

Abstract
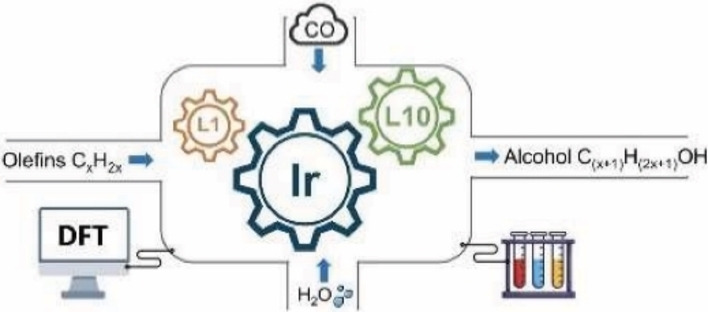
A novel one‐pot iridium‐catalyzed domino hydroxymethylation of olefins, which relies on using two different ligands at the same time, is reported. DFT computation reveals different activities for the individual hydroformylation and hydrogenation steps in the presence of mono‐ and bidentate ligands. Whereas bidentate ligands have higher hydrogenation activity, monodentate ligands show higher hydroformylation activity. Accordingly, a catalyst system is introduced that uses dual ligands in the whole domino process. Control experiments show that the overall selectivity is kinetically controlled. Both computation and experiment explain the function of the two optimized ligands during the domino process.
Keywords: density functional calculations, dual ligands, hydroxymethylation, iridium, water-gas shift reaction
Going to plan: Olefins have been hydroxymethylated by using an iridium catalyst and the synergy of two ligands. DFT calculations provided a chance to develop new catalytic systems before empirical studies were undertaken. Then the iridium‐catalyzed domino hydroformylation/reduction of olefins to alcohols under water–gas shift reaction conditions was studied as an example. Two different ligands (L1 and L10) applied at the same time improved alcohol yields and verified the computational model.
Introduction
Homogeneous catalysis research has thrived for nearly a century, and the resulting catalysts are important for almost every aspect of our daily life. Nevertheless, to develop the future potential in this area and to increase the sustainability of chemical processes, it is crucial to continue to invent and improve new molecularly defined catalytic systems. In general, this is achieved by the synthesis of new ligands and their organometallic complexes, which has been a most popular approach so far. Other approaches based on screening reaction conditions (acid, base, temperature, solvent, additives and so on) are common ways to advance a given transformation. As a more sophisticated concept, the development of bimetallic and/or multifunctional catalytic systems became popular in recent years. [1]
Despite the vast mechanistic information available for many catalytic transformations, most methodology improvements are empirically driven, and serendipity continues to play a crucial role for the discovery of new reactions. Obviously, predictive modeling offers the possibility to a more rational and potentially faster development of homogeneous catalysts. [2] To demonstrate the feasibility of this concept, we selected the important domino transformation of olefins to alcohols. Aliphatic alcohols are central bulk chemicals and feedstock in research and industry. [3] In general, the industrial way to transform olefins to alcohols follows two steps: a) catalytic hydroformylation of olefins to aldehydes and b) subsequent catalytic hydrogenation of aldehydes to alcohols. Clearly, it would be more efficient and economical to combine both steps in one process. Thus, in past decades, the so‐called hydroxymethylation process, [4] a one‐pot reaction from alkenes to alcohols using synthesis gas received considerable attention in industry and academia. [5]
Originally, cobalt carbonyl complexes in the presence of aliphatic phosphines have been used to prepare alcohols from olefins as early as 1968; [6] however, drastic reaction conditions are required for this process and the substrate scope is very limited until today. More recently, using a Rh‐catalyzed hydroformylation‐reduction sequence with the assistance of a tertiary diamine ligand, Alper et al., [7] reported the synthesis of alcohols from aromatic olefins, the products were obtained in excellent isolated yields with high iso/n ratios. Notably, neither aldehyde from hydroformylation as intermediate nor alkane from olefine hydrogenation as side products were detected. Following that work, Hapiot et al., [8] reported the hydroxymethylation of internal C=C double bonds in triglycerides by tertiary amine‐based Rh catalyst. As an interesting method, Takahashi et al., [9] reported a Rh−Ru dual system in the hydroxymethylation of dec‐1‐ene to undecanol with Xantphos/[Rh(acac)(CO)2] responsible for the hydroformylation of olefine to aldehyde and Shvo catalyst for the hydrogenation of aldehyde to alcohol. Following the same idea of such dual Rh−Ru catalyst system, some of us, [10] reported that monophosphite rhodium complex in combination with the Shvo catalyst can convert not only terminal, di‐ and tri‐substituted but also tetra‐substituted alkenes to the desired linear alcohols.
In addition to Co‐ and Rh‐based catalysts, there is a growing interest using other metals for such domino transformations. For example, our group reported an efficient and regioselective Ru catalyst with 2‐phosphino‐substituted imidazole ligands for the hydroxymethylation of internal alkenes to terminal alcohols, [11] while Drent et al., [12] reported the transformation of internal alkenes to linear alcohols in high selectivity using halide anion promoted palladium catalyst.
Based on our long‐standing interest in new carbonylation catalysts, we wondered whether Ir‐catalyzed hydroxymethylation reactions are in principle feasible. While few examples of Ir‐catalyzed hydroformylation of olefins using synthesis gas were reported by Alper, [13] Faraone, [14] Pakkanen, [15] Dmitri [16] and us, [17] none of these catalysts was active for the further reduction of aldehydes to alcohols.
As a poor catalyst for hydroformylation using synthesis gas, Fe(CO)5 can be transferred into an active catalyst at relatively mild temperatures and pressures under Reppe's conditions using CO and H2O, [18] where H2 is produced in situ in a water‐gas shift reaction (WGSR). This phenomenon is also observed for other noble metal carbonyl complexes, for example, Rh6(CO)16, Ru3(CO)12 and H4Ru4(CO)12 as well as Ir4(CO)12.[ 19 , 20 ] We therefore supposed that the WGSR conditions facilitate the reductive addition of CO to an olefin. Inspired by all these results, here we suggest a new concept for a general one‐pot domino hydroformylation and hydrogenation reactions of olefines to alcohols under WGRS conditions. Predictive modelling has done firstly to check the activity of iridium complexes with PPh3 or/and DPPE ligands. Exploratory experiments expand this to iridium complexes with various mono‐ and bidentate ligands as well as their mixture. Key for such methodology is the in situ generation of different kinds of active iridium complexes and tuning the alcohol yield in a limited time by changing the ratio of mono‐ and bidentate ligand. The respective and synergetic function of the two types of ligands during the domino process is revealed by close interaction of theoretical modelling and catalytic experiments.
Results and Discussion
Prior to experimental studies, we computed the structure and stability of Ir complexes with PPh3 and DPPE (1,2‐bis(diphenylphosphino)ethane) ligands and their catalytic activity for hydroformylation as alkenes and the hydrogenation of alkenes and aldehyde. All computational details are given in the Supporting Information, where the performance of different functional was assessed based on the available experimental data (Figure DFT−S1 and Table DFT−S1). It was found that all tested methods give similar trends on reactivity while M062X‐GD3‐SMD gave quantitative results of stability, verified by FTIR and NMR experiments. [20] Thus, the results of M062X‐GD3‐SMD at 413 K are used for discussion.
The stability of different iridium hydride complexes was compared based on the computed substitution Gibbs free energies of CO ligand in HIr(CO)4 by different ligands (Scheme 1). As expected, DPPE has stronger coordination than PPh3 (Figure DFT−S1). For DPPE coordination, the DPPE‐EA isomer (−24.5 kcal mol−1) with one P center at the axial position and one P center at the equatorial position is more stable than the DPPE‐EE isomer with two P centers at the equatorial positions by 4.2 kcal mol−1. For one PPh3 substitution, the 1Ph3‐A isomer (−12.9 kcal mol−1) with PPh3 at the axial position is more stable than the 1Ph3‐E isomer with PPh3 at the equatorial positions by 4.2 kcal mol−1. For two PPh3 substitution, the 2Ph3‐EA complex, with one PPh3 at the axial position and one PPh3 at the equatorial position, has very close substitution Gibbs free energy to that of 1Ph3‐A complex (−13.2 kcal mol−1) and is more stable than the 2Ph3‐EE isomer with two PPh3 both at the equatorial position by 3.8 kcal mol−1. However, the 3Ph3‐EEA complex, with one PPh3 at the axial position and two PPh3 at the equatorial positions is less stable. These results show the mono‐ and bis‐PPh3‐coordinated complexes have comparative stability and could be shifted to the other under different [PPh3]:[Ir] ratios or CO pressure, in agreement with the experimentally reported results. [20] The mixed DPPE and PPh3 complex DPPE‐EA‐1Ph3‐E isomer (−23.0 kcal mol−1) has very close substitution Gibbs free energy to that of DPPE‐EA isomer.
Scheme 1.
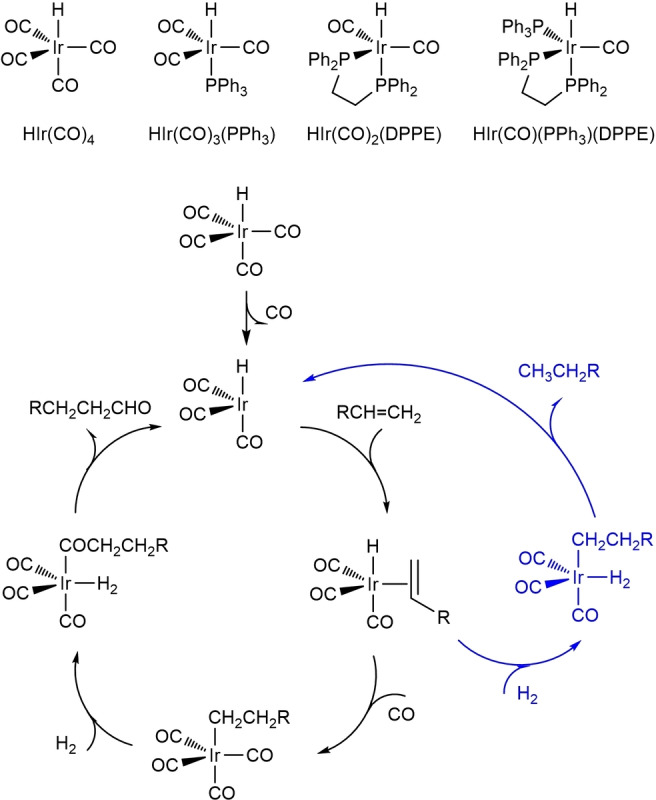
Pre‐catalysts and the reaction mechanisms of hydroformylation and hydrogenation (same cycle for alkene and aldehyde).
Considering the high CO pressure (40 bar) and high temperature (140 °C) as well as the fixed ligand‐to‐metal ratios in planned experiment, the real catalysts should be HIr(CO)3(PPh3) and HIr(CO)2(DPPE), while HIr(CO)2(PPh3)2, HIr(CO)(DPPE) (PPh3) and HIr(CO)4 were not stable under such conditions (Figure DFT−S3). Therefore, the catalytic activity of HIr(CO)3(PPh3) and HIr(CO)2(DPPE) were systematically studied and that of HIr(CO)4 and HIr(CO)2(PPh3)2 was included for comparison. The catalytic cycle has been computed on the basis of the typically accepted reaction mechanism of hydroformylation [21] and subsequent hydrogenation shown in Scheme 1 for HIr(CO)4. Using but‐1‐ene as substrate, we computed only the linear aldehyde and alcohols. The full potential energy surfaces (PES) of different catalysts are shown in Figure DFT−S4–S6. It should be mentioned that the real catalytic cycle starts with the formation of the coordinatively unsaturated active catalysts via CO dissociation from the pre‐catalysts and this is the so‐called initiation step in general. Notably, the DPPE‐EE isomer needs considerably higher energy (33.6 vs. 10.0 kcal mol−1) in the initiation step towards CO dissociation than the EA isomer, and the reaction possibility along the EE pathway should be rather low. Therefore, the following discussion of DPPE ligand refers only to the EA isomer. Based on this initiation mechanism, the effective barriers of the formation of aldehyde, alkane and alcohol using different catalysts are listed in Table 1 (more detailed information see Supporting Information).
Table 1.
Effective barriers to the formation of aldehyde, alkane and alcohol of different catalysts at M062X‐D3 level (including solvation correction and Gibs free energy correction at 413K) with respect to the real active species.
|
Ligand |
Barrier [kcal mol−1] |
||
|---|---|---|---|
|
|
Aldehyde |
Alkane |
Alcohol |
|
CO |
22.4 |
25.9 |
46.1 |
|
1Ph3‐A |
25.7 |
27.5 |
41.3 |
|
1Ph3‐E |
26.0 |
26.0 |
40.7 |
|
2Ph3‐EA |
26.5 |
26.5 |
31.3 |
|
2Ph3‐EE |
26.8 |
26.8 |
35.9 |
|
DPPE‐EA |
23.8 |
26.8 |
36.3 |
|
L1‐A |
26.2 |
27.1 |
41.3 |
|
L1‐E |
23.8 |
25.6 |
40.6 |
|
L10‐EA |
24.8 |
30.2 |
38.4 |
It is found that aldehyde formation has a lower barrier than alkane formation for HIr(CO)4 (22.4 vs. 25.9 kcal mol−1). Apparently, aldehyde has higher kinetic selectivity than alkane. However, alkane formation is more thermodynamically favored than aldehyde formation (−18.7 vs. −13.8 kcal mol−1). Based on these low apparent barriers, a switch of chemoselectivity simply by raising the temperature can be expected. According to the Eyring–Polanyi equation higher temperature affects the reaction with higher barrier more strongly than that with lower barrier; thus, the yield of alkane increases. Indeed, this trend has been found in our control experiments for different iridium precursors without phosphine ligands in NMP (N‐methylpyrrolidone) solution under synthesis gas and WGSR conditions (Tables S1 and S2). Notably, alcohol formation has a significantly higher barrier (46.1 kcal mol−1) and consequently in all cases no alcohols should be observed. It is noted that WGSR conditions give much more internal olefines than using synthesis gas. This might be due to the lower H2 content under WGSR conditions.
For HIr(CO)3(PPh3), aldehyde formation is slightly more favored than alkane generation (25.7 vs. 26.0 kcal mol−1), while alcohol formation has the highest barrier (40.7 kcal mol−1). Control experiments (Table 2, entries 1–3, Table S3, entry 1) under WGSR conditions as well as previous studies in the presence of synthesis gas [17] also showed the preferred formation of aldehyde over alkane in NMP solution, and these supported our computed results. The same trend is observed for HIr(CO)2(DPPE). Compared to HIr(CO)4 and HIr(CO)3(PPh3), however, HIr(CO)2(DPPE) allows hydrogenation of the aldehyde due to the lower barrier for alcohol formation (36.6 kcal mol−1). Interestingly, for the HIr(CO)2(PPh3)2 complex, it is found that the formation of aldehyde and alkane has an equal barrier (26.5 kcal mol−1), and the formation of alcohol has a low barrier (31.3 kcal mol−1). This indicates that HIr(CO)2(PPh3)2 does not show any selectivity of aldehyde and alkane but has relatively high selectivity for the respective alcohol. However, considering the low stability of HIr(CO)2(PPh3)2, it is hard to achieve a high yield of alcohol simply by using this complex.
Table 2.
Ir‐catalyzed hydroformylation/reduction of oct‐1‐ene: Ligand variation.[a]
|
| |||||
|---|---|---|---|---|---|
|
|
Ligand |
Yield [%][b] |
|||
|
2 (n:iso)[c] |
3 (n:iso) |
4 |
5 |
||
|
1[d] |
PPh3 |
n.d. |
81 (67 : 33) |
19 |
<1 |
|
2[e] |
PPh3 |
n.d. |
92 (71 : 29) |
8 |
<1 |
|
3[f,g] |
PPh3 |
n.d. |
94 (70 : 30) |
5 |
<1 |
|
4[f] |
P(p‐CH3OC6H4)3 |
n.d. |
91 (74 : 26) |
6 |
3 |
|
5[f] |
L1 |
n.d. |
96 (72 : 28) |
2 |
1 |
|
6[f] |
L2 |
n.d. |
81 (65 : 35) |
9 |
8 |
|
7[f] |
L3 |
n.d. |
89 (50 : 50) |
9 |
1 |
|
8[f] |
Ph2PPy (L4) |
n.d. |
88 (64 : 36) |
11 |
<1 |
|
9[f,g] |
PCy3 |
n.d. |
94 (75 : 25) |
6 |
<1 |
|
10 |
L5 |
n.d. |
74 (65 : 35) |
17 |
8 |
|
11 |
L6 |
n.d. |
72 (67 : 33) |
18 |
9 |
|
12 |
L7 |
n.d. |
79 (64 : 36) |
14 |
7 |
|
13 |
L8 |
n.d. |
84 (67 : 33) |
10 |
5 |
|
14 |
DPPE (L9) |
1 |
47 (58 : 42) |
36 |
15 |
|
15 |
L10 |
47 (66 : 33) |
26 (46 : 54) |
18 |
3 |
|
16[h] |
L10 |
62 (63 : 37) |
16 (32 : 68) |
19 |
3 |
|
17 |
L11 |
21 (57 : 43) |
43 (46 : 54) |
34 |
2 |
|
18 |
L12 |
17 (50 : 50) |
44 (44 : 56) |
26 |
13 |
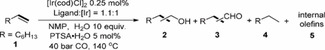

[a] Rection conditions: 1.0 mmol of oct‐1‐ene, 0.25 mol % of [Ir(cod)Cl]2 (0.5 mol % of [Ir]), 0.55 mol % of ligand (ligand:[Ir]=1.1 : 1), NMP 1.0 mL, CO 40 bar, 10 equiv. of H2O, 5 mol % of PTSA⋅H2O, 140 °C, 20 h. [b] Determined by GC using isooctane (57 mg) as internal standard. [c] n:iso is the ratio of linear product to all branched products. [d] Synthesis gas 40 bar (CO:H2=1 : 1) and without acid. [e] Synthesis gas 40 bar (CO:H2=3 : 1) and without acid. [f] 1.1 mol % of ligand (ligand:[Ir]=2.2 : 1). [g] 48 h. [h] 1 mol % of [Ir] and 1.1 mol of L10.
Next, we carried out further experimental studies to confirm these computational results. Thus, carbonylation reactions of oct‐1‐ene were performed in the presence of Ir‐complexes of monodentate and bidentate phosphine ligands (Table 2). Initially, this model reaction was carried out with 0.25 mol % of [Ir(cod)Cl]2, 0.55 mol % of PPh3 in NMP under synthesis gas conditions (140 °C, 40 bar, CO:H2 1 : 1, 20 h), similar with those previously applied in Ir‐catalyzed hydroformylation. [17] Apart from the expected aldehydes (81 %), alkane is also formed (19 %; Table 2, entry 1). Lowering H2 partial pressure (CO:H2=3 : 1) suppresses alkane formation (8 %), while promotes the formation of aldehydes (92 %; Table 2, entry 2). To further improve this effect, reaction using H2 generated in situ from WGSR was tested, and PTSA⋅H2O (p‐toluenesulfonic acid) was used to promote WGSR. Notably, base promoters did not improve the yields (Table S3). Using 40 bar of CO in the presence of 10 equivalents H2O and 5 mol % PTSA⋅H2O under analogous conditions for 48 h, octane 4 formation is suppressed (5 %), whereas the desired C9‐aldehydes 3 (94 %; Table 2, entry 3) are promoted.
To improve the selectivity, different phosphine ligands were tested under WGSR condition (Table 2, entries 4–18). Compared to PPh3, arylphosphines with electron‐donating or ‐withdrawing substituents (Table 2, entries 4–7) showed reactivity increase and best results are obtained for bis(4‐methoxyphenyl)phenyl‐phosphine L1, which gives 3 in excellent yield (96 %; Table 2, entry 5). For the well‐known 2‐pyridyldiphenylphosphine ligand L4 (Table 2, entry 8), which was found to be more active for alkyne carbonylations, [22] aldehydes 3 are formed (88 %). Applying electron‐rich and sterically hindered PCy3 gave similar result (94 %, Table 2, entry 9). Employing own prepared ligands L5– [11] led to slightly lower selectivity of aldehydes (Table 2, entries 10–13). Notably, complexes with monodentate phosphine ligands ended up with the formation of aldehydes and no further reduction of aldehydes to alcohols is observed; however, they produced alkane from olefine hydrogenation. All these results agree with the computationally proposed preference of aldehyde generation instead of alkane formation.
In addition to monodentate phosphines, bidentate phosphine ligands were also tested. Using DPPE as a ligand (Table 2, entry 14), aldehydes (47 %), octane (36 %) and internal octenes (15 %) are formed, whereas in the presence of modified DPPE derivatives such as L10–L12, the desired alcohols (17–47 %) are formed. Notably, the chemoselectivity of this reaction is also influenced by solvent, which allows to further increase the alcohol yield (Table S4). Among the tested solvents, NMP gave the lowest amount of undesired alkane and internal olefines (18 and 3 %, Table 2, entry 15). Increasing iridium loading improved the selectivity of alcohols (62 %, Table 2, entry 16) and lowered the selectivity of aldehyde (16 %), indicating that increasing metal loading can promote the hydrogenation of aldehydes to alcohols.
We further tested several iridium precursors in the presence of L10 (Table S5). No alcohols are obtained for Ir(acac)(CO)2 and Ir4(CO)12, and [Ir(cod)2]BF4 does not show improved performance. IrCl3⋅nH2O gives slightly better results than [Ir(cod)Cl]2 with lower cost. Because LiCl as additive was reported to suppress the hydrogenation of olefines when using synthesis gas,[ 11a , 15 , 23 ] we tested it and found overall reduced reactivity (Table S6). We further investigated the effects of CO pressure (Table S7), temperature (Table S8) and water content (Table S9). Summarizing the results above gives the best conditions using 1.0 mol % of IrCl3 ⋅ n H2O, 1.0 mol % of L10, NMP solvent, 10 equivalent of H2O, 40 bar CO, 140 °C and 20 h reaction time. Additionally, applying such high CO pressure aim to promote H2 formation in the WGSR (Table S7, entry 4).
Due to the better performance of L1 and L10, further computational studies based on these two ligands were carried out. The stability of the L1/L10 coordinated Iridium hydride complexes (Figure DFT−S2) shows that the two isomers of L10 coordinated bidentate complexes, L10‐EA and L10‐EE, are much more stable than the two isomers of L1 coordinated monodentate complexes L1‐E and L1‐A (−23.8 vs. −14.4 and −20.2 vs. −10.6 kcal mol−1). It can be found that both L1 and L10 coordinated isomers at the axial positions (L1‐A and L10‐EA) are more stable than those at only equatorial positions (L1‐E and L10‐EE). Furthermore, complex 2 L10 has a lower substitution Gibbs free energy than L10‐EA by 2.3 kcal mol−1. Surprisingly, the L1 and L10 containing complex L10‐EA‐L1‐E (−25.2 kcal mol−1) is more stable than the complexes with only one kind of ligand by 1.4 and 10.8 kcal mol−1, respectively. Under our reaction conditions with high CO pressure and fixed ligand‐to‐metal ratio, however, the formation of L10‐EA‐L1‐E is less likely (Figure DFT−S3). The relative stability of the L1/L10 coordinated iridium hydride complex is comparable to that of PPh3/DPPE, and the real catalysts under our reaction conditions should be HIr(CO)3(L1) and HIr(CO)2(L10) (Tables DFT−S3).
Considering the similar coordination mode between PPh3 and L1 as well as between DPPE and L10 ligands, it is reasonably to suppose that L1/L10 have similar PES with those of PPh3/DPPE in the whole hydroformylation and hydrogenation process, and only the possible rate‐determining transition states of L1/L10 coordinated Iridium hydride complexes were considered. The obtained minimum effective barriers of the formation of aldehyde, alkane, and alcohol are listed in Table 1 (detailed in Table DFT−S2/DFT−S3), and the full PES are shown in Figures DFT−S4–S6. The pathway along L10‐EE is also ignored in the following discussion due to the high initiation energy.
The effective barrier for the formation of aldehyde/alkane in the presence of HIr(CO)3(L1) and HIr(CO)2(L10) is 23.8/25.6, 24.8/30.2 kcal mol−1, respectively. Both L1 and L10 ligands increase the barrier difference between the formation of aldehyde and alkane compared with PPh3 (1.8 vs. 0.3 kcal mol−1) and DPPE (5.4 vs. 3.0 kcal mol−1) (Table DFT−S3 and Figure DFT−S6). Thus, L1 and L10 can promote the formation of aldehydes and suppress the formation of alkane than PPh3 and DPPE, respectively. However, L1 has lower barriers for the formation of aldehyde and alkane than L10, indicating the faster aldehyde production rate of L1. In addition, L10 has lower barrier for the formation of alcohols than that of L1 (38.4 vs. 40.6 kcal mol−1), therefore L10 can give alcohols easier than L1.
Based on all the computational results, we can deduce that mono‐phosphine iridium complexes have higher aldehyde selectivity and low alcohol selectivity, whereas bis‐mono‐phosphine and bidentate phosphine iridium complexes have higher alcohol selectivity. In the presence of both mono‐ and bidentate phosphine ligands, under high CO pressure, the iridium complex with both ligands co‐coordinated has a low stability.
Having these proposals in mind, we carried out further experiments using 1 mol % of IrCl3 ⋅ n H2O as precursors in the presence of L1 and L10 in different ratios to combine the positive effects of the different catalyst species (Table 3).
Table 3.
Iridium‐catalyzed hydroformylation/reduction of oct‐1‐ene: influence of ligand concentrations.[a]
|
| ||||||
|---|---|---|---|---|---|---|
|
|
Mol % |
Yield [%][b] |
||||
|
x |
y |
2 (n:iso)[c] |
3 (n:iso) |
4 |
5 |
|
|
1 |
0 |
1.0 |
69 (63 : 37) |
11 (27 : 73) |
18 |
<1 |
|
2 |
1.0 |
0 |
n.d. |
96 (72 : 28) |
2 |
1 |
|
3 |
0 |
2.0 |
15 (79 : 21) |
55 (63 : 37) |
12 |
9 |
|
4 |
2.0 |
0 |
n.d. |
96 (72 : 28) |
2 |
1 |
|
5 |
1.0 |
1.0 |
26 (77 : 23) |
65 (68 : 32) |
6 |
<1 |
|
6 |
0.1 |
0.9 |
77 (69 : 31) |
8 (44 : 56) |
10 |
1 |
|
7 |
0.2 |
0.8 |
67 (70 : 30) |
16 (48 : 52) |
9 |
4 |
|
8 |
0.3 |
0.7 |
35 (72 : 28) |
44 (50 : 50) |
7 |
4 |
|
9 |
0.5 |
0.5 |
16 (75 : 25) |
75 (71 : 29) |
5 |
1 |
|
10 |
0.7 |
0.3 |
11 (73 : 27) |
77 (68 : 32) |
7 |
3 |
|
11[d] |
0 |
1.0 |
77 (73 : 27) |
2 (2 : 98) |
16 |
3 |
|
12[d] |
0.1 |
0.9 |
80 (72 : 28) |
2 (5 : 95) |
12 |
<1 |
|
13[d] |
0.2 |
0.8 |
79 (73 : 27) |
5 (10 : 90) |
8 |
1 |
|
14[d] |
0.3 |
0.7 |
65 (69 : 31) |
12 (11 : 89) |
8 |
<1 |
|
15[d] |
0.5 |
0.5 |
49 (77 : 23) |
35 (42 : 58) |
6 |
2 |
|
16[d] |
0.7 |
0.3 |
27 (70 : 30) |
57 (57 : 43) |
6 |
<1 |
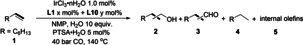
[a] Each result is the average of three experiments. Rection conditions: 1.0 mmol of oct‐1‐ene, 1.0 mol % of IrCl3 ⋅ n H2O, x mol % of L1, y mol % of L10, NMP 1.0 mL, CO 40 bar, 10 equiv. of H2O, 5 mol % of PTSA⋅H2O, 140 °C, 20 h. [b] Determined by GC using isooctane (57 mg) as internal standard. [c] n:iso is the ratio of linear product to all branched products. [d] 48 h.
Only using L10 (Table 3, entry 1), the reaction gives not only alcohols (69 %) and aldehydes (11 %) but also alkane (18 %) and internal olefins (<1%), while in the presence of only L1 (Table 3, entry 2) the reaction gives mainly aldehydes (96 %), slight alkane (2 %) and internal olefins (1 %) and no alcohols are found. This shows that L1 prefers the formation of aldehydes and L10 can prefers hydroformylation and hydrogenation of aldehyde, in line with the computed results.
Next, different metal‐to‐ligand ratios were experimentally tested. With increasing ligand concentration ([Ir]:[P]=1 : 4) the activity dropped dramatically and only 15 % of 2 is obtained along with 55 % aldehydes (Table 3, entry 3). In line with our previous test (Table 2, entry 5), no alcohols are detected for using L1 even at higher catalyst concentration (Table 3, entry 4). In the presence of both ligands L1 and L10 ([Ir]:[P]=1 : 6), low activity and low yields of the corresponding aldehydes are observed (Table 3, entry 5). Obviously, the ligand loading is crucial for the activity of the catalysts, and therefore, we combined L1 and L10 in different ratios (L10/L1+L10=0, 0.3, 0.5, 0.7, 0.8, 0.9 and 1.0, [Ir]:[L1+L10]=1 : 1) for 20 h for evaluating the activity and selectivity. With the increase of L10 content (Table 3, entries 1, 2, 6–10, Figure 1a), the yield of aldehydes decreases, while the yield of alcohols increases. The same trend is found for reaction for 48 h (Table 3, entries 11–16, Figure 1b), however, at a given L10 content, longer reaction time gives higher yield of alcohols and lower yield of aldehydes. This reveals that simply mixing L1 and L10 ligand in one system can improve the hydroxymethylation process to a certain extent, and such reaction is kinetically controlled. According to Table 3, the best result is obtained with a combination of L1:L10=1 : 9. Obviously, the reaction rate is mainly determined by L10 amount.
Figure 1.
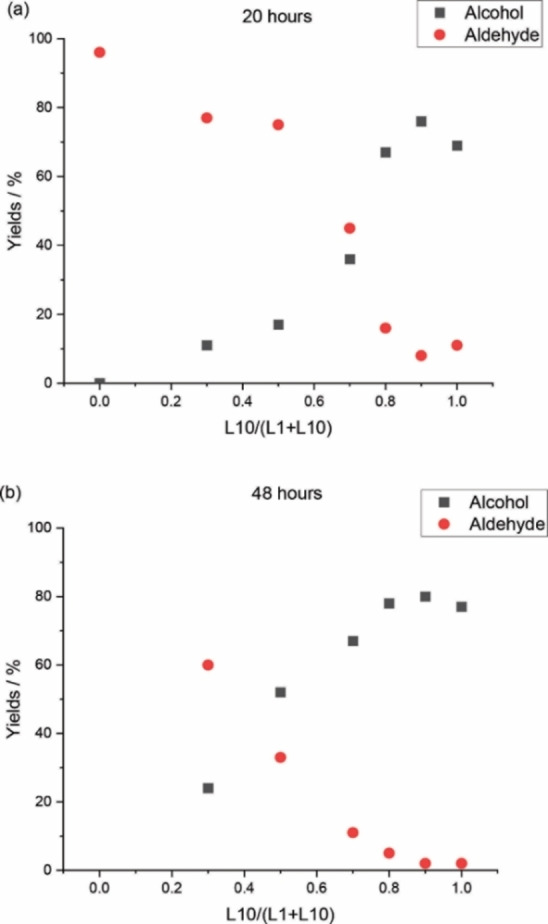
Charts of Table 3: a) Entries 1, 2, and 6–10; b) entries 11–16.
To confirm the previous conclusions, control experiments with olefine and aldehyde were performed with L1 and L10 at 100, 120 and 140 °C (Table S10). For L1 (Figure 2a), the yield of aldehydes increases with the increase of temperature, meanwhile the formed internal olefines are also converted to aldehydes, and alcohols are not formed. For L10 (Figure 2b), low temperature gives mainly aldehydes and alkane at low conversion. At higher temperature the yield of alcohols, aldehydes and alkane increases, and aldehydes can be further hydrogenated to alcohols at elongated reaction time. Next, further control experiments using nonanal as substrate with different L10 concentrations were done (Table S11). With lower concentration of L10, the nonanal conversion is decreased. Although prolonging the reaction time led to higher alcohol yields, the undesired condensation of aldehyde also increased.
Figure 2.

Control experiments in different temperature: 1.0 mmol of substrate, 1.0 mL of NMP, 1.0 mol % of IrCl3 ⋅ n H2O, 1.1 mol % of ligand, 5 mol % of PTSA⋅H2O, 10 equiv. of H2O, 40 bar CO. a) Hydroformylation/reduction of oct‐1‐ene only using L1 in different temperature; b) Hydroformylation/reduction of oct‐1‐ene only using L10 in different temperature; c) Reduction of nonanal only using L1 in different temperature (ca. 30 % of nonanal condensed); d) Reduction of nonanal only using L10 in different temperature (ca. 30 % of nonanal condensed).
With nonanal as the substrate, alcohol is not formed for L1 at low or high temperature (Figure 2c), whereas for L10 (Figure 2d), alcohol is not formed at 100 °C, but is at 120 °C, and the yield increases at 140 °C. The mixture of aldehyde and the alcohol formed at 140 °C (Figure 2b, d) show that the hydrogenation of aldehyde to alcohol has higher barrier than the hydroformylation of olefine to aldehydes, in full agreement with computation.
Having an appropriate catalyst system in hand, we tested the scope of substrates. Initially some industrially relevant aliphatic alkenes (1 a–1 g) were tested. The alcohol products were isolated by chromatography and isolated yields are given in Scheme 2. As expected, linear terminal olefins gave similar results as model substrate oct‐1‐ene (Scheme 2, 2 a–c). The alcohol yields of 73 % (Scheme 2, 2 c) to 79 % (Scheme 2, 2 b) are obtained, and the n:i ratios decreased with shorter carbon chain. Alkenes with hinder and strong electron‐donating group (1 d, 1 e) achieved high selectivity of alcohols and only n‐alcohol products are detected. Internal olefins were also tested (1 f–1 g) and they also provided relatively high alcohol yields. Notably, internal olefins are also isomerized during the reaction and then a part of terminal products were observed. Aromatic and cyclo‐alkenes were also tested (Scheme 3, 1 h–1 o) and similar trends compared to aliphatic olefins are observed. However, in these cases the reactivity was somewhat lower under the model conditions and branched products dominated for the aromatic substrates (2 h–). Nevertheless, the alcohol yields could be improved when the loading of metal and ligands is increased (2 i, a). Likewise, for 1k, the alcohol yield is improved at higher catalyst loading.
Scheme 2.
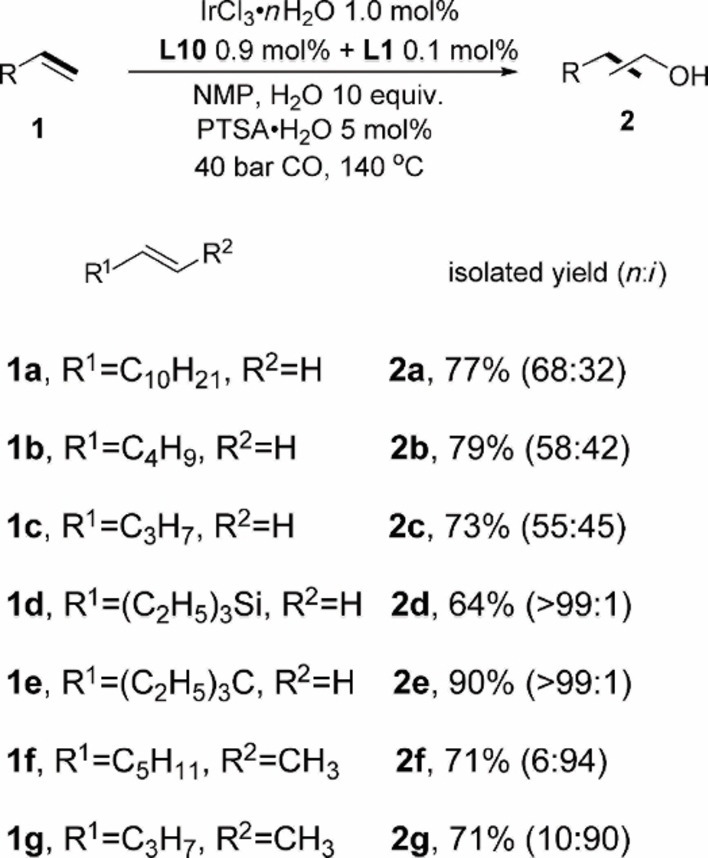
Iridium‐catalyzed hydroformylation/reduction of olefins: aliphatic terminal and internal olefins. Rection conditions: 2.0 mmol of olefin, 1.0 mol % of IrCl3 ⋅ n H2O, 0.9 mol % of L10, 0.1 mol % of L1, NMP 2.0 mL, CO 40 bar, 10 equiv. of H2O, 5 mol % of PTSA⋅H2O, 140 °C, 24 h. Linear and branch products ratios (n:i) were determined by GC and NMR spectroscopy.
Scheme 3.

Iridium‐catalyzed hydroformylation/reduction of olefins: aromatic and cycloalkenes. Rection conditions: 2.0 mmol of olefin, 1.0 mol % of IrCl3 ⋅ n H2O, 0.9 mol % of L10, 0.1 mol % of L1, NMP 2.0 mL, CO 40 bar, 10 equiv. of H2O, 5 mol % of PTSA⋅H2O, 140 °C, 24 h. Linear and branch product ratios (n:i) were determined by GC and NMR spectroscopy. [a] 2.0 mol % of IrCl3⋅nH2O, 1.8 mol % of L10, 0.2 mol % of L1. [b] 3.0 mol % of IrCl3⋅nH2O, 2.7 mol % of L10, 0.3 mol % of L1. [c] ratio of carbonylated positions 1 and 2.
Notably, less‐reactive cyclo‐olefins (1 l–1 o) provided moderate‐to‐good yields of alcohol when the amount of catalyst was increased threefold. For example, 4‐methylcyclohexene (1 m) and cyclo‐octene (1 n) are converted to the desired products in 79–81 % yield. Finally, norbornene (1 o) was transformed into an alcohol in high yield (77 %) with no extra amount of catalyst, albeit a mixture of exo and endo products is obtained.
Conclusion
A novel iridium catalyst system based on two different ligands for the one‐pot hydroxymethylation of olefins to alcohols is presented. Theoretical investigations predict a high hydroformylation rate of iridium‐monophosphine complexes, and a relatively high aldehyde reduction rate of iridium‐bidentate‐phosphine complexes. Mixing these two types of ligand in one system can keep the advantage of each catalyst, especially under high CO pressure. Based on modeling studies, a novel catalyst system is introduced that uses both mono‐ and bidentate phosphine ligands at the same time in a domino process. Good‐to‐very‐good alcohol yields were obtained from aliphatic and aromatic olefins, including cyclic and internal ones. With an appropriate ligand ratio (L1/L10), the desired alcohol yield could be further improved. Control experiments show that the overall reaction is kinetically dominated, and the final yield mainly depends on the amount of L10 and the reaction time.
Conflict of interest
The authors declare no conflict of interest.
1.
Supporting information
As a service to our authors and readers, this journal provides supporting information supplied by the authors. Such materials are peer reviewed and may be re‐organized for online delivery, but are not copy‐edited or typeset. Technical support issues arising from supporting information (other than missing files) should be addressed to the authors.
Supporting Information
Acknowledgements
We are grateful to Drs. Yang Li, Jiawang Liu, Yao Ge and Ji Yang for assistance and advice. We are also grateful to the analytic team at LIKAT. Open Access funding enabled and organized by Project DEALand the National Natural Science Foundation of China (21903049). Open Access funding enabled and organized by Projekt DEAL.
W. Huang, X. Tian, H. Jiao, R. Jackstell, M. Beller, Chem. Eur. J. 2022, 28, e202104012.
Contributor Information
Prof. Haijun Jiao, Email: haijun.jiao@catalysis.de.
Dr. Ralf Jackstell, Email: ralf.jackstell@catalysis.de.
Prof. Dr. Matthias Beller, Email: matthias.beller@catalysis.de.
Data Availability Statement
The data that support the findings of this study are available in the supplementary material of this article.
References
- 1.
- 1a. Zhang X., Tian X., Shen C., Xia C., He L., ChemCatChem 2019, 11, 1986; [Google Scholar]
- 1b. Arifin K., Majlan E. H., Daud W. R. W., Kassim M. B., Int. J. Hydrogen Energy 2012, 37, 3066. [Google Scholar]
- 2.
- 2a. Maldonado A. G., Rothenberg G., Chem. Soc. Rev. 2010, 39, 1891; [DOI] [PubMed] [Google Scholar]
- 2b. Landman I. R., Paulson E. R., Rheingold A. L., Grotjahn D. B., Rothenberg G., Catal. Sci. Technol. 2017, 7, 4842; [Google Scholar]
- 2c. Durand D. J., Fey N., Chem. Rev. 2019, 119, 6561. [DOI] [PubMed] [Google Scholar]
- 3.
- 3a. Zhao S., Mankad N. P., Angew. Chem. Int. Ed. 2018, 57, 5867; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2018, 130, 5969; [Google Scholar]
- 3b. Tang X., Gan L., Zhang X., Huang Z., Sci. Adv. 2020, 6, eabc6688; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3c. Jin X., Fu H. C., Wang M. Y., Huang S., Wang Y., He L. N., Ma X., Org. Lett. 2021, 23, 4997; [DOI] [PubMed] [Google Scholar]
- 3d. Gorbunov D., Nenasheva M., Naranov E., Maximov A., Rosenberg E., Karakhanov E., Appl. Catal. A 2021, 623, 118266. [Google Scholar]
- 4.“Alcohols, Aliphatic”, Falbe J., Bahrmann H., Lipps W., Mayer D., Frey G. D., in Ullmann's Encyclopedia of Industrial Chemistry, Wiley-VCH, Weinheim, 2013. [Google Scholar]
- 5. Torres G. M., Frauenlob R., Franke R., Börner A., Catal. Sci. Technol. 2015, 5, 34. [Google Scholar]
- 6. Slaugh L. H., Mullineaux R. D., J. Organomet. Chem. 1968, 13, 469. [Google Scholar]
- 7. Cheung L. L., Vasapollo G., Alper H., Adv. Synth. Catal. 2012, 354, 2019. [Google Scholar]
- 8. Vanbésien T., Monflier E., Hapiot F., Green Chem. 2016, 18, 6687. [Google Scholar]
- 9. Takahashi K., Yamashita M., Ichihara T., Nakano K., Nozaki K., Angew. Chem. Int. Ed. 2010, 49, 4488; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2010, 122, 4590. [Google Scholar]
- 10. Rodrigues F. M., Kucmierczyk P. K., Pineiro M., Jackstell R., Franke R., Pereira M. M., Beller M., ChemSusChem 2018, 11, 2310. [DOI] [PubMed] [Google Scholar]
- 11.
- 11a. Fleischer I., Dyballa K. M., Jennerjahn R., Jackstell R., Franke R., Spannenberg A., Beller M., Angew. Chem. Int. Ed. 2013, 52, 2949; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2013, 125, 3021; [Google Scholar]
- 11b. Wu L., Fleischer I., Jackstell R., Profir I., Franke R., Beller M., J. Am. Chem. Soc. 2013, 135, 14306. [DOI] [PubMed] [Google Scholar]
- 12. Konya D., Almeida Leñero K. Q., Drent E., Organometallics 2006, 25, 3166. [Google Scholar]
- 13. Crudden C. M., Alper H., J. Org. Chem. 1994, 59, 3091. [Google Scholar]
- 14. Franciò G., Scopelliti R., Arena C. G., Bruno G., Drommi D., Faraone F., Organometallics 1998, 17, 338. [Google Scholar]
- 15. Moreno M. A., Haukka M., Pakkanen T. A., J. Catal. 2003, 215, 326. [Google Scholar]
- 16. Musa S., Filippov O. A., Belkova N. V., Shubina E. S., Silantyev G. A., Ackermann L., Gelman D., Chem. Eur. J. 2013, 19, 16906. [DOI] [PubMed] [Google Scholar]
- 17. Piras I., Jennerjahn R., Jackstell R., Spannenberg A., Franke R., Beller M., Angew. Chem. Int. Ed. 2011, 50, 280; [DOI] [PubMed] [Google Scholar]; Angew. Chem. Int. Ed. 2011, 123, 294. [Google Scholar]
- 18.
- 18a. Reppe W., Vetter H., Liebigs Ann. Chem. 1953, 552, 133; [Google Scholar]
- 18b. Kutepov N., Kindler H., Angew. Chem. 1960, 72, 802; [Google Scholar]
- 18c. Chini P., Inorg. Chim. Acta Rev. 1968, 2, 31; [Google Scholar]
- 18d. Massoudi R., Kim J. H., King R. B., King A. D. Jr., J. Am. Chem. Soc. 1987, 109, 7428. [Google Scholar]
- 19.
- 19a. Kang H. C., Mauldin C. H., Cole T., Slegeir W., Cann K., Pettit R., J. Am. Chem. Soc. 1977, 99, 8323; [Google Scholar]
- 19b. Kaneda K., Imanaka T., Teranishi S., Chem. Lett. 1983, 12, 1465. [Google Scholar]
- 20. Kubis C., Baumann W., Barsch E., Selent D., Sawall M., Ludwig R., Neymeyr K., Hess D., Franke R., Börner A., ACS Catal. 2014, 4, 2097. [Google Scholar]
- 21. Börner A., Franke R., Hydroformylation: Fundamentals, Processes, and Applications in Organic Synthesis, Wiley-VCH, Weinheim, 2016. [Google Scholar]
- 22. Drent E., Arnoldy P., Budzelaar P. H. M., J. Organomet. Chem. 1993, 455, 247. [Google Scholar]
- 23. Behr A., Kämper A., Nickel M., Franke R., Appl. Catal. A 2015, 505, 243. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
As a service to our authors and readers, this journal provides supporting information supplied by the authors. Such materials are peer reviewed and may be re‐organized for online delivery, but are not copy‐edited or typeset. Technical support issues arising from supporting information (other than missing files) should be addressed to the authors.
Supporting Information
Data Availability Statement
The data that support the findings of this study are available in the supplementary material of this article.