the hallmarks of being that species, and therefore represents an extraordinary discovery and a key finding for subsequent sweetpotato studies.
Author contributions
RWS, JRIW, PM‐R, TW and TC conceived the project; PM‐R and TW conducted the analyses; NLA and RLJ contributed material and information about its provenance; PM‐R, TW, RWS, JRIW and TC wrote the manuscript. PM‐R and TW contributed equally to this work.
Supporting information
Fig. S1 Ipomoea aequatoriensis specimen PI 355830/K300/CH71.3.
Fig. S2 Ipomoea aequatoriensis specimen K500/CH80.3.
Fig. S3 Ipomoea aequatoriensis specimen PI 561248/CIP 403553.
Fig. S4 Ipomoea aequatoriensis specimen PI 561258.
Fig. S5 Ipomoea tabascana specimen PI 518479/CIP 460824 and Ipomoea batatas var. apiculata specimen PI 518474/CIP 403953.
Fig. S6 trnL‐rpl32 chloroplast DNA barcode phylogeny.
Fig. S7 Nuclear phylogenies of Ipomoea Clade A3 indicating the position of the Ecuadorian tetraploids and the modern hybrids.
Fig. S8 Nuclear phylogenies of Ipomoea Clade A3 indicating the position of the Ecuadorian tetraploids and the modern hybrids. Phylogenies inferred including IUPAC characters for heterozygous sites.
Fig. S9 Additional principal component analyses.
Fig. S10 Scientific illustration of Ipomoea aequatoriensis T. Wells & P. Muñoz.
Methods S1 Preliminary analysis of the trnL‐rpl32 chloroplast DNA region.
Methods S2 K‐mer analysis of putative hybrid tetraploids.
Methods S3 Additional phylogenetic analysis of nuclear probes.
Methods S4 Additional principal component analyses.
Methods S5 K‐mer analysis of putative hybrid tetraploids.
Notes S1 Modern hybrids closely related to Ipomoea batatas.
Notes S2 K‐mer analyses diagrams.
Notes S3 Description and additional information for Ipomoea aequatoriensis.
Notes S4 Hybrid specimens in other studies.
Table S1 Tetraploid accessions in previous studies, indicating past and present identifications.
Table S2 Passport data of all samples included in morphological analyses.
Table S3 Passport data of all samples included in phylogenetic analyses.
Table S4 Statistics of the putative single copy nuclear regions used in phylogenetic analysis.
Table S5 Patterns of nucleotide heterozygosity in k‐mer spectra of sequencing reads (k = 21).
Please note: Wiley Blackwell are not responsible for the content or functionality of any Supporting Information supplied by the authors. Any queries (other than missing material) should be directed to the New Phytologist Central Office.
Acknowledgements
This project was funded by a BBSRC research grant to RWS (T001445/1). TW was funded by an Interdisciplinary DTP BBSRC scholarship. PM‐R was also funded by an Interdisciplinary DTP BBSRC scholarship at the early stages of this project. The authors thank botanical artist Rosemary Wise for the illustration of Ipomoea aequatoriensis. The authors thank all herbarium curators and germplasm managers for providing access to their material, especially Masaru Tanaka at NARO – Japan, as well as the people who did fieldwork and collected the specimens in this study.
See also the Commentary on this article by Särkinen et al., 234: 1107–1108.
Data availability
Raw reads from the 2018 study and newly generated data are available in the Sequence Repository Archive, BioProjects PRJNA453382 and PRJNA796763 respectively. Original and edited files with morphological and molecular analyses and scripts are available via the Oxford Research Archive (https://ora.ox.ac.uk/objects/uuid:055e2f01‐bbb1‐4a69‐a3ae‐dac009db31d1). Any other information required to re‐analyse the data is available from the lead contact upon request.
References
- Austin DF. 1977. Hybrid polyploids in Ipomoea section Batatas . Journal of Heredity 68: 259–260. [Google Scholar]
- Austin DF. 1978. The Ipomoea batatas complex‐I. Taxonomy. Bulletin of the Torrey Botanical Club 105: 114–129. [Google Scholar]
- Austin DF. 1982. Flora of Ecuador. 165. Convolvulaceae. Lund, Sweden: Gleerup; Stockholm, Sweden: Publishing House of the Swedish Research Council. [Google Scholar]
- Austin DF. 1988. The taxonomy, evolution, and genetic diversity of the sweet potato and its wild relatives. In: International Potato Center , ed. Exploration, maintenance and utilization of sweet potato genetic resources. Lima, Peru: International Potato Center, 27–60. [Google Scholar]
- Austin DF, Jarret RL, Tapia C, de la Puente F. 1993. Collecting tetraploid I. batatas (L.) Lam. in Ecuador. FAO/IBPGR Plant Genetic Resources Newsletter 91: 33–35. [Google Scholar]
- Austin DF, de la Puente F, Contreras J. 1991. Ipomoea tabascana, an endangered tropical species. Economic Botany 45: 435. [Google Scholar]
- Bandelt HJ, Forster P, Röhl A. 1999. Median‐joining networks for inferring intraspecific phylogenies. Molecular Biology and Evolution 16: 37–48. [DOI] [PubMed] [Google Scholar]
- Bohac JR, Austin DF, Jones A. 1993. Discovery of wild tetraploid sweetpotatoes. Economic Botany 47: 193–201. [Google Scholar]
- Bolger AM, Lohse M, Usadel B. 2014. Trimmomatic: a flexible trimmer for Illumina sequence data. Bioinformatics 30: 2114–2120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Borowiec ML. 2016. Amas: a fast tool for alignment manipulation and computing of summary statistics. PeerJ 4: e1660. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Castresana J. 2000. Selection of conserved blocks from multiple alignments for their use in phylogenetic analysis. Molecular Biology and Evolution 17: 540–552. [DOI] [PubMed] [Google Scholar]
- Chang CC, Chow CC, Tellier LC, Vattikuti S, Purcell SM, Lee JJ. 2015. Second‐generation Plink: rising to the challenge of larger and richer datasets. GigaScience 4: 7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Danecek P, Auton A, Abecasis G, Albers CA, Banks E, DePristo MA, Handsaker RE, Lunter G, Marth GT, Sherry ST et al. 2011. The variant call format and VCFtools. Bioinformatics 27: 2156–2158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Danecek P, Bonfield JK, Liddle J, Marshall J, Ohan V, Pollard MO, Whitwham A, Keane T, McCarthy SA, Davies RM et al. 2021. Twelve years of SAMtools and BCFtools . GigaScience 10: giab008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dodson CH, Gentry AH. 1978. Flora of the Rio Palenque Science Center: Los Rios Province, Ecuador. Selbyana 4: 301–302. [Google Scholar]
- Dodson CH, Gentry AH, Valverde FM. 1985. La flora de Jauneche, Los Ríos, Ecuador. Quito, Ecuador: Banco Central del Ecuador. [Google Scholar]
- Gao M, Ashu GM, Stewart L, Akwe WA, Njiti V, Barnes S. 2011. Wx intron variations support an allohexaploid origin of the sweetpotato [Ipomoea batatas (L.) Lam]. Euphytica 177: 111–133. [Google Scholar]
- Huaman Z, ed. 1991. Descriptors for sweet potato. Lima, Peru: CIP, AVRDC and IBPGR. [Google Scholar]
- Isobe S, Shirasawa K, Hirakawa H. 2019. Current status in whole genome sequencing and analysis of Ipomoea . Plant Cell Reports 38: 1365–1371. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jarret RL, Gawel N, Whittemore AT. 1992. Phylogenetic relationships of the sweetpotato [Ipomoea batatas (L.) Lam.]. Journal of the American Society for Horticultural Science 117: 633–637. [Google Scholar]
- Jin J‐J, Yu W‐B, Yang J‐B, Song Y, dePamphilis CW, Yi T‐S, Li D‐Z. 2018. GetOrganelle: a fast and versatile toolkit for accurate de novo assembly of organelle genomes. Genome Biology 21: 241. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jones A. 1965. Cytological observations and fertility measurements of sweet potato (Ipomoea batatas (L.) Lam.). Proceedings of the American Society of Horticultural Science 86: 527–537. [Google Scholar]
- Jones A. 1967. Should Nishiyama’s K123 (Ipomoea trifida) be designated I. batatas? Economic Botany 21: 163–166. [Google Scholar]
- Kalyaanamoorthy S, Minh BQ, Wong TKF, von Haeseler A, Jermiin LS. 2017. ModelFinder: fast model selection for accurate phylogenetic estimates. Nature Methods 14: 587–589. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Katoh K, Standley DM. 2013. Mafft multiple sequence alignment software v.7: improvements in performance and usability. Molecular Biology and Evolution 30: 772–780. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lê S, Josse J, Husson F. 2008. factominer: an R package for multivariate analysis. Journal of Statistical Software 25: 1–18. [Google Scholar]
- Magoon ML, Krishnan R, Vijaya BK. 1970. Cytological evidence on the origin of sweet potato. Theoretical and Applied Genetics 40: 360–366. [DOI] [PubMed] [Google Scholar]
- Marçais G, Kingsford C. 2011. A fast, lock‐free approach for efficient parallel counting of occurrences of k‐mers. Bioinformatics 27: 764–770. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martin FW, Jones A. 1972. The species of Ipomoea closely related to the sweet potato. Economic Botany 26: 201–215. [Google Scholar]
- Martin FW, Ruberté RM, Jones A. 1974. A wild Ipomoea species closely related to the sweet potato. Economic Botany 28: 287–292. [Google Scholar]
- McDonald JA, Austin DF. 1990. Changes and additions in Ipomoea sect. Batatas . Brittonia 42: 116–120. [Google Scholar]
- Muñoz‐Rodríguez P, Carruthers T, Wood JRI, Williams BRM, Weitemier K, Kronmiller B, Ellis D, Anglin NL, Longway L, Harris SA et al. 2018. Reconciling conflicting phylogenies in the origin of sweet potato and dispersal to Polynesia. Current Biology 28: 1246–1256. [DOI] [PubMed] [Google Scholar]
- Muñoz‐Rodríguez P, Carruthers T, Wood JRI, Williams BRM, Weitemier K, Kronmiller B, Goodwin Z, Sumadijaya A, Anglin NL, Filer D et al. 2019. A taxonomic monograph of Ipomoea integrated across phylogenetic scales. Nature Plants 5: 1136–1144. [DOI] [PubMed] [Google Scholar]
- Nguyen L‐T, Schmidt HA, von Haeseler A, Minh BQ. 2015. Iq‐Tree: a fast and effective stochastic algorithm for estimating maximum‐likelihood phylogenies. Molecular Biology and Evolution 32: 268–274. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nishiyama I. 1971. Evolution and domestication of the sweet potato. Botanical Magazine 84: 377–387. [Google Scholar]
- Nishiyama I, Miyazaki T, Sakamoto S. 1975. Evolutionary autoploidy in the sweet potato (Ipomoea batatas (L.) Lam.) and its progenitors. Euphytica 24: 197–208. [Google Scholar]
- Orjeda G, Freyre R, Iwanaga M. 1991. Use of Ipomoea trifida germ plasm for sweet potato improvement. 3. Development of 4x interspecific hybrids between Ipomoea batatas (L.) Lam. (2n=6x=90) and I. trifida (H.B.K) G. Don. (2n=2x=30) as storage‐root initiators for wild species. Theoretical and Applied Genetics 83: 159–163. [DOI] [PubMed] [Google Scholar]
- Privé F, Luu K, Blum MGB, McGrath JJ, Vilhjálmsson BJ. 2020. Efficient toolkit implementing best practices for principal component analysis of population genetic data. Bioinformatics 36: 4449–4457. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Purcell S. 2021. Plink v. 2.00a3LM AVX2. [WWW document] URL https://www.cog‐genomics.org/plink/2.0/ [accessed 1 October 2021]. [Google Scholar]
- Ranallo‐Benavidez TR, Jaron KS, Schatz MC. 2020. GenomeScope 2.0 and Smudgeplot for reference‐free profiling of polyploid genomes. Nature Communications 11: 1432. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roullier C, Duputié A, Wennekes P, Benoit L, Fernández Bringas VM, Rossel G, Tay D, McKey D, Lebot V. 2013. Disentangling the origins of cultivated sweet potato (Ipomoea batatas (L.) Lam.). PLoS ONE 8: e62707. [DOI] [PMC free article] [PubMed] [Google Scholar]
- RStudio Team . 2021. Rstudio: integrated development for R, v.1.4.1717. Boston, MA, USA: RStudio, PBC. [Google Scholar]
- Schindelin J, Arganda‐Carreras I, Frise E, Kaynig V, Longair M, Pietzsch T, Preibisch S, Rueden C, Saalfeld S, Schmid B et al. 2012. Fiji: an open‐source platform for biological‐image analysis. Nature Methods 9: 676–682. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shiotani I, Kawase T. 1987. Synthetic hexaploids derived from wild species related to sweet potato. Japanese Journal of Breeding 37: 367–376. [Google Scholar]
- Srisuwan S, Sihachakr D, Siljak‐Yakovlev S. 2006. The origin and evolution of sweet potato (Ipomoea batatas Lam.) and its wild relatives through the cytogenetic approaches. Plant Science 171: 424–433. [DOI] [PubMed] [Google Scholar]
- Talavera G, Castresana J. 2007. Improvement of phylogenies after removing divergent and ambiguously aligned blocks from protein sequence alignments. Systematic Biology 56: 564–577. [DOI] [PubMed] [Google Scholar]
- Tange O. 2011. GNU Parallel – the command‐line power tool. The USENIX Magazine 36: 42–47. [Google Scholar]
- Thiers B. 2018. Index Herbariorum: a global directory of public herbaria and associated staff. New York Botanical Garden’s Virtual Herbarium. [WWW document] URL http://sweetgum.nybg.org/science/ih/ [accessed 10 November 2021]. [Google Scholar]
- Ting YC, Kehr AE. 1953. Meiotic studies in the sweet potato: (Ipomoea batatas Lam.). Journal of Heredity 44: 207–211. [Google Scholar]
- Ting YC, Kehr AE, Miller JC. 1957. A cytological study of the sweet potato plant Ipomoea batatas (L.) Lam. and its related species. The American Naturalist 91: 197–203. [Google Scholar]
- Venables WN, Ripley BD, Venables WN. 2002. Modern applied statistics with S. New York, NY, USA: Springer. [Google Scholar]
- Wickham H. 2016. ggplot2: elegant graphics for data analysis. New York, NY, USA: Springer‐Verlag. [Google Scholar]
- Wickham H, Averick M, Bryan J, Chang W, McGowan L, François R, Grolemund G, Hayes A, Henry L, Hester J et al. 2019. Welcome to the tidyverse . Journal of Open Source Software 4: 1686. [Google Scholar]
- Wood JRI, Muñoz‐Rodríguez P, Williams BRM, Scotland RW. 2020. A foundation monograph of Ipomoea (Convolvulaceae) in the New World. PhytoKeys 143: 1–823. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yan M, Li M, Moeinzadeh M‐H, Quispe‐Huamanquispe DG, Fan W, Nie H, Wang Z, Heider B, Jarret R, Kreuze J et al. 2021. Haplotype‐based phylogenetic analysis uncovers the tetraploid progenitor of sweet potato. Research Square. doi: 10.21203/rs.3.rs-750500/v1. [DOI] [Google Scholar]
- Yang J, Moeinzadeh M‐H, Kuhl H, Helmuth J, Xiao P, Haas S, Liu G, Zheng J, Sun Z, Fan W et al. 2017. Haplotype‐resolved sweet potato genome traces back its hexaploidization history. Nature Plants 3: 696–703. [DOI] [PubMed] [Google Scholar]
- Zhang C, Rabiee M, Sayyari E, Mirarab S. 2018. Astral‐III: polynomial time species tree reconstruction from partially resolved gene trees. BMC Bioinformatics 19: 153. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Fig. S1 Ipomoea aequatoriensis specimen PI 355830/K300/CH71.3.
Fig. S2 Ipomoea aequatoriensis specimen K500/CH80.3.
Fig. S3 Ipomoea aequatoriensis specimen PI 561248/CIP 403553.
Fig. S4 Ipomoea aequatoriensis specimen PI 561258.
Fig. S5 Ipomoea tabascana specimen PI 518479/CIP 460824 and Ipomoea batatas var. apiculata specimen PI 518474/CIP 403953.
Fig. S6 trnL‐rpl32 chloroplast DNA barcode phylogeny.
Fig. S7 Nuclear phylogenies of Ipomoea Clade A3 indicating the position of the Ecuadorian tetraploids and the modern hybrids.
Fig. S8 Nuclear phylogenies of Ipomoea Clade A3 indicating the position of the Ecuadorian tetraploids and the modern hybrids. Phylogenies inferred including IUPAC characters for heterozygous sites.
Fig. S9 Additional principal component analyses.
Fig. S10 Scientific illustration of Ipomoea aequatoriensis T. Wells & P. Muñoz.
Methods S1 Preliminary analysis of the trnL‐rpl32 chloroplast DNA region.
Methods S2 K‐mer analysis of putative hybrid tetraploids.
Methods S3 Additional phylogenetic analysis of nuclear probes.
Methods S4 Additional principal component analyses.
Methods S5 K‐mer analysis of putative hybrid tetraploids.
Notes S1 Modern hybrids closely related to Ipomoea batatas.
Notes S2 K‐mer analyses diagrams.
Notes S3 Description and additional information for Ipomoea aequatoriensis.
Notes S4 Hybrid specimens in other studies.
Table S1 Tetraploid accessions in previous studies, indicating past and present identifications.
Table S2 Passport data of all samples included in morphological analyses.
Table S3 Passport data of all samples included in phylogenetic analyses.
Table S4 Statistics of the putative single copy nuclear regions used in phylogenetic analysis.
Table S5 Patterns of nucleotide heterozygosity in k‐mer spectra of sequencing reads (k = 21).
Please note: Wiley Blackwell are not responsible for the content or functionality of any Supporting Information supplied by the authors. Any queries (other than missing material) should be directed to the New Phytologist Central Office.
Data Availability Statement
Raw reads from the 2018 study and newly generated data are available in the Sequence Repository Archive, BioProjects PRJNA453382 and PRJNA796763 respectively. Original and edited files with morphological and molecular analyses and scripts are available via the Oxford Research Archive (https://ora.ox.ac.uk/objects/uuid:055e2f01‐bbb1‐4a69‐a3ae‐dac009db31d1). Any other information required to re‐analyse the data is available from the lead contact upon request.