

Abstract
Background
Rare inherited thrombocytopenias are caused by alterations in genes involved in megakaryopoiesis, thrombopoiesis and/or platelet release. Diagnosis is challenging due to poor specificity of platelet laboratory assays, large numbers of culprit genes, and difficult assessment of the pathogenicity of novel variants.
Objectives
To characterize the clinical and laboratory phenotype, and identifying the underlying molecular alteration, in a pedigree with thrombocytopenia of uncertain etiology.
Patients/Methods
Index case was enrolled in our Spanish multicentric project of inherited platelet disorders due to lifelong thrombocytopenia and bleeding. Bleeding score was recorded by ISTH‐BAT. Laboratory phenotyping consisted of blood cells count, blood film, platelet aggregation and flow cytometric analysis. Genotyping was made by whole‐exome sequencing (WES). Cytoskeleton proteins were analyzed in resting/spreading platelets by immunofluorescence and immunoblotting.
Results
Five family members displayed lifelong mild thrombocytopenia with a high number of enlarged platelets in blood film, and mild bleeding tendency. Patient's platelets showed normal aggregation and granule secretion response to several agonists. WES revealed a novel nonsense variant (c.322C>T; p.Gln108*) in TPM4 (NM_003290.3), the gene encoding for tropomyosin‐4 (TPM4). This variant led to impairment of platelet spreading capacity after stimulation with TRAP‐6 and CRP, delocalization of TPM4 in activated platelets, and significantly reduced TPM4 levels in platelet lysates. Moreover, the index case displayed up‐regulation of TPM2 and TPM3 mRNA levels.
Conclusions
This study identifies a novel TPM4 nonsense variant segregating with macrothrombocytopenia and impaired platelet cytoskeletal remodeling and spreading. These findings support the relevant role of TPM4 in thrombopoiesis and further expand our knowledge of TPM4‐related thrombocytopenia.
Keywords: inherited platelet disorders, macrothrombocytopenia, TPM4, tropomyosin‐4, whole‐exome sequencing
Essentials.
A novel nonsense variant in TPM4 was identified in a family with mild bleeding and mild macrothrombocytopenia.
The TPM4 p.Gln108* variant displayed a deleterious effect on platelet cytoskeleton remodeling.
This alteration leads to the delocalization of tropomyosin‐4 in the spreading structures.
This new pedigree with TPM4‐RT expands the genetic and clinical spectrum and supports TPM4 as a Tier 1 gene.
1. INTRODUCTION
Inherited thrombocytopenias (ITs) are a large heterogeneous group of rare platelet disorders characterized by low platelet counts which mainly lead to an increased bleeding tendency. 1 , 2 ITs are caused by defects in genes involved in megakaryocyte (Mk) differentiation, maturation and migration, but also in proplatelet formation and/or platelet release into blood. 2 , 3 Their prevalence has been estimated to be around 2.7 of 100 000 individuals but it is likely that this is an underestimate. 1 Diagnostic algorithms for ITs consist of a stepwise process based on clinical data and several laboratory tests, including the use of whole blood electronic counters to evaluate platelet count and size, blood film analysis with or without immunofluorescence staining, and platelet function assays. 4 , 5 Nowadays, the appropriateness of using next generation sequencing (NGS) procedures in the mainstream of diagnosis of inherited platelet disorders is well established. 6 Since 2010, both whole‐exome and whole‐genome sequencing (WES/WGS) approaches have allowed the identification of up to 15 new genes involved in ITs. 5 , 7 According to the International Society on Thrombosis and Haemostasis (ISTH) Scientific and Standardization Committee (SSC) for Genetics in Thrombosis and Haemostasis (GinTH), there are 71 Tier1 genes related to inherited platelet disorders 8 , 9 (https://www.isth.org/page/GinTh_GeneLists accessed 20th November 2021).
The most common ITs are macrothrombocytopenia (MCT), characterized by reduced platelet count and increased platelet size or mean platelet volume (MPV). These MCTs are mainly caused by genetic defects affecting early megakaryopoiesis and proplatelet formation. 7 , 10 In particular, proplatelet formation is highly dependent on cytoskeletal proteins, including myosin IIA, actin filaments and tubulins. 2 , 7 , 10 , 11 Thus, genetic changes affecting genes of the actomyosin cytoskeleton, such as MYH9, ACTN1, or in recent years, FLNA, DIAPH1 or TUBB1 associate with MCT. 6 , 12 In 2017, the first five patients from two unrelated pedigrees with mild bleeding and TPM4‐related thrombocytopenia (TPM4‐RT) due to the nonsense variant p.Arg69* (NM_003290.3) were identified. 13 Recently, Stapley, et al. have just reported two new TPM4‐RT cases carrying novel missense variants; p.Arg182Cys and p.Ala183Val (NM_001145160.2) related to relevant bleeding, platelet dysfunction, and mild or absent thrombocytopenia. 14 At the 2021 ISTH Meeting, we advanced the identification of a Spanish pedigree with TPM4‐RT, 15 which clinical, laboratory and molecular characterization is formally reported here. Acknowledging our new family with TPM4‐RT prompted the ISTH SSC‐GinTH to include TPM4 in the Tier1 genes for Inherited bleeding, thrombotic, and platelet disorders (BTPD). 16
2. METHODS
2.1. Patients and blood sampling
This study involved patients with suspected ITs recruited in the Spanish multicenter project “Functional and Molecular Characterization of Patients with Inherited Platelet Disorders” coordinated by Grupo Español de Alteraciones Plaquetarias Congénitas (GEAPC). Investigations abided by the Helsinki Declaration and were approved by the Ethics Committee of the Instituto de Investigación Biomédica de Salamanca. All patients gave written informed consent. Bleeding symptoms were scored using ISTH‐BAT questionnaire. 17 Venous blood samples were drawn into 7.5% K3 EDTA (blood cells count, blood film, nucleic acid purification and protein lysates) and buffered 0.105 M sodium citrate for platelet functional studies. 18 Platelet (P) count and mean platelet volume (MPV) were performed using a Advia 2120i hematological counter (Siemens). To measure platelet dimensions on May–Grünwald–Giemsa‐stained blood smears, 100 platelets were evaluated and classified as normal (diameter <3 µm), large (3–4 µm) or giant (>4 µm). 4
2.2. Platelet functional tests
Platelet functional characterization was performed in the index case [II.2] in parallel with a healthy control. Detailed platelet phenotyping was made as described. 12 , 19 Briefly, platelet‐rich‐plasma (PRP) was obtained by centrifugation at low speed (100×g, 10 min). Light transmission aggregometry (LTA) was performed using an TA‐8V Aggregometer (Stago) and was recorded during 5 min upon PRP stimulation with agonists.
Platelet expression of major glycoproteins (GPs) was assessed in diluted whole blood (1:10 in PBS) by flow cytometry (FC) with specific antibodies (anti‐CD42a*PE, anti‐CD42b*PE, anti‐GPVI*FITC, anti‐CD61*PE and anti‐CD41*APC) (BD Biosciences). Platelets were stained at room temperature (RT) for 30 min under static conditions. For platelet function analysis, PRP was stimulated for 30 min with ADP, CRP and TRAP6, and fibrinogen‐binding (fibrinogen*AF488, Invitrogen), α‐granule (anti‐CD62*PE antibody), and δ‐granule secretion (anti‐CD63*PE antibody) (BD Biosciences) were evaluated by FC using an Accuri C6TM cytometer (BD). Mean Fluorescence Intensity (MFI) or the percentage of positive platelets were analyzed using FlowJo software (vX.0.7. TreeStar).
Platelets were washed in modified Tyrode's buffer and platelet lysates were prepared as previously described. 12 Tropomyosin‐4 (TPM4) (Invitrogen), β‐tubulin, actinin‐1, filamin A, Nonmuscle Myosin Heavy Chain II‐A (NMHC‐IIA) and β‐actin (Cell Signaling), were detected by immunoblotting in platelet lysates, and by immunofluorescence (IF) under resting and spreading conditions using washed platelets in fibrinogen‐coated coverslips (Sigma‐Aldrich). Samples were visualized using a Leica TCS‐SP8 confocal microscope, and images were analyzed using ImageJ software (National Institutes of Health). Platelet spreading assays in the proband and in a healthy control were performed essentially as reported. 12 We evaluated the increase in the mean platelet surface between resting and agonist (TRAP6 and CPR) stimulated conditions, upon evaluation of 50 platelets per condition.
Platelet RNA was extracted using RLT‐plus lysis buffer (RNeasy kit, Qiagen), and tropomyosin mRNA levels in the patients were quantified by qPCR (Applied Biosystems) and compared to those in a healthy control using the 2−ΔΔ CT method. GAPDH was used as the housekeeping gene. Three different qPCR experiments, each one in triplicate, to obtain a mean average Ct value, were performed in patients and in the control for each gen.
2.3. Molecular analysis by exome sequencing based on candidate gene panel
Genomic DNA was extracted from peripheral blood samples using a DNeasy blood and tissue kit, following the manufacturer's protocol (Qiagen). The propositus (II.2), and both siblings (II.3, II.4) (Figure 1A) were analyzed by WES after capture and library preparation using an academic protocol that included enrichment with the xGEN® Exome Research Panel v2 (Integrated Technologies). 20 , 21 Sequencing was performed with an S1 Flow Cell on the NovaSeq 6000 system with a mean target coverage of 50× at the sequencing service MLL Münchner Leukämielabor GmbH. Variant was confirmed and segregated in the rest of family members (I.1, I.2 and II.1, Figure 1A) by Sanger sequencing.
FIGURE 1.
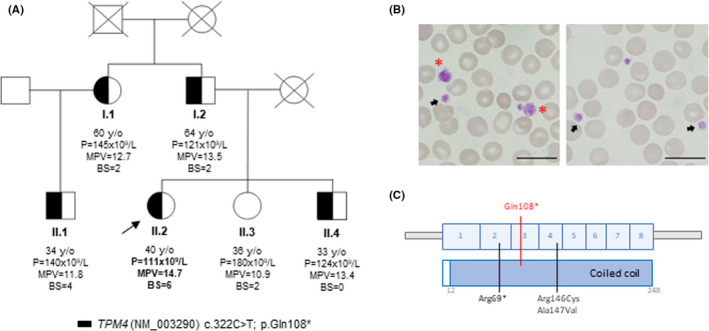
Molecular alteration and clinical and laboratory parameters of a family with tropomyosin‐4 related macrothrombocytopenia (TPM4‐RT). (A) Pedigree of a family with lifelong dominant inherited macrothrombocytopenia and bleeding tendency. The index case is indicated with a black arrow. Partially filled black symbols indicate heterozygosis for the indicated TPM4 variant. (B) Representative peripheral blood film from II.2 patient (propositus) after May‐Grunwald Giemsa staining (×100). Variable platelet size was observed with large (black arrows) and giant (red asterisk) platelets. Bar: 20 µm (C) Schematic representation of the TPM4 protein, which contains 248 amino acids and a unique coiled‐coil domain. Figure shows the previously reported variants (in black), p.Arg69*, 13 p.Arg146Cys and p.Ala147Val, 14 and the novel genetic change p.Gln108* (in red) found in the pedigree reported here. All genetic alterations are numbered according to positions in the NM_003290.3 transcript for TPM4. Variants p.Arg146Cys and p.Ala147Val correspond to described p.Arg182Cys and p.Ala183Val (NM_001145160.2) 14
We followed the guidelines of the American College of Medical Genetics and Genomics and Association for Molecular Pathology (ACMG) to qualify variant pathogenicity. 22 General information about variants was obtained using the Varsome web tool. 23
3. RESULTS AND DISCUSSION
A 40 year‐old female (propositus, II.2) (Figure 1A) was referred due to lifelong mild macrothrombocytopenia (p = 111 × 109/L [Normal range: p = 150–400 × 109/L] and MPV 14.7fL [7.2–11.1 fL]). She has a lifelong mild bleeding tendency (ISTH‐BAT = 6) characterized by hematomas, gum bleeding, and menorrhagia which needed iron and hormonal therapy. Several family members (I.1, I.2, II.1, II.4), which were either asymptomatic or have a mild bleeding tendency, also displayed mild thrombocytopenia (Figure 1A). Peripheral blood film revealed giant (31%) and enlarged platelets (39%) in the propositus (II.2) (Figure 1B).
DNA analysis by WES identified a novel nonsense variant (c.322C>T; p.Gln108*) in TPM4 (NM_003290.3) in all affected family members (Figure 1A). This variant was located in exon 3, affecting the coiled coil domain (Figure 1C). Following the ACMG/AMP guidelines, it was classified as likely pathogenic variant (PVS1, PM2). 22
Carrier of the variant displayed normal platelet aggregation with several agonists, including low doses of TRAP‐6 and collagen. A mild impairment and delay in the platelet aggregation response to low dose of ADP and epinephrine was observed (Figure 2A). Indeed, we observed slightly increased levels of major GPs, in accordance with the increased platelet size (Figure 2B), and normal fibrinogen‐binding and α and δ‐granule secretion upon platelet activation with by TRAP‐6, ADP, and CRP (Figure 2C). A mild bleeding tendency and an almost unaffected platelet phenotype was previously observed in carriers of the TPM4 p.Arg69* (NM_003290.3) variant. 13 In contrast, carriers of the missense variants p.Arg182Cys and p.Ala183Val (NM_001145160.2) have been recently described to have relevant bleeding, significantly impaired platelet secretion and aggregation, and normal or slightly reduced platelet count and TPM4 levels. 14 The reasons for such differences in bleeding and platelet phenotype in these TPM4‐RT patients are still unknown. We speculate that heterozygous TPM4 nonsense variants, such as p.Arg69* 13 and p.Gln108*, reported here, mainly cause haploinsufficiency, which is suggested by a reduction in tropomyosin‐4 levels (Figure 2D), while missense variants could present a genetic negative dominant effect exacerbating TPM4 dysfunction and leading to major platelet disorder. Variably mild thrombocytopenia with minor, if any, associated bleeding tendency also happens in other inherited thrombocytopenias caused by mutations in structural platelet proteins. 12 Indeed, as previously described, 13 proplatelet formation is TPM4 dose‐dependent, which could justify that both nonsense variants (p.Arg69* and p.Gln108*) are associated with thrombocytopenia, while the missense variants are not. Alternatively, additional platelet or hemostatic abnormalities yet undiscovered, and beyond TPM4, may be present in the patients reported by Stapley, et al. 14
FIGURE 2.
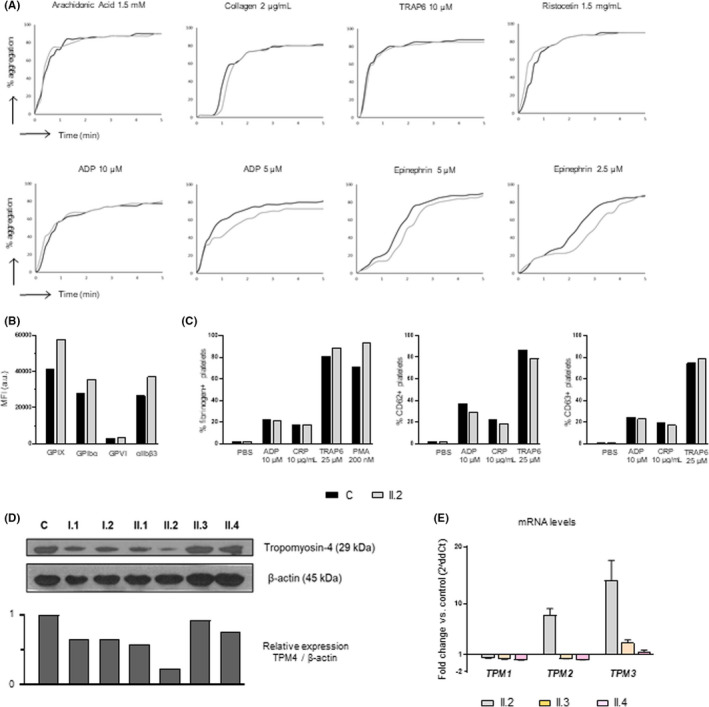
Platelet functional phenotyping performed by light transmission aggregometry and flow cytometry (propositus) and immunoblotting (all family members). (A) Platelet aggregation, with several agonists, was evaluated in control and in the propositus. (B) Glycoproteins (GPs) expression profiles were assessed by flow cytometry with indicated fluorescently labeled antibodies. The mean fluorescence intensity (MFI) is represented. (C) Fibrinogen binding and alpha and dense granules secretion were evaluated by FC in the propositus and healthy control. (D) Western blot of tropomyosin‐4 levels in platelet lysates from family members and a healthy control. β‐actin was used as internal control. (E) TPM1, TPM2 and TPM3 mRNA levels in platelets from cases II.2, II.3 and II.4, relative to those in a healthy control, were assessed by qRT‐PCR using the 2ddCt method and using GAPDH as housekeeping gen. For each gene, three different qPCR experiments, each one in triplicate, to obtain a mean average Ct value were performed in patients and in the control. Histogram represents the mean ±standard error of the 2ddCt values in the three qPCR experiments
Interestingly, we observed that TPM2 and TPM3 mRNA levels in the propositus platelets were 8 and 14 times higher, respectively, than those in control platelets (Figure 2E). The fact that mutant TPM4 associates with macrothrombocytopenia despite an increased level of other tropomyosins, further supports a highly specific and key effect of TPM4 in megakaryopoiesis and platelet biogenesis. This finding is in agreement with previous reports suggesting that other tropomyosin isoforms have non‐redundant functions with TPM4, 13 since TPM1 and TPM2 are mainly expressed in skeletal muscle, and TPM3 in both muscle and non‐muscle cells. 24 Although TPM3 has an important role in cytoskeleton, overexpression of the isoform does not restore TPM4 function in Mks. Besides the important involvement of TPM4 in the cytoskeleton, it is also expressed in the striated and smooth muscle, 25 however, no associated myopathies have been reported in any TPM4‐RT patient.
Finally, platelet spreading studies, revealed a significance reduction in the formation of filipodia and lamellipodia, as well as severe reduction of full‐spreading structures (Figure 3A) in the propositus vs. control platelets, leading to an impaired spreading function, evaluated as the increased platelet area, upon stimulation with TRAP‐6 and CRP for different times (Figure 3B). These results are consistent with the previously reported finding in other TPM4‐RT cases. 13 , 14 In addition, we first show that while TPM4 in control platelets is homogeneously distributed throughout the cytoplasm along with actin filaments, Gln108* mutant TPM4 is accumulated mainly in the center of the patient platelets, and the protein is reduced in filopodia/lamellipodia in spread platelets, as shown in the fluorescence diagram of distribution (Figure 3C). Thus, this genetic alteration leads to a mild reduction in the localization of TPM4 with other proteins of the cytoskeleton (10%–20%) (Figure 3A). The moderate reduction of TPM4 in the spreading structures could account for the defect in cytoskeleton remodeling. However, no alterations in the level (data not shown) and localization of other cytoskeleton proteins (β‐actin, actinin‐1, β1‐tubulin, filamin A, and NMHC‐IIA) were found (Figure 3A). These data were unexpected, since previous studies in Tpm4plt53 mutant mice demonstrated increased levels of degraded filamin A and actinin‐1. 13 Finally, immunoblotting of platelet lysates showed a reduction of TPM4 levels in all p.Gln108* carriers, which was more pronounced in the index case (II.2, 80% reduction vs. level of TPM4 in control platelets) (Figure 2D). Thus, patients with TPM4‐RT showed variable phenotypic expression, indicating differences in genetic penetrance of the different TPM4 variants, and/or the contribution of other unrecognized factors in some affected patients.
FIGURE 3.
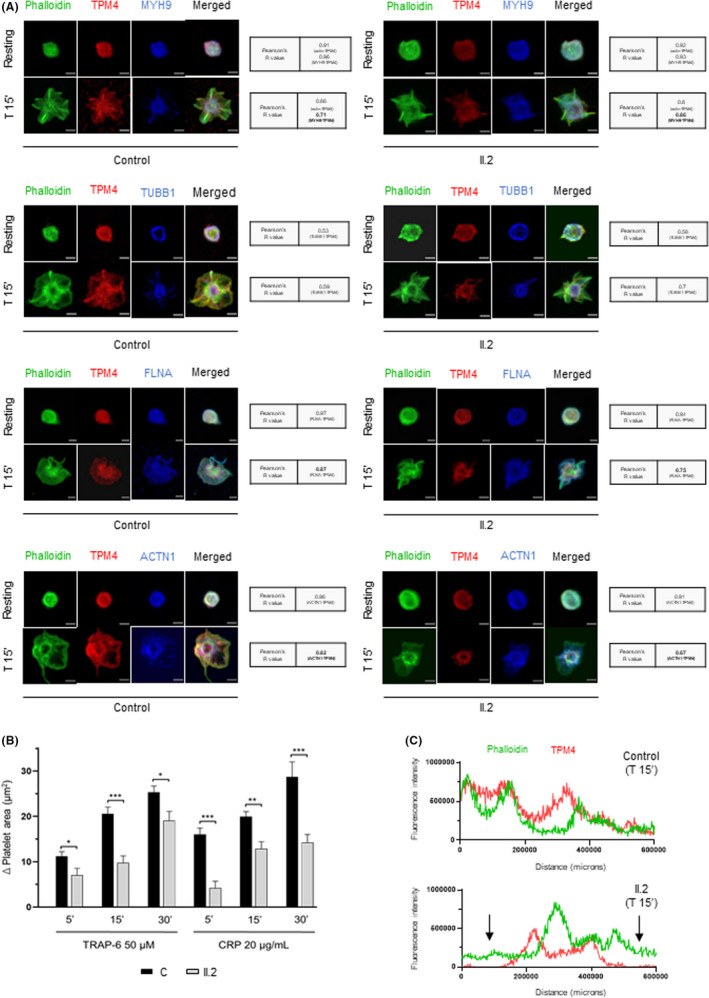
Deleterious effect of p.Gln108* TPM4 variant in cytoskeleton remodeling. Platelet spreading on fibrinogen coverslips was analyzed in washed platelets from control and II.2 patient. (A) Representative immunofluorescence image of platelet spreading under no treatment (resting), and at 15 min from activation with 50 µM TRAP‐6 (T 15´). Platelets were labeled Oregon Green 514 Phalloidin (actin, green), anti‐TPM4_568 (red), and anti‐MYH9_647 (blue) or TUBB1_647 (blue) or FLNA_647 (blue) or ACTN1_647 (blue). Co‐localization of TPM4 with other proteins of the cytoskeleton was determined with Pearson's R value (Coster p value =1). Images were acquired in a Leica TCS‐SP8 confocal microscope, with a 100× objective lens, and images were analyzed using ImageJ software. Scale bars are 2 μm. (B) Platelet spreading assays were performed in the proband and in a healthy control. Histogram bars correspond to the increase (mean ±standard error, n = 50) in the area (μm2) of agonist (TRAP6 and CPR) stimulated platelets vs. the area of resting platelets (50 platelets evaluated for each condition). *p<.05, **p<.01 and ***p<.001 for patient platelets vs. control platelets. (C) The line profile plots represent the fluorescence intensity distribution of channels: actin (green) and TPM4 (red) at 15 min from TRAP‐6 50 µM treatment in control and II.2 platelets. Black arrows indicate the protein distribution in the spreading structures
Although no patients with TPM4‐RT receiving thrombopoietin receptor agonists have been reported to date, the effectiveness of agents have been shown in other inherited thrombocytopenias with defects of the platelet cytoskeleton, such as MYH9 or DIAPH1. 10 , 26
In summary, we have identified and characterized a novel nonsense variant in TPM4, c.322C>T [p.Gln108*], which is associated with mild macrothrombocytopenia and a deleterious effect on platelet cytoskeleton remodeling. Our findings reinforce the key role of TPM4 in thrombopoiesis and expand the phenotype and genotype spectrum of TPM4‐RT. This new TPM4‐RT pedigree guarantees the inclusion of TPM4 as a Tier1 list of gene involved in ITs.
CONFLICT OF INTEREST
The authors state that they have no conflict of interest.
AUTHOR CONTRIBUTION
JMB, JMHR and JRGP designed the research; AMQ, CFI, VPB, JR and EV performed the functional experiments. RB and JMB conducted the whole exome sequencing analysis. AMQ, IGT, JRGP and JMB analyzed and interpretated the results; AMQ, JR and JMB wrote the paper. All authors reviewed the results and approved the final version of the manuscript.
ACKNOWLEDGEMENTS
We thank Sandra Santos‐Mínguez and Cristina Miguel‐García for preparing DNA samples for sequencing; Sara González Briones and Irene Rodríguez Iglesias for technical support with experiments; the CIC‐IBMCC Microscopy and Cytometry Service for technical assistance with the confocal immunofluorescence studies; María Díez‐Campelo, Félix López‐Cadenas, María de los Ángeles Manrique Gonzalo, Nuria Vicente Holgado, Mercedes Rodríguez Martín, Isabel Ramos Sevillano, María del Mar Cambronero Estévez and Beatriz Oreja Martín for blood samples collection, hemograms and blood smears.
Marín‐Quílez A, Vuelta E, Díaz‐Ajenjo L, et al. A novel nonsense variant in TPM4 caused dominant macrothrombocytopenia, mild bleeding tendency and disrupted cytoskeleton remodeling. J Thromb Haemost. 2022;20:1248–1255. doi: 10.1111/jth.15672
José Rivera and José María Bastida authors share senior authorship
Manuscript Handled by: Wolfgang Bergmeier
Final decision: Wolfgang Bergmeier, 14 February 2022
Funding information
This work was partially supported by grants from Instituto de Salud Carlos III (ISCIII) and Feder (PI17/01966, PI20/00926), Gerencia Regional de Salud (GRS2061A/19, GRS2135/A/2020, GRS2314/A/2021), Fundación Mutua Madrileña (FMM, AP172142019) and Sociedad Española de Trombosis y Hemostasia (SETH‐FETH; Premio López Borrasca 2019 and Ayuda a Grupos de Trabajo en Patología Hemorrágica 2020 and 2021). The author's research on Inherited Platelet Disorders is conducted in accordance with the aims of the Functional and Molecular Characterization of Patients with Inherited Platelet Disorders Project, which is supported by “Grupo Español de Alteraciones Plaquetarias Congénitas (GEAPC)”. AMQ and CFI hold a predoctoral grant from the Junta de Castilla y León.
REFERENCES
- 1. Balduini CL, Melazzini F, Pecci A. Inherited thrombocytopenias—recent advances in clinical and molecular aspects. Platelets. 2017;28(1):3‐13. [DOI] [PubMed] [Google Scholar]
- 2. Nurden AT, Nurden P. Inherited thrombocytopenias: history, advances and perspectives. Haematologica. 2020;105(8):2004‐2019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Rodeghiero F, Pecci A, Balduini CL. Thrombopoietin receptor agonists in hereditary thrombocytopenias. J Thromb Haemost. 2018;16(9):1700‐1710. [DOI] [PubMed] [Google Scholar]
- 4. Zaninetti C, Greinacher A. Diagnosis of inherited platelet disorders on a blood smear. J Clin Med. 2020;9(2):539. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Bastida JM, Benito R, Lozano ML, et al. Molecular diagnosis of inherited coagulation and bleeding disorders. Semin Thromb Hemost. 2019;45(7):695‐707. [DOI] [PubMed] [Google Scholar]
- 6. Bastida JM, Lozano ML, Benito R, et al. Introducing high‐throughput sequencing into mainstream genetic diagnosis practice in inherited platelet disorders. Haematologica. 2018;103(1):148‐162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Bury L, Falcinelli E, Gresele P. Learning the ropes of platelet count regulation: inherited thrombocytopenias. J Clin Med. 2021;10(3):533. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Megy K, Downes K, Simeoni I, et al. Curated disease‐causing genes for bleeding, thrombotic, and platelet disorders: communication from the SSC of the ISTH. J Thromb Haemost. 2019;17(8):1253‐1260. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Megy K, Downes K, Morel‐Kopp MC, et al. GoldVariants, a resource for sharing rare genetic variants detected in bleeding, thrombotic, and platelet disorders: communication from the ISTH SSC subcommittee on genomics in thrombosis and hemostasis. J Thromb Haemost. 2021;19(10):2612‐2617. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Bastida JM, Gonzalez‐Porras JR, Rivera J, Lozano ML. Role of thrombopoietin receptor agonists in inherited thrombocytopenia. Int J Mol Sci. 2021;22(9):4330. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Collins J, Astle WJ, Megy K, Mumford AD, Vuckovic D. Advances in understanding the pathogenesis of hereditary macrothrombocytopenia. Br J Haematol. 2021;195(1):25‐45. [DOI] [PubMed] [Google Scholar]
- 12. Palma‐Barqueros V, Bury L, Kunissima S, et al. Expanding the genetic spectrum of TUBB1‐ related thrombocytopenia. Blood Adv. 2021;5(24):5453‐5467. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Pleines I, Woods J, Chappaz S, et al. Mutations in tropomyosin 4 underlie a rare form of human macrothrombocytopenia. J Clin Invest. 2017;127(3):814‐829. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Stapley RJ, Poulter NS, Khan AO, et al. Rare missense variants in Tropomyosin‐4 (TPM4) are associated with platelet dysfunction, cytoskeletal defects and excessive bleeding. J Thromb Haemost. 2021;20(2):478‐485. [DOI] [PubMed] [Google Scholar]
- 15. Marín‐Quílez A, Fernández‐Infante C, Manrique Gonzalo MÁ, et al. Characterization of a new family with lifelong macrothrombocytopenia caused by a novel nonsense variant in TPM4. Res Pr Thromb Haemost. 2021;5(suppl 2):1. [Google Scholar]
- 16. Freson K. Hemostatic phenotypes and genetic disorders. Res Pr Thromb Haemost. 2021;5(5):e12532. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Rodeghiero F, Tosetto A, Abshire T, et al. ISTH/SSC bleeding assessment tool: a standardized questionnaire and a proposal for a new bleeding score for inherited bleeding disorders. J Thromb Haemost. 2010;8(9):2063‐2065. [DOI] [PubMed] [Google Scholar]
- 18. Frelinger AL, Rivera J, Connor DE, et al. Consensus recommendations on flow cytometry for the assessment of inherited and acquired disorders of platelet number and function: communication from the ISTH SSC subcommittee on platelet physiology. J Thromb Haemost. 2021;19(12):3193‐3202. [DOI] [PubMed] [Google Scholar]
- 19. Marín‐Quílez A, García‐Tuñón I, Fernández‐Infante C, et al. Characterization of the platelet phenotype caused by a germline RUNX1 variant in a CRISPR/Cas9‐generated murine model. Thromb Haemost. 2021;121(9):1193‐1205. [DOI] [PubMed] [Google Scholar]
- 20.xGen Exome Research Panel v2 targets. 2020. Accessed November 20, 2021. https://sfvideo.blob.core.windows.net/sitefinity/docs/default-source/flyer/idt_xgen-exome-research-panel-v2.pdf?sfvrsn=fda61707_10.
- 21. Tilleman L, Heindryckx B, Deforce D, Van Nieuwerburgh F. Pan‐cancer pharmacogenetics: targeted sequencing panels or exome sequencing? Pharmacogenomics. 2020;21(15):1073‐1084. [DOI] [PubMed] [Google Scholar]
- 22. Richards S, Aziz N, Bale S, et al. Standards and guidelines for the interpretation of sequence variants: a joint consensus recommendation of the American college of medical genetics and genomics and the association for molecular pathology. Genet Med. 2015;17(5):405‐424. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Kopanos C, Tsiolkas V, Kouris A, et al. VarSome: the human genomic variant search engine. Bioinformatics. 2019;35(11):1978‐1980. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Lin JJC, Eppinga RD, Warren KS, McCrae KR. Human tropomyosin isoforms in the regulation of cytoskeleton functions. Adv Exp Med Biol. 2008;644:201‐222. [DOI] [PubMed] [Google Scholar]
- 25. Abouhamed M, Reichenberg S, Robenek H, Plenz G. Tropomyosin 4 expression is enhanced in dedifferentiating smooth muscle cells in vitro and during atherogenesis. Eur J Cell Biol. 2003;82(9):473‐482. [DOI] [PubMed] [Google Scholar]
- 26. Pecci A, Gresele P, Klersy C, et al. Eltrombopag for the treatment of the inherited thrombocytopenia deriving from MYH9 mutations. Blood. 2010;116(26):5832‐5837. [DOI] [PubMed] [Google Scholar]