

Abstract
Background and Purpose
Analogues of fibroblast growth factor 21 (FGF21) demonstrate diverse metabolic benefits in preclinical models of type 2 diabetes, dyslipidaemia and non‐alcoholic steatohepatitis (NASH), but clinical responses with different analogues are inconsistent. Efruxifermin is an Fc‐FGF21 fusion protein with prolonged half‐life and enhanced receptor affinity compared with native human FGF21. Efruxifermin is in clinical trials for the treatment of non‐alcoholic steatohepatitis.
Experimental Approach
Efruxifermin was administered weekly to male and female Sprague Dawley rats for 4 or 26 weeks. Body and organ weights, macroscopic and microscopic pathology, clinical chemistry, blood cytology and serum and urine biomarkers were analysed to characterize the pharmacodynamics of efruxifermin and to investigate potential non‐clinical toxicities following chronic administration of supra‐pharmacological doses of efruxifermin.
Key Results
Efruxifermin significantly reduced body weight gain after 4 and 26 weeks, despite increasing food intake. Changes in tissue pathology, clinical chemistry and serum biomarkers generally appeared to be associated with weight loss, except for a significant decrease in urine volume in both males and females without perturbed electrolyte balance. Markers of sympathetic activation, urinary corticosterone and ratio of adrenal‐to‐body weight were unchanged.
Conclusion and Implications
Efruxifermin attenuated body weight gain, consistent with other FGF21 analogues. In contrast to at least one other FGF21 analogue, efruxifermin decreased rather than increased urine volume. The absence of an increase in sympathetic tone in rats mirrors the unchanged salivary cortisol and systemic blood pressure following efruxifermin treatment in humans.
Keywords: electrolyte balance, FGF21, fibroblast growth factor 21, hypothalamic–pituitary–adrenal axis, kidney, translational pharmacology
Abbreviations
- CTX‐1
type 1 collagen cross‐linked C‐terminal telopeptide
- PEG
polyethylene glycol
What is already known
Antidyslipidaemic effects of FGF21 analogues have translated from preclinical models to humans with metabolic disease.
Urine parameter and BP changes across FGF21 analogues are inconsistent in preclinical species and humans.
What does this study add
Unlike some FGF21 analogues, efruxifermin does not increase urine output or sympathetic tone in rats.
What is the clinical significance
Effects of efruxifermin in rats are consistent with two clinical studies in metabolic disease patients.
1. INTRODUCTION
Fibroblast growth factor 21 (FGF21) is an endocrine hormone that regulates lipid, glucose and protein homeostasis as well as energy metabolism (Tillman & Rolph, 2020). Preclinical studies have demonstrated the ability of FGF21 to reduce body weight (Kharitonenkov et al., 2005), ameliorate dyslipidaemia (Badman et al., 2007; Inagaki et al., 2007), increase insulin sensitivity (Berglund et al., 2009) and reduce hepatic steatosis (Xu et al., 2009) and fibrosis (Jimenez et al., 2018), suggesting potential as a therapy for metabolic diseases including non‐alcoholic steatohepatitis (NASH).
However, therapeutic application of native FGF21 is limited by a short half‐life in circulation (Kharitonenkov et al., 2007), as a result of its susceptibility to rapid cleavage and inactivation by a circulating endopeptidase, fibroblast activation protein alpha (Dunshee et al., 2016; Zhen et al., 2016). Fibroblast activation protein activity removes 10 C‐terminal amino acid residues from human FGF21. This abolishes FGF21 high‐affinity binding to its obligate coreceptor, β‐Klotho, in turn preventing interaction of the N‐terminus of FGF21 with, and signalling through, canonical FGF receptors 1c, 2c or 3c (Ogawa et al., 2007). Truncation of residues from the N‐terminus of FGF21 does not impair its β‐Klotho affinity but abolishes FGFR signalling. Such a truncation may functionally antagonize native FGF21 signalling (Micanovic et al., 2009; Yie et al., 2009). Using this knowledge of the structure–function relationship for FGF21, various strategies have sought to prolong FGF21 pharmacodynamic effects in vivo by reducing clearance, including Fc‐fusion (Stanislaus et al., 2017), PEGylation (Mu et al., 2012), and conjugation to an antibody scaffold (Huang et al., 2013).
Non‐clinical and clinical development of these different FGF21 analogues has uncovered differential pharmacology. For example, PF‐05231023 (CVX‐343), an antibody‐conjugated FGF21 analogue, has been observed to raise heart rate and blood pressure in rats and humans but not in non‐human primates (Kim et al., 2017). Interestingly, while PF‐05231023 decreased body weight in rodents (Huang et al., 2013) and non‐human primates (Talukdar et al., 2016), it decreased body weight in one human study (Talukdar et al., 2016) while slightly increasing body weight in another (Kim et al., 2017). PF‐05231023 also increased water intake, urine output and sympathetic activity in rats, consistent with the finding of elevated blood pressure (Turner et al., 2018). Although designed as a long‐acting FGF21 analogue, the C‐terminus of the FGF21 moiety of PF‐05231023, necessary for binding to β‐Klotho, undergoes 10‐fold more rapid degradation than the N‐terminal, FGFR‐interacting portion of the molecule (Talukdar et al., 2016; Weng et al., 2015). These different rates of degradation may underlie the divergent pharmacology seen across species and observed with weekly compared with twice‐weekly dosing intervals in humans (Kim et al., 2017; Talukdar et al., 2016).
In contrast to PF‐05231023, the half‐lives of intact N‐ and C‐termini of the FGF21 moiety of efruxifermin (AKR‐001; AMG 876; Fc‐FGF21[RGE]), a 92‐kDa FGF21 analogue comprising a homodimer of two IgG1 Fc‐FGF21 variant polypeptide chains, are both extended (Stanislaus et al., 2017), enabling sustained pharmacology across a once‐weekly interdose interval (Kaufman et al., 2020). Weekly administration of efruxifermin reduced body weight and improved markers of glucose and lipid metabolism in patients with non‐alcoholic steatohepatitis (Harrison et al., 2021). Efruxifermin also improved lipoprotein profile and markers of glycaemic control in patients with type 2 diabetes, without increasing blood pressure (Kaufman et al., 2020). This lack of effect on blood pressure is consistent with the association of a putative loss‐of‐function FGF21 allele with a slight increase in systolic blood pressure in humans (Frayling et al., 2018). The pharmacodynamic effects of efruxifermin in humans recapitulate improvements in glycaemic control and in lipid and lipoprotein profiles seen in both mice and non‐human primates (Stanislaus et al., 2017).
Several other FGF21 analogues are currently in clinical trials for patients with non‐alcoholic steatohepatitis. Some are based on PEGylation to extend half‐life, while others are antibody‐based agonists of the β‐Klotho/FGFR1c complex. The most extensively studied PEGylated FGF21 analogue, pegbelfermin, which lacks engineered protection of its N‐ or C‐terminal regions, has a half‐life of 19–24 h for its intact C‐terminus (Charles et al., 2016). Pegbelfermin has been reported to reduce liver fat and decrease markers of liver fibrogenesis (Sanyal et al., 2019) without altering blood pressure (Charles et al., 2016, 2019; Sanyal et al., 2019). Similarly, BFKB8488A, which selectively activates the FGFR1/β‐Klotho coreceptor complex, has shown robust FGF21‐like pharmacodynamic effects in patients with metabolic diseases including obesity (Baruch et al., 2020), type 2 diabetes (Wong et al., 2019),and non‐alcoholic fatty liver disease (Kunder et al., 2019), with no reports of increased blood pressure.
This study explores the pharmacology and toxicology of efruxifermin administered at high doses over 4 or 26 weeks to male and female Sprague Dawley rats. The analyses described herein focus on those pharmacological effects that have been reported to translate inconsistently from preclinical to clinical studies, which may arise from differences in stability of the N‐ and C‐terminal regions of the respective FGF21 analogues.
2. METHODS
2.1. Ethics statement/ethical review
Animal studies are reported in compliance with the ARRIVE guidelines (Percie du Sert et al., 2020) and with the recommendations made by the British Journal of Pharmacology (Lilley et al., 2020). Further, animals in both studies also adhered to the USDA Animal Welfare Act (9CFR, Parts 1, 2 and 3) and the Guide for the Care and Use of Laboratory Animals (ILAR publication, 1996, National Academy Press). The study protocol was approved by the Testing Facility Institutional Animal Care and Use Committee.
2.2. Animals
The Sprague Dawley rat was chosen as a well‐characterized strain for preclinical toxicity testing. Its pharmacological responses to efruxifermin are representative of rodent species. The total number of animals, including group sizes and number of groups, was the minimum required to characterize the toxicity and toxico‐kinetics of efruxifermin.
The 4‐week study was conducted at Charles River Laboratories Preclinical Services (Reno, NV, USA). Eighty male and 80 female Sprague Dawley Crl:CD®IGS rats (Charles River Laboratories, Hollister, CA, USA) were 9 weeks old on Day 1 of the study. Males and females ranged in weight from 249 to 285 g and from 180 to 204 g, respectively, at Day −4 of the study. The 26‐week study was conducted at Charles River Laboratories Montreal ULC (Senneville, QC, Canada). One hundred twenty male and 120 female Sprague Dawley Crl:CD(SD) rats (Charles River Canada, St. Constant, QC, Canada) were 7 weeks old on Day 1 of the study. Males and females ranged in body weight from 226 to 332 g and from 187 to 273 g, respectively, on Day 1 of the study.
2.3. Housing and husbandry
Animals were housed individually in stainless steel cages (4‐week study) or by group (up to three animals of the same sex and dosing group together) in polycarbonate cages (26‐week study). They were provided with suitable environmental enrichment, except during study procedures. Study animals were acclimated to their environment for 7–14 days prior to the first day of dosing (Day 1). Room temperature was within the range of 18°C to 29°C, with relative humidity of 30% to 70%. Animals were maintained on a 12‐h light/12‐h dark photoperiod, except when interrupted by study procedures. PMI Nutritional International Certified Rodent Chow® No. 5002 and Lab Diet Certified CR Rodent Diet 5CR4 were provided ad libitum, except during designated procedures.
2.4. Experimental procedures
For the 4‐week and the 26‐week studies, animals were assigned to groups by a computer randomization scheme, with males and females randomized separately.
All animals were observed for signs of morbidity and/or mortality twice daily without removal from cage. For detailed clinical observation, animals were removed from their cage once daily during the study. Food consumption was quantified for all animals. Body weight was measured before dosing on Day 1 then weekly thereafter on the day before dosing.
Urine was collected on ice in a cage pan overnight (at least 8 h) in metabolic caging. Food was withdrawn for urine collection, but water was provided ad libitum. For clinical pathology, blood was collected by jugular venepuncture from animals fasted overnight (for at least 8 h). Blood collection was rotated across dose groups to minimize confounding effects of collection time differences. Urine specific gravity was measured with a Clinitek 500 refractometer. Urinary corticosterone was measured using a commercial radioimmunoassay kit (MP Biomedicals Cat # 07120102). The following serum chemistry analytes were measured on either an Olympus AU640e or a Cobas 6000 analyser: sodium, potassium and chloride (ion selective electrode‐based indirect measurement); urea nitrogen (urease kinetic UV); and creatinine (Jaffe kinetic colorimetric). Haematocrit was measured on an Advia 120 analyser.
Efruxifermin serum concentration was measured by a validated ELISA method using capture and detection antibodies developed by Amgen, Inc. (Thousand Oaks, CA, USA) (Kaufman et al., 2020). Toxicokinetic analysis of efruxifermin serum concentration–time data was conducted using either WinNonlin® Enterprise (Pharsight Corp., Mountain View, CA, USA) or Phoenix pharmacokinetic software version 1.4 (Certara, USA) to estimate noncompartmental parameters.
2.5. Data and statistical analysis
The data and statistical analysis comply with the recommendations on experimental design and analysis in pharmacology (Curtis et al., 2018). Data from the 4‐ and 26‐week studies were analysed independently. Male and female animals were analysed independently unless noted. Declared group size (n) refers to the number of independent values rather than technical replicates, and statistical analyses were undertaken only where group size was at least n = 5. Outliers were included in data analysis and presentation. Statistical tests were conducted at the 5% significance level, and comparisons are reported with P values corrected for multiple hypothesis testing.
In both studies, groups were compared using ANOVA or Kruskal–Wallis test as described below. Post hoc tests were conducted only if F in ANOVA achieved P < 0.05. P < 0.05 was considered the threshold for statistical significance. Where parametric tests were used, residuals were examined to confirm underlying assumptions. All reported means are arithmetic. Statistical analyses were conducted using either SAS software version 9.2 (Cary, NC, USA) or GraphPad Prism software version 9.2.0 (GraphPad Software, San Diego, CA, USA). Data analysis was performed blinded to study groups.
In the 4‐week study, differences in body weight for each dose group compared with control were assessed by two‐way repeated measures ANOVA with dose and study week as factors, followed by Dunnett's test (Figure 1). Urine volume and urine specific gravity were assessed by two‐way ANOVA with dose and sex as factors, followed by Dunnett's test (Figure 2). Serum chemistry and haematology were assessed by one‐way ANOVA followed by Dunnett's test (Table 2).
FIGURE 1.
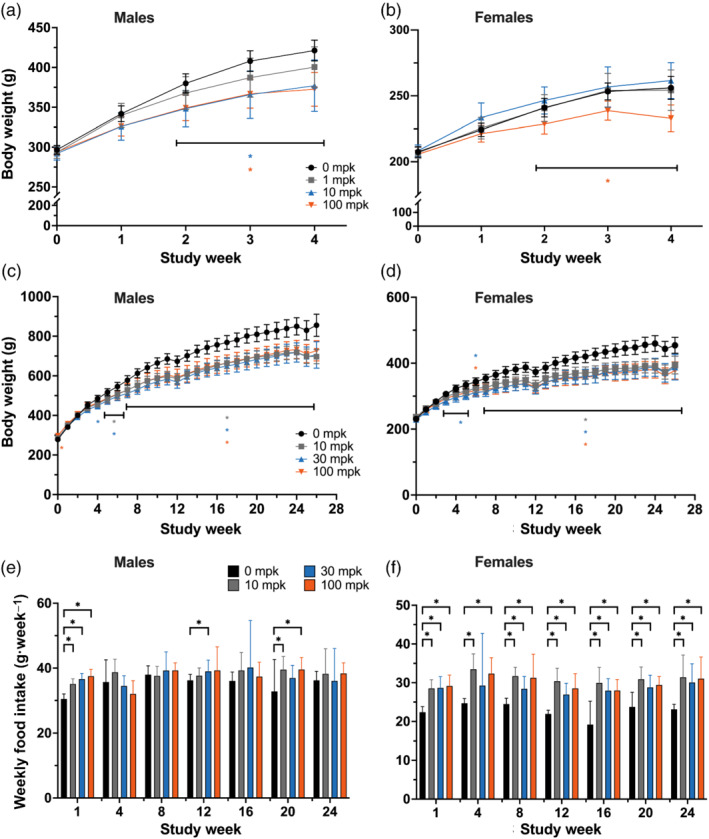
Efruxifermin attenuates body weight gain but increases food intake in rats. Efruxifermin was administered once weekly for (a,b) 4 or (c–f) 26 weeks to (a,c,e) male or (b,d,f) female rats fed a standard chow diet. (a–d) Points and error bars represent mean and 95% confidence interval. Food intake is presented as mean + SD. Statistical significance was tested by (a,b) two‐way (dose, week) ANOVA or (c–f) mixed effects model for repeated measures followed by Dunnett's multiple comparisons test. * P < 0.05 versus 0 mg·kg−1, horizontal brackets in (c) and (d) indicate range of weeks referred to by set of significance annotations. No annotation denotes lack of statistical significance
FIGURE 2.
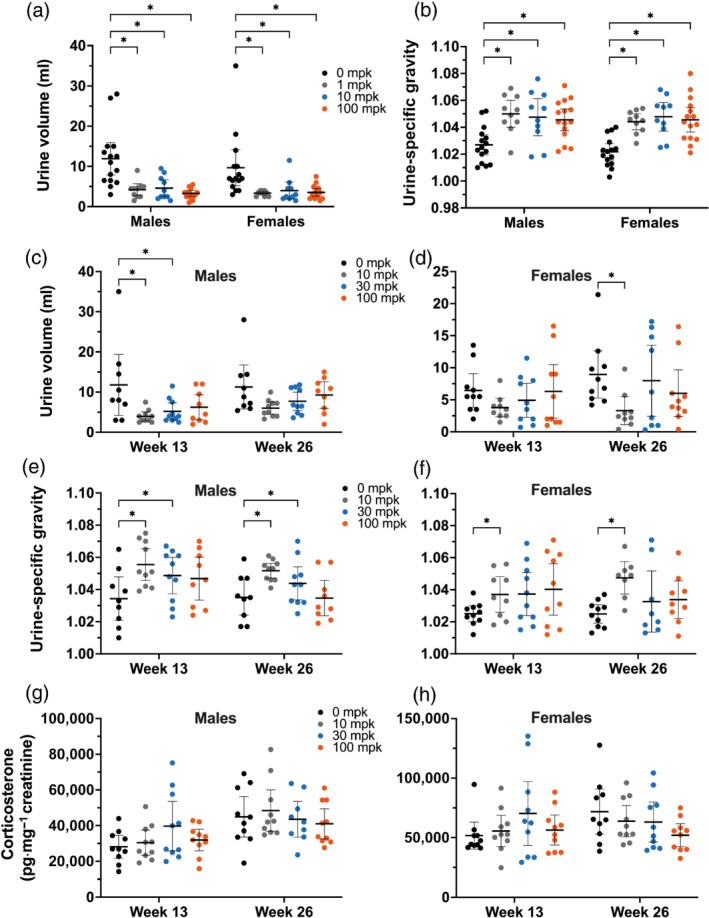
Urinalysis of rats dosed with efruxifermin once weekly for 4 (a,b, measurement at Week 4) or (c–h) 26 weeks to (a,c,e,g) males or (b,d,f,h) females fed a standard chow diet. Lines representing mean and 95% confidence interval are overlaid on points representing individual animal measurements. Statistical significance was tested by two‐way (a,b: dose, sex; c–h: dose, week) ANOVA followed by Dunnett's multiple comparisons test. * P < 0.05 versus 0 mg·kg−1. No annotation indicates lack of statistical significance
TABLE 2.
Serum electrolytes, markers of renal function and haematocrit
| EFX (mg·kg−1) | n | Sodium (mM) | Potassium (mM) | Chloride (mM) | Creatinine (mg·dl−1) | BUN (mg·dl−1) | Haematocrit (%) | |
|---|---|---|---|---|---|---|---|---|
| M | 0 | 15 | 145.0 (2.2) | 5.65 (0.49) | 104.9 (1.3) | 0.367 (0.062) | 16.1 (2.5) | 53.2 (2.5) |
| 1 | 10 | 143.8 (1.0) | 5.27 (0.46) | 103.8 (1.4) | 0.340 (0.052) | 15.2 (2.6) | 52.5 (2.1) | |
| 10 | 10 | 144.5 (1.2) | 5.44 (0.32) | 104.6 (1.6) | 0.370 (0.048) | 14.7 (1.8) | 52.5 (2.7) | |
| 100 | 15 | 144.2 (1.2) | 5.27 (0.38) | 104.4 (1.7) | 0.340 (0.051) | 14.5 (2.2) | 52.8 (2.3) | |
| F | 0 | 15 | 143.4 (1.6) | 5.19 (0.42) | 105.3 (1.9) | 0.447 (0.064) | 15.2 (2.0) | 50.0 (2.6) |
| 1 | 10 | 142.4 (1.6) | 5.28 (0.36) | 104.5 (1.6) | 0.390 (0.057) * | 15.6 (2.3) | 50.0 (2.1) | |
| 10 | 10 | 141.8 (1.4) | 5.23 (0.30) | 104.2 (1.9) | 0.350 (0.053) * | 12.6 (1.5) * | 49.5 (3.1) | |
| 100 | 15 | 142.1 (1.6) | 5.32 (0.40) | 103.9 (1.4) | 0.373 (0.059) | 14.7 (2.1) | 49.5 (3.2) a | |
Note: Male (M) and female (F) rats were administered efruxifermin once weekly for 4 weeks. Values represent mean (SD). Statistical significance was assessed within each sex for each treatment group relative to control (0 mg·kg−1), by one‐way ANOVA followed by Dunnett's or Dunn's post‐test.
Abbreviations: BUN, blood urea nitrogen; EFX, efruxifermin; mg·kg−1, mg efruxifermin per kg body weight.
n = 14.
P < 0.05.
In the 26‐week study, to account for missing values, differences in body weight and food intake for each dose group compared with control were assessed by a mixed effects model for repeated measures using restricted maximum likelihood, with dose and week as factors, followed by Dunnett's test (Figure 1). Urine volume, specific gravity and corticosterone concentration were assessed by mixed effects model for repeated measures with dose and week as factors, followed by Dunnett's test (Figure 2). Urine electrolytes, fractional excretion and creatinine differences were assessed by one‐way ANOVA followed by Dunnett's test or, for non‐parametric analysis, by Kruskal–Wallis followed by Dunn's post‐test (Table 3). Adrenal weights and adrenal‐to‐body weight ratios were assessed by one‐way ANOVA followed by Dunnett's or Dunn's post‐test (Table 4).
TABLE 3.
Concentration of urinary electrolytes and creatinine and fractional excretion of electrolytes
| Week | EFX (mg·kg−1) | n | Sodium | Potassium | Chloride | Creatinine | ||||
|---|---|---|---|---|---|---|---|---|---|---|
| mM | F.E. (%) | mM | F.E. (%) | mM | F.E. (%) | mg·dl−1 | ||||
| 13 | M | 0 | 9 | 64.7 (33.0) | 0.15 (0.08) | 186.0 (100.2) | 10.9 (2.6) | 88.3 (46.9) | 0.30 (0.14) | 147.2 (87.1) |
| 10 | 10 | 95.1 (34.5) | 0.14 (0.06) | 260.6 (85.3) | 8.8 (2.3) | 119.0 (52.6) | 0.25 (0.13) | 227.6 (69.6) | ||
| 30 | 10 | 75.7 (28.4) | 0.13 (0.05) | 218.4 (76.1) | 9.1 (3.0) | 98.5 (38.7) | 0.25 (0.10) | 190.1 (68.3) | ||
| 100 | 9 | 72.1 (15.4) | 0.16 (0.10) a | 209.5 (84.0) | 9.1 (2.9) a | 98.7 (21.7) | 0.30 (0.18) a | 181.7 (81.6) | ||
| F | 0 | 10 | 55.0 (22.9) | 0.24 (0.085) | 115.6 (38.5) | 13.3 (2.8) | 50.0 (18.6) | 0.32 (0.13) | 94.5 (37.4) | |
| 10 | 9 | 89.3 (25.0) | 0.34 (0.075) | 182.0 (97.0) | 14.3 (2.8) | 90.4 (41.0) | 0.46 (0.09) | 100.4 (40.4) | ||
| 30 | 10 | 81.2 (33.8) | 0.33 (0.17) | 138.1 (71.2) | 11.8 (3.8) | 76.3 (37.9) | 0.42 (0.22) | 100.4 (61.3) | ||
| 100 | 10 | 101 (62.2) | 0.36 (0.15) | 171.7 (117.5) | 12.7 (4.2) b | 121.9 (73.6) * | 0.60 (0.20) * | 108.5 (71.6) | ||
| 26 | M | 0 | 9 | 48.0 (23.0) | 0.13 (0.10) | 172.2 (69.9) | 9.6 (2.4) | 82.2 (40.1) | 0.28 (0.16) | 168.5 (81.1) |
| 10 | 10 | 79.0 (22.8) * | 0.13 (0.03) | 228.9 (42.7) | 8.3 (1.8) | 106.6 (50.6) | 0.25 (0.09) | 188.8 (37.3) | ||
| 30 | 10 | 63.4 (29.0) | 0.14 (0.06) | 214.2 (74.3) | 10.7 (2.6) | 104.9 (35.7) | 0.33 (0.11) | 159.1 (60.3) | ||
| 100 | 9 | 49.3 (22.7) | 0.11 (0.04) | 151.6 (73.6) | 7.8 (1.6) | 60.8 (32.4) | 0.19 (0.08) | 152.7 (74.0) | ||
| F | 0 | 10 | 52.3 (19.3) | 0.24 (0.06) | 119.9 (38.3) | 15.8 (4.3) | 70.2 (33.8) | 0.45 (0.14) | 84.8 (30.2) | |
| 10 | 8 | 115.1 (29.6) | 0.32 (0.09) | 225.1 (88.4) | 13.6 (3.4) | 143.1 (38.9) | 0.54 (0.15) | 146.3 (44.7) | ||
| 30 | 8 | 82.1 (72.5) | 0.27 (0.11) | 139.8 (118.2) | 12.2 (4.8) | 95.9 (75.8) | 0.46 (0.19) | 120.7 (112.7) | ||
| 100 | 10 | 79.2 (53.7) | 0.28 (0.14) | 133.2 (71.5) | 11.4 (2.1) | 92.4 (59.1) | 0.44 (0.21) | 101.0 (48.9) | ||
Note: Male (M) and female (F) rats were administered efruxifermin once weekly for 26 weeks. Values represent mean (SD). Statistical significance was assessed relative to control (0 mg·kg−1) by one‐way ANOVA followed by Dunnett's test or Kruskal–Wallis followed by Dunn's post‐test.
Abbreviations: EFX, efruxifermin; F.E., fractional excretion; mg·kg−1, mg efruxifermin per kg body weight.
n = 8.
n = 6.
P < 0.05.
TABLE 4.
Adrenal weight
| EFX (mg·kg−1) | n | Adrenal weight (mg) | Adrenal‐to‐body weight ratio (mg·g−1) | |
|---|---|---|---|---|
| M | 0 | 9 | 56.9 (9.5) | 0.067 (0.014) |
| 10 | 10 | 56.0 (10.1) | 0.081 (0.016) | |
| 30 | 10 | 58.4 (10.1) | 0.085 (0.018) | |
| 100 | 9 | 58.2 (11.0) | 0.081 (0.016) | |
| F | 0 | 10 | 63.7 (9.7) | 0.146 (0.029) |
| 10 | 10 | 60.0 (14.6) | 0.161 (0.048) | |
| 30 | 10 | 63.9 (10.4) | 0.169 (0.022) | |
| 100 | 10 | 62.2 (8.5) | 0.170 (0.036) | |
Note: Male (M) and female (F) rats were administered the indicated dose of efruxifermin once weekly for 26 weeks before terminal necropsy. Values are mean (SD). Statistical significance was assessed for each sex independently relative to control (0 mg·kg−1) by one‐way ANOVA followed by Dunnett's (adrenal weight) or Dunn's (adrenal‐to‐body weight ratio) post‐test.
Abbreviations: EFX, efruxifermin; mg·kg−1, mg efruxifermin per kg body weight.
2.6. Materials
Efruxifermin was synthesized as previously described (Stanislaus et al., 2017). Vehicle control was 10‐mM Tris base (Sigma‐Aldrich, Inc), pH 7.8, with 2.32% l‐lysine (Sigma‐Aldrich, Inc) and 0.008% polysorbate 20 (A&C American Chemicals LTD). To ensure stability, formulated efruxifermin and vehicle control were prepared from frozen stock solutions weekly and thawed at 2°C to 8°C prior to subcutaneous injection. Efruxifermin and control solutions were administered after equilibration to ambient temperature and were used for dosing within 3 h after removal from refrigeration. Accurate concentration of efruxifermin was confirmed multiple times during the studies for each dosing solution.
2.7. Nomenclature of targets and ligands
Key protein targets and ligands in this article are hyperlinked to corresponding entries in the IUPHAR/BPS Guide to PHARMACOLOGY http://www.guidetopharmacology.org and are permanently archived in the Concise Guide to PHARMACOLOGY 2021/22 (Alexander et al., 2021).
3. RESULTS
Once‐weekly administration of efruxifermin for 4 or 26 weeks resulted in dose‐related increases in exposure, with minimal accumulation (Table 1). In the 4 week study, efruxifermin reduced body weight gain over the course of the study in a dose‐dependent manner, with statistically significant differences from controls in male rats at Weeks 2, 3 and 4 in the 10 and 100‐mg·kg−1 dose groups (Figure 1a) and in female rats at Weeks 2, 3 and 4 in the 100 mg·kg−1 dose group only (Figure 1b). In the 26‐week study, the magnitude of reduction in body weight gain generally increased, relative to control, over approximately the first 12 weeks of dosing, after which the groups gained weight at similar rates (Figure 1c,d), although food intake was either significantly increased (females) or trended higher (males) relative to control (Figure 1e,f).
TABLE 1.
Mean serum toxicokinetic parameters for male (M) and female (F) rats following once‐weekly subcutaneous injection of efruxifermin for 4 or 26 weeks
| 4‐week study | 26‐week study | ||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Day 1 | Day 22 | Day 1 | Day 176 | ||||||||||
| EFX (mg·kg−1) | 1 | 10 | 100 | 1 | 10 | 100 | 10 | 30 | 100 | 10 | 30 | 100 | |
| n | 4 | 4 | 4 | 4 | 4 | 4 | 3 | 3 | 3 | 3 | 3 | 3 | |
| C max (μg·ml−1) | M | 1.47 | 8.94 | 93.9 | 2.12 | 13.4 | 218 | 9.4 | 18.7 | 83.8 | 8.0 | 35.2 | 64.4 |
| F | 1.18 | 17.7 | 136 | 1.57 | 21.3 | 273 | 7.8 | 33.8 | 98.1 | 10.0 | 41.0 | 141 | |
| AUC0−t (day * μg·ml−1) | M | 2.72 | 29 | 284 | 4.46 | 42.2 | 576 | 19.7 | 39.4 | 170 | 18.0 | 84.6 | 174 |
| F | 2.58 | 45.7 | 401 | 3.94 | 62.4 | 761 | 18.8 | 66.7 | 242 | 22.9 | 89.2 | 350 | |
Abbreviations: AUC0−t , area under the concentration–time curve during the interdose interval; C max, maximum observed concentration; EFX, efruxifermin; mg·kg−1, mg efruxifermin kg body weight; n, number of animals per group.
Once‐weekly administration of efruxifermin to male and female rats for 4 weeks was associated with an approximately 50% decrease in 24‐h urine volume at all doses tested (Figure 2a). Consistent with lower urine volumes, urine specific gravity was significantly higher (Figure 2b). Despite these changes, serum electrolyte concentrations, markers of renal function (creatinine and blood urea nitrogen) and blood volume (based on haematocrit) were maintained (Table 2). Urinary electrolytes were not measured in this 4‐week study.
In the 26‐week study, there was a trend towards decreased 24‐h urine volume in both males (Figure 2c) and females (Figure 2d). At Week 13, but not at Week 26, 10 and 30 mg·kg−1 doses were associated with a statistically significant decrease in urine output, while mean urine output was numerically decreased in all efruxifermin dose groups at both Weeks 13 and 26 (Figure 2c). At Week 26, females administered 10 mg·kg−1 had significantly lower urine output than controls, consistent with the trend towards decreased urine output observed at Week 13 (Figure 2d).
Consistent with the pattern in the 4 week study, after dosing for 13 and 26 weeks, efruxifermin‐treated male and female rats generally exhibited increased urine specific gravity compared with control (Figure 2e,f); however, in male rats administered 100 mg·kg−1, the increase was not significant at Week 13 and was absent at Week 26 (Figure 2e). Similarly, in female rats receiving more than 10 mg·kg−1, urine specific gravity was numerically but not statistically significantly higher at Weeks 13 and 26 (Figure 2f). Consistent with the increase in urine specific gravity following efruxifermin administration, urinary electrolyte concentrations were generally numerically higher than controls at Week 26 (Table 3). Renal function as indicated by fractional excretion rates of sodium, potassium and chloride was generally maintained, compared with control, with the exception of higher rates of chloride excretion at 100 mg·kg−1 in females after 13 weeks, which was not apparent after 26 weeks (Table 3). In addition, the kidneys appeared microscopically normal in all dose groups after 4 or 26 weeks of efruxifermin treatment.
To explore whether change in urine volume may have been centrally mediated by sympathetic nervous system activation (Guyton, 1990), urinary corticosterone, a marker of hypothalamic–pituitary–adrenal axis activation (Brennan et al., 2000; Buhl et al., 2007), was measured in the 26‐week study. In male and female rats, doses of up to 100‐mg·kg−1 efruxifermin did not significantly alter urinary corticosterone at either Week 13 or 26 (Figure 2g,h). Consistent with an apparent lack of long‐term activation of the sympathetic nervous system, adrenal‐to‐body weight ratio was unchanged in all dose groups after 26 weeks (Table 4). Also consistent with a lack of stress‐induced sustained corticosterone elevation (Hardy et al., 2018), no significant effect on bone remodelling was observed by histology or circulating CTX‐1 (a marker of bone resorption).
4. DISCUSSION
Efruxifermin, a 92‐kDa FGF21 analogue comprising a homodimer of two IgG1 Fc‐FGF21 variant fusion polypeptide chains, demonstrated a dose‐dependent decrease in body weight gain relative to control‐treated rats, consistent with the effects of other FGF21 analogues in rodents (Huang et al., 2013; Kharitonenkov et al., 2013; Wu et al., 2011). In the two studies reported herein, urine volume was reduced and urine concentration was generally increased by efruxifermin relative to control, although blood volume, electrolyte balance and kidney function appeared unaltered. While body weight gain was significantly reduced in efruxifermin‐treated rats, food intake was increased or unchanged, consistent with an increase in energy expenditure mediated by FGF21 in rats and with browning of white adipose tissue (Gehring et al., 2020; Sarruf et al., 2010; Yilmaz et al., 2018). In clinical trials with efruxifermin dosed weekly up to 70 mg, for up to 16 weeks, urine electrolyte concentration, blood pressure and salivary cortisol level were also unchanged (Harrison et al., 2021; Kaufman et al., 2020).
These preclinical and clinical findings with efruxifermin differ notably from the reported pharmacology of PF‐05231023, an antibody‐conjugated FGF21 analogue (Huang et al., 2013). In rats, PF‐05231023 acutely increased both blood pressure and urine output. A concomitant increase in water intake was proposed as a mechanism to compensate for the hypertension‐induced diuresis (Turner et al., 2018). In contrast, although blood pressure was not measured in the two independent studies reported here, one of similar duration to that with PF‐05231023, efruxifermin decreased or did not change urine output in rats.
Consistent with preclinical findings, PF‐05231023 was associated with a significant increase in blood pressure in one clinical study at doses eliciting similar changes in adiponectin levels (a marker of agonism of FGFR1c) to those observed with efruxifermin (Harrison et al., 2021; Kaufman et al., 2020; Kim et al., 2017). The researchers proposed that PF‐05231023 may increase sympathetic tone by acting directly on the central nervous system, as no evidence for renal or endocrine mediation of fluid homeostasis was uncovered (Turner et al., 2018). In contrast, efruxifermin did not increase systolic blood pressure in patients with type 2 diabetes (Kaufman et al., 2020), nor was there evidence of hypothalamic–pituitary–adrenal axis activation in patients with non‐alcoholic steatohepatitis (Harrison et al., 2021). In the rat study described herein, efruxifermin did not appear to increase sympathetic tone, as direct (urinary corticosterone) and indirect (adrenal weight) measures of hypothalamic–pituitary–adrenal axis activation were unchanged, though a comprehensive physiological assessment to rule out sympathetic activation was not performed.
While there appear to be notable differences in the preclinical and clinical effects of efruxifermin and PF‐05231023, two other analogues of FGF21 modified to extend half‐life, pegbelfermin (via PEGylation) and LY2405319 (via amino acid substitutions), have been reported not to increase blood pressure in either patients with obesity and type 2 diabetes (Charles et al., 2019; Gaich et al., 2013) or patients with biopsy‐confirmed non‐alcoholic steatohepatitis (Sanyal et al., 2019). The absence of changes in blood pressure in clinical trials with efruxifermin, pegbelfermin and LY2405319 appear to be consistent with preclinical studies in rodents which report no increase, however in some models a decrease in blood pressure upon FGF21 administration was observed (He et al., 2016; Pan et al., 2018).
In the context of the reported preclinical (Huang et al., 2013; Kharitonenkov et al., 2013; Wu et al., 2011) and clinical effects of FGF21 analogues (Baruch et al., 2020; Charles et al., 2019; Gaich et al., 2013; Harrison et al., 2021; Kaufman et al., 2020; Sanyal et al., 2019), the apparently different preclinical and clinical pharmacology of PF‐05231023 appears molecule‐specific and unrepresentative of the class. A basis for the different pharmacology of PF‐05231023 among FGF21 analogues remains to be explained. Differences in structure resulting in up to 10‐fold more rapid proteolytic degradation of the C‐terminus relative to the N‐terminus of the FGF21 moiety may alter pharmacology (Talukdar et al., 2016; Weng et al., 2015). For example, truncation of C‐terminal residues generates species with reduced affinity for β‐Klotho and lower potency as agonists of FGF21 receptors (Yie et al., 2009), while clipping of N‐terminal residues can result in species that may act as functional antagonists of FGF21 signalling in vivo (Micanovic et al., 2009; Yie et al., 2009). In contrast, the N‐terminus of the FGF21 moiety of efruxifermin is protected against cleavage by being fused to IgG1 Fc, while substitution of Pro for a Gly at position 171 (relative to mature human FGF21) greatly reduces fibroblast activation protein‐mediated cleavage and inactivation of the C‐terminus (Stanislaus et al., 2017). These structural modifications underlie efruxifermin's sustained agonism of FGF21's cognate FGFRs (Kaufman et al., 2020).
While changes in urine volume, urine concentration and blood pressure have not been reported for all FGF21 analogues developed to date, the pharmacological profile of efruxifermin appears consistent with other members of the class, with the exception of PF‐05231023.
CONFLICT OF INTEREST
EJT is an employee and shareholder of Akero Therapeutics. WJB is a consultant to Akero Therapeutics. TR is a co‐founder, employee and shareholder of Akero Therapeutics.
AUTHOR CONTRIBUTIONS
WJB and TR were responsible for the conception and design of study. EJT, WJB and TR were responsible for the data analysis and interpretation. EJT and TR wrote the first draft of the manuscript. EJT, WJB and TR contributed to the final draft of the manuscript.
DECLARATION OF TRANSPARENCY AND SCIENTIFIC RIGOUR
This Declaration acknowledges that this paper adheres to the principles for transparent reporting and scientific rigour of preclinical research as stated in the BJP guidelines for Design & Analysis and Animal Experimentation, and as recommended by funding agencies, publishers and other organisations engaged with supporting research.
ACKNOWLEDGEMENTS
The authors thank Reshma Shringarpure and Kerry af Forselles for critical review of the manuscript. This work was supported by Akero Therapeutics.
Tillman, E. J. , Brock, W. J. , & Rolph, T. (2022). Efruxifermin, a long‐acting Fc‐fusion FGF21 analogue, reduces body weight gain but does not increase sympathetic tone or urine volume in Sprague Dawley rats. British Journal of Pharmacology, 179(7), 1384–1394. 10.1111/bph.15725
Funding information Akero Therapeutics
DATA AVAILABILITY STATEMENT
The data that support the findings of this study are available from the corresponding author upon reasonable request.
REFERENCES
- Alexander, S. P. , Fabbro, D. , Kelly, E. , Mathie, A. , Peters, J. A. , Veale, E. L. , Armstrong, J. F. , Faccenda, E. , Harding, S. D. , Pawson, A. J. , Southan, C. , Davies, J. A. , Beuve, A. , Brouckaert, P. , Bryant, C. , Burnett, J. C. , Farndale, R. W. , Friebe, A. , Garthwaite, J. , … Waldman, S. A. (2021). THE CONCISE GUIDE TO PHARMACOLOGY 2021/22: Catalytic receptors. British Journal of Pharmacology, 178(S1), S264–S312. 10.1111/bph.15541 [DOI] [PubMed] [Google Scholar]
- Badman, M. K. , Pissios, P. , Kennedy, A. R. , Koukos, G. , Flier, J. S. , & Maratos‐Flier, E. (2007). Hepatic fibroblast growth factor 21 is regulated by PPARalpha and is a key mediator of hepatic lipid metabolism in ketotic states. Cell Metabolism, 5, 426–437. 10.1016/j.cmet.2007.05.002 [DOI] [PubMed] [Google Scholar]
- Baruch, A. , Wong, C. , Chinn, L. W. , Vaze, A. , Sonoda, J. , Gelzleichter, T. , Chen, S. , Lewin‐Koh, N. , Morrow, L. , Dheerendra, S. , Boismenu, R. , Gutierrez, J. , Wakshull, E. , Wilson, M. E. , & Arora, P. S. (2020). Antibody‐mediated activation of the FGFR1/Klothoβ complex corrects metabolic dysfunction and alters food preference in obese humans. Proceedings of the National Academy of Sciences, 117(46), 28992–29000. 10.1073/pnas.2012073117 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Berglund, E. D. , Li, C. Y. , Bina, H. A. , Lynes, S. E. , Michael, M. D. , Shanafelt, A. B. , Kharitonenkov, A. , & Wasserman, D. H. (2009). Fibroblast growth factor 21 controls glycemia via regulation of hepatic glucose flux and insulin sensitivity. Endocrinology, 150, 4084–4093. 10.1210/en.2009-0221 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brennan, F. X. , Ottenweller, J. E. , Seifu, Y. , Zhu, G. , & Servatius, R. J. (2000). Persistent stress‐induced elevations of urinary corticosterone in rats. Physiology & Behavior, 71, 441–446. 10.1016/S0031-9384(00)00365-6 [DOI] [PubMed] [Google Scholar]
- Buhl, E. S. , Neschen, S. , Yonemitsu, S. , Rossbacher, J. , Zhang, D. , Morino, K. , Flyvbjerg, A. , Perret, P. , Samuel, V. , Kim, J. , Cline, G. W. , & Petersen, K. F. (2007). Increased hypothalamic‐pituitary‐adrenal axis activity and hepatic insulin resistance in low‐birth‐weight rats. American Journal of Physiology. Endocrinology and Metabolism, 293, E1451–E1458. 10.1152/ajpendo.00356.2007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Charles, E. D. , Morrow, L. , Hompesch, M. , Luo, Y. , Wu, C. K. , & Christian, R. (2016). A phase 1 study of BMS‐986036 (pegylated FGF21) in healthy obese subjects. Hepatology, 64, 546A, AASLD, The Liver Meeting Poster 1082. [Google Scholar]
- Charles, E. D. , Neuschwander‐Tetri, B. A. , Pablo Frias, J. , Kundu, S. , Luo, Y. , Tirucherai, G. S. , & Christian, R. (2019). Pegbelfermin (BMS‐986036), PEGylated FGF21, in patients with obesity and type 2 diabetes: Results from a randomized phase 2 study. Obesity (Silver Spring), 27, 41–49. 10.1002/oby.22344 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Curtis, M. J. , Alexander, S. , Cirino, G. , Docherty, J. R. , George, C. H. , Giembycz, M. A. , Hoyer, D. , Insel, P. A. , Izzo, A. A. , Ji, Y. , MacEwan, D. J. , Sobey, C. G. , Stanford, S. C. , Teixeira, M. M. , Wonnacott, S. , & Ahluwalia, A. (2018). Experimental design and analysis and their reporting II: Updated and simplified guidance for authors and peer reviewers. British Journal of Pharmacology, 175(7), 987–993. 10.1111/bph.14153 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dunshee, D. R. , Bainbridge, T. W. , Kljavin, N. M. , Zavala‐Solorio, J. , Schroeder, A. C. , Chan, R. , Corpuz, R. , Wong, M. , Zhou, W. , Deshmukh, G. , Ly, J. , Sutherlin, D. P. , Ernst, J. A. , & Sonoda, J. (2016). Fibroblast activation protein cleaves and inactivates fibroblast growth factor 21. The Journal of Biological Chemistry, 291, 5986–5996. 10.1074/jbc.M115.710582 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Frayling, T. M. , Beaumont, R. N. , Jones, S. E. , Yaghootkar, H. , Tuke, M. A. , Ruth, K. S. , Casanova, F. , West, B. , Locke, J. , Sharp, S. , Ji, Y. , Thompson, W. , Harrison, J. , Etheridge, A. S. , Gallins, P. J. , Jima, D. , Wright, F. , Zhou, Y. , Innocenti, F. , … Wood, A. R. (2018). A common allele in FGF21 associated with sugar intake is associated with body shape, lower total body‐fat percentage, and higher blood pressure. Cell Reports, 23, 327–336. 10.1016/j.celrep.2018.03.070 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gaich, G. , Chien, J. Y. , Fu, H. , Glass, L. C. , Deeg, M. A. , Holland, W. L. , Kharitonenkov, A. , Bumol, T. , Schilske, H. K. , & Moller, D. E. (2013). The effects of LY2405319, an FGF21 analog, in obese human subjects with type 2 diabetes. Cell Metabolism, 18, 333–340. 10.1016/j.cmet.2013.08.005 [DOI] [PubMed] [Google Scholar]
- Gehring, J. , Azzout‐Marniche, D. , Chaumontet, C. , Piedcoq, J. , Tomé, D. , & Even, P. (2020). Plasma FGF21 levels in rats are dependent on dietary proteins but not on dietary carbohydrates or fats. Current Developments in Nutrition, 4, 627. 10.1093/cdn/nzaa049_020 [DOI] [Google Scholar]
- Guyton, A. C. (1990). Long‐term arterial pressure control: An analysis from animal experiments and computer and graphic models. The American Journal of Physiology, 259, R865–R877. 10.1152/ajpregu.1990.259.5.R865 [DOI] [PubMed] [Google Scholar]
- Hardy, R. S. , Zhou, H. , Seibel, M. J. , & Cooper, M. S. (2018). Glucocorticoids and bone: Consequences of endogenous and exogenous excess and replacement therapy. Endocrine Reviews, 39, 519–548. 10.1210/er.2018-00097 [DOI] [PubMed] [Google Scholar]
- Harrison, S. A. , Ruane, P. J. , Freilich, B. L. , Neff, G. , Patil, R. , Behling, C. A. , Hu, C. , Fong, E. , de Temple, B. , Tillman, E. J. , Rolph, T. P. , Cheng, A. , & Yale, K. (2021). Efruxifermin, a long‐acting Fc‐FGF21 fusion protein, in non‐alcoholic steatohepatitis: The randomized, 16‐week, phase 2a BALANCED trial. Nature Medicine, 27, 1262–1271. [DOI] [PubMed] [Google Scholar]
- He, J.‐L. , Zhao, M. , Xia, J.‐J. , Guan, J. , Liu, Y. , Wang, L.‐Q. , Song, D. X. , Qu, M. Y. , Zuo, M. , Wen, X. , Yu, X. , Huo, R. , Pan, Z. W. , Ban, T. , Zhang, Y. , Zhu, J. X. , Shou, W. , Qiao, G. F. , & Li, B. Y. (2016). FGF21 ameliorates the neurocontrol of blood pressure in the high fructose‐drinking rats. Scientific Reports, 6, 29582. 10.1038/srep29582 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang, J. , Ishino, T. , Chen, G. , Rolzin, P. , Osothprarop, T. F. , Retting, K. , Li, L. , Jin, P. , Matin, M. J. , Huyghe, B. , Talukdar, S. , Bradshaw, C. W. , Palanki, M. , Violand, B. N. , Woodnutt, G. , Lappe, R. W. , Ogilvie, K. , & Levin, N. (2013). Development of a novel long‐acting antidiabetic FGF21 mimetic by targeted conjugation to a scaffold antibody. The Journal of Pharmacology and Experimental Therapeutics, 346, 270–280. 10.1124/jpet.113.204420 [DOI] [PubMed] [Google Scholar]
- Inagaki, T. , Dutchak, P. , Zhao, G. , Ding, X. , Gautron, L. , Parameswara, V. , Li, Y. , Goetz, R. , Mohammadi, M. , Esser, V. , Elmquist, J. K. , Gerard, R. D. , Burgess, S. C. , Hammer, R. E. , Mangelsdorf, D. J. , & Kliewer, S. A. (2007). Endocrine regulation of the fasting response by PPARα‐mediated induction of fibroblast growth factor 21. Cell Metabolism, 5, 415–425. 10.1016/j.cmet.2007.05.003 [DOI] [PubMed] [Google Scholar]
- Jimenez, V. , Jambrina, C. , Casana, E. , Sacristan, V. , Muñoz, S. , Darriba, S. , Rodó, J. , Mallol, C. , Garcia, M. , León, X. , Marcó, S. , Ribera, A. , Elias, I. , Casellas, A. , Grass, I. , Elias, G. , Ferré, T. , Motas, S. , Franckhauser, S. , … Bosch, F. (2018). FGF21 gene therapy as treatment for obesity and insulin resistance. EMBO Molecular Medicine, 10, e8791. 10.15252/emmm.201708791 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kaufman, A. , Abuqayyas, L. , Denney, W. S. , Tillman, E. J. , & Rolph, T. (2020). AKR‐001, an Fc‐FGF21 analog, showed sustained pharmacodynamic effects on insulin sensitivity and lipid metabolism in type 2 diabetes patients. Cell Reports Medicine, 1, 100057. 10.1016/j.xcrm.2020.100057 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kharitonenkov, A. , Beals, J. M. , Micanovic, R. , Strifler, B. A. , Rathnachalam, R. , Wroblewski, V. J. , Li, S. , Koester, A. , Ford, A. M. , Coskun, T. , Dunbar, J. D. , Cheng, C. C. , Frye, C. C. , Bumol, T. F. , & Moller, D. E. (2013). Rational design of a fibroblast growth factor 21‐based clinical candidate, LY2405319. PLoS ONE, 8, e58575. 10.1371/journal.pone.0058575 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kharitonenkov, A. , Shiyanova, T. L. , Koester, A. , Ford, A. M. , Micanovic, R. , Galbreath, E. J. , Sandusky, G. E. , Hammond, L. J. , Moyers, J. S. , Owens, R. A. , Gromada, J. , Brozinick, J. T. , Hawkins, E. D. , Wroblewski, V. J. , Li, D. S. , Mehrbod, F. , Jaskunas, S. R. , & Shanafelt, A. B. (2005). FGF‐21 as a novel metabolic regulator. The Journal of Clinical Investigation, 115, 1627–1635. 10.1172/JCI23606 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kharitonenkov, A. , Wroblewski, V. J. , Koester, A. , Chen, Y.‐F. , Clutinger, C. K. , Tigno, X. T. , Hansen, B. C. , Shanafelt, A. B. , & Etgen, G. J. (2007). The metabolic state of diabetic monkeys is regulated by fibroblast growth factor‐21. Endocrinology, 148, 774–781. 10.1210/en.2006-1168 [DOI] [PubMed] [Google Scholar]
- Kim, A. M. , Somayaji, V. R. , Dong, J. Q. , Rolph, T. P. , Weng, Y. , Chabot, J. R. , Gropp, K. E. , Talukdar, S. , & Calle, R. A. (2017). Once‐weekly administration of a long‐acting fibroblast growth factor 21 analogue modulates lipids, bone turnover markers, blood pressure and body weight differently in obese people with hypertriglyceridaemia and in non‐human primates. Diabetes, Obesity & Metabolism, 19, 1762–1772. 10.1111/dom.13023 [DOI] [PubMed] [Google Scholar]
- Kunder, R. , Frederickson, J. , Yeh, F. L. , Chinn, L. , Dash, A. , Lewin‐Koh, N. , Kim, N. , Yoshida, K. , Chen, S. , Wilson, M. & Wong, C. (2019). Multiple doses of an anti‐FGFR1/KLB bispecific antibody (BFKB8488A) are associated with a decrease in hepatic fat in patients in NAFLD. AASLD, The Liver Meeting, Poster #LP8.
- Lilley, E. , Stanford, S. C. , Kendall, D. E. , Alexander, S. P. H. , Cirino, G. , Docherty, J. R. , George, C. H. , Insel, P. A. , Izzo, A. A. , Ji, Y. , Panettieri, R. A. , Sobey, C. G. , Stefanska, B. , Stephens, G. , Teixeira, M. , & Ahluwalia, A. (2020). ARRIVE 2.0 and the British Journal of Pharmacology: Updated guidance for 2020. British Journal of Pharmacology, 177(16), 3611–3616. 10.1111/BPH.15178 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Micanovic, R. , Raches, D. W. , Dunbar, J. D. , Driver, D. A. , Bina, H. A. , Dickinson, C. D. , & Kharitonenkov, A. (2009). Different roles of N‐ and C‐termini in the functional activity of FGF21. Journal of Cellular Physiology, 219, 227–234. 10.1002/jcp.21675 [DOI] [PubMed] [Google Scholar]
- Mu, J. , Pinkstaff, J. , Li, Z. , Skidmore, L. , Li, N. , Myler, H. , Dallas‐Yang, Q. , Putnam, A. M. , Yao, J. , Bussell, S. , Wu, M. , Norman, T. C. , Rodriguez, C. G. , Kimmel, B. , Metzger, J. M. , Manibusan, A. , Lee, D. , Zaller, D. M. , Zhang, B. B. , … Axelrod, D. W. (2012). FGF21 analogs of sustained action enabled by orthogonal biosynthesis demonstrate enhanced antidiabetic pharmacology in rodents. Diabetes, 61, 505–512. 10.2337/db11-0838 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ogawa, Y. , Kurosu, H. , Yamamoto, M. , Nandi, A. , Rosenblatt, K. P. , Goetz, R. , Eliseenkova, A. V. , Mohammadi, M. , & Kuro‐o, M. (2007). BetaKlotho is required for metabolic activity of fibroblast growth factor 21. Proceedings of the National Academy of Sciences, 104, 7432–7437. 10.1073/pnas.0701600104 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pan, X. , Shao, Y. , Wu, F. , Wang, Y. , Xiong, R. , Zheng, J. , Tian, H. , Wang, B. , Wang, Y. , Zhang, Y. , Han, Z. , Qu, A. , Xu, H. , Lu, A. , Yang, T. , Li, X. , Xu, A. , Du, J. , & Lin, Z. (2018). FGF21 prevents angiotensin II‐induced hypertension and vascular dysfunction by activation of ACE2/angiotensin‐(1–7) axis in mice. Cell Metabolism, 27, 1323–1337.e5. 10.1016/j.cmet.2018.04.002 [DOI] [PubMed] [Google Scholar]
- Percie du Sert, N. , Hurst, V. , Ahluwalia, A. , Alam, S. , Avey, M. T. , Baker, M. , Browne, W. J. , Clark, A. , Cuthill, I. C. , Dirnagl, U. , Emerson, M. , Garner, P. , Holgate, S. T. , Howells, D. W. , Karp, N. A. , Lazic, S. E. , Lidster, K. , MacCallum, C. J. , Macleod, M. , … Würbel, H. (2020). The ARRIVE guidelines 2.0: updated guidelines for reporting animal research. PLoS Biology, 18(7), e3000410. 10.1371/journal.pbio.3000410 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sanyal, A. , Charles, E. D. , Neuschwander‐Tetri, B. A. , Loomba, R. , Harrison, S. A. , Abdelmalek, M. F. , Lawitz, E. J. , Halegoua‐DeMarzio, D. , Kundu, S. , Noviello, S. , Luo, Y. , & Christian, R. (2019). Pegbelfermin (BMS‐986036), a PEGylated fibroblast growth factor 21 analogue, in patients with non‐alcoholic steatohepatitis: A randomised, double‐blind, placebo‐controlled, phase 2a trial. Lancet, 392, 2705–2717. 10.1016/S0140-6736(18)31785-9 [DOI] [PubMed] [Google Scholar]
- Sarruf, D. A. , Thaler, J. P. , Morton, G. J. , German, J. , Fischer, J. D. , Ogimoto, K. , & Schwartz, M. W. (2010). Fibroblast growth factor 21 action in the brain increases energy expenditure and insulin sensitivity in obese rats. Diabetes, 59, 1817–1824. 10.2337/db09-1878 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stanislaus, S. , Hecht, R. , Yie, J. , Hager, T. , Hall, M. , Spahr, C. , Wang, W. , Weiszmann, J. , Li, Y. , Deng, L. , Winters, D. , Smith, S. , Zhou, L. , Li, Y. , Véniant, M. M. , & Xu, J. (2017). A novel Fc‐FGF21 with improved resistance to proteolysis, increased affinity toward β‐Klotho, and enhanced efficacy in mice and cynomolgus monkeys. Endocrinology, 158, 1314–1327. 10.1210/en.2016-1917 [DOI] [PubMed] [Google Scholar]
- Talukdar, S. , Zhou, Y. , Li, D. , Rossulek, M. , Dong, J. , Somayaji, V. , Weng, Y. , Clark, R. , Lanba, A. , Owen, B. M. , Brenner, M. B. , Trimmer, J. K. , Gropp, K. E. , Chabot, J. R. , Erion, D. M. , Rolph, T. P. , Goodwin, B. , & Calle, R. A. (2016). A long‐acting FGF21 molecule, PF‐05231023, decreases body weight and improves lipid profile in non‐human primates and type 2 diabetic subjects. Cell Metabolism, 23, 427–440. 10.1016/j.cmet.2016.02.001 [DOI] [PubMed] [Google Scholar]
- Tillman, E. J. , & Rolph, T. (2020). FGF21: An emerging therapeutic target for non‐alcoholic steatohepatitis and related metabolic diseases. Front Endocrinol (Lausanne), 11, 601290. 10.3389/fendo.2020.601290 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Turner, T. , Chen, X. , Zahner, M. , Opsahl, A. , DeMarco, G. , Boucher, M. , Goodwin, B. , & Perreault, M. (2018). FGF21 increases water intake, urine output and blood pressure in rats. PLoS ONE, 13, e0202182. 10.1371/journal.pone.0202182 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weng, Y. , Chabot, J. , Bernardo, B. , Yan, Q. , Zhu, Y. , Brenner, M. , Vage, C. , Logan, A. , Calle, R. , & Talukdar, S. (2015). Pharmacokinetics (PK), pharmacodynamics (PD) and integrated PK/PD modeling of a novel long acting FGF21 clinical candidate PF‐05231023 in diet‐induced obese and leptin‐deficient obese mice. PLoS ONE, 10, e0119104. 10.1371/journal.pone.0119104 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wong, C. , Yeh, F. L. , Chinn, L. , Dash, A. , Lewin‐Koh, N. , Yoshida, K. , Chen, S. , Wilson, M. & Arora, P. (2019). A bispecific antibody to FGFR1/KLB, BFKB8488A, improves lipid profile, markers of fibrogenesis and liver health in patients with type 2 diabetes—Preliminary results from a phase 1b study. AASLD, The Liver Meeting, Poster #2310.
- Wu, A.‐L. , Kolumam, G. , Stawicki, S. , Chen, Y. , Li, J. , Zavala‐Solorio, J. , Phamluong, K. , Feng, B. , Li, L. , Marsters, S. , Kates, L. , van Bruggen, N. , Leabman, M. , Wong, A. , West, D. , Stern, H. , Luis, E. , Kim, H. S. , Yansura, D. , … Sonoda, J. (2011). Amelioration of type 2 diabetes by antibody‐mediated activation of fibroblast growth factor receptor 1. Science Translational Medicine, 3, 113ra126. 10.1126/scitranslmed.3002669 [DOI] [PubMed] [Google Scholar]
- Xu, J. , Lloyd, D. J. , Hale, C. , Stanislaus, S. , Chen, M. , Sivits, G. , Vonderfecht, S. , Hecht, R. , Li, Y. S. , Lindberg, R. A. , Chen, J. L. , Young Jung, D. , Zhang, Z. , Ko, H. J. , Kim, J. K. , & Veniant, M. M. (2009). Fibroblast growth factor 21 reverses hepatic steatosis, increases energy expenditure, and improves insulin sensitivity in diet‐induced obese mice. Diabetes, 58, 250–259. 10.2337/db08-0392 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yie, J. , Hecht, R. , Patel, J. , Stevens, J. , Wang, W. , Hawkins, N. , Steavenson, S. , Smith, S. , Winters, D. , Fisher, S. , Cai, L. , Belouski, E. , Chen, C. , Michaels, M. L. , Li, Y. S. , Lindberg, R. , Wang, M. , Véniant, M. , & Xu, J. (2009). FGF21 N‐ and C‐termini play different roles in receptor interaction and activation. FEBS Letters, 583, 19–24. 10.1016/j.febslet.2008.11.023 [DOI] [PubMed] [Google Scholar]
- Yilmaz, E. , Tekin, S. , Demir, M. , Cigremis, Y. , & Sandal, S. (2018). Effects of central FGF21 infusion on the hypothalamus‐pituitary‐thyroid axis and energy metabolism in rats. The Journal of Physiological Sciences, 68, 781–788. 10.1007/s12576-018-0595-7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhen, E. Y. , Jin, Z. , Ackermann, B. L. , Thomas, M. K. , & Gutierrez, J. A. (2016). Circulating FGF21 proteolytic processing mediated by fibroblast activation protein. The Biochemical Journal, 473, 605–614. 10.1042/BJ20151085 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
The data that support the findings of this study are available from the corresponding author upon reasonable request.