

Abstract
Anionic, metal‐centered nucleophiles are emerging compounds with unique reactivities. Here, we describe the isolation and full characterization of the first tetraamido tin(II) dianion, its behavior as ligand towards transition metals, and its reactivity as a tin‐centered nucleophile. Experimental values such as the Tolman electronic parameter (TEP) and computations attest tin‐located σ‐donor ability exceeding that of carbenes or electron‐rich phosphines. Against transition metals, the stannate(II) can act as η 1‐ or η 5‐type ligand. With aldehydes, it reacts by hydride substitution to give valuable acyl stannates. The reductive dehalogenation of iodobenzene indicates facile redox pathways mediated by halogen bond interaction. Calix[4]pyrrolato stannate(II) represents the first example of this macrocyclic ligand in low‐valent p‐block element chemistry.
Keywords: Calix[4]Pyrrole, Dianions, Low-Valent, Tin, σ-Donor
The macrocyclic calix[4]pyrrole enables the synthesis of the first tetraamido tin(II) dianion. The compound is a very strong metal‐centered σ‐donor with a Tolman electronic parameter (TEP) value exceeding that of carbenes and electron‐rich phosphines. The coordinative and nucleophilic features are demonstrated with transition metals and organic substrates.

Introduction
N‐Heterocyclic carbenes (NHCs; e.g. A in Figure 1) and cyclic alkyl amino carbenes (CAACs, e.g. B) became indispensable tools in transition metal catalysis or for the stabilization of reactive p‐block element centers. [1] Their pronounced σ‐donor ability is of most vital impact, usually expressed by the energy of the carbon‐centered, highest occupied molecular orbital (HOMO, A=−5.74 eV, B=−5.04 eV, all values in Figure 1 computed at a consistent, solvation corrected level of theory). [2] In contrast to the lighter tetrylenes, the donor ability of stannylenes drops significantly by the high s‐character of the Sn‐centered electron lone pair. [3] Accordingly, the HOMO of N‐heterocyclic stannylene C shows delocalized π‐character, but only the lower‐lying HOMO−2 (−5.94 eV) is of SnII lone pair type.[ 3e , 4 ] Among the strategies to raise the σ‐donor strength of tetrylenes, [5] the coordination of additional ligands to SnR2 appears particularly effective (e.g. amidinato stannylene D in Figure 1).[ 4 , 6 ] Coordination of anionic ligands to SnII may lead to an even more pronounced increase of the σ‐donicity, as attested in the class of nucleophilic anionic stannates(II), R3Sn−. [7] For instance, the tridentate amido ligand in Gade's amido stannate(II) [E]− causes a solvation‐corrected HOMO energy of −3.93 eV. Accordingly, this [8] and related compounds proved expedient ligands in a range of transition metal complexes. [9] We surmised that the coordination of a fourth anionic ligand to SnII might push the σ‐donor strength even farther. SnII‐based dianions are scarce, [10] and molecular compounds are limited to [F]2− [11] and [G]2− , [12] which are stabilized by charge delocalization into the carborate ([F]2− ) or the fluorenyl moiety ([G]2− ). Given the substantial π‐donor ability of nitrogen and the unique role of amido substituents in low valent tin chemistry, [13] the formation of a yet elusive tetraamido SnII dianion, [Sn(NR2)4]2−, appeared of particular relevance. However, a strong Pauli‐ and Coulomb repulsion between the anionic, electron‐rich [Sn(NR2)3]− and NR2 − fragments renders the formation of such high‐energy species highly challenging. Thus, a macrocyclic strategy was sought. The meso‐octaalkyl calix[4]pyrroles have been intensively investigated as macrocyclic ligands in transition and rare‐earth metal chemistry. [14] Owing to their flexible coordination behavior, these ligands enabled the isolation of many d‐ and f‐block metal complexes.[ 14d , 15 ]
Figure 1.
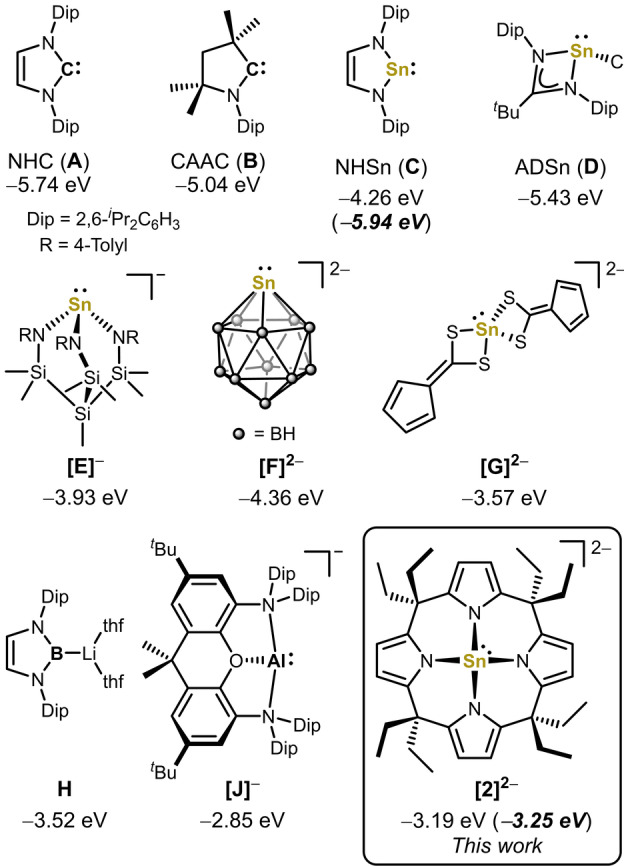
Solvation‐corrected HOMO energy levels for the NHC (A), CAAC (B), the N‐heterocyclic stannylene (NHSn) (C), the amidinato stannylene (ADSn) (D), the anionic stannate(II) [E]− , the dianionic stannates(II) [F]2− , and [G]2− , boryl H and the aluminyl anion [J]− , and [2]2− (B97M‐D3(BJ)/def2‐TZVPP CPCM(THF)//B97M‐D3(BJ)/def2‐TZVPP). For C and [2]2− the HOMO and HOMO−1 are ligand‐centered and the energies of their tin electron lone pairs centered HOMO−2 are given in parentheses.
More recently, the calix[4]pyrrole ligand enabled the synthesis of planar, tetracoordinated AlIII[16] and SiIV[17] complexes and transferred concepts like element ligand cooperativity, ligand‐to‐metal‐charge‐transfer and C−H agostic interactions into the p‐block. [18] However, no complexes of calix[4]pyrroles with low‐valent p‐block elements have been described so far. Herein we present the synthesis of calix[4]pyrrolato stannate(II) [2]2− as the first dianionic, tetraamido SnII, and its chemistry as a nucleophile and strong σ‐donor. Compared to previous carbenes, stannylenes, and (di)anionic stannates, [2]2− possess a drastically increased tin‐centered electron lone pair energy (Figure 1). Strikingly, these hallmarks are approaching that of seminal (semi)metal‐centered σ‐donors such as boryl (e.g. H in Figure 1) [19] or aluminyl anions (e.g. [J]− in Figure 1). [20]
Results and Discussion
Reaction of the lithium salt of meso‐octaethylcalix[4]pyrrolate [Li4(thf)3][1] with one equivalent tin(II) chloride in tetrahydrofuran (THF) at room temperature completed within minutes, forming a clear, colorless reaction solution. Workup yielded the lithium salt of the dianionic stannate(II) [Li2(thf) x ][2] with varying THF content in excellent yields and at a multigram scale (e.g. 80 % yield at a 2.0 g scale) (Figure 2A). 1H, 13C, 119Sn NMR spectroscopy revealed the incorporation of a tin(II) center into a C 4v symmetric calix[4]pyrrole species. In THF‐d8, [2]2− exhibits two diastereotopic sets of ethyl residues (syn and anti to the SnII center) and one singlet for the pyrrolyl β‐hydrogen atoms at 5.85 ppm. One sharp 7Li NMR resonance at −2.12 ppm indicated solvated lithium cations. The comparatively high‐field shifted 119Sn NMR resonance at −481.3 ppm disclosed a highly coordinated tin(II) atom with increased electron density compared to related species. [21] Even though [Li2(thf) x ][2] can be seen as a lithium salt of a dianion, it rendered surprisingly soluble in non‐polar organic solvents such as Et2O, benzene and toluene. Interestingly, NMR spectroscopy in C6D6 showed symmetry lowering for [2]2− and the appearance of shifted resonances in the 7Li NMR spectrum indicating the coordination of lithium cations to the complex [2]2− (Figure S4). This picture was confirmed by SCXRD of [Li2(thf) x ][2] (Figure S43), resembling the structure of other anionic calix[4]pyrrolato complex–alkali metal salts in non‐polar media.[ 14b , 14c , 22 ]
Figure 2.

A) Synthesis of the dianionic calix[4]pyrrolato stannate(II) as the lithium salt [Li2(thf) x ][2] and B) subsequent salt exchange reactions with PPh4Cl and NBu4Cl forming the phosphonium salt [(PPh4)2(oDFB)0.5][2] and the ammonium salt [(NBu4)2][2]. C) Solid‐state molecular structure of [(NBu4)2][2]. Displacement ellipsoids are drawn at 50 % probability level. Hydrogens and NBu4 + counter cations are omitted for clarity. Selected bond lengths [pm] and angels [°]: Sn1−N1 232.27(16), Sn1−N2 230.61(15), Sn1−N3 230.73(15), Sn1−N4 232.11(15), Sn1−N4‐plane 95.8, N1−Sn1−N4 133.23(5), N2−Sn1−N3 129.10(5), cis−N−Sn−N between 79.64(5) and 80.85(5). D) Occupied frontier molecular orbitals calculated at the B97M‐D3(BJ)/def2‐TZVPP CPCM(THF)//B97M‐D3(BJ)/def2‐TZVPP level of theory. E) Effects of Lewis base coordination (i) and distortion into the calix[4]pyrrole coordination geometry (ii) on the energy of the lone pair (lp) containing orbital at tin(II) for the hypothetical bis(pyrrolato)stannylene K, tetrakis(pyrrolato)stannate(II) [L]2− , and [2]2− .
Metathesis of [Li2(thf) x ][2] with two equivalents of the respecting phosphonium and ammonium chlorides yielded the PPh4 + and NBu4 + salts of [2]2− in up to 92 % yield (Figure 2B). Whereas the NBu4 + salt is soluble in less polar solvents such as 1,2‐ortho‐difluorobenzene (oDFB), the PPh4 + salt only dissolves in solvent mixtures containing MeCN. NMR spectroscopy and elemental analysis confirmed the bulk purity of both salts. The Li+ and PPh4 + salts of [2]2− possess comparable 119Sn NMR shifts in THF‐d 8 at −481.32 ppm and −486.17 ppm, respectively, but the NBu4 + salt exhibits a significantly more high‐field shifted 119Sn NMR resonance at −517.83 ppm. The 119Sn shift of free [2]2− was DFT‐computed at −509.38 ppm (PBE0/SO‐ZORA‐TZ2P), supporting the “naked” dianion character in THF. Indeed, the solid‐state molecular structure of [(NBu4)2][2] contains a free tin(II) calix[4]pyrrolato dianion with two independent NBu4 + cations in the asymmetric unit (Figure 2C). The absence of associated cations results in a highly symmetric, calix[4]pyrrolato stannate(II) dianion possessing a nearly perfect square‐pyramidal coordination geometry and only minor differences between the Sn−N bond lengths (Sn−Navg=231 pm), comparable to previously reported N4‐coordinated, neutral tin(II) complexes. [23] In contrast, the solid‐state molecular structure of [(PPh4)2(oDFB)0.5][2] shows a saddle‐shaped calix[4]pyrrolato ligand with alternating orientations of the bridging meso‐carbon atoms, highlighting the structural flexibility of the calix[4]pyrrolato ligand backbone in the coordination sphere of tin (Figure S45). All salts of [2]2− are stable under inert gas at room temperature for several months, but decompose under oxygen atmosphere or with traces of humidity. Their temperature stability in aprotic solvents depends on the respective counter cation, e.g., with [Li2(thf) x ][2] being stable in THF at 80 °C for several hours.
The electronic structure of [2]2− was analyzed by DFT calculations and bonding analysis tools. [2]2− possess an energetically high lying HOMO−2 with a major contribution of the tin‐centered lone pair (Figure 2D and S55, B97M‐D3(BJ)/def2‐TVZPP CPCM(THF)). NBO calculation revealed a diminished positive charge at the tin (+1.13) compared to trisamido stannate [E]− (+1.23), and a decreased s‐character of the tin‐centered lone pair (79 %) compared to all other stannylenes (>88 %, Figure S51 and Table S12). To elucidate the influence of structure and electronics imparted by the calix[4]pyrrolato ligand, [2]2− was compared with hypothetical bis(pyrrolato)stannylene K and tetrakis(pyrrolato)stannate(II) [L]2− (Figure 2E and S52). The coordination of two additional anionic pyrrolato ligands to the neutral bis(pyrrolato)stannylene K (step i) raises the energy of the lone pair containing HOMO by 3.10 eV and decreases the positive charge located at the tin(II) center from +1.41 to +1.15. The tethering of four pyrrolato ligands into a macrocyclic ligand and the concomitant distortion of the coordination geometry around tin(II) in [2]2− raises the lone pair containing HOMO by an additional 0.38 eV and decreases the atomic charge at tin to +1.13 (step ii, an additional effect might result from the pyrrole alkyl substitution). ETS‐NOCV bonding analysis showed that the nearly coplanar arrangement of the four pyrrolato units in the calix[4]pyrrole backbone of [2]2− enables the ligand to act as both a σ‐donor towards the Sn(5s,p) orbital and as π‐donor towards the Sn(5pz ) orbital. Especially, interaction of these latter orbitals contributes to the high lying HOMO−2, and constitutes the highly directional and high‐energy lone pair at the tin(II) center. Overall, these theoretical considerations strongly suggested that [2]2− should act as a tin‐centered σ‐donor that was thus probed experimentally.
The Tolman electronic parameter (TEP) [24] and 77Se{1H} NMR spectroscopy [25] have become standard techniques to evaluate the electronic properties of a variety of ligands. Thus, to gain experimental insights into the coordination chemistry and scale the electron donor and acceptor properties of [2]2− , metal carbonyl complexes and its selenium adduct were synthesized. Reaction of [(PPh4)2(oDFB)0.5][2] with W(CO)5(thf) at room temperature over 36 hours, and subsequent workup yielded the tungsten complex [(PPh4)2][W(η 1‐2)(CO)5] in acceptable yield (Figure 3B). The respective Ni(CO)3 adduct [(NBu4)2][Ni(η 1‐2)(CO)3] was accessed via the reaction of [(NBu4)2][2] with Ni(COD)2 (COD=1,5‐cyclooctadiene) and carbon monoxide at room temperature. 1H NMR spectroscopy revealed C 4v symmetry for both compounds at room temperature in solution, indicative for symmetrical η 1‐coordinations of the tin(II) lone pair to the transition metal carbonyl fragments. The coordination of tin(II) to W and Ni causes coordination chemical shifts Δδ(119Sn)=δ(119Sn)coord.−δ(119Sn)free compared to free [2]2− which are more pronounced for [(NBu4)2][Ni(η 1‐2)(CO)3] (Δδ(119Sn)=183 ppm) than for [(PPh4)2][W(η 1‐2)(CO)5] (Δδ(119Sn)=35 ppm) being in line with previous observations.[ 26 , 27 ] The solid‐state molecular structure of [(PPh4)2][W(η 1‐2)(CO)5] verifies [2]2− acting as a η 1 σ‐donor ligand via the tin(II) electron lone pair (Figure 4A). Coordination of the tin(II) to tungsten causes a shortening of the Sn−N bonds and a decreased pyramidalization of [2]2− as shown by increased trans‐N−Sn−N valence angles compared to free [2]2− (cf. Figure 2C). Both effects align well with the transfer of electron density from tin(II) to the Lewis acidic W(CO)5 fragment. Interestingly, the Sn1−W1 bond is slightly longer than the W−Sn bonds found for a variety of tin(II) ligands (283.44(6) ppm vs. an average of 274.2 ppm). [21] As can be seen by the bending of the tungsten bound carbonyl ligands, this bond lengthening can be attributed to the steric pressure imparted by the meso‐octaethylcalix[4]pyrrolato backbone. Indeed, a solution ATIR spectrum of [(NBu4)2][Ni(η 1‐2)(CO)3] in THF delivered a TEP (A 1 symmetrical C−O stretching band) of 2032 cm−1 (Figure S49). Remarkably, this value lies below that of all hitherto described carbenes (including CAACs) [2b] and the most electron‐rich phosphine ligands, [28] corroborating the σ‐donor and weak π‐acceptor properties of [2]2− .
Figure 3.

Synthesis of A) the lithium stanna(II) selenido complex [Li2(thf) x ][2‐Se], the η 1‐coordinated metal carbonyl complexes [(PPh4)2][W(η 1‐2)(CO)5] (B) and [(NBu4)2][Ni(η 1‐2)(CO)3] (C) and the η 5‐coordinated ruthenium(II) sandwich complex Ru(η 5‐2)(p‐cymene) (D).
Figure 4.
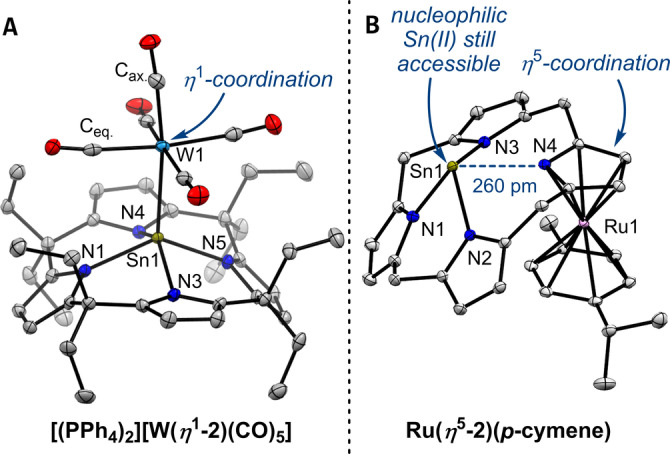
Solid‐state molecular structure of A) the η 1‐complex [(PPh4)2][W(η 1‐2)(CO)5] and B) the η 5‐complex Ru(η 5‐2)(p‐cymene). Displacement ellipsoids are drawn at 50 % probability level. Hydrogens, counter cations, solvent molecules, and meso‐ethyl residues in B) are omitted for clarity. Selected bond lengths [pm] and angles [°] for [(PPh4)2][W(η 1‐2)(CO)5]: Sn1−N1 222.3(2), Sn1−N3 225.7(2), Sn1−N4 224.6(2), Sn1−N5 222.5(2), W1−Sn1 283.44(6), W1−Cax. 196.9(3), W1−Ceq. between 202.0(3) and 204.4(3), Sn1−N4‐plane 83.2, N1−Sn1−N4 133.41(9), N3−Sn1−N4 139.25(9), cis−N−Sn−N between 81.54(8) and 82.53(8); and Ru(η 5‐2)(p‐cymene): Sn1−N1 219.80(11), Sn1−N2 232.01(11), Sn1−N3 233.01(12), Sn1−N4 259.95(12), Sn1−N4‐plane 103.3, Ru1‐pyrrole 183.7, Ru1‐cymene 171.8, N1−Sn1−N4 126.45(4), N2−Sn1−N3 128.10(4), cis−N−Sn−N between 74.76(4) and 83.13(4).
Reaction of [Li2(thf) x ][2] with excess selenium powder in THF‐d 8 at room temperature completed within minutes, quantitatively forming the lithium salt of the stanna(II) selenido complex [Li2(thf) x ][2‐Se] (Figure 3A). Depending on the π‐accepting properties of stannylenes, the tin‐selenium bond can be described via two resonance forms: i) as a Sn=Se double bond (strong π‐acceptor, deshielded selenium, downfield shift) and ii) as a Sn+−Se− single bond with partial charges (weak π‐acceptor, shielded selenium, upfield shift). [Li2(thf) x ][2‐Se] shows a 77Se NMR shift of −391.99 ppm, lying in a range comparable to a tetracoordinated but neutral, bis amidinato stanna(II) selenido complex—thus no notable strong upfield shift. [29] An explanation for this seemingly contrasting result with the TEP number was required. In 2020, Bertrand and co‐workers reported carbene selenide adducts with stronger than expected downfield 77Se NMR chemical shifts caused by C(sp 3 )H−Se non‐classical hydrogen bonding. [30] Indeed, as indicated by 1H and 2D 77Se,1H‐HMBC NMR spectroscopy (Figure S16) and evidenced by NBO and ETS‐NOCV calculations (Table S11, S12 and Figure S56) on [2‐Se]2− , significant interactions exist between the selenium and the methylene hydrogen atoms of the meso‐ethyl residues in [2‐Se]2− , shifting the 77Se NMR signals potentially downfield (cf. Figure 3A). To correct the experimental 77Se NMR shifts for these secondary interactions, we calculated the 77Se NMR chemical shifts of [2‐Se]2− and of the hypothetical meso‐unsubstituted derivative [2H‐Se]2− (PBE0/SO‐ZORA‐TZ2P COSMO(THF)//B97M‐D3(BJ)/def2‐TZVPP). The computed 77Se shift for [2‐Se]2− lies at −449.37 ppm. Notably, the absence of meso‐alkyl residues in [2H‐Se]2− induces a 77Se NMR resonance strongly upfield shifted to −638.57 ppm. NBO calculations revealed that the removal of the meso‐substituents do not change the direct Sn(lp)→Se(pz) bonding interactions, supporting the 77Se NMR shift of [2H‐Se]2− as a meaningful, value corrected for the absence of C(sp 3 )H−Se hydrogen bonding. Hence, a notably negative selenium shift in comparison with 77Se NMR shifts of other literature‐known stanna(II) selenido complexes[ 29 , 31 ] additionally confirms [2]2− as a strong σ‐donor ligand with weak π‐acceptor properties (Table S13 and Figure S57). The absent π‐acceptor property is further illustrated by ETS‐NOCV and NBO calculations of [2‐Se]2− , showing negligible contributions of a Se(lp)→π*(Sn−N) back donation to the total orbital interaction between [2]2− and Se (Table S11, S12 and Figure S56).
Next, alternative coordination modes of [2]2− against transition metals were probed. A rapid reaction of [Li2(thf) x ][2] with 0.5 equivalents [{Ru(Cl)(p‐cymene)}2(μ‐Cl)2] at room temperature provided neutral Ru(η 5‐2)(p‐cymene) at excellent yield and bulk purity (Figure 3D). The structure of Ru(η 5‐2)(p‐cymene) was confirmed by SCXRD (Figure 4B). η 5‐coordination to a ruthenium(II) p‐cymene fragment decreases the electron density in one of the pyrrolato units in [2]2− , causing an elongated Sn1‐N4 bond in contrast to the other Sn−N bonds. However, 119Sn NMR spectroscopy still shows a high‐field resonance at −424.83 ppm as expected for a tetra‐coordinated tin(II) atom. Owing to the energetically nearly degenerated HOMO, HOMO−1 and HOMO−2 (Figure 2D), [2]2− can either act as a η 1‐coordinating 2e− donor with 16 VE fragments like W(CO)5 and Ni(CO)3 or as a η 5‐coordinating 6e− donor with the 12 VE fragment [Ru(p‐cymene)]2+. Interestingly, dianionic calix[4]pyrrole complexes of NiII and CuII were shown to act as η 5‐coordinating 6e− donors solely via their pyrrolato units forming polymetallic sandwich‐type complexes in their second coordination sphere, whereas the σ‐donor mode is unique for [2]2− .[ 14d , 32 ]
Oxidative addition across carbon‐halogen bonds displays a common reactivity mode for low‐valent main group element species, including classical stannylenes. [33] However, since the tin(II) center in [2]2− possesses a high‐energy lone pair but no π‐accepting character at tin, a reactivity closer to R3Sn− nucleophiles should be expected.[ 7a , 34 ] Indeed, in toluene‐d 8 the lithium salt [Li2(thf) x ][2] readily substitutes the chloride in benzoyl chloride forming acyl stannate(IV) [Li(thf)2][3‐COPh] (Figure 5A) as shown by NMR spectroscopy and high‐resolution ESI(−) MS. Cooling of the reaction solution to −30 °C yielded single crystals suitable for X‐ray diffractometry, validating the nucleophilic substitution at the carbonyl C atom (Figure 5C). Compared to free [2]2− and the η 1‐complex [W(η 1‐2)(CO)5]2− , the acyl stannate(IV) [3‐COPh]− possess significantly shortened Sn‐N bond lengths and increased trans−N−Sn−N valence angles, being in line with formal oxidation of the tin(II) center to a tin(IV). It represents a rare example of an acyl stannate characterized in the solid‐state and, to the best of our knowledge, the first example with a pentacoordinated tin. To our surprise, the respective acyl stannates(IV) were also obtained upon reacting [Li2(thf) x ][2] with 4‐CF3‐benzaldehyde and benzaldehyde, alongside the precipitation of lithium hydride. Whereas for benzaldehyde, the product could only be detected by HRMS ESI(−) measurements, the more reactive 4‐CF3‐benzaldehyde yielded [3‐COC6H4(CF3)]− at around 45 % yield. In contrast to monoanionic tin nucleophiles such as n Bu3SnMgBr and n Bu3SnLi, which need specialized oxidants as hydride acceptors to form neutral acyl stannates(IV) from the alcoholato stage, the dianionic nucleophile [2]2− enables a direct hydride substitution. [35] Possibly, the missing π‐acceptor ability and the high electron richness of [2]2− destabilizes the tetrahedral alcoholato intermediate to an extend that hydride expulsion becomes a viable process. Noteworthily, the corresponding dianionic calix[4]pyrrole complexes of NiII react both with acyl chlorides or aldehydes by nucleophilic attack of the pyrrole‐backbone, once again underscoring the unique metal‐centered nucleophilicity of [2]2− . [36] In the reaction of [(NBu4)2][2] with benzoyl chloride, CH2Cl2 or benzyl chloride, 1H, 13C and 119Sn NMR spectroscopy showed near quantitative conversion of the starting materials into new species, which were identified as the nucleophilic substitution products [NBu4][3‐R] (R=CH2Cl, CH2Ph, COPh; Figure 5B). No reaction was observed with benzaldehydes, rendering the precipitation of lithium hydride as an additional factor for the hydride substitution.
Figure 5.

Reactivity of A) [Li2(thf) x ][2] and B) [(NBu4)2][2] with electrophiles and D) reaction of [(NBu4)2][2] with iodobenzene. Yields where determined by NMR spectroscopy. Oxidation potentials were determined by cyclic voltammetry of [(NBu4)2][2] (10−3 M) and Bu4NPF6 (0.01 M) in 1,2‐difluorobenzene at a scan rate of 0.05 V s−1. C) Solid state molecular structure of [Li(thf) x ][3‐COPh]. Displacement ellipsoids are drawn at 50 % probability level. Hydrogens and toluene solvent molecules are omitted for clarity. Selected bond lengths [pm] and angles [°]: Sn1−N1 208.11(10), Sn1−N2 210.25(10), Sn1−N3 211.02(12), Sn1−N4 211.31(11), Sn1−N4‐plane 49.4, Sn1−C9 220.94(13), C9−O3 123.20(14), N1−Sn1−N4 152.56(4), N2−Sn1−N3 153.09(4), cis−N−Sn−N between 84.37(4) and 89.43(4).
Besides being a nucleophile, [2]2− also rendered as reductant, showing two irreversible one‐electron oxidations at −0.42 V and 0.04 V and quasi‐reversible two‐electron oxidation at 0.55 V (vs. AgCl/Ag/KCl, Figure S54). Since electron donors can engage in radical‐nucleophilic aromatic substitutions, [37] [(NBu4)2][2] was reacted with one equivalent iodobenzene. Mixing for 48 hours at room temperature in THF‐d 8, the near quantitative reduction of iodobenzene to benzene could be observed alongside the clean formation of the tin(IV) bis‐(thf) complex 3‐(thf)2 . [38] Following literature reports, the NBu4 + cation was assumed as the proton source. [39] Interestingly, the reduction potential of [(NBu4)2][2] should not allow a direct reduction of iodobenzene (≈−2.24 V vs. AgCl/Ag/KCl). [40] Instead, computations revealed the exergonic formation of a precursor complex via Sn→I σ‐hole halogen bond interaction, potentially reducing the activation barrier of an inner sphere electron transfer (Figure S53). Generally, this observation adds a promising facet to the features of [2]2− , such as selective reductant or for the participation in frustrated radical pair chemistry. [41]
Conclusion
Amido ligands hold a unique role in the chemistry of low‐valent p‐block element compounds. [13] With the present work, we extend this substance class by the first tetraamido tetrylene dianion, [2]2− . The substantial repulsion that usually would prohibit the formation of such high‐energy species is surpassed by taking advantage of the macrocyclic effect of the calix[4]pyrrole ligand. Consequently, by coordinating four anionic nitrogen π‐donors, the energetically buried tin(II) electron lone pair is pushed to extreme σ‐donor capacity. A TEP value of 2032 cm−1 attests donor strength exceeding that of all previously described carbene or phosphine ligands, offering potential as an electron‐rich ligand in transition metal catalysis (Figure 6). [42]
Figure 6.
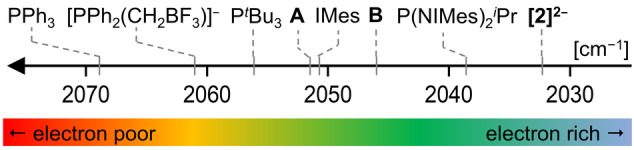
Comparison between the TEP values of [2]2− and other literature‐known carbene and phosphine ligands. Absolute values and references are given in Table S9.
The coordinative properties of [2]2− are exemplified in structurally diverse complexes with tungsten and ruthenium. The η 5‐coordination behavior suggests the possibility of placing a transition metal center near a nucleophilic tin(II) atom, expedient for cooperative activation modes. [43] Spontaneous hydride substitution reaction with benzaldehydes and the straightforward reaction of [2]2− with acyl chlorides grants access to the yet underexplored class of acyl metalloids, e.g., of interest as visible‐light photoinitiators. [44] Of note, [2]2− represents the first foray of low‐valent p‐block element chemistry with the versatile calix[4]pyrrole ligand.
Conflict of interest
The authors declare no conflict of interest.
1.
Supporting information
As a service to our authors and readers, this journal provides supporting information supplied by the authors. Such materials are peer reviewed and may be re‐organized for online delivery, but are not copy‐edited or typeset. Technical support issues arising from supporting information (other than missing files) should be addressed to the authors.
Supporting Information
Supporting Information
Supporting Information
Supporting Information
Supporting Information
Supporting Information
Supporting Information
Acknowledgements
We thank Prof. H.‐J. Himmel for his constant support. Financial support was provided by the European Research Council (ERC) under the European Union's Horizon 2020 research and innovation program (grant agreement No948708) and the Foundation of German Business (sdw, H.R.). The BWFor/BWUniCluster, funded by the DFG, are acknowledged for computational resources. Open Access funding enabled and organized by Projekt DEAL.
H. Ruppert, L. Greb, Angew. Chem. Int. Ed. 2022, 61, e202116615; Angew. Chem. 2022, 134, e202116615.
Data Availability Statement
The data that support the findings of this study are available in the supplementary material of this article.
References
- 1.
- 1a. Doddi A., Peters M., Tamm M., Chem. Rev. 2019, 119, 6994–7112; [DOI] [PubMed] [Google Scholar]
- 1b. Hopkinson M. N., Richter C., Schedler M., Glorius F., Nature 2014, 510, 485–496; [DOI] [PubMed] [Google Scholar]
- 1c. Nesterov V., Reiter D., Bag P., Frisch P., Holzner R., Porzelt A., Inoue S., Chem. Rev. 2018, 118, 9678–9842; [DOI] [PubMed] [Google Scholar]
- 1d. Bellotti P., Koy M., Hopkinson M. N., Glorius F., Nat. Chem. Rev. 2021, 5, 711–725; [DOI] [PubMed] [Google Scholar]
- 1e. Melaimi M., Jazzar R., Soleilhavoup M., Bertrand G., Angew. Chem. Int. Ed. 2017, 56, 10046–10068; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2017, 129, 10180–10203; [Google Scholar]
- 1f. Jazzar R., Soleilhavoup M., Bertrand G., Chem. Rev. 2020, 120, 4141–4168. [DOI] [PubMed] [Google Scholar]
- 2.
- 2a. Nelson D. J., Nolan S. P., Chem. Soc. Rev. 2013, 42, 6723–6753; [DOI] [PubMed] [Google Scholar]
- 2b. Huynh H. V., Chem. Rev. 2018, 118, 9457–9492. [DOI] [PubMed] [Google Scholar]
- 3.
- 3a. Álvarez-Rodríguez L., Cabeza J. A., García-Álvarez P., Polo D., Coord. Chem. Rev. 2015, 300, 1–28; [Google Scholar]
- 3b. Parvin N., Pal S., Rojisha V. C., De S., Parameswaran P., Khan S., ChemistrySelect 2016, 1, 1991–1995; [Google Scholar]
- 3c. So C.-W., Roesky H. W., Magull J., Oswald R. B., Angew. Chem. Int. Ed. 2006, 45, 3948–3950; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2006, 118, 4052–4054; [Google Scholar]
- 3d. Arp H., Baumgartner J., Marschner C., Zark P., Müller T., J. Am. Chem. Soc. 2012, 134, 10864–10875; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3e. Nguyen T. A. N., Frenking G., Chem. Eur. J. 2012, 18, 12733–12748; [DOI] [PubMed] [Google Scholar]
- 3f. Kutzelnigg W., Angew. Chem. Int. Ed. Engl. 1984, 23, 272–295; [Google Scholar]; Angew. Chem. 1984, 96, 262–286; [Google Scholar]
- 3g. Nechaev M. S., Organometallics 2021, 40, 3408–3423. [Google Scholar]
- 4. Hahn F. E., Zabula A. V., Pape T., Hepp A., Tonner R., Haunschild R., Frenking G., Chem. Eur. J. 2008, 14, 10716–10721. [DOI] [PubMed] [Google Scholar]
- 5.
- 5a. Mohapatra C., Scharf L. T., Scherpf T., Mallick B., Feichtner K.-S., Schwarz C., Gessner V. H., Angew. Chem. Int. Ed. 2019, 58, 7459–7463; [DOI] [PMC free article] [PubMed] [Google Scholar]; Angew. Chem. 2019, 131, 7537–7541; [Google Scholar]
- 5b. Takahashi S., Sekiguchi J., Ishii A., Nakata N., Angew. Chem. Int. Ed. 2021, 60, 4055–4059; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2021, 133, 4101–4105. [Google Scholar]
- 6.
- 6a. Angermund K., Jonas K., Krüger C., Latten J. L., Tsay Y.-H., J. Organomet. Chem. 1988, 353, 17–25; [Google Scholar]
- 6b. Drost C., Hitchcock P. B., Lappert M. F., Angew. Chem. Int. Ed. 1999, 38, 1113–1116; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 1999, 111, 1185–1187; [Google Scholar]
- 6c. Wagner M., Dorogov K., Schürmann M., Jurkschat K., Dalton Trans. 2011, 40, 8839–8848; [DOI] [PubMed] [Google Scholar]
- 6d. Cabeza J. A., Garcia-Alvarez P., Polo D., Eur. J. Inorg. Chem. 2016, 10–22. [Google Scholar]
- 7.
- 7a. Capperucci A., Degl′Innocenti A., Faggi C., Reginato G., Ricci A., Dembech P., Seconi G., J. Org. Chem. 1989, 54, 2966–2968; [Google Scholar]
- 7b. Riviere P., Castel A., Riviere-Baudet M., in The Chemistry of Organic Germanium, Tin and Lead Compounds, Vol. 2 (Ed.: Rappoport Z.), Wiley, Hoboken, 2002, pp. 653–748; [Google Scholar]
- 7c. Lee V. Y., Sekiguchi A., in Organometallic Compounds of Low-Coordinate Si, Ge, Sn and Pb: From Phantom Species to Stable Compounds, Wiley, Hoboken, 2010, pp. 89–138; [Google Scholar]
- 7d. Wang D.-Y., Wang C., Uchiyama M., J. Am. Chem. Soc. 2015, 137, 10488–10491; [DOI] [PubMed] [Google Scholar]
- 7e. Davies A. G., in Organotin Chemistry , 2nd ed., Wiley, Hoboken, 2004, pp. 311–332. [Google Scholar]
- 8.
- 8a. Memmler H., Kauper U., Gade L. H., Stalke D., Lauher J. W., Organometallics 1996, 15, 3637–3639; [Google Scholar]
- 8b. Kilian M., Wadepohl H., Gade L. H., Dalton Trans. 2008, 582–584. [DOI] [PubMed] [Google Scholar]
- 9.
- 9a. Gade L. H., Eur. J. Inorg. Chem. 2002, 1257–1268; [Google Scholar]
- 9b. Lutz M., Haukka M., Pakkanen T. A., Gade L. H., Inorg. Chem. 2003, 42, 2798–2804; [DOI] [PubMed] [Google Scholar]
- 9c. Kilian M., Wadepohl H., Gade L. H., Organometallics 2007, 26, 3076–3078; [Google Scholar]
- 9d. Kilian M., Wadepohl H., Gade L. H., Eur. J. Inorg. Chem. 2008, 1892–1900. [Google Scholar]
- 10.
- 10a. Elder R. C., Heeg M. J., Deutsch E., Inorg. Chem. 1978, 17, 427–431; [Google Scholar]
- 10b. Schiemenz B., Huttner G., Angew. Chem. Int. Ed. Engl. 1993, 32, 297–298; [Google Scholar]; Angew. Chem. 1993, 105, 295–296; [Google Scholar]
- 10c. Renner G., Kircher P., Huttner G., Rutsch P., Heinze K., Eur. J. Inorg. Chem. 2001, 973–980; [Google Scholar]
- 10d. Kircher P., Huttner G., Zsolnai L., Driess A., Angew. Chem. Int. Ed. 1998, 37, 1666–1668; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 1998, 110, 1756–1758. [Google Scholar]
- 11.
- 11a. Wesemann L., Z. Anorg. Allg. Chem. 2004, 630, 1349–1356; [Google Scholar]
- 11b. Chapman R. W., Kester J. G., Folting K., Streib W. E., Todd L. J., Inorg. Chem. 1992, 31, 979–983. [Google Scholar]
- 12. Vollgraff T., Michel N., Sundermeyer J., Eur. J. Inorg. Chem. 2021, 3852–3860. [Google Scholar]
- 13.
- 13a. Lappert M. F., Power P. P., Sanger A. R., Srivastava R. C., Metal and Metalloid Amides: Synthesis, Structures, and Physical and Chemical Properties, Ellis Horwood-Wiley, Chichester, 1980; [Google Scholar]
- 13b. Lappert M. F., Power P. P., Protchenko A., Seeber A. Metal Amide Chemistry, Wiley, Hoboken„: 2009; [Google Scholar]
- 13c. Veith M., Angew. Chem. Int. Ed. Engl. 1987, 26, 1–14; [Google Scholar]; Angew. Chem. 1987, 99, 1–14; [Google Scholar]
- 13d. Mizuhata Y., Sasamori T., Tokitoh N., Chem. Rev. 2009, 109, 3479–3511; [DOI] [PubMed] [Google Scholar]
- 13e. Asay M., Jones C., Driess M., Chem. Rev. 2011, 111, 354–396. [DOI] [PubMed] [Google Scholar]
- 14.
- 14a. Jacoby D., Floriani C., Chiesi-Villa A., Rizzoli C., J. Chem. Soc. Chem. Commun. 1991, 220–222; [Google Scholar]
- 14b. Floriani C., Floriani-Moro R., in The Porphyrin Handbook, Vol. 3 (Eds.: Kadish K. M., Smith K. M., Guilard R.), Academic Press, San Diego, 2000, pp. 405–420; [Google Scholar]
- 14c. Floriani C., Floriani-Moro R., in The Porphyrin Handbook, Vol. 3 (Eds.: Kadish K. M., Smith K. M., Guilard R.), Academic Press, San Diego, 2000, pp. 385–402; [Google Scholar]
- 14d. Cuesta L., Sessler J. L., Chem. Soc. Rev. 2009, 38, 2716–2729; [DOI] [PubMed] [Google Scholar]
- 14e. Bachmann J., Nocera D. G., J. Am. Chem. Soc. 2004, 126, 2829–2837; [DOI] [PubMed] [Google Scholar]
- 14f. Bachmann J., Nocera D. G., J. Am. Chem. Soc. 2005, 127, 4730–4743; [DOI] [PubMed] [Google Scholar]
- 14g. Buranaprasertsuk P., Tangsakol Y., Chavasiri W., Catal. Commun. 2007, 8, 310–314; [Google Scholar]
- 14h. Mohebbi S., Rayati S., Transition Met. Chem. 2007, 32, 1035–1038. [Google Scholar]
- 15.
- 15a. Deacon G. B., Guo Z., Junk P. C., Wang J., Angew. Chem. Int. Ed. 2017, 56, 8486–8489; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2017, 129, 8606–8609; [Google Scholar]
- 15b. Mukthar N. F. M., Schley N. D., Ung G., Dalton Trans. 2020, 49, 16059–16061. [DOI] [PubMed] [Google Scholar]
- 16.
- 16a. Ebner F., Wadepohl H., Greb L., J. Am. Chem. Soc. 2019, 141, 18009–18012; [DOI] [PubMed] [Google Scholar]
- 16b. Ebner F., Sigmund L. M., Greb L., Angew. Chem. Int. Ed. 2020, 59, 17118–17124; [DOI] [PMC free article] [PubMed] [Google Scholar]; Angew. Chem. 2020, 132, 17266–17272; [Google Scholar]
- 16c. Sigmund L. M., Greb L., Chem. Sci. 2020, 11, 9611–9616; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16d. Ebner F., Mainik P., Greb L., Chem. Eur. J. 2021, 27, 5120–5124; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16e. Sigmund L. M., Ehlert C., Enders M., Graf J., Gryn′ova G., Greb L., Angew. Chem. Int. Ed. 2021, 60, 15632–15640; [DOI] [PMC free article] [PubMed] [Google Scholar]; Angew. Chem. 2021, 133, 15761–15769. [Google Scholar]
- 17.
- 17a. Ebner F., Greb L., J. Am. Chem. Soc. 2018, 140, 17409–17412; [DOI] [PubMed] [Google Scholar]
- 17b. Ebner F., Greb L., Chem 2021, 7, 2151–2159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Ruppert H., Sigmund L. M., Greb L., Chem. Commun. 2021, 57, 11751–11763. [DOI] [PubMed] [Google Scholar]
- 19.
- 19a. Segawa Y., Yamashita M., Nozaki K., Science 2006, 314, 113–115; [DOI] [PubMed] [Google Scholar]
- 19b. Segawa Y., Suzuki Y., Yamashita M., Nozaki K., J. Am. Chem. Soc. 2008, 130, 16069–16079; [DOI] [PubMed] [Google Scholar]
- 19c. Weber L., Eur. J. Inorg. Chem. 2017, 3461–3488; [Google Scholar]
- 19d. Protchenko A. V., Vasko P., Fuentes M. Á., Hicks J., Vidovic D., Aldridge S., Angew. Chem. Int. Ed. 2021, 60, 2064–2068; [DOI] [PMC free article] [PubMed] [Google Scholar]; Angew. Chem. 2021, 133, 2092–2096. [Google Scholar]
- 20.
- 20a. Hicks J., Vasko P., Goicoechea J. M., Aldridge S., Nature 2018, 557, 92–95; [DOI] [PubMed] [Google Scholar]
- 20b. Liu Y., Li J., Ma X., Yang Z., Roesky H. W., Coord. Chem. Rev. 2018, 374, 387–415; [Google Scholar]
- 20c. Hicks J., Vasko P., Goicoechea J. M., Aldridge S., J. Am. Chem. Soc. 2019, 141, 11000–11003; [DOI] [PubMed] [Google Scholar]
- 20d. Hicks J., Vasko P., Goicoechea J. M., Aldridge S., Angew. Chem. Int. Ed. 2021, 60, 1702–1713; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2021, 133, 1726–1737. [Google Scholar]
- 21. Agustin D., Ehses M., C. R. Chim. 2009, 12, 1189–1227. [Google Scholar]
- 22.Deposition numbers 2125915, 2125916, 2125917, 2125918, 2125919 and 2125920 contain the supplementary crystallographic data for this paper. These data are provided free of charge by the joint Cambridge Crystallographic Data Centre and Fachinformationszentrum Karlsruhe Access Structures service.
- 23.
- 23a. Atwood D. A., Atwood V. O., Cowley A. H., Atwood J. L., Roman E., Inorg. Chem. 1992, 31, 3871–3872; [Google Scholar]
- 23b. Kim S. B., Sinsermsuksakul P., Hock A. S., Pike R. D., Gordon R. G., Chem. Mater. 2014, 26, 3065–3073; [Google Scholar]
- 23c. Ungpittagul T., Wongmahasirikun P., Phomphrai K., Dalton Trans. 2020, 49, 8460–8471; [DOI] [PubMed] [Google Scholar]
- 23d. Samii R., Zanders D., Fransson A., Bacic G., Barry S. T., Ojamae L., Kessler V., Pedersen H., O'Brien N. J., Inorg. Chem. 2021, 60, 12759–12765. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Tolman C. A., Chem. Rev. 1977, 77, 313–348. [Google Scholar]
- 25. Liske A., Verlinden K., Buhl H., Schaper K., Ganter C., Organometallics 2013, 32, 5269–5272. [Google Scholar]
- 26. Mont W.-W. D., Kroth H.-J., Z. Naturforsch. B 1980, 35, 700–702. [Google Scholar]
- 27.Even via prolonged 119Sn NMR measurements of [(PPh4)2][W(η 1-2)(CO)5] no 1JSn-W coupling constant could be obtained, which might be related to the structural flexibility of the calix[4]pyrrole ligand causing broadened 119Sn resonances.
- 28. Kelty M. L., McNeece A. J., Filatov A. S., Anderson J., ChemRxiv. 2021, 10.33774/chemrxiv-2021-4jd82-v2. This content is a preprint and has not been peer-reviewed. [DOI] [Google Scholar]
- 29. Ahmet I. Y., Thompson J. R., Johnson A. L., Eur. J. Inorg. Chem. 2018, 1670–1678. [Google Scholar]
- 30. Junor G. P., Lorkowski J., Weinstein C. M., Jazzar R., Pietraszuk C., Bertrand G., Angew. Chem. Int. Ed. 2020, 59, 22028–22033; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2020, 132, 22212–22217. [Google Scholar]
- 31.
- 31a. Leung W.-P., Kwok W.-H., Zhou Z.-Y., Mak T. C. W., Organometallics 2000, 19, 296–303; [Google Scholar]
- 31b. Mairychová B., Dostál L., Růžička A., Fulem M., Růžička K., Lyčka A., Jambor R., Organometallics 2011, 30, 5904–5910; [Google Scholar]
- 31c. Saito M., Tokitoh N., Okazaki R., J. Am. Chem. Soc. 2004, 126, 15572–15582; [DOI] [PubMed] [Google Scholar]
- 31d. Park J.-H., Kang S. G., Lee Y. K., Chung T.-M., Park B. K., Kim C. G., Inorg. Chem. 2020, 59, 3513–3517. [DOI] [PubMed] [Google Scholar]
- 32.
- 32a. Bonomo L., Solari E., Martin G., Scopelliti R., Floriani C., Chem. Commun. 1999, 2319–2320; [Google Scholar]
- 32b. Bonomo L., Solari E., Latronico M., Scopelliti R., Floriani C., Chem. Eur. J. 1999, 5, 2040–2047; [Google Scholar]
- 32c. Cuesta L., Gross D., Lynch V. M., Ou Z., Kajonkijya W., Ohkubo K., Fukuzumi S., Kadish K. M., Sessler J. L., J. Am. Chem. Soc. 2007, 129, 11696–11697. [DOI] [PubMed] [Google Scholar]
- 33.
- 33a. Chu T., Nikonov G. I., Chem. Rev. 2018, 118, 3608–3680; [DOI] [PubMed] [Google Scholar]
- 33b. Lappert M. F., Misra M. C., Onyszchuk M., Rowe R. S., Power P. P., Slade M. J., J. Organomet. Chem. 1987, 330, 31–46. [Google Scholar]
- 34. Reich H. J., Borst J. P., Dykstra R. R., Organometallics 1994, 13, 1–3. [Google Scholar]
- 35.
- 35a. Marshall J. A., in Organometallics in Synthesis: A Manual, 2nd ed. (Ed.: Schlosser M.), Wiley, Hoboken, 2001, pp. 353–464; [Google Scholar]
- 35b. Marshall J. A., Welmaker G. S., Gung B. W., J. Am. Chem. Soc. 1991, 113, 647–656. [Google Scholar]
- 36. Bonomo L., Solari E., Floriani C., Chiesi-Villa A., Rizzoli C., J. Am. Chem. Soc. 1998, 120, 12972–12973. [Google Scholar]
- 37.
- 37a. Bunnett J. F., Kim J. K., J. Am. Chem. Soc. 1970, 92, 7463–7464; [Google Scholar]
- 37b. Rossi R. A., Guastavino J. F., Budén M. E., in Arene Chemistry: Reaction Mechanisms and Methods for Aromatic Compounds (Ed.: Mortier J.), Wiley, Hoboken, 2015, pp. 243–268. [Google Scholar]
- 38.
- 38a. Benech J.-M., Bonomo L., Solari E., Scopelliti R., Floriani C., Angew. Chem. Int. Ed. 1999, 38, 1957–1959; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 1999, 111, 2107–2109; [Google Scholar]
- 38b. Fulmer G. R., Miller A. J. M., Sherden N. H., Gottlieb H. E., Nudelman A., Stoltz B. M., Bercaw J. E., Goldberg K. I., Organometallics 2010, 29, 2176–2179. [Google Scholar]
- 39. Alvarado de la Torre R., Sease J. W., J. Am. Chem. Soc. 1979, 101, 1687–1690. [Google Scholar]
- 40. Pause L., Robert M., Savéant J.-M., J. Am. Chem. Soc. 1999, 121, 7158–7159. [Google Scholar]
- 41.
- 41a. Liu L. L., Stephan D. W., Chem. Soc. Rev. 2019, 48, 3454–3463; [DOI] [PubMed] [Google Scholar]
- 41b. Holtrop F., Jupp A. R., Kooij B. J., Van Leest N. P., De Bruin B., Slootweg J. C., Angew. Chem. Int. Ed. 2020, 59, 22210–22216; [DOI] [PMC free article] [PubMed] [Google Scholar]; Angew. Chem. 2020, 132, 22394–22400. [Google Scholar]
- 42. Wünsche M. A., Mehlmann P., Witteler T., Buß F., Rathmann P., Dielmann F., Angew. Chem. Int. Ed. 2015, 54, 11857–11860; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2015, 127, 12024–12027. [Google Scholar]
- 43. Somerville R. J., Campos J., Eur. J. Inorg. Chem. 2021, 3488–3498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.
- 44a. Eibel A., Radebner J., Haas M., Fast D. E., Freißmuth H., Stadler E., Faschauner P., Torvisco A., Lamparth I., Moszner N., Stueger H., Gescheidt G., Polym. Chem. 2018, 9, 38–47; [Google Scholar]
- 44b. Radebner J., Eibel A., Leypold M., Jungwirth N., Pickl T., Torvisco A., Fischer R., Fischer U. K., Moszner N., Gescheidt G., Stueger H., Haas M., Chem. Eur. J. 2018, 24, 8281–8285; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44c. Püschmann S. D., Frühwirt P., Pillinger M., Knöchl A., Mikusch M., Radebner J., Torvisco A., Fischer R. C., Moszner N., Gescheidt G., Haas M., Chem. Eur. J. 2021, 27, 3338–3347; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44d. Holownia A., Apte C. N., Yudin A. K., Chem. Sci. 2021, 12, 5346–5360. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
As a service to our authors and readers, this journal provides supporting information supplied by the authors. Such materials are peer reviewed and may be re‐organized for online delivery, but are not copy‐edited or typeset. Technical support issues arising from supporting information (other than missing files) should be addressed to the authors.
Supporting Information
Supporting Information
Supporting Information
Supporting Information
Supporting Information
Supporting Information
Supporting Information
Data Availability Statement
The data that support the findings of this study are available in the supplementary material of this article.