

Abstract
Immunotherapy sheds new light to cancer treatment and is satisfied by cancer patients. However, immunotoxicity, single‐source antibodies, and single‐targeting stratege are potential challenges to the success of cancer immunotherapy. A huge number of promising lead compounds for cancer treatment are of natural origin from herbal medicines. The application of natural products from herbal medicines that have immunomodulatory properties could alter the landscape of immunotherapy drastically. The present study summarizes current medication for cancer immunotherapy and discusses the potential chemicals from herbal medicines as immune checkpoint inhibitors that have a broad range of immunomodulatory effects. Therefore, this review provides valuable insights into the efficacy and mechanism of actions of cancer immunotherapies, including natural products and combined treatment with immune checkpoint inhibitors, which could confer an improved clinical outcome for cancer treatment.
Keywords: cancer immunotherapy, herbal medicine, immune response, immune‐checkpoint blockade, natural products
1. INTRODUCTION
Millions of people with cancer die each year without receiving efficacious treatments from traditional therapies. Since the late 19th century, scientists have attempted to develop generalizable strategies to boost the immune system and balance the dysfunction to eliminate cancer cells. To date, immunotherapy has yielded dramatic and durable remission for many types of advanced cancer patients, including adoptive cellular therapy, immune checkpoint blockade (ICB), and exosome therapy. 1 , 2 A novel principle for cancer immunotherapy by inhibiting immune checkpoints to regulate negative immune and activate anticancer T cells was discovered by James Allison (cytotoxic T lymphocyte‐associated protein 4 [CTLA‐4]) and Tasuku Honjo (programmed cell death protein 1 [PD‐1]), who won the Nobel Prize (Physiology or Medicine) in 2018. Therefore, there is now considerable emphasis on the development and application of novel ways to further improve treatment response with satisfied efficacy and decrease the side effects with a specific target of these revolutionary cancer therapies.
Cancer immunotherapy, a shining concept for cancer treatment, aims to restore the anticancer immune response in tumors through modifying immune cells (e.g., macrophage cells; T lymphocytes; dendritic cells [DCs]) and tumor microenvironment (TME). Cytotoxic T lymphocytes (CTLs) are activated by DCs to enable tumor‐specific CTLs to initiate durable anticancer immunity. DCs are activated by inflammatory stimuli and the immune signals transmit from CD4+ T to CD8+ T lymphocytes to maximize the CTL response in cancer immunotherapy. 3 Besides, tumor‐infiltrating myeloid cells (TIMs), including macrophages, monocytes, and neutrophil, play as major regulators to promote or limit tumor outgrowth. 4
The immunosuppressive TME, a complex ecosystem, can be demolished to overcome immune evasion through the activation of effector T (Teff) cells via suppressing oxidative and glycolytic metabolism using a glutamine antagonist. 5 Gene expression profiles have proven that tertiary lymphoid structures and B cells enriched in the TME can promote responses of immunotherapy in cancer patients with a good prognosis. 6
Cancer immunotherapy faces unique challenges because those cancers present biodiversity in different patients based on different clonality of the tumor cells themselves and the multifaceted role of the TME. 7 Countless cancer patients have clinical benefits by immunotherapy, however many patients have experienced a minimal response to immunotherapeutic intervention, presumably owing to different subclasses of TME or tumor immune microenvironment (TIME), different metabolic characteristics, and adverse effects. 8 Hence, the diversity of TME or TIME and the complexity of cellular metabolism should be explored deeply. Several clinical trials are underway to develop the interventions targeting the “metabolic circuits” of TIME to enhance immunotherapy. 9 Increasing evidence claims that multidrug resistance (MDR) is an obstacle that impedes the anticancer efficacy of natural products, including paclitaxel and vincristine. Interestingly, immunotherapy is also a potential strategy to help combat against MDR with ICB treatment via targeting the overexpressed ATP‐binding cassette (ABC) transporters.
Until now, clinical practices and strategies are focused on primary immune escape and maximizing the immune response of the T‐cell compartment, however, only a few studies have investigated on secondary immune escape or other immune subtypes in response to ICB treatment. 10 Moreover, some patients with melanoma, urothelial cancer, Merkel cell carcinoma, or lung cancer have benefited from ICB treatment, which targets single molecular abnormalities with monoclonal antibodies (mAbs). To improve treatment options for cancer patients and achieve satisfied efficacies in ICB treatment, we should explore novel therapeutic agents, combinations or clinical trial approaches on the immune system across various types of cancers. 11
This review was summarized novel studies and viewpoints in advance of cancer immunotherapy and the use of natural products collected from Web of Science (http://www.webofknowledge.com), Medline (https://www.medline.com), PubMed (https://www.ncbi.nlm.nih.gov/pubmed), Scopus (https://www.scopus.com), and ClinicalTrials (https://clinicaltrials.gov). This key work provides an insight into the efficacy and mechanism of actions of cancer immunotherapies, including natural products and combined treatment with ICB, which confer an improved clinical outcome for cancer treatment.
2. CANCER IMMUNOTHERAPY
2.1. Adoptive cellular therapy
Adoptive cellular therapy aims to adoptively transfer lymphocytes to acquire antitumor effector function(Figure 1). This includes the use of chimeric antigen receptor (CAR)‐T cells, T‐cell receptor (TCR)‐T cells, and tumor‐infiltrating lymphocytes (TILs).
Figure 1.
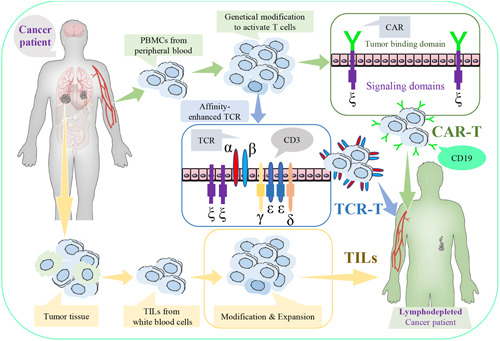
Adoptive cellular therapy. Adoptive cellular therapy includes the use of chimeric antigen receptor (CAR)‐T cells, T‐cell receptor (TCR), T cells, or tumor‐infiltrating lymphocytes (TILs) [Color figure can be viewed at wileyonlinelibrary.com]
CAR‐T cells are mainly derived from the peripheral blood of own patient or healthy donor and are rarely derived from donated umbilical cord blood and genetically modified to generate artificial TCRs as “living drugs” for cancer therapy. CAR‐T‐cell therapy has promising clinical efficacy against hematologic malignancies rather than solid tumors, this was mainly due to antigenic mismatch and an inhibitory microenvironment. 12 CAR‐T‐cell function can be enhanced by several novel strategies, including the promotion of donor cells through their CARs, discovery of specific antigens by tumor‐targeting adenoviruses, and enrichment of nonviral aptamer‐T cells in the TME using the sugar N‐azidomannosamine for metabolic labeling in solid tumors. 13 , 14 Moreover, the U.S. Food and Drug Administration (FDA) has approved to conduct a Phase I clinical trial for evaluating the safety and anticancer efficacy of a γ‐secretase inhibitor combined with B‐cell maturation antigen‐specific CAR‐T‐cell therapy against multiple myeloma. 15 Importantly, YESCARTA® and KYMRIAH® are U.S. FDA‐approved CAR‐T‐cell therapies modified by empowering the immune system via targeting B‐lymphocyte antigen CD19 to recognize and destroy non‐Hodgkin lymphoma cells or acute lymphoblastic leukemia cells. 16 In 2021, ABECMA®, first BCMA‐targeted CAR‐T‐cell therapy, has also been approved by U.S. FDA to treat relapsed or refractory multiple myeloma. However, CAR‐T‐cell therapy is not free from toxicities but can cause severe side effects, including neurologic toxicities and cytokine release syndrome, which limit its clinical application.
TCR‐T cells are generated from peripheral blood and engineered to recognize a tumor‐specific protein fragment/major histocompatibility complex (MHC) combination within cells in adoptive TCR‐T‐cell therapy. A robust clinical response leading to cancer cell destruction can be initiated by engineering autologous TCR‐T cells with the expression of immunogenic cancer‐testis antigen (CTA), thereby enhancing its affinity with synthetic biology. 17 For example, TCR New York Esophageal Squamous Cell Carcinoma‐1 (NY‐ESO‐1) and L‐antigen‐1 (LAGE‐1) are CTAs that are overexpressed in several types of cancer cells. The clinical trials at Phase I/II demonstrated that NY‐ESO‐1‐specific TCR‐T‐cell therapy is safe and displays promising clinical responses in multiple myeloma by recognizing NY‐ESO‐1 and LAGE‐1. 18 As different T lymphocytes can use the same functionally distinct TCR, sequencing of MHC and TCR α/β genes can expedite the analyses of T‐cell responses and specific ligands to understand the “immune landscape” in cancers. 19 , 20
TILs are white blood cells collected from the tumor tissue, which can be modified or expanded greatly and then infused back into the body to attack cancer cells. Cancer patients normally receive TIL therapy after chemotherapy and/or radiotherapy. Moreover, the combined therapies of TILs with interleukin‐2 (IL‐2) have elicited significant clinical responses to metastatic melanoma, skin cancer, metastatic head and neck cancer, and pleural mesothelioma in clinical trials. 21 Studies have suggested that CD8+‐enriched TILs are more therapeutically beneficial than unselected immature TILs, so future investigations should focus on exploring the use of adoptive transfer with TILs. 22
2.2. ICB therapy
ICB therapy targeting immune checkpoints has revolutionized cancer immunotherapy. Anticancer immunity can be restored by blocking CTLA‐4, PD‐1, and programmed death‐ligand 1 (PD‐L1) pathways (Table 1). Since 2011, the U.S. FDA has approved seven mAbs as immune checkpoint inhibitors for the clinical treatments of melanoma, lung cancer, urothelial cancer, Hodgkin lymphoma, cutaneous squamous cell carcinoma, head and neck squamous cell carcinoma, hepatocellular carcinoma, renal cell carcinoma, Merkel cell carcinoma, and breast cancer, including Atezolizumab (Tecentriq®, targeting PD‐L1), Avelumab (Bavencio®, targeting PD‐L1), Cemiplimab (Libtayo®, targeting PD‐1), Durvalumab (Imfinzi®, targeting PD‐L1), Ipilimumab (Yervoy®, targeting CTLA‐4), Nivolumab (Opdivo®, targeting PD‐1), and Pembrolizumab (Keytruda®, targeting PD‐1). The use of these drugs has been shown satisfactory responses in cancer patients via targeting the immune checkpoints of CTLA‐4, PD‐1, and PD‐L1 (Table 2). 23
Table 1.
Immune checkpoints in cancer
| Immune checkpoints | Expression in tumor or other cells | Mechanism of action | References |
|---|---|---|---|
| CTLA‐4 | FoxP3+ Treg cells or activated conventional T cells | CTLA‐4 of activated effector T cell (Teff) interacts with higher affinity to the ligand B7 than the receptor CD28, resulting in blocking T‐cell immune response | 24 |
| PD‐1 | Lymphocytes (T cells, B cells, monocytes, natural killer T cells, and macrophages) | PD‐1 interacts with PD‐L1 or PD‐L2 of cancer cells to generate immunosuppressive effect and suppress T‐cell activation through clustering with TCR, recruiting the inhibitory phosphatase SHP2 via ITIM, and inhibiting proximal TCR signaling | 25 |
| PD‐L1 | Tumor cells and lymphocytes | In addition to bind to PD‐1 and CD80 heterodimerize in cis to activate the costimulatory receptor CD28 while repress the inhibitory PD‐1 and CTLA‐4 pathways | 26 , 27 |
Abbreviations: CTLA‐4, cytotoxic T lymphocyte‐associated protein 4; ITIM, immunoreceptor tyrosine‐based inhibition motif; PD‐1, programmed cell death protein 1; PD‐L1, programmed death‐legend 1; SHP2, Src homology 2 domain‐containing tyrosine phosphatase 2; TCR, T‐cell receptor.
Table 2.
Anticancer drugs approved by U.S. FDA for immunotherapy in clinical practice
| Generic name | Brand/manufacturer | FDA approval year | Target | Combination | Indication(s) | References |
|---|---|---|---|---|---|---|
| Atezolizumab | TECENTRI® Genentech | 2016 | PD‐L1 | Bevacizumab, paclitaxel, and carboplatin; paclitaxel protein‐bound and carboplatin; paclitaxel protein‐bound; carboplatin and etoposide; bevacizumab | Metastatic urothelial carcinoma (UC); metastatic non‐small‐cell lung cancer (NSCLC); unresectable locally advanced or metastatic triple‐negative breast cancer; extensive‐stage small‐cell lung cancer (SCLC); unresectable or metastatic hepatocellular carcinoma (HCC) | https://www.fda.gov/ |
| Avelumab | BAVENCIO® Merck and Pfizer Inc. | 2017 | PD‐L1 | Axitinib | Metastatic Merkel cell carcinoma (MCC); locally advanced or metastatic UC; advanced renal cell carcinoma (RCC) | https://www.fda.gov/ |
| Cemiplimab | LIBTAYO® REGENERON and SANOFI | 2018 | PD‐1 | Locally advanced or metastatic cutaneous squamous cell carcinoma (CSCC) | https://www.fda.gov/ | |
| Durvalumab | IMFINZI® AstraZeneca | 2017 | PD‐L1 | Etoposide plus either carboplatin or cisplatin | Metastatic UC; unresectable Stage III NSCLC; extensive SCLC | https://www.fda.gov/ |
| Ipilimumab | YERVOY® Bristol‐Myers Squibb | 2011 | CTLA‐4 | Nivolumab; nivolumab and 2 cycles of platinumdoublet chemotherapy | Unresectable or metastatic melanoma; advanced RCC; microsatellite instability‐high (MSI‐H) or mismatch repair deficient (dMMR) metastatic colorectal cancer (CRC); HCC; metastatic NSCLC | https://www.fda.gov/ |
| Nivolumab | OPDIVO® Bristol‐Myers Squibb | 2014 | PD‐1 | Ipilimumab; ipilimumab and two cycles of platinum‐doublet chemotherapy | Unresectable or metastatic melanoma; metastatic NSCLC; metastatic SCLC; advanced RCC; classical Hodgkin lymphoma (cHL); recurrent or metastatic squamous cell carcinoma of the head and neck (HNSCC); advanced or metastatic UC; MSI‐H or dMMR metastatic CRC; HCC; unresectable advanced, recurrent or metastatic esophageal squamous cell carcinoma (ESCC) | https://www.fda.gov/ |
| Pembrolizumab | KEYTRUD® Merck & Co | 2014 | PD‐1 | Pemetrexed and platinum chemotherapy; carboplatin and either paclitaxel or paclitaxel protein‐bound; axitinib; lenvatinib | Unresectable or metastatic melanoma; metastatic NSCLC; metastatic SCLC; metastatic or with unresectable, recurrent HNSCC; cHL; primary mediastinal large B‐cell lymphoma; locally advanced or metastatic UC; MSI‐H or dMMR CRC; MSI‐H or dMMR cancer; recurrent locally advanced or metastatic gastric or gastroesophageal junction adenocarcinoma; locally advanced or metastatic ESCC; recurrent or metastatic cervical cancer; HCC; MCC; advanced RCC; advanced endometrial carcinoma; tumor mutational burden‐high cancer; recurrent or metastatic CSCC; early triple‐negative breast cancer | https://www.fda.gov/ |
Abbreviations: CTLA‐4, cytotoxic T lymphocyte‐associated protein 4; FDA, Food and Drug Administration; PD‐1, programmed cell death protein 1; PD‐L1, programmed death‐legend 1.
3. CANCER IMMUNITY AND IMMUNE CHECKPOINTS
3.1. Dysregulation of immune checkpoints in cancer
Immune checkpoints operate as brake to prevent autoimmune response and are expressed on both the surfaces of cancer cells and T cells. The binding of immune checkpoints between cancer cells and T cells inhibits the activation of T cells, which results in weakening the immune attack to recognize cancer cells. 28 In TIMs, DCs, and antigen‐presenting cells recognize the cytokines released from cancer cells and immune cells, including adenosine, colony‐stimulating factor‐1, IL‐10, prostaglandin E2, transforming growth factor‐β (TGF‐β), and vascular endothelial growth factor. 29 The ligand B7 of DCs recognizes the receptor CD28 of T cells to activate the proliferation and migration of CTLs towards tumors. Conversely, CTLA‐4 of activated Teff cells, an inhibitory receptor of the immune response, interacts with a higher affinity to ligand B7 than that of receptor CD28, resulting in blockade of the T‐cell immune response. 30 CTLA‐4 is highly expressed in some types of cancers but is downregulated in other types of cancers, which results in different clinical outcomes. 31 For example, overexpression of CTLA‐4 indicates poor prognosis in patients with melanoma, mesothelioma, non‐small‐cell lung cancer (NSCLC), and nasopharyngeal carcinoma, but confers positive clinical outcomes in patients with glioma and B‐cell chronic lymphocytic leukemia. 32 , 33
Unlike the inhibitory receptor CTLA‐4, expression of which is exclusive to activated T cells, PD‐1 is also exerted in myeloid and B cells, acting as a secondary immune response and requiring transcriptional activation. PD‐1 interacts with PD‐L1 or PD‐L2 in cancer cells to suppress T‐cell activation by clustering with TCRs, thereby recruiting the inhibitory phosphatase Src homology 2 domain‐containing tyrosine phosphatase 2 via the immunoreceptor tyrosine‐based inhibition motif, and inhibiting proximal TCR signaling. 34 In low‐risk endometrial carcinoma, there is a significant increase in the expression and immunophenoscore of the immune checkpoints, including PD‐1, PD‐L1, and PD‐L2. In contrast, the low expression of PD‐1 in cytotoxic CD8+ TILs confers a specific TIME in NSCLC patients with a good prognosis. A significantly worse prognosis has been documented in patients with hepatocellular carcinoma with overexpression of PD‐L1 and PD‐L2. 35 , 36 The expression or prognostic value involved in PD‐L1 and PL‐L2 are quite distinct depending on microenvironmental stimuli and different types of cancer. For example, PD‐L2 has an overlapping function with PD‐L1 as a second ligand for PD‐1 and brakes T‐cell activation, but engagement with PD‐1 does not inhibit T‐cell proliferation at high antigen concentrations. 37 As PD‐L2 exerts a key functional role in T‐helper cell 2 (Th2) response regardless of Th1 response, it seems that targeting PD‐L2 alone may not be sufficient to mediate anticancer immunity. In fact, PD‐L2 status is also a valuable prognostic factor that can provide clinical benefits to patients with anti‐PD‐1 therapy, because the clinical response was shown to be greater in both PD‐L1‐ and PD‐L2‐positive cancer patients (27.5%) than alone PD‐L1‐positive cancer patients (11.4%). 38 Therefore, it is a sufficient rationale to further develop the novel strategies to enhance the therapeutic efficacy by combining the blockades of CTLA‐4, PD‐1, PD‐L1, and PD‐L2 for cancer therapy.
3.2. The regulatory factors of immune checkpoints in TME
The efficacy of cancer immunotherapy is affected by the TME, TIME, tumor mutational burden (TMB), TILs, and intestinal bacteria. TME is the specific environment around a tumor, and it is built with TILs, fibroblasts, blood vessels, exosomes, and extracellular matrix, as well as shaped by acidity, hydrogen peroxide, adenosine triphosphate (ATP), TGF‐β, hypoxia‐inducible factor (HIF)‐1, and IL‐38. 39 , 40 Increasing evidence supports the notion that TME manipulation can enhance the therapeutic efficacy of ICB. Given the fact that TME shows partiality for the ineffective Th2 immune response in the immune milieu, this allows cancer cells to evade the immune system. 41 Adenosine, an immunosuppressive factor, is released in the TME owing to the degradation of extracellular ATP in the P2 purinergic pathway. In contrast, ATP and adenosine are generated in higher levels at the tumor sites. 42 In addition, combined PD‐1 and CTLA‐4 blockade increase Teff infiltration, which leads to a highly advantageous percentage of Teff to regulatory T (Treg) cells in the tumor, thereby shifting the TME from being immunosuppressive to becoming inflammatory status. 43 Based on the exploration of TME, enhanced immune cell infiltration and TMB are found in low‐risk endometrial carcinoma, thereby indicating a better prognosis. 44 TMB indirectly represents the tumor antigenicity encoded by somatic mutations in the tumor, whereas T‐cell‐inflamed gene expression profile and PD‐L1 expression are inflammatory indicators of the T‐cell‐inflamed TME, which can help predict therapeutic response to anti‐PD‐1 immunotherapy. 45 Nivolumab, an anti‐PD‐1 mAb, induces functional expansion of mutation‐associated and neoantigen‐specific T cells from the primary tumor to peripheral blood. It was shown to induce a pathological response to 45% of resected lung cancer cells as predicted by the TMB with few side effects, regardless of PD‐L1 expression. 46
TILs are white blood cells and traffic towards tumor beds. This confers different and distinct immune cell populations, including leukocytes, macrophages, DCs, and mast cells, with specific immunity against cancer cells. 47 The molecules, populations, and subsets of TILs are highly associated with the tumor prognosis and clinical outcomes of cancer immunotherapy. High levels of CD4+ and CD8+ TILs are independent and good prognostic biomarkers in cancer patients, which are significantly associated with improved overall survival (OS). 48 Besides, the density of tumor‐infiltrating CD45RO+ cells linked to microsatellite instability is a good prognostic factor of longer survival in patients with colorectal cancer (CRC) that is independent of pathological, clinical, and molecular features. 49 A discrete population of PD‐1‐high tumor‐infiltrating CD8+ T lymphocytes in patients with gastric cancer and hepatocellular carcinoma, who might benefit from combined ICB treatment. 50 , 51 However, PD‐1‐negative cytotoxic CD8+ TILs confer an immune‐privileged microenvironment with a positive prognosis and satisfactory response to immunotherapy in advanced NSCLC. 36 These effector‐ and memory‐TILs rather than naive‐precursor‐like TILs contain tumor antigen‐specific cells. Those subsets of CD8+ TILs exhibit proliferative and effector capacities in response to different types of ICB therapies across distinct tumor models. 52 Furthermore, CD39 expression of CD8+ TILs is phenotypically distinct that aids to identify bystander T cells and can help to predict diagnostic clinical parameters. 53 The population of CD39‐high tumor‐infiltrating CD8+ T lymphocytes is increased with tumor growth towards an exhausted phenotype, which results in an immunosuppressive TME that may promote PD‐L1 upregulation in tumors. 54 Moreover, tumor‐infiltrating T and B lymphocytes play a role in functional interaction due to close proximity to enhance local immune activation, thereby leading to a better prognosis for cancer patients. 55
The host immune system involves the exchange and balance of the microbial community in the gut, known as the gut microbiome, even for cancer patients receiving ICB treatment. Cancer patients with promising anticancer immunity to PD‐1 immunotherapy have a favorable gut microbiome owing to high alpha diversity and a commensal microbiome. 56 For example, patients with metastatic melanoma who are undergoing ICB treatment have been shown to have longer OS for carriership of Streptococcus parasanguinis and longer progression‐free survival (PFS) for carriers of Bacteroides massiliensis, but carriership of the family Peptostreptococcaceae predicts shorter OS and PFS. 57 Moreover, abundant bacterial species in treatment responders, including Bifidobacterium longum, Collinsella aerofaciens, and Enterococcus faecium, can enhance the anticancer efficacy to PD‐L1 immunotherapy by improving tumor control via augmenting T‐cell immune responses in patients with metastatic melanoma. 58 The relative abundance of Akkermansia muciniphila could restore the efficacy of PD‐1 immunotherapy by increasing the enrichment of CCR9+CXCR3+CD4+ T cells into the epithelial tumor sites depending on IL‐12 secretion from DCs. 59 The efficacy of CTLA‐4 immunotherapy favors the microbiota composition of Bacteroides fragilis and/or Bacteroides thetaiotaomicron and Burkholderiales with IL‐12‐dependent Th1 immunity, thereby sparing intestinal integrity and leading to tumor control in patients with metastatic melanoma or NSCLC. 60 Therefore, the precise ability to predict the immune response by identifying the unique classes and subsets of the TME and TIME in cancer patients will improve the efficacy to ICB.
3.3. The cytokines and pathways involved in ICB
Cytokines, small cell‐signaling proteins, play a critical role in the integrative regulation of cancer immunity and ICB. A blockade of CTLA‐4 or PD‐1/PD‐L1 can increase interferon γ (IFN‐γ) expression in response to enhanced chemokine‐driven immune cell infiltration in tumor cells. Hence, IFN‐γ deficiency in TME is closely associated with primary resistance to ICB therapy. 61 , 62 , 63 A blockade of anti‐CTLA‐4 results in a greater amount of CD4+ inducible costimulatoryhi T lymphocytes and enhanced IFN‐γ release in tumor and lymph nodes. 64 , 65 PD‐1 expression on CD8+ T lymphocytes can be decreased by alone blockade of IFN‐γ or PD‐1, as well as combined blockade of IFN‐γ and PD‐1. 66 A successful response to PD‐1 blockade requires IL‐12 to be released from DCs to sensitize IFN‐γ produced from T cells. 67 Moreover, combined treatment with α‐CTLA‐4 and α‐PD‐1 blockade eradicates tumor synergistically by upregulating IL‐7Rα expression on tumor‐infiltrating T lymphocytes depending on IFN‐γ/IFN‐γ R pathways. 68 Notably, IFN‐γ stimulation increases the basal level or exosomal level of PD‐L1 to induce immunosuppression and facilitate tumor growth. However, the impairment of IFN‐γ signaling is not responsible for MHC‐I reduction and the anticancer sensitivity to PD‐L1 blockade. 56 , 69 , 70 It comes as no surprise that other key regulators also play important roles in ICB.
Chemokine (C‐X‐C motif) receptor 3 (CXCR3), a chemokine receptor, is overexpressed on Teff. CXCR3 and its ligand, chemokine (C‐X‐C motif) ligand (CXCL)9, can facilitate the interactions of CD8+ T cells or CD103+ DCs within the TME to promote a clinical response to PD‐1 blockade. 71 CXCL10 and CXCL11, other key members of chemokines, are the ligands of CXCR3 stimulated by IFN‐γ to induce T‐cell recruitment and enhance immune response. 72 , 73 TGF‐β is a crucial “enforcer” that promotes tumor emergence and is an immunosuppressive factor within the TME, which results in immune evasion and a poor outcome from cancer immunotherapy. 74 A blockade of phosphoinositide 3‐kinase (PI3K) in DCs suppresses the expressions of TGF‐β and IL‐10 induced by the Toll‐like receptor 5 (TLR‐5) ligand flagellin. The latter can be combined with a TLR agonist to induce IFN‐γ+IL‐17+ polyfunctional T lymphocytes to strengthen the immune response to cancer therapy. 75 , 76 A blockade of TLR7/8 expression can improve resistance to cancer immunotherapy by modulating the functional polarization of tumor‐associated macrophages towards an antitumorigenic M1 phenotype in TIMs. 77
Several other important transcription factors and protein kinases are also involved in cancer immunity and immunotherapy. The transcription factor T‐cell factor 1 (TCF‐1) is a useful biomarker for the adaptive immune response to cancer and is essential for the stem‐like functions of TCF1+PD‐1+CD8+ T cells in ICB. 78 An immunotherapy resistance program has been identified using single‐cell RNA sequencing from NSCLC and melanoma tumors. A blockade of cyclin‐dependent kinase 4/6 (CDK 4/6) expression may repress this program to promote immunotherapeutic efficacy and overcome resistance to cancer immunotherapy in vivo. 79 Alterations in serine/threonine kinase 11 (STK11)/liver kinase B1 (LKB1) expression were identified to be the most genomic resistance drivers in anti‐PD‐1 immunotherapy. This action leads to suppression of the expression of stimulator of IFN genes (STING) in aggressive and PD‐L1− Kirsten rat sarcoma viral oncogene homolog‐mutant lung adenocarcinoma. 80 , 81 Emerging therapeutic strategies to restore the expressions of LKB1 or STING may promote antitumor immune response in cancers with resistance to ICB treatment. Overall, the exact mechanisms by which immune checkpoints act against cancers and the suppressive factors of the TME are incompletely understood (Figure 2).
Figure 2.
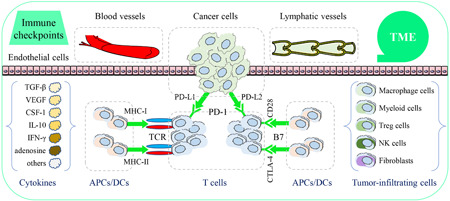
Immune checkpoints and tumor immune environment (TME). Immune checkpoints are expressed in cancer cells or immune cells and include programmed cell death protein 1 (PD‐1), the programmed death‐ligand 1/2 (PD‐L1/2), and the cytotoxic T lymphocyte‐associated protein 4 (CTLA‐4). TME is a specific environment and shaped around cancer cells, T cells, tumor‐infiltrating cells, fibroblasts, blood vessels, lymphatic vessels, exosomes, and extracellular matrix [Color figure can be viewed at wileyonlinelibrary.com]
3.4. The relationship between immune checkpoints and MDR
A major drawback for successful chemotherapy is the development of cellular resistance to multiple anticancer drugs that are structurally unrelated, in which the phenomenon is termed as MDR. Therefore, this cellular resistance can dramatically reduce the efficacy of cancer treatments. Although several mechanisms involved in MDR were identified, the most common mechanism is through a family of energy‐dependent transporters, known as ABC transporters. These transporters increase the efflux of hydrophobic cytotoxic drugs. 82 Several ABC transporters overexpressed in MDR were first described in the 1970s, such as P‐glycoprotein (P‐gp, also known as ABCB1 or MDR1) and multidrug resistance protein 1 (also known as ABCC1). In fact, targeting a single pathway is hard to tackle chemoresistance due to the multifaceted MDR, thus overcoming MDR has been a major goal for cancer biologists by using novel and multiple strategies during the past 50 years. 83
Cancer immunotherapy could be a vital adjunct strategy to circumvent and overcome MDR, as resistance to immunotherapy is generally unrelated to the mechanisms of resistance in response to cytotoxic agents. 84 In this regard, novel approaches are applied to overcome tumor MDR with immunotherapy, including (1) antibody strategies 85 ; (2) antibody‐directed toxins 86 ; (3) adoptive cell therapy 87 ; (4) cytokine‐based strategies 88 ; (5) cancer stem cells or cancer‐initiating cells 89 , 90 ; (6) immune checkpoint inhibitors 91 ; (7) cancer vaccines 92 ; (8) oncolytic viruses. 93 Currently, the strategy of ICB is regarded as an emerging tool for cancer treatment, which activates T cells to attack tumor cells by blocking the negative regulatory signal. Recent studies reveal that the expression of immune checkpoints is induced by chemotherapy in some types of cancers, thus leading to MDR. For example, chemotherapy induces several immunological changes in ovarian cancers, including PD‐L1 upregulation. 94 PD‐L1 or PD‐1 expression is positively associated with P‐gp expression in breast and gastric cancers, respectively. Moreover, PD‐L1 or PD‐1 expression is upregulated in line with P‐gp expression in drug‐resistant breast cancer and gastric cancer. 95 , 96 , 97 Interestingly, 5‐carboxy‐8‐hydroxyquinoline, a histone demethylase inhibitor, in combination with doxorubicin does not only decrease P‐gp expression but also inhibits PD‐L1 expression in cancer cells, thereby promoting T‐cell infiltration and suppressing tumor growth in vivo. 98 Indeed, novel drug delivery system (DDS), including versatile nanoparticles and liposomal formulation combined with PD‐1/PD‐L1 or antibody‐free blockade may activate immune responses and inhibit P‐gp expression in cancer therapy. 99 To the best of our knowledge, few studies have been performed to investigate the relationship between CTLA‐4 and ABC transporters. Thus, new therapeutic modalities targeting immune checkpoints combined with the inhibitors of ABC transporters used to overcome MDR, are urged to develop as complement chemotherapy.
3.5. The application of approved immune checkpoint inhibitors and their disadvantages
Until now, some mAbs targeting PD‐1 (e.g., cemiplimab, dostarlimab, nivolumab, and pembrolizumab), PD‐L1 (e.g., atezolizumab, avelumab, and durvalumab), and CTLA‐4 (e.g., ipilimumab) have been approved by the U.S. FDA as immune checkpoint inhibitors for immunotherapy or monoclonal therapy of several malignancies in the United States. Although these mAbs display favorable therapeutic effects in clinical practices, disadvantages, such as nonresponsiveness, immune‐related adverse events, immunogenicity, difficult manufacture, and high production costs, have been observed and restrictions are imposed for their clinical uses. 84 , 100 Sufficient evidence indicates that ICB with mAbs induces severe immune‐related adverse events, such as myocarditis, 101 acute kidney injury, 102 liver injury, 103 neurological toxicity, 104 and inflammatory arthritis. 105 Moreover, these mAbs have poor delivery and is permeability limited into the tumor tissues (i.e., high interstitial fluid pressure) due to their large molecular size (∼150 kD), which reduces their therapeutic efficacies, and it is urged to improve the DDS for monoclonal therapy. 106 The relatively long biological half‐life of these mAbs enhances the time for drug elimination, thereby leading to severe adverse effects. 107
The use of mAbs can result in severe adverse effects, but the rechallenge of ICB with mAbs is acceptable for cancer patients who are suffering from immune‐related adverse events. 108 Alternatively, small molecule inhibitors can penetrate into the tumor and have favorable oral bioavailability. These small molecule inhibitors have other advantages, including fewer adverse effects, that are better for self‐administration and less expensive than mAbs and have a shorter biological half‐life. Therefore, this has attracted great interest to pharmaceutical companies. However, most of these small molecule inhibitors targeting immune checkpoints are still in the preclinical stage. 109 At present, clinical studies have suggested that small molecule inhibitors are biosafe and can block tumor growth potently when compared to mAbs. Some synthetic small molecules from Bristol‐Myers Squibb (e.g., BMS‐1166 and BMS‐202) and Curis Inc. (i.e., CA‐170: Phase I) display potent antitumor activities as orally bioavailable inhibitors via interrupting PD‐1/PD‐L1 interactions in advanced tumors and lymphomas. 110 However, there are only several reports and clinical studies focusing on the use of natural products as small molecule inhibitors. Therefore, increasing interest has been driven towards novel potential modulators from natural products for tumor immunotherapy, a worldwide hot research topic. Biological resources for the discovery of antitumor drugs from nature remain abundant, and persistent search may identify new chemical entities with antitumor activity. Plants, a major source of complex and highly structurally and functionally diverse phytochemicals, such as phenols, polyphenols, polysaccharides, tannins, peptides, terpenes, and alkaloids, may serve as a source of lead compounds for the development of novel immune checkpoint inhibitors with improved pharmacological effects that can be used as adjuvant therapy to enhance the potency of chemotherapeutic drugs against cancers.
4. POTENTIAL NATURAL PRODUCTS FROM HERBAL MEDICINES WITH IMMUNOMODULATORY EFFECTS ON IMMUNE CHECKPOINTS
Herbal medicines have been widely used as adjuvant agents for cancer treatment and can also enhance the sensitivity of chemotherapeutic resistance. Moreover, several naturally bioactive compounds isolated from herbal medicines have been shown to modulate immune checkpoints in tumors, as well as promoting anticancer immunity (Table 3). As herbal medicines target multiple pathways, they do not modulate immune checkpoints only, but can also modulate the activities of several types of immune cells that contribute to anticancer immunity, including T lymphocytes, B lymphocytes, Treg cells, DCs, and natural killer (NK) cells, thereby modulating the TME. In addition, herbal medicines have low toxicity and fewer side effects than that of mAb blockers, so naturally bioactive compounds from medicinal herbs could be potential agents for the treatment of cancers. The chemical structures of the natural products from medicinal herbs with immunomodulatory effects on immune checkpoints are presented in Figure 3.
Table 3.
The lead compounds derived from natural products in cancer immunotherapy
| Natural compounds | Immune checkpoint | Cancer or immune system | Experimental models | Mechanisms | Pharmacological effects | References |
|---|---|---|---|---|---|---|
| Apigenin | PD‐L1 |
Breast cancer; lung cancer; melanoma; pancreatic cancer |
MDA‐MB‐468, SK‐BR‐3, 4T1 cells; H460, H358, A549 cells; A‐375, A2058, RPMI‐7951 cells; Panc02 cells; genetically engineered KRASLA2 mice; B16‐F10 xenograft mice; Panc02 xenograft mice. In vitro dose: 10–60 µM; in vivo dosage: 10–150 mg/kg |
Reduce PD‐L1 expression in the tumor and DCs; inhibit IFN‐γ‐ and IFN‐β‐induced PD‐L1 upregulation; reduce Treg cell population; promote T‐cell proliferation; promote CD4+ and CD8+ T‐cell infiltration into the tumor; enhance IL‐2 secretion in the coculture of A375 cells and Jurkat T cells |
Induce cancer cell apoptosis and cell cycle arrest at G2/M phase; induce T‐cell‐mediated cell death when cocultured with Jurkat T cells; activate T‐cell antitumor immunity and inhibit immunosuppression; suppress tumor growth and induce apoptosis; promote T‐cell‐mediated cancer cell killing; increase mouse survival rates and reduced tumor burden; restore T‐cell homeostasis and antitumor immune response |
111 , 112 , 113 , 114 |
| Berberine | PD‐L1 |
Breast cancer; leukemia; lung cancer |
HL‐60 cells; A549, H175, H460, H1299, H1975, H358, HCC827, Lewis cells; urethane‐induced mouse lung cancer model; H22 xenograft mice; Lewis lung xenograft mice. In vitro dose: 5–10 µM; in vivo dosage: 4–10 mg/kg |
Downregulate PD‐L1 expression; trigger PD‐L1 degradation through ubiquitin/proteasome‐dependent pathway by directly binding to CSN5 and inhibiting its activity; reduce PD‐L1 activity, thus activating T cells; reduce serum PD‐L1 levels; decreases the populations of MDSCs and Treg cells in the tumor; enhance T‐cell infiltration into the tumor and activated cytotoxic T cells |
Regulate neutrophil phenotypes to reverse doxorubicin‐induced cancer cell resistance and carcinogenesis; suppress tumor growth; enhance the sensitivity of tumor cells when cocultured with T cells; promote the sensitivity of cancer cells to T‐cell killing |
115 , 116 |
| Chrysophanol | PD‐L1 | T‐cell acute lymphoblastic leukemia |
CCRF‐SB, Loucy, Jurkat T cells, TALL‐104 cells; Jurkat T‐cell xenograft mice. In vitro dose: 10–40 µM; in vivo dosage: 20–40 mg/kg |
Upregulate miR‐9 to target PD‐L1 by binding to its 3ʹ‐untranslated region |
Inhibit cancer cell proliferation, cell migration and invasion; induce cancer cell apoptosis; suppress tumor growth and metastasis |
117 |
| Curcumin |
PD‐1; PD‐L1; PD‐L2 |
Cervical and uterine cancer; Ehrlick's ascites carcinoma; head and neck cancer; hepatocellular carcinoma; Lewis lung carcinoma; melanoma; tongue squamous cell carcinoma |
EAC cells; SNU1041 and SCC15 cells; 3LL cells; A‐375, A2058, RPMI‐7951 cells; Cal 27 and FaDu cells; Hep3B and CSQT‐2 cells; MC38 murine tumor model; 4‐nitroquinoline‐oxide‐induced tongue carcinoma mice; Lewis lung carcinoma xenograft mice; 3LL xenograft mice. In vitro dose: 5–80 µM; in vivo dosage: 50–100 mg/kg |
Reduce PD‐1, PD‐L1, PD‐L2, TIM‐3, and galectin‐9 expressions; inhibit IFN‐γ‐induced STAT1 phosphorylation; restore DC cells by inhibiting directly STAT3; enhance CD8+ T‐cell infiltration into the tumor, IFN‐γ secretion from CD3+ T cells, and IL‐2 secretion in the coculture of A‐375 cells and Jurkat T cells; decrease the populations of CD4+CD25+Foxp3+ Treg cells and MDSCs, and IL‐6 levels in the tumor and serum; inhibit tumor‐induced depletion of T cells, and increase CD4+ and CD8+ T‐cell infiltration into the tumor; restore effector and memory T‐cell populations in the tumor; block Treg cell suppressive activity; reduce the population of MDSCs in the spleen and tumor and the secretion of TGF‐β and IL‐10 in Treg cells |
Inhibit cancer cell proliferation; induce cancer cell apoptosis and cell cycle arrest at G2/M phase; suppress tumor growth and increase mouse survival rates; enhance tumor‐specific cytotoxic T‐cell proliferation and lymphocyte proliferation; regulate immune checkpoint blockade and T‐cell dysfunction; modulate TME; induce T cell‐mediated cell death when cocultured with Jurkat T cells |
112 , 118 , 119 , 120 , 121 , 122 |
| CTLA‐4 |
CD4+CD25+ Treg cells; CD4+CD25− T cells |
T cells isolated from mouse splenocytes. In vitro dose: 5–20 µM; in vivo dosage: 50–100 mg/kg |
Reduce CTLA‐4 and Foxp3 expressions in CD4+CD25+ Treg cells; prevent p65 and c‐Rel nuclear translocation; decrease TGF‐β1 secretion and IL‐2 production; increase IL‐4 expression and decrease IFN‐γ secretion in the coculture of CD4+CD25+ Treg cells and CD4+CD25− T cells |
Inhibit cell–cell contract; block the suppressive activity of CD4+CD25+ Treg cells; impart immunosuppression |
123 | |
| β‐Elemene | PD‐L1 | Esophageal cancer |
TE‐1 and KYSE‐150 cells; TE‐1 xenograft mice; KYSE‐150 xenograft mice. In vitro dose: 10–50 µg/ml; in vivo dosage: 100 mg/kg |
Reduce PD‐L1 expression |
Suppress cancer cell proliferation, migration and invasion; induce cancer cell apoptosis and cell cycle arrest at G1 phase; inhibit tumor growth |
124 |
| Epigallocatechin gallate (EGCG) |
PD‐L1; PD‐L2 |
Lung cancer; Melanoma |
A549 and H1299 cells; Lu99 cells; TC‐1 xenograft mice. In vitro dose: 10–50 µM; in vivo dosage: 0.1–2.5 mg/kg |
Inhibit IFN‐γ‐induced upregulation of PD‐L1 or PD‐L2 via inhibiting Jak2/STAT1 signaling; inhibit EGF‐induced PD‐L1 upregulation; increase the population of CD8+ T cells in the spleen |
Induce tumor cell apoptosis; suppress tumor growth; enhance antitumor immune responses |
125 , 126 |
| Gallic acid | PD‐L1 |
Colorectal cancer; lung cancer |
HCT 116 and HT‐29 cells; A549 and H292 cells. In vitro dose: 20–906.3 µM. |
Decrease PD‐L1 expression; inhibit the phosphorylation of EGFR, PI3K, and AKT; activate p53; upregulate miR‐34a |
Inhibit cancer cell proliferation; induce cancer cell apoptosis; activate T‐cell‐mediated immune response |
127 , 128 |
| Ginsenoside Rg3 |
PD‐1; PD‐L1 |
Breast cancer; lung cancer |
MDA‐MB‐231 and BT‐549 cells; A549 and A549/DDP cells; MDA‐MB‐231 xenograft nude mice. In vitro dose: 2–160 µg/ml |
Decrease IFN‐γ‐induced PD‐L1 upregulation and cisplatin‐induced PD‐L1 upregulation; reduce PD‐1 expression in activated T cells; stimulate the production of IFN‐γ, IL‐2, IL‐9, IL‐10, IL‐22, and IL‐23 |
Suppress cancer cell proliferation; induce cancer cell apoptosis; resume immune; potentiate CD8+ T‐cell cytotoxicity when cocultured with A549/DDP cells; attenuate MDA‐MB‐231 breast cancer cell growth in vivo |
129 , 130 |
| Ginsenoside Rh2 | PD‐L1 |
Melanoma; lung cancer |
B16‐F10 cells; A549 and H1299 cells; B16‐F10 xenograft mice. In vivo dosage: 0.2–0.5 mg/kg |
Inhibit cisplatin‐induced PD‐L1 upregulation via superoxide; enhance CD4+ and CD8+ T‐cell infiltration into the tumor |
Enhance cisplatin‐induced cancer cell apoptosis/cyototoxicity in the tumor and T‐cell cytotoxicity in the spleen; decrease cisplatin‐induced autophagy; increase mouse survival rates; suppress tumor growth |
131 , 132 |
| Ginsenoside Rh4 | PD‐L1 | Esophageal cancer |
Eca109 and KYSE‐150 cells; Eca109 xenograft mice. In vitro dose: 20–100 µM; in vivo dosage: 20–40 mg/kg |
Inhibit PD‐L1 expression via AKT/mTOR pathway |
Suppress cancer cell growth, colony formation, and aerobic glycolysis; induce cell cycle arrest at G1 phase; inhibit tumor growth |
133 |
| Ginsenoside Rk1 | PD‐L1 | Lung cancer |
A549 and PC9 cells; A549 xenograft mice. In vitro dose: 50–150 µM; in vivo dosage: 10–20 mg/kg |
Reduce PD‐L1 expression by inhibiting NF‐κB and caspase‐dependent pathways |
Inhibit cancer cell proliferation and colony formation; block tumor growth; induce cell cycle arrest at G1 phase; induce cancer cell apoptosis |
134 |
| β‐Glucans | PD‐L1 |
Lung cancer; breast cancer; melanoma; osteosarcoma |
Lewis xenograft mice; EO771 xenograft mice; B16‐F10 xenograft mice; K7 M2‐Luc2 xenograft mice. In vitro dose: 50–100 µg/ml; in vivo dosage: 800 µg/mouse, 25 mg/kg |
Upregulate PD‐L1 mRNA levels in mouse peritoneal macrophages and tumor; downregulate PD‐L1 expression in tumor‐educated DCs; upregulate CD40, CD80, CD86, and MHC‐II in tumor‐educated DCs; activate dectin‐1; stimulate the secretion of TNF‐α and IL‐12p70 in tumor‐educated DCs; alter tumor‐associated macrophage phenotype towards M1 phenotype; polarize M2 phenotype into M1 phenotype in bone marrow–derived macrophages; enhance T‐cell priming; increase the population of IFN‐γ producing CD4+ T cells in the spleen and tumor; increase the populations of CD11b+F4/80+ macrophages and CD11b+Gr‐1+ granulocytes in the tumor; increase the populations of effector memory, central memory T cells, and CD11c+CD8+DCs in the tumor draining lymph nodes; increase CD4+ and CD8+ T‐cell infiltration into the tumor; increase mRNA levels of CD8α, CXCL9, CXCL10, granzyme B, IFN‐γ, IL‐12p40, iNOS, IRF‐1, and TNF‐α in the tumor; increase the mRNA levels of IL‐6, IL‐12, and TNF‐α in tumor‐associated macrophages; increase the mRNA levels of IL‐6, IL‐12p40, iNOS, and TNF‐αin tumor‐educated DCs; decrease the population of CD4+Foxp3+ Treg cells in the spleen and tumor; decrease Treg cell differentiation; decrease the mRNA levels of arginase, TGF‐β, IL‐17, and Foxp3 in the tumor; decrease the mRNA levels of arginase and IL‐10 in tumor‐associated macrophages; decrease the mRNA levels of TGF‐β and arginase in tumor‐educated DCs |
Decrease tumor burden; inhibit tumor growth; enhance mouse survival rates; modulate macrophage polarization and immunosuppressive TAM conversion; regulate macrophages, DCs, T cells, and NK cells |
135 , 136 , 137 , 138 |
| Resveratrol |
PD‐1; PD‐L1 |
Melanoma; breast cancer; renal cell carcinoma; leukemia; lymphoma; colon carcinoma |
B16‐F10 cells; 4T1, MDA‐MB‐231, and JIMT‐1 cells; L1210 cells; 4T1 xenograft mice; Renca xenograft mice; L1210 xenograft mice; EG7 xenograft mice; CT‐26 xenograft mice; B16‐F10 xenograft mice; cyclophosphamide‐induced immunosuppressive mice. In vitro dose: 0.1–100 µM; in vivo dosage: 12.5–100 mg/kg |
Downregulate PD‐1 expression in T cells; upregulate PD‐L1 expression in cancer cells; interact with the inner surface of PD‐L1; decrease the population of Treg cells is in the tumor; decrease the populations of tBreg and Treg cells in the spleen and lymph nodes; decrease the population of CD4+CD25+Treg cells in the spleen; increase the population of IFN‐γ‐expressing CD8+ T cells in the lymph nodes; increase the populations of CD3+ and CD4+ T cells in the peripheral blood; increase CD4/CD8 ratios in the peripheral blood T cells; enhance spleen lymphocyte proliferation; enhance CD8+ T‐cell infiltration into the tumor; enhance T‐cell‐mediated tumor cell killing; increase NK cell activity; increase the mRNA levels of IFN‐γ and FoxO1 in the tumor; increase NF‐κB p65 expression in the spleen; convert macrophages to M1 phenotype in the tumor; inhibit TGF‐β secretion from tBreg cells and spleenocytes; reduce serum IL‐2 and NF‐κB levels; decrease the mRNA levels of IL‐6 and IL‐10 in the tumor |
Induce cancer cell cytotoxicity and apoptosis; inhibit cancer cell proliferation; induce cell cycle arrest at G0/G1 phase; suppress tumor growth and metastasis; promote antitumor T‐cell immunity; increase mouse survival rates; suppress triple‐negative breast cancer lung metastasis by elevating local antitumor immunity |
139 , 140 , 141 , 142 , 143 , 144 , 145 , 146 , 147 |
| Silibinin | PD‐L1 |
Lung cancer; nasopharyngeal carcinoma |
A549, H292, and H460 cells; C666‐1 cells; human tumor explants. In vitro dose: 25–200 µM |
Downregulate PD‐L1 expression; inhibit STAT5/PD‐L1 complex; reduce HIF‐1α expression |
Decrease cancer cell viability; enhance aerobic glycolysis. Induce cell apoptosis and cell cycle arrest at G0/G1 phase; inhibit tumor angiogenesis, migration, and invasion |
148 , 149 |
| Triptolide | PD‐L1 |
Breast cancer; glioma; leukemia; melanoma; oral squamous cell carcinoma |
MDA‐MB‐435S, MDA‐MB‐468, and MCF‐7 cells; U251‐MG, U87‐MG, A172, LN229, LN18, and T98G cells; WEHI‐3 cells; B16‐F10 cells; SAS cells; B16‐F10 xenograft mice; WHEI‐3 xenograft mice; PDTX xenograft mice. In vitro dose: 5–200 nM; in vivo dosage: 0.02–0.3 mg/kg |
Decrease PD‐L1 expression; inhibit IFN‐γ‐induced PD‐L1 upregulation; decrease the population of Treg cells in the spleen and axillary lymph nodes; inhibit CD4+ and CD8+ T‐cell proliferation; enhance B‐ and T‐cell proliferation; promote macrophage phagocytosis from PBMC and NK cell activity; increase ROS and Ca2+ productions; increase CD19 level in the blood; induce LC3‐II accumulation; decrease the mRNA level of Foxp3 in the spleen and axillary lymph nodes; decrease the mRNA levels of IL‐10 and TGF‐β in the spleen; decrease the production of IFN‐γ, IL‐10, and TGF‐β in the serum; decrease CD3 level in the blood; enhance the secretion of IFN‐γ and IL‐2 in glioma cells |
Decrease cancer cell viability and mitochondrial membrane potential; induce DNA damage, autophagy, cell apoptosis, and cell cycle arrest at G1 phase; inhibit tumor growth |
150 , 151 , 152 , 153 , 154 |
Abbreviations: AKT, protein kinase B; CTLA‐4, cytotoxic T lymphocyte‐associated protein 4; DC, dendritic cell; EGF, epidermal growth factor; EGFR, epidermal growth factor receptor; HIF‐1α, hypoxia‐inducible factor 1α; IFN‐γ, interferon γ; IL, interleukin; MDSC, myeloid‐derived suppressor cell; MHC, major histocompatibility complex; mRNA, messenger RNA; mTOR, mammalian target of rapamycin; NF‐κB, nuclear factor kappa‐light‐chain‐enhancer of activated B cells; NK, natural killer; PBMC, peripheral blood mononuclear cell; PD‐L1, programmed death‐legend 1; PI3K, phosphoinositide 3‐kinase; ROS, reactive oxygen species; RPMI, Roswell Park Memorial Institute; tBreg, tumor‐evoked regulatory B; TGF‐β, transforming growth factor β; TME, tumor microenvironment; TNF‐α, tumor necrosis factor α; Treg, Teff to regulatory T.
Figure 3.
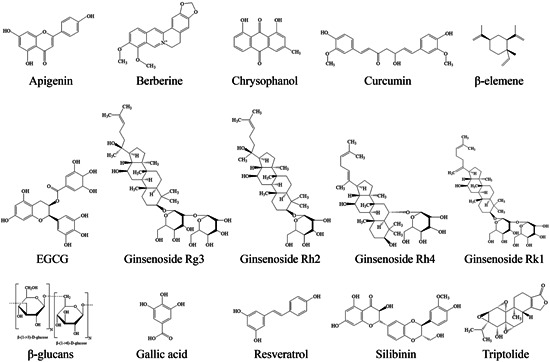
The chemical structure of the natural products with immunomodulatory potential for immune checkpoints from herbal medicines. Apigenin, berberine, chrysophanol, curcumin, β‐elemene, EGCG, gallic acid, ginsenoside Rg3, ginsenoside Rh2, ginsenoside Rh4, ginsenoside Rk1, β‐glucans, resveratrol, silibinin, and triptolide. EGCG, epigallocatechin gallate
4.1. Apigenin
Apigenin, a dietary flavonoid, is derived from many fruits, vegetables, and beverages, including onions, oranges, and chamomile tea. 155 It can modulate T‐cell immunity in breast, lung, and pancreatic cancer, and regulate DCs in melanoma. 111 , 112 , 113 , 156 Apigenin can attenuate PD‐L1 upregulation induced by IFN‐γ in breast cancer (MDA‐MB‐468, SK‐BR‐3, and 4T1) and melanoma (A375, A2058, and RPMI‐7951) cells. 111 , 112 This inhibition occurs through blocking the STAT1 pathway in breast cancer (MDA‐MB‐468) cells and melanoma (A375, A2058, and RPMI‐7951) cells. Similarly, apigenin also dismisses PD‐L1 expression induced by IFN‐β in breast cancer (MDA‐MB‐468) cells, but it does not affect constitutive PD‐L1 expression, 111 whereas it reduces PD‐L1 expression in the tumor tissues of melanoma (B16‐F10)‐bearing mice. 112 Moreover, apigenin can stimulate the proliferation of T lymphocytes and induce T‐cell‐mediated cell death in breast cancer (MDA‐MB‐468) and melanoma (A375) cells cocultured with Jurkat T cells, respectively. 111 , 112 This suggests that T‐cell response is associated with apigenin‐mediated PD‐L1 downregulation and cancer cell death. Furthermore, apigenin can enhance CD4+ and CD8+ T‐cell infiltration in the tumor tissues of melanoma (B16‐F10)‐bearing mice, and reduce PD‐L1 expression in DCs, thereby activating T‐cell immunity by downregulating PD‐L1 expression in DCs. 112 Similarly, it also increases the populations of CD4+ and CD8+ T lymphocytes and decreases the population of Treg cells in pancreatic cancer (Panc02)‐bearing mice. 113 This suggests that apigenin inhibits immunosuppression and activates T‐cell immunity to exert its anticancer effects.
Vaccination with E7‐HSP70 DNA enhances T‐cell‐mediated immune response to prevent tumor growth in NSCLC (TC‐1)‐bearing mice. 157 Apigenin combined with E7‐HSP70 DNA vaccine can produce greater amounts of primary and memory E7‐specific CD8+ T lymphocytes, as well as stimulating memory‐recall response in NSCLC (TC‐1)‐bearing mice. 156 Taken together, we suggest that apigenin can be used as an immune checkpoint inhibitor for cancer therapy.
Anti‐PD‐1 mAb alone treatment cannot effectively inhibit tumor growth in the Lewis lung carcinoma model, but the combined treatment of apigenin and anti‐PD‐1 mAb has a synergistic effect through decreasing tumor volume and lung lesions by increasing the populations of CD8+ T lymphocytes and generation of tumor necrosis factor α (TNF‐α), IFN‐γ, and granzyme B. 114 Clinical trials could be conducted to explore the combination of apigenin with ICB therapy in future.
4.2. Berberine
Berberine, an isoquinoline alkaloid, is mainly derived from many medicinal plants including Coptidis chinensis Franch and Phellodendron chinense Schneid. 158 It is employed widely for the treatment of cancer and inflammatory diseases. 159 , 160 Berberine exerts immunomodulatory effects in breast cancer cells, NSCLC cells, HL‐60‐differentiated neutrophils, and T lymphocytes. 115 , 116 , 161 Besides, it has been identified as a negative regulator of PD‐LI expression, as it reduces PD‐L1 expression and diminishes PD‐L1 upregulation induced by IFN‐γ in NSCLC cells. This downregulation is through enhancing its degradation via ubiquitin/proteasome‐dependent pathway. Moreover, berberine activates tumor‐infiltrating T lymphocytes, and reduces immunosuppressive Treg cells and myeloid‐derived suppressor cells (MDSCs), thereby inducing antitumor activity in NSCLC tumor. 115 Interestingly, the anticancer effect of berberine is abolished in Lewis lung carcinoma‐bearing T‐cell deficient mice, suggesting that berberine‐induced anticancer activity can be attributed to the T‐cell activation in the immune response.
MDSCs and Treg cells have been identified as potent immunosuppressive cells that contribute to immune blockade and escape in the immunosuppressive TME by suppressing T‐cell immunity. 162 , 163 Berberine reduces the populations of activated MDSCs and Treg cells, which indicates that berberine switches the TME from immunosuppression to immunoactivation. Furthermore, berberine can regulate neutrophil phenotypes to reverse doxorubicin‐induced cancer cell resistance. 116 Continuous treatment of doxorubicin has been shown to induce a shift towards the N2 phenotype in differentiated HL‐60 neutrophils. However, combined treatment of berberine and doxorubicin can reverse this effect and polarize the cells towards the N1 phenotype. In the mice models of lung and liver cancers, berberine treatment can ameliorate lung carcinogenesis, and its combined treatment with doxorubicin induces neutrophil polarization to the N1 phenotype, reduces PD‐L1 levels, or expression in serum and cell surface of splenic T cells, respectively. 116 Taken together, berberine is suggested to be developed as an immune checkpoint inhibitor for immunotherapy against cancers.
4.3. Chrysophanol
Chrysophanol, an anthraquinone compound, is derived from the rhizomes of Rheum palmatum L. Chrysophanol exerts anticancer effects against T‐cell acute lymphoblastic leukemia (TALL) by reducing cell proliferation and metastasis, as well as inducing cell apoptosis in TALL (Jurkat and TALL‐104) cells in vitro. 117 However, a microRNA‐9 inhibitor and a PD‐L1 inhibitor, atezolizumab, block this effect, suggesting that chrysophanol may exert anticancer effects via the microRNA‐9/PD‐L1 pathway in TALL cells. More evidence should be collected to confirm the effect of chrysophanol on immune checkpoints and TME in vitro and in vivo.
4.4. Curcumin
Curcumin, a phytopolylphenol pigment, derived from the rhizomes of Acorus calamus L., Curcuma longa, and Curcuma zedoaria. 118 It exhibits immunomodulatory effects by regulating B cells, DCs, macrophages, MDSCs, and T cells in CRC, head and neck squamous cell carcinoma, lung cancer, melanoma, and tongue squamous cell carcinoma. 112 , 118 , 119 , 120 , 121 , 123 , 164 Curcumin reduces PD‐L1 protein expression in carcinogen‐induced oral tumorigenesis mice and in melanoma (B16‐F10)‐bearing mice. A combination of curcumin and sildenafil can also decrease PD‐L1 expression in CRC (CT26, HCT 116, and HT‐29) cells, and liver cancer (HuH7) cells. 112 , 118 , 164 Similarly, it can also inhibit PD‐L1 upregulation induced by IFN‐γ via the STAT1 pathway in melanoma cells, and prevent IFN‐γ‐induced cell death in melanoma (A375) cells cocultured with TALL (Jurkat) cells. 112 Besides, curcumin can inhibit the immunosuppressive response by decreasing the populations of MDSCs and Treg cells, as well as increasing the populations of CD8+ T lymphocytes in the immunosuppressive TME. Hence, curcumin can activate the T‐cell immune response and improve tumor‐induced immunosuppressive TME in tongue carcinoma. 118
Development of advanced tumor always results in immune dysfunction, for example, a loss of Teff and memory T‐cell population, and enlargement of Treg cell population. 165 Curcumin prevents this loss of T‐cell population in Ehrlich's ascites carcinoma‐bearing mice, and enhances the activity of T lymphocytes to attack cancer cells. 119 It can also enhance CD4+ and CD8+ T‐cell infiltration in the tumor tissues of melanoma (B16‐F10)‐bearing mice. 112 Curcumin can also inhibit Treg‐mediated immunosuppression by reducing TGF‐β and IL‐10 secretion, thereby alleviating tumor‐induced immunosuppressive response. 119 , 123 Besides, it can also downregulate CTLA‐4 expression, a protein that is important for the immunosuppressive activity by Treg cells, to mediate immunosuppressive activity in CD4+CD25+ Treg cells. 123 It can also enhance IL‐2 secretion but reduce IFN‐γ secretion in a coculture of Treg and T cells, whereas it upregulated IL‐4 expression. Curcumin can also inhibit the accumulation and immunosuppressive function of MDSCs and IL‐6 levels in the tumor tissues and spleen of Lewis lung carcinoma‐bearing mice, thus leading to an inhibition of tumor growth. 120 Interestingly, a study demonstrates that a low dose of curcumin increases T‐cell population in tumors derived from Lewis lung carcinoma (3LL)‐bearing mice, leading to retardation of tumor growth. 121 In contrast, a high dose of curcumin reduces T‐cell population. Besides, curcumin can also enhance IFN‐γ secretion from CD3+ T lymphocytes and cannot suppress tumor growth in T‐cell‐deficient mice. These findings suggest that curcumin is a potential immune activator and T lymphocytes play a critical role in the anticancer immunity response to the action of curcumin.
Growing evidence shows that curcumin can augment the anticancer effect of anti‐PD‐1/PD‐L1 mAb through the activation of antitumor immunity in CRC, heptatocellular carcinoma (HCC), and cervical and uterine cancer. 166 , 167 Curcumin cannot only decrease PD‐1 expression, but also combine with anti‐PD‐1 mAb to exert a synergistic anticancer effect on cell growth, lymphocyte activation, and TME improvement in HCC in vitro or in vivo. 122 , 168 In clinical trials, a phase II study has completed to discover the action of a novel combination of pembrolizumab (PD‐1 blockade) and curcumin in recurrent cervical and uterine cancer. 169 Curcumin exerts a powerful effect on immune checkpoints via multiple targets (e.g., PD‐1, PD‐L1, PD‐L2, and CLTA‐4) with in vitro, in vivo, and in clinical evidence, which indicates that curcumin have potent to be developed as natural ICB from herbal medicines.
4.5. β‐Elemene
β‐Elemene, a volatile terpene, is derived from many herbal medicines including Curcuma wenyujin Y. H. Chen et C. Ling and Curcuma Zedoary. 170 It can inhibit cell migration and invasion in esophageal squamous cell carcinoma (ESCC) (TE‐1 and KYSE‐150) cells in vitro, and suppress tumor growth in ESCC (TE‐1 and KYSE‐150)‐bearing mice by regulating protein kinase B (AKT) signaling, thereby modulating PD‐L1 expression. 124 Therefore, β‐elemene can reduce PD‐L1 protein expression in the tumor tissues of ESCC (TE‐1 and KYSE‐150)‐bearing mice. The underlying of action of β‐elemene on immune checkpoints and TME should be further explored in the future.
4.6. Epigallocatechin gallate
Epigallocatechin gallate (EGCG), a polyphenol, is derived from Camellia sinensis (green tea). It plays a role in modulating immunity in NSCLC (A549, H1299, and Lu99) cells, and coculture of melanoma (B16‐F10) cells expressing ovalbumin (F10‐OVA) and tumor‐specific CD3+ T cells. 125 Besides, the combination of EGCG and Sig/E7/LAMP‐1 DNA vaccine exerts immunomodulatory effects in NSCLC (TC‐1) ‐bearing mice. 126 EGCG has also been shown to attenuate PD‐L1 upregulation‐induced by IFN‐γ at both protein and messenger RNA (mRNA) levels in NSCLC (A549 and H1299) cells, and this inhibition is through suppressing the JAK2/STAT1 pathway. 125 Similarly, EGCG can also prevent PD‐L1 upregulation induced by EGF via the AKT pathway in NSCLC (Lu99) cells. Moreover, EGCG can decrease PD‐L1 cell surface protein and mRNA expressions in F10‐OVA cocultured with CD3+ T lymphocytes isolated from F10‐OVA immunized mice, and increase the number of CD3+ T cells and the levels of IL‐2 mRNA, suggesting that it can restore T‐cell immune response by inhibiting PD‐1/PD‐L1 pathway. EGCG is also shown to increase the population of E7‐specific CD8+ T lymphocytes in NSCLC (TC‐1)‐bearing mice. 126 In addition, the combination of EGCG and DNA vaccine (Sig/E7/LAMP‐1) is effective in treating large and bulky tumors. 126 DNA vaccine (Sig/E7/LAMP‐1) is designed to enhance E7‐specific T‐cell immune responses, which can induce potential anticancer effects in E7‐positive tumors. 171 Combined EGCG and DNA vaccine can increase the population of IFN‐γ‐secreting E7‐specific CD8+ T lymphocytes and activate the immune response of CD4+ and CD8+ T lymphocytes, along with a decreased tumor volume in NSCLC (TC‐1)‐bearing mice. 126 Otherwise, tumor growth is not inhibited in CD8+ T‐cell‐deficient tumor‐bearing mice, this indicates that CD8+ T lymphocytes exert an important role in the anticancer effects of EGCG. EGCG is a safe natural product, but the combination of EGCG with ICB therapy has not been well justified.
4.7. Gallic acid
Gallic acid, a polyphenol, is derived from many natural plants, fruits, and green tea. 172 It exerts anticancer effects through modulating tumor immunity in NSCLC and CRC. 127 , 128 It can strongly reduce PD‐L1 at both protein and mRNA expressions by inhibiting its binding to epidermal growth factor receptor in NSCLC (A549 and H292) cells, thereby suppressing the phosphorylation of PI3K and AKT and activating p53. 127 Similarly, gallic acid can also decrease PD‐L1 protein expression in CRC (HT‐29 and HCT 116) cells. 128 Taken together, we suggest that gallic acid inhibits immune checkpoint to exert anticancer effects in NSCLC and CRC.
In addition, combined treatment of gallic acid and anti‐PD‐1 mAb exerts synergistic anticancer effect through the activation of T‐cell‐mediated immune response via decreasing PD‐L1 expression and enhancing IFN‐γ production in NSCLC. 127 It is urgent to explore the combination of gallic acid with ICB therapy in clinical trials.
4.8. Ginsenosides
Ginsenosides, a group of dammarane triterpenoids, are derived from the rhizomes of Panax ginseng, Panax notoginseng (Burk.) F. H. Chen, and Cinnamomum cassia Presl. 173 They possess immunomodulatory effects by regulating T cells in breast cancer, ESCC, lung cancer, and melanoma. 129 , 131 , 132 , 133 , 134 , 174 Ginsenoside Rg3 and Rh2 have been shown to reduce cisplatin‐induced PD‐L1 upregulation in NSCLC cells, whereas ginsenoside Rh4 can downregulate PD‐L1 protein expression via the AKT/mammalian target of rapamycin pathway in ESCC (Eca109 and KYSE‐150) cells, and tumor (Eca109)‐bearing mice. 129 , 132 , 133 Similarly, ginsenoside Rk1 can decrease PD‐L1 expression in NSCLC (A549 and PC9)‐bearing mice, through inhibiting the nuclear factor kappa‐light‐chain‐enhancer of activated B cells (NF‐κB) pathway. 134 Moreover, ginsenoside Rh2 can enhance CD4+ and CD8a+ T‐cell infiltration in the tumor tissues of melanoma (B16‐F10)‐bearing mice, and ginsenoside Rg1 can increase CD4+ T‐cell proliferation and activity in mice. 131 , 174 Importantly, carbon nanotubes‐loaded ginsenoside Rg3 can reduce PD‐1 expression in activated T cells and attenuate PD‐L1 expression in triple‐negative breast cancer cells, thereby stimulating the production of IFN‐γ, IL‐2, IL‐9, IL‐10, IL‐22, and IL‐23 from the activated T cells and attenuating the MDA‐MB‐231 breast cancer cell growth in vivo. 130 However, it is not clear whether ginsenosides target CTLA‐4.
4.9. β‐Glucans
β‐Glucans, natural polysaccharides, derived from plants, as well as the cell walls of yeast, fungi, and bacteria. 175 They can stimulate immunomodulatory effects in tumor‐associated macrophages and DCs and significantly prolong survival prognosis in Lewis lung cancer, melanoma, and osteosarcoma. 135 , 136 , 137 , 138 Particulate β‐glucans can upregulate PD‐L1 at mRNA and protein levels in cultured mouse peritoneal macrophages that can interact with PD‐1 on activated T lymphocytes to send downregulatory signals to T cells. 136 Besides, treatment with oat‐derived β‐glucans has been shown to alter TME by enhancing the infiltrating DCs, Teff, and memory T cells (CD4+ and CD8+ T lymphocytes), activating M1 macrophages, as well as generating proinflammatory cytokines (TNF‐α, IFN‐γ, and IL‐2) in the tumor tissues of melanoma (B16‐F10)‐bearing mice. 137 This treatment also upregulates PD‐L1 expression to stimulate antitumor immunity in the tumor tissues. These findings suggest that T lymphocytes, macrophages, DCs, and NK cells are all necessary factors for the therapeutic effects of β‐glucans in melanoma. Similarly, yeast‐derived β‐glucans also induce tumor‐educated DC maturation to promote antitumor response. 138 They can suppress Treg cell differentiation in tumor‐educated DCs cocultured with naïve CD4+ T cells in vitro and promote CD8+ T‐cell proliferation and differentiation into teff cells. The combination of β‐glucan treatment and tumor‐educated DCs enhances the infiltration of CD11b+F4/80+ macrophages and CD11b+Gr‐1+ granulocytes in the tumor tissues of Lewis lung carcinoma‐bearing mice, whereas it decreases the populations of CD11c+TIM‐3+ DCs, CD4+Foxp3+ Treg cells, and CD4+PD‐1+ T lymphocytes in the tumor tissues and draining lymph nodes. These data suggest that β‐glucans alter immunity to exert anticancer effects in lung carcinoma.
In Lewis lung carcinoma, breast cancer (EO771), and melanoma (B16‐F10)‐bearing mice, a significant increase in F40/80+ macrophage infiltration into the tumor tissues with a typical M2 phenotype and immunosuppressive function have been observed. 135 Yeast‐derived particulate β‐glucans can downregulate M2 marker genes, whereas they upregulate M1 marker genes in bone marrow–derived M2 macrophages. They also attenuate M2 macrophage‐driven immunosuppressive property involved in the proliferation of CD4+ and CD8+ T lymphocytes. Besides, β‐glucans suppress tumor growth with an altered tumor‐associated macrophage phenotype in Lewis lung carcinoma‐bearing mice, and macrophage depletion in these mice reverse these effects, suggesting that the anticancer effects are mediated via modulating these effects on macrophages. Moreover, β‐glucans can also reduce the population of CD4+Foxp3+ Treg cells in the spleen and tumor tissues of breast cancer (EO771)‐bearing mice, but increase the population of CD4+ T cells. Therefore, targeting tumor‐associated macrophages by β‐glucans can improve the efficacy of cancer immunotherapies. Taken together, we suggest that β‐glucans modulate different immune cells to mediate antitumor immune response and anticancer effects.
4.10. Resveratrol
Resveratrol, a nonflavonoid polyphenol, derived from Polygonum cuspidatum Sieb. et Zucc. as well as other natural resources including grapes, peanuts, soy, and berries. 176 It possesses antitumor immunomodulatory effects by regulating NK cells, T cells, Treg cells, and tumor‐evoked regulatory B (tBreg) cells in breast cancer, leukemia, melanoma, and renal cell carcinoma. 139 , 140 , 141 , 142 , 143 , 144 , 145 , 177 It has been shown to act as a direct inhibitor of α‐glucosidase and α‐mannosidase, glyco‐PD‐L1‐processing enzymes, and to promote anticancer T‐cell immunity by releasing the brake of immune checkpoint via occupying the inner surface of PD‐L1. 144 Besides, resveratrol can promote infiltrating CD8+ T lymphocytes into the tumor tissues of renal cell carcinoma Renca tumor‐bearing mice to enhance anticancer immunity. However, this effect is abolished in CD8+ T‐cell‐deficient mice, suggesting that CD8+ T lymphocytes is an important factor contributed to resveratrol‐induced anticancer immunity in renal cell carcinoma. 140 In contrast, it reduces Treg cell population in the tumor tissues of Renca tumor‐bearing mice and spleen of melanoma (B16‐F10)‐bearing mice, and decreases Foxp3+CD4+CD25+ Treg cell population and TGF‐β secretion in the spleen of lymphoma (EG7)‐bearing mice. 140 , 141 , 143 It also increases IL‐10 and IL‐6 at mRNA levels but decreases IFN‐γ at mRNA level in the tumor tissues of Renca tumor‐bearing mice. 140 Moreover, resveratrol can also enhance the proliferation of T lymphocytes and the activity of NK cells in leukemia (L1210)‐bearing mice. 141
tBreg cells play a critical role in breast cancer lung metastasis by converting from non‐Treg CD4+ T lymphocytes into metastasis‐promoting Foxp3+ Treg cells, thereby inducing metastasis. 178 Some studies demonstrate that resveratrol blocks the generation and function of tBreg cells, downregulates PD‐1 expression on T cells, and promotes macrophages toward M1 phenotype to induce antitumor immune response in breast cancer (4T1)‐bearing mice, thereby attenuating lung metastasis from triple‐negative breast cancer. 139 , 146 Furthermore, resveratrol can also enhance immune function in immunosuppressive mice. 145 It increases the population of CD3+ and CD4+ T lymphocytes in the peripheral blood and lymphocyte proliferation in the spleen, and reduces serum IL‐2 and NF‐κB levels. In contrast, resveratrol increases NF‐κB p65 expression in the spleen of the immunosuppressive mice. These data suggest that resveratrol can recover from immunosuppression via the NF‐κB pathway. Interestingly, repeated administration of resveratrol for 28 days is safe and well‐tolerated in healthy Japanese subjects, and it increases the population of circulating γδ T cells and Treg cells. 177 Similarly, resveratrol also enhances the growth of γδ T lymphocytes and Treg cells in cultured peripheral blood mononuclear cells from healthy subjects. Even though resveratrol fails to affect CTLA‐4 expression, we also suggest that resveratrol exerts a potent anticancer immune response via the PD‐1/PD‐L1 axis in cancer treatment. Future clinical trials may be conducted to explore the synergetic effects of resveratrol with ICB therapy.
4.11. Silibinin
Silibinin, a flavonoid, is derived from Silybum marianum L. Gaertn. It possesses potent anticancer effects against nasopharyngeal, lung, and pancreatic carcinoma. 148 , 179 , 180 It also exerts immunomodulatory effects in nasopharyngeal carcinoma. 148 In particular, silibinin has been shown to reduce PD‐L1 expression in nasopharyngeal carcinoma (C666‐1) cells and primary tumors isolated from patients with nasopharyngeal carcinoma. 148 This reduction in expression of PD‐L1 is mediated by inhibiting its transcription factor, HIF‐1α. In addition, silibinin inhibits the STAT5 activation and disrupts the STAT5/PD‐L1 complex. 149 Therefore, silibinin restores anticancer immunity via the HIF‐1α, STAT5, and PD‐L1 pathways. The effect of silibinin on other immune checkpoints (PD‐1, PD‐L2, and CTLA‐4) should be further validated in the future.
4.12. Triptolide
Triptolide, a diterpenoid epoxide, is derived from the roots of Tripterygium wilfordii Hook. F. It has been shown to exert immunomodulatory effects by regulating B cells, macrophages, monocytes, T lymphocytes, and Treg cells in breast cancer, glioma, melanoma, oral cancer. 150 , 151 , 152 , 153 , 154 Triptolide has been shown to reduce the upregulation of PD‐L1 induced by IFN‐γ in breast cancer (MDA‐MB‐468 and MCF‐7) cells, and glioma (A172, LN18, LN229, U251, U87, and T98G) cells. 151 , 152 Similarly, it can downregulate the PD‐L1 expression induced by IFN‐γ in OSCC (SAS) cells through the JAK/STAT signaling pathway. 153 Besides, triptolide treatment has been shown to decrease PD‐L1 expression in oral cancer patient‐derived tumor‐bearing mice and OSCC (SAS)‐bearing mice. 153 Unexpectedly, triptolide can also decrease constitutive expression of PD‐L1 protein in NSCLC (H460) cells. 115 Moreover, triptolide can reverse IFN‐γ‐induced immunosuppressive CD4+ and CD8+ T lymphocytes in glioma cells cocultured with T lymphocytes, as well as enhancing IFN‐γ and IL‐2 secretion, and decreasing IL‐10 secretion. 152 In addition to cancer cells, triptolide can promote the proliferation of T and B cells in leukemic mice. 154 It can also decrease the population of Treg cells and the levels of Foxp3 mRNA in the spleen and axillary lymph nodes of melanoma (B16‐F10)‐bearing mice, as well as the levels of TGF‐β and IL‐10 mRNA in the spleen and the production of TGF‐β and IL‐10 cytokines in the serum. 150 Continuing studies should be performed to explore the effect of triptolide on other immune checkpoints (PD‐1, PD‐L2, and CTLA‐4) and TME in the future.
Taken together, apigenin, berberine, chrysophanol, curcumin, β‐elemene, EGCG, gallic acid, ginsenoside Rg3, ginsenoside Rh2, ginsenoside Rh4, ginsenoside Rk1, β‐glucans, resveratrol, silibinin, and triptolide could improve immune response to induce cancer cell death through activating anticancer T‐cell immunity via targeting immune checkpoints and modulating the TME via regulating cytokine secretion, so we suggest that natural products from herbal medicines can act as immune checkpoint inhibitors and TME modulators for cancer immunotherapy (Figure 4).
Figure 4.
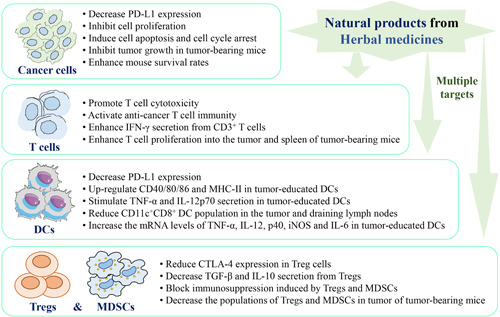
The immunomodulatory potential of natural products from herbal medicines as immune checkpoints inhibitors to fight against cancer via multiple targets. Natural products from herbal medicines act on different cells, including cancer cells, T cells, dendritic cells (DCs), regulatory T cells (Tregs), and myeloid‐derived suppressor cells (MDSCs), to present anticancer activities via multiple targets [Color figure can be viewed at wileyonlinelibrary.com]
5. CONCLUSION AND PERSPECTIVES
Until now, single target, immune resistance, and adverse reactions are the major challenges in the clinical use of ICB treatment for cancer immunotherapy, so the researchers aim to develop novel agents, improve targeting responses, and reduce side effects for cancer immunotherapy. Genetic mutations and epigenetics are complicated and reversible factors that are valuable indicators for the success of ICB treatment and merit further exploration. Although the experience with ICB treatment in cancer therapy is limited, promising lead compounds derived from herbal medicines could be important in cancer immunotherapy. However, the research and development of immune checkpoint inhibitors from herbal medicines are very challenging, as herbal medicines is mainly used as an adjuvant therapy in cancer treatment. Further studies are urgent to test whether these lead compounds from herbal medicines are potent enough to be used alone as immune checkpoint inhibitors. Besides, the toxicities of lead compounds are also needed to be tested to check whether these lead compounds have any effects on healthy tissues. Despite these challenges, lead compounds from herbal medicines remain to be novel agents as immune checkpoint inhibitors for fighting against cancers.
CTLA‐4 blockade is also a promising strategy to modulate the TME, but few studies have demonstrated the relationship between CTLA‐4 and natural products from herbal medicines in cancer studies. Future studies are warranted to discover the bioactive compounds that can target CTLA‐4, as well as the underlying mechanisms. As few clinical trials were performed on immune checkpoints for natural products from herbal medicines compared to ICB mAbs, there is still a gap in the research of the underlying mechanisms of natural products for improving ICB therapy. A chemical library of natural products from herbal medicines could aid the discovery of promising lead compounds targeting immune checkpoints in cancer immunotherapy.
ACKNOWLEDGMENTS
This study was partially supported by Wong's donation (Grant No. 200006276), a donation from the Gaia Family Trust of New Zealand (Grant No. 200007008), the Research Grants Committee (RGC) of Hong Kong, HKSAR (Grant No. 17121419), the Health and Medical Research Fund (Project Codes 16172751), the China Postdoctoral Science Foundation Funded Project (2017M622811), the Natural Science Foundation of Guangdong Province, China (2018A030310226 and 2020A1515010922), and the Macao Science and Technology Development Fund (FDCT 007/2020/ALC). This research is also funded by the Hong Kong Scholars Program.
Zhong Z, Vong CT, Chen F, et al. Immunomodulatory potential of natural products from herbal medicines as immune checkpoints inhibitors: helping to fight against cancer via multiple targets. Med Res Rev. 2022;42:1246‐1279. 10.1002/med.21876
Zhangfeng Zhong and Chi Teng Vong contributed equally to this study.
Contributor Information
Yitao Wang, Email: ytwang@um.edu.mo.
Yibin Feng, Email: yfeng@hku.hk.
DATA AVAILABILITY STATEMENT
The data used to support the findings of this study are available from the corresponding author upon request.
REFERENCES
- 1. June CH, Sadelain M. Chimeric antigen receptor therapy. N Engl J Med. 2018;379(1):64‐73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. June CH, Riddell SR, Schumacher TN. Adoptive cellular therapy: a race to the finish line. Sci Transl Med. 2015;7(280):280‐287. [DOI] [PubMed] [Google Scholar]
- 3. Borst J, Ahrends T, Babala N, Melief CJM, Kastenmuller W. CD4(+) T cell help in cancer immunology and immunotherapy. Nat Rev Immunol. 2018;18(10):635‐647. [DOI] [PubMed] [Google Scholar]
- 4. Zilionis R, Engblom C, Pfirschke C, et al. Single‐cell transcriptomics of human and mouse lung cancers reveals conserved myeloid populations across individuals and species. Immunity. 2019;50(5):1317‐1334. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Leone RD, Zhao L, Englert JM, et al. Glutamine blockade induces divergent metabolic programs to overcome tumor immune evasion. Science. 2019;366(6468):1013‐1021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Petitprez F, de Reynies A, Keung EZ, et al. B cells are associated with survival and immunotherapy response in sarcoma. Nature. 2020;577(7791):556‐560. [DOI] [PubMed] [Google Scholar]
- 7. Parsons BL. Multiclonal tumor origin: evidence and implications. Mutat Res. 2018;777:1‐18. [DOI] [PubMed] [Google Scholar]
- 8. Binnewies M, Roberts EW, Kersten K, et al. Understanding the tumor immune microenvironment (TIME) for effective therapy. Nat Med. 2018;24(5):541‐550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Li X, Wenes M, Romero P, Huang SC, Fendt SM, Ho PC. Navigating metabolic pathways to enhance antitumour immunity and immunotherapy. Nat Rev Clin Oncol. 2019;16(7):425‐441. [DOI] [PubMed] [Google Scholar]
- 10. Helmink BA, Reddy SM, Gao J, et al. B cells and tertiary lymphoid structures promote immunotherapy response. Nature. 2020;577(7791):549‐555. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Hegde PS, Chen DS. Top 10 challenges in cancer immunotherapy. Immunity. 2020;52(1):17‐35. [DOI] [PubMed] [Google Scholar]
- 12. Tang X, Li Y, Ma J, et al. Adenovirus‐mediated specific tumor tagging facilitates CAR‐T therapy against antigen‐mismatched solid tumors. Cancer Lett. 2020;487:1‐9. [DOI] [PubMed] [Google Scholar]
- 13. Ma L, Dichwalkar T, Chang JYH, et al. Enhanced CAR‐T cell activity against solid tumors by vaccine boosting through the chimeric receptor. Science. 2019;365(6449):162‐168. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Liu CG, Wang Y, Liu P, et al. Aptamer‐T cell targeted therapy for tumor treatment using sugar metabolism and click chemistry. ACS Chem Biol. 2020;15(6):1554‐1565. [DOI] [PubMed] [Google Scholar]
- 15. Pont MJ, Hill T, Cole GO, et al. γ‐Secretase inhibition increases efficacy of BCMA‐specific chimeric antigen receptor T cells in multiple myeloma. Blood. 2019;134(19):1585‐1597. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Zheng PP, Kros JM, Li J. Approved CAR T cell therapies: ice bucket challenges on glaring safety risks and long‐term impacts. Drug Discov Today. 2018;23(6):1175‐1182. [DOI] [PubMed] [Google Scholar]
- 17. Morgan RA, Chinnasamy N, Abate‐Daga D, et al. Cancer regression and neurological toxicity following anti‐MAGE‐A3 TCR gene therapy. J Immunother. 2013;36(2):133‐151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Rapoport AP, Stadtmauer EA, Binder‐Scholl GK, et al. NY‐ESO‐1‐specific TCR‐engineered T cells mediate sustained antigen‐specific antitumor effects in myeloma. Nat Med. 2015;21(8):914‐921. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Glanville J, Huang H, Nau A, et al. Identifying specificity groups in the T cell receptor repertoire. Nature. 2017;547(7661):94‐98. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Han A, Glanville J, Hansmann L, Davis MM. Linking T‐cell receptor sequence to functional phenotype at the single‐cell level. Nature Biotechnol. 2015;33(2):210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Saint‐Jean M, Knol AC, Volteau C, et al. Adoptive cell therapy with tumor‐infiltrating lymphocytes in advanced melanoma patients. J Immunol Res. 2018;2018:3530148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Dudley ME, Gross CA, Somerville RPT, et al. Randomized selection design trial evaluating CD8(+)‐enriched versus unselected tumor‐infiltrating lymphocytes for adoptive cell therapy for patients with melanoma. J Clin Oncol. 2013;31(17):2152‐2159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Hargadon KM, Johnson CE, Williams CJ. Immune checkpoint blockade therapy for cancer: an overview of FDA‐approved immune checkpoint inhibitors. Int Immunopharmacol. 2018;62:29‐39. [DOI] [PubMed] [Google Scholar]
- 24. Rowshanravan B, Halliday N, Sansom DM. CTLA‐4: a moving target in immunotherapy. Blood. 2018;131(1):58‐67. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Zak KM, Grudnik P, Magiera K, Domling A, Dubin G, Holak TA. Structural biology of the immune checkpoint receptor PD‐1 and its ligands PD‐L1/PD‐L2. Structure. 2017;25(8):1163‐1174. [DOI] [PubMed] [Google Scholar]
- 26. Guerrouahen BS, Maccalli C, Cugno C, Rutella S, Akporiaye ET. Inflammatory cytokines IL‐17 and TNF‐α up‐regulate PD‐L1 expression in human prostate and colon cancer cells. Immunol Lett. 2017;184:7‐14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Leach DR, Krummel MF, Allison JP. PD‐L1:CD80 Cis‐heterodimer triggers the co‐stimulatory receptor CD28 while repressing the inhibitory PD‐1 and CTLA‐4 pathways. Immunity. 2019;51(6):1059‐1736. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Ramsay AG. Immune checkpoint blockade immunotherapy to activate anti‐tumour T‐cell immunity. Br J Haematol. 2013;162(3):313‐325. [DOI] [PubMed] [Google Scholar]
- 29. Guerrouahen BS, Maccalli C, Cugno C, Rutella S, Akporiaye ET. Reverting immune suppression to enhance cancer immunotherapy. Front Oncol. 2019;9:1554. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Leach DR, Krummel MF, Allison JP. Enhancement of antitumor immunity by CTLA‐4 blockade. Science. 1996;271(5256):1734‐1736. [DOI] [PubMed] [Google Scholar]
- 31. Gao Q, Wang XY, Qiu SJ, et al. Comprehensive analysis of CTLA‐4 in the tumor immune microenvironment of 33 cancer types. Int Immunopharmacol. 2020;85:106633‐106979. [DOI] [PubMed] [Google Scholar]
- 32. Zhao YH, Yang W, Huang YY, Cui RJ, Li XY, Li BJ. Evolving roles for targeting CTLA‐4 in cancer immunotherapy. Cell Physiol Biochem. 2018;47(2):721‐734. [DOI] [PubMed] [Google Scholar]
- 33. Liu FK, Huang J, Liu XM, Cheng Q, Luo CK, Liu ZX. CTLA‐4 correlates with immune and clinical characteristics of glioma. Cancer Cell Int. 2020;20(1):7‐8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Buchbinder EI, Desai A. CTLA‐4 and PD‐1 pathways similarities, differences, and implications of their inhibition. American Journal of Clinical Oncology‐Cancer Clinical Trials. 2016;39(1):98‐106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Tang Z, Liu Y, He M, Bu W. Overexpression of PD‐L1 significantly associates with tumor aggressiveness and postoperative recurrence in human hepatocellular carcinoma. Clin Cancer Res. 2009;15(3):971‐979. [DOI] [PubMed] [Google Scholar]
- 36. Yang L, Pang Y, Moses HL. Low PD‐1 expression in cytotoxic CD8(+) tumor‐infiltrating lymphocytes confers an immune‐privileged tissue microenvironment in NSCLC with a prognostic and predictive value. Clin Cancer Res. 2018;24(2):407‐419. [DOI] [PubMed] [Google Scholar]
- 37. Rozali EN, Hato SV, Robinson BW, Lake RA, Lesterhuis WJ. PD‐L2 is a second ligand for PD‐I and inhibits T cell activation. Nature Immunol. 2001;2(3):261‐268. [DOI] [PubMed] [Google Scholar]
- 38. Di Virgilio F, Sarti AC, Falzoni S, De Marchi E, Adinolfi E. PD‐L2 expression in human tumors: relevance to anti‐PD‐1 therapy in cancer. Clin Cancer Res. 2017;23(12):3158‐3167. [DOI] [PubMed] [Google Scholar]
- 39. Tang ZM, Liu YY, He MY, Bu WB. Chemodynamic therapy: tumour microenvironment‐mediated Fenton and Fenton‐like reactions. Angew Chem Int Ed Engl. 2019;58(4):946‐956. [DOI] [PubMed] [Google Scholar]
- 40. Yang L, Pang YL, Moses HL. TGF‐β and immune cells: an important regulatory axis in the tumor microenvironment and progression. Trends Immunol. 2010;31(6):220‐227. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Rozali EN, Hato SV, Robinson BW, Lake RA, Lesterhuis WJ. Programmed death ligand 2 in cancer‐induced immune suppression. Clin Dev Immunol. 2012;2012:656340. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Di Virgilio F, Sarti AC, Falzoni S, De Marchi E, Adinolfi E. Extracellular ATP and P2 purinergic signalling in the tumour microenvironment. Nat Rev Cancer. 2018;18(10):601‐618. [DOI] [PubMed] [Google Scholar]
- 43. Curran MA, Montalvo W, Yagita H, Allison JP. PD‐1 and CTLA‐4 combination blockade expands infiltrating T cells and reduces regulatory T and myeloid cells within B16 melanoma tumors. Proc Natl Acad Sci U S A. 2010;107(9):4275‐4280. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Liu J. Exploration of a novel prognostic risk signatures and immune checkpoint molecules in endometrial carcinoma microenvironment. Genomics. 2020;112(5):3117‐3134. [DOI] [PubMed] [Google Scholar]
- 45. Nosho K, Baba Y, Tanaka N, et al. Pan‐tumor genomic biomarkers for PD‐1 checkpoint blockade‐based immunotherapy. Science. 2018;362(6411):197‐366. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Kim HD, Song GW, Park S, et al. Neoadjuvant PD‐1 blockade in resectable lung cancer. N Engl J Med. 2018;378(21):1976‐1986. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Shen Y, Teng Y, Lv Y, et al. Role of tumor‐infiltrating lymphocytes in patients with solid tumors: can a drop dig a stone? Cell Immunol. 2019;343:103753. [DOI] [PubMed] [Google Scholar]
- 48. Kurtulus S, Madi A, Escobar G, et al. Tumor infiltrating lymphocytes and survival in patients with head and neck squamous cell carcinoma. Head Neck. 2016;38(7):1074‐1084. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Simoni Y, Becht E, Fehlings M, et al. Tumour‐infiltrating T‐cell subsets, molecular changes in colorectal cancer, and prognosis: cohort study and literature review. J Pathol. 2010;222(4):350‐366. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Canale FP, Ramello MC, Nunez N, et al. Association between expression level of PD1 by tumor‐infiltrating CD8(+) T cells and features of hepatocellular carcinoma. Gastroenterology. 2018;155(6):1936‐1950. [DOI] [PubMed] [Google Scholar]
- 51. Garnelo M, Tan A, Her Z, et al. PD‐1 does not mark tumor‐infiltrating CD8+ T cell dysfunction in human gastric cancer. J Immunother Cancer. 2020;8(2):e000422‐351. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Chen G, Huang AC, Zhang W, et al. Checkpoint blockade immunotherapy induces dynamic changes in PD‐1(‐)CD8(+) tumor‐infiltrating T cells. Immunity. 2019;50(1):181‐194. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Wind TT, Gacesa R, Vich Vila A, et al. Bystander CD8(+) T cells are abundant and phenotypically distinct in human tumour infiltrates. Nature. 2018;557(7706):575‐579. [DOI] [PubMed] [Google Scholar]
- 54. Matson V, Fessler J, Bao R, et al. CD39 expression defines cell exhaustion in tumor‐infiltrating CD8(+) T cells. Cancer Res. 2018;78(1):115‐128. [DOI] [PubMed] [Google Scholar]
- 55. Routy B, Le Chatelier E, Derosa L, et al. Interaction between tumour‐infiltrating B cells and T cells controls the progression of hepatocellular carcinoma. Gut. 2017;66(2):342‐351. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Vetizou M, Pitt JM, Daillere R, et al. Exosomal PD‐L1 contributes to immunosuppression and is associated with anti‐PD‐1 response. Nature. 2018;560(7718):382‐386. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Gao J, Shi LZ, Zhao H, et al. Gut microbial species and metabolic pathways associated with response to treatment with immune checkpoint inhibitors in metastatic melanoma. Melanoma Res. 2020;30(3):235‐246. [DOI] [PubMed] [Google Scholar]
- 58. Watari K, Konnai S, Maekawa N, et al. The commensal microbiome is associated with anti‐PD‐1 efficacy in metastatic melanoma patients. Science. 2018;359(6371):104‐108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Peng W, Liu C, Xu C, et al. Gut microbiome influences efficacy of PD‐1‐based immunotherapy against epithelial tumors. Science. 2018;359(6371):91‐97. [DOI] [PubMed] [Google Scholar]
- 60. Chen H, Liakou CI, Kamat A, et al. Anticancer immunotherapy by CTLA‐4 blockade relies on the gut microbiota. Science. 2015;350(6264):1079‐1084. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Paradis TJ, Floyd E, Burkwit J, et al. Loss of IFN‐γ pathway genes in tumor cells as a mechanism of resistance to anti‐CTLA‐4 therapy. Cell. 2016;167(2):397‐404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Ding G, Shen T, Yan C, Zhang M, Wu Z, Cao L. Immune inhibitory function of bovine CTLA‐4 and the effects of its blockade in IFN‐γ production. BMC Vet Res. 2019;15(1):380. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Garris CS, Arlauckas SP, Kohler RH, et al. PD‐1 blockade enhances T‐cell migration to tumors by elevating IFN‐γ inducible chemokines. Cancer Res. 2012;72(20):5209‐5218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Shi LZ, Fu T, Guan B, et al. Anti‐CTLA‐4 therapy results in higher CD4(+)ICOS(hi) T cell frequency and IFN‐γ levels in both nonmalignant and malignant prostate tissues. Proc Natl Acad Sci U S A. 2009;106(8):2729‐2734. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. Vackova J, Piatakova A, Polakova I, Smahel M. The anti‐tumor activity of anti‐CTLA‐4 is mediated through its induction of IFN gamma. Cancer Immunol Immunother. 2001;50(3):125‐133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66. Ding GP, Shen T, Chen Y, Zhang MJ, Wu ZR, Cao LP. IFN‐γ down‐regulates the PD‐1 expression and assist nivolumab in PD‐1‐blockade effect on CD8+T‐lymphocytes in pancreatic cancer. BMC Cancer. 2019;19(1):1053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67. Chow MT, Ozga AJ, Servis RL, et al. Successful anti‐PD‐1 cancer immunotherapy requires T cell‐dendritic cell crosstalk involving the cytokines IFN‐γ and IL‐12. Immunity. 2018;49(6):1148‐1161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68. Chu Y, Yang X, Xu W, Wang Y, Guo Q, Xiong S. Interdependent IL‐7 and IFN‐γ signalling in T‐cell controls tumour eradication by combined α‐CTLA‐4+α‐PD‐1 therapy. Nat Commun. 2016;7:12335‐1549. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69. Vackova J, Piatakova A, Polakova I, Smahel M. Abrogation of IFN‐γ signaling may not worsen sensitivity to PD‐1/PD‐L1 blockade. Int J Mol Sci. 2020;21(5):1806‐1444. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70. Batlle E, Massague J. IFN‐γ‐induced PD‐L1 expression in melanoma depends on p53 expression. J Exp Clin Cancer Res. 2019;38(1):397‐940. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71. Marshall NA, Galvin KC, Corcoran AMB, Boon L, Higgs R, Mills KHG. Intratumoral activity of the CXCR3 Chemokine system is required for the efficacy of anti‐PD‐1 therapy. Immunity. 2019;50(6):1498‐1512. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72. Chu YW, Yang XL, Xu W, Wang Y, Guo Q, Xiong SD. In situ expression of IFN‐γ‐inducible T cell α chemoattractant in breast cancer mounts an enhanced specific anti‐tumor immunity which leads to tumor regression. Cancer Immunol Immunother. 2007;56(10):1539‐1549. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73. Yang XL, Chu YW, Wang Y, Zhang RH, Xiong SD. Targeted in vivo expression of IFN‐γ‐inducible protein 10 induces specific antitumor activity. J Leukoc Biol. 2006;80(6):1434‐1444. [DOI] [PubMed] [Google Scholar]
- 74. Batlle E, Massague J. Transforming growth factor‐β signaling in immunity and cancer. Immunity. 2019;50(4):924‐940. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75. Marshall NA, Galvin KC, Corcoran AMB, Boon L, Higgs R, Mills KHG. Immunotherapy with PI3K inhibitor and Toll‐like receptor agonist induces IFN‐γ+IL‐17(+) polyfunctional T cells that mediate rejection of murine tumors. Cancer Res. 2012;72(3):581‐591. [DOI] [PubMed] [Google Scholar]
- 76. Skoulidis F, Goldberg ME, Greenawalt DM, et al. TLR4/IFNγ pathways induce tumor regression via NOS II‐dependent NO and ROS production in murine breast cancer models. Oncoimmunology. 2016;5(5):e1123369‐e1123835. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77. Kitajima S, Ivanova E, Guo S, et al. TLR7/8‐agonist‐loaded nanoparticles promote the polarization of tumour‐associated macrophages to enhance cancer immunotherapy. Nat Biomed Eng. 2018;2(8):578‐588. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78. Hofmann S, Januliene D, Mehdipour AR, et al. Intratumoral Tcf1(+)PD‐1(+)CD8(+) T Cells with stem‐like properties promote tumor control in response to vaccination and checkpoint blockade immunotherapy. Immunity. 2019;50(1):195‐211. [DOI] [PubMed] [Google Scholar]
- 79. Szakacs G, Paterson JK, Ludwig JA, Booth‐Genthe C, Gottesman MM. A cancer cell program promotes T cell exclusion and resistance to checkpoint blockade. Cell. 2018;175(4):984‐997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80. Pérez‐Ruiz E, Melero I, Kopecka J, Sarmento‐Ribeiro AB, García‐Aranda M, De Las Rivas J. STK11/LKB1 mutations and PD‐1 inhibitor resistance in KRAS‐mutant lung adenocarcinoma. Cancer Discov. 2018;8(7):822‐835. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81. Gatouillat G, Odot J, Balasse E, et al. Suppression of STING associated with LKB1 loss in KRAS‐driven lung cancer. Cancer Discov. 2019;9(1):34‐45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82. Matsui H, Takeshita A, Naito K, et al. Conformation space of a heterodimeric ABC exporter under turnover conditions. Nature. 2019;571(7766):580‐583. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83. Szakács G, Paterson JK, Ludwig JA, Booth‐Genthe C, Gottesman MM. Targeting multidrug resistance in cancer. Nat Rev Drug Discov. 2006;5(3):219‐234. [DOI] [PubMed] [Google Scholar]
- 84. Perez‐Ruiz E, Melero I, Kopecka J, Sarmento‐Ribeiro AB, Garcia‐Aranda M, De Las Rivas J. Cancer immunotherapy resistance based on immune checkpoints inhibitors: Targets, biomarkers, and remedies. Drug Resist Updat. 2020;53:100718. [DOI] [PubMed] [Google Scholar]
- 85. Gupta PB, Chaffer CL, Weinberg RA. Immunization with liposome‐anchored pegylated peptides modulates doxorubicin sensitivity in P‐glycoprotein‐expressing P388 cells. Cancer Lett. 2007;257(2):165‐171. [DOI] [PubMed] [Google Scholar]
- 86. Visvader JE, Lindeman GJ. Reduced effect of gemtuzumab ozogamicin (CMA‐676) on P‐glycoprotein and/or CD34‐positive leukemia cells and its restoration by multidrug resistance modifiers. Leukemia. 2002;16(5):813‐819. [DOI] [PubMed] [Google Scholar]
- 87. Mizukoshi E, Honda M, Arai K, Yamashita T, Nakamoto Y, Kaneko S. Expression of multidrug resistance‐associated protein 3 and cytotoxic T cell responses in patients with hepatocellular carcinoma. J Hepatol. 2008;49(6):946‐954. [DOI] [PubMed] [Google Scholar]
- 88. Miao L, Zhang Y, Huang L. TRAIL sensitize MDR cells to MDR‐related drugs by down‐regulation of P‐glycoprotein through inhibition of DNA‐PKcs/Akt/GSK‐3β pathway and activation of caspases. Mol Cancer. 2010;9:199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89. Gupta PB, Chaffer CL, Weinberg RA. Cancer stem cells: mirage or reality? Nature Med. 2009;15(9):1010‐1012. [DOI] [PubMed] [Google Scholar]
- 90. Visvader JE, Lindeman GJ. Cancer stem cells in solid tumours: accumulating evidence and unresolved questions. Nat Rev Cancer. 2008;8(10):755‐768. [DOI] [PubMed] [Google Scholar]
- 91. Wang X, Guo G, Guan H, Yu Y, Lu J, Yu J. Challenges and potential of PD‐1/PD‐L1 checkpoint blockade immunotherapy for glioblastoma. J Exp Clin Cancer Res. 2019;38(1):87. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92. Miao L, Zhang Y, Huang L. mRNA vaccine for cancer immunotherapy. Mol Cancer. 2021;20(1):41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93. Liu S, Chen S, Yuan W, et al. The emerging role of viruses in the treatment of solid tumours. Cancer Treat Rev. 2011;37(8):618‐632. [DOI] [PubMed] [Google Scholar]
- 94. Liu J, Zhao Z, Qiu N, et al. Chemotherapy induces programmed cell death‐ligand 1 overexpression via the nuclear factor‐κB to Foster an immunosuppressive tumor microenvironment in ovarian cancer. Cancer Res. 2015;75(23):5034‐5045. [DOI] [PubMed] [Google Scholar]
- 95. Jiang W, Su L, Ao M, et al. Anticancer effects of asiatic acid against doxorubicin‐resistant breast cancer cells via an AMPK‐dependent pathway in vitro . Phytomedicine. 2021;92:153737. [DOI] [PubMed] [Google Scholar]
- 96. D'souza M, Nielsen D, Svane IM, et al. PD‐1/PD‐L1 enhanced cisplatin resistance in gastric cancer through PI3K/AKT mediated P‐gp expression. Int Immunopharmacol. 2021;94:107443. [DOI] [PubMed] [Google Scholar]
- 97. Liu S, Chen S, Yuan W, et al. PD‐1/PD‐L1 interaction up‐regulates MDR1/P‐gp expression in breast cancer cells via PI3K/AKT and MAPK/ERK pathways. Oncotarget. 2017;8(59):99901‐99912. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98. Cortazar FB, Marrone KA, Troxell ML, et al. Co‐delivery of IOX1 and doxorubicin for antibody‐independent cancer chemo‐immunotherapy. Nat Commun. 2021;12(1):2425‐2647. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99. Zen Y, Yeh MM. Amplified antitumor efficacy by a targeted drug retention and chemosensitization strategy‐based “combo” nanoagent together with PD‐L1 blockade in reversing multidrug resistance. J Nanobiotechnology. 2021;19(1):200‐973. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100. Dubey D, David WS, Reynolds KL, et al. The risk of cardiac events in patients receiving immune checkpoint inhibitors: a nationwide Danish study. Eur Heart J. 2021;42(16):1621‐1631. [DOI] [PubMed] [Google Scholar]
- 101. Braaten TJ, Brahmer JR, Forde PM, et al. Cardiovascular megnetic resonance in immune checkpoint Inhibitor‐associated myocarditis. Eur Heart J. 2020;41(18):1733‐1743. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102. Subbotin V, Fiksel G. Clinicopathological features of acute kidney injury associated with immune checkpoint inhibitors. Kidney Int. 2016;90(3):638‐647. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103. Zen Y, Yeh MM. Hepatotoxicity of immune checkpoint inhibitors: a histology study of seven cases in comparison with autoimmune hepatitis and idiosyncratic drug‐induced liver injury. Mod Pathol. 2018;31(6):965‐973. [DOI] [PubMed] [Google Scholar]
- 104. Simonaggio A, Michot JM, Voisin AL, et al. Severe neurological toxicity of immune checkpoint inhibitors: growing spectrum. Ann Neurol. 2020;87(5):659‐669. [DOI] [PubMed] [Google Scholar]
- 105. Wang T, Wu X, Guo C, et al. Immune checkpoint inhibitor‐induced inflammatory arthritis persists after immunotherapy cessation. Ann Rheum Dis. 2020;79(3):332‐338. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106. Subbotin V, Fiksel G. Modeling multi‐needle injection into solid tumor. Am J Cancer Res. 2019;9(10):2209‐2215. [PMC free article] [PubMed] [Google Scholar]
- 107. Keizer RJ, Huitema AD, Schellens JH, Beijnen JH. Clinical pharmacokinetics of therapeutic monoclonal antibodies. Clin Pharmacokinet. 2010;49(8):493‐507. [DOI] [PubMed] [Google Scholar]
- 108. Coombs MRP, Harrison ME, Hoskin DW. Evaluation of readministration of immune checkpoint inhibitors after immune‐related adverse events in patients with cancer. JAMA Oncol. 2019;5(9):1310‐1317. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109. Xu L, Zhang Y, Tian K, et al. Development of inhibitors of the programmed cell death‐1/programmed cell death‐ligand 1 signaling pathway. J Med Chem. 2019;62(4):1715‐1730. [DOI] [PubMed] [Google Scholar]
- 110. Nelson N, Szekeres K, Iclozan C, et al. Immune functions as a ligand or a receptor, cancer prognosis potential, clinical implication of VISTA in cancer immunotherapy. Semin Cancer Biol. 2021;12:e0170197. [DOI] [PubMed] [Google Scholar]
- 111. Coombs MR, Harrison ME, Hoskin DW. Apigenin inhibits the inducible expression of programmed death ligand 1 by human and mouse mammary carcinoma cells. Cancer Lett. 2016;380(2):424‐433. [DOI] [PubMed] [Google Scholar]
- 112. Xu L, Zhang Y, Tian K, et al. Apigenin suppresses PD‐L1 expression in melanoma and host dendritic cells to elicit synergistic therapeutic effects. J Exp Clin Cancer Res. 2018;37(1):261. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113. Jiang ZB, Wang WJ, Xu C, et al. Apigenin: selective CK2 inhibitor increases Ikaros expression and improves T cell homeostasis and function in murine pancreatic cancer. PLoS One. 2017;12(2):e0170197‐48. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114. Wang K, Feng X, Chai L, Cao S, Qiu F. Luteolin and its derivative apigenin suppress the inducible PD‐L1 expression to improve anti‐tumor immunity in KRAS‐mutant lung cancer. Cancer Lett. 2021;515:36‐48. [DOI] [PubMed] [Google Scholar]
- 115. Zhang C, Sheng J, Li G, et al. Berberine diminishes cancer cell PD‐L1 expression and facilitates antitumor immunity via inhibiting the deubiquitination activity of CSN5. Acta Pharm Sin B. 2020;10(12):2299‐2312. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116. Habtemariam S. Berberine maintains the neutrophil N1 phenotype to reverse cancer cell resistance to doxorubicin. Front Pharmacol. 2019;10:1658‐599. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 117. Kim S, You D, Jeong Y, et al. Chrysophanol suppresses growth and metastasis of T cell acute lymphoblastic leukemia via miR‐9/PD‐L1 axis. Naunyn‐Schmiedeberg's Arch Pharmacol. 2020;393(2):273‐286. [DOI] [PubMed] [Google Scholar]
- 118. Liao F, Liu L, Luo E, Hu J. Curcumin enhances anti‐tumor immune response in tongue squamous cell carcinoma. Arch Oral Biol. 2018;92:32‐37. [DOI] [PubMed] [Google Scholar]
- 119. Zhang S, Zhou L, Zhang M, et al. Curcumin reverses T cell‐mediated adaptive immune dysfunctions in tumor‐bearing hosts. Cell Mol Immunol. 2010;7(4):306‐315. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120. Tanaka A, Sakaguchi S. Inhibition of curcumin on myeloid‐derived suppressor cells is requisite for controlling lung cancer. Int Immunopharmacol. 2016;39:265‐272. [DOI] [PubMed] [Google Scholar]
- 121. Luo F, Song X, Zhang Y, Chu Y. Low‐dose curcumin leads to the inhibition of tumor growth via enhancing CTL‐mediated antitumor immunity. Int Immunopharmacol. 2011;11(9):1234‐1240. [DOI] [PubMed] [Google Scholar]
- 122. Yin J, Yin Q, Liang B, et al. The effect of curcumin on multi‐level immune checkpoint blockade and T cell dysfunction in head and neck cancer. Phytomedicine. 2021;92:153758. [DOI] [PubMed] [Google Scholar]
- 123. Liao F, Liu L, Luo E, Hu J. Curcumin inhibits suppressive capacity of naturally occurring CD4+CD25+ regulatory T cells in mice in vitro . Int Immunopharmacol. 2012;14(1):99‐106. [DOI] [PubMed] [Google Scholar]
- 124. Liang Y, Li S, Zheng G, Zhang L. β‐Elemene suppresses the malignant behavior of esophageal cancer cells by regulating the phosphorylation of AKT. Acta Histochem. 2020;122(4):151538. [DOI] [PubMed] [Google Scholar]
- 125. Zhao G, Lu Z, Tang L, et al. Green tea catechin is an alternative immune checkpoint inhibitor that inhibits PD‐L1 expression and lung tumor growth. Molecules. 2018;23(8):2071‐2106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126. Dent P, Booth L, Roberts JL, Poklepovic A, Hancock JF. Epigallocatechin‐3‐gallate enhances CD8+ T cell‐mediated antitumor immunity induced by DNA vaccination. Cancer Res. 2007;67(2):802‐811. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127. Liu D, You M, Xu Y, et al. The inhibitory mechanisms of tumor PD‐L1 expression by natural bioactive gallic acid in non‐small‐cell lung cancer (NSCLC) cells. Cancers. 2020;12(3):727‐272. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128. Mazewski C, Kim MS, Gonzalez de Mejia E. Anthocyanins, delphinidin‐3‐O‐glucoside and cyanidin‐3‐O‐glucoside, inhibit immune checkpoints in human colorectal cancer cells in vitro and in silico . Sci Rep. 2019;9(1):11560. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129. Ohm JE, Carbone DP. Ginsenoside Rg3 attenuates cisplatin resistance in lung cancer by downregulating PD‐L1 and resuming immune. Biomed Pharmacother. 2017;96:378‐383. [DOI] [PubMed] [Google Scholar]
- 130. Luo X, Wang H, Ji D. Carbon nanotubes (CNT)‐loaded ginsenosides Rb3 suppresses the PD‐1/PD‐L1 pathway in triple‐negative breast cancer. Aging. 2021;13(13):17177‐17189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131. Xiao Z, Su Z, Han S, Huang J, Lin L, Shuai X. Ginsenoside Rh2 enhances the antitumor immunological response of a melanoma mice model. Oncol Lett. 2017;13(2):681‐685. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 132. Chen Y, Zhang Y, Song W, Zhang Y, Dong X, Tan M. Ginsenoside Rh2 improves the cisplatin anti‐tumor effect in lung adenocarcinoma A549 cells via superoxide and PD‐L1. Anti Cancer Agents Med Chem. 2020;20(4):495‐503. [DOI] [PubMed] [Google Scholar]
- 133. Liu L, Lim MA, Jung SN, et al. Ginsenoside Rh4 suppresses aerobic glycolysis and the expression of PD‐L1 via targeting AKT in esophageal cancer. Biochem Pharmacol. 2020;178:114038. [DOI] [PubMed] [Google Scholar]
- 134. Tuyaerts S, Van Nuffel A, Naert E, et al. Ginsenoside Rk1 induces apoptosis and downregulates the expression of PD‐L1 by targeting the NF‐κB pathway in lung adenocarcinoma. Food Funct. 2020;11(1):456‐471. [DOI] [PubMed] [Google Scholar]
- 135. Xie Q, Li F, Fang L, Liu W, Gu C. Dectin‐1 activation by a natural product β‐glucan converts immunosuppressive macrophages into an M1‐like phenotype. J Immunol. 2015;195(10):5055‐5065. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 136. Hunter KW Jr., DuPre S, Redelman D. Microparticulate β‐glucan upregulates the expression of B7.1, B7.2, B7‐H1, but not B7‐DC on cultured murine peritoneal macrophages. Immunol Lett. 2004;93(1):71‐78. [DOI] [PubMed] [Google Scholar]
- 137. Rawangkan A, Wongsirisin P, Namiki K, et al. Systemic administration of β‐glucan of 200 kDa modulates melanoma microenvironment and suppresses metastatic cancer. Oncoimmunology. 2018;7(2):e1387347. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 138. Kang TH, Lee JH, Song CK, et al. β‐Glucan restores tumor‐educated dendritic cell maturation to enhance antitumor immune responses. Int J Cancer. 2016;138(11):2713‐2723. [DOI] [PubMed] [Google Scholar]
- 139. Kim TW, Hung CF, Ling M, et al. Inhibition of breast cancer metastasis by resveratrol‐mediated inactivation of tumor‐evoked regulatory B cells. J Immunol. 2013;191(8):4141‐4151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 140. Chen L, Yang S, Liao W, Xiong Y. Modification of antitumor immunity and tumor microenvironment by resveratrol in mouse renal tumor model. Cell Biochem Biophys. 2015;72(2):617‐625. [DOI] [PubMed] [Google Scholar]
- 141. Li T, Fan GX, Wang W, Li T, Yuan YK. Resveratrol induces apoptosis, influences IL‐6 and exerts immunomodulatory effect on mouse lymphocytic leukemia both in vitro and in vivo . Int Immunopharmacol. 2007;7(9):1221‐1231. [DOI] [PubMed] [Google Scholar]
- 142. Yang Y, Paik JH, Cho D, Cho JA, Kim CW. Resveratrol induces the suppression of tumor‐derived CD4+CD25+ regulatory T cells. Int Immunopharmacol. 2008;8(4):542‐547. [DOI] [PubMed] [Google Scholar]
- 143. Guan H, Singh NP, Singh UP, Nagarkatti PS, Nagarkatti M. Resveratrol prevents endothelial cells injury in high‐dose interleukin‐2 therapy against melanoma. PLoS One. 2012;7(4):e35650‐e35659. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 144. Wang M, Yan SJ, Zhang HT, et al. Resveratrol targets PD‐L1 glycosylation and dimerization to enhance antitumor T‐cell immunity. Aging. 2020;12(1):8‐34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 145. Jiang Z, Yang Y, Yang Y, et al. The enhancement of immune function and activation of NF‐κB by resveratrol‐treatment in immunosuppressive mice. Int Immunopharmacol. 2016;33:42‐47. [DOI] [PubMed] [Google Scholar]
- 146. Han X, Zhao N, Zhu W, Wang J, Liu B, Teng Y. Resveratrol attenuates TNBC lung metastasis by down‐regulating PD‐1 expression on pulmonary T cells and converting macrophages to M1 phenotype in a murine tumor model. Cell Immunol. 2021;368:104423‐104503. [DOI] [PubMed] [Google Scholar]
- 147. Deng X, Zhao J, Qu L, et al. Resveratrol induces PD‐L1 expression through snail‐driven activation of Wnt pathway in lung cancer cells. J Cancer Res Clin. 2021;147(4):1101‐1113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 148. Hu M, Yang J, Qu L, et al. Silibinin down‐regulates PD‐L1 expression in nasopharyngeal carcinoma by interfering with tumor cell glycolytic metabolism. Arch Biochem Biophys. 2020;690:108479. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 149. Lee E, Ko E, Lee J, et al. Silibinin regulates tumor progression and tumorsphere formation by suppressing PD‐L1 expression in non‐small cell lung cancer (NSCLC) cells. Cells. 2021;10(7):1632. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 150. Luo X, Wang H, Ji D. Triptolide downregulates Treg cells and the level of IL‐10, TGF‐β, and VEGF in melanoma‐bearing mice. Planta Med. 2013;79(15):1401‐1407. [DOI] [PubMed] [Google Scholar]
- 151. Liang M, Fu J. Triptolide inhibits interferon‐γ‐induced programmed death‐1‐ligand 1 surface expression in breast cancer cells. Cancer Lett. 2008;270(2):337‐341. [DOI] [PubMed] [Google Scholar]
- 152. Zhang L, Yu JS. Triptolide reverses helper T cell inhibition and down‐regulates IFN‐γ induced PD‐L1 expression in glioma cell lines. JNO. 2019;143(3):429‐436. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 153. Kuo C, Yang C, Lin C, Lin G, Sytwu H, Chen Y. Triptolide suppress oral cancer cell PD‐L1 expression in the interferon‐γ‐modulated microenvironment in vitro, in vivo and in clinical patients. Biomed Pharmacother. 2021;133:111057. [DOI] [PubMed] [Google Scholar]
- 154. Zhang M, Chun L, Sandoval V, et al. Triptolide induced cell death through apoptosis and autophagy in murine leukemia WEHI‐3 cells in vitro and promoting immune responses in WEHI‐3 generated leukemia mice in vivo . Environ Toxicol. 2017;32(2):550‐568. [DOI] [PubMed] [Google Scholar]
- 155. Shukla S, Gupta S. Apigenin: a promising molecule for cancer prevention. Pharm Res. 2010;27(6):962‐978. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 156. Chuang CM, Monie A, Wu A, Hung CF. Combination of apigenin treatment with therapeutic HPV DNA vaccination generates enhanced therapeutic antitumor effects. J Biomed Sci. 2009;16(1):49. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 157. Lee‐Chang C, Bodogai M, Martin‐Montalvo A, et al. Enhancement of DNA vaccine potency by linkage of antigen gene to an HSP70 gene. Cancer Res. 2000;60(4):1035‐1042. [PubMed] [Google Scholar]
- 158. Wang K, Feng X, Chai L, Cao S, Qiu F. The metabolism of berberine and its contribution to the pharmacological effects. Drug Metab Rev. 2017;49(2):139‐157. [DOI] [PubMed] [Google Scholar]
- 159. Li T, Fan GX, Wang W, Li T, Yuan YK. Effects of berberine and its derivatives on cancer: a systems pharmacology review. Front Pharmacol. 2019;10:1461. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 160. Habtemariam S. Berberine and inflammatory bowel disease: a concise review. Pharmacol Res. 2016;113(Pt A):592‐599. [DOI] [PubMed] [Google Scholar]
- 161. Guan H, Singh NP, Singh UP, Nagarkatti PS, Nagarkatti M. Berberine down‐regulates IL‐8 expression through inhibition of the EGFR/MEK/ERK pathway in triple‐negative breast cancer cells. Phytomedicine. 2018;50:43‐49. [DOI] [PubMed] [Google Scholar]
- 162. Tanaka A, Sakaguchi S. Regulatory T cells in cancer immunotherapy. Cell Res. 2017;27(1):109‐118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 163. Kumar V, Patel S, Tcyganov E, Gabrilovich DI. The nature of myeloid‐derived suppressor cells in the tumor microenvironment. Trends Immunol. 2016;37(3):208‐220. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 164. Dent P, Booth L, Roberts JL, Poklepovic A, Hancock JF. (Curcumin+sildenafil) enhances the efficacy of 5FU and anti‐PD1 therapies in vivo . J Cell Physiol. 2020;235(10):6862‐6874. [DOI] [PubMed] [Google Scholar]
- 165. Ohm JE, Carbone DP. Immune dysfunction in cancer patients. Oncology. 2002;16(1 Suppl 1):11‐18. [PubMed] [Google Scholar]
- 166. Hayakawa T, Yaguchi T, Kawakami Y. Enhanced anti‐tumor effects of the PD‐1 blockade combined with a highly absorptive form of curcumin targeting STAT3. Cancer Sci. 2020;111(12):4326‐4335. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 167. Xiao Z, Su Z, Han S, Huang J, Lin L, Shuai X. Dual pH‐sensitive nanodrug blocks PD‐1 immune checkpoint and uses T cells to deliver NF‐κB inhibitor for antitumor immunotherapy. Sci Adv. 2020;6(6):eaay7785‐117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 168. Guo L, Li HB, Fan TL, Ma YL, Wang LL. Synergistic efficacy of curcumin and anti‐programmed cell death‐1 in hepatocellular carcinoma. Life Sci. 2021;279:119359. [DOI] [PubMed] [Google Scholar]
- 169. Amirsaadat S, Pilehvar‐Soltanahmadi Y, Zarghami F, Alipour S, Ebrahimnezhad Z, Zarghami N. PRIMMO study protocol: a phase II study combining PD‐1 blockade, radiation and immunomodulation to tackle cervical and uterine cancer. BMC Cancer. 2019;19(1):506. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 170. Xie Q, Li F, Fang L, Liu W, Gu C. The antitumor efficacy of β‐elemene by changing tumor inflammatory environment and tumor microenvironment. BioMed Res Int. 2020;2020:6892961. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 171. Liu B, Zhang H, Li J, et al. Enhancing DNA vaccine potency by coadministration of DNA encoding antiapoptotic proteins. J Clin Invest. 2003;112(1):109‐117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 172. Abdelwahed A, Bouhlel I, Skandrani I, et al. Study of antimutagenic and antioxidant activities of gallic acid and 1,2,3,4,6‐pentagalloylglucose from Pistacia lentiscus. Confirmation by microarray expression profiling. Chem Biol Interact. 2007;165(1):1‐13. [DOI] [PubMed] [Google Scholar]
- 173. He NW, Zhao Y, Guo L, Shang J, Yang XB. Antioxidant, antiproliferative, and pro‐apoptotic activities of a saponin extract derived from the roots of Panax notoginseng (Burk.) F.H. Chen. J Med Food. 2012;15(4):350‐359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 174. Kuo CS, Yang CY, Lin CK, Lin GJ, Sytwu HK, Chen YW. Ginsenoside Rg1 enhances CD4(+) T‐cell activities and modulates Th1/Th2 differentiation. Int Immunopharmacol. 2004;4(2):235‐244. [DOI] [PubMed] [Google Scholar]
- 175. Chan GC, Chan WK, Sze DM. The effects of β‐glucan on human immune and cancer cells. J Hematol Oncol. 2009;2:25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 176. Varoni EM, Lo Faro AF, Sharifi‐Rad J, Iriti M. Anticancer molecular mechanisms of resveratrol. Front Nutr. 2016;3:8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 177. Espinoza JL, Trung LQ, Inaoka PT, et al. The repeated administration of resveratrol has measurable effects on circulating T‐cell subsets in humans. Oxid Med Cell Longevity. 2017;2017:6781872. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 178. Olkhanud PB, Damdinsuren B, Bodogai M, et al. Tumor‐evoked regulatory B cells promote breast cancer metastasis by converting resting CD4⁺ T cells to T‐regulatory cells. Cancer Res. 2011;71(10):3505‐3515. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 179. Nambiar D, Prajapati V, Agarwal R, Singh RP. In vitro and in vivo anticancer efficacy of silibinin against human pancreatic cancer BxPC‐3 and PANC‐1 cells. Cancer Lett. 2013;334(1):109‐117. [DOI] [PubMed] [Google Scholar]
- 180. Amirsaadat S, Pilehvar‐Soltanahmadi Y, Zarghami F, Alipour S, Ebrahimnezhad Z, Zarghami N. Silibinin‐loaded magnetic nanoparticles inhibit hTERT gene expression and proliferation of lung cancer cells. Artif Cells Nanomed Biotechnol. 2017;45(8):1649‐1656. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
The data used to support the findings of this study are available from the corresponding author upon request.