

Abstract
Aim
To assess the evolution of bulbar function in nusinersen‐treated spinal muscular atrophy type 1 (SMA1).
Method
This single‐centre retrospective study identified 24 patients (14 females and 10 males) with SMA1, treated with nusinersen between 2017 and 2020. We adapted and validated the Paediatric Functional Oral Intake Scale (p‐FOIS), which is an outcome measure to assess bulbar function. Analysis considered SMA1 subtype, nutritional support, and Children's Hospital of Philadelphia Infant Test of Neuromuscular Disorders (CHOP INTEND) and p‐FOIS scores at initiation of nusinersen treatment (baseline) and at 6, 12, and 24 months after initiation.
Results
The median age at baseline was 11 months (range 1 month–7 years 6 months). Median age at initiation of tube feeding was 8 months (range 0–2 years 2 months). Fourteen patients were tube fed at baseline. The median p‐FOIS score was 3 at baseline and 2 at 12 and 24 months. Four patients, all with type 1c SMA, remained orally fed at 24 months. Median CHOP INTEND scores increased from 32 at baseline to 42 at 12 and 24 months.
Interpretation
Impaired bulbar function persisted as a significant complication in most nusinersen‐treated patients with SMA1, in contrast to the improvement in motor abilities demonstrated in the majority. p‐FOIS allows for tracking of bulbar function progression and treatment response. Larger, prospective studies investigating the longer‐term impacts of nusinersen on bulbar function are warranted.
Abbreviations
- CHOP INTEND
Children's Hospital of Philadelphia Infant Test of Neuromuscular Disorders
- NIV
non‐invasive ventilation
- p‐FOIS
Paediatric Functional Oral Intake Scale
- SMA
spinal muscular atrophy
- SMA1
Spinal muscular atrophy type 1
- SMN
survival motor neuron
What this paper adds.
Feeding/swallowing difficulties persist and tend to deteriorate in nusinersen‐treated spinal muscular atrophy type 1 (SMA1).
The need for tube feeding increased in our cohort despite treatment with nusinersen.
SMA1 with the least severe phenotype, type 1c, is more likely to maintain bulbar function.
Spinal muscular atrophy (SMA) is caused by degeneration of alpha motor neurons in the spinal cord and is characterized by hypotonia, weakness, and muscle wasting. SMA is an autosomal recessive disorder caused by deletion, or less frequently other mutations, of the SMN1 gene, resulting in deficiency of the survival motor neuron (SMN) protein. Patients with SMA rely on the second form of the gene, SMN2, to produce sufficient SMN protein for survival. SMN2 differs from SMN1 by a single nucleotide, which results in the exclusion of exon 7 during transcription, producing approximately 10% of functional SMN protein. 1 , 2 , 3
Paediatric‐onset SMA is classically defined into three main subtypes, types 1 to 3, based on maximal achieved motor milestones and age at onset. SMA type 1 (SMA1), the most severe type, is defined as onset between birth and 6 months, and as patients who are never able to sit without support. SMA1 is further divided into subtypes 1a (neonatal presentation), 1b (symptom onset before 3 months), and 1c (onset between 3 months and 6 months of age). 4 However, recent therapeutic developments have resulted in significant improvement in outcomes, thereby altering these natural history phenotypes.
Nusinersen was the first approved disease‐modifying treatment for SMA. It is an antisense oligonucleotide drug, which modifies pre‐mRNA splicing to promote inclusion of SMN2 exon 7, leading to increased production of full‐length SMN protein. 5 In clinical trials, nusinersen was demonstrated to provide significant improvement in motor function and overall survival. 6 , 7
Among the several comorbidities that can affect patients with SMA, feeding and swallowing difficulties play a significant role, and regular assessments of nutrition and swallow form part of the standards of care. In patients with SMA1 a proactive approach is recommended if there are growth difficulties or an abnormal swallow due to bulbar dysfunction, with placement of tube feeding to prevent aspiration, respiratory infections, and poor weight gain. 8 The natural history of nutritional support requirements for SMA1 have been well established. 1 , 9 , 10 However, since the evolution of SMA1 phenotypes with the development of treatments such as nusinersen, there is increasing need for better understanding of the bulbar function trajectories for these patients. Although swallowing and feeding difficulties have been demonstrated to persist in infants with SMA1 treated with nusinersen, despite the improvements seen in motor function and survival, studies on the progress of bulbar function in children with SMA1 treated with nusinersen are still limited. 11 , 12 , 13 This is due to both the lack of validated tools to assess swallow function in SMA1 and to the limited availability of longitudinal data.
We modified the Functional Oral Intake Scale (FOIS), which is a tool to assess change in functional eating abilities in adult patients with stroke, so that it could be used in children of all ages, and assessed the interrater reliability and consensual and face validity of the adapted tool, termed the Paediatric Functional Oral Intake Scale (p‐FOIS). 14 In this study we aimed primarily to investigate the evolution of bulbar function in SMA1 treated with nusinersen by using the p‐FOIS, along with other aspects, including nutritional, motor, and respiratory outcomes.
METHOD
This was a single‐centre retrospective study of children with SMA1 treated with nusinersen between 2017 and 2020 at Great Ormond Street Hospital for Children, London, UK. Children received nusinersen as part of the Early Access Programme, and/or Managed Access Agreement, or compassionate use. The p‐FOIS, adapted from the FOIS for use in the paediatric population, was applied to this population with SMA1. The methods and results of the adaptation and validation of the p‐FOIS are reported in Appendix S1. Patients who had at least 24 months of nusinersen treatment were included. No presymptomatically treated children were included in this study.
Outcome measures
Medical records of included patients were reviewed for the following characteristics: date of birth, age at symptom onset, SMA type 1 subtype, SMN2 copy number and age at initiation of nusinersen treatment, non‐invasive ventilation (NIV) requirement, including NIV hours of use (as needed, nocturnal [including for naps], or >16 hours/day), and type of feeding (oral/nasogastric tube/gastrostomy). p‐FOIS (Table 1) was applied retrospectively to patients with SMA1 at baseline, and 6, 12, and 24 months after initiation of nusinersen treatment.
TABLE 1.
Paediatric Functional Oral Intake Scale (p‐FOIS)
|
CHOP INTEND scores
To measure functional motor outcomes, we used the Children's Hospital of Philadelphia Infant Test of Neuromuscular Disorders (CHOP INTEND). 15 CHOP INTEND scores were collected at baseline and at 6, 12, and 24 months after initiation of nusinersen treatment.
Statistical analysis
Groupwise medians and ranges were calculated from the data. The individual trajectories for p‐FOIS, motor function, and feeding characteristics were plotted.
Ethics approval
This study was reviewed and registered by the Clinical Audit Department at Great Ormond Street Hospital for Children. It was assessed that the work was in line with the criteria outlined by the NHS Health Research Authority for determining whether work was audit, service evaluation, or research, and therefore did not require any approval from research for ethics. It was confirmed that the work was conducted with sound principles in line with our hospital clinical audit policy.
RESULTS
Characteristics
We identified 24 infants with SMA type 1 treated with nusinersen for at least 24 months at our centre: three with type 1a, nine with type 1b, and 12 with type 1c. Patients' characteristics are summarized in Table 2. No deaths were reported in the cohort during the study period.
TABLE 2.
Patients' characteristics
| All patients | SMA type 1a | SMA type 1b | SMA type 1c | |
|---|---|---|---|---|
| Number of patients, n | 24 | 3 | 9 | 12 |
| Median age at initiation of nusinersen, year:month (range) | 0:11 (0:1–7:6) | 2:2 (0:1–4:3) | 0:4 (0:2–1:4) | 1:7 (0:8–7:6) |
| Median age at initiation of tube feeding, year:month (n, range) | 0:8 (20, 0–2:2) | 0:7 (3, 0–0:9) | 0:7 (9, 0:2–1:1) | 0:11 (8, 0:5–2:2) |
| Median age at initiation of NIV, year:month (n, range) | 0:8 (21, 0:2–3:6) | 0:11 (3, 0:8–1:0) | 0:6 (8, 0:2–0:7) | 1:6 (10, 0:6–3:6) |
| SMN2 copy, number, n | 23 | 3 | 9 | 11 |
| Two copies a | 17 (74) | 2 (67) | 9 (100) | 6 (55) |
| Three copies a | 6 (26) | 1 (33) | 0 (0) | 5 (45) |
| Baseline, n | 24 | 3 | 9 | 12 |
| Tube fed a | 14 (58) | 3 (100) | 5 (56) | 6 (50) |
| Nasogastric tube a | 11 (46) | 3 (100) | 4 (44) | 4 (33) |
| Gastrostomy a | 3 (13) | 0 (0) | 1 (11) | 2 (17) |
| 12 months after initiation, n | 24 | 3 | 9 | 12 |
| Tube fed a | 20 (83) | 3 (100) | 9 (100) | 8 (67) |
| Nasogastric tube a | 12 (50) | 3 (100) | 4 (44) | 5 (42) |
| Gastrostomy a | 8 (33) | 0 (0) | 5 (56) | 3 (25) |
| 24 months after initiation, n | 24 | 3 | 9 | 12 |
| Tube fed a | 20 (83) | 3 (100) | 9 (100) | 8 (67) |
| Nasogastric tube a | 5 (21) | 2 (67) | 0 (0) | 3 (25) |
| Gastrostomy a | 15 (63) | 1 (33) | 9 (100) | 5 (42) |
| Baseline, n | 24 | 3 | 9 | 12 |
| NIV use a | 13 (54) | 2 (67) | 5 (56) | 6 (50) |
| None a | 11 (46) | 1 (33) | 4 (44) | 6 (50) |
| As needed a | 0 (0) | 0 (0) | 0 (0) | 0 (0) |
| Nocturnal use a | 7 (29) | 0 (0) | 3 (33) | 4 (33) |
| >16 hours/day a | 6 (25) | 2 (67) | 2 (22) | 2 (17) |
| 12 months after initiation, n | 24 | 3 | 9 | 12 |
| NIV use a | 20 (83) | 3 (100) | 8 (89) | 9 (75) |
| None a | 4 (17) | 0 (0) | 1 (11) | 3 (25) |
| As needed a | 1 (4) | 0 (0) | 0 (0) | 1 (8) |
| Nocturnal use a | 13 (54) | 1 (33) | 6 (67) | 6 (50) |
| >16 hours/day a | 6 (25) | 2 (67) | 2 (22) | 2 (17) |
| 24 months after initiation, n | 24 | 3 | 9 | 12 |
| NIV use a | 21 (88) | 3 (100) | 8 (89) | 10 (83) |
| None a | 3 (13) | 0 (0) | 1 (11) | 2 (17) |
| As needed a | 2 (8) | 0 (0) | 0 (0) | 2 (17) |
| Nocturnal use a | 14 (58) | 1 (33) | 7 (78) | 6 (50) |
| >16 hours/day a | 5 (21) | 2 (67) | 1 (11) | 2 (17) |
Data are n (%) unless otherwise stated.
Abbreviation: NIV, non‐invasive ventilation.
Expressed as percentages of total number of available assessments at the specified time points.
Feeding characteristics
Fourteen patients, consisting of three out of three with type 1a, five out of nine with type 1b, and six out of 12 with type 1c, were tube fed at baseline. Twenty of the 24 patients were tube fed at 24 months after initiation: all patients with type 1a and 1b SMA and eight out of 12 patients with type 1c. Four patients, all with type 1c (two with two SMN2 copies and two with three SMN2 copies), remained orally fed at 24 months after initiation. Table 2 shows the feeding route by SMA1 subtype at baseline, and at time points after initiation of nusinersen.
Age at initiation of nusinersen treatment
Patients were grouped into the following age ranges when nusinersen treatment was initiated: younger than 6 months, 6–12 months, and older than 12 months.
Seven patients were started on nusinersen younger than 6 months of age: one with type 1a SMA and six with type 1b. Of these, four were orally fed at baseline and all were fed by gastrostomy at 24 months follow‐up. Seven patients, two with type 1b and five with type 1c, started on nusinersen between the ages of 6 and 12 months; all were tube fed at 24 months follow‐up. Ten patients were started on nusinersen older than 12 months of age: seven with type 1c, one with type 1b, and two with type 1a; all these patients were born more than 12 months before nusinersen approval in the UK. At 24‐months follow‐up, four of these patients remained fully orally fed, while four had gastrostomy and two had nasogastric tubes. Figure 1 shows feeding routes by SMA1 subtype and by age at initiation of nusinersen treatment.
FIGURE 1.
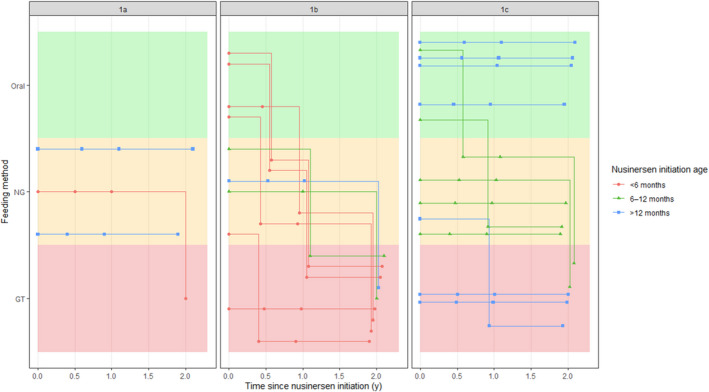
Progression of feeding characteristics by spinal muscular atrophy type 1 (SMA1) subtype and age at nusinersen initiation Abbreviations: GT, gastrostomy; NG, nasogastric tube.
Videofluoroscopy
Videofluoroscopy swallow study reports were available for eight patients: four with type 1b SMA and four with type 1c. For all patients these were performed after starting nusinersen and at variable, unspecified time points. All patients with type 1b and two with type 1c were reported as being at risk of aspiration on all consistencies. Another with type 1c was at risk of aspiration on residue but had strategies to adapt to their overall weakness. The remaining patient with type 1c had a weak swallow and reduced pharyngeal clearance; no aspiration was seen but there was extensive residue on thick puree consistencies, placing them at increased risk of aspiration on thick/sticky consistencies. No follow‐up videofluoroscopy swallow assessments were available for any of the eight patients.
p‐FOIS longitudinal scores
The median p‐FOIS declined from baseline (three at baseline, one at 6 months after initiation of nusinersen, two at 12 months, and two at 24 months). Median p‐FOIS for type 1a remained at 1 from baseline and at all follow‐up points. Median p‐FOIS for type 1b declined from 3 at baseline to 1 at 6 months, and 2 at 12 and 24 months respectively. For patients with type 1c, median p‐FOIS was 5 at baseline, 2 at 6 months after initiation of nusinersen, and 3 at 12 and 24 months. Six patients had a 1‐point improvement in their p‐FOIS score: five had an improvement of 1 to 2; one patient with type 1c had an improvement of 2 to 3. Figure 2 shows p‐FOIS scores for SMA1 subtypes at baseline and at time points after initiation of nusinersen.
FIGURE 2.
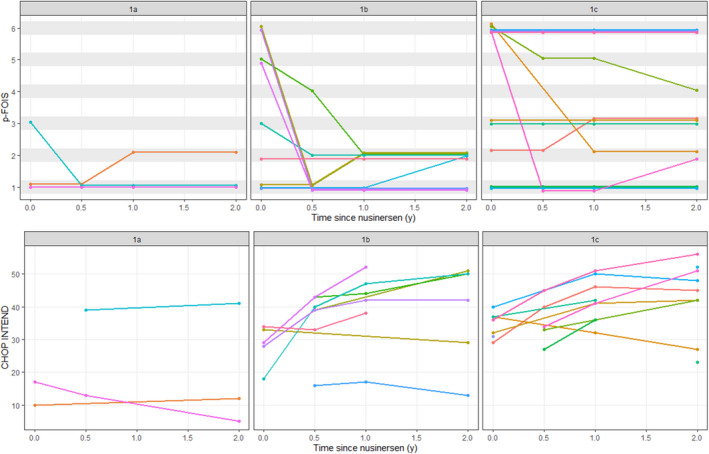
Paediatric Functional Oral Intake Scale (p‐FOIS) and Children's Hospital of Philadelphia Infant Test of Neuromuscular Disorders (CHOP INTEND) scores over time for individual patients by spinal muscular atrophy type 1 subtype
NIV characteristics
Median age at the start of NIV was 11 months for those with type 1a SMA, 6 months for type 1b, and 1 year 6 months for type 1c. Thirteen patients required NIV at baseline: seven required nocturnal use and six more than 16 hours per day. At 24 months after initiation of nusinersen 21 patients required NIV: 14 required nocturnal use, five required more than 16 hours per day, and two required ‘as needed’ use. Three patients, two with type 1c and one with type 1b, remained without any NIV use at 24 months after initiation of nusinersen. Hours of use per subtype at specified time points are listed in Table 2.
CHOP INTEND characteristics
CHOP INTEND scores were available for 14, 14, 15, and 18 patients at baseline, 6, 12, and 24 months after initiation of nusinersen respectively. Median CHOP INTEND scores across all available patients' observations increased from 32 at baseline, to 39, 42, and 42 at 6, 12, and 24 months after initiation of nusinersen respectively.
CHOP INTEND scores improved for two out of three patients with type 1a SMA. Six out of nine patients with type 1b had an improvement in their CHOP INTEND scores, and two showed minimal decline (16 at 6 months to 13 at 24 months for one patient; 33 at baseline to 29 at 24 months for the second patient). No CHOP INTEND data were available for one patient with type 1b SMA. Eight out of 12 patients with type 1c had an improvement in their CHOP INTEND scores. Three only had one CHOP INTEND score available so we were unable to assess change in score. One patient with type 1c had a decline in their CHOP INTEND score from 37 at baseline to 27 at 24 months after start of treatment.
DISCUSSION
Our study shows that feeding and swallowing difficulties persist and worsen in the majority of symptomatic patients with SMA1 treated with nusinersen. We found that there was an increase in the need for tube feeding with only a very small proportion of patients (n = 4), all having type 1c, remaining exclusively orally fed 24 months after treatment started. SMA standard of care recommends a proactive approach of tube feeding for those with growth failure or a failed swallow study. The fact that all patients were assessed and managed by the same speech and language therapy team, in line with the most updated standard of care, makes it more likely that the increased need for tube feeding demonstrated in our study reflects a real deterioration due to disease progression, rather than an effect of a more proactive management with time. 8 Although early treatment with nusinersen has been associated with reduced need for permanent assisted ventilation and improved motor function, there is limited evidence on the impact of early treatment on bulbar outcomes. 6 , 16 , 17 In this small cohort we did not find that younger age at initiation of nusinersen had an impact on maintaining bulbar function; however, of the seven patients who started treatment before 6 months of age, all were at the most severe end of the spectrum, type 1a and 1b, so it is difficult to draw clear conclusions. Our study did not include any presymptomatically treated children but preliminary evidence from the Nurture Study (NCT02386553) suggests that swallowing abilities are preserved among patients initiating nusinersen in the presymptomatic stage, with only a small proportion with two SMN2 copies requiring tube feeding. 18 This further highlights the urgency to implement neonatal screening programmes to identify affected patients and start treatment presymptomatically.
We used the 6‐point graded p‐FOIS which had excellent interrater reliability and validity, similar to previous studies in adults and paediatric cohorts and enabling tracking of functional abilities using a single measure across all time points. 14 , 19 , 20 Of note, we evaluated the p‐FOIS in children aged 0 to 16 years in the validation process, reflecting the full age range of SMA assessed in most paediatric settings.
Although the importance of regular nutritional assessments for children with SMA is well acknowledged, there is currently a lack of systematically collected agreed outcome measures of swallow and bulbar function for the population with SMA, thereby making multi‐centre collaborative studies more difficult. 8 An outcome tool, such as the p‐FOIS, enables monitoring of disease progression and treatment response. Another tool has recently been developed to assess swallowing abilities in patients with SMA type 1 and has been applied retrospectively to historical untreated patients with SMA1. However, this study did not assess bulbar function progression in treated patients with SMA1 and requires further validation. 21
We have shown that, overall, the p‐FOIS declines or remains static in SMA1 treated with nusinersen, reflecting worsening or lack of improvement in bulbar function, despite the majority showing improved motor abilities. Only six children had improvements in their p‐FOIS score of 1 point which does not reflect a substantial functional change in swallowing, as all remained tube fed. More importantly, we found that only a small proportion of patients with the least severe phenotype, 1c, were able to maintain a p‐FOIS score of 6, which encompasses total, age‐appropriate oral intake with no restrictions. An Australian study has demonstrated that all (n = 4) newly diagnosed patients with two SMN2 copy numbers (all type 1b) required gastrostomy after a median nusinersen treatment time of 30 months, while all newly diagnosed patients (n = 3) with three SMN2 copies (two type 1c, one type 1b) remained orally fed after a median treatment time of 28 months. 11 However, SMN2 copy number does not always predict response to treatment, as in our study two out of four patients with type 1c who remained completely orally fed had only two copies of SMN2. 2 , 12
Similar to previous studies, we observed that our population had an increase in the number of patients requiring nocturnal‐only NIV after 12 months of treatment, but this was followed by a stability with no additional patients requiring more than 16 hours of ventilator support. 22 , 23
Despite the observed effects on motor and respiratory function, nusinersen‐treated patients with SMA1 remain vulnerable to the significant comorbidity of bulbar impairment. Our findings of persistent feeding difficulties in nusinersen‐treated infants with SMA1 are consistent with other studies. 11 , 13 A comparison of natural history SMA1 controls to nusinersen‐treated patients with SMA1 would allow more conclusive evidence of the effect of nusinersen on bulbar function. However, given the significantly shortened life expectancy of historical untreated patients with SMA1, it is difficult to gather sufficient retrospective data on bulbar function for this cohort; therefore this was not performed in this study. Recent findings suggest that motor function benefits can continue to be obtained after the first year of nusinersen treatment, and even up to 3 years of treatment. 12 , 24 Therefore, longer follow‐up studies in larger SMA nusinersen‐treated cohorts may capture additional findings of nuisnersen's impact on bulbar function to those demonstrated in this study.
Post‐mortem studies have shown invariable and early brainstem motor nuclei abnormalities in SMA. Neuropathological studies have also documented that, similar to spinal cord motor neurons, high levels of SMN protein are developmentally expressed at early stages of fetal and postnatal development in brainstem nuclei, where they are required for their function and survival. 25 Additionally, magnetic resonance imaging abnormalities in bulbar muscles have been demonstrated. 26 , 27 Bulbar denervation alongside head posture due to weak neck muscles probably lead to the dysphagia and weak suck/swallow seen in SMA. 27 , 28 Interestingly, nusinersen concentrations have been reported as higher in the caudal than rostral regions in autopsy findings of infants with SMA1, which resulted in higher SMN protein levels in the lumbar motor neurons compared with the cervical and bulbar motor neurons. A combination of selectively high and early vulnerability of brainstem nuclei and a lower bioavailability of the medication at this level in treated patients may explain the limited effects seen on bulbar function compared with its benefits for limb motor function. 29 , 30 Of note, a recent study on two patients with SMA1 who passed away during a clinical trial with the intravenous administration of the AAV9‐mediated gene therapy onasemnogene abeparvovec showed widespread biodistribution of vector genomes and transgenes throughout the central nervous system, including pons, medulla, and upper cervical spine. 31 This may suggest that systemic treatment could be more effective in preserving bulbar function; however, longitudinal real‐world data will be needed to evaluate whether this corresponds to a clinical benefit for treated patients.
Awareness of persistent feeding difficulties in nusinersen‐treated patients with SMA1 helps guide discussions with families, including prognosis and treatment, as well as informing healthcare resource allocation and standards of care. Despite treatment with nusinersen, the need for close attention to nutritional status of children with SMA1 remains as outlined in standards of care. 8
Our study indicates that the p‐FOIS can be adapted for children across a wide age range and with different feeding disorders, while maintaining the excellent interrater reliability that is seen in the adult version of the tool. 14 This suggests the p‐FOIS is applicable for use in the paediatric population with SMA; however, larger studies investigating its use in this population will be needed to confirm its psychometric properties.
CONCLUSION
To our knowledge, this is the first study of nusinersen‐treated patients with SMA1 that has quantified bulbar dysfunction using a validated tool and compared between the SMA1 subtypes. The p‐FOIS is a six‐point, ordinal, clinician‐rated outcome measurement scale for use with a paediatric feeding‐disordered population. International collaboration to achieve consensus on a single adaptation of the tool is required, as well as studies to further validate this tool in SMA. A validated outcome measure for children with SMA that demonstrates change over time in swallowing abilities will enable monitoring of individual progress and tailoring of treatment. This will help provide guidance and comparison with those patients treated with other systemically distributed novel treatments, such as the gene‐replacement therapy onasemnogene abeparvovec and the oral SMN2 splicing modifier risdiplam, recently approved by both the US Food and Drug Administration and the European Medicines Agency.
Conflict of interest
GB has received speaker and consulting fees from Biogen, Novartis Gene Therapies (AveXis), and Roche, and has worked as principal investigator of SMA studies sponsored by Novartis Gene Therapies and Roche. FM has received speaker and consulting fees from Biogen, Novartis, and Roche, and has worked as principal investigator of SMA clinical trials sponsored by Biogen, Novartis, and Roche. He is also the principal investigator of an investigator‐initiated registry study funded by Biogen (SMA REACH UK). HW has worked as the sub‐investigator of SMA clinical trials sponsored by Biogen, Novartis, and Roche. MS has received speaker and consulting honoraria from Biogen, Novartis gene therapy (Avexis), and Roche, and has worked as principal investigator and sub‐investigator for SMA studies sponsored by Biogen and Novartis gene therapy. EJ has acted as a paid consultant for Roche and has collected data for SMA studies sponsored by Novartis gene therapy.
Supporting information
Appendix S1: Adaptation and validation of p‐FOIS.
Acknowledgements
Members of the SMA p‐FOIS Working Group are as follows: Adnan Manzur, Pinki Munot, Marion Main, and Claudia Kate Au‐Yeung.
GB, GS, and FM are supported by a grant from the Great Ormond Street Hospital Children's Charity and Muscular Dystrophy UK. All research at Great Ormond Street Hospital NHS Foundation Trust and UCL Great Ormond Street Institute of Child Health is made possible by the NIHR Great Ormond Street Hospital Biomedical Research Centre. The views expressed are those of the author(s) and not necessarily those of the NHS, the NIHR or the Department of Health. FM is partly supported by the National Institute for Health Research Centre at Great Ormond Street Institute of Child Health and Great Ormond Street Hospital for Children. The authors also thank the SMA reach grant from Biogen for its support to the Great Ormond Street team involved in SMA research.
Weststrate H, Stimpson G, Thomas L, Scoto M, Johnson E, Stewart A, et al. Evolution of bulbar function in spinal muscular atrophy type 1 treated with nusinersen. Dev Med Child Neurol. 2022;64:907–914. 10.1111/dmcn.15171
Giovanni Baranello and Eleanor Conway are joint senior authors.
*Members of the SMA p‐FOIS Working Group are listed in the Acknowledgements.
Contributor Information
Giovanni Baranello, Email: g.baranello@ucl.ac.uk.
SMA p‐FOIS Working Group*:
Adnan Manzur, Pinki Munot, Marion Main, and Claudia Kate Au‐Yeung
DATA AVAILABILITY STATEMENT
Research data are not shared.
REFERENCES
- 1. Kolb SJ, Coffey CS, Yankey JW, Krosschell K, Arnold WD, Rutkove SB, et al. Natural history of infantile‐onset spinal muscular atrophy. Ann Neurol. 2017;82(6):883–91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Kolb SJ, Kissel JT. Spinal muscular atrophy. Neurol Clin. 2015;33(4):831–46. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Wirth B, Karakaya M, Kye MJ, Mendoza‐Ferreira N. Twenty‐five years of spinal muscular atrophy research: from phenotype to genotype to therapy, and what comes next. Annu Rev Genomics Hum Genet. 2020;21(1):231–61. [DOI] [PubMed] [Google Scholar]
- 4. Finkel R, Bertini E, Muntoni F, Mercuri E. 209th ENMC international workshop: outcome measures and clinical trial readiness in spinal muscular atrophy 7–9 November 2014, Heemskerk, The Netherlands. Neuromuscul Disord. 2015;25(7):593–602. [DOI] [PubMed] [Google Scholar]
- 5. Passini MA, Bu J, Richards AM, Kinnecom C, Sardi SP, Stanek LM, et al. Antisense oligonucleotides delivered to the mouse CNS ameliorate symptoms of severe spinal muscular atrophy. Sci Transl Med. 2011;3(72):72ra18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Finkel RS, Mercuri E, Darras BT, Connolly AM, Kuntz NL, Kirschner J, et al. Nusinersen versus sham control in infantile‐onset spinal muscular atrophy. N Engl J Med. 2017;377(18):1723–32. [DOI] [PubMed] [Google Scholar]
- 7. Mercuri E, Darras BT, Chiriboga CA, Day JW, Campbell C, Connolly AM, et al. Nusinersen versus sham control in later‐onset spinal muscular atrophy. N Engl J Med. 2018;378(7):625–35. [DOI] [PubMed] [Google Scholar]
- 8. Mercuri E, Finkel RS, Muntoni F, Wirth B, Montes J, Main M, et al. Diagnosis and management of spinal muscular atrophy: Part 1: recommendations for diagnosis, rehabilitation, orthopedic and nutritional care. Neuromuscul Disord. 2018;28(2):103–15. [DOI] [PubMed] [Google Scholar]
- 9. Bertoli S, De Amicis R, Mastella C, Pieri G, Giaquinto E, Battezzati A, et al. Spinal muscular atrophy, types I and II: what are the differences in body composition and resting energy expenditure? Clin Nutr. 2017;36(6):1674–80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Hurst Davis R, Godshall BJ, Seffrood E, Marcus M, LaSalle BA, Wong B, et al. Nutritional practices at a glance. J Child Neurol. 2014;29(11):1467–72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Chen K‐A, Widger J, Teng A, Fitzgerald DA, D'Silva A, Farrar M. Real‐world respiratory and bulbar comorbidities of SMA type 1 children treated with nusinersen: 2‐Year single centre Australian experience. Paediatr Respir Rev. 2021;39:54–60. [DOI] [PubMed] [Google Scholar]
- 12. Pane M, Coratti G, Sansone VA, Messina S, Catteruccia M, Bruno C, et al. Type I SMA “new natural history”: long‐term data in nusinersen‐treated patients. Ann Clin Transl Neurol. 2021;8(3):548–57. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. van der Heul AMB, Cuppen I, Wadman RI, Asselman F, Schoenmakers M, van de Woude DR, et al. Feeding and swallowing problems in infants with spinal muscular atrophy type 1: an observational study. J Neuromuscul Dis. 2020;7:323–30. [DOI] [PubMed] [Google Scholar]
- 14. Crary MA, Mann GDC, Groher ME. Initial psychometric assessment of a Functional Oral Intake Scale for dysphagia in stroke patients. Arch Phys Med Rehabil. 2005;86(8):1516–20. [DOI] [PubMed] [Google Scholar]
- 15. Glanzman AM, Mazzone E, Main M, Pelliccioni M, Wood J, Swoboda K, et al. The Children’s Hospital of Philadelphia Infant Test of Neuromuscular Disorders (CHOP INTEND): test development and reliability. Neuromuscul Disord. 2010;20(3):155–61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Dangouloff T, Servais L. Clinical evidence supporting early treatment of patients with spinal muscular atrophy: current perspectives. Ther Clin Risk Manag. 2019;15:1153–61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Pane M, Coratti G, Sansone VA, Messina S, Bruno C, Catteruccia M, et al. Nusinersen in type 1 spinal muscular atrophy: twelve‐month real‐world data. Ann Neurol. 2019;86(3):443–51. [DOI] [PubMed] [Google Scholar]
- 18. Swoboda KJSV, De Vivo DC, Bertini E, Hwu WL, Makepeace C, Bohn J, et al. Preserved Swallowing Function in Infants Who Initiated Nusinersen Treatment in the Presymptomatic Stage of SMA: Results From the NURTURE Study. Cure SMA Virtual Research and Clinical Care Meeting June 9–11, 2021. Biogen; 2021.
- 19. Yi YG, Shin H‐I. Psychometrics of the Functional Oral Intake Scale for Infants. Frontiers. Pediatrics. 2019;7:1–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Yi YG, Shin H‐I. Psychometrics of the Functional Oral Intake Scale for children with dysphagia. J Pediatr Gastroenterol Nutr. 2020;71(5):686–91. [DOI] [PubMed] [Google Scholar]
- 21. Berti B, Fanelli L, De Sanctis R, Onesimo R, Palermo C, Leone D, et al. Oral and swallowing abilities tool (OrSAT) for Type 1 SMA patients: development of a new module. J Neuromuscul Dis. 2021;8(4):589–601. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Lavie M, Diamant N, Cahal M, Sadot E, Be'er M, Fattal‐Valevski A, et al. Nusinersen for spinal muscular atrophy type 1: Real‐world respiratory experience. Pediatr Pulmonol. 2021;56(1):291–8. [DOI] [PubMed] [Google Scholar]
- 23. Sansone VA, Pirola A, Albamonte E, Pane M, Lizio A, D'Amico A, et al. Respiratory needs in patients with type 1 spinal muscular atrophy treated with nusinersen. J Pediatr. 2020;219:223–8.e4. [DOI] [PubMed] [Google Scholar]
- 24. Darras BT, Chiriboga CA, Iannaccone ST, Swoboda KJ, Montes J, Mignon L, et al. Nusinersen in later‐onset spinal muscular atrophy. Long‐term results from the phase 1/2 studies. Neurology. 2019;92(21):e2492–e506. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Giavazzi A, Setola V, Simonati A, Battaglia G. Neuronal‐specific roles of the survival motor neuron protein: evidence from survival motor neuron expression patterns in the developing human central nervous system. J Neuropathol Exp Neurol. 2006;65(3):267–77. [DOI] [PubMed] [Google Scholar]
- 26. Byers RK, Banker BQ. Infantile muscular atrophy. Arch Neurol. 1961;5(2):140–64. [DOI] [PubMed] [Google Scholar]
- 27. Wadman RI, Van Bruggen HW, Witkamp TD, Sparreboom‐Kalaykova SI, Stam M, van den Berg LH, et al. Bulbar muscle MRI changes in patients with SMA with reduced mouth opening and dysphagia. Neurology. 2014;83(12):1060–6. [DOI] [PubMed] [Google Scholar]
- 28. Van Den Engel‐Hoek L, Erasmus CE, Van Bruggen HW, de Swart BJ, Sie LT, Steenks MH, et al. Dysphagia in spinal muscular atrophy type II: More than a bulbar problem? Neurology. 2009;73(21):1787–91. [DOI] [PubMed] [Google Scholar]
- 29. Ramos DM, D'Ydewalle C, Gabbeta V, Dakka A, Klein SK, Norris DA, et al. Age‐dependent SMN expression in disease‐relevant tissue and implications for SMA treatment. Journal of Clinical Investigation. 2019;129(11):4817–31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Sumner CJ, Crawford TO. Two breakthrough gene‐targeted treatments for spinal muscular atrophy: challenges remain. Journal of Clinical Investigation. 2018;128(8):3219–27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Thomsen G, Burghes AHM, Hsieh C, Do J, Chu BTT, Perry S, et al. Biodistribution of onasemnogene abeparvovec DNA, mRNA and SMN protein in human tissue. Nat Med. 2021;27(10):1701–11. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Appendix S1: Adaptation and validation of p‐FOIS.
Data Availability Statement
Research data are not shared.