

Abstract
Cefiderocol is a siderophore cephalosporin for the treatment of infections caused by gram‐negative bacteria including carbapenem‐resistant strains. The aim of this study was to develop an intrapulmonary pharmacokinetic (PK) model of cefiderocol and assess the PK profile in lungs. An intrapulmonary PK model of cefiderocol was developed using the concentration data in plasma and epithelial lining fluid (ELF) from 7 patients with pneumonia requiring mechanical ventilation and 20 healthy subjects. Subsequently, the model was applied to assess the ELF exposure of 125 patients with nosocomial pneumonia. Monte Carlo simulations were performed to calculate the probability of target attainment for the percentage of time for which free ELF concentrations exceed the minimum inhibitory concentration (MIC) over the dosing interval (%fT>MIC,ELF). The developed model adequately described ELF concentrations and suggested the delayed distribution in ELF for patients with pneumonia compared to healthy subjects. Lung penetration ratio of cefiderocol in patients with pneumonia was calculated to be 34%, which was 1.4‐fold that in healthy subjects. The estimated %fT>MIC,ELF was 100% in most of patients with nosocomial pneumonia, and no PK/pharmacodynamic relationship with %fT>MIC,ELF was found for microbiological or clinical outcome. The probability of target attainment for 100% fT>MIC,ELF was ≥ 99.5% against MICs ≤2 μg/mL and ≥87.0% against MICs ≤4 μg/mL regardless of renal function. The median of simulated ELF trough concentrations at steady state was >4 μg/mL regardless of renal function. These results reveal the adequacy of cefiderocol exposure in plasma and ELF at the recommended dosing regimens adjusted on the basis of renal function in critically ill patients with pneumonia.
Keywords: cefiderocol, intrapulmonary pharmacokinetics, modeling and simulation, patients with pneumonia, siderophore cephalosporin
Cefiderocol is a parenteral siderophore cephalosporin with antibacterial activity against carbapenem‐resistant gram‐negative bacteria including Enterobacterales, Pseudomonas aeruginosa, and Acinetobacter baumannii. 1 , 2 , 3 , 4 , 5 Cefiderocol has been approved for the treatment of hospital‐acquired bacterial pneumonia, ventilator‐associated bacterial pneumonia, and complicated urinary tract infections, including pyelonephritis, caused by gram‐negative microorganisms in adults in the United States. 6 Cefiderocol was also approved for the treatment of infections (regardless of infection site) due to aerobic gram‐negative organisms in adults with limited treatment options in Europe. 7
Cefiderocol is mainly excreted via the kidneys, and renal function is the most influential factor for the pharmacokinetics. 8 , 9 , 10 , 11 The approved standard dosing regimen of cefiderocol is 2 g administered as a 3‐hour infusion every 8 hours, and it is adjusted on the basis of renal function (creatinine clearance, <60 mL/min or ≥120 mL/min). 6 , 7 The percentage of time for which free drug concentrations exceed minimum inhibitory concentration (MIC) over the dosing interval (%fT>MIC) in plasma was shown to be the pharmacokinetic/pharmacodynamic (PK/PD) parameter that best correlated with efficacy in murine thigh infection models. 5 In the lung infection model, the mean plasma %fT>MIC required for a 1‐log10 reduction against Enterobacterales, P aeruginosa, A baumannii, and Stenotrophomonas maltophilia were 64%, 70%, 88%, and 54%, respectively. 5
While the plasma profile of the β‐lactams has historically been used to predict efficacy in patients with pneumonia because this matrix is easily attainable and integrated with available MIC data, 11 , 12 , 13 the assessment of drug exposure at the pulmonary target site (ie, epithelial lining fluid [ELF], interstitial extracellular space) is of increasing interest to ensure adequate exposure. Penetration ratios are variable even among antimicrobial agents in the same class (eg, β‐lactams such as cefiderocol, ceftazidime, and cefepime), 14 , 15 and the variability may result in subtherapeutic drug exposure.
As such, the assessment of lung disposition is important for consideration of antimicrobial efficacy against increasingly challenging nosocomial pathogens. During the development of cefiderocol, the intrapulmonary PK assessment was conducted first in healthy subjects where parallel plasma and ELF exposure were observed. 16 Recently, a phase 1b study assessing the lung penetration in hospitalized subjects with bacterial pneumonia requiring mechanical ventilation was completed (NCT03862040). 17 Furthermore, the efficacy and safety of cefiderocol treatment in patients with nosocomial pneumonia were assessed in 2 phase 3 studies, the CREDIBLE‐CR study (NCT02714595) and the APEKS‐NP study (NCT03032380). 18 , 19
The aim of this study was to develop an intrapulmonary PK model of cefiderocol using plasma and ELF concentration data from patients with pneumonia and healthy subjects. Monte Carlo simulation was performed in consideration of PK variability for calculation of probability of target attainment (PTA) for the identified target %fT>MIC in ELF (%fT>MIC,ELF) to assess the dosing regimens of cefiderocol. In addition, PK/PD relationships of %fT>MIC,ELF with microbiological outcome, clinical outcome, and vital status were evaluated in patients with nosocomial pneumonia from the phase 3 studies.
Methods
Data for Analyses
Concentration data of cefiderocol in plasma and ELF were collected from 7 patients with pneumonia with mechanical ventilation in a phase 1b study (NCT03862040) 17 and 20 healthy subjects in a phase 1 study 16 as shown in Table S1. A total of 168 plasma concentrations and 27 ELF concentrations from the 20 healthy subjects and 7 patients with pneumonia were used to develop an intrapulmonary PK model of cefiderocol.
Data for plasma concentrations, MIC of causative gram‐negative pathogens, and microbiological or clinical outcome after cefiderocol administration from patients with nosocomial pneumonia in the phase 3 studies were used for PK/PD analysis. The number of isolated pathogens were 42 from 28 patients in the CREDIBLE‐CR study (NCT02714595) 18 and 122 from 97 patients in the APEKS‐NP study (NCT03032380), 19 and their MICs ranged from ≤0.03 to 64 (median, 0.25) μg/mL.
Bioanalytical Method
A detail of the bioanalytical method was shown in the previous reports. 10 , 16 , 17 Briefly, blood and bronchoalveolar lavage (BAL) fluid were collected at specified sampling time points as shown in Table S1. Cefiderocol concentrations in plasma and BAL were determined by liquid chromatography–tandem mass spectrometry. Apparent volume of ELF was estimated using urea concentrations in plasma and BAL, and ELF concentrations were calculated on the basis of BAL concentrations and volume of BAL and ELF.
Intrapulmonary PK Modeling
A model structure for cefiderocol PK in plasma and ELF is shown in Figure 1. The mass balance for each compartment is given by the following equations:
Figure 1.
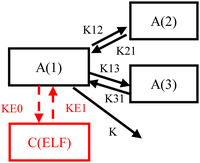
Model structure for cefiderocol pharmacokinetics in plasma and epithelial lining fluid (ELF). A(1) is the drug amount in the central compartment, A(2) and A(3) are the drug amounts in peripheral compartments, and C(ELF) is the ELF concentration of the drug. K is the first‐order rate constant of elimination, and K12, K21, K13, K31, KE0, and KE1 are first‐order transfer rate constants between compartments.
where A(1) is the drug amount in the central compartment, A(2) and A(3) are the drug amounts in the peripheral compartments, and C(ELF) is the ELF concentration of the drug. K is the first‐order rate constant of elimination and K12, K21, K13, K31, KE0, and KE1 are the first‐order transfer rate constants between compartments. It was assumed that distribution of cefiderocol into ELF would not affect plasma concentrations since the volume of ELF (20‐40 mL) 20 is considered extremely smaller than V1 (7.78 L). 10
To calculate the fraction of ELF to plasma concentration at steady state and to describe the delayed distribution in ELF for patients with pneumonia, the following equations were used:
where FRC is the fraction of ELF to total plasma concentration at steady state, FRCHV is FRC in healthy subjects, and EFRC is the effect of patients with pneumonia on FRC. PT is an identification variable of pneumonia patients on FRC (PT = 0 for healthy subjects and PT = 1 for pneumonia patients). Total ELF concentrations were regarded as free ELF concentrations in this study since albumin concentrations in ELF were expected to be low. 21 , 22 , 23
In the study for healthy subjects, 16 ELF concentration profile was parallel to plasma concentration profile (instantaneous equilibrium). Therefore, C(ELF) in healthy subjects was calculated by the following equation as dC(ELF)/dt could be assumed to be 0 at steady state.
The interindividual variability for FRC was assumed to follow a log‐normal distribution and could be modeled with an exponential error model. As ELF concentrations were obtained at 1 time point from each patient, the intraindividual variability could not be estimated and was fixed to an extremely small value of 0.00001.
The plasma PK model previously developed 10 was applied in this modeling. Individual post hoc plasma PK parameters in patients with pneumonia and healthy subjects were calculated with empirical Bayesian estimation, and then ELF PK parameters (KE1, FRCHV, EFRC, and interindividual variability for FRC) were estimated using the plasma PK parameters. The PK parameters were estimated using NONMEM (ICON plc, Dublin, Ireland) with the first‐order conditional estimation method with interaction.
Model Evaluation
The developed intrapulmonary PK model was evaluated by the goodness‐of‐fit plots. Predictive performance of the developed model was also evaluated by a prediction‐corrected visual predictive check with 200 simulations. 24 The robustness of the developed model was confirmed by a bootstrap technique. 25 The 300 bootstrap data sets were generated by resampling from the original data set, and the medians and 95% confidence intervals (CIs) of the bootstrap estimates were compared to the parameter estimate for the developed model.
Penetration Ratio Based on Post Hoc Estimate of Area Under the Concentration‐Time Curve
Area under the concentration‐time curve (AUC) based on concentrations in ELF (AUCELF) and free plasma concentrations (fAUCplasma) were calculated using individual post hoc plasma and ELF PK parameters with empirical Bayesian estimation. The AUCELF was calculated on the basis of simulated ELF concentrations at steady state every 0.25 hours by using the linear trapezoidal method. The fAUCplasma was calculated as dose divided by the total clearance using the unbound fraction of 0.422 in plasma. 26 The penetration ratio of AUCELF to fAUCplasma was calculated in patients with pneumonia and healthy subjects.
Monte Carlo Simulation and Probability of Target Attainment
Monte Carlo simulation was conducted to calculate the PTA for either 75% fT>MIC,ELF and 100% fT>MIC,ELF against an MIC range of 0.25 to 16 μg/mL. One thousand virtual patients with pneumonia with different renal functions were generated and the PTA for target %fT>MIC,ELF was calculated by renal function group. In addition, the PTA integrated with all renal function groups was calculated on the basis of a distribution of creatinine clearance in the phase 3 CREDIBLE‐CR and APEKS‐NP studies. 10 The dosing regimens adjusted on the basis of renal function 10 were used for this simulation. The simulated trough concentrations in ELF at steady state were also summarized.
Sensitivity analysis for uncertainty of the estimated KE1 was performed due to the large relative standard error of KE1 (63.4%) and the limited number of ELF data. The effects of KE1 on the PTA and trough concentration in ELF were examined using an upper limit of 95%CI of the bootstrap estimate of KE1 (0.557 h–1), which corresponded to faster elimination in ELF.
Pharmacokinetic/Pharmacodynamic Analysis
For the 125 patients with nosocomial pneumonia enrolled in the phase 3 studies, %fT>MIC,ELF against MIC of the isolated pathogens in the phase 3 studies were calculated using the developed PK models. In the calculation, individual plasma concentrations were predicted using the post hoc PK parameters from observed plasma concentration data with empirical Bayesian estimation. As ELF concentrations were not measured in the phase 3 studies, individual ELF concentrations corresponding to the individual plasma concentrations were predicted using the population mean values of ELF PK parameters. The PK/PD relationships of %fT>MIC,ELF with microbiological outcome, clinical outcome, and vital status were examined. The details of the outcomes were shown in the previous report. 10
Software
Model building and Monte Carlo simulations were performed using NONMEM (version 7.3.0; ICON plc), 27 Perl‐speaks NONMEM (version 4.2.0; Uppsala University, Uppsala, Sweden), 28 and Pirana (version 2.9.4; Certara, Princeton, New Jersey). 29 R (version 3.5.1; R Foundation for Statistical Computing, Vienna, Austria) 30 was used to calculate the PTA.
Results
The intrapulmonary PK model of cefiderocol was developed using 168 plasma concentration data and 27 ELF concentration data from 7 patients with pneumonia and 20 healthy subjects. Their background characteristics are summarized in Table 1. The parameter estimates and model code are shown in Table 2 and Table S2, respectively. The fraction of ELF to total plasma concentration at steady state in patients with pneumonia was calculated to be 0.143 (0.103 × 1.39), which was 1.4‐fold that in healthy subjects. Considering unbound fraction of 0.422 in plasma, 26 the concentration ratios of ELF to free plasma were calculated as 0.339 in patients with pneumonia and 0.244 in healthy subjects. Interindividual variability for fraction of ELF to total plasma concentration at steady state was 34.6%.
Table 1.
Background Characteristics of Subjects Used for Intrapulmonary Pharmacokinetic Modeling
| Healthy Subjects | Patients With Pneumonia | |
|---|---|---|
| (N = 20) | (N = 7) | |
| Age, y | 25 (21‐36) | 70 (19‐78) |
| Body weight, kg | 61.9 (54.9‐72.3) | 88.1 (69.4‐113.0) |
| CrCL, mL/min | 125 (95‐148) | 78 (44‐275) |
| Albumin concentration, g/dL | 4.7 (4.3‐4.9) | 2.8 (1.5‐3.0) |
| Sex (male:female) a | 20 (100%): 0(0%) | 3 (42.9%):4 (57.1%) |
| Race (Asian:White:Black:Others) a | 20 (100%):0 (0%):0 (0%):0 (0%) | 0 (0%):5 (71.4%):1 (14.3%):1 (14.3%) |
CrCL, creatinine clearance calculated by Cockcroft‐Gault equation.
Median (range).
Number of subjects (percentage of all subjects).
Table 2.
Parameter Estimates of Cefiderocol for Intrapulmonary Pharmacokinetic Model
| Bootstrap Estimates | ||||
|---|---|---|---|---|
| Pharmacokinetic Parameters | Estimates | %RSE | Median | 95% CI |
| FRC in healthy subjects | 0.103 | 6.2 | 0.102 | 0.0907‐0.117 |
| Effect of pneumonia patients on FRC | 1.39 | 19.6 | 1.41 | 0.890‐2.02 |
| KE1, h–1 | 0.151 | 63.4 | 0.163 | 0.0206‐0.557 |
| Interindividual variability | ||||
| FRC (CV%) | 34.6 | 34.9 | 32.9 | 19.7‐44.0 |
CI, confidence interval; CV, coefficient of variation; FRC, fraction of epithelial lining fluid (ELF) to total plasma concentration; KE1, first‐order transfer rate constant from epithelial lining fluid to drug amount in central compartment; %RSE, relative standard error in percent.
The goodness‐of‐fit plots for the developed model indicated that population predicted data corresponded to observed data with the line of unity (Figure S1). The prediction‐corrected visual predictive check demonstrated that the model well captured the observed ELF concentration data (Figure 2). The bootstrap median estimates were comparable to the parameter estimates for the developed model (Table 2), suggesting model robustness.
Figure 2.
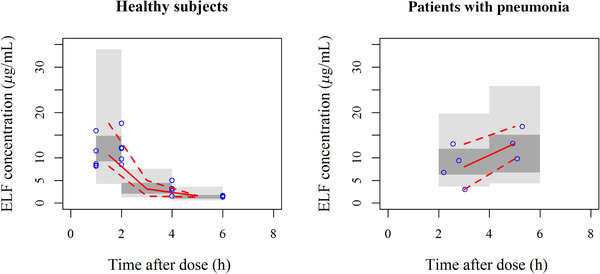
Prediction‐corrected visual predictive check for intrapulmonary pharmacokinetic model to describe cefiderocol concentrations in epithelial lining fluid (ELF).
The results for 200 simulations. Solid line: observed median. Dashed line: observed 2.5th and 97.5th percentiles. Dark gray shaded area: model predicted 95% confidence interval (CI) of median. Gray shaded area: model predicted 95% CIs of 2.5th and 97.5th percentiles.
Based on the estimated AUC, the medians of the penetration ratios (AUCELF to fAUCplasma) were 0.340 (range, 0.176‐0.576) in patients with pneumonia and 0.263 (range, 0.122‐0.416) in healthy subjects.
Simulated ELF concentration profiles at steady state in patients with nosocomial pneumonia are shown by renal function group in Figure S2. The PTA for 75% fT>MIC,ELF was ≥99.6% against MICs ≤2 μg/mL and ≥87.7% against MICs ≤ 4 μg/mL regardless of renal function based on the Monte Carlo simulation (Table 3). Even the PTA for 100% fT>MIC,ELF was ≥87.0% against MICs ≤4 μg/mL regardless of renal function. The PTA integrated with all renal function groups for 75% fT>MIC,ELF and 100% fT>MIC,ELF were 93.1% and 92.4%, respectively, against MICs ≤4 μg/mL, as shown in Figure 3, in which MIC distributions in 3 consecutive multinational surveillance studies in 2014 to 2016 are also presented. 31
Table 3.
Probability of target attainment for 75% and 100% of time for which free concentrations of cefiderocol in epithelial lining fluid exceed the minimum inhibitory concentration (MIC) over dosing interval (fT>MIC,ELF) by renal function group
| Probability of Target Attainment | |||||||||
|---|---|---|---|---|---|---|---|---|---|
| MIC, μg/mL | |||||||||
| Target | Renal Function Group | Dose Regimens With 3‐h Infusion | 0.25 | 0.5 | 1 | 2 | 4 | 8 | 16 |
| 75% fT>MIC,ELF | Augmented renal function | 2 g every 6 h | 100 | 100 | 100 | 99.8 | 91.8 | 54.0 | 10.2 |
| Normal renal function | 2 g every 8 h | 100 | 100 | 100 | 99.6 | 87.7 | 42.9 | 6.2 | |
| Mild renal impairment | 2 g every 8 h | 100 | 100 | 100 | 99.8 | 93.8 | 59.8 | 14.9 | |
| Moderate renal impairment | 1.5 g every 8 h | 100 | 100 | 100 | 100 | 95.9 | 66.0 | 17.5 | |
| Severe renal impairment | 1 g every 8 h | 100 | 100 | 100 | 99.9 | 97.7 | 74.6 | 24.8 | |
| ESRD | 0.75 g every 12 h | 100 | 100 | 100 | 99.9 | 94.3 | 63.1 | 20.8 | |
| 100% fT>MIC,ELF | Augmented renal function | 2 g every 6 h | 100 | 100 | 100 | 99.8 | 91.8 | 53.9 | 10.2 |
| Normal renal function | 2 g every 8 h | 100 | 100 | 100 | 99.5 | 87.0 | 41.8 | 6.0 | |
| Mild renal impairment | 2 g every 8 h | 100 | 100 | 100 | 99.7 | 93.1 | 58.8 | 14.4 | |
| Moderate renal impairment | 1.5 g every 8 h | 100 | 100 | 100 | 100 | 95.8 | 65.6 | 17.5 | |
| Severe renal impairment | 1 g every 8 h | 100 | 100 | 100 | 99.9 | 97.7 | 74.5 | 24.7 | |
| ESRD | 0.75 g every 12 h | 100 | 100 | 100 | 99.9 | 93.8 | 61.9 | 20.1 | |
CV, coefficient of variation; ESRD, end‐stage renal disease; fT>MIC,ELF, percentage of time for which free epithelial lining fluid concentrations exceed MIC over the dosing interval; MIC, minimum inhibitory concentration.
Pharmacokinetics at steady state was assumed. Probability of target attainment is shown in percent (%).
Augmented: creatinine clearance (CrCL) > 120 mL/min (120 to < 150 = 50%; > 150 = 50%). Normal: CrCL 90 to < 120 mL/min. Mild: CrCL 60 to < 90 mL/min. Moderate: CrCL 30 to < 60 mL/min. Severe: CrCL 15 to < 30 mL/min. End stage of renal disease (ESRD): CrCL 5 to < 15 mL/min.
1000 simulated patients in each simulation scenario.
Body weight was assumed to be log‐normal distributed with mean of 72.6 kg and CV of 30%.
Albumin was assumed to be log‐normal distributed with mean of 2.8 g/dL and CV of 30%.
Figure 3.
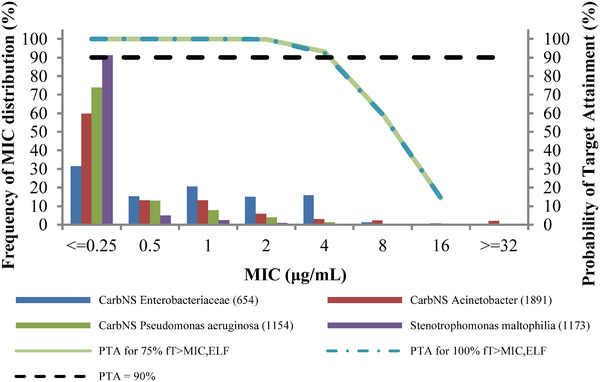
Probability of target attainment (PTA) integrated with all renal function groups for 75% and 100% of time for which free concentrations of cefiderocol in epithelial lining fluid exceed the minimum inhibitory concentration (MIC) over dosing interval (fT>MIC,ELF). The bars present minimum inhibitory concentration distributions of carbapenem nonsusceptible (CarbNS) Enterobacteriaceae, CarbNS Acinetobacter, CarbNS Pseudonomas aeruginosa, and Stenotrophomonas maltophilia in order from left to right.
Sensitivity analysis for KE1 on the PTA and trough concentration in ELF was performed using an upper limit of 95%CI of the bootstrap estimate (0.557 hr–1). The PTA for 75% fT>MIC,ELF and 100% fT>MIC,ELF calculated using the high KE1 were both ≥95.9% against MICs ≤2 μg/mL regardless of renal function (Table S3). The PTA for 75% fT>MIC,ELF and 100% fT>MIC,ELF against an MIC of 4 μg/mL were 80% to 97% and 71% to 96%, respectively (Table S3). The median of the simulated ELF trough concentrations at steady state was >4 μg/mL in all renal function groups even assuming faster elimination of ELF concentrations (Table 4).
Table 4.
Simulated Trough Concentrations of Cefiderocol at Steady State in ELF
| Trough Concentrations at Steady State in ELF | |||
|---|---|---|---|
| KE1 Value for Simulation | Renal Function Group | Dose Regimens With 3‐h Infusion | Median (90% Prediction Interval) |
| 0.151 h–1 | Augmented renal function | 2 g every 6 h | 8.47 (3.57‐19.3) |
| (Population mean estimate) | Normal renal function | 2 g every 8 h | 7.19 (3.12‐16.7) |
| Mild renal impairment | 2 g every 8 h | 8.98 (3.70‐21.3) | |
| Moderate renal impairment | 1.5 g every 8 h | 9.94 (4.16‐22.8) | |
| Severe renal impairment | 1 g every 8 h | 11.3 (4.69‐26.7) | |
| ESRD | 0.75 g every 12 h | 9.49 (3.81‐26.6) | |
| 0.557 h‐1 | Augmented renal function | 2 g every 6 h | 7.25 (2.89‐17.8) |
| (Assumed faster elimination of ELF concentrations) | Normal renal function | 2 g every 8 h | 5.43 (2.16‐13.7) |
| Mild renal impairment | 2 g every 8 h | 7.29 (2.67‐18.8) | |
| Moderate renal impairment | 1.5 g every 8 h | 8.69 (3.40‐20.8) | |
| Severe renal impairment | 1 g every 8 h | 10.3 (4.24‐24.9) | |
| ESRD | 0.75 g every 12 h | 8.55 (3.27‐24.4) |
CV, coefficient of variation; ELF, epithelial lining fluid; ESRD, end‐stage renal disease; KE1, first‐order transfer rate constant from ELF to drug amount in central compartment; MIC, minimum inhibitory concentration.
Augmented: creatinine clearance (CrCL) > 120 mL/min (120 to < 150 = 50%; > 150 = 50%). Normal: CrCL 90 to < 120 mL/min. Mild: CrCL 60 to < 90 mL/min. Moderate: CrCL 30 to < 60 mL/min. Severe: CrCL 15 to < 30 mL/min. End stage of renal disease (ESRD): CrCL 5 to < 15 mL/min.
1000 simulated patients in each simulation scenario.
Body weight was assumed to be log‐normal distributed with mean of 72.6 kg and CV of 30%.
Albumin was assumed to be log‐normal distributed with mean of 2.8 g/dL and CV of 30%.
For the patients with nosocomial pneumonia enrolled in the phase 3 studies, the %fT>MIC,ELF was 100% in 89% (25/28) and 98% (95/97) of the patients in the CREDIBLE‐CR and APEKS‐NP study, respectively. No clear PK/PD relationships of %fT>MIC,ELF with microbiological and clinical outcomes and vital status were found due to high %fT>MIC,ELF in most of the patients (Figure 4).
Figure 4.
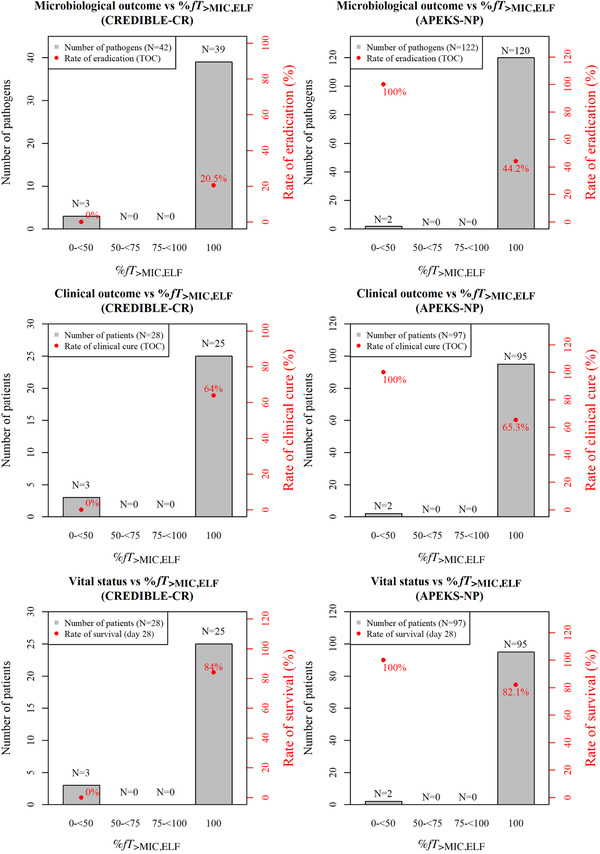
Relationships of percentage of time for which free concentrations of cefiderocol in epithelial lining fluid exceed the minimum inhibitory concentration over dosing interval (%fT>MIC,ELF) with microbiological outcome at test of cure (TOC), clinical outcome at TOC, and vital status at day 28 for patients with pneumonia in CREDIBLE‐CR and APEKS‐NP studies.
Discussion
Lung penetration of antibiotics is considered an important PK/PD parameter for characterizing the potential antibacterial effect on lung infections. In the guidelines/guidance from agencies in the United States, Europe, and Japan, 32 , 33 , 34 collection of drug concentrations in ELF from infected patients are recommended to define the dosing regimen that achieves concentrations sufficient to exert the effect at the site of infection. This intrapulmonary PK modeling of cefiderocol ELF concentrations from patients with pneumonia provides useful information to support the current dosing recommendations of cefiderocol for treatment of nosocomial pneumonia. 6 , 7
As shown in Figure S3, the observed cefiderocol concentration ratios of ELF to free plasma at 2 hours after the end of infusion (5 hours after the start of infusion) in patients with pneumonia were higher than those at the end of infusion in patients with pneumonia and healthy subjects. This result suggests a delayed distribution and/or delayed elimination of cefiderocol in ELF in patients with pneumonia, in contrast to the parallel PK for plasma and ELF observed in healthy subjects. 16 , 17 For ceftolozane of ceftolozane/tazobactam, another cephalosporin, a delayed distribution into ELF was observed in patients with pneumonia (time to Cmax of 1 and 6 hours in plasma and ELF, respectively), 35 while no delayed distribution of ceftolozane was observed in healthy subjects. 36 These findings suggest the difference in distribution of these antibiotics into ELF between mechanically ventilated patients with pneumonia and nonventilated healthy subjects may be derived from the difference in physiologic conditions in lung such as inflammation. 14 This delayed distribution in ELF appears to enhance the overall target site exposure of these new agents and further optimizes the time above the MIC as the driver of efficacy. There have been reports for modeling to address lung distribution of antivirals. 37 , 38 Our findings suggest that it may be useful to consider lung distribution in pneumonia patients for further modeling of antivirals.
In the intrapulmonary PK modeling, the fraction of ELF to free plasma concentration at steady state in patients with pneumonia (0.339) was 1.4‐fold that in healthy subjects (0.244). The post hoc estimates of AUC ratio (AUCELF to fAUCplasma) in patients with pneumonia (0.340) was 1.3‐fold that in healthy subjects (0.263). From these results, the penetration ratios calculated on the basis of model parameters (concentration ratio at steady state) and AUC were similar and estimated as 34% in patients with pneumonia. There was no ELF concentration data of cefiderocol in the elimination phase; however, an increase in the lung penetration of cefiderocol in patients with pneumonia compared to healthy subjects was suggested on the basis of the observed concentrations and intrapulmonary PK modeling. Previous reports have shown variability of lung penetration of cephalosporins. 14 , 15 The cefepime concentration ratio in ELF to total plasma in healthy subjects was 0.39, 39 while cefepime ELF concentrations in critically ill patients were similar to the total plasma concentrations. 40 The concentration ratios of ELF to total plasma of ceftazidime were 21% to 44% in patients with pneumonia 41 , 42 and 31% to 32% in healthy volunteers. 43 As for ceftolozane, the penetration ratios of AUCELF to fAUCplasma were 50% in patients with pneumonia 35 and 61% in healthy volunteers. 36 The lung penetration ratio of cefiderocol in patients with pneumonia was similar to or higher than that of these other cephalosporins.
The PTA for 75% fT>MIC,ELF and 100% fT>MIC,ELF were both ≥90% against MICs ≤4 μg/mL except for patients with normal renal function, where it was >87%. (Table 3). The target 75% fT>MIC,ELF was selected as the mean value of the estimated %fT>MIC achieving 1‐log10 reduction activity in the neutropenic murine lung infection model, and 100% fT>MIC,ELF was used as a very conservative target to address the variability of %fT>MIC among some pathogens. 5 In the phase 3 studies, the %fT>MIC,ELF was 100% in 89% to 98% of the patients with nosocomial pneumonia. The PTA for 100% fT>MIC,ELF calculated using the high KE1 was 71% to 96% against an MIC of 4 μg/mL regardless of renal function, which is a pretty conservative scenario considering the variations of %fT>MIC estimates in the animal infection model 5 and the upper limit of CI for the estimate of KE1 (assuming faster elimination of ELF concentrations). Furthermore, the median of simulated ELF trough concentrations was >4 μg/mL in all renal function groups regardless of KE1 value (Table 4). These results suggested that the dosing regimens of cefiderocol adjusted on the basis of renal function would provide adequate exposure in lungs up to an MIC of 4 μg/mL in critically ill patients with pneumonia including those with augmented renal function.
A protein binding of cefiderocol in ELF was not considered in this study. The albumin concentration in ELF in critically ill ventilated patients was reported to be 0.32 g/dL, 21 which was much lower than that in plasma (mean, 2.7 and 3.0 g/dL in the CREDIBLE‐CR and APEKS‐NP studies, respectively). The protein‐binding ratio of cefiderocol is 57.8%. 26 Craig and Suh 22 reported that for antimicrobial agents with ≈60% of protein binding (sulfamethoxazole and trimethoprim), the protein binding was decreased to ≤20% in the expected range of albumin concentration in ELF (5%‐10% of normal albumin concentration in plasma [5%‐10% of 4.2 g/dL]). A review paper also reported that protein binding of antibiotics is expected to be negligible at low albumin concentrations. 23 Therefore, ELF concentrations were regarded as free ELF concentrations in this study.
One of the limitations of this study was the limited information on ELF concentration data at only 2 sampling time points (ie, at the end of infusion and 2 hours after the end of infusion) with 1 ELF datum per subject from 7 patients with pneumonia. Therefore, since it was difficult to construct an intrapulmonary PK model based on the data only in patients with pneumonia, intrapulmonary modeling was conducted by using the integrated data with healthy subjects, which provided more ELF sampling time points and more subjects per time point. Consequently, the model parameters were estimated precisely with <35% relative standard error except for KE1. Regarding the uncertainty of the KE1 estimate, the sensitivity analysis was conducted considering the CI of the KE1 estimate, and the results showed 71% to 96% PTA in ELF for an MIC of 4 μg/mL in all renal function groups even using the conservative target 100% fT>MIC and higher KE1.
Conclusions
The developed intrapulmonary PK model adequately described ELF cefiderocol concentrations in patients with pneumonia and healthy subjects. The lung penetration ratio of cefiderocol in patients with pneumonia was 34%, which is 1.4‐fold that in healthy subjects. In the phase 3 study with patients with nosocomial pneumonia, the estimated %fT>MIC,ELF was 100% in most patients, and no clear PK/PD relationship for the %fT>MIC,ELF was found for any of the outcomes or vital status. The ELF concentration‐time profile of cefiderocol derived in this current analysis is consistent with the observations from phase 3 data and is predictive of adequate ELF trough concentrations to treat a gram‐negative pathogen with MICs ≤4 μg/mL. These study results support the current dosing regimens of cefiderocol adjusted on the basis of renal function in patients with nosocomial pneumonia, including those with augmented renal clearance.
This study was funded and conducted by Shionogi & Co., Ltd. (Osaka, Japan) and its subsidiary Shionogi Inc. (Florham Park, New Jersey) and Shionogi Europe (London, UK).
Conflicts of Interest
N.K. and T.K. are employees of Shionogi & Co., Ltd., and T.W. was an employee of Shionogi & Co., Ltd. at the time of this research. R.E. is a consultant for Shionogi Inc. and received a consultancy fee. D.P.N. has acted as a primary investigator and consultant and is on the speaker bureau of Shionogi Inc.
Supporting information
Supporting Information
Acknowledgments
Selected results from this study were previously presented as a poster presentation at IDWeek 2020.
We thank all patients, investigators, and study personnel who participated in this study.
References
- 1. Kohira N, West J, Ito A, et al. In vitro antimicrobial activity of a siderophore cephalosporin, S‐649266, against Enterobacteriaceae clinical isolates, including carbapenem‐resistant strains. Antimicrob Agents Chemother. 2016;60:729‐734. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Ito A, Kohira N, Bouchillon SK, et al. In vitro antimicrobial activity of S‐649266, a catechol‐substituted siderophore cephalosporin, when tested aginst non‐fermenting gram‐negative bacteria. J Antimicrob Chemother. 2016;71:670‐677. [DOI] [PubMed] [Google Scholar]
- 3. Ito A, Sato T, Ota M, et al. In vitro antibacterial paropoerties of cefiderocol, a novel siderophore cephalosporin, against gram‐negative bacteria. Antimicrob Agents Chemother. 2018;62:e01454‐17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Sato T, Yamawaki K. Cefiderocol: discovery, chemistry, and in vivo profiles of a novel siderophore cephalosporin. Clin Infect Dis. 2019;69(suppl 7):S538‐S543. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Nakamura R, Ito‐Horiyama T, Takemura M, et al. In vivo pharmacodynamic study of cefiderocol, a novel parenteral siderophore cephalosporin, in murine thigh and lung infection models. Antimicrob Agents Chemother. 2019;63:e02031‐18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Fetroja® (cefiderocol) injection for intravenous use. Prescribing Information. Florham Park, NJ: Shionogi Inc.; 2020. [Google Scholar]
- 7. European Medicines Agency Fetcroja® (cefiderocol). Product information. https://www.ema.europa.eu/documents/product-information/fetcroja-epar-product-information_en.pdf. Published April 29, 2020. Accessed October 22, 2021.
- 8. Katsube T, Echols R, Arjona Ferreira JC, et al. Cefiderocol, a siderophore cephalosporin for gram‐negative bacterial infections: pharmacokinetics and safety in subjects with renal impairment. J Clin Pharmacol. 2017;57:584‐591. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Katsube T, Wajima T, Ishibashi T, et al. Pharmacokinetic/pharmacodynamic modeling and simulation of cefiderocol, a parenteral siderophore cephalosporin, for dose adjustment based on renal function. Antimicrob Agents Chemother. 2017;61:e01381‐16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Kawaguchi N, Katsube T, Echols R, et al. Population pharmacokinetic and pharmacokinetic/pharmacodynamic analyses of cefiderocol, a parenteral siderophore cephalosporin, in patients with pneumonia, bloodstream infection/sepsis, or complicated urinary tract infection. Antimicrob Agents Chemother. 2021;65:e01437‐17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Crandon JL, Bulik CC, Kuti JL, et al. Clinical pharmacodynamics of cefepime in patients infected with Pseudomonas aeruginosa . Antimicrob Agents Chemother. 2010;54:1111‐1116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. MacVane SH, Kuti JL, Nicolau DP. Clinical pharmacodynamics of antipseudomonal cephalosporins in patients with ventilator‐associated pneumonia. Antimicrob Agents Chemother. 2014;58:1359‐1364. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Crandon JL, Luyt CE, Aubry A, et al. Pharmacodynamics of carbapenems for the treatment of P. aeruginosa ventilator associated pneumonia, associations with clinical outcome and recurrence. J Antimicrob Chemother. 2016;71:2534‐2537. [DOI] [PubMed] [Google Scholar]
- 14. Rodvold KA, George JM, Yoo L. Penetration of anti‐infective agents into pulmonary epithelial lining fluid: focus on antibacterial agents. Clin Pharmacokinet. 2011;50:637‐664. [DOI] [PubMed] [Google Scholar]
- 15. Rodvold KA, Hope WW, Boyd SE. Considerations for effect site pharmacokinetics to estimate drug exposure: concentrations of antibiotics in the lung. Curr Opin Pharmacol. 2017;36:114‐123. [DOI] [PubMed] [Google Scholar]
- 16. Katsube T, Saisho Y, Shimada J, et al. Intrapulmonary pharmacokinetics of cefiderocol, a novel siderophore cephalosporin, in healthy adult subjects. J Antimicrob Chemother. 2019;74:1971‐1974. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Katsube T, Nicolau D, Rodvold K, et al. Intrapulmonary pharmacokinetic profile of cefiderocol in mechanically ventilated patients with pneumonia. J Antimicrob Chemother. 2021;76:2902‐2905. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Bassetti M, Echols R, Matsunaga Y, et al. Efficacy and safety of cefiderocol or best available therapy for the treatment of serious infections caused by carbapenem‐resistant Gram‐negative bacteria (CREDIBLE‐CR): a randomised, open‐label, multicentre, pathogen‐focused, descriptive, phase 3 trial. Lancet Infect Dis. 2021;21:226‐240. [DOI] [PubMed] [Google Scholar]
- 19. Wunderink RG, Matsunaga Y, Ariyasu M, et al. Cefiderocol versus high‐dose, extended‐infusion meropenem for the treatment of Gram‐negative nosocomial pneumonia (APEKS‐NP): a randomised, double‐blind, phase 3, non‐inferiority trial. Lancet Infect Dis. 2021;21:213‐225. [DOI] [PubMed] [Google Scholar]
- 20. Renard SI, Baset G, Lecossier D, et al. Estimation of volume of epithelial lining fluid recovered by lavage using urea as marker of dilution. J Appl Physiol. 1986;60:532‐538. [DOI] [PubMed] [Google Scholar]
- 21. Lamer C, De Beco V, Soler P, et al. Analysis of vancomycin entry into pulmonary lining fluid by bronchoalveolar lavage in critically ill patients. Antimicrob Agents Chemother. 1993;37(2):281‐286. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Craig WA, Suh B. Protein binding and the antimicrobial effects: methods for the determination of protein binding. In: Lorian V, ed. Antibiotics in Laboratory Medicine. Baltimore, MD: Williams & Wilkins; 1986; 477‐513. [Google Scholar]
- 23. Kiem S, Schentag JJ. Interpretation of antibiotic concentration ratios measured in epithelial lining fluid. Antimicrob Agents Chemother. 2008;52:2436. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Bergstrand M, Hooker AC, Wallin JE, et al. Prediction‐corrected visual predictive checks for diagnosing nonlinear mixed‐effects models. AAPS J. 2011;13:143‐151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Ette EI. Stability and performance of a population pharmacokinetic model. J Clin Pharmacol. 1997;37:486‐495. [DOI] [PubMed] [Google Scholar]
- 26. Matsumoto S, Singley CM, Hoover J, et al. Efficacy of cefiderocol against carbapenem‐resistant gram‐negative bacilli in immunocompetent‐rat respiratory tract infection models recreating human plasma pharmacokinetics. Antimicrob Agents Chemother. 2017;61:e00700‐17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Beal SL, Sheiner LB, Boeckmann AJ. NONMEM Users Guide. Ellicott City, MD: Icon Development Solutions; 1989‐2006. [Google Scholar]
- 28. Lindbom, L , Pihlgren P, Jonsson EN. PsN‐Toolkit – a collection of computer intensive statistical methods for non‐linear mixed effect modeling using NONMEM. Comput Methods Programs Biomed. 2005;79:241–57. [DOI] [PubMed] [Google Scholar]
- 29. Keizer RJ, Karlsson MO, Hooker A. Modeling and simulation workbench for NONMEM: tutorial on Pirana, PsN, and Xpose. CPT Pharmacometrics Syst Pharmacol. 2015;2:e50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. R Core Team . R: A Language and Environment for Statistical Computing. Vienna, Austria: R Foundation for Statistical Computing; 2013. [Google Scholar]
- 31. Yamano Y. In vitro activity of cefiderocol against a broad range of clinically important gram‐negative bacteria. Clin Infect Dis. 2019;69(suppl 7):S544‐51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. US Food and Drug Administration . Hospital‐Acquired Bacterial Pneumonia and Ventilator‐Associated Bacterial Pneumonia: Developing Drugs for Treatment Guidance for Industry. Silver Spring, MD: US Department of Health and Human Services Food and Drug Administration Center for Drug Evaluation and Research; 2020. [Google Scholar]
- 33. European Medicines Agency . Guideline on the Use of the Pharmacokinetics and Pharmacodynamics in the Development of Antimicrobial Medicinal Products. Amsterdam, the Netherlands: Committee for Medical Products for Human Use; 2016. [Google Scholar]
- 34. Ministry of Health, Labor and Welfare . Guidelines for Clinical Evaluation of Antibacterial Drugs. Tokyo, Japan: Evaluation and Licensing Division, Pharmaceutical and Food Safety Bureau; 2017. [Google Scholar]
- 35. Caro L, Nicolau DP, De Waele JJ, et al. Lung penetration, bronchopulmonary pharmacokinetic/pharmacodynamic profile and safety of 3 g of ceftolozane/tazobactam administered to ventilated, critically ill patients with pneumonia. J Antimicrob Chemother. 2020;75:1546‐1553. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Chandorkar G, Huntington JA, Gotfried MH, et al. Intrapulmonary penetration of ceftolozane/tazobactam and piperacillin/tazobactam in healthy adult subjects. J Antimicrob Chemother. 2012;67:2463‐2469. [DOI] [PubMed] [Google Scholar]
- 37. Yao X, Ye F, Zhang M, et al. In vitro antiviral activity and projection of optimized dosing design of hydroxychloroquine for the treatment of severe acute respiratory syndrome coronavirus 2 (SARS‐CoV‐2). Clin Infect Dis. 2020;71:732‐739. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Fan J, Zhang X, Liu J, et al. Connecting hydroxychloroquine in vitro antiviral activity to in vivo concentration for prediction of antiviral effect: a critical step in treating patients with coronavirus disease 2019. Clin Infect Dis. 2020;71:3232‐3236. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Rodvold KA, Gotfried MH, Chugh R, et al. Plasma and intrapulmonary concentrations of cefepime and zidebactam following intravenous administration of WCK 5222 to healthy adult subjects. Antimicrob Agents Chemother. 2018;62:e00682‐18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Boselli E, Breilh D, Duflo F, et al. Steady‐state plasma and intrapulmonary concentrations of cefepime administered in continuous infusion in critically ill patients with severe nosocomial pneumonia. Crit Care Med. 2003;31:2012‐2016. [DOI] [PubMed] [Google Scholar]
- 41. Boselli E, Breilh D, Rimmele T, et al. Plasma and lung concentrations of ceftazidime administered in continuous infusion to critically ill patients with severe nosocomial pneumonia. Intensive Care Med. 2004;30:989‐991. [DOI] [PubMed] [Google Scholar]
- 42. Cousson J, Floch T, Guillard T, et al. Lung concentrations of ceftazidime administered by continuous versus intermittent infusion in patients with ventilator‐associated pneumonia. Antimicrob Agents Chemother. 2015;59:1905‐1909. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Nicolau DP, Siew L, Armstrong J, et al. Phase 1 study assessing the steady‐state concentration of ceftazidime and avibactam in plasma and epithelial lining fluid following two dosing regimens. J Antimicrob Chemother. 2015;70:2862‐2869. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supporting Information