

Abstract
Although the number of countries participating in pivotal trials submitted to enable drug registration has nearly doubled over the past 25 years, there has not been a substantial increase in the diversity of clinical trial populations. In parallel, our understanding of factors that influence medicine response and variability has continued to evolve. The notion of intrinsic and extrinsic sources of variability has been embedded into different regulatory guidelines, including the recent guideline on the importance of enhancing the diversity of clinical trial populations.
In addition to presenting the clinical and scientific reasons for ensuring that clinical trial populations represent the demographics of patient populations, this overview outlines the efforts of regulatory agencies, patient advocacy groups and clinical researchers to attain this goal through strategies to meet representation in recruitment targets and broaden eligibility criteria. Despite these efforts, challenges to participation in clinical trials remain, and certain groups continue to be underrepresented in development programmes. These challenges are amplified when the representativeness of specific groups may vary across countries and regions in a global clinical programme.
Whilst enhanced trial diversity is a critical step towards ensuring that results will be representative of patient populations, a concerted effort is required to characterise further the factors influencing interindividual and regional differences in response for global populations. Quantitative clinical pharmacology principles should be applied to allow extrapolation of data across groups or regions as well as provide insight into the effect of patient‐specific characteristics on a medicine's dose rationale and efficacy and safety profiles.
Keywords: age, diversity, ethnicity, recruitment, sex
1. INTRODUCTION
Modern medicine has evolved beyond anecdotal observations and is now guided by scientific standards. Over many centuries, medicines have been prescribed to a diverse spectrum of patients: men and women of different ages and ethnic groups resident in different geographic regions, without careful consideration of the potential contribution of these characteristics to interindividual differences and variability in treatment response.
The evolution of our understanding of the factors that influence variability in pharmacokinetics (PK), pharmacodynamics (PD), and the efficacy and safety profile of a medicine has guided the patient characteristics to be evaluated in clinical research programmes. Currently the approval of a medicine relies on evidence from a sample of the overall target patient population (Figure 1). Following drug approval, risk management and pharmacovigilance activities focus on further characterisation of the safety profile across the wider population. Such an approach relies on the expectation that the data evaluated arise from a representative sample of the patient population that resembles the population from which they were drawn in all the ways that are important for the medicine and its indication. It also assumes that the results can be generalised, and consequently will provide information on a group larger than the sample originally studied.
FIGURE 1.

The smaller cube represents a fraction of the general patient population enrolled in clinical trials. Despite strict inclusion and exclusion criteria, this sample is assumed to be representative of the wider patient population, for whom the medicine may be indicated after marketing authorisation. Due to the limited sample size of the clinical trial population, safety data are collected during the postmarketing phase to ensure that the benefit–risk profile of the medicine remains accurate
Indeed, an important contribution of clinical pharmacology during drug development is to characterise the factors influencing variation in the PK, PD, efficacy and safety of a medicine, and, when appropriate, to determine dosing regimens tailored to relevant patient characteristics. Specifically, the influence of intrinsic factors (e.g. age, sex/gender, race/ethnicity, comorbidities [renal and hepatic dysfunction]) and extrinsic factors (e.g. food, drug interactions) should be investigated. 1 , 2 Clinical pharmacology principles also provide the basis for extrapolation and/or generalisation of findings, so that accurate recommendations regarding the dose rationale, and overall efficacy and safety profile can be applied to the broader patient population. To achieve this goal without substantial uncertainty, patient population diversity is not only desirable, but also scientifically appropriate.
International (e.g. International Council for Harmonisation of Technical Requirements for Pharmaceuticals for Human Use [ICH]) and local (e.g. European Medicines Association [EMA] in the EU, Food and Drug Administration [FDA] in the USA) guidelines, which provide a framework for drug development, specify that the patients included in clinical trials should be representative of the population for whom the medicine will be indicated in clinical practice, as defined in the final label or summary of product characteristics. However, clinical drug development programmes have enrolled a subset of patients that has not reflected the epidemiological profile of that indication. 3 , 4 , 5 The clinical relevance of patient characteristics not assessed during the development programme has been a matter for postmarketing activities. However, this situation is changing. Various initiatives have been proposed to ensure that patient populations in clinical trials reflect the patient population to be treated. However, the ongoing lack of clinically representative trial populations has prompted the US FDA to develop a new guidance entitled Enhancing the Diversity of Trial Populations – Eligibility Criteria, Enrolment Practices, and Trial Designs Guidance for Industry, which was issued in November 2020. 6
This overview outlines several topics pertinent to diversity in clinical trials and seeks to provide the clinical pharmacologist with insights into the current challenges and strategies being adopted to increase participant diversity. Awareness of these perspectives is essential for clinical pharmacologists to design suitable clinical studies and identify alternative, complementary approaches, including modelling, simulation and extrapolation to ensure the impact of patient diversity is evaluated during the clinical development and approval of new medicines. We conclude with some perspectives on the implications of these principles for global development programmes, where regional differences in participant diversity must also be considered. Attention is given to the role of modelling and simulation as a basis for the extrapolation of PK, efficacy and safety results, and assessment of dosing recommendations, across different patient populations. It is notable that the definitions of demographic variables used in clinical trials and clinical practice can vary significantly, creating a barrier to the pooling of results across studies or extrapolation to additional patient populations. 7 , 8
2. THE SCIENCE BEHIND THE INCLUSION OF DIVERSE POPULATIONS
Whilst clinical and disease specific characteristics may ultimately determine how medicines should be used, demographic differences can play an important role in interindividual variability in the PK, PD, and efficacy and safety profile of a medicine. Several ICH guidelines address the need to explore the potential effect of demographic differences on dose–exposure–response (E4, M4E) and the safety profile of a medicine (E8, M3). The demographic factors of age, race/ethnicity and sex/gender are specifically mentioned in some guidelines, based on their established associations with response for some drugs. 9 However, the range of factors that can influence medicine response extends beyond age, sex/gender and race/ethnicity, with the ICH E17 guideline 2 advocating for the intrinsic and extrinsic factors important for each new chemical or biological entity to be established early during clinical development (Figure 2).
FIGURE 2.
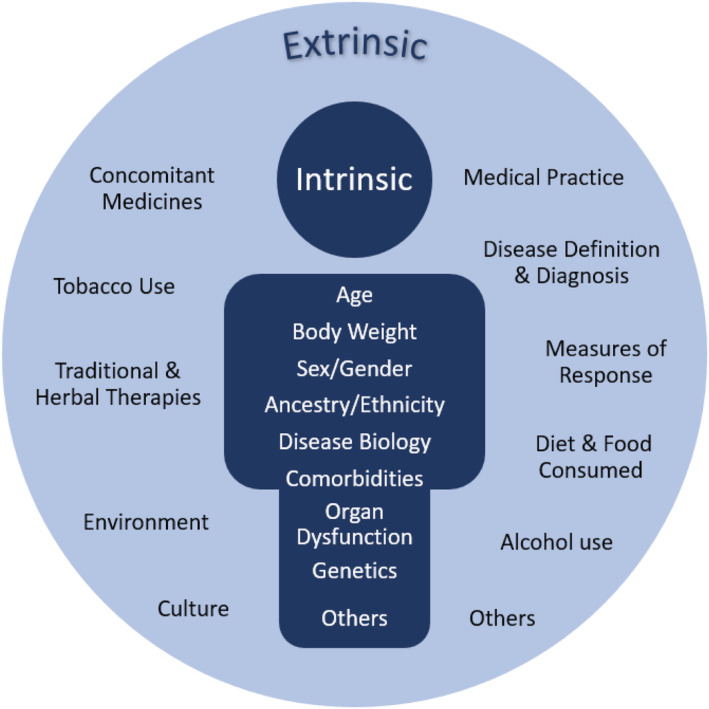
Intrinsic and extrinsic factors contributing to interindividual variability in pharmacokinetics and pharmacodynamics, which may lead to clinically relevant differences in treatment response (adapted from Liu et al., 2021) 101
Some additional factors may only be observed in later phase studies or after a drug has been used in broader patient populations postmarketing. For example, socioeconomic factors, including level of education and income, often correlate with disease prevalence and severity 10 and can also be associated with medicine responses. 11 , 12 , 13 Individuals who are less well educated or who have lower incomes are generally not well represented in clinical trials. 14 Furthermore, these additional factors can overlap with sex/gender and race/ethnicity 15 and should also be considered as contributing to the variation in response when interpreting clinical trial results.
Ensuring that the demographics of clinical trial populations reflects the age, race/ethnicity and sex/gender distribution of the patient population with the relevant disease is the focus of the recent FDA guidance. Obtaining this information is important when assessing the benefit: risk balance for each individual patient based on the generalisability and representativeness of the clinical results observed.
2.1. Age
Older adults represent a major demographic of relevance for drug development. In 2019, it was estimated that 9% of the global population was aged 65 years or older. 16 The burden of disease and the age distribution of affected patients varies substantially between countries. For example, in 2019, the proportion of the population aged ≥65 years in China was 11.5% compared with 18.8% in Europe and 28.0% in Japan. Many populations are ageing, and it is anticipated that by 2050 >1 in every 4 people in China (26.1%), Europe (28.1%) and Japan (37.7%) will be ≥65 years. 16
The importance of including older adults in clinical trials to ensure that representative populations are investigated has been recognised by regulatory agencies. Both the ICH E7 guideline Studies in Support of Special Populations: Geriatrics 17 and the US FDA Guideline for the Study of Drugs Likely to Be Used in the Elderly published in 1989, 18 highlight that individuals aged >65 years represent an important group for inclusion in clinical trials. The ICH E7 guideline defines geriatrics as “patients aged 65 years or older”, with this age reflecting the standard retirement age at that time, 17 and this definition also used in the US FDA Guideline for the Study of Drugs Likely to Be Used in the Elderly. 18
It is also critical that drug development programmes consider appropriate doses for patients aged <18 years. Paediatric considerations are now embedded into the regulatory processes in the USA and EU, which ensures a systematic assessment of the requirement for evidence from clinical trials, or alternative approaches, to support the dose rationale as well as the efficacy and safety profile of a medicine for the paediatric population. 19 , 20 , 21 Details on this group are, therefore, beyond the scope of this overview.
An important point to consider is that increasing age is associated with co‐morbidities and polypharmacy. 22 , 23 Older adults consume the majority of prescribed medicines, with the number of medicines related to the number of medical diagnoses. 23 Consequently, although the elderly usually take >3 medications, 22 there will be considerable polypharmacy in some groups. For example, older residents in US Nursing Homes receive slightly more than 8 oral medications/d. 23 Most clinical trials report that a high proportion of participants aged >65 years receive 3–5 concomitant medications, although estimates vary. 22 , 24
Age‐associated changes in organ function (e.g. renal function) and body composition, declining homeostatic reserve, and comorbid diseases contribute to changes in PK with advancing age. 25 , 26 PD changes have also been reported. 27 Characteristics associated with frailty such as sarcopenia and the increased prevalence of comorbidities and polypharmacy also increase the potential for drug–drug interactions 5 and other adverse drug reactions in older relative to younger adults. 25 Factors with the potential to alter the PK or PD of many drugs have been extensively reviewed and include characteristics summarised in Table 1 for PK and Table S1 for PD. Therefore, the changes with age in physiology, frequency of chronic diseases and high potential for drug–drug interactions can all increase the potential for variation in treatment response in older populations.
TABLE 1.
Physiological changes of older age and potential influences on pharmacokinetics (adapted from Saeed et al., 2015) 27
| PK process | Age‐related change | Impact | Clinical consequences |
|---|---|---|---|
| Absorption | Increased gastric pH | Slightly decreased absorption | May impact maximum plasma concentration and time to maximum plasma concentration |
| Delayed gastric emptying | |||
| Reduced splanchnic blood flow | |||
| Decreased absorption surface area | |||
| Decreased gastrointestinal motility | |||
| Distribution | Increased body fat | Altered volume of distribution | Increased volume of distribution and half‐life of lipophilic drugs |
| Reduced lean body mass | |||
| Reduced body total water | Increased plasma concentration of water‐soluble drugs | ||
| Reduced serum albumin | Increased free‐fraction of highly protein‐bound acidic drugs | ||
| Increased α1‐acid glycoprotein | Decreased free‐fraction of basic drugs | ||
| Decreased cerebrovascular P‐glycoprotein functionality | Altered blood–brain barrier permeability | Excessive levels and prolonged residence of drugs and xenobiotics in the brain | |
| Metabolism | Reduced hepatic blood flow and overall liver mass | Less effective first‐pass and phase I metabolism | Increased bioavailability of drugs undergoing extensive first‐pass metabolism or reduced bioavailability of drugs which need to be activated in the liver |
| Excretion | Reduced renal blood flow | Impaired renal elimination of water‐soluble drugs/metabolites | Increased volume of distribution for water soluble drugs and enhanced risk of ADRs especially for drugs with a narrow therapeutic index (e.g. digoxin, aminoglycosides) |
| Reduced glomerular filtration rate | |||
| Increased filtration fraction |
Despite the prevalence of diseases and established polypharmacy in older populations, this demographic subgroup is frequently under‐represented in clinical trials conducted by academics or sponsored by industry, 22 , 28 , 29 , 30 as indicated for the simvastatin example in Figure 3. A further example is oncology with individuals aged >70 years comprising approximately 50% of cancer patients, yet historically only representing 13% of oncology trial participants. 24 Osteoarthritis is another example, with the prevalence higher in women aged >85 years (>50%) than in women aged 45–65 years (20%). 31 Consistent with this profile, the average age of osteoarthritis patients prescribed selective cyclooxygenase‐2 inhibitors is 79 years. 31 However, an extensive review of osteoarthritis intervention trials reported that the average age of participants was 63 years. 31 The failure to include sufficient older (>65 y) and very old (>75 y) participants in osteoarthritis trials clearly compromises the relevance of the results of these trials to the patients who are the most likely to receive these medicines in clinical practice. Physicians are therefore making prescribing decisions based on suboptimal evidence. 31 For drugs approved by the US FDA in 2019, 36% of pivotal clinical trial participants were aged ≥65 years. 32
FIGURE 3.
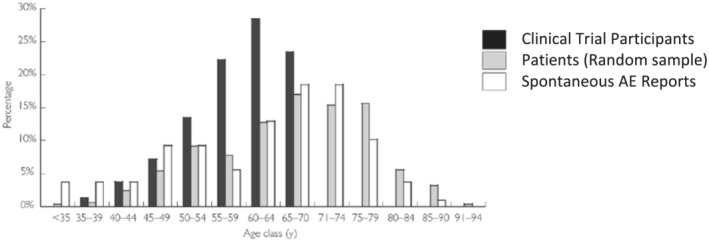
Age distribution of 3 populations providing information on the safety of simvastatin: the participants in clinical trials for simvastatin approval in France (n = 2221), a random sample of reimbursed simvastatin prescriptions in France (n = 500) and spontaneous report cases for 20 mg simvastatin in the French Pharmacovigilance database (n = 112; reproduced with permission from Martin et al., 2004) 102
In practice, older individuals often fail to meet other inclusion criteria for clinical trials, precisely because of their increased potential for comorbidities and potential for drug–drug interactions. Both the ICH and US FDA guidelines emphasise that investigators should ensure older age participants are included in clinical trials, in particular individuals aged >75 years. The more recent draft US FDA guidance Inclusion of Older Adults in Cancer Clinical Trials Guidance for Industry (Draft Guidance) 33 also emphasises the importance of studying individuals aged >75 years and indicates that sponsors may consider conducting targeted trials designed to recruit only elderly participants, rather than relying on subgroup analyses of elderly participants from trials including a broader age range. However, this approach has not been widely adopted. 34 , 35 The results of studies in the healthy elderly may not be generalisable to the frail or those with multiple comorbidities and taking numerous medications. Clearly, alternative and complementary methods are required to ensure the impact of age on treatment response and dose rationale for these patients is accurately characterised. 27
Above all, the safety of clinical trial participants must be the primary concern of sponsors and academic investigators, although this objective should be balanced against the ethical need for clinical trials to provide information of relevance for the frail and elderly patients who will ultimately receive the new medicines developed. Despite the challenges involved, there is evidence to suggest that careful selection of inclusion criteria and judicious, rather than arbitrary, selection of exclusion criteria related to organ function, concomitant therapies and comorbid conditions, can increase the proportion of older participants enrolled in trials without compromising safety. 24 For example, Mariano et al. analysed adverse event data from 46 breast cancer clinical trials and reported that the frequency of meaningful toxicity (defined as grade 3 or 4 adverse events, events leading to dose reduction or delay, or premature discontinuation of therapy) was similar between older (≥65 y) and younger patients. 36
2.2. Race and ethnicity
Population differences in the PK and PD of a medicine as well as the pathophysiology of disease and its local clinical management, can all contribute to an interethnic difference in the efficacy or safety profile reported for a medicine. 1 , 37 Nevertheless, for the majority of medicines, the labels approved by the US FDA and EMA do not recommend dose adjustments for specific race or ethnic groups. However, for some molecules, clinically significant interethnic differences in efficacy or safety have been observed, which can be of a magnitude to warrant ethnic group‐specific dosing recommendations. 38
Terminology related to race and ethnicity is complex and not consistently applied in the literature. Consequently, the definitions used for these important demographic variables are discussed in the Supporting Information. In brief, clinical studies in global drug development programmes use the criteria defined by the US FDA guidance for the Collection of Race and Ethnicity Data in Clinical Trials 39 and describe each clinical trial participant according to specified Race and Ethnicity categories. However, there are limitations to the interpretation of results reported using these categories as they do not reflect the diversity of populations included in global clinical trials, have been acknowledged to not be scientifically based 39 and do not always align with terminology used in the scientific literature. Consequently, it is critical to consider the terminology used when evaluating clinical trial results or literature in this area.
Since 2006, only 1 medicine approved in the USA, the thrombopoietin receptor agonist eltrombopag, has a different dose recommended for a specific ethnic group at first approval. For eltrombopag, 2‐fold higher plasma concentrations (area under the curve) are observed in patients with immune thrombocytopenia of East Asian than European ancestry. 40 Therefore, to avoid steep increases in the platelet counts, the recommended initial dose of eltrombopag in East Asian patients (25 mg) is half the initial dose in patients of other ancestries (50 mg once daily). 41 With tailoring of subsequent eltrombopag doses to the platelet count response, a favourable long‐term benefit: risk ratio can be achieved in East Asian patients, despite the interethnic difference in PK and consequent platelet response. 42
Interethnic differences in the target can have important consequences for the choice of medicine to treat a patient or select the appropriate clinical dose. These examples are usually identified when the medicine is used in larger patient populations. For example, ancestry is 1 of the factors that influences the response to warfarin, principally reflecting population differences in the frequency of polymorphisms in the gene encoding the warfarin target, VKORC1. In a cohort of patients in the USA (Figure 4), a target international normalised ratio of 3–4 was achieved at lower weekly doses in Asian Americans, than Americans of European ancestry, and even higher doses were required in African Americans. 43 In oncology, differences in tumour biology across ethnic groups can determine the medicine selected to treat a specific indication. The example of gefitinib for the treatment of non‐small cell lung cancer is presented in Figure 5.
FIGURE 4.
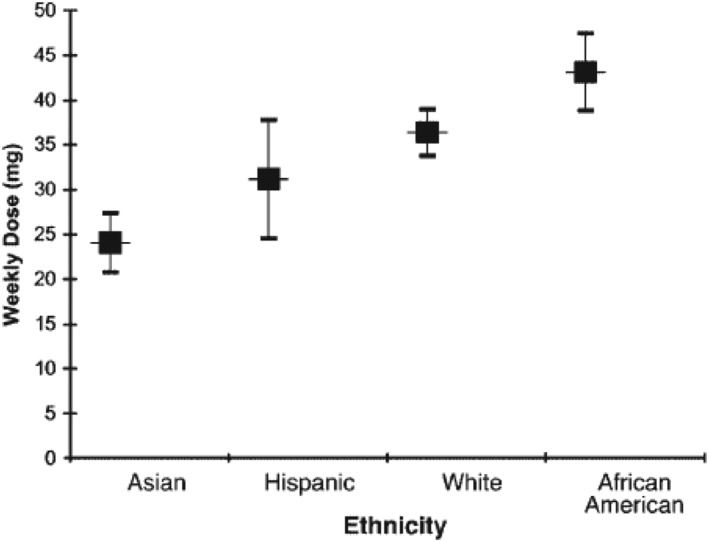
Adjusted mean weekly warfarin dose (95% confidence interval) in 345 patients in the USA of different ethnic groups resulting in a goal international normalised ratio of 3 to 4 (reproduced with permission from Dang et al., 2005) 43
FIGURE 5.
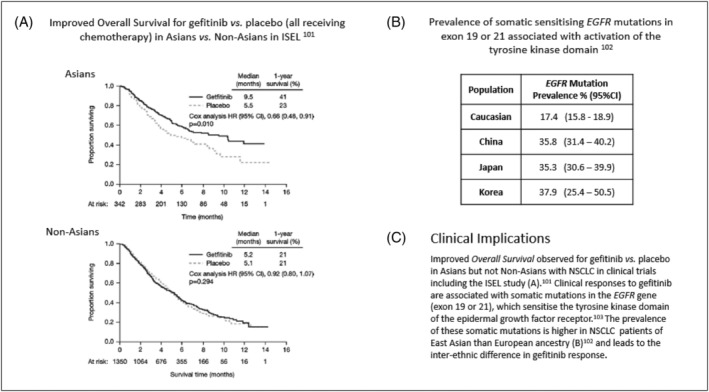
Gefitinib response in Asian and non‐Asian patients with non‐small cell lung cancer 103 and the prevalence of somatic EGFR mutations 104 associated with tyrosine kinase responses. 105 (Figure reproduced with permission from Chang et al., 2006) 103
Factors that can contribute to interethnic differences in response are discussed in the ICH E5 guideline Ethnic Factors in the Acceptability of Foreign Clinical Data 1 and relevant literature. 44 , 45 Interethnic differences in response to a medicine are observed when there are population differences in the factors leading to interindividual variation in the response measured. Consequently, it is important to understand the drivers of a medicines' PK and PD as well as the efficacy and safety reported, and the distribution of these factors across populations. Both intrinsic factors related to an individual (e.g. body weight, drug metabolising enzymes, genetics) and extrinsic factors related to their environment (e.g. medical practice including concomitant drugs, rating scales used to measure drug response) can be important and are identified and profiled across populations of interest when the ethnic sensitivity of a medicine is assessed. This process is critical, as whenever possible, associations need to be disentangled from causal correlations. For example, for many monoclonal antibodies, interethnic differences in exposure can be attributed to population differences in body weight distribution rather than any differences in biology. 46
Given the large number of diverse populations worldwide, establishing populations with a similar spectrum of intrinsic and extrinsic factors and anticipated medicine response, could support pooling data from prespecified ethnic groups. Larger subject numbers would provide more meaningful assessments of efficacy and safety than smaller subgroups 2 and could facilitate obtaining clinical insights for a broader spectrum of ancestry groups world‐wide. However, understanding sources of variability may be facilitated by conducting larger clinical studies, or multiple smaller targeted studies, along with in silico modelling. Simulation and extrapolation approaches also play an important, complementary role in characterising interethnic differences.
2.3. Sex and gender
The sex (determined by genetic and other biological factors) and gender (comprising the social, environmental, cultural, and behavioural factors and choices that influence a person's self‐identity) of an individual have also been associated with variability in medicine response. Such a variation may be explained by differences in underlying pathophysiology as well as PK and PD. Sex‐related differences in PK have been extensively reviewed and are attributable to factors including the influence of lower average body weight, lower glomerular filtration rate, gut physiology, different body composition (e.g. lower lean body mass, higher adipose tissue) as well as sex hormone effects in women relative to men. 47 , 48 , 49 , 50 , 51 These factors can result in sex‐related differences in volume of distribution or clearance and consequently exposure of a medicine as well as efficacy and safety.
Many diseases have well established differences in prevalence, severity or spectrum of symptoms in women relative to men, which may also lead to differences in the response to treatment. For example, immune responses are generally higher in females than males, contributing to the 2–10‐fold higher incidence and severity of many autoimmune conditions in women relative to men. 48 , 52 Similarly, for several cardiovascular conditions such as acute coronary syndrome and non‐ST‐elevation myocardial infarction, there are well‐established differences in prevalence, symptomatic presentation and disease management, which are influenced by both sex (underlying cardiovascular physiology) and gender (socioeconomic status, physician perception). 53 , 54
An important sex/gender difference is the nearly 2‐fold greater risk of women than men exhibiting adverse events across all medicine classes. Furthermore, women are significantly more likely to be hospitalised secondary to an adverse drug reaction. 50 , 55 The trend to higher rates of adverse events is not restricted to any particular category of adverse events, or to any specific class of therapy. 50 The higher incidence of drug‐related adverse events in women relative to men may be explained, at least in part, by higher rates of prescribing to women, 50 or adverse events that are unique to women (e.g. increased vaginal bleeding associated with anticoagulants). 49
The higher rates of adverse drug reactions in women than men, highlights the importance of appropriate sex or gender representation in clinical trials. Historically women have been underrepresented in clinical trials, partially due to US guidelines (now superseded), which explicitly excluded women of childbearing potential from early phase clinical trials. 56 Based on the most recent data, women do not appear to be under‐represented in clinical trials. Specifically, of the 48 novel drugs approved by the FDA in 2019, 72% of participants in the pivotal clinical trials were female. 32 However, this 2019 statistic is exceptional, reflecting the relatively high proportion of approvals (6 of 49) for female‐only indications (e.g. postmenopausal osteoporosis, postpartum depression). More broadly, participation of females in both academic and industry‐sponsored clinical trials has progressively increased in recent years. 57 , 58 Nevertheless, female participants are under‐represented in early phase trials 58 , 59 and in later phase trials of certain indications relative to the global prevalence in women (e.g. HIV, chronic kidney disease). 57
Whilst progress has been made regarding the contribution of sex‐related differences to variability in treatment response, there are still important gaps to be addressed with regard to gender‐related effects. For example, despite increasing interest in transgender healthcare research 60 the ICH and US FDA do not currently provide guidelines addressing transgender or intersex individuals. Furthermore, pregnancy and lactation can influence drug disposition; however, pregnant and lactating women have generally been excluded from clinical trials. Therefore, at approval, there is usually a paucity of information to guide appropriate dosing of a new drug in pregnant or lactating women. 61
The Sex and Gender Equity in Research Guidelines 62 published in 2016 with representation from 8 countries provide an international set of guidelines intended to encourage a more systematic approach to the reporting of sex and gender in research across scientific disciplines. Country‐specific guidelines are also in place. 62 The international guidelines endorse the reporting of both sex and gender in scientific research and that the method by which sex is assigned in a trial should be reported (e.g. genetic test, self‐reported). 62 Further detail on sex and gender definitions is provided in the Supporting Information. In many clinical trials, it is not appropriate/ethical to conduct genetic evaluations to determine the sex of a participant. In practice, when asked to report “sex assigned at birth”, the percentage of individuals with inaccurate reporting relative to genetic testing will probably be low, allowing self‐reporting of sex to be an acceptable surrogate. 63
3. GLOBAL DRUG DEVELOPMENT
3.1. Clinical pharmacology perspectives
Clinical pharmacology studies are aimed at establishing the dose rationale taking into account the impact of factors known to cause variability in treatment response. Therefore, the clinical pharmacology programme offers an opportunity for efficient data generation, providing insight into the underlying exposure–response relationship along with intrinsic and extrinsic sources of variability, which may affect treatment response and consequently result in dose adjustment. Identifying factors that determine interindividual variability occurs throughout clinical development from first‐in‐human studies up to Phase 3, when large patient cohorts are exposed to a new treatment at the anticipated therapeutic dosing regimen. Initiatives to increase efficiency through global programmes have highlighted questions for early clinical development: are the demographics of participants in early phase studies representative of the patient population and are the findings generalisable to other populations?
Historically, the scope of Phase I trials has been to assess the PK, safety and tolerability of a new molecule and today's consensus has shifted to include PD as well as evidence of target engagement. Whilst study protocols are usually too small to understand drivers of variation using standard trial designs, this should not preclude inclusion of diverse populations. Efforts are needed to ensure data arising from different groups in the population are evaluated in an integrated manner throughout the development programme. Stratification and randomisation techniques can be applied to ensure balanced data collection across dose levels and treatment arms, so that baseline demographic and clinical factors such as age, geographic ancestry, sex/gender, genotype or phenotype can be evaluated and explored during (covariate) analyses. Further research investigating personalised drug dosing and responses, based on these and additional factors, will inform selection of the population characteristics to guide dosing and be investigated in future clinical studies.
It is worth noting that 2 ICH guidelines are of particular relevance when considering diversity in global clinical trials: ICH E5 and ICH E17. 1 , 2 The ICH E5 guideline Ethnic Factors in the Acceptability of Foreign Clinical Data, describes the importance of determining the intrinsic and extrinsic factors that can influence drug response and lead to population differences in response to therapy. Even though this guideline was finalised in 1998 and knowledge of factors influencing PK and PD has evolved since that time, the basic principles outlined by ICH E5 are still applicable. As concentrations are important drivers of variability in the dose–response relationship, 64 identifying the intrinsic and extrinsic factors driving absorption, distribution, metabolism and excretion such as drug metabolising enzymes and transporters, and knowledge of their relative activity across ethnic groups, can indicate subgroups warranting further investigation (Figure 1). Moreover, understanding the important intrinsic and extrinsic factors influencing PK can also indicate whether age and sex differences in exposure may be observed.
More recently, ICH E17, General Principles for Planning and Design of Multi‐regional Clinical Trials, 2 has been adopted. It provides guidance for performing multiregional clinical trials (MRCTs) including considerations related to potential population differences in response. ICH E17 advocates that the early phase of clinical development identifies factors that may contribute to variation in PK and PD across populations, which can then be evaluated in larger later phase clinical trials in diverse populations. However, understanding the clinical relevance of the effect of diversity on the overall treatment response requires an iterative approach, with a mix of hypothesis‐generating and hypothesis‐testing leading to ongoing identification and quantification of sources of interindividual variation. 65 Implementation of such an iterative approach implies early understanding, or assumption of the role of, potential influential factors across populations that would justify further investigation to confirm their clinical relevance. 66 , 67
ICH E17 indicates that treatment effects across all regions of an MRCT be evaluated, along with the consistency of treatment effects among the regions. Structured analyses should also be performed to confirm the contribution of specific influential factors to any regional differences identified. These principles signify the importance of the accurate characterisation of factors influencing variability in medicine response early in development and highlight the importance of clinical trial participant diversity.
The statistical methods applied for the assessment of consistency are the subject of many scientific papers. The Guideline on the Investigation of Subgroups in Confirmatory Clinical Trials 68 issued by the EMA in 2019 explicitly addresses issues related to subgroup analyses and nominates age, gender and ethnicity as appropriate categories to evaluate. In the USA, marketing authorisation applications to the FDA are required to include effectiveness and safety results not only for the overall population but also stratified by sex/gender, age and racial subgroups in order to identify any modifications of dose or dose interval needed for specific subgroups. 69
Therefore, clinical trial diversity is essential to allow identification of potential differences in drug response between subpopulations. However, it is important to note that not all differences identified will be of sufficient magnitude to be of clinical relevance. Specifically, interindividual differences may or may not require a change in dose, dosing regimen or clinical management of patients in a specific subpopulation. In fact, Ramamoorthy et al. report that approximately 1/5 of the new molecular entities approved by the US FDA between 2008 and 2013 reported an interethnic or racial difference in the Product Information. Most differences reported were PK and in nearly all cases the magnitude observed did not require dose adjustment. 38 These findings are in line with the ICH E17 guideline, which indicates that interpretation of any population differences should be based on clinical rather than statistical significance. 2
3.2. Inclusiveness and appropriate participant representation
Whereas the inclusion of predefined cohorts of diverse participants during Phase I can offer insight into potential influential factors, attention has primarily been given to Phase 2/3 trials, which should ensure enrolment of participants representative of the demographics of the patients who will ultimately use the therapy under investigation. 6 As many medical conditions are more common in specific subgroups (e.g. older age) the demographics of the clinical trial participants should reflect the patient profile rather than overall demographics of the population. For example, in the USA, multiple myeloma (MM) is more prevalent in men and the elderly 70 and 20% of MM patients are of African American ancestry, whereas census data indicate that Black or African American individuals represent approximately 13% of the US population. 71 However, African Americans are under‐represented in MM clinical trials. 72
A review of submissions to the US FDA's Division of Haematology Products between 2003 and 2017 reported that the median percentage of Blacks enrolled in pivotal MM trials was 4.5%. 72 In trials that had low enrolment in the USA, the median proportion of Blacks was 1.8%, whereas in trials with high enrolment in the USA, the median representation was 10.5%. 72 This observation reflects the reality that the national population distribution by race/ethnicity, as well as by age and sex/gender, is not the same in every country. The demographic distribution of patients who are most likely to use a medicine in 1 country is unlikely to be identical to the demographic distribution of patients in another country. In the context of the design of MRCT, this raises the question of defining the reference population to be enrolled in order to ensure that the patients recruited reflect the population most likely to use the therapy.
For example, if an MM trial recruits participants only in the USA, ideally 20% of participants should be of African American ancestry to reflect the demographics of MM patients in the USA. 70 However, if an MRCT is planned, countries will be included with much lower proportions of patients of African ancestry such as in Europe (<2%), 73 , 74 Japan (<1%) 75 and China (<1%). 75 Consequently, 2 options for the target population of the MRCT can be considered, as illustrated in Figure 6.
FIGURE 6.

Regional participation in a hypothetical multiregional clinical trial enrolling 1000 participants allocated by geographic region (USA n = 200, Europe n = 400, China n = 200, Japan n = 200). The aim is to ensure 20% of participants are of African American ancestry
Option 1 ensures that the proportion of African American participants in the entire trial reflects the proportion of African Americans in the USA. Option 2 ensures that the proportion of participants in the US subset of the trial reflects the proportion of African American patients in the USA. In Option 1, patients of African ancestry will represent 20% of the total trial population and could constitute the entire cohort of participants recruited in the USA. Treatment effects will not be studied in participants of non‐African ancestry in the USA. Therefore if a difference in treatment response (efficacy or safety) is observed in the US cohort relative to the patients studied outside the USA, it will be challenging to determine if this reflects differences due to ancestry or local medical practice in US vs. non‐US regions. 76 In Option 2, the proportion of participants of African ancestry reflects the US profile (20% of the US allocation) but only 4% of the total trial population, potentially limiting interpretation of results in the African American subgroup.
In theory, a third option to achieve appropriate representation of participants of African ancestry would enrol MM patients in Africa. However, there are additional issues that must be considered if individuals resident in sub‐Saharan Africa are to be considered representative of African Americans in the USA. There is great diversity among the populations of sub‐Saharan Africa, with differences in the intrinsic and extrinsic factors that can influence response, including the genetics of ADME genes. 77
This example of African American representation in MM clinical trials has been simplified to highlight the challenges involved when considering just 1 of the many factors to consider when planning clinical trials to reflect diversity targets. In practise, individuals from multiple, overlapping subgroups of race/ethnicity, sex/gender and age represent a potential clinical phenotype and should be considered when defining the rationale for appropriate representation of the target patient population in a clinical trial. From a scientific perspective, decisions on the target population required to attain diversity targets in a Phase 3 MRCT can also be informed by the knowledge of factors identified in early phase trials to influence PK and PD and in turn the potential to pool results in prespecified populations. 2 This approach should be complemented by quantitative clinical pharmacology methods, which can be used to explore the implications of baseline clinical and demographic characteristics on the PK, PD, safety and efficacy of new molecules. 27
The challenge to ensure adequate representation remains a clinical and scientific rather than a regulatory endeavour. Local differences in epidemiology and clinical management will need to be considered during drug development as well as after drug approval. In addition to potential enrichment approaches, regulatory and clinical decision making on questions regarding the implications of intrinsic and/or extrinsic factors influencing treatment response can be complemented by alternative methods. Evolving technologies, such as clinical trial simulation can be performed including virtual patient cohorts with the relevant characteristics to explore treatment performance, considering not only individual covariates, but also clinical scenarios.
In addition to these challenges, the planning of a clinical trial must balance the appropriate patient representation against other practical considerations such as the speed of enrolment in different geographic regions and local regulatory requirements. Some regulatory agencies request representation of nominated proportions of participants from their local population in pivotal MRCTs. For example, the Pharmaceuticals and Medical Devices Agency guidance presents methods to calculate the proportion of participants in Japan required to assess the consistency of efficacy results in the Japan subgroup relative to the other regions studied. 78 If there is a concern that efficacy or safety responses have not been adequately characterised in a particular subgroup of a Phase 3 clinical trial, a smaller trial specifically targeted to enrich the subgroup of concern could be conducted. Postapproval commitments to conduct clinical trials in specific populations (e.g. African Americans) or the establishment of a registry to monitor outcomes in patients prescribed a new medicine after approval may be alternate paths to provide information on the efficacy and safety profile and appropriate doses in specific populations.
4. DIVERSE CLINICAL TRIAL REPRESENTATION: BARRIERS AND OPPORTUNITIES
Research, systematic reviews, surveys and consortia conducted over the past several decades, have offered not only a good understanding of the barriers to the inclusion of diverse populations in clinical trials but also approaches that have successfully increased participant diversity. 79 To ensure appropriate representation of relevant race/ethnic groups as well as sex/gender ratios and to increase the inclusion of older patients in clinical trials, a range of strategies have been proposed. These strategies focus on the sponsor/academic investigator, the clinical trial site (e.g. staff, referring health care providers) and potential trial participants. Ongoing commitment from all stakeholders is required to ensure each trial and clinical trial programme provides information that is generalisable to the targeted patient population.
4.1. Barriers to enrolment
Important barriers to the enrolment of diverse populations in clinical trials from the perspective of the individual participant, clinical trial sites and the sponsor/academic investigator are summarised in Table 2. In addition to similar factors having been identified as barriers to clinical trial participation for ethnic minorities, women and the elderly, 80 there are also factors that are more relevant for specific groups such as child care access for women who are primary care givers, or accommodating mobility and health issues for the elderly. 81 , 82 A recent literature review, along with key stakeholder input, has reported that critical barriers to diverse participation could be categorised as mistrust, lack of comfort with the clinical trial process, lack of information about clinical trials, time and resource constraints associated with participation and lack of awareness of the existence of clinical trials. 11 , 83 Mistrust is important in older populations as they can feel vulnerable to potential exploitation. 82 From the perspective of ancestry, in the USA it has been reported that mistrust relates to a fear of purposeful mistreatment (e.g. US Public Health Service Tuskegee Syphilis Experiment conducted in the USA and Guatemala), unknown research procedures, and unintended consequences. 83 , 84 Poor communication and cultural barriers have also been identified as limiting clinical trial participation. 11 , 82
TABLE 2.
Barriers to demographically diverse and clinically relevant clinical trial enrolment in relation to the participants, clinical trial sites and the Sponsor/Academic Investigator
| Barriers | ||
|---|---|---|
| Participants | Clinical trial sites | Sponsor/academic investigator |
| Lack of trust in pharmaceutical industry and medical researchers, and fears of exploitation | Limited commitment and effort | Limited commitment and effort |
| Practical obstacles to participation and inconvenience | Unconscious bias | Low willingness to work with research naïve sites/investigators |
| Lack of awareness/information about disease and trials | Lack of culturally or racially/ethnically diverse staff | Limited understanding of what a potential participant wants or needs to enrol |
| Low health literacy | Lack of effective referral basis/ health care providers fear of losing patient | Negative attitudes about minority willingness |
| Lack of access to clinical trial sites | Lack of community engagement | Assumptions that diverse enrolment would conflict with trial efficiency |
| Language barriers | Lack of knowledge of cultural differences leading to ineffective communication | Costs and potential time delays associated with engagement |
4.2. Opportunities to enhance participation
4.2.1. Participant involvement
As mistrust has been reported to be a common barrier to clinical trial participation, building trust is important to facilitate the enrolment and retention of diverse participants in clinical trials. Community outreach including engaging community and patient advocacy groups or advisory boards has been demonstrated to be beneficial in building trust, and it is recommended to ensure these relationships are maintained beyond the time period relevant for a specific clinical trial. 85 Communication and education including information sessions, whether culturally or age appropriate, can build awareness and address scepticism of the clinical research process. 79 While mistrust and fear of exploitation persists, it can be overcome, as indicated by the high proportion of respondents from a survey of diverse populations in the USA who would be likely to participate in a clinical trial if asked. 86 , 87
4.2.2. Diversity as best practice at clinical trial sites
Clinical trial sites that have successfully recruited diverse populations have been surveyed to build awareness of best practices. 85 , 88 Commitment to diversity by the trial site leadership is of primary importance. 84 , 88 Some of the initiatives associated with successful enrolment of diverse participant populations 11 , 82 in relation to important barriers to participation are presented in Table 3.
TABLE 3.
Initiatives associated with successful enrolment of diverse participant populations to address barriers identified 11 , 82
| Barrier | Initiatives associated with successful enrolment |
|---|---|
| Mistrust |
|
| Lack of comfort with the process |
|
| Lack of information |
|
| Time and resource constraints |
|
| Lack of awareness |
|
4.2.3. Referring clinicians
Clinicians have an important role to play alerting patients of clinical trials for which they may be eligible. Indeed, a public poll in the USA indicated that 72% of Americans would be to participate in a clinical trial if recommended by their doctor, but that a doctor or other health care professional had only mentioned clinical research to 22% of the cohort. 89 However, some qualified patients may not be provided the opportunity to participate in a clinical trial due to unconscious bias by referring clinicians. 90 To address this barrier to clinical trial participation, assumptions relating to availability to participate (e.g. mobility) must be overcome. 82
4.2.4. Sponsor or academic investigator
The sponsor of a clinical trial or an academic investigator can contribute to improving the diversity of clinical trial participants by addressing issues related to the protocol and clinical trial site selection as well as their commitment to inclusive research.
Protocol
The inclusion and exclusion criteria nominated in the protocol of a clinical trial may limit the diversity of the participants enrolled. For example, arbitrary maximum ages for participation and exclusion of comorbidities and associated concomitant medications can limit inclusion of older participants. Actively questioning and broadening inclusion criteria could enable more older participants to enrol in clinical trials. 91
Ensuring that patient perspectives are considered during protocol development can result in trial designs that facilitate participation of diverse populations. Patient representatives, advocates and caregivers can provide insight on how a disease affects an individual and their treatment challenges from a diverse range of patient perspectives. The level of participant burden associated with a proposed protocol design can be reviewed in order to optimise the frequency of trial visits and assessments. Electronic resources to facilitate remote data capture and trial site interactions could also enable participation of patients who do not reside close to a clinical trial site or whose symptoms may limit travel.
Protocols can include targets for sex/gender representation, participant age distribution as well as inclusion of participants from diverse ancestry groups and geographic regions. This approach must be supported by a strong commitment to diversity, as standard approaches may result in quicker recruitment but a less diverse demographic profile of clinical trial participants.
Site selection
The sponsor/academic investigator has a critical role in selecting the clinical sites where a trial will be conducted. For early phase clinical pharmacology trials, clinical trial units can be selected with experience in recruiting participants with the specified demographic characteristics. However, in larger Phase 2/3 trials, which provide efficacy and safety data explored during exposure–response analyses, recruiting participants with specific demographic characteristics can be challenging.
While continuing with sites that have a record of clinical trial delivery assures an expected level of recruitment and retention efficiencies, they will deliver the same patient demographics as historically unless prompted to change. Therefore, increasing the diversity of clinical trial participants will often require expanding the pool of trial sites beyond that usually considered during assessment of site feasibility. Geospatial capabilities can be used to identify new clinical trial sites with ethnically diverse or elderly communities. Working with clinical sites and physicians with no clinical trial experience may indeed be effective, but may require additional time and cost to engage with the clinical trial team and potentially provide training.
Communication of recruitment goals for desired demographics and sharing of best practices by sponsors/academic investigators will build awareness and capabilities of the clinical trial site staff. In addition, the sponsor can discuss the recruitment plans, engagement with the local community, staff training in cultural competency and the provision of culturally, linguistically and age‐appropriate clinical trial information. This sponsor engagement is reported to be a driver for more successful recruitment of diverse populations. 11
Commitment to inclusive research
Prioritising an increase in the diversity of clinical trial populations requires commitment from all involved in clinical drug development or academic research. Leadership by each organisation is critical as the increased time and resources initially required to meet diversity targets could increase costs and potentially delay trial completion and regulatory submissions. It is an aspiration that, over time, with appropriate awareness raising, trial planning and strategy implementation, the inclusion of diverse populations will become part of standard practise. The ultimate goal is that diverse clinical trial populations could be enrolled without additional costs or delays to trial completion.
5. WHAT CAN WE DO NOW?
Early phase clinical pharmacology trials can be conducted to determine the contribution of demographic factors such as age, sex/gender and race/ethnicity to interindividual variation in drug PK and PD. Clinical pharmacology units can be selected with experience in enrolling participants with specific demographic profiles.
For Phase 2/3 trials, proactively considering the demographic profile of the patient population requiring treatment can provide a target to ensure a representative population is enrolled and retained. This can be achieved by initially understanding the epidemiology of the disease including its prevalence and incidence across age, sex/gender and geographic ancestry/ethnic subgroups. Real world evidence can provide important insights on additional factors to consider. For MRCTs, the potential to pool results in participant groups with similar profiles of intrinsic and extrinsic factors should be evaluated.
With the demographic goals for a clinical trial determined, trial sites can be selected to enrol participants in alignment with the representative patient population profile. Appropriate techniques to evaluate the dose–concentration–response relationship in the diverse subgroups of interest must also be selected. Assigning a clinical trial team member to champion attainment of diversity targets can reinforce the participant inclusion goals during trial planning, implementation and result evaluation.
To characterise responses in defined populations and confirm appropriate doses during the focussed timelines of a development programme can be a challenge, especially considering the diverse populations world‐wide. Registries and post marketing surveillance can collect information on efficacy and safety outcomes in local populations receiving a new medicine within the full spectrum of local intrinsic and extrinsic factors particular to that population. This approach can provide reassurance that global doses are appropriate for local populations or defined subgroups as well as indicate when dose adjustment may be required.
In summary, although it would be ideal to include all recognised patient subgroups and global populations in efficacy and safety clinical trials, this is clearly an impractical proposition. However, a commitment to address the following 3 steps provides a more realistic approach to balancing the ethical, scientific and practical considerations:
Invest early in trials characterising the drivers of PK and PD to help predict and plan for population subgroups that may respond differently from the average
Prioritise enrolment of diverse participants in clinical trials throughout the development process to allow for identification of subgroups with unanticipated differences in response
If signals of subgroup differences are identified in the larger sample sizes included in later phase trials, commit to investigate these subgroups further during the postmarketing phase.
5.1. Modelling, simulation and extrapolation
Whilst efforts to ensure data generation in representative populations are critical to overcome the evidence gap due to the lack of diversity in the data supporting marketing authorisations, obtaining this clinical data will often be fraught with operational challenges and sample size considerations. Therefore, results at the time of approval may not be easily generalisable to a broader population, especially if the global market is considered.
Modelling and simulation (M&S) can be used as a basis to assess the impact of patient diversity in a given indication and its clinical implications and used to help ensure the selection of appropriate dose(s)/regimen(s) in the target patient population. While a detailed overview of the application of M&S in drug development is out‐of‐scope, there are various publications providing relevant background information. 92 , 93 , 94 Here we emphasise the need to consider the concepts of bridging, extrapolation, and subgroup analysis to help generate appropriate information for safe/effective use of medicines across the overall patient population.
In this context, M&S can be used to bring together from a range of sources and disciplines (i.e. evidence synthesis) the available evidence on PK, PD, safety and efficacy (Figure 7). It also allows for systematic evaluation of uncertainty when data availability is limited. Model predictions can then be confirmed by conducting prospective (exploratory) bridging studies or, if deemed necessary, by investigating observational cohorts after drug approval. Such an approach does not only tackle the issues regarding population diversity, but also ensures treatment personalisation, where appropriate. The term extrapolation can be described as considering the use of information in 1 or more source populations (e.g. adult females) to be relevant to a target population (e.g. older females), in a way that can be quantified and used as a basis for clinical and regulatory decision making. 95 Ongoing debates on the importance of enhancing patient diversity in clinical trials should not overlook important lessons learned from the use of inferential, model‐based approaches for evidence synthesis in paediatric indications. 19 , 20
FIGURE 7.
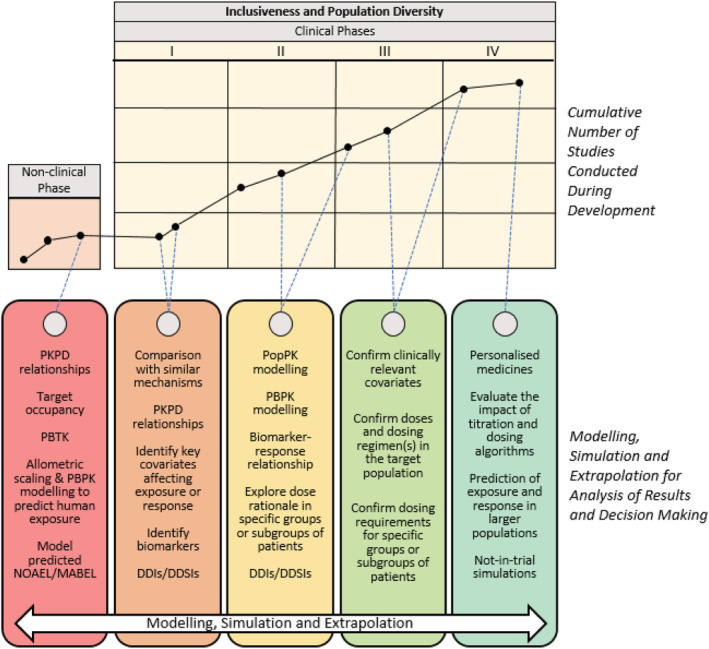
Illustration of an integrated drug development framework based on modelling, simulation and extrapolation concepts. In the upper section, each black circle within the nonclinical and clinical matrix represents a study conducted in a given phase of development. The solid black lines connecting the circles highlight that the data generated from each preceding study may be used to support and plan later studies, allowing for the creation of a comprehensive and contemporary body of evidence as the cumulative number of studies providing data grows. The dotted lines connecting the upper nonclinical/clinical matrix to the modelling and simulation tools described in the lower section indicate how such a framework can be used at each phase to analyse the emerging data and support decision making in subsequent stages of development (Adapted from Saeed et al. 27 ). DDIs, drug–drug interactions; DDSIs, drug–disease interactions; MABEL, minimal anticipated biological effect level; NOAEL, no observed adverse effect level; PBPK, physiologically based pharmacokinetics; PopPK, population pharmacokinetics; PBTK, physiology‐based toxicology; PKPD, pharmacokinetic–pharmacodynamic
5.1.1. Covariate effects and dosing recommendations
In addition to generating data that reflect population diversity, further insight can be obtained from extrapolation and simulation scenarios, including relevant covariates (intrinsic and extrinsic factors). The challenge is that influential covariates, which may affect the PKPD relationship or overall efficacy/safety profile of a given medicine, may not be identified in small clinical studies. This can be even more challenging when multiple factors contribute to variability and some may be correlated with each other (e.g. body weight and ethnicity, age and height). By contrast, the use of quantitative pharmacology methods may guide study design optimisation and support the characterisation of potentially important covariates influencing response. Insight into the magnitude of the effect of a given covariate can subsequently be obtained in confirmatory trials or during Phase 4. Simulated responses to treatment in subgroups of interest can support the planning of future Phase 2/3 trials in diverse populations and ensure that the clinical relevance of earlier observations can be confirmed. Furthermore, modelling allows exploration of patient characteristics, such as variability in age, sex and ethnicity/geographic ancestry, and can provide additional insights that it may not be possible to assess in a randomised controlled trial. 96 Similarly, physiologically based PK modelling may be used as a tool to simulate a variety of clinical scenarios and explore the influence of multiple factors (e.g. body weight and disease severity) in some subgroups. These simulations can identify key population characteristics requiring further focus and inform the design of later phase trials to confirm the anticipated effect of characteristics in key population subgroups. 64
5.1.2. Clinical trial simulations and extrapolation
Simulations, and in particular clinical trial simulations (CTS), are a critical component of a model‐based framework for extrapolation. In addition to providing insight into the interaction between drug‐specific properties and patient‐related characteristics, CTS allow for the evaluation of treatment context, including trial design, confounding (drop outs, inclusion vs. exclusion criteria) and other relevant factors which are known to affect treatment performance. 97 , 98 Most importantly, it allows the evaluation of multiple interacting factors in a systematic manner, which can be valuable to establish clinical relevance of interindividual differences due to diversity and heterogeneity in the patient population. 99 Consequently, CTS can be used as a tool for the exploration of suitable uncertainty mitigation methods for specific situations. Whilst there may be interest in a given group or individuals, diversity involves a matrix of characteristics, which may not always be represented in a study (e.g. Participant 1 may be male, Igbo [Western African], body mass index >30 kg/m2, age >65 y; Participant 2 may be female, African American, body mass index >30 kg/m2, age >75 y). Based on a predefined extrapolation plan, CTS offer the opportunity to explore PK, PD, safety and overall treatment performance in settings for which data availability is limited or lacking completely at the time of marketing authorisation. 100
6. CONCLUSION
There is clear evidence that demographic characteristics such as age, sex/gender and geographic ancestry/ethnicity have the potential to influence response to therapy. These demographics may be a surrogate for underlying factors, or clusters of factors, such as organ function, phenotype or genotype, that ultimately influence drug response. Nevertheless, in clinical practise, for a physician faced with making prescribing decisions for an individual patient of known age, sex and ancestry, an understanding of the clinical relevance of these demographics may be beneficial. It is therefore surprising, in this era of personalised medicine, how few clinical trials enrol an appropriately diverse range of participants, limiting the ability to establish the clinical relevance of interindividual differences in a given population or subgroups of patients who could require a different dose, dosing regimen or even intervention.
Increasing the diversity of clinical trial populations to reflect the patient population should be best practice in clinical drug development. Representative populations will allow a broader range of intrinsic and extrinsic factors to be explored, provide more precise estimates of the clinical relevance of covariate effects and the benefit: risk profile to be better characterised for relevant groups of the target population. These richer datasets will establish the need for tailored dosing and consequently recommendations for specific patient groups in the prescribing information.
In the context of global drug development and the ethical perspective of providing timely access to new therapies, representativeness can play a critical role in the implementation of MRCTs, as diversity will be interpreted differently across regions and countries. Clearly, the potential to pool data across regions in populations with similar characteristics further reinforces the need to consider diversity early in development.
It is imperative that industry sponsors and academic investigators embrace diversity in research and development to address the variability of individual patients treated. Clinical pharmacology has a fundamental role to play, from basic PK/PD profiling through to modelling and simulation and clinical trial design, to support the development of medicines with evidence to support appropriate dosing of all patients. This goal can be achieved by working together with patient populations traditionally underrepresented in clinical trials to break down barriers to clinical trial participation. It also requires consideration of strategic resources, such as the creation of integrated databases including relevant epidemiological and clinical characteristics for relevant diseases in diverse populations. Such data will enable potential discrepancies in patient representation in randomised clinical trials and real‐world settings to be identified.
COMPETING INTERESTS
The authors have no conflicts of interest to disclose in addition to their affiliation with GSK.
Supporting information
TABLE S1 Physiological changes of older age and potential impact on pharmacodynamics and clinical consequences (Adapted from Saeed et al., 2015). 1
TABLE S2 The minimum categories of Ethnicity and Race recommended to be collected for each clinical trial participant, by the US FDA Guidance on Collection of Race and Ethnicity Data in Clinical Trials, 2016.4 The definitions provided are also presented.
ACKNOWLEDGEMENTS
The contribution of Carwyn Davies to discussions on topics covered in this manuscript and the assistance of Nicki Kyriacou in manuscript preparation are acknowledged with thanks. Open access publishing facilitated by The University of Sydney, as part of the Wiley ‐ The University of Sydney agreement via the Council of Australian University Librarians.
Gross AS, Harry AC, Clifton CS, Della Pasqua O. Clinical trial diversity: An opportunity for improved insight into the determinants of variability in drug response. Br J Clin Pharmacol. 2022;88(6):2700-2717. doi: 10.1111/bcp.15242
REFERENCES
- 1. International Conference on Harmonisation of Technical Requirements for Registration of Pharmaceuticals for Human Use . ICH Harmonised Tripartite Guideline. ICH E5 (R1): Ethnic Factors in the Acceptability of Foreign Clinical Data. 1998. Accessed August 5, 2020. https://database.ich.org/sites/default/files/E5_R1__Guideline.pdf
- 2. International Council on Harmonisation of Technical Requirements for Pharmaceuticals for Human Use . ICH Harmonised Guidelines. General Principles for Planning and Design of Multi‐Regional Clinical Trials. ICH E17. 2017. Accessed October 6, 2020. https://database.ich.org/sites/default/files/E17EWG_Step4_2017_1116.pdf
- 3. Polo AJ, Makol BA, Castro AS, Colón‐Quintana N, Wagstaff AE, Guo S. Diversity in Randomized Clinical Trials of Depression: A 36‐Year Review. Clin Psychol Rev. 2019;67:22‐35. doi: 10.1016/j.cpr.2018.09.004 [DOI] [PubMed] [Google Scholar]
- 4. Khan MS, Shahid I, Siddiqi TJ, et al. Ten‐Year Trends in Enrollment of Women and Minorities in Pivotal Trials Supporting Recent US Food and Drug Administration Approval of Novel Cardiometabolic Drugs. J Am Heart Assoc. 2020;9(11):e015594. doi: 10.1161/jaha.119.015594 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Ruiter R, Burggraaf J, Rissmann R. Under‐Representation of Elderly in Clinical Trials: An Analysis of the Initial Approval Documents in the Food and Drug Administration Database. Br J Clin Pharmacol. 2019;85(4):838‐844. doi: 10.1111/bcp.13876 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. US Department of Health and Human Services Food and Drug Administration (US FDA) . Enhancing the Diversity of Clinical Trial Populations ‐ Eligibility Criteria, Enrollment Practices, and Trial Designs Guidance for Industry. 2020. Accessed November 5, 2021. https://www.fda.gov/media/127712/download
- 7. Mersha TB, Abebe T. Self‐Reported Race/Ethnicity in the Age of Genomic Research: Its Potential Impact on Understanding Health Disparities. Hum Genomics. 2015;9(1):1. doi: 10.1186/s40246-014-0023-x [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Institute of Medicine . Exploring the Biological Contributions to Human Health: Does Sex Matter? Washington, DC: The National Academies Press; 2001. doi: 10.17226/10028 [DOI] [PubMed] [Google Scholar]
- 9. Franconi F, Campesi I. Sex and Gender Influences on Pharmacological Response: An Overview. Expert Rev Clin Pharmacol. 2014;7(4):469‐485. doi: 10.1586/17512433.2014.922866 [DOI] [PubMed] [Google Scholar]
- 10. Mackenbach JP, Kulhánová I, Bopp M, et al. Variations in the Relation between Education and Cause‐Specific Mortality in 19 European Populations: A Test of the "Fundamental Causes" Theory of Social Inequalities in Health. Soc Sci Med. 2015;127:51‐62. doi: 10.1016/j.socscimed.2014.05.021 [DOI] [PubMed] [Google Scholar]
- 11. Clark LT, Watkins L, Piña IL, et al. Increasing Diversity in Clinical Trials: Overcoming Critical Barriers. Curr Probl Cardiol. 2019;44(5):148‐172. doi: 10.1016/j.cpcardiol.2018.11.002 [DOI] [PubMed] [Google Scholar]
- 12. Inman M, Daneman D, Curtis J, et al. Social Determinants of Health Are Associated with Modifiable Risk Factors for Cardiovascular Disease and Vascular Function in Pediatric Type 1 Diabetes. J Pediatr. 2016;177:167‐172. doi: 10.1016/j.jpeds.2016.06.049 [DOI] [PubMed] [Google Scholar]
- 13. Shahu A, Herrin J, Dhruva SS, et al. Disparities in Socioeconomic Context and Association with Blood Pressure Control and Cardiovascular Outcomes in Allhat. J Am Heart Assoc. 2019;8(15):e012277. doi: 10.1161/jaha.119.012277 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Napoles A, Cook E, Ginossar T, Knight KD, Ford ME. Chapter Four ‐ Applying a Conceptual Framework to Maximize the Participation of Diverse Populations in Cancer Clinical Trials. In: Ford ME, Watson DK, eds. Adv Canver Res. Academic Press; 2017:77‐94. doi: 10.1016/bs.acr.2016.08.004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. de Brey C, Musu L, McFarland J, et al. Status and Trends in the Education of Racial and Ethnic Groups 2018 (NCES 2019–038). U.S. Department of Education. Washington, DC: National Center for Education Statistics; 2019. Accessed October 11, 2021. https://nces.ed.gov/pubs2019/2019038.pdf [Google Scholar]
- 16. United Nations Department of Economic and Social Affairs Population Division . World Population Ageing 2019 (ST/ESA/SER.A/444). 2020. Accessed August 31, 2020. https://www.un.org/en/development/desa/population/publications/pdf/ageing/WorldPopulationAgeing2019-Report.pdf
- 17. International Conference of Harmonisation of Technical Requirements for Registration of Pharmaceuticals for Human Use . Final Concept Paper E7(R1): Studies in Support of Special Populations: Geriatrics (Revision of the ICH E7 Guideline). 2008. Accessed August 5, 2020. https://database.ich.org/sites/default/files/E7_Q%26As_Concept_Paper.pdf
- 18. US Department of Health and Human Services Food and Drug Administration (US FDA) . Guidance for Industry: Guideline for the Study of Drugs Likely to Be Used in the Elderly. 1989. Accessed August 5, 2020. https://www.fda.gov/media/71114/download
- 19. Bellanti F, Della PO. Modelling and Simulation as Research Tools in Paediatric Drug Development. Eur J Clin Pharmacol. 2011;67(Suppl 1):75‐86. doi: 10.1007/s00228-010-0974-3 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Harnisch L, Shepard T, Pons G, Della PO. Modeling and Simulation as a Tool to Bridge Efficacy and Safety Data in Special Populations. CPT: Pharmacomet Syst Pharmacol. 2013;2(2):e28. doi: 10.1038/psp.2013.6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Ollivier C, Thomson A, Manolis E, et al. Commentary on the EMA Reflection Paper on the Use of Extrapolation in the Development of Medicines for Paediatrics. Br J Clin Pharmacol. 2019;85(4):659‐668. doi: 10.1111/bcp.13883 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Vitale C, Fini M, Spoletini I, Lainscak M, Seferovic P, Rosano GM. Under‐Representation of Elderly and Women in Clinical Trials. Int J Cardiol. 2017;232:216‐221. doi: 10.1016/j.ijcard.2017.01.018 [DOI] [PubMed] [Google Scholar]
- 23. Moore KL, Patel K, Boscardin WJ, Steinman MA, Ritchie C, Schwartz JB. Medication Burden Attributable to Chronic Co‐Morbid Conditions in the Very Old and Vulnerable. PLoS One. 2018;13(4):e0196109. doi: 10.1371/journal.pone.0196109 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Buechel M, McGinnis A, Vesely SK, Wade KS, Moore KN, Gunderson CC. Consideration of Older Patients for Enrollment in Phase 1 Clinical Trials: Exploring Treatment Related Toxicities and Outcomes. Gynecol Oncol. 2018;149(1):28‐32. doi: 10.1016/j.ygyno.2017.11.021 [DOI] [PubMed] [Google Scholar]
- 25. Mangoni AA, Jansen PA, Jackson SH. Under‐Representation of Older Adults in and Pharmacodynamic Studies: A Solvable Problem? Expert Rev Clin Pharmacol. 2013;6(1):35‐39. doi: 10.1586/ecp.12.75 [DOI] [PubMed] [Google Scholar]
- 26. McLachlan AJ, Hilmer SN, Le Couteur DG. Variability in Response to Medicines in Older People: Phenotypic and Genotypic Factors. Clin Pharmacol Ther. 2009;85(4):431‐433. doi: 10.1038/clpt.2009.1 [DOI] [PubMed] [Google Scholar]
- 27. Saeed MA, Vlasakakis G, Della PO. Rational Use of Medicines in Older Adults: Can We Do Better During Clinical Development? Clin Pharmacol Ther. 2015;97(5):440‐443. doi: 10.1002/cpt.87 [DOI] [PubMed] [Google Scholar]
- 28. Crome P, Lally F, Cherubini A, et al. Exclusion of Older People from Clinical Trials: Professional Views from Nine European Countries Participating in the Predict Study. Drugs Aging. 2011;28(8):667‐677. doi: 10.2165/11591990-000000000-00000 [DOI] [PubMed] [Google Scholar]
- 29. Beers E, Moerkerken DC, Leufkens HGM, Egberts TCG, Jansen PAF. Participation of Older People in Preauthorization Trials of Recently Approved Medicines. J Am Geriatr Soc. 2014;62(10):1883‐1890. doi: 10.1111/jgs.13067 [DOI] [PubMed] [Google Scholar]
- 30. van Marum RJ. Underrepresentation of the Elderly in Clinical Trials, Time for Action. Br J Clin Pharmacol. 2020;86(10):2014‐2016. doi: 10.1111/bcp.14539 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Liberopoulos G, Trikalinos NA, Ioannidis JP. The Elderly Were under‐Represented in Osteoarthritis Clinical Trials. J Clin Epidemiol. 2009;62(11):1218‐1223. doi: 10.1016/j.jclinepi.2008.12.009 [DOI] [PubMed] [Google Scholar]
- 32. US Department of Health and Human Services Food and Drug Administration (US FDA) . 2019 Drug Trials Snapshots Summary Report. 2020. Accessed August 5, 2020. https://www.fda.gov/media/135337/download
- 33. US Department of Health and Human Services Food and Drug Administration (US FDA) . Inclusion of Older Adults in Cancer Clinical Trials Guidance for Industry (Draft Guidance). 2020. Accessed August 5, 2020. https://www.fda.gov/media/135804/download
- 34. Bourgeois FT, Olson KL, Tse T, Ioannidis JP, Mandl KD. Prevalence and Characteristics of Interventional Trials Conducted Exclusively in Elderly Persons: A Cross‐Sectional Analysis of Registered Clinical Trials. PLoS One. 2016;11(5):e0155948. doi: 10.1371/journal.pone.0155948 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Broekhuizen K, Pothof A, de Craen AJ, Mooijaart SP. Characteristics of Randomized Controlled Trials Designed for Elderly: A Systematic Review. PLoS One. 2015;10(5):e0126709. doi: 10.1371/journal.pone.0126709 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Mariano C, Francl M, Pope J, Wong L, Lim HJ, Lohrisch C. Comparison of Toxicity Experienced by Older Versus Younger Patients Enrolled in Breast Cancer Clinical Trials. Clin Breast Cancer. 2015;15(1):73‐79. doi: 10.1016/j.clbc.2014.09.002 [DOI] [PubMed] [Google Scholar]
- 37. O'Donnell PH, Dolan ME. Cancer Pharmacoethnicity: Ethnic Differences in Susceptibility to the Effects of Chemotherapy. Clin Cancer Res. 2009;15(15):4806‐4814. doi: 10.1158/1078-0432.Ccr-09-0344 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Ramamoorthy A, Pacanowski MA, Bull J, Zhang L. Racial/Ethnic Differences in Drug Disposition and Response: Review of Recently Approved Drugs. Clin Pharmacol Ther. 2015;97(3):263‐273. doi: 10.1002/cpt.61 [DOI] [PubMed] [Google Scholar]
- 39. US Department of Health and Human Services Food and Drug Administration (US FDA) . Collection of Race and Ethnicity Data in Clinical Trials, Guidance for Industry and Food and Drug Administration Staff. October, 2016. Accessed August 5, 2020. https://www.fda.gov/media/75453/download
- 40. Hayes S, Ouellet D, Zhang J, Wire MB, Gibiansky E. Population PK/PD Modeling of Eltrombopag in Healthy Volunteers and Patients with Immune Thrombocytopenic Purpura and Optimization of Response‐Guided Dosing. J Clin Pharmacol. 2011;51(10):1403‐1417. doi: 10.1177/0091270010383019 [DOI] [PubMed] [Google Scholar]
- 41. US Department of Health and Human Services Food and Drug Administration (US FDA) . Promacta (Eltrombopag) Prescribing and Labeling Information. 2008. Accessed October 6, 2021. https://www.accessdata.fda.gov/drugsatfda_docs/label/2008/022291lbl.pdf
- 42. Yang R, Li J, Jin J, et al. Multicentre, Randomised Phase III Study of the Efficacy and Safety of Eltrombopag in Chinese Patients with Chronic Immune Thrombocytopenia. Br J Haematol. 2017;176(1):101‐110. doi: 10.1111/bjh.14380 [DOI] [PubMed] [Google Scholar]
- 43. Dang MT, Hambleton J, Kayser SR. The Influence of Ethnicity on Warfarin Dosage Requirement. Ann Pharmacother. 2005;39(6):1008‐1012. doi: 10.1345/aph.1E566 [DOI] [PubMed] [Google Scholar]
- 44. Huang SM, Temple R. Is This the Drug or Dose for You? Impact and Consideration of Ethnic Factors in Global Drug Development, Regulatory Review, and Clinical Practice. Clin Pharmacol Ther. 2008;84(3):287‐294. doi: 10.1038/clpt.2008.144 [DOI] [PubMed] [Google Scholar]
- 45. Phan VH, Tan C, Rittau A, Xu H, McLachlan AJ, Clarke SJ. An Update on Ethnic Differences in Drug Metabolism and Toxicity from Anti‐Cancer Drugs. Expert Opin Drug Metab Toxicol. 2011;7(11):1395‐1410. doi: 10.1517/17425255.2011.624513 [DOI] [PubMed] [Google Scholar]
- 46. Chiba K, Yoshitsugu H, Kyosaka Y, et al. A Comprehensive Review of the Pharmacokinetics of Approved Therapeutic Monoclonal Antibodies in Japan: Are Japanese Phase I Studies Still Needed? J Clin Pharmacol. 2014;54(5):483‐494. doi: 10.1002/jcph.231 [DOI] [PubMed] [Google Scholar]
- 47. Anderson GD. Sex and Racial Differences in Pharmacological Response: Where Is the Evidence? Pharmacogenetics, Pharmacokinetics, and Pharmacodynamics. J Womens Health (Larchmt). 2005;14(1):19‐29. doi: 10.1089/jwh.2005.14.19 [DOI] [PubMed] [Google Scholar]
- 48. Soldin OP, Mattison DR. Sex Differences in Pharmacokinetics and Pharmacodynamics. J Clin Pharmacol. 2009;48(3):143‐157. doi: 10.2165/00003088-200948030-00001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Ferner R, Aronson J. Susceptibility to Adverse Drug Reactions. Br J Clin Pharmacol. 2019;85(10):2205‐2212. doi: 10.1111/bcp.14015 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Zucker I, Prendergast BJ. Sex Differences in Pharmacokinetics Predict Adverse Drug Reactions in Women. Biol Sex Differ. 2020;11:32. doi: 10.1186/s13293-020-00308-5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Freire AC, Basit AW, Choudhary R, Piong CW, Merchant HA. Does Sex Matter? The Influence of Gender on Gastrointestinal Physiology and Drug Delivery. Int J Pharm. 2011;415(1–2):15‐28. doi: 10.1016/j.ijpharm.2011.04.069 [DOI] [PubMed] [Google Scholar]
- 52. Fischinger S, Boudreau CM, Butler AL, Streeck H, Alter G. Sex Differences in Vaccine‐Induced Humoral Immunity. Semin Immunopathol. 2019;41(2):239‐249. doi: 10.1007/s00281-018-0726-5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Humphries KH, Izadnegahdar M, Sedlak T, et al. Sex Differences in Cardiovascular Disease ‐ Impact on Care and Outcomes. Front Neuroendocrinol. 2017;46:46‐70. doi: 10.1016/j.yfrne.2017.04.001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Mouat MA, Coleman JLJ, Smith NJ. Gpcrs in Context: Sexual Dimorphism in the Cardiovascular System. Br J Pharmacol. 2018;175(21):4047‐4059. doi: 10.1111/bph.14160 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. de Vries ST, Denig P, Ekhart C, et al. Sex Differences in Adverse Drug Reactions Reported to the National Pharmacovigilance Centre in the Netherlands: An Explorative Observational Study. Br J Clin Pharmacol. 2019;85(7):1507‐1515. doi: 10.1111/bcp.13923 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. US Department of Health and Human Services Food and Drug Administration (US FDA) . Guideline for the Study and Evaluation of Gender Differences in the Clinical Evaluation of Drugs. 1993. Accessed August 5, 2020. https://www.fda.gov/media/71107/download
- 57. Feldman S, Ammar W, Lo K, Trepman E, van Zuylen M, Etzioni O. Quantifying Sex Bias in Clinical Studies at Scale with Automated Data Extraction. JAMA Netw Open. 2019;2(7):e196700. doi: 10.1001/jamanetworkopen.2019.6700 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Labots G, Jones A, de Visser SJ, Rissmann R, Burggraaf J. Gender Differences in Clinical Registration Trials: Is There a Real Problem? Br J Clin Pharmacol. 2018;84(4):700‐707. doi: 10.1111/bcp.13497 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Tannenbaum C, Day D. Age and Sex in Drug Development and Testing for Adults. Pharmacol Res. 2017;121:83‐93. doi: 10.1016/j.phrs.2017.04.027 [DOI] [PubMed] [Google Scholar]
- 60. Collin L, Reisner SL, Tangpricha V, Goodman M. Prevalence of Transgender Depends on the "Case" Definition: A systematic Review. J Sex Med. 2016;13(4):613‐626. doi: 10.1016/j.jsxm.2016.02.001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Illamola SM, Bucci‐Rechtweg C, Costantine MM, Tsilou E, Sherwin CM, Zajicek A. Inclusion of Pregnant and Breastfeeding Women in Research ‐ Efforts and Initiatives. Br J Clin Pharmacol. 2018;84(2):215‐222. doi: 10.1111/bcp.13438 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Heidari S, Babor TF, De Castro P, Tort S, Curno M. Sex and Gender Equity in Research: Rationale for the SAGER Guidelines and Recommended Use. Res Integr Peer Rev. 2016;1:2. doi: 10.1186/s41073-016-0007-6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Clayton JA, Tannenbaum C. Reporting Sex, Gender, or Both in Clinical Research? Jama. 2016;316(18):1863‐1864. doi: 10.1001/jama.2016.16405 [DOI] [PubMed] [Google Scholar]
- 64. Huang S, Rowland M. The Role of Physiologically Based Pharmacokinetic Modeling in Regulatory Review. Clin Pharmacol Ther. 2012;91(3):542‐549. doi: 10.1038/clpt.2011.320 [DOI] [PubMed] [Google Scholar]
- 65. Stronks K, Wieringa NF, Hardon A. Confronting Diversity in the Production of Clinical Evidence Goes Beyond Merely Including under‐Represented Groups in Clinical Trials. Trials. 2013;14(1):177. doi: 10.1186/1745-6215-14-177 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66. Knepper TC, McLeod HL. When Will Clinical Trials Finally Reflect Diversity? Nature. 2018;557(7704):157‐159. doi: 10.1038/d41586-018-05049-5 [DOI] [PubMed] [Google Scholar]
- 67. Xu XS, Yuan M, Zhu H, et al. Full Covariate Modelling Approach in Population Pharmacokinetics: Understanding the Underlying Hypothesis Tests and Implications of Multiplicity. Br J Clin Pharmacol. 2018;84(7):1525‐1534. doi: 10.1111/bcp.13577 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68. European Medicines Association (EMA) Committee for Medicinal Products for Human Use . Guideline on the Investigation of Subgroups in Confirmatory Clinical Trials. 2019. Accessed August 5, 2020. https://www.ema.europa.eu/en/documents/scientific-guideline/guideline-investigation-subgroups-confirmatory-clinical-trials_en.pdf
- 69. US Department of Health and Human Services Food and Drug Administration (US FDA) . Content and Format of a New Drug Application, 21 CFR §314.50. 2021. Accessed November 5, 2021. https://www.govinfo.gov/content/pkg/CFR-2021-title21-vol5/pdf/CFR-2021-title21-vol5-sec314-50.pdf
- 70.Howlader N, Noone AM, Krapcho M, et al. (eds). SEER Cancer Statistics Review, 1975‐2018. Bethesda, MD: National Cancer Institute. 2021. Accessed September 5, 2021. https://seer.cancer.gov/csr/1975_2018/, based on November 2020 SEER data submission, posted to the SEER web site.
- 71. Rastogi S, Johnson T, Hoeffel E, Drewery M. The Black Population: 2010, U.S. Census Bureau 2011. Accessed August 5, 2020. https://www.census.gov/content/dam/Census/library/publications/2011/dec/c2010br-06.pdf
- 72. Bhatnagar V, Gormley N, Kazandjian D, et al. FDA Analysis of Racial Demographics in Multiple Myeloma Trials. Blood. 2017;130(Supplement 1):4352. doi: 10.1182/blood [DOI] [Google Scholar]
- 73. House of Representatives 389 112th US congress . Recognizing Persons of African Descent in Europe During the International Year for People of African Descent. 2011. Accessed August 5, 2020. https://www.congress.gov/bill/112th-congress/house-resolution/389?s=1&r=18
- 74. Gauci J. People for Change Foundation. Racism in Europe ENAR Shadow Report 2010‐2011. Brussels: European Network against Racism (ENAR); 2012. Accessed August 5, 2020. https://ec.europa.eu/migrant-integration/librarydoc/racism-in-europe-enar-shadow-report-2010-2011 [Google Scholar]
- 75. Central Intelligence Agency . The World Factbook 2021. Accessed November 5, 2021. https://www.cia.gov/the-world-factbook/
- 76. Mahaffey KW, Wojdyla DM, Carroll K, et al. Ticagrelor Compared with Clopidogrel by Geographic Region in the Platelet Inhibition and Patient Outcomes (Plato) Trial. Circulation. 2011;124(5):544‐554. doi: 10.1161/CIRCULATIONAHA.111.047498 [DOI] [PubMed] [Google Scholar]
- 77. da Rocha JEB, Othman H, Botha G, et al. The Extent and Impact of Variation in ADME Genes in Sub‐Saharan African Populations. Front Pharmacol. 2021;12:634016. doi: 10.3389/fphar.2021.634016 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78. Ministry of Health Labour and Welfare of Japan . Basic Principles on Global Clinical Trials. 2007. Accessed August 5, 2020. https://www.pmda.go.jp/files/000153265.pdf
- 79. Bierer B.E., White S.A., Meloney L.G., Ahmed H.R., Strauss D.H., Clark L.T. Achieving Diversity, Inclusion, and Equity in Clinical Research Guidance Document Version 1.2. Cambridge and Boston, MA: Multi‐Regional Clinical Trials Center of Brigham and Women's Hospital and Harvard (MRCT Center). 2021. Accessed October 12, 2021. https://mrctcenter.org/diversity-in-clinical-research/wp-content/uploads/sites/11/2021/09/MRCT-Center-Diversity-Guidance-Document-Version-1.2.pdf
- 80. Ford JG, Howerton MW, Lai GY, et al. Barriers to Recruiting Underrepresented Populations to Cancer Clinical Trials: A Systematic Review. Cancer. 2008;112(2):228‐242. doi: 10.1002/cncr.23157 [DOI] [PubMed] [Google Scholar]
- 81. Liu KA, Mager NAD. Women's Involvement in Clinical Trials: Historical Perspective and Future Implications. Pharm Pract (Granada). 2016;14(1):708. doi: 10.18549/PharmPract.2016.01.708 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82. Hughson JA, Woodward‐Kron R, Parker A, et al. A Review of Approaches to Improve Participation of Culturally and Linguistically Diverse Populations in Clinical Trials. Trials. 2016;17(1):263. doi: 10.1186/s13063-016-1384-3 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83. George S, Duran N, Norris K. A Systematic Review of Barriers and Facilitators to Minority Research Participation among African Americans, Latinos, Asian Americans, and Pacific Islanders. Am J Public Health. 2014;104(2):e16‐e31. doi: 10.2105/ajph.2013.301706 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84. Durant RW, Wenzel JA, Scarinci IC, et al. Perspectives on Barriers and Facilitators to Minority Recruitment for Clinical Trials among Cancer Center Leaders, Investigators, Research Staff, and Referring Clinicians: Enhancing Minority Participation in Clinical Trials (EMPaCT). Cancer. 2014;120(Supplement 7):1097‐1105. doi: 10.1002/cncr.28574 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85. Regnante JM, Richie N, Fashoyin‐Aje L, et al. Operational Strategies in US Cancer Centers of Excellence That Support the Successful Accrual of Racial and Ethnic Minorities in Clinical Trials. Contemp Clin Trials Commun. 2020;17:100532. doi: 10.1016/j.conctc.2020.100532 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86. Research!America . Clinical Trials: Survey Data of Minority Populations. 2017. Accessed August 5, 2020. https://www.researchamerica.org/sites/default/files/July2017ClinTrialMinorityOversamplesPressReleaseSlidesFINAL_0.pdf
- 87. Garza MA, Quinn SC, Li Y, et al. The Influence of Race and Ethnicity on Becoming a Human Subject: Factors Associated with Participation in Research. Contemp Clin Trials Commun. 2017;7:57‐63. doi: 10.1016/j.conctc.2017.05.009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88. Schmotzer GL. Barriers and Facilitators to Participation of Minorities in Clinical Trials. Ethn Dis. 2012;22(2):226‐230. [PubMed] [Google Scholar]
- 89. Research!America . Poll: Majority of Americans Would Participate in Clinical Trials If Recommended by Doctor. July 31, 2013. Accessed August, 2020. https://www.elsevier.com/connect/poll‐majority‐of‐americans‐would‐participate‐in‐clinical‐trials‐if‐recommended‐by‐doctor#:~:text=More%20than%20two‐thirds%20%2872%25%29%20of%20Americans%20say%20it%27s,new%20national%20public%20opinion%20poll%20commissioned%20by%20Research%21America
- 90. Hamel LM, Penner LA, Albrecht TL, Heath E, Gwede CK, Eggly S. Barriers to Clinical Trial Enrollment in Racial and Ethnic Minority Patients with Cancer. Cancer Control. 2016;23(4):327‐337. doi: 10.1177/107327481602300404 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91. Sedrak MS, Mohile SG, Sun V, et al. Barriers to Clinical Trial Enrollment of Older Adults with Cancer: A Qualitative Study of the Perceptions of Community and Academic Oncologists. J Geriatr Oncol. 2020;11(2):327‐334. doi: 10.1016/j.jgo.2019.07.017 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92. Duffull SB, Wright DFB. What Do We Learn from Repeated Population Analyses? Br J Clin Pharmacol. 2015;79(1):40‐47. doi: 10.1111/bcp.12233 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93. Wright DFB, Winter HR, Duffull SB. Understanding the Time Course of Pharmacological Effect: A PKPD Approach. Br J Clin Pharmacol. 2011;71(6):815‐823. doi: 10.1111/j.1365-2125.2011.03925.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94. Marshall SF, Burghaus R, Cosson V, et al. Good Practices in Model‐Informed Drug Discovery and Development: Practice, Application, and Documentation. CPT: Pharmacomet Syst Pharmacol. 2016;5(3):93‐122. doi: 10.1002/psp4.12049 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95. European Medicines Agency Science Medicines Health . Reflection Paper on the Use of Extrapolation in the Development of Medicines for Paediatrics. 2018. Accessed May 10, 2021. https://www.ema.europa.eu/en/documents/scientific-guideline/adopted-reflection-paper-use-extrapolation-development-medicines-paediatrics-revision-1_en.pdf
- 96. Rowland A, van Dyk M, Hopkins AM, et al. Physiologically Based Pharmacokinetic Modeling to Identify Physiological and Molecular Characteristics Driving Variability in Drug Exposure. Clin Pharmacol Ther. 2018;104(6):1219‐1228. doi: 10.1002/cpt.1076 [DOI] [PubMed] [Google Scholar]
- 97. Santen G, Horrigan J, Danhof M, Della PO. From Trial and Error to Trial Simulation. Part 2: An Appraisal of Current Beliefs in the Design and Analysis of Clinical Trials for Antidepressant Drugs. Clin Pharmacol Ther. 2009;86(3):255‐262. doi: 10.1038/clpt.2009.107 [DOI] [PubMed] [Google Scholar]
- 98. D'Agate S, Chavan C, Manyak M, et al. Impact of Early Vs. Delayed Initiation of Dutasteride/Tamsulosin Combination Therapy on the Risk of Acute Urinary Retention or BPH‐Related Surgery in LUTS/BPH Patients with Moderate‐to‐Severe Symptoms at Risk of Disease Progression. World J Urol. 2021;39(7):2635‐2643. doi: 10.1007/s00345-020-03517-0 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99. Deng J, Vozmediano V, Rodriguez M, Cavallari LH, Schmidt S. Genotype‐Guided Dosing of Warfarin through Modeling and Simulation. Eur J Pharm Sci. 2017;109(Supplement):S9‐S14. doi: 10.1016/j.ejps.2017.05.017 [DOI] [PubMed] [Google Scholar]
- 100. Ahmed MA, Okour M, Brundage R, Kartha RV. Orphan Drug Development: The Increasing Role of Clinical Pharmacology. J Pharmacokinet Pharmacodyn. 2019;46(5):395‐409. doi: 10.1007/s10928-019-09646-3 [DOI] [PubMed] [Google Scholar]
- 101. Liu Q, Ahadpour M, Rocca M, Huang S‐M. Clinical Pharmacology Regulatory Sciences in Drug Development and Precision Medicine: Current Status and Emerging Trends. AAPS J. 2021;23(3):54. doi: 10.1208/s12248-021-00563-3 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102. Martin K, Bégaud B, Latry P, Miremont‐Salamé G, Fourrier A, Moore N. Differences between Clinical Trials and Postmarketing Use. Br J Clin Pharmacol. 2004;57(1):86‐92. doi: 10.1046/j.1365-2125.2003.01953.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103. Chang A, Parikh P, Thongprasert S, et al. Gefitinib (IRESSA) in Patients of Asian Origin with Refractory Advanced Non‐Small Cell Lung Cancer: Subset Analysis from the ISEL Study. J Thorac Oncol. 2006;1(8):847‐855. doi: 10.1097/01243894-200610000-00014 [DOI] [PubMed] [Google Scholar]
- 104. Zhang YL, Yuan JQ, Wang KF, et al. The Prevalence of EGFR Mutation in Patients with Non‐Small Cell Lung Cancer: A Systematic Review and Meta‐Analysis. Oncotarget. 2016;7(48):78985‐78993. doi: 10.18632/oncotarget.12587 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105. Chung C. Tyrosine Kinase Inhibitors for Epidermal Growth Factor Receptor Gene Mutation‐Positive Non‐Small Cell Lung Cancers: An Update for Recent Advances in Therapeutics. J Oncol Pharm Pract. 2016;22(3):461‐476. doi: 10.1177/1078155215577810 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
TABLE S1 Physiological changes of older age and potential impact on pharmacodynamics and clinical consequences (Adapted from Saeed et al., 2015). 1
TABLE S2 The minimum categories of Ethnicity and Race recommended to be collected for each clinical trial participant, by the US FDA Guidance on Collection of Race and Ethnicity Data in Clinical Trials, 2016.4 The definitions provided are also presented.