

Abstract
Amelogenins, the principal proteins in the developing enamel microenvironment, self-assemble into supramolecular structures to govern the remodeling of a proteinaceous organic matrix into longitudinally ordered hydroxyapatite nanocrystal arrays. Extensive in vitro studies using purified native or recombinant proteins have revealed the potential of N-terminal amelogenin on protein self-assembly and its ability to guide the mineral deposition. We have previously identified a 14-aa domain (P2) of N-terminal amelogenin that can self-assemble into amyloid-like fibrils in vitro. Here we investigated how this domain affects the ability of amelogenin self-assembling and stability of enamel matrix protein scaffolding in an in vivo animal model. Mice harboring mutant amelogenin lacking P2 domain had a hypoplastic, hypomineralized and aprismatic enamel. In vitro, the mutant recombinant amelogenin without P2 had a reduced tendency to self-assemble and was prone to accelerated hydrolysis by MMP20, the prevailing metalloproteinase in early developing enamel matrix. A reduced amount of amelogenins and a lack of elongated fibrous assemblies in the development enamel matrix of mutant mice were evident as compared to that in the wild type mouse enamel matrix. Our study is the first to demonstrate that a subdomain (P2) at the N-terminus of amelogenin controls amelogenin’s assembly into a transient protein scaffold that resists rapid proteolysis during enamel development in an animal model. Understanding the building blocks of fibrous scaffold that guides the longitudinal growth of hydroxyapatites in enamel matrix sheds light on protein-mediated enamel bioengineering.
Keywords: amelogenin, amyloid forming peptide, enamel matrix formation, MMP20 hydrolysis, scaffold
Introduction:
The functions of proteins, such as enzymes and substrates, are largely defined by their higher-order conformation and interaction partners in a given microenvironment. However, the primary amino acid sequences and functional groups of the monomer are building blocks that contribute to protein conformational dynamics and ability to form spatial and temporal functional protein complexes [1].
Enamel is the hardest tissue in mammals and the only nonregenerative mineralized tissue in our body. While mature enamel consists of 95% of minerals packed as longitudinally ordered hydroxyapatite nanocrystal arrays, its formation is controlled by proteins. Understanding the processes by which proteins create a transient scaffold to build an orderly aligned mineral structure is of considerable interest, both from a developmental biology standpoint, and from the standpoint of protein-based enamel bioengineering.
Enamel formation begins when enamel-forming ameloblasts synthesize and secrete large amounts of enamel matrix proteins (EMPs) and proteinases to build up an organic matrix scaffold onto which hydroxyapatite mineral is deposited [2]. Amelogenins, composed of more than 90% of EMPs, are indispensable for the formation of the orderly hydroxyapatite nanocrystals and an enamel tissue of normal thickness [3, 4]. Normal enamel development requires both amelogenin assembly into organized supramolecular structures, and their timely degradation during the mineralization process. However, the molecular mechanisms allowing amelogenins to function as key protein scaffolds and to sustain degradation in the secretory stage of enamel matrix have not been well elucidated.
Amelogenin gene contains up to 7 exons in humans and 9 exons in rodents [5–10]. During development, alternative splicing results in at least sixteen amelogenin variants [9, 11]. The most abundant native amelogenins in the secretory stage human enamel matrix are the 175-residue full-length amelogenin and a 56-residue amelogenin also called leucine-rich amelogenin protein (LRAP) [8, 12]. In the developing enamel microenvironment, the primary matrix metalloproteinase MMP20 processes amelogenins by removing its C-terminus to generate a set of partially degraded amelogenins of 164, 148 and 45 residues, also named as tyrosine rich amelogenin polypeptide (TRAP) [13].
Various native amelogenins and their cleavage products are believed to play manifold roles in amelogenesis [14]. However, most of amelogenin derivatives in the developing enamel matrix contain the hydrophobic N-terminus and/or the hydrophilic C-terminus. Both regions are highly conserved among mammal species while the central region of amelogenin shows some degree of variability [15]. Amelogenesis imperfecta-associated mutations that occur at the C-terminus of amelogenin are linked with hypoplastic enamel [16, 17]. Recombinant amelogenin with a C-terminus deletion possesses reduced affinity bound to hydroxyapatite crystals, strongly indicating that the highly charged C-terminus contains mineral binding domains [17–20]. Additional function of the amelogenin C-terminus involves in promoting crystal growth [21].
Full-length amelogenin, LRAP, TRAP and many other derivatives contain the first 33 residues of the N-terminus of amelogenin. The significance of this N-terminal segment has caught considerable attention of enamel researchers. One notable factor is that the sole phosphorylation site of amelogenin, serine 16, localizes at the N-terminus [22–26]. Previous research has demonstrated that phosphorylation on Ser-16 is essential for stabilizing amorphous calcium phosphate minerals, subsequently regulating the shape and orientation of hydroxyapatite crystals [27, 28].
Amelogenin is an intrinsically disordered enamel matrix protein [29]. Previous studies have intensively focused on the supramolecules (nanosphere, microribbons and nanochains) assembled by recombinant amelogenin completely in in vitro model systems [30, 31] [32–34]. Habelitz and Li’s groups have recently found that nanoribbons can be assembled when the recombinant full-length human amelogenin (rH174) or amelogenin without C-terminus (rH146) is incubated in calcium phosphate solutions. What are the key subdomains at the N-terminus of amelogenin mediating the assembling property of amelogenin? A number of bioinformatics approaches were applied to analyze the full-length amelogenin sequence, and all predicted that a domain spanning residues 11-18 at the N-terminus of amelogenin had the greatest propensity to form ß-sheet aggregates or amyloid-like fibrils. ß-sheet aggregates may serve as structural basis of those previously described nanoribbons in the developing enamel matrix. Using a library of tiled 14-residue peptides spanning the amelogenin protein sequence, we have identified a peptide, called 14P2, corresponding to the residues from 8 to 21 of amelogenin that can self-assemble into amyloid-like fibrils in vitro. Intriguingly, the self-assembly kinetics of 14P2 is more robust than that of recombinant full length amelogenin (rH174), and phosphorylated 14P2 is considerably more effective in forming dense fiber-like bundles than unphosphorylated 14P2 [35]. These data encouraged us to investigate whether P2 is necessary for the enamel matrix development in an in vivo model system.
However, 14P2 contains eleven residues from amelogenin’s exon 3 and three residues from exon 5, spanning one splice site. We therefore wondered whether a shorter peptide entirely encoded by exon 3 would have similar biochemical properties. This eleven-residue peptide (11P2), corresponding to residues 8 to 18 at the N-terminus of amelogenin, has the same number of tyrosines as 14P2. Tyrosine, processing a phenolic functional group, plays roles in conformation control and efficient charge (or proton) transport during redox reaction [36, 37]. Here we demonstrate that 11P2 possesses a comparable ability to form amyloid-like fibrils in calcium phosphate solution as 14P2 does. We then deleted 11P2 from amelogenin in in vivo and in vitro models. These models allow us to characterize the developmental and phenotypic consequence of 11P2 deletion from mouse amelogenin genome, and explore the molecular mechanism how 11P2 stabilizes the amelogenin supramolecular scaffold in the early developing mouse enamel matrix to support the growth of enamel crystals.
Methods:
Peptide self-assembly and atomic force microcopy (AFM) analysis:
Peptide 11P2 (GHPGYINLSYE) corresponding to the N-terminal sequences (residues 8-18) of human amelogenin was synthesized by ELIM Biopharmaceuticals Inc, and reconstituted in 1 mM HCl. The peptide was diluted with CaCl2 and KH2PO4 resulting in the final concentration of 1 mg/ml peptide, 3.3 mMCaCl2, and 2.1 mM KH2PO4 at pH 5.5. Peptide solution was incubated at 37°C for 48 hours prior to AFM analysis.
As previously described [35], 15 μL of peptide solution was placed onto mica substrate and incubated for 1 hour on a wet cell for immobilization. Samples were then rinsed with 100 μL of autoclaved deionized water and immediately dried with compressed air. AFM measurements were performed in ambient conditions, using a Bruker Multimode 8 with Nanoscope V controller (Bruker, Santa Barbara, CA) and silicon cantilevers (Bruker ScanAsyst Air-HR, Bruker AFM probes, Santa Barbara, CA, USA). The images were analyzed using Nanoscope Analysis software (v1.80).
Generation of a mouse model harboring a deletion of 33 base pairs (bp) DNAs within amelogenin exon 3 that encode 11P2 by the CRISPR-Cas9 genome editing approach:
Guide RNA complementary to the DNA sequences within amelogenin exon 3 was designed using Benchling software. Guide RNA (gRNA), tracrRNA and a 200bp single-stranded unmodified oligodeoxynucleotide (ssODN) donor were synthesized by Integrated DNA Technologies. This donor ssODN contains 100bp genome sequences immediately upstream and downstream of P2 sequences respectively. Super-ovulated female C57BL/6J mice were mated to C57BL/6J males. Fertilized zygotes were collected from oviducts and injected with Cas9 protein (30 ng/μl), gRNA and tracrRNA (20 ng/μl for each), and donor ssODN (10 ng/μl) into pronucleus of fertilized zygotes by Transgenic Core Facility at Gladstone Institutes. Injected zygotes were implanted into oviducts of two pseudopregnant CD1 female mice.
The tails of P7 offspring from these two CD1 female mice were snipped for genome DNA extraction. The genotyping PCR was conducted using forward primer recognized the intron 2 of amelogenin gene (5’- TAAAGCTGCTGGTGGGAACT) and reverse primer complementary to the intron 3 of amelogenin gene (5’- ATGGGGTTCCTCCTTCACAG). PCR reaction produced a 400bp amplicon from wild-type amelogenin allele, and a 367bp amplicon from mutant amelogenin allele with the deletion of 11P2 sequences. Both amplicons were then subjected to DNA sequencing analysis. The confirmed founders (Amelx11P2−/−) were backcrossed with wild type C57BL/6J mice for three generation to avoid unintended genomic modifications before phenotypes were evaluated. Other than enamel defect, Amelx11P2−/− mice have developed normally. We used female mice intentionally to avoid any potential compensatory amelogenin expression that may derive from Y-chromosome amelogenin gene.
All animals were maintained in the UCSF animal care facility, which is a barrier facility, accredited by Association for Assessment and Accreditation of Laboratory Animal Care (AAALAC). All experimental procedures associated with this established mouse model were approved by the Institutional Animal Care and Use Committee (IACUC) under the protocol AN179159-01B.
Cloning, expression and purification of recombinant human wild-type amelogenin and mutant amelogenin without 11P2:
A site-directed mutagenesis kit (Takara Bio., Mountain View, CA) was employed to delete 33bp 11P2 coding DNA from human wild-type amelogenin cDNA. After the site specific deletion was confirmed by sequencing analysis, wild-type human amelogenin (rH174) and the mutant amelogenin with 33bp of P2 deletion (rH174-P2) was expressed in T7 Express Competent Escherichia coli (New England Biolabs, Ipswich,MA) respectively and purified using an acid/heat treatment method described previously [38]. The purified proteins were further characterized by SDS-PAGE, mass spectrometry, and western blot through incubation either with rabbit anti-amelogenin or with rabbit anti-11P2 IgG. IgG was purified using protein-A affinity chromatography from antisera of rabbits immunized either with recombinant rH174 or synthesized 11P2 peptide.
Recombinant amelogenin protein (1mg/ml) in 3.3 mMCaCl2 and 2.1 mM KH2PO4 at pH 5.5 was incubated at 37°C for 72 hours prior to AFM analysis.
Characterization of the enamel matrix and adjacent ameloblasts of Amelx11P2−/− mouse model:
Wild-type C57BL/6J mice (Amelx11P2+/+) and 11P2 deletion mutant amelogenin mice (Amelx11P2−/−) at postnatal seven weeks old (P7W) were carbon dioxide euthanized following standard IACUC protocols. Mouse hemimandibles were dissected and air-dried prior to imaging with a Leica M165C digital stereo microscope and LAS V4.2 software. Hemimandibles were next subjected to X-ray micro-tomography scans using a SkyScan 1275 microCT system at Micro Photonics in Allentown, PA. Briefly, scans were performed at 10W of power with no filter at 40kV/250uA of voltage with an isotropic voxel size of 8μm. Projection images were reconstructed into transaxial slice images utilizing Bruker NRecon (version 1.7.5.2) software. Reconstructed images were visualized using Bruker’s DataViewer (version 1.5.6.3) and CTVox (version 3.3.0) visualization software programs.
To characterize the histological phenotype of ameloblast/enamel matrix of Amelx11P2+/+ and Amelx11P2−/− mice, mouse incisors were examined because they possess all developmental stages of ameloblasts and enamel matrix. Briefly, mice at P7W were anesthetized with 240mg/kg tribromoethanol (Sigma-Aldrich, St. Louis, MO), and perfuse-fixed with 4% paraformaldehyde (PFA). The hemimandibles were dissected and post-fixed with 4% PFA for 24 hours at 4°C, then followed by decalcification in 8% EDTA at 4°C for 3 weeks with agitation. The hemimandibles were then processed, embedded and sectioned at the sagittal plane. Sections were stained with hematoxylin&eosin (H&E) for ameloblast and enamel matrix morphological examination.
For immunostaining, the sagittal sections were boiled in 10 mM citrate buffer (pH 6.0) for 20 minutes to retrieve antigens. Next, the sections were incubated with 10% swine and 5% goat sera followed by incubation with rabbit anti-amelogenin antibody or rabbit anti-amyloid fibrils OC antibody (Millipore Sigma, Hayward, CA) overnight at 4°C. The latter is designed to selectively recognize amyloid fibrils, but not random-coil monomers or pre-fibrillar oligomers, in a conformation dependent manner [39, 40]. Sections were then incubated with fluorescein- conjugated anti-rabbit IgG for 1 hour at RT, followed by counterstained with 1ug/ml Hoechst for 5 minutes (Life Technologies). The slides were imaged using either a high-resolution and high-speed Leica TCS SP5 spectrum confocal microscope or a W1-SoRA spinning disk super resolution confocal microscope.
Western blot analyses:
Enamel matrix at the secretory stage was dissected from incisors underlying the second molars according to incisal enamel stage reference line published previously [41]. Then the matrix was incubated in 1ml 0.17N HCl/0.95% formic acid for 2 hours at 4°C. After centrifugation at 3500g at 4°C, the supernatant was filtered through Amnicon Ultra filter (UFC500324, EMD Millipore) and proteins bound to the filter were eluted with 0.01% formic acid. Ten micrograms of the total enamel matrix proteins were loaded to a 15% SDS-PAGE gel. After separation on SDS-PAGE gel, proteins were transferred to PVDF membrane, which was then incubated with either rabbit anti-amelogenin IgG or rabbit anti-11P2 IgG for overnight at 4°C. Following washing, PVDF was secondly incubated with IRDye 680RD conjugated anti-rabbit IgG for 1 hour at RT. The membranes were scanned using Odyssey Imaging System.
Scanning electron microscopy (SEM):
Three sets of hemimandibles from either at postnatal seven weeks old (P7W) Amelx11P2+/+ or Amelx11P2−/− mice were dissected, embedded in epoxy. A series of SiC paper and diamond polishing suspension were used to polish the hemimandible starting from incisor tip and stopping when mesial angle of the first molar was revealed. Subsequently specimens were etched with 10% HCl for 30 second. Specimens were sputter-coated with Au-Pd, and incisal enamel thickness and crystal rods was imaged at 20kV using a Scanning Electron Microscope, Quanta 3D FEG (FEI) as described previously [42].
Assessment of expression levels of genes of interest by semi-quantitative PCR and RNAscope:
After pups were euthanized with carbon dioxide asphyxiation followed by cervical dislocation, mandibles were dissected from P5 Amelx11P2+/+ and Amelx11P2−/− mice. Enamel epithelium was collected from the surface of the first molars, which have enamel primarily at the secretory stage of development [43]. The total RNA was then purified using Qiagen RNeasy Micro Kit and mRNA was served as templates to synthesize cDNA libraries with SuperScriptTM III First-Strand Synthesis System (Life Technologies). Semi-quantitative PCR was performed to quantify the relative expression levels of target genes in those Amelx11P2+/+ and Amelx11P2−/− mouse secretory ameloblasts. The sequences of primers used to amplify mouse endogenous GAPDH and target genes were listed as follows: GAPDH sense- 5’-TGGCCTTCCGTGTTCCTAC-3’, antisense- 5’-GAGTTGCTGTTGAAGTCGCA-3’; amelogenin sense- 5’-GGGACCTGGATTTTGTTTGCC-3’, antisense- 5’-TTCAAAGGGGTAAGCACCTCA;-3’ enamelin sense- 5’-GCTTTGGCTCCAATTCAAA-3’, antisense- 5’-AGGACTTTCAGTGGGTGT-3’, ameloblastin sense- 5’-CTGTTACCAAAGGCCCTGAA-3’, antisense- 5’-GCCATTTGTGAAAGGAGAGC-3’; MMP20 sense- 5’-TCCAAGCATTATACGGACCCC-3’, antisense- 5’-GTCACTGCATCAAAGGACGAG-3’. To determine the relative expression levels of these target genes, the comparative threshold cycle method was used as previously published [44].
Hemimandibles were fixed in 4% paraformaldehyde (PFA) at 4°C overnight, then decalcified for 2 weeks in 0.5 M EDTA pH 7.5. After processing and embedding hemimandibles in paraffin, samples were processed into 5 μm-thick sagittal sections. RNAscope in situ hybridization 2.5 HD Brown Detection Kit and mouse amelogenin probe (Advanced Cell Diagnostics, ACDbio.) were used by following the manufacturer’s instructions. Briefly, after dewaxing, the sections were boiled in the target retrieval buffer at 100°C for 15 minutes and then incubated in the protease plus solution at 40°C for 15 minutes. The sections were next incubated with probe for 2 hours. After incubating with a serial of amplification reagents, the sections were incubated the HRP and substrate to reveal the positive mRNA signal.
Characterization of the role of 11P2 on recombinant amelogenin hydrolysis by MMP20:
First, we used SDS-PAGE to access the hydrolysis of recombinant amelogenin (rH174) and mutant amelogenin (rH174-P2) by MMP20 (Enzo Life Sciences, Inc, Famingdale, NY). Briefly, we incubated 3 μg of rH174 or rH174-P2 with 3 ng of MMP20 in a solution containing 50 mM Tris-HCl (pH 7.5), 10 mM CaCl2, 150 mM NaCl, and 10 nM ZnCl2 at 37°C for 2 and 4 hours. The digested samples were supplied with loading buffer, boiled and then loaded on a Bio-Rad 8-16% Mini-protein gel. After Coomassie staining, the gel was air-dried and scanned for band intensity analysis. The intensity of each protein band was measured with ImageJ software.
For LC-MS/MS analysis, we incubated 1 μg of rH174 or rH174-P2 with 1ng of MMP20 in the same reaction buffer as above mentioned for 4 hours. Then the protein samples were separated and fractioned by ESI-MS in MS Center at Stanford University with the Agilent 1260 HPLC. Each fraction was characterized and mass spectrum was generated by Bruker MicroTOF-Q II.
We also assessed whether 11P2 affects the assembly of amelogenins under the situation with and without the presence of MMP20 using in situ AFM as previously described and turbidity analysis. We prepared 1 μg of rH174 or rH174-P2 without or with 1 ng of MMP20 in 50 mM Tris-HCl (pH 7.5), 10 mM CaCl2, 150 mM NaCl, and 10 nM ZnCl2 solution at RT. For turbidity assessment, one hundred microliter reaction solution in triplicate was added into each well of a 96-well clear flat bottom plate. The absorbance of each well was recorded for 8 hours at a 2-minute interval using Molecular Devices SpectraMax iD3 plate reader.
Results:
11P2 is required for the self-assembly of full-length wild-type amelogenin into nanofibrils in vitro.
Our previous studies have demonstrated that the 14P2 peptide (GHPGYINFSYEVLT) corresponding to the N-terminus of amelogenin can form a ß-sheet and aggregate into amyloid-like fibrous structures [45]. Corresponding DNA sequences of 14P2 within amelogenin gene are illustrated in Fig.1A. We then characterized the self-assembly of an 11aa peptide at the N-terminus of 14P2, which was designated as 11P2. This 11P2 motif is solely encoded by one exon (exon 3), will not be possibly interrupted by any alternative splicing. Our AFM analysis showed that 11P2 could self-assemble into nanofibrils micrometers in length (see Fig.1B), resembling the self-assembled nanostructures previously formed by 14P2 [35].
Fig.1.
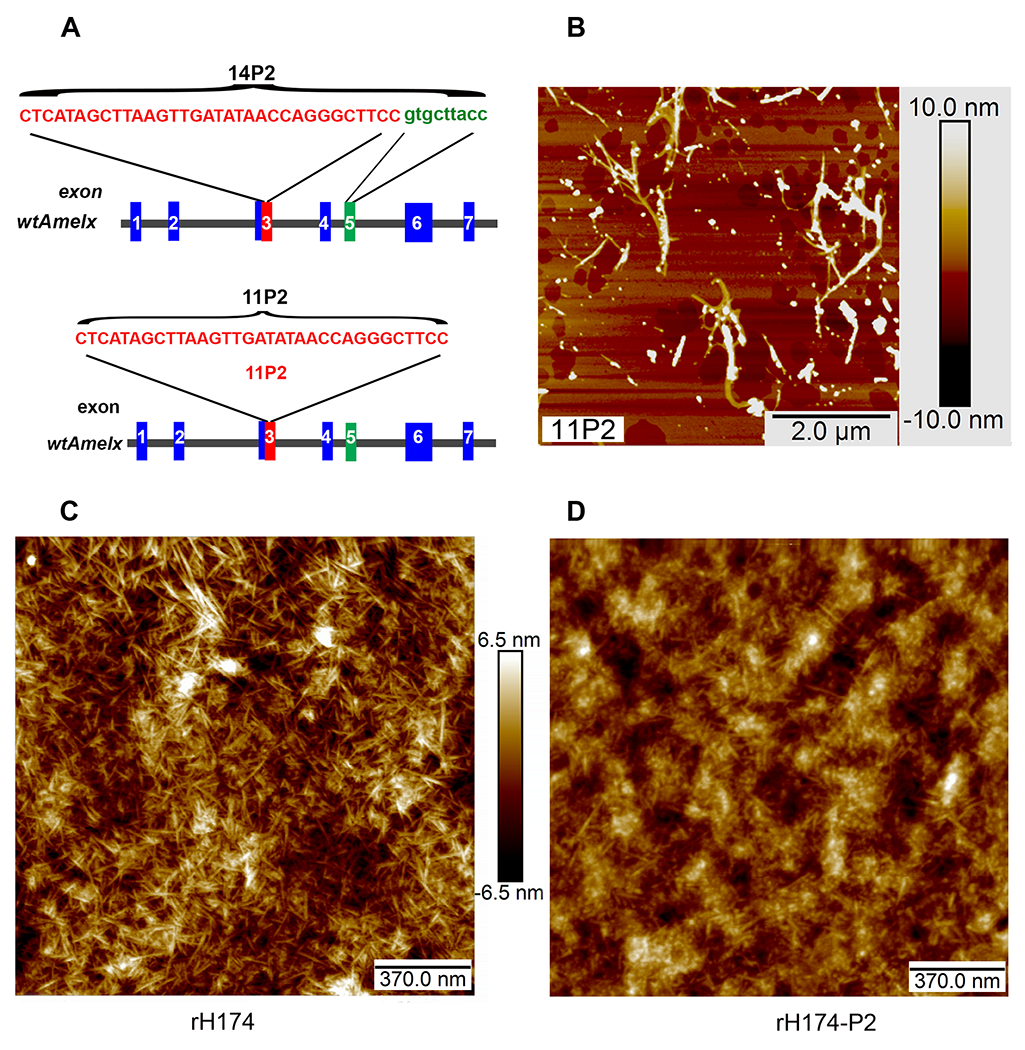
Characterization of the self-assembly propensity of 11P2. A) Schematic representation of nucleotide sequences of 14P2 and 11P2, and their location within human amelogenin gene: Peptide 14P2 is encoded by 33 nucleotides (letters in red) in exon 3 (box in red) and 9 nucleotides(letters in green) in exon 5 (box in green) of wild-type X-linked amelogenin gene (wtAmelx); Peptide 11P2 is encoded by 33 nucleotides (letter in red) in exon 3 (box in red). B) Self-assemblies formed by 11P2 were analyzed using AFM. The average length and width of nanoribbons are about 1.5±0.68 microns and 6±2.53 nm respectively. C) AFM analysis shows the uniform nanofibrils self-assembled by recombinant human full length amelogenin (rH174) in a solution supplemented with 3.3 mM calcium and 2.1 mM phosphate ions. D) Under the same condition, rH163 self-assemblies were composed of a mix of nanospheres and a few nanofibers.
We then assessed whether the absence of 11P2 would affect self-assembly of full-length recombinant amelogenin rH174, which can form uniform nanofibrils in 3.3 mMCaCl2 and 2.1 mM KH2PO4 in pH5.5 [35]. Indeed, we saw that rH174 consistently self-assembled into interwoven fibrils of several hundred nanometers in length (Fig.1C). Under the same condition, mutant amelogenin rH174-P2, which lacks the 11P2 motif, displayed a different self-assembly behavior: most of self-assembled rH174-P2 supramolecules observed under AFM were spherical, and only a small percentage of supramolecular structures were nanofibrils. These nanospheres were predisposed to form aggregates with greater height and width (Fig.1D). This observation demonstrates that the 11P2 motif is essential for the self-assembly of recombinant full-length amelogenin into uniform nanofibrils.
11P2 is critical for the formation of normal enamel tissue.
To determine whether the 11P2 motif was also indispensable for healthy enamel development in vivo, we deleted the 33bp 11P2 encoding sequences from exon 3 of amelogenin gene in a CRISPR-mediated genome editing mouse model (see Fig.2A). The site-specific deletion was confirmed by genotyping PCR (Fig.2B) and sequencing (see supplement 1). The expression of mutant amelogenin lacking 11P2 was confirmed by western blot and immunostaining (Fig.2C and D). The deletion of 11P2 resulted in significant changes in the appearance of incisors and molars. As revealed by a stereo microscope, the enamel overlying the incisors of wild-type Amelx11P2+/+ mice was characteristically translucent, smooth and pigmented. By contrast, the enamel on the Amelx11P2−/− mouse incisors was whitish and rough with numerous nodules while it appeared dark brown on molars. Molar cusps were also noticeably worn down on Amelx11P2−/− mice as compared to the well-developed molar cusps of Amelx11P2+/+ (see Fig.2E).
Fig.2.
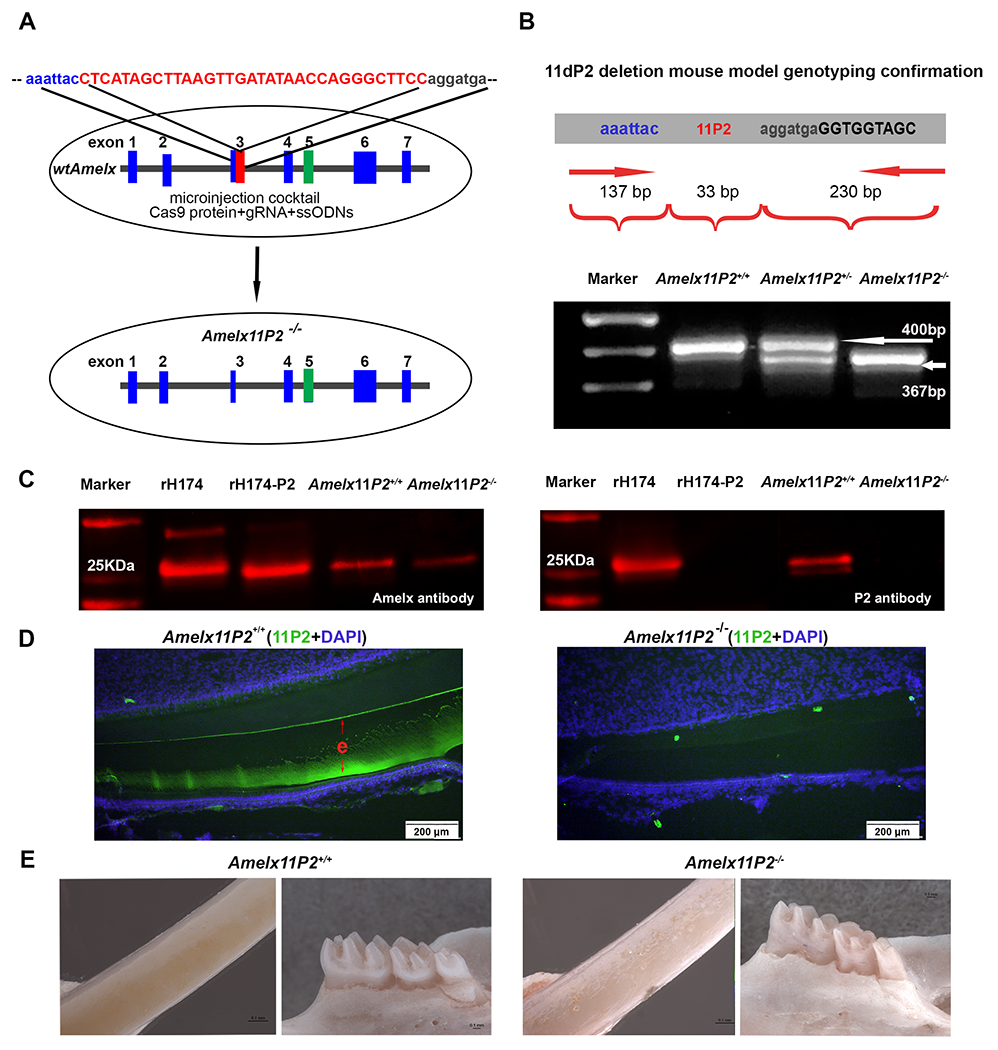
Construction and analysis of mouse model harboring amelogenin without 11P2. A) Schematic representation of the approach using CRISPR-Cas9 mediated genome editing to delete 11P2. The 33 deoxynucleotides encoding 11P2 is highlighted in red within exon 3 of X-linked amelogenin gene. Cocktail composed of cas9 protein, guide gRNA and ssODN donor was microinjected into zygotes. ssODN was designed to replace 11P2 flanking chromosome sequences through homologous recombination triggered by gRNA/cas9 generated site-specific double-strand DNA break.
B) Genotyping PCR primers were designed to analyze the presence of 11P2 coding sequences in X-linked amelogenin gene. The primers target to amplify sequences from −137bp of 11P2 (in intron 2) to +230 bp of 11P2 (in intron 3). Based on the electrophoresis analysis on a 2% agarose gel, a 400bp PCR product was amplified from wild-type X-linked amelogenin allele (Amelx11P2+/+), a 367bp from 11P2 deleted X-linked amelogenin allele (Amelx11P2−/−), both 400bp and 367bp from heterozygote (Amelx11P2+/−).
C) Secretory stage enamel matrix from Amelx11P2+/+ and Amelx11P2−/− mice was subjected to western blot assessment for the presence of 11P2. Recombinant rH174 and rH174-P2 (without P2) proteins served as controls. The left panel shows that amelogenin polyclonal antibody detected the presence of amelogenins in enamel proteins from both mouse models. The size of amelogenin in Amelx11P2−/− mouse matrix indicates that 11P2 deletion did not interfere with the reading frame of edited amelogenin gene. The right panel shows that polyclonal antibody purified from sera of 11P2 peptide immunized rabbit recognized full length recombinant and native amelogenins, but not 11P2 deleted amelogenin mutants.
D) Positive 11P2 immunostaining signal (in green) was detected in the enamel matrix (e) on Amelx11P2w+/+ mouse incisor sagittal sections (left panel). No positive 11P2 immunostaining signal was detected in Amelx11P2−/− enamel matrix (right panel).
E) Stereo microscopic analysis shows that Amelx11P2+/+ mouse incisor has smooth and brown pigmented enamel, and molar cusps are sharp and well-developed (left two panels), while Amelx11P2−/− mouse incisor enamel is whitish and rough, molar cusps are heavily stained and worn out (right two panels).
The relative mineralization state of enamel on Amelx11P2+/+ and Amelx11P2−/− mouse teeth was assessed by X-ray microtomography (μCT). The reconstituted 3D μCT hemimandible images clearly showed that the enamel layer (see Fig.3A, pointed by red arrows) on both Amelx11P2+/+ molars and incisor was radiopaque and distinguishable from the underlying dentin (pointed by blue arrows) and surrounding alveolar bone (pointed by black arrows). In contrast, the enamel layer on both Amelx11P2−/− molars and incisor was less radiopaque and distinguishable from dentin and alveolar bone, and thinner than that of Amelx11P2+/+ (see Fig.3C). As compared to the smooth surface of Amelx11P2+/+ enamel (see Fig.3B), Amelx11P2−/− enamel featured with a dull incisal tip and increased surface roughness, observed as nodules. These nodules on enamel surface were radiopaque (see Fig.3D).
Fig.3.

Assessment of enamel property of Amelx11P2−/− mice. A) MicroCT images of mouse hemimandibles show an intense radiopaque layer covering incisor and molars of Amelx11P2+/+ mouse. This enamel layer (indicated by red arrows) is clearly contrast to the underlaying dentin (indicated by blue arrows) and surrounding alveolar bone (indicated by black arrows). B) The enamel surface of Amelx11P2+/+ incisor is smooth. Enamel layer is distinguishable from dentin layer. C) MicroCT images show that radiopaque of enamel, dentin and alveolar bone remains similar in Amelx11P2−/− mouse hemimandible. D) Radiopaque nodules were detected on the rough surface of incisal enamel.
E) Under SEM, the interplay between enamel and dentin (DEJ) is well defined (indicated by a pink dotted line). The average thickness of Amelx11P2+/+ centrolabial mandibular incisal enamel layer is about 116±9.7 uM. E1) High magnified SEM image shows the decussating pattern of enamel rods and interrods. The elongated enamel crystals grew uniformly in the shape of filaments. F) The average thickness of Amelx11P2−/− centrolabial mandibular incisal enamel layer is about 35±2.3 uM. The DEJ (indicated by a pink dotted line) is less distinguishable. F1) There is no enamel rod or filament-like structure that can be identified in the magnified SEM images of Amelx11P2−/− incisor enamel. Minerals grew in the shape of particulates and flakes. G) The average thickness of Amelx−/− mandibular incisal enamel layer is about 12.3±1.2 uM. G1) In Amelx−/− mouse enamel, short crystals could be identified, but grew in the shape of fans.
The effects of the 11P2 deletion on the shape and arrangement of enamel crystals were investigated by scanning electron microscope (SEM). This analysis showed that the thickness of Amelx11P2+/+ incisor centrolabial enamel underlying the mesial angle of the first molar was about 116±9.7 uM (see Fig.3E), Amelx11P2−/− enamel at the same plane was about 35±2.3 uM (See Fig.3F), amelogenin null (Amelx−/− ) mouse enamel was about 12.3±1.2 uM (see Fig.3G). The dentin-enamel junction was clearly recognizable in Amelx11P2+/+ mouse teeth (see Fig.3E), but not in Amelx11P2−/− teeth (see Fig.3F). The well-oriented crystal rod structures and crystal’s decussation pattern was detected in Amelx11P2+/+ mouse enamel (See Fig.3E1), the minerals in Amelx11P2−/− mouse enamel was disorganized, failing to form elongated fibril-like crystals (see Fig.3F1). When amelogenin was completely absent, Amelx−/− mouse enamel was comprised of a layer of octacalcium phosphate like fans that developed on top of a thin layer of prismless enamel at the DEJ (see Fig.3G1).
H&E histological analyses of Amelx11P2+/+ mice revealed the presence of organic matrix typical of secretory stage enamel (see Fig.4A, e, indicated by red arrows) that gradually expanded and developed to its full thickness. At the end of the maturation stage, enamel tissue was absent as a normal consequence of the decalcification process. By contrast, the red-stained matrix of secretory stage enamel (e, indicated by red arrows) in Amelx11P2−/− mice was remarkably thinner, but remained until the end of the maturation stage (see Fig.4B). Both secretory and maturation stage ameloblasts (SAB and MAB) were significantly shorter in Amelx11P2−/− than control mice (see Fig.4A1, 4A2, 4B1, 4B2). Tomes’ processes (TPs), the characteristic cytoplasmic projections at the apical surface of wild-type secretory ameloblasts (SAB) (see Fig.4A1), were absent in Amelx11P2−/− secretory ameloblasts (SAB) (see Fig.4B1). Additionally, we consistently found that the enamel layer in Amelx11P2−/− mouse teeth was prone to detaching from the dentin matrix (see Fig.4B and 4B2).
Fig.4.
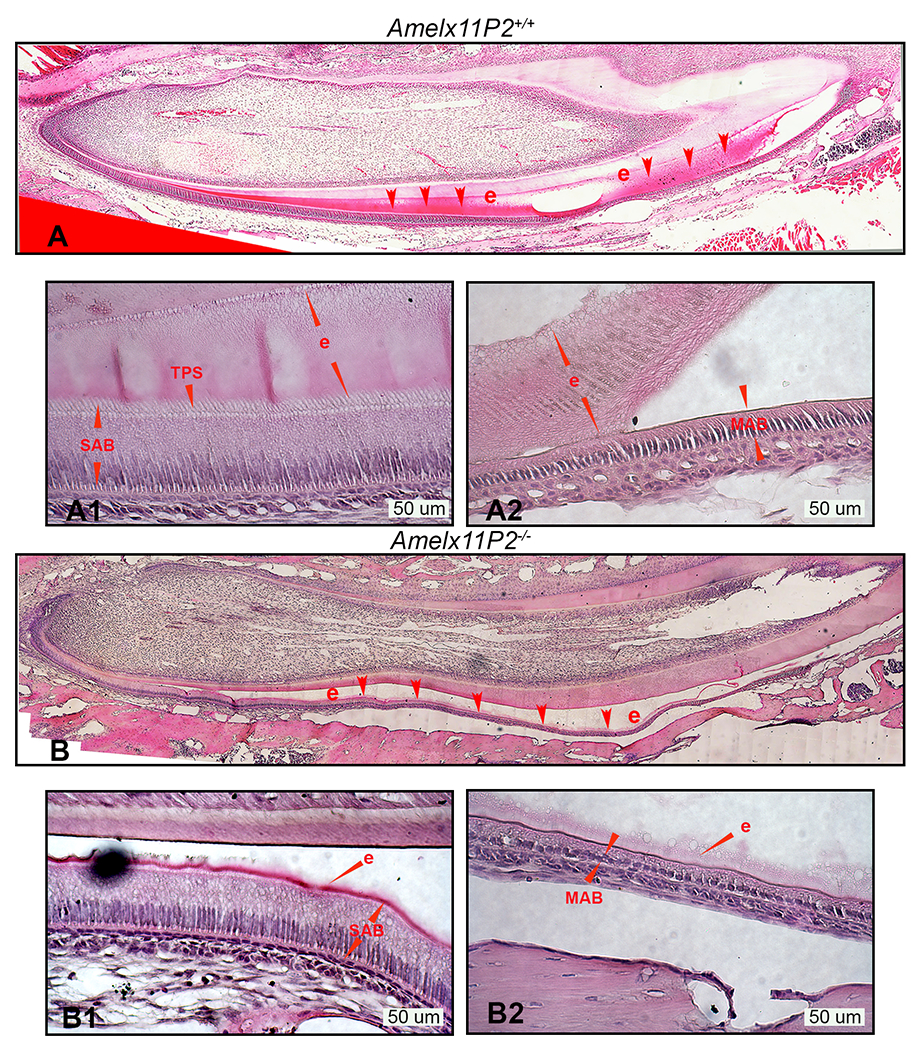
Histological assessment of Amelx11P2−/− mouse ameloblasts and developing enamel matrix. A) H&E analysis shows that Amelx11P2+/+ enamel (e, indicated by red arrows) stained in pink grew thick through secretory stage of development and then disappeared at the end of maturation stage as a consequence of decalcification processing. A1) Enlarged image shows the properly differentiated secretory ameloblasts (SAB) with elongated cell body and “picket-fence” appearance Tomes’ processes (TPS) at the their apical pole, and organic enamel matrix (e) adhered to ameloblasts and dentin matrix. A2) Enlarged image shows the wild-type maturation stage ameloblasts (MAB) and the full thickness of mineralizing enamel matrix (e). B) Histological image shows that the enamel layer (e) in Amelx11P2−/− mouse incisor is thinner and prone to detach from dentin matrix. B1) In Amelx11P2−/− mouse incisor sagittal section, SAB is shorter and enamel layer (e) is thinner. B2) Both MAB and enamel matrix (e) are thinner as compared to that in Amelx11P2+/+ mouse incisor.
These observations consistently demonstrated an indispensable role of 11P2 in the formation of normal enamel.
A reduced amount of amelogenin is found in the enamel matrix of Amelx11P2−/− mice.
To understand why enamel layer was thinner in Amelx11P2−/− mice, we first evaluated whether 11P2 deletion affected the expression levels of amelogenins and other major enamel matrix proteins. We collected dental epithelial cells at the secretory stage and measured the relative expression levels of genes encoding the major enamel matrix proteins (EMPs) and MMP20 by RT-PCR. Although amelogenin expression appeared lower in Amelx11P2−/− vs wild-type ameloblasts, the difference was not significant based on Student’s t-test. However, a significant downregulation of other EMP genes (Enam, Ambn, and Mmp20) was detected in Amelx11P2−/− SABs as compared to wild-type controls, with P< 0.05 by Student’s t-test (see supplement 2) . In situ hybridization RNAscope analysis was used to visualize the amelogenin mRNA in the progressively differentiated ameloblasts, which showed that amelogenin mRNA remarkably upregulated in the presecretory ameloblasts (PSA) (see Fig.5A and 5C) and even more in the secretory ameloblasts (SAB) (Fig.5B and 5D) in both Amelx11P2+/+ and Amelx11P2−/− mouse incisors. There was no significant difference in the expression levels of amelogenin between Amelx11P2+/+ and Amelx11P2−/− presecretory and secretory ameloblasts, although Amelx11P2−/− ameloblasts were shorter than control ameloblasts. Other cells in those incisal sections were stained negatively for amelogenin mRNA.
Fig.5.
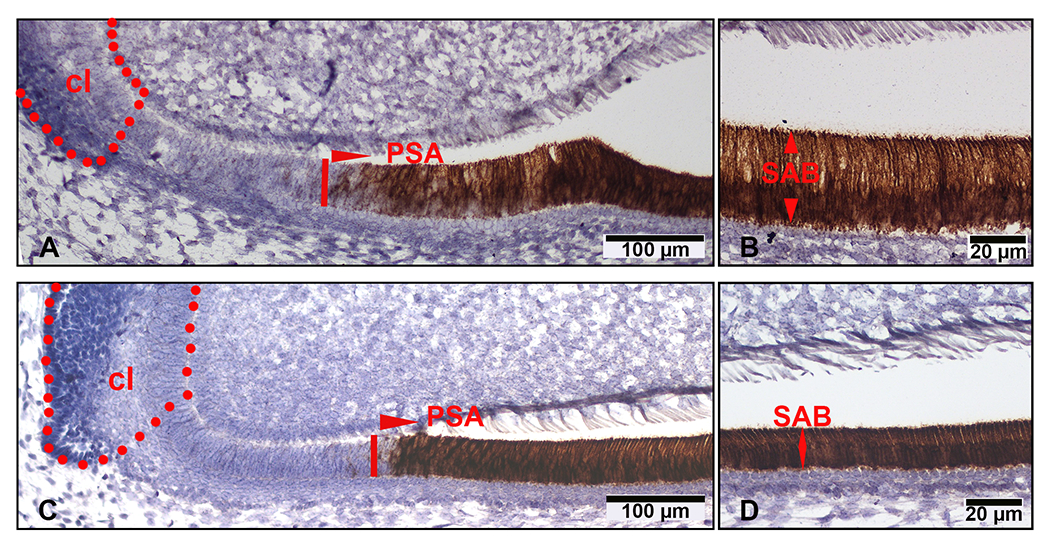
Assessment of amelgenin mRNA levels in Amelx11P2−/− mouse ameloblasts by in situ RNAscope. On the sagittal section of Amelx11P2+/+ mouse hemimandible, amelogenin mRNA positive signal (in brown) started to raise in presecretory ameloblasts (PSA) (A), then continuously increased in secretory ameloblasts (SAB) (B). The intensity of amelogenin mRNA positive signal (in brown) in Amelx11P2 −/− mouse presecretory ameloblasts (PSA) (C) and secretory ameloblasts (SAB) (D) was comparable with that in wild-type control ameloblasts. There was no positive signal in the dental epithelial cells resided in cervical loop region (cl) and mesenchymal cells in pulp tissue.
We also found that MMP20 extracted from the same amount of Amelx11P2+/+ and Amelx11P2−/− mouse enamel matrix exhibited similar ability to hydrolyze gelatin (see supplement 3). Thus, the reduced amount of amelogenin in Amelx11P2−/− enamel matrix was not the consequence of altered amelogenin gene expression or MMP20 efficiency.
To assess the impact of 11P2 deletion on amelogenin property in developing enamel, we microdissected the enamel matrix of incisors at the secretory stage of development from six Amelx11P2+/+ and six Amelx11P2−/− mice, and resolved it on SDS-PAGE under reducing conditions. As expected from the multiple splicing forms of amelogenin RNAs, protein post-translational modification and from enzymatic hydrolysis, multiple protein bands were detected in the enamel matrix of Amelx11P2+/+ mice (see Fig.6A). The most abundant band in Amelx11P2+/+ enamel sample migrated slightly faster than that of recombinant full-length rH174 which reflects the fact that the wild-type full-length amelogenin is phosphorylated. Several other intense bands appeared with molecular weights ranging from 20 to 23kDa. Those could be the hydrolysis products resulting from the removal of C-terminus 11aa or 25aa residues from full-length amelogenin. Overall, fewer proteins were detected in Amelx11P2−/− incisor enamel matrix. In particular, the band corresponding in size to recombinant mutant amelogenin rH174-P2 was absent. A 15kDa band was found in the enamel matrix from both Amelx11P2+/+ and Amelx11P2−/− mice (see Fig.6A). We assumed that this band resulted from the removal of C- and N-termini by MMP20, which we further confirmed by LC-MS/MS (data not shown).
Fig.6.
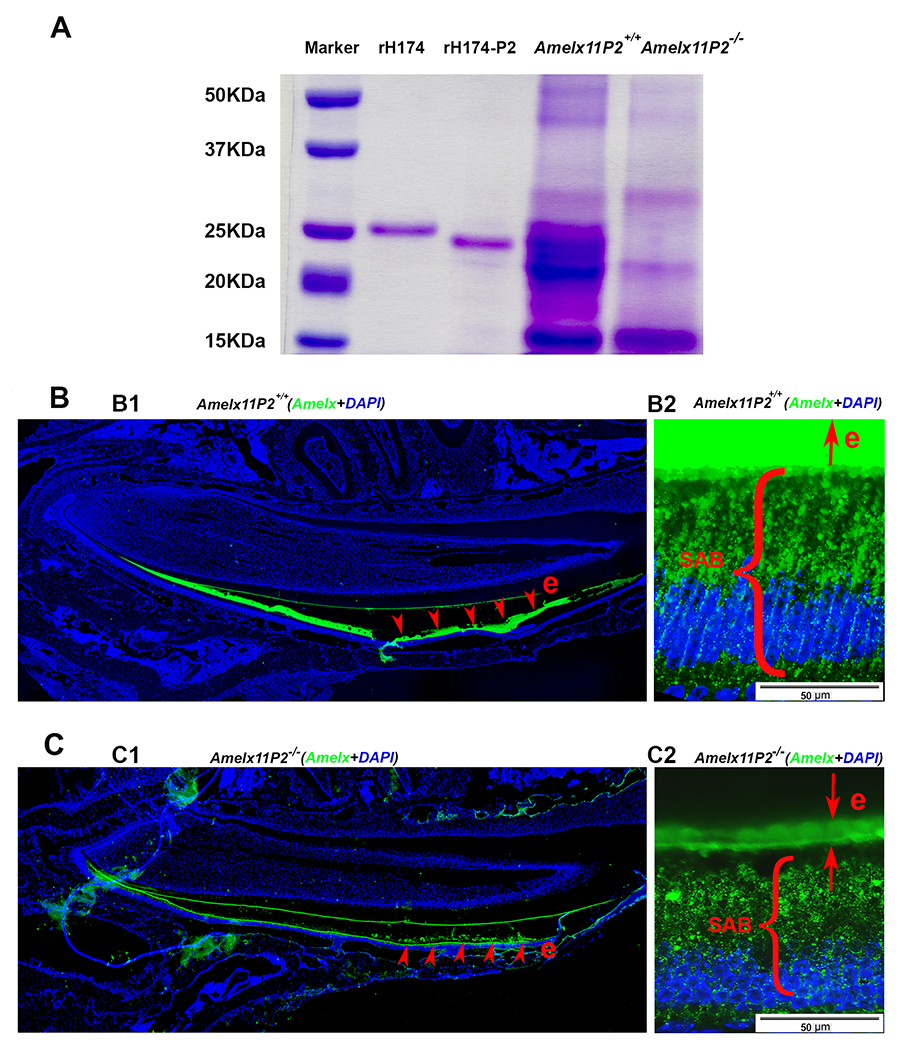
Assessment of effects of 11P2 deletion on the amelogenin proteins present in the developing enamel organ. A) SDS-PADG analysis shows that there was more protein content in Amelx11P2+/+ secretory stage enamel matrix as compared to Amelx11P−/− enamel matrix. Most of Amelx11P2+/+ matrix proteins migrated as proteins at size between 20-25 kDa. There was a heavy band about 22 kDa in Amelx11P2−/− enamel matrix. A band at about 14 kDa was revealed in both Amelx11P2+/+ and Amelx11P−/− enamel matrix. B1) Amelogenin immunostaining analysis on Amelx11P2+/+ mouse sagittal section shows that amelogenin signal (in green) was detected in the secretory and maturation stage of enamel matrix (e, pointed by the red arrows), but significantly reduced at the end of maturation stage. B2) Amelogenin immunostaining signal was very intense in Amelx11P2+/+ organic enamel matrix (e), and directionally distributed along the long-axis of secretory ameloblasts (SABs). C1) Amelogenin immunostaining analysis on Amelx11P2−/− mouse sagittal section shows that amelogenin signal (in green) was significantly reduced in the secretory and maturation stage of enamel matrix (e, pointed by the red arrows). C2) Amelogenin was immune localized in the thin Amelx11P−/− enamel matrix and in the cytosols in a disordered manner.
Next, we examined the sagittal sections of Amelx11P2+/+ and Amelx11P2−/− mouse hemimandibles by immunostaining for amelogenin. As compared to the full thickness of wild-type enamel matrix (see Fig.6B1), enamel matrix in Amelx11P2−/− mouse incisor was thinner although was stained positively for amelogenin (see Fig.6C1). Aside from the intense signal in the enamel matrix (e), amelogenin positive immunostaining puncta that lined up along the long axis of Amelx11P2+/+ secretory ameloblasts (SAB) (see Fig.6B2). However, amelogenin puncta were scarce in the mutant enamel matrix (e) and randomly dispersed in the cytosol of Amelx11P2−/− SAB (see Fig.6C2).
The absence of 11P2 accelerates MMP20-mediated amelogenin hydrolysis.
The results in which 11P2 deletion did not affect the expression of amelogenin in ameloblasts, but less amelogenin proteins were detected in enamel matrix, led us focus on the hydrolysis and stability of mutant amelogenin. MMP20 is the most dominant extracellular proteinase in the secretory stage of enamel matrix [46]. To test the sensitivity of the mutant amelogenin to MMP20, we incubated recombinant protein rH174 or rH174-P2 with MMP20 and assessed hydrolysis at multiple time points by SDS-PAGE under reducing conditions. Significant reduction of rH174-P2 was notable beginning at the 2-hour incubation timepoint when only a small amount of rH174 was hydrolyzed (see Fig.7A). Based on ImageJ densitometric analysis, about 12% of rH174 was hydrolyzed, while 55% of rH174-P2 was hydrolyzed. Hydrolysis of rH174-P2 amelogenin resulted in a protein less than 22 kDa corresponding to rH174-P2 lacking its C-terminus.
Fig.7.
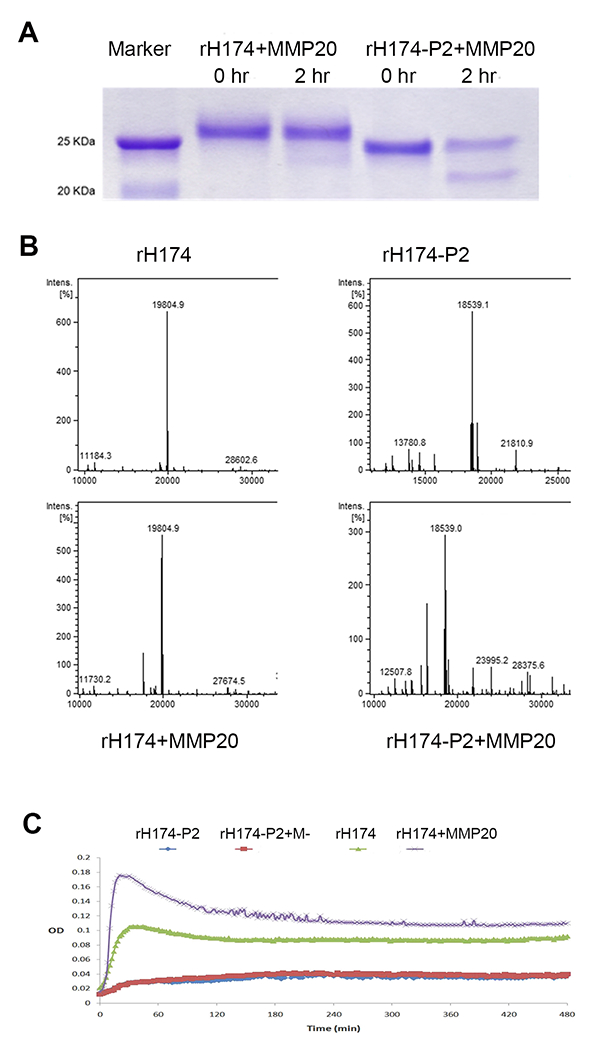
Assessment of the effects of 11P2 on MMP20-mediated amelogenin hydrolysis. A) After two hours of MMP20 hydrolysis, SDS-PAGE analysis shows that only a small amount of rH174 was hydrolyzed by MMP20, while about 50% of rH174-P2 was hydrolyzed resulting in a clear band at about 22 kDa. B) LC-MS/MS analysis shows that after incubation with MMP20, the peak of remaining rH174 reduced 15% compared to that of initial peak of rH174. However, the peak of remaining rH174-P2 was about 50% of initial rH174-P2’s. C) Turbidity analysis shows that rH714 in 50 mM Tris-HCl/10 mM CaCl2/150 mM NaCl solution was prone to self-assemble and form supramolecules that increase the turbidity of solution. MMP20 hydrolysis enhanced the aggregation of supramolecules. rH174-P2 adapted different conformation, which had reduced propensity to self-assemble to form large size supramolecules with and without MMP20 hydrolysis.
This observation motivated us to compare the products of rH174 and rH174-P2 hydrolysis by MMP20 using a quantitative LC-MS/MS approach. The data showed that the intensity of the remaining full length amelogenin (rH174 at predicted m/z 19,805 dalton) had minor change (15% reduction, intensity changed from 650 to 550) after MMP20 digestion, but mutant amelogenin (rH174-P2 at predicted m/z 18,540 dalton) had about 50% reduction (intensity changed from 600 to 300) as a consequence of MMP20 digestion (Fig.7B). After MMP20 digestion, MS spectra showed extra peaks at lower m/z (17,589.7 dalton for rH174, and 16323.6 dalton for rH174-P2), which is the hydrolyzed amelogenins with the removal of C-termini by MMP20 (see Fig.7B). Both SDS-PAGE and LC-MS/MS data demonstrated that human full-length recombinant amelogenin (rH174) is more resistant (or stable) to MMP20 hydrolysis than recombinant amelogenin lacking 11P2 (rH174-P2).
We also utilized AFM to assess the assembling ability of rH174 and rH174-P2 in response to MMP20 hydrolysis. There was apparently more spacing between supramolecular assemblies in rH174-P2 hydrolysis condition, consistent with our observation that rH174-P2 was hydrolyzed and consumed more rapidly than rH174. In addition, we saw fewer finely assembled fibrils but more nanospheres in the rH174-P2 than that in rH174 (see supplement 4 ), implying that domains other than 11P2 might be present in amelogenin to collaboratively assist the self-assembly behavior during amelogenesis.
Previous studies have shown that full-length amelogenin can self-assemble to form aggregates/or supramolecules in an aqueous conditions in vitro [47–49]. We tested whether this aggregation property of amelogenin would be affected by 11P2 deletion through turbidity assay. Our analyses showed that rH174 was prone to form aggregates, and MMP20 hydrolysis of rH174 (likely through removal of C-terminus) facilitated the formation of larger size and more persistent supramolecules, indicating by the increased turbidity. In contrast, rH174-P2 could not effectively form aggregates under the same condition incubated with and without MMP20 (see Fig.7C).
This data indicates that 11P2 can mediate the amelogenin self-assembly to render supramolecular amelogenins less accessible to MMP20 docking, and a removal of the hydrophilic C-terminus by MMP20 further stabilizes the supramolecular amelogenins likely through increased intermolecular hydrophobic forces. The aggregation of the amelogenin (maybe through amyloid assembly initially) is important in enamel matrix development at the secretory stage as it may shield amelogenin from rapid hydrolysis, allowing amelogenin template hydroxyapatite growth and scaffold matrix expansion.
Deletion of 11P2 disturbs the assembly of amelogenin and amyloid-like fibrils in mouse developing enamel matrix.
In the secretory stage of enamel matrix of Amelx11P2+/+ and Amelx11P2−/− mouse incisors stained by amelogenin antibody, through a W1-SoRA spinning disk super resolution confocal microscope, we found a remarkable difference in amelogenin assembly (see Fig.8). Amelogenin in Amelx11P2+/+ enamel matrix assembled to form highly ordered architecture, either surrounding hexagon-shape objects, like sheath capsulates enamel rods (indicated by red circle in Fig.8A2) or lining up as filaments across the full thickness of enamel layer (indicated by red dotted line) (Fig. 8A2). In the thin Amelx11P2−/− enamel layer, amelogenin staining was irregularly scattered and no enamel rods were recognized (Fig.8B1 and B2).
Fig.8.
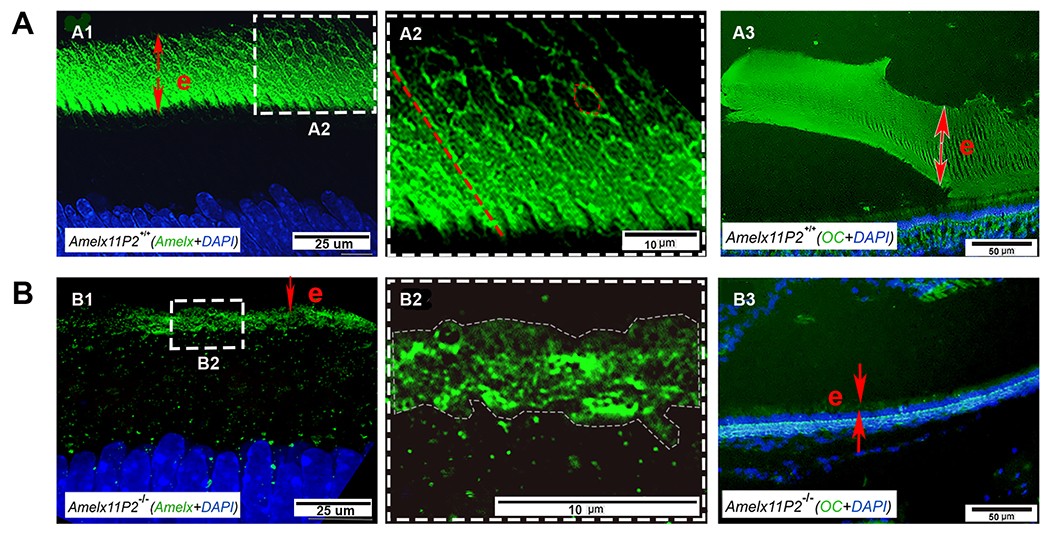
Assessment of the effects of 11P2 on amelogenin assembly in the developing enamel matrix. Super-resolution confocal microscopic image shows that amelogenin was detected in the secretory stage enamel matrix (e) (A1). Amelogenin positive signal either distributed along the long-axis of enamel rods like fibrils (indicated by the dotted red line) or encompassed the objects in the shape of hexagon (indicated by dotted red hexagon) in Amelx11P2+/+ enamel matrix (A2). A3) OC antibody immunostaining analysis shows the positive signal (in green) colocalized with the aligned enamel rods in enamel matrix (e). B1) There is less amelogenin immunostaining signal in the thin Amelx11P2−/− enamel matrix (e). B2) Enlarged image shows that amelogenin distributed in enamel matrix without a pattern. B3) No OC positive immunostaining signal was found in Amelx11P2−/− mouse enamel matrix (e).
At the maturation stage of enamel matrix, we found that the positively immunostained amyloid-like fibrils, indicated by OC antibody, align parallelly to the longitudinal enamel rods in Amelx11P2+/+ mice (Fig.8A3). These fibrils are definitely sustainable in the secretory stage of enamel development. Positively immunostained amyloid-like fibrils in maturation Amelx11P2−/− mouse enamel matrix were not identified (Fig.8B3).
Discussion:
Enamel is the hardest tissue in vertebrates because it is composed of more than 95% highly organized hydroxyapatite crystals by weight. These crystals initially form and grow longitudinally in a protein-rich matrix during the secretory stage of amelogenesis [50]. Amelogenins, the most abundant proteins in the enamel matrix, can self-assemble into higher-order supramolecules to guides the directional growth of hydroxyapatite crystals [35, 38, 47, 49, 51–53]. The evolutionarily conserved and hydrophobic N-terminus of amelogenin ( 1-33 aa) has been proposed to account for amelogenin’s self-assembling ability [34]. Habelitz and Li have recently narrowed down a subdomain (14P2) of the N-terminus of amelogenin that is capable of self-assembling into ß-sheets and possibly mediates the formation of amyloid-like fibrils by recombinant amelogenins [35]. In this study, we provided in vivo data for the first time to demonstrate that the core motif of this peptide, which we named 11P2, is essential for the proper enamel matrix development, and in vitro evidence to shed light on the physiological roles of the11P2 in stabilizing amelogenin fibrous supramolecules from enzymatic degradation. Our findings elucidate how this key motif contributes to amelogenin’s roles in templating the crystal growth and framing the formation of the full thickness of enamel matrix in both in vitro and in vivo model systems.
To investigate the physiological roles of 11P2 in enamel development, CRISPR-Cas9 mediated genome editing approach allowed us to delete 33bp from amelogenin exon 3 without compromising the reading frame of the rest of amelogenin gene and the immediate splicing sites at the upstream and downstream of amelogenin exon 3. This site-specific deletion provided us a mouse model harboring a full-length amelogenin without residue 8-18 (GSPGYINFSYE) within the wild-type amelogenin gene. We characterized the mutant amelogenin (amelogenin without 11P2) expression through RT-PCR and in situ hybridization using probes recognizing a large portion of exon 6, and the proteins from enamel matrix through SDS-PAGE and western blot. Analyses from both mRNA and protein levels demonstrated that 11P2 deletion did not interfere with the expression of the rest of amelogenin in this mouse model. Nevertheless, rough surfaced, remarkably thin, hypomineralized and aprismatic enamel layer was formed on both incisors and molars of Amelx11P2−/− mice. In line with remarkable thin enamel matrix layer at both secretory and maturation stages, we detected reduced amount of amelogenin proteins and no organized amelogenin fibrous nanostructures in the thin layer of Amelx11P2−/− enamel.
We also compared the enamel of Amelx11P2−/− mice to that of amelogenin null (Amelx−/− ) mice. With the deletion of all amelogenins, the thickness of Amelx−/− enamel layer was about 10% of normal enamel matrix, while Amelx11P2−/− was about 30% of normal enamel. In Amelx11P2−/− mouse, enamel developed particulate- and flake-like crystals, but no fibril- or ribbon-like crystals. It seems that the crystallites in Amelx11P2−/− mouse enamel matrix had been held together by proteins during their development, but no clear guidance had been given to orient the crystals’ growth direction. While in Amelx−/− mouse enamel, a layer of octacalcium phosphate crystals grew like fan-like structures that developed on top of a thin layer of prismless enamel at the dentin-enamel junction, completely in line with what Simmer et al. have previously described [54]. The structures of crystals in Amelx−/− enamel suggested that there was no sufficient proteins in matrix to cushion and confine the development of enamel rods. This comparison also suggest the roles of amelogenin as a scaffold protein and its N-termini are critical for amelogenin scaffold assembly.
Although 11P2 deletion had no significantly effect on the expression level of amelogenin in Amelx11P2−/− presecretory and secretory ameloblasts, there was nearly 40% reduction of enamelin (Enam) and ameloblastin (Ambn) in Amelx11P2−/− SABs compared to Amelx11P2+/+ SABs. This reduction could be a secondary effect of thinner enamel matrix or impaired secretion of ameloblasts. Genes Enam and Ambn localize in the same gene cluster on mouse chromosome 5 [55], and may therefore be coregulated. We also detected a down-regulation of MMP20,although MMP20 hydrolysis efficiency remained unchanged in Amelx11P2−/− enamel matrix in comparison to that in controls as indicated by gelatin zymography assays. Thus, we concluded that this reduced amount of amelogenin in Amelx11P2−/− enamel matrix is neither a consequence of amelogenin downregulation nor increased MMP20 expression/hydrolysis efficiency. This led us to investigate the impact of 11P2 on MMP20-mediated amelogenin hydrolysis.
It is well known that amelogenin and MMP20 interaction is necessary for the normal enamel formation [4, 46, 56, 57]. When incubated with MMP20, recombinant mutant amelogenin rH174-P2 was processed more rapidly than recombinant wild-type amelogenin rH174. The results raised the question of whether the difference in the primary amino acid sequences between rH174 and rH174-P2 led to a conformation change, and whether this conformation change alters hydrolysis by MMP20.
Previous studies have shown that amelogenins are capable of self-assembling to nanospheres/aggregates or ß-sheet and nanoribbons, depending on the ionic compositions in assembling solution [26]. Using an aqueous solution (3.3 mMCaCl2 and 2.1 mM KH2PO4) that has been reported to facilitate nanoribbon formation, we did see that rH174 effectively assembled into fibrils of up to hundred micrometers in length, while rH174-P2 assembled into a mixture of nanospheres and shorter nanoribbons. During in-situ MMP20 hydrolysis experiments, we observed more space, less ribbons and more larger-size aggregates developing in the cocktail of rH174-P2+MMP20 as compared to rH174-P2 alone or rH174+MMP20, indicating that amelogenin conformation does affect MMP20 hydrolysis, as assembled amelogenin ribbons are more resistant to MMP20 hydrolysis. In another solution (50 mM Tris-HCl, 10 mM CaCl2, 150 mM NaCl, and 10 nM ZnCl2), rH174 rapidly formed supramolecular nanospheres/aggregates in suspension and displayed increased turbidity under spectrometer. MMP20 co-incubation with rH174 generated even more large-sized aggregates presenting with greater turbidity. However, under the same condition, the average turbidity of both rH174-P2 and rH174-P2+MMP20 solution was relatively decreased. Regardless of the composition in solution, rH174-P2 had a lower tendency to self-assemble into higher order nanoscale assemblies, presumably leading to an increased accessibility of amelogenin to MMP20 docking and hydrolysis.
Interestingly, in the normal developing mouse enamel we found an organized amelogenin distribution pattern and orderly aligned nanofibers, which coexist with secretory stage specific proteinase MMP20. Evidence for the presence of amyloid-like assemblies in the normal maturation stage enamel matrix is physiologically significant. What we have learned from those neurodegenerative amyloid-like fibrils is that this type of structure is generally stable and resistant to enzymatic degradation [58, 59]. The existence of amyloid-like fibrils in the normal mature enamel matrix indicate that this type of assembly allows some amelogenin domains to remain between enamel crystals and provide support for hydroxyapatite crystals throughout the lifetime of the tooth. The residues GHPGYINFSYE have been found in human tooth fossils [60] and its homolog GSPGYINFSYE were identified in the erupted wild-type mouse enamel by mass spectrometry (unpublished data from our laboratory), both indicating that the amyloid-like fibrils formed by 11P2 motifs are resistant to MMP20, KLK4 hydrolysis, and even proteinases present in saliva. KLK4 is a less selective but more potent proteinase prevailing in the maturation stage enamel matrix [61]. Identification of 11P2 residues in the erupted enamel provides compelling evidence to highlight the physiological functions of amyloid-like fibril assemblies in normal development.
Ser-16, the only phosphorylation site of amelogenin [23, 62], is one of the residues within 11P2 motif. Researchers has done exceptional investigations to unfold the roles of Ser-16 on enamel formation. Using in vitro systems, Margolis and coworkers have demonstrated that phosphorylation of amelogenin at Ser-16 stabilizes amorphous calcium phosphate (ACP), organizes the initial mineral clusters and regulates interactions of amelogenin with hydroxyapatites [25, 27, 63, 64]. These are all necessary preceding events for the formation of ordered and aligned HA crystals. Furthermore, mice harboring the amelogenin gene with Ser-16 replaced with Ala-16 lack enamel rods within a greatly reduced layer of enamel. This defect is associated with a significant acceleration of ACP-to-apatitic crystal transformation [26]. We also found that phosphorylation of Ser-16 is critical for amyloid-like fibril assembly of 14P2 [35]. Together, we speculate that Ser-16 ,as an element of 11P2, is important not only for the formation of hydroxyapatite crystal arrays/enamel rods, but also for the regulation of amelogenin self-assembly into protein ribbons and the construction of a sustainable scaffold in the developing enamel matrix. Consistent with this speculation, Amelx11P2−/− mice had a significantly reduced amelogenin content in the thin and disorganized enamel matrix.
Even in the absence of 11P2, amelogenin rH174-P2 still has some degree of self-assembly capacity based on our AFM analysis, suggesting that although 11P2 is critical, additional assembly domains might also exist to participate in hierarchical self-assembly of amelogenin. Previous studies have identified a motif Y/F-X-Y/L/F-X-Y/F common to both amelogenin and ameloblastin, which mediates EMPs self-assembly into supramolecular structures [29]. There are two Y/F-X-Y/L/F-X-Y/F motifs at the N-terminus of amelogenin. One is YINFSY that localizes within 11P2 (or 14P2) GHPGYINFSYE, the second one is YTSYGY, residue 34 to 39 of full length human amelogenin. Further in vivo studies are warranted that will examine the physiological functions of motif YTSYGY, and the potential interaction between these two domains in enamel matrix construction. Deletion of 11P2 may also affect the overall conformation of amelogenin, therefore change amelogenin’s self-assembling behavior.
In addition to the enamel matrix phenotype, we also found that secretory ameloblasts in Amelx11P2−/− mice were shorter and had lost Tomes’ processes, which might be a consequence of decreased thickness of enamel matrix, or dysregulation of amelogenin post-translational modification in the absence of Ser-16. Posttranslational modifications including protein phosphorylation are critical for proper protein folding and secretion [65].
Taking together, our finding demonstrate that the physiological functions of 11P2, a subdomain of N-terminus of amelogenin, include mediating self-assembly of amelogenin into ribbons and stabilizing amelogenin assemblies from hydrolysis by MMP20 and KLK4. This study aids our understanding how amelogenins template the longitudinal growth of hydroxyapatite crystals and strengthen the mechanical frameworks for enamel matrix development.
Enamel is a non-regenerative tissue since ameloblasts commit to apoptosis before teeth erupt. Keen understanding of the physiological significance of amyloid-like fibril assembly motif of amelogenin and of the major protein elements templating hydroxyapatite crystallization, will pave the way to develop strategies for biomimetic peptide-mediated enamel regeneration.
Supplementary Material
Supplement 2: Semi-quantitative PCR shows the relative expression levels of amelogenin (Amlex), enamelin (Enam), ameloblastin (Ambn), and MMP20 (Mmp20) in Amelx11P2−/− mouse SABs as compared to Amelx11P2+/+ SABs. The expression levels of Enam, Ambn and Mmp20 were reduced in Amelx11P2−/− SABs vs Amelx11P2+/+ SABs, n=4, P<0.05 by Student t-test using SPSS Statistics package 19.
Supplement 3: 11P2 deletion does not affect the enzymatic activity of MMP20 in the developing enamel matrix.
Supplement 4: Recombinant protein rH174-P2 (without P2) is hydrolyzed faster by MMP20 and forms less fibril-like structures as compared to protein rH174 (with P2).
Acknowledgement:
Thanks to Dr. Junli Zhang in Transgenic Core Facility at Gladstone Institutes for technique support to generate Amelx11P2−/− mouse model; to Dr. Seth Hogg at Micro Photonics for X-ray micro-tomography scans.
This research was funded by NIDCR grants R01 DE025709-03 To Habelitz and Li and R01 DE027076 to YZ.
Footnotes
All authors have no conflict of interest to declare. Data were analyzed at least by two individuals, one was aware of the group allocation, others were not.
References
- 1.Panchenko AR and Babu MM, Editorial overview: Linking protein sequence and structural changes to function in the era of next-generation sequencing. Curr Opin Struct Biol, 2015. 32: p. viii–x. [DOI] [PubMed] [Google Scholar]
- 2.Ruan Q and Moradian-Oldak J, Amelogenin and Enamel Biomimetics. J Mater Chem B, 2015. 3: p. 3112–3129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Robinson C, et al. , Crystal growth in dental enamel: the role of amelogenins and albumin. Adv Dent Res, 1996. 10(2): p. 173–9; discussion 179-80. [DOI] [PubMed] [Google Scholar]
- 4.Gibson CW, et al. , Amelogenin-deficient mice display an amelogenesis imperfecta phenotype. J Biol Chem, 2001. 276(34): p. 31871–5. [DOI] [PubMed] [Google Scholar]
- 5.Gibson CW, et al. , DNA sequences of amelogenin genes provide clues to regulation of expression. Eur J Oral Sci, 1998. 106 Suppl 1: p. 292–8. [DOI] [PubMed] [Google Scholar]
- 6.Gibson CW, et al. , Bovine amelogenin message heterogeneity: alternative splicing and Y-chromosomal gene transcription. Biochemistry, 1992. 31(35): p. 8384–8. [DOI] [PubMed] [Google Scholar]
- 7.Fincham AG, et al. , Amelogenins. Sequence homologies in enamel-matrix proteins from three mammalian species. Biochem J, 1983. 211(1): p. 149–54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Hu CC, et al. , Cloning, cDNA sequence, and alternative splicing of porcine amelogenin mRNAs. J Dent Res, 1996. 75(10): p. 1735–41. [DOI] [PubMed] [Google Scholar]
- 9.Sire JY, et al. , Evolutionary story of mammalian-specific amelogenin exons 4, “4b”, 8, and 9. J Dent Res, 2012. 91(1): p. 84–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Li W, et al. , Identification of two additional exons at the 3’ end of the amelogenin gene. Arch Oral Biol, 1998. 43(6): p. 497–504. [DOI] [PubMed] [Google Scholar]
- 11.Li R, Li W, and DenBesten PK, Alternative splicing of amelogenin mRNA from rat incisor ameloblasts. J Dent Res, 1995. 74(12): p. 1880–5. [DOI] [PubMed] [Google Scholar]
- 12.Pandya M and Diekwisch TGH, Enamel biomimetics-fiction or future of dentistry. Int J Oral Sci, 2019. 11(1): p. 8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Yamakoshi Y, Porcine Amelogenin : Alternative Splicing, Proteolytic Processing, Protein - Protein Interactions, and Possible Functions. J Oral Biosci, 2011. 53(3): p. 275–283. [PMC free article] [PubMed] [Google Scholar]
- 14.Gibson CW, Kulkarni AB, and Wright JT, The use of animal models to explore amelogenin variants in amelogenesis imperfecta. Cells Tissues Organs, 2005. 181(3-4): p. 196–201. [DOI] [PubMed] [Google Scholar]
- 15.Delgado S, Ishiyama M, and Sire JY, Validation of amelogenesis imperfecta inferred from amelogenin evolution. J Dent Res, 2007. 86(4): p. 326–30. [DOI] [PubMed] [Google Scholar]
- 16.Hart PS, et al. , Amelogenesis imperfecta phenotype-genotype correlations with two amelogenin gene mutations. Arch Oral Biol, 2002. 47(4): p. 261–5. [DOI] [PubMed] [Google Scholar]
- 17.Pugach MK, et al. , The amelogenin C-terminus is required for enamel development. J Dent Res, 2010. 89(2): p. 165–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Aoba T, et al. , Selective adsorption of porcine-amelogenins onto hydroxyapatite and their inhibitory activity on hydroxyapatite growth in supersaturated solutions. Calcif Tissue Int, 1987. 41(5): p. 281–9. [DOI] [PubMed] [Google Scholar]
- 19.Moradian-Oldak J, et al. , Analysis of self-assembly and apatite binding properties of amelogenin proteins lacking the hydrophilic C-terminal. Matrix Biol, 2002. 21(2): p. 197–205. [DOI] [PubMed] [Google Scholar]
- 20.Shaw WJ and Ferris K, Structure, orientation, and dynamics of the C-terminal hexapeptide of LRAP determined using solid-state NMR. J Phys Chem B, 2008. 112(51): p. 16975–81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Pandya M, et al. , Posttranslational Amelogenin Processing and Changes in Matrix Assembly during Enamel Development. Front Physiol, 2017. 8: p. 790. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Takagi T, et al. , Complete amino acid sequence of amelogenin in developing bovine enamel. Biochem Biophys Res Commun, 1984. 121(2): p. 592–7. [DOI] [PubMed] [Google Scholar]
- 23.Fincham AG and Moradian-Oldak J, Amelogenin post-translational modifications: carboxy-terminal processing and the phosphorylation of bovine and porcine “TRAP” and “LRAP” amelogenins. Biochem Biophys Res Commun, 1993. 197(1): p. 248–55. [DOI] [PubMed] [Google Scholar]
- 24.Kjoelby M, et al. , Degradation of the dental basement membrane during mouse tooth development in vitro. Int J Dev Biol, 1994. 38(3): p. 455–62. [PubMed] [Google Scholar]
- 25.Yamazaki H, et al. , Protein Phosphorylation and Mineral Binding Affect the Secondary Structure of the Leucine-Rich Amelogenin Peptide. Front Physiol, 2017. 8: p. 450. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Shin NY, et al. , Amelogenin phosphorylation regulates tooth enamel formation by stabilizing a transient amorphous mineral precursor. J Biol Chem, 2020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Le Norcy E, et al. , Effect of phosphorylation on the interaction of calcium with leucine-rich amelogenin peptide. Eur J Oral Sci, 2011. 119 Suppl 1: p. 97–102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Wiedemann-Bidlack FB, et al. , Effects of phosphorylation on the self-assembly of native full-length porcine amelogenin and its regulation of calcium phosphate formation in vitro. J Struct Biol, 2011. 173(2): p. 250–60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Wald T, et al. , Intrinsically disordered proteins drive enamel formation via an evolutionarily conserved self-assembly motif. Proc Natl Acad Sci U S A, 2017. 114(9): p. E1641–E1650. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Bromley KM, et al. , Dissecting amelogenin protein nanospheres: characterization of metastable oligomers. J Biol Chem, 2011. 286(40): p. 34643–53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Du C, et al. , Supramolecular assembly of amelogenin nanospheres into birefringent microribbons. Science, 2005. 307(5714): p. 1450–4. [DOI] [PubMed] [Google Scholar]
- 32.Zhang X, et al. , Amelogenin supramolecular assembly in nanospheres defined by a complex helix-coil-PPII helix 3D-structure. PLoS One, 2011. 6(10): p. e24952. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Paine ML, et al. , Enamel biomineralization defects result from alterations to amelogenin self-assembly. J Struct Biol, 2000. 132(3): p. 191–200. [DOI] [PubMed] [Google Scholar]
- 34.Dissanayake SSM, et al. , Identification of Key Functional Motifs of Native Amelogenin Protein for Dental Enamel Remineralisation. Molecules, 2020. 25(18). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Carneiro KM, et al. , Amyloid-like ribbons of amelogenins in enamel mineralization. Sci Rep, 2016. 6: p. 23105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Lee J, et al. , Tyrosine-Rich Peptides as a Platform for Assembly and Material Synthesis. Adv Sci (Weinh), 2019. 6(4): p. 1801255. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Warren JJ, Winkler JR, and Gray HB, Redox properties of tyrosine and related molecules. FEBS Lett, 2012. 586(5): p. 596–602. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Bonde J and Bulow L, Random mutagenesis of amelogenin for engineering protein nanoparticles. Biotechnol Bioeng, 2015. 112(7): p. 1319–26. [DOI] [PubMed] [Google Scholar]
- 39.Kayed R, et al. , Fibril specific, conformation dependent antibodies recognize a generic epitope common to amyloid fibrils and fibrillar oligomers that is absent in prefibrillar oligomers. Mol Neurodegener, 2007. 2: p. 18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Kayed R, et al. , Common structure of soluble amyloid oligomers implies common mechanism of pathogenesis. Science, 2003. 300(5618): p. 486–9. [DOI] [PubMed] [Google Scholar]
- 41.Smith CE, et al. , Relationships between protein and mineral during enamel development in normal and genetically altered mice. Eur J Oral Sci, 2011. 119 Suppl 1: p. 125–35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Habelitz S, Materials engineering by ameloblasts. J Dent Res, 2015. 94(6): p. 759–67. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Smith CE and Nanci A, A method for sampling the stages of amelogenesis on mandibular rat incisors using the molars as a reference for dissection. Anat Rec, 1989. 225(3): p. 257–66. [DOI] [PubMed] [Google Scholar]
- 44.Pfaffl MW, A new mathematical model for relative quantification in real-time RT-PCR. Nucleic Acids Res, 2001. 29(9): p. e45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Cannan WJ and Pederson DS, Mechanisms and Consequences of Double-Strand DNA Break Formation in Chromatin. J Cell Physiol, 2016. 231(1): p. 3–14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Bartlett JD and Simmer JP, Proteinases in developing dental enamel. Crit Rev Oral Biol Med, 1999. 10(4): p. 425–41. [DOI] [PubMed] [Google Scholar]
- 47.Fincham AG, et al. , Evidence for amelogenin “nanospheres” as functional components of secretory-stage enamel matrix. J Struct Biol, 1995. 115(1): p. 50–9. [DOI] [PubMed] [Google Scholar]
- 48.Moradian-Oldak J, et al. , A review of the aggregation properties of a recombinant amelogenin. Connect Tissue Res, 1995. 32(1–4): p. 125–30. [DOI] [PubMed] [Google Scholar]
- 49.Fang PA, et al. , Hierarchical self-assembly of amelogenin and the regulation of biomineralization at the nanoscale. Proc Natl Acad Sci U S A, 2011. 108(34): p. 14097–102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Warshawsky H, Organization of crystals in enamel. Anat Rec, 1989. 224(2): p. 242–62. [DOI] [PubMed] [Google Scholar]
- 51.Fincham AG, Moradian-Oldak J, and Simmer JP, The structural biology of the developing dental enamel matrix. J Struct Biol, 1999. 126(3): p. 270–99. [DOI] [PubMed] [Google Scholar]
- 52.Margolis HC, Beniash E, and Fowler CE, Role of macromolecular assembly of enamel matrix proteins in enamel formation. J Dent Res, 2006. 85(9): p. 775–93. [DOI] [PubMed] [Google Scholar]
- 53.Bai Y, et al. , Protein nanoribbons template enamel mineralization. Proc Natl Acad Sci U S A, 2020. 117(32): p. 19201–19208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Smith CE, et al. , Ultrastructure of early amelogenesis in wild-type, Amelx(−/−), and Enam(−/−) mice: enamel ribbon initiation on dentin mineral and ribbon orientation by ameloblasts. Mol Genet Genomic Med, 2016. 4(6): p. 662–683. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Kawasaki K and Weiss KM, Mineralized tissue and vertebrate evolution: the secretory calcium-binding phosphoprotein gene cluster. Proc Natl Acad Sci U S A, 2003. 100(7): p. 4060–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Caterina JJ, et al. , Enamelysin (matrix metalloproteinase 20)-deficient mice display an amelogenesis imperfecta phenotype. J Biol Chem, 2002. 277(51): p. 49598–604. [DOI] [PubMed] [Google Scholar]
- 57.Fukae M and Tanabe T, Degradation of enamel matrix proteins in porcine secretory enamel. Connect Tissue Res, 1998. 39(1-3): p. 123–9; discussion 141-9. [DOI] [PubMed] [Google Scholar]
- 58.Rambaran RN and Serpell LC, Amyloid fibrils: abnormal protein assembly. Prion, 2008. 2(3): p. 112–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Chen GF, et al. , Amyloid beta: structure, biology and structure-based therapeutic development. Acta Pharmacol Sin, 2017. 38(9): p. 1205–1235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Castiblanco GA, et al. , Identification of proteins from human permanent erupted enamel. Eur J Oral Sci, 2015. 123(6): p. 390–5. [DOI] [PubMed] [Google Scholar]
- 61.Scully JL, et al. , Enamel matrix serine proteinase 1: stage-specific expression and molecular modeling. Connect Tissue Res, 1998. 39(1-3): p. 111–22; discussion 141-9. [DOI] [PubMed] [Google Scholar]
- 62.Strawich E and Glimcher MJ, Synthesis and degradation in vivo of a phosphoprotein from rat dental enamel. Identification of a phosphorylated precursor protein in the extracellular organic matrix. Biochem J, 1985. 230(2): p. 423–33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Connelly C, et al. , Dynamic interactions of amelogenin with hydroxyapatite surfaces are dependent on protein phosphorylation and solution pH. Colloids Surf B Biointerfaces, 2016. 148: p. 377–384. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Fang PA, et al. , CryoTEM study of effects of phosphorylation on the hierarchical assembly of porcine amelogenin and its regulation of mineralization in vitro. J Struct Biol, 2013. 183(2): p. 250–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Caporaso GL, et al. , Protein phosphorylation regulates secretion of Alzheimer beta/A4 amyloid precursor protein. Proc Natl Acad Sci U S A, 1992. 89(7): p. 3055–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplement 2: Semi-quantitative PCR shows the relative expression levels of amelogenin (Amlex), enamelin (Enam), ameloblastin (Ambn), and MMP20 (Mmp20) in Amelx11P2−/− mouse SABs as compared to Amelx11P2+/+ SABs. The expression levels of Enam, Ambn and Mmp20 were reduced in Amelx11P2−/− SABs vs Amelx11P2+/+ SABs, n=4, P<0.05 by Student t-test using SPSS Statistics package 19.
Supplement 3: 11P2 deletion does not affect the enzymatic activity of MMP20 in the developing enamel matrix.
Supplement 4: Recombinant protein rH174-P2 (without P2) is hydrolyzed faster by MMP20 and forms less fibril-like structures as compared to protein rH174 (with P2).