

Abstract
The largest group of viruses in the Baltimore classification system comprises viruses with a positive‐sense, single‐stranded RNA genome. Once the viral genome is released into the cytoplasm of a specific host cell following virus entry, it functions directly as an mRNA, and the virus‐encoded proteins that are essential for genome replication are produced by the translation apparatus of the host cell. The positive‐sense genome is replicated in two stages, initially the positive strand is copied to make a negative‐sense RNA, which then functions as the template for transcription of many new positive‐sense genomes. Virus infections can be detected at different stages throughout the infection cycle for diagnostic and scientific purposes. Here, the advantages and disadvantages of some of the relevant methods for genome detection will be briefly reviewed with special emphasis on techniques allowing strand‐specific RNA detection. Furthermore, tools of the future are considered.
Keywords: Plus‐stranded RNA viruses, methods, genome, replication, RT‐PCR, strand‐specific detection
Viruses are small parasitic particles found in many shapes and sizes. The virus particle, also known as a virion, comprises genomic material, in the form of single‐stranded (ss) or double‐stranded (ds) RNA or DNA, surrounded by a coat of virus‐encoded proteins called a capsid. Some viruses are enveloped, meaning that the genome is surrounded by a lipid membrane with viral proteins embedded. In order to propagate, viruses depend on infecting specific host cells to exploit their metabolic and biosynthetic machineries for the production of virus‐encoded proteins, to achieve viral genome replication and for the assembly of new virions. Protein production requires translation of the positive‐sense mRNA, but because viruses do not all have the same type of genomic material, the production of these mRNAs happens in different ways. The mode of mRNA synthesis is the basis of the Baltimore virus classification system, which has seven classes, I‐VII [1]. Viruses, belonging to class IV, have a single‐stranded positive‐sense RNA ((+)‐RNA) genome, which can be translated directly as mRNA or used to produce more mRNA via a negative‐sense RNA ((−)‐RNA) intermediate. This is the largest group of viruses, comprising both bacteriophages, like Qβ and MS2 [2], and eukaryotic viruses such as hepatitis C virus (HCV), poliovirus (PV) and the coronaviruses, including the recently discovered severe acute respiratory syndrome coronavirus 2 (SARS‐CoV‐2) [3, 4, 5].
The life cycle of (+)‐RNA viruses
In order to establish a successful infection, each type of virus has specific requirements from a potential host cell. On the cell surface, specific protein structures or receptors are necessary for virus entry, while intracellular host cell factors may be required for the production of new virus particles. Some viruses use additional co‐receptors for entry, further increasing the host specificity of the virus. Some types of virus have a broader range in host organisms than others. For example, SARS‐CoV‐2 requires expression of the specific host cell surface receptor angiotensin‐converting enzyme 2 (ACE2) and co‐receptor NRP1, which are both highly conserved among mammals [6, 7], whereas the Qβ bacteriophage is specific for Escherichia coli strains that have surface‐exposed F‐pili that the phage can attach to [8]. Viruses, specific for animal host cells, can enter the cell via different mechanisms of endocytosis, often mediated through binding of the virus particle to a surface receptor on the host cell [9, 10, 11].
After successful entry of a virus into a host cell, the viral RNA genome needs to be released (Fig. 1, step 1). This can happen when the lipid envelope of enveloped viruses like dengue virus (DENV) and HCV fuse with the endosomal host membrane, creating a pore, through which the genome gains access to the cytosol [12, 13]. In contrast, (+)‐RNA viruses without a lipid envelope must rely on a strategy that does not involve membrane fusion for genome liberation. PV is a non‐enveloped virus that enters the animal cell via clathrin‐dependent endocytosis like some of the enveloped viruses, but the PV genome is then uncoated in several steps through conformational changes resulting in expansion of the capsid induced by binding to the poliovirus receptor [14].
Fig. 1.

Life cycle of positive‐sense RNA viruses. After virus entry into the host cell, the viral genome is released (blue bar, step 1) and virus‐encoded proteins, including the RdRp, are produced by translation of the RNA (different colours and shapes, step 2). The genome also functions as template for transcription of a complementary negative‐sense RNA molecule (red bar, step 3), from which many new positive‐sense genomes are made (step 4). These genomes are either used for further protein production (step 5) or packaged into new virions (step 6).
The genomes of positive‐sense RNA viruses function as mRNA for translation in addition to acting as templates for genome replication. Thus, these viruses each encode their own viral RNA‐dependent RNA polymerase (RdRp), which must be produced, before genome replication is possible [15]. The viral genome is translated by the host translation apparatus into one or more polyproteins, which are subsequently processed into mature products, generally via cleavage by proteases. As an example, the PV genome is translated into a single polyprotein, which is cleaved into multiple structural and non‐structural proteins mainly by viral proteases, whereas the translation of the larger genomes of coronaviruses, like SARS‐CoV‐2, is more complex and involves ribosome frameshifts between different open reading frames and production of subgenomic RNA transcripts [16, 17].
When the necessary proteins have been produced (Fig. 1, step 2) and the viral replication machinery assembled, genome replication can start. Often, host proteins form part of the viral RdRp complex to assist various steps during replication [18]. In eukaryotic host cells, the genome replication takes place at intracellular membrane compartments remodelled from host organelles, including the Golgi apparatus and the endoplasmic reticulum (ER) [19]. During replication, the (+)‐stranded viral genome is first transcribed into a complementary (−)‐strand replicative intermediate (Fig. 1, step 3), from which, a large number of new (+)‐RNA strands can be produced in 10‐100 times excess [18] (Fig. 1, step 4). These are initially used for additional protein production and later packaged into new virions [20] (Fig. 1, step 5+6).
The viral capsids usually only contain one or a few different structural proteins, and the assembly of new virions involves encapsidation of the newly synthesized (+)‐RNA genomes, which can happen via different mechanisms. As an example, icosahedral capsids are formed by self‐assembly of the viral proteins during infection by the non‐enveloped PV [21]. The genomes of the enveloped coronaviruses are packaged by nucleocapsid proteins, which bind to the RNA, oligomerize and form a helical nucleocapsid [22]. Assembly of enveloped virions is facilitated by the membrane compartments of the eukaryotic host cell, in which the viral envelope proteins are embedded. This is at the Golgi apparatus and ER for coronaviruses and HCV, respectively [19].
Release of the progeny virions usually happens through exocytosis, budding or lysis of the host cell, and each type of virus is not restricted to use just one pathway. Non‐enveloped viruses, like PV, are usually categorized as lytic viruses, because the new virions primarily exit the host cells by inducing rupture of the cell. This is, however, not always the case. Nonlytic mechanisms of virus exit have been observed, for example enteroviruses PV and A71 using the secretory autophagy pathway [23]. Enveloped viruses, like DENV and coronaviruses, can be trafficked between host cell membrane compartments and either exit by exocytosis or budding directly from the plasma membrane [24, 25].
VIRAL GENOME DETECTION
Detection of viral infections is used in many different settings, hence, a variety of methods for this purpose are available. It is widely employed in research and diagnostic laboratories in order to get a better understanding of the infection process of different viruses, but it is also applied in commercial laboratories, for example for production of viral vectors, which are often used in gene therapy and vaccine development. During the ongoing SARS‐CoV‐2 pandemic, it has become increasingly clear to everyone just how important the development of accurate and reproducible tools for detection of viral infections are in a diagnostic setting, and why extensive knowledge about a virus, gained through research, can help development of vaccines and antiviral drugs.
Viruses can be detected at different steps during their infection cycle. A common way of estimating the infectivity of a virus is quantification of viral particles using a plaque assay. Here, the sample containing the virus is spread onto a monolayer of appropriate host cells on a plate, and the number of plaque‐forming units (PFUs), caused by individual infectious particles in adequately diluted samples, is counted. In general, this is a very sensitive method for detection of infectious particles, because a single infectious particle should give rise to one PFU [26]. Enzyme‐linked immunosorbent assay (ELISA) is another widely used technique for detection of viruses, where a combination of virus‐specific primary antibodies and enzyme‐linked secondary antibodies are used to determine the presence of viral proteins [27]. Virus‐specific antibodies are also often used in immunofluorescence assays (IFA), for example for visualization of virus antigens in different tissues by microscopy [28]. ELISA and IFA are characterized by a high specificity, but sometimes suffer from limited sensitivity depending on the binding affinity between antibody and antigen among others. Although very useful, the methods mentioned above do not provide specific information about the genome replication process, which is a characteristic property of an active, viral infection. For this purpose, methods detecting the RNA genome directly can be used.
RT‐PCR
The current golden standard for detection of a viral (+)‐RNA genome is reverse transcription polymerase chain reaction (RT‐PCR)‐based assays, which have been in use since the late 1980s [29]. RT‐PCR techniques are often used in both research and clinical settings for RNA virus detection, but it is also a popular method in gene expression analyses, where the level of the different mRNAs in a cell can be monitored and quantified [30].
Very briefly, the extracted RNA in a sample is first subjected to first‐strand synthesis of the complementary DNA (cDNA) strand by reverse transcription (RT) using an RNA‐dependent DNA polymerase, also known as reverse transcriptase. Priming of the reverse transcription can be done with random hexamers, when strand‐specificity of the assay is not needed. In this case, first‐strand cDNA copies are produced from the total extracted RNA, while the specificity of the assay is achieved in the subsequent exponential cDNA amplification by polymerase chain reaction (PCR) using specifically designed primers [31]. These primers can be designed in different ways, depending on the scope of the analysis. For example, when using RT‐PCR for diagnosing viral infections, the PCR primer pair is designed to anneal to a specific region of the viral genome. If this region is conserved among different variants of a virus, the method detects multiple variants without discrimination. If, however, the primers are designed to amplify a variable genomic region, it is possible to only detect a specific variant of the virus [32]. When using RT‐PCR for diagnosing infection by novel viruses like SARS‐CoV‐2, it can be important to target more than one conserved region to avoid losing sensitivity of the assay, if the virus changes in the target region [33].
Due to the exponential amplification of the starting material that happens during the PCR step, RT‐PCR is considered a very sensitive method for viral RNA detection. This, however, is not only an advantage, since it can result in more false‐positive tests because even a small amount of contamination of the sample (in principle as little as one molecule), could be amplified and cause a positive result. This means that primer specificity and the avoidance of sample contamination are two critical aspects of the RT‐PCR procedure. Combining the RT‐PCR with a nested PCR, where a second round of PCR is performed with a new primer pair amplifying an internal region of the amplicon of the first round, is a widely used strategy in research settings to increase both specificity and sensitivity of the assay [34, 35]. However, this technique is not usually applied in clinical diagnostics, because it requires more handling of the samples, which increases the risk of cross‐contamination and false‐positive results.
RT‐PCR is not in itself considered quantitative, but only a means to amplify the target RNA to a detectable level. However, if an assessment of gene expression levels via mRNA or the amount of viral RNA is required, the reverse transcription can be combined with a quantitative PCR (qPCR), also known as real‐time qPCR, using a dye or DNA probes for detection. This could, for example, be a non‐specific fluorescent dye like SYBR green, or specifically designed fluorescent TaqMan probes, which can increase the specificity of the assay [36]. In the clinical laboratory, a standard panel of well‐characterized reference material is included to improve quantitation [37].
When RT‐qPCR is used to detect viral infections in clinical samples, the samples need to be pre‐treated in order to get rid of sample components that could potentially inhibit the PCR. The proteins immunoglobulin G and haemoglobin in blood samples have, for example, been shown to inhibit PCR [38]. Removal of these components can be both time‐consuming and labour‐intensive, which is especially a problem when rapid and effective diagnosis is essential for controlling a virus outbreak in areas with limited resources. With this in mind, direct RT‐qPCR assays (dirRT‐qPCR) have been developed specifically to avoid this pre‐treatment. As an example, Li et al. developed an optimized dirRT‐qPCR assay for detection of ZIKV [39]. The authors tested several DNA polymerases to see which one was the least affected by the PCR inhibitors in the clinical sample, and what kinds of PCR enhancers like dithiothreitol (DTT) and KCl were the most effective. They ended up with an assay that is able to diagnose a ZIKV infection, in different biological samples, within 2 h [39]. Similar assays have been developed for detection of SARS‐CoV‐2 during the COVID‐19 pandemic, where supplies were suddenly limited worldwide and fast and accurate diagnostic tools were critical [40].
STRAND‐SPECIFIC GENOME DETECTION
Most RT‐PCR assays used for RNA detection are not designed to be strand‐specific, and therefore do not distinguish between the (+)‐RNA genome of the virus and the complementary negative‐sense (−)‐RNA. Yet, the presence of (−)‐RNA is considered a hallmark of an active virus infection, because it is specifically produced during the replication of the viral genome, while virus particles only contain the (+)‐stranded genome. Strand‐specific genome detection by RT‐PCR is therefore widely used to study viral replication and infectivity. Achieving this specificity, however, is not trivial, since the mere use of a strand‐specific primer for reverse transcription is rarely sufficient [41]. One of the main problems, when aiming for specific detection of the (−)‐strand, is referred to as ‘false’ priming events, where non‐specific negative‐strand cDNA molecules are produced during reverse transcription in the absence of RT primers. These events could be caused by contaminating nucleic acids functioning as primers or self‐priming due to RNA secondary structures. A model for a possible self‐priming event during detection of DENV genome was proposed, where the RNA genome template folds upon itself creating a double‐stranded region suitable for reverse transcription initiation [41, 42]. Because the (+)‐strand is present at much higher levels than the (−)‐strand during a viral infection, false priming events affect specific detection of the latter more significantly.
Strand‐specific RT‐PCR
A very popular approach to circumvent this problem and improve the strand‐specificity of RT‐PCR‐based assays is referred to as tagged RT‐PCR (Fig. 2A). Here, the use of a single strand‐specific RT primer (marked in green in Fig. 2A) with a 5′ tag‐sequence, unrelated to the viral genome, results in tagged first‐strand cDNA molecules that can subsequently be amplified during PCR with a primer pair consisting of one tag‐specific and one genome‐specific primer (marked in orange in Fig. 2A). This ensures that any cDNA molecules resulting from ‘false’ priming cannot be amplified and cause a false‐positive detection result, because they do not contain the tag‐sequence [42]. An important aspect of this strategy is to avoid carry‐over of the tagged RT primer to the PCR amplification step, since that could result in unspecific, tagged cDNA fragments again causing false‐positive results or, when doing a qPCR, an overestimation of the amount of (−)‐strand [42]. Although both silica‐based purification of cDNA and treatments with exonuclease I prior to PCR have proven beneficial, it is just as important to limit the amount of RT primer to what is absolutely necessary, because it can be hard to remove unincorporated primers completely [43, 44]. Tagged RT‐PCR assays have been developed for many different (+)‐RNA viruses, with just a few examples being human enteroviruses, ZIKV and SARS‐CoV‐2 [42, 45, 46]. Further improvement of strand‐specificity of tagged RT‐PCR assays has been observed by using a thermostable reverse transcriptase, for example the recombinant Thermus thermophilus (rTth) DNA polymerase or SuperScript RT‐III, for cDNA amplification. This improvement is likely due to a higher reaction temperature limiting potential RNA secondary structures, which could otherwise give rise to self‐priming events [43, 44].
Fig. 2.
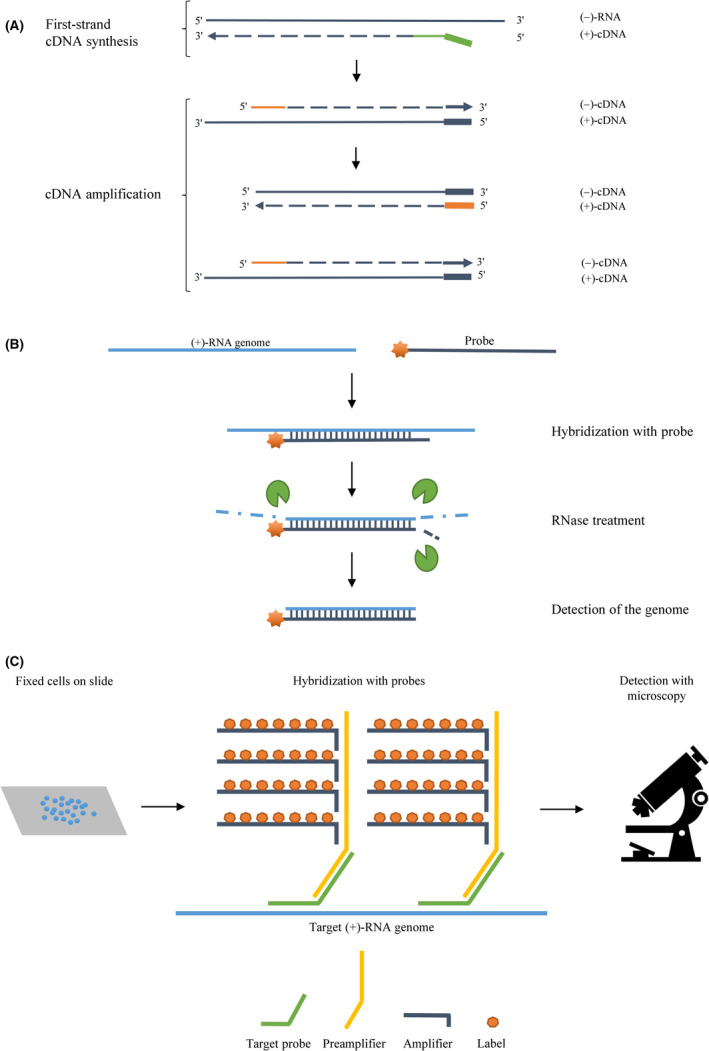
Illustrations of the principles behind strand‐specific tagged RT‐PCRs, RNase protection assays and in situ hybridization assays with branched DNA probes. (A) During tagged RT‐PCR, first‐strand cDNA synthesis is carried out using a strand‐specific reverse transcription primer (green) with a non‐viral 5′ tag‐sequence (bold). One tag‐specific and one sequence‐specific primer (both in orange; tag‐specific primer in bold) are used for subsequent cDNA amplification by PCR. Dashed lines indicate newly synthesized DNA, and the sense of the nucleic acids strands is shown on the right. (B) In an RNase protection assay, a fluorescently or radioactively labelled probe (dark blue with an orange star) hybridizes to the target RNA (light blue). RNases (green) degrade single‐stranded regions, while the labelled double‐stranded region is detected. (C) During in situ hybridization with branched DNA probes, cells are fixed on slides, and the intracellular target RNA (light blue) is first hybridized with the target probe (green) and subsequently the preamplifier (yellow). Lastly, amplifiers with multiple labels (dark blue with orange heptagons) are added to hybridize with the preamplifier and visualization is possible using the appropriate type of microscopy.
RNase protection assay
The RNase protection assay (RPA) is an example of another method for strand‐specific genome detection (Fig. 2B). Here, a strand‐specific probe (marked in dark blue in Fig. 2B) is designed to bind the single‐stranded genome (marked in light blue in Fig. 2B) and thus form a region of double‐stranded RNA. Subsequent treatment with RNases degrades the single‐stranded RNA, and the protected RNA can be visualized via a fluorescent or radioactive label attached to the probe (marked with an orange star in Fig. 2B). Novak and Kirkegaard [47] developed an improved version of this method, which was shown to allow sensitive, strand‐specific RNA detection, even when the complementary strand is in large excess. This is very important for detection of the (−)‐strand replication intermediate during active infection by (+)‐RNA viruses, since the (+)‐strand is present at higher levels. A round of hybridization of cytoplasmic RNA without probes was included in the beginning of the assay, resulting in all (−)‐strands hybridizing to the complementary (+)‐strands. They subsequently removed all excess (+)‐strand by RNase digestion, which left an equal amount of (+)‐ and (−)‐strands and allowed the radioactive probe added later to hybridize to the (−)‐strand without significant interference by the (+)‐strand [47].
In situ hybridization assay
In situ hybridization (ISH) techniques are commonly used for strand‐specific in situ visualization of RNA in cells, tissue samples, cytological preparations and even whole organisms. For this technique, a detectable strand‐specific probe can be designed as used for RPAs. The most used techniques are fluorescent in situ hybridization (FISH), where the probes are fluorescently labelled and visualized in a fluorescence microscope, and chromogenic in situ hybridization (CISH), in which the probes are usually labelled with biotin or digoxigenin and visualized by bright‐field microscopy [48]. For specific detection of an RNA species with relatively low abundance, like the (−)‐strand, branched DNA (bDNA) probes have been used to increase the signal (Fig. 2C). A set of bDNA probes consist of several nucleic acid fragments (marked in different colours in Fig. 2C) of which the first one, called the target (or capture) probe, specifically binds the target RNA sequence. The second probe, called the preamplifier, binds the target probe and then multiple fluorescently labelled probes, called amplifiers, bind the preamplifier, which results in a stronger localized signal [49]. Liu et al. combined this technique with immunofluorescence staining of viral proteins to examine viral RNA and proteins simultaneously during HCV infection using confocal microscopy. This allowed the authors to test the effect of antiviral drugs on the replication of HCV RNA [20].
FUTURE PERSPECTIVES
Even though, the RNA detection strategies described here have been very useful and optimized in many ways, they still have their limitations, for example by being labour‐intensive and/or requiring specialized instrumentation as well as trained personnel. New methods are still being developed, especially aiming for faster and simpler diagnostic tools, which are becoming increasingly necessary with the emergence of novel viral outbreaks as seen worldwide with SARS‐CoV‐2.
Zhou et al. have recently developed a viral RNA detection assay utilizing specifically designed DNA nanoswitches, where the DNA binds viral RNA and undergoes a conformational change, which is detected by gel electrophoresis due to a shift in migration pattern [50]. This assay can work without the use of any enzymes, which would usually increase both cost and complexity of assays due to strict conditions for use and storage. Even though the assay showed promising results when tested on ZIKV, DENV and SARS‐CoV‐2, further optimization is needed to increase sensitivity without an RNA extraction step or even an isothermal preamplification step, which would require the use of enzymes [50]. In another line of advancements, the clustered regularly interspaced palindromic repeats (CRISPR)/CRISPR‐associated enzyme (Cas) system, has been used as a basis for newly developed RNA detection methods [51]. One of these methods is called specific high‐sensitivity enzymatic reporter unlocking (SHERLOCK). Briefly, a CRISPR RNA (crRNA) is designed to guide the Cas13 enzyme to cleave a virus‐specific RNA sequence, but because Cas13 has promiscuous RNase activity, nearby RNA molecules are also cleaved by the activated enzyme. This is exploited by designing reporter RNA probes with a fluorophore in one end and a quencher in the other. Hence, a fluorescent signal is detected when the reporter molecule is cleaved to separate the fluorophore from the quencher [52]. An optimized version of the method, called SHERLOCK version 2 (SHERLOCKv2), was developed with increased sensitivity and adapted for lateral flow detection, which gives it great potential in diagnostic settings, where access to special laboratory equipment is limited [53]. The CRISPR‐based methods also have the potential to be optimized for strand‐specific detection in the future, allowing specific detection of actively replicating viruses.
The continued development of new strategies can improve virus detection in clinical settings as well as in research environments, where the techniques are still needed for further advancing the knowledge of both well‐known and novel viruses, which is crucial for the development of potential antiviral drugs and vaccines.
CONFLICT OF INTEREST
All authors declare no potential conflict of interest.
We would like to express our special thanks to professor Graham J. Belsham, Department of Veterinary and Animal Sciences at University of Copenhagen, Denmark, for his continuous support and for his valuable comments on this manuscript.
Warncke SR, Knudsen CR. Detection methods targeting the positive‐ and negative‐sense RNA transcripts from plus‐stranded RNA viruses. APMIS. 2022; 130: 284–292.
REFERENCES
- 1. Baltimore D. Expression of animal virus genomes. Bacteriol Rev. 1971;35(3):235–41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Callanan J, Stockdale SR, Shkoporov A, Draper LA, Ross RP, Hill C. RNA phage biology in a metagenomic era. Viruses. 2018;10(7):386. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Choo QL, Kuo G, Weiner AJ, Overby LR, Bradley DW, Houghton M. Isolation of a cDNA clone derived from a blood‐borne non‐A, non‐B viral hepatitis genome. Science. 1989;244(4902):359–62. [DOI] [PubMed] [Google Scholar]
- 4. Racaniello VR, Baltimore D. Cloned poliovirus complementary DNA is infectious in mammalian cells. Science. 1981;214(4523):916–9. [DOI] [PubMed] [Google Scholar]
- 5. Wenjie T, Xiang Z, Xuejun M, Wenling W, Peihua N, Wenbo X, et al. A novel coronavirus genome identified in a cluster of pneumonia cases—Wuhan, China 2019–2020. China CDC Weekly. 2020;2(4):61–2. [PMC free article] [PubMed] [Google Scholar]
- 6. Damas J, Hughes GM, Keough KC, Painter CA, Persky NS, Corbo M, et al. Broad host range of SARS‐CoV‐2 predicted by comparative and structural analysis of ACE2 in vertebrates. Proc Natl Acad Sci USA. 2020;117(36):22311–22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Sun K, Gu L, Ma L, Duan Y. Atlas of ACE2 gene expression reveals novel insights into transmission of SARS‐CoV‐2. Heliyon. 2021;7(1):e05850. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Loeb T. Isolation of a bacteriophage specific for the F plus and Hfr mating types of Escherichia coli K‐12. Science. 1960;131(3404):932–3. [DOI] [PubMed] [Google Scholar]
- 9. Richard AS, Zhang A, Park SJ, Farzan M, Zong M, Choe H. Virion‐associated phosphatidylethanolamine promotes TIM1‐mediated infection by Ebola, dengue, and West Nile viruses. Proc Natl Acad Sci USA. 2015;112(47):14682–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Raj VS, Mou H, Smits SL, Dekkers DHW, Müller MA, Dijkman R, et al. Dipeptidyl peptidase 4 is a functional receptor for the emerging human coronavirus‐EMC. Nature. 2013;495(7440):251–4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Wang H, Yang P, Liu K, Guo F, Zhang Y, Zhang G, et al. SARS coronavirus entry into host cells through a novel clathrin‐ and caveolae‐independent endocytic pathway. Cell Res. 2008;18(2):290–301. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. van der Schaar HM, Rust MJ, Chen C, van der Ende‐Metselaar H, Wilschut J, Zhuang X, et al. Dissecting the cell entry pathway of dengue virus by single‐particle tracking in living cells. PLoS Pathog. 2008;4(12):e1000244. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Blanchard E, Belouzard S, Goueslain L, Wakita T, Dubuisson J, Wychowski C, et al. Hepatitis C virus entry depends on clathrin‐mediated endocytosis. J Virol. 2006;80(14):6964–72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Butan C, Filman DJ, Hogle JM. Cryo‐electron microscopy reconstruction shows poliovirus 135S particles poised for membrane interaction and RNA release. J Virol. 2014;88(3):1758–70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Salonen A, Ahola T, Kääriäinen L. Viral RNA replication in association with cellular membranes. Curr Top Microbiol Immunol. 2005;285:139–73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Toyoda H, Nicklin MJH, Murray MG, Anderson CW, Dunn JJ, Studier FW, et al. A second virus‐encoded proteinase involved in proteolytic processing of poliovirus polyprotein. Cell. 1986;45(5):761–70. [DOI] [PubMed] [Google Scholar]
- 17. Kim D, Lee JY, Yang JS, Kim JW, Kim VN, Chang H. The architecture of SARS‐CoV‐2 transcriptome. Cell. 2020;181(4):914–21 e10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Ahlquist P, Noueiry AO, Lee WM, Kushner DB, Dye BT. Host factors in positive‐strand RNA virus genome replication. J Virol. 2003;77(15):8181–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Zhang Z, He G, Filipowicz NA, Randall G, Belov GA, Kopek BG, et al. Host lipids in positive‐strand RNA virus genome replication. Front Microbiol. 2019;10:286. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Liu D, Tedbury PR, Lan S, Huber AD, Puray‐Chavez MN, Ji J, et al. Visualization of positive and negative sense viral RNA for probing the mechanism of direct‐acting antivirals against hepatitis C virus. Viruses. 2019;11(11). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Adeyemi OO, Sherry L, Ward JC, Pierce DM, Herod MR, Rowlands DJ, et al. Involvement of a nonstructural protein in poliovirus capsid assembly. J Virol. 2019;93(5). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Chang CK, Chen CM, Chiang MH, Hsu YL, Huang TH. Transient oligomerization of the SARS‐CoV N protein–implication for virus ribonucleoprotein packaging. PLoS One. 2013;8(5):e65045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Su W, Huang S, Zhu H, Zhang B, Wu X. Interaction between PHB2 and enterovirus A71 VP1 induces autophagy and affects EV‐A71 infection. Viruses. 2020;12(4). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Savidis G, McDougall WM, Meraner P, Perreira JM, Portmann JM, Trincucci G, et al. Identification of zika virus and dengue virus dependency factors using functional genomics. Cell Rep. 2016;16(1):232–46. [DOI] [PubMed] [Google Scholar]
- 25. Ng ML, Tan SH, See EE, Ooi EE, Ling AE. Proliferative growth of SARS coronavirus in Vero E6 cells. J Gen Virol. 2003;84(Pt 12):3291–303. [DOI] [PubMed] [Google Scholar]
- 26. Baer A, Kehn‐Hall K. Viral concentration determination through plaque assays: using traditional and novel overlay systems. J Vis Exp. 2014;93:e52065. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Clark MF, Adams AN. Characteristics of the microplate method of enzyme‐linked immunosorbent assay for the detection of plant viruses. J Gen Virol. 1977;34(3):475–83. [DOI] [PubMed] [Google Scholar]
- 28. Zhou J, Chu H, Li C, Wong B‐Y, Cheng Z‐S, Poon V‐M, et al. Active replication of Middle East respiratory syndrome coronavirus and aberrant induction of inflammatory cytokines and chemokines in human macrophages: implications for pathogenesis. J Infect Dis. 2014;209(9):1331–42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Gama RE, Horsnell PR, Hughes PJ, North C, Stanway G, Bruce CB, et al. Amplification of rhinovirus specific nucleic acids from clinical samples using the polymerase chain reaction. J Med Virol. 1989;28(2):73–7. [DOI] [PubMed] [Google Scholar]
- 30. Etienne W, Meyer MH, Peppers J, Meyer RA Jr. Comparison of mRNA gene expression by RT‐PCR and DNA microarray. Biotechniques. 2004;36(4):618–20, 22, 24‐6. [DOI] [PubMed] [Google Scholar]
- 31. Zheng Z, Ke X, Wang M, He S, Li Q, Zheng C, et al. Human microRNA hsa‐miR‐296‐5p suppresses enterovirus 71 replication by targeting the viral genome. J Virol. 2013;87(10):5645–56. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Nunes ARD, Alves BEB, Pereira HWB, Nascimento YM, Morais IC, Fernandes JV, et al. Improved reverse transcription‐polymerase chain reaction assay for the detection of flaviviruses with semi‐nested primers for discrimination between dengue virus serotypes and Zika virus. Mem Inst Oswaldo Cruz. 2018;113(5):e170393. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Peñarrubia L, Ruiz M, Porco R, Rao SN, Juanola‐Falgarona M, Manissero D, et al. Multiple assays in a real‐time RT‐PCR SARS‐CoV‐2 panel can mitigate the risk of loss of sensitivity by new genomic variants during the COVID‐19 outbreak. Int J Infect Dis. 2020;97:225–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Feronato C, Leme RA, Diniz JA, Agnol AMD, Alfieri AF, Alfieri AA. Development and evaluation of a nested‐PCR assay for Senecavirus A diagnosis. Trop Anim Health Prod. 2018;50(2):337–44. [DOI] [PubMed] [Google Scholar]
- 35. Pfeffer M, Linssen B, Parke MD, Kinney RM. Specific detection of chikungunya virus using a RT‐PCR/nested PCR combination. J Vet Med B Infect Dis Vet Public Health. 2002;49(1):49–54. [DOI] [PubMed] [Google Scholar]
- 36. Plaskon NE, Adelman ZN, Myles KM. Accurate strand‐specific quantification of viral RNA. PLoS One. 2009;4(10):e7468. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Jorgensen PA, Neuwald PD. Standardized hepatitis C virus RNA panels for nucleic acid testing assays. J Clin Virol. 2001;20(1–2):35–40. [DOI] [PubMed] [Google Scholar]
- 38. Al‐Soud WA, Jönsson LJ, Râdström P. Identification and characterization of immunoglobulin G in blood as a major inhibitor of diagnostic PCR. J Clin Microbiol. 2000;38(1):345–50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Li L, He J‐A, Wang W, Xia Y, Song LI, Chen Z‐H, et al. Development of a direct reverse‐transcription quantitative PCR (dirRT‐qPCR) assay for clinical Zika diagnosis. Int J Infect Dis. 2019;85:167–74. [DOI] [PubMed] [Google Scholar]
- 40. Byrnes SA, Gallagher R, Steadman A, Bennett C, Rivera R, Ortega C, et al. Multiplexed and extraction‐free amplification for simplified SARS‐CoV‐2 RT‐PCR tests. Anal Chem. 2021;93(9):4160–5. [DOI] [PubMed] [Google Scholar]
- 41. Tuiskunen A, Leparc‐Goffart I, Boubis L, Monteil V, Klingstrom J, Tolou HJ, et al. Self‐priming of reverse transcriptase impairs strand‐specific detection of dengue virus RNA. J Gen Virol. 2010;91(Pt 4):1019–27. [DOI] [PubMed] [Google Scholar]
- 42. Bessaud M, Autret A, Jegouic S, Balanant J, Joffret ML, Delpeyroux F. Development of a Taqman RT‐PCR assay for the detection and quantification of negatively stranded RNA of human enteroviruses: evidence for false‐priming and improvement by tagged RT‐PCR. J Virol Methods. 2008;153(2):182–9. [DOI] [PubMed] [Google Scholar]
- 43. Craggs JK, Ball JK, Thomson BJ, Irving WL, Grabowska AM. Development of a strand‐specific RT‐PCR based assay to detect the replicative form of hepatitis C virus RNA. J Virol Methods. 2001;94(1–2):111–20. [DOI] [PubMed] [Google Scholar]
- 44. Chatterjee SN, Devhare PB, Lole KS. Detection of negative‐sense RNA in packaged hepatitis E virions by use of an improved strand‐specific reverse transcription‐PCR method. J Clin Microbiol. 2012;50(4):1467–70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Huits R, De Smet B, Grard G, Eggermont K, Minto‐Bain C, Jess N, et al. Detection of Zika virus replication in human semen by reverse‐transcription polymerase chain reaction targeting of antisense ribonucleic acid. J Infect Dis. 2020;222(2):319–23. [DOI] [PubMed] [Google Scholar]
- 46. Liao M, Wu J, Dai M, Li H, Yan N, Yuan R, et al. Rapid detection of SARS‐CoV‐2, replicating or non‐replicating, using RT‐PCR. Int J Infect Dis. 2021;104:471–3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Novak JE, Kirkegaard K. Improved method for detecting poliovirus negative strands used to demonstrate specificity of positive‐strand encapsidation and the ratio of positive to negative strands in infected cells. J Virol. 1991;65(6):3384–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Pfankuche V, Hahn K, Bodewes R, Hansmann F, Habierski A, Haverkamp A‐K, et al. Comparison of different in situ hybridization techniques for the detection of various RNA and DNA viruses. Viruses. 2018;10(7):384. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Player AN, Shen LP, Kenny D, Antao VP, Kolberg JA. Single‐copy gene detection using branched DNA (bDNA) in situ hybridization. J Histochem Cytochem. 2001;49(5):603–12. [DOI] [PubMed] [Google Scholar]
- 50. Zhou L, Chandrasekaran AR, Punnoose JA, Bonenfant G, Charles S, Levchenko O, et al. Programmable low‐cost DNA‐based platform for viral RNA detection. Sci Adv. 2020;6(39). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Ran FA, Hsu PD, Wright J, Agarwala V, Scott DA, Zhang F. Genome engineering using the CRISPR‐Cas9 system. Nat Protoc. 2013;8(11):2281–308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Gootenberg JS, Abudayyeh OO, Lee JW, Essletzbichler P, Dy AJ, Joung J, et al. Nucleic acid detection with CRISPR‐Cas13a/C2c2. Science. 2017;356(6336):438–42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Gootenberg JS, Abudayyeh OO, Kellner MJ, Joung J, Collins JJ, Zhang F. Multiplexed and portable nucleic acid detection platform with Cas13, Cas12a, and Csm6. Science. 2018;360(6387):439–44. [DOI] [PMC free article] [PubMed] [Google Scholar]