

Abstract
Hepatocyte Growth Factor (HGF), the ligand for the MET receptor tyrosine kinase, is a tumor-promoting factor that is abundant in the tumor microenvironment. Proteolytic activation of inactive pro-HGF by one or more of the serine endopeptidases matriptase, hepsin, and HGF activator (HGFA) is the rate-limiting step in HGF/MET signaling. Herein, we have rationally designed a novel class of sidechain cyclized macrocyclic peptide inhibitors. The new series of cyclic tripeptides have superior metabolic stability and significantly improved pharmacokinetics in mice relative to corresponding linear peptides. We identified lead compound VD2173 which potently inhibits matriptase and hepsin which was tested in parallel alongside acyclic inhibitor ZFH7116 using both in vitro and in vivo models of lung cancer. We demonstrated that both compounds block pro-HGF activation, abrogate HGF-mediated wound healing and overcome resistance to EGFR and MET-targeted therapy in lung cancer models. Furthermore, VD2173 inhibited HGF-dependent growth of lung cancer tumors in mice.
Graphical Abstract
1. INTRODUCTION
Recent advances in the targeted treatment of lung cancer have improved the outcome of lung cancer patients1. However, the five-year survival rates remain low at 57% for patients with localized disease and 5% for patients diagnosed with metastatic disease in the USA1. Therapies targeting programmed death-ligand 1 (PD-L1), epidermal growth factor receptor (EGFR) mutations, anaplastic lymphoma kinase (ALK) rearrangements, ROS1 rearrangements, and B-Raf protein (BRAF) mutations have been developed. Although many patients initially respond to these therapies, inevitably almost all patients develop resistance2, 3. EGFR mutations are found in 10-20% of all non-small-cell lung cancer (NSCLC) patients and 19-25% of patients with lung adenocarcinoma 4. Although about 60-80% of NSCLC patients with EGFR activating mutations respond to the first and/or second-generation EGFR inhibitors, almost all patients develop resistance to EGFR kinase inhibitors within 6-14 months 5, 6. Approximately half of the patients with acquired resistance to EGFR inhibition develop a secondary mutation in EGFR (T790M) 2, 5, 7. Third-generation EGFR inhibitors, such as Osimertinib, which targets the T790M mutation, have been shown to improve the survival of advanced lung cancer patients8. However, patients ultimately also develop resistance to Osimertinib9.
In addition to the alterations in the cancer cells, factors in the tumor microenvironment also regulate the response to anti-cancer therapy, including targeted therapy10, 11. Several growth factors, such as Hepatocyte Growth Factor (HGF) have been shown to counter the activity of anti-cancer agents12, 13. Indeed, aberrant activation of the HGF/MET pathway has been associated with primary and acquired resistance to targeted therapy in lung cancer patients2, 14. HGF is the only known ligand for the MET receptor tyrosine kinase; binding of HGF to MET receptor results in MET phosphorylation, receptor dimerization, and activation of downstream signaling pathways, including Rat sarcoma (RAS)/ extracellular-signal-regulated kinase (ERK)/ mitogen activated protein kinase (MAPK), Phosphoinositide 3-Kinase PI3K/ Akt serine threonine kinase (AKT), Signal transducer and activator of transcription 3 (STAT3), and Wingless-related integration site (Wnt)/β-catenin signaling. Activation of MET in cancer cells has been shown to induce cancer cell proliferation, survival, cell scattering, epithelial-to-mesenchymal transition (EMT), migration, and primary and acquired resistance to anti-cancer therapy3, 15-23. Although HGF is mostly expressed by cancer-associated fibroblasts and activates MET receptor on tumor cells in a paracrine fashion, tumor cells have also been shown to express HGF24-26.
Ligand-independent MET activation is triggered by MET mutation, MET amplification, or MET overexpression, which are frequently found in lung cancer patients2. 2-3% of lung cancer patients have MET exon 14 splice mutations, resulting in constitutive MET signaling due to limited receptor degradation27. MET amplification is observed in 2-5% of NSCLC patients, whereas MET overexpression is observed in 15-70% of NSCLC patients, depending on the assay used to detect MET3. Both MET and HGF overexpression are correlated with tumor stage and decreased survival in NSCLC and small cell lung cancer (SCLC)28-33. Furthermore, MET amplification and HGF expression are associated with primary and acquired resistance to EGFR-targeting agents. MET amplification is detected in 5-10% and HGF overexpression in 61% of patients that have acquired resistance to EGFR tyrosine kinase inhibitors (TKIs) in NSCLC9, 14, 34-36. Accordingly, increased HGF expression has been associated with primary and acquired resistance to anti-EGFR therapy in lung cancer patients36, 37.
Because of the importance of HGF/MET signaling in tumor progression and resistance to targeted therapies, both HGF and MET have emerged as important therapeutic targets. Various small molecule inhibitors of MET, and anti-MET and anti-HGF antibodies have been developed and tested in clinical trials either as single agents or combined with other therapies. The efficacy of MET and HGF targeting agents in clinical trials has been disappointing3, 38. However, most trials were conducted in unselected patients. Recently, capmatinib and tepotinib, a specific MET kinase inhibitors, have been recently approved for lung cancer patients with MET exon 14 alterations39.
HGF is released from cells as an inactive single-chain pro-factor, pro-HGF, which can bind the MET receptor but cannot activate MET signaling40. Proteolytic activation of pro-HGF is a rate-limiting step in HGF/MET signaling25, 41. One of the three serine proteases, matriptase, hepsin, or HGFA, are required for site-specific proteolytic processing of pro-HGF into an active protein, capable of binding and activating the receptor25. The HGF-activating proteases are upregulated in lung cancer tissue compared to non-cancerous lung tissue25, 42, 43. Activation of these proteases is kept in check by the endogenous inhibitors of these proteases, HGF activator inhibitors (HAI-1 and HAI-2), whose expression is often reduced in cancer tissues25, 43-45.
During the past several years, we have developed small-molecule inhibitors as well as peptidomimetic inhibitors of matriptase, hepsin, and HGFA46-50, the three physiologically relevant HGF-activating proteases. These inhibitors closely mimic the activity of HAI-1 and HAI-2 and thus we refer to them as synthetic HAIs (sHAIs). We have previously shown that sHAIs block HGF activation and prevent HGF-mediated MET activation in colon and lung cancer cells and prevent EMT and migration of cancer cells42, 51. Furthermore, we have shown that sHAIs overcome HGF-mediated primary and acquired resistance to EGFR inhibition in colon cancer cells and overcome resistance to anti-MET therapy in MET-amplified NSCLC cells42, 51.
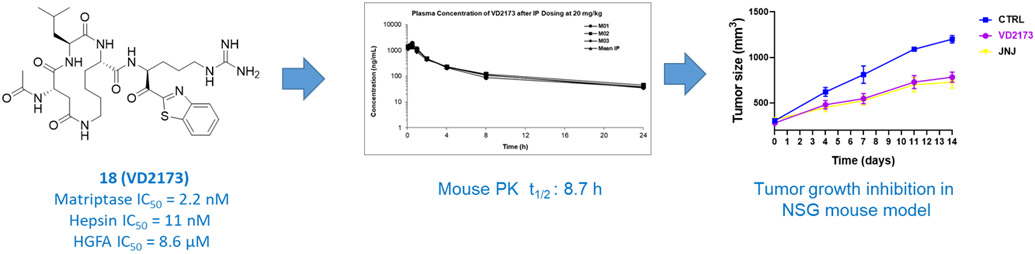
Here we report on the rational design of new macrocyclic peptide inhibitors of HGFA, matriptase and hepsin. We discuss their metabolic stability, selectivity, pharmacokinetic (PK) properties, and the anti-cancer activity of two advanced compounds, the macrocyclic inhibitor VD2173 (compound 18), a dual inhibitor of matriptase and hepsin, We compared its activity to ZFH7116 (compound 31), a previously reported48 triplex inhibitor of matriptase, hepsin, and HGFA. We demonstrate that both VD2173 and ZFH7116 overcome HGF-mediated resistance to EGFR and MET inhibitors and prevent HGF-mediated wound healing in NSCLC cell lines. The cyclic inhibitor VD2173 has significantly improved PK properties compared to ZFH7116, with a longer half-life and AUC. Both compounds prevented the HGF-driven growth of METEx14-deleted H596 lung cancer cells in human HGF-knock-in (hHGF-KI) mice. Thus, these newly developed small-molecule inhibitors of HGF activation (sHAIs) represent a unique therapeutic strategy for inhibiting the tumor-promoting activity of HGF, including overcoming resistance to targeted therapy, and preventing tumor progression and metastasis in lung cancer.
2. INITIAL DESIGN OF MACROCYCLES AND BENEFITS TO DRUG DISCOVERY
Peptides are challenging to develop as therapeutics for multiple reasons including poor metabolic stability, low or no cellular permeability and corresponding lack of oral bioavailability52, 53. The highly polar nature of peptides, taken together with the high molecular weight and large number of both hydrogen bond acceptors and donors contributes to the lackluster drug-like properties of peptides. Therefore, to develop suitable drug candidates, it is necessary to convert them into a non-peptide molecule or minimize the peptide-like characteristics of lead inhibitors derived from initial drug discovery54, 55. However, this approach is highly challenging, unpredictable and has not been successful in most protease inhibitor drug discovery programs. The use of unnatural amino acids is one strategy to potentially increase the potency and target selectivity as well as decrease the metabolic liability of peptide therapeutics56 but typically will not improve other important properties for drug discovery including cell permeability and oral bioavailability. Another approach to facilitate peptide-based drug development is to use cyclic peptides (Figure 1) which have been successfully utilized to increase potency, selectivity, metabolic stability, and cellular permeability of acyclic peptides in multiple drug discovery applications57, 58. In this manuscript we describe a strategy to significantly improve protease inhibitor drug discovery and development by employing the use of versatile cyclic peptide templates as conformationally restricted scaffolds which fuse two alternating amino acid sidechains of the peptide.
Figure 1. Design of macrocyclic inhibitors.

Cyclization is performed via connection of alternating P2 and P4 amino acid sidechains using a covalent linker PDB code: 2WUC.
Macrocyclization of linear peptides is an established way of increasing binding affinity of protease inhibitors while at the same time reducing the undesired characteristics of acyclic/linear peptides in drug discovery. This strategy locks the peptide backbone into an anti-parallel β-sheet, the conformation in which proteases recognize and bind their substrates. This conformational restriction coupled with functional group (sidechain) preorganization, can significantly enhance pharmacological activity by minimizing entropic loss upon inhibitor binding55, 59. Carefully designed macrocyclic peptides have been used effectively to produce robust selective interactions with targeted proteins. These macrocycles can also enhance cellular permeability, proteolytic stability, and in some cases even oral bioavailability60-63. Based on these considerations, we developed our strategy for development of HGFA, matriptase and hepsin inhibitors via macrocyclization of linear tripeptides by tethering the P2 and P4 alternating side chains branching the S2-S4 pockets (Figure 1). We explored a large number of structural variants focused on linker directionality and size as well as modification of the central P3 residue, which binds in the S3 pocket affording opportunities for favorable interactions on the opposite side of the P2-P4 linker (Figure 2).
Figure 2.

A) X-ray of Ac-KQLR-CMK covalently linked to HGFA serine protease, PDB code: 2WUC and B) overlaid with Ac-SKLR-kbt (yellow) and Ac-cyclo(KLD)-R-CHO (green) computationally docked (Schrödinger/GLIDES) in the active site of HGFA.
Shown in Figure 3, structures A and B are generic templates for our intramolecular macrolactamization approach using P2 and P4 sidechain amines (Lys) and a carboxylic acid (Glu or Asp) in amide coupling reactions. We selected the desired P3 residues based on our existing SAR and computationally directed rational design for HGFA matriptase and hepsin.
Figure 3. Structural design of P4-P2 cycloamide linked macrocyclic ketobenzothiazole (kbt) inhibitors of serine proteases.

Cyclization is performed via a P4 Asp or Glu to a P2 Lys (A) or a Lys to an Asp or Glu (B).
3. SYNTHESIS AND EVALUATION OF PEPTIDYL MACROCYCLIC ARGININE KETOBENZOTHIAZOLE INHIBITORS
We previously published methodology for the synthesis of acyclic ketobenzothiazole (kbt) inhibitors in solution phase as well as using solid phase peptide synthesis (SPPS)49-48. With regards to intramolecular cyclization reactions, they need to be carried out under dilution (concentrations less than 2.0 mM) conditions to prevent intermolecular reaction and the polymerization of linear peptides. To limit the use of solvent and multiple purification steps associated with solution phase synthesis, we have employed solid phase peptide synthesis (SPPS) for the construction of the cyclic tripeptide precursors followed by solution phase incorporation of the Arg-kbt warhead. Shown in Scheme 1A, the synthesis begins with commercially available Fmoc-protected Asp(tBu) or Glu(tBu) on Wang resin as the P2 amino acid which is first deprotected with piperidine and then reacted with the Fmoc-protected P3 amino acid using HBTU. After Fmoc removal, the P4 residue Fmoc-Lys(Boc) was next coupled to the P3-P2 dipeptide. To explore SAR of directionality relating to the amide bond of the linker, we generated matched pair analogs by installing Lys as the P2 residue and Asp or Glu as the P4 residue (Scheme 1B). Following dual deprotection of the Boc and t-Butyl ester with 4N HCl, the intramolecular cyclization reaction was performed effectively on resin using standard peptide coupling reagents such as EDC/HOBt or HATU without having to increase volume of the solvent as required in solution phase. The resultant cycloamide tripeptide intermediates 1-14 were obtained in good yields via cleavage from the resin with a TFA:water:thioanisole (95:2.5:2.5 % v/v) cocktail followed by purification on silica gel. Next, the macrocyclic acids 1-14 were coupled with H-Arg(Pbf)-kbt as previously reported 48 giving the final target compounds 15-30 after final deprotection and purification on prep HPLC.
Scheme 1.

Synthesis of macrocyclic compounds using SPPS approach using Wang-resin A) P4(Lys)-P2 (Asp/Glu) B) P4 (Asp/Glu)-P2 (Lys)
4. RESULTS AND DISCUSSION
4.1. Enzymatic activity of macrocyclic inhibitors.
We tested all compounds for their inhibitory activity using a kinetic enzyme inhibition assay for HGFA, matriptase, and hepsin as well as for Factor Xa and Thrombin using flurogenic peptide substrates as published earlier 47. All tested compounds exhibited potent nanomolar activity for matriptase and hepsin but surprisingly only relatively weak inhibition inhibitory activity for HGFA (Table 1 and 2). In an attempt to improve the HGFA activity, we modified the central P3 residue of the cycloamide with several different amino acids but found that the HGFA activity only improved to the micromolar range. This can be exemplified with inhibitors having macrocyclic linkers connected though a P4 Lys and P2 Asp/Glu (compounds 15-17). Analogue 15 (cyclo Lys (P4)-Asp (P2)) was not active against HGFA up to 20 μM but showed excellent potency for matriptase (IC50 = 20 nM) and hepsin (IC50 = 25 nM). When the P2 Asp was replaced with Glu (compound 16) improved activity resulted not only for HGFA (IC50 = 4.5 μM), but also for matriptase (IC50 = 4.4 nM), and hepsin (IC50 = 1.7 nM). We also investigated a different P3 residue to further improve the activity, When Leu was replaced with Arg (17) this increased activity only for matriptase (IC50 = 0.13 nM) and hepsin (IC50 = 0.08 nM) but with no significant change in HGFA inhibitory activity (IC50 = 5.05 μM)
Table 1.
Enzyme inhibition activity of cycloamide ketobenzothiazole (kbt) inhibitors.
| Compd | Structure | HGFA IC50 (nM) |
Matriptase IC50 (nM) |
Hepsin IC50 (nM) |
Factor Xa IC50 (nM) |
Thrombin IC50 (nM) |
|---|---|---|---|---|---|---|
| 15 | Ac-cyclo[Lys-Leu-Asp]-Arg-kbt | >20,000 | 20 | 25 | 1161 | >20,000 |
| 16 | Ac-cyclo[Lys-Leu-Glu]-Arg-kbt | 4,466 | 4.4 | 1.7 | 723 | >20,000 |
| 17 | Ac-cyclo[Lys-Arg-Glu]-Arg-kbt | 5046 | 0.13 | 0.08 | 155 | >20,000 |
| 18 | Ac-cyclo[Asp-Leu-Lys]-Arg-kbt | 8610 | 2.2 | 11 | 2048 | 8140 |
| 19 | Ac-cyclo[Glu-Leu-Lys]-Arg-kbt | >20,000 | 3.1 | 8.4 | 1714 | >20,000 |
| 20 | Ac-cyclo[Asp-Leu-Lys]-Arg-kbt-[Val]-amide | 5378 | 11 | 5.4 | 118 | 34 |
| 21 | Ac-cyclo[Asp-Leu-Lys]-Arg-kt | >20,000 | 41 | 136 | 1363 | 7939 |
Table 2.
SAR of cyclo[Asp-P3-Lys] peptides based on 18 with P3 group variations.
| Compd | Ac-cyclo[Asp-P3-Lys]- Arg-kbt |
HGFA IC50 (nM) |
Matriptase IC50 (nM) |
Hepsin IC50 (nM) |
Factor Xa IC50 (nM) |
Thrombin IC50 (nM) |
|---|---|---|---|---|---|---|
| 18 | Leu | 8610 | 2.2 | 11 | 2048 | 8140 |
| 22 | Gly | 4,312 | 6.3 | 5.9 | 6183 | >20,000 |
| 23 | Ala | 13,705 | 2.5 | 8.8 | >20,000 | >20,000 |
| 24 | Val | 1,832 | 3.2 | 7.8 | 1514 | >20,000 |
| 25 | Asn | 15,837 | 3.8 | 10 | 4653 | >20,000 |
| 26 | Gln | 16,108 | 7.7 | 22 | >20,000 | >20,000 |
| 27 | Thr | 11,777 | 3.7 | 5.8 | 13341 | >20,000 |
| 28 | Met | 3,240 | 1.0 | 5.9 | 1395 | 18456 |
| 29 | Phe | 3,173 | 3.4 | 17 | 1530 | 1090 |
| 30 | Tyr | >20,000 | 6.1 | 44 | 16962 | >20,000 |
| 32 | Ac-cyclo[Asp-Leu-Lys(N-Me)]-Arg-kbt | 12,000 | 1.1 | 15 | 678 | 17100 |
We found that switching the P4 residue from Lys to Asp or Glu and the P2 residue to Lys resulted in increased activity relative to the reversed linker as seen in 15-17 (Table 1). For example, Compound 18, the matched pair to compound 15 has increased activity towards matriptase (IC50 = 2.2 nM) and hepsin (IC50 = 11 nM) and showed some inhibition of HGFA (IC50 = 8610 nM) in contrast to 15. We earlier found that ketobenzothiazole-[Val]-amide (kbt V amide) warhead can improve the HGFA activity and ketothiazole (kt) warheads can show differential protease selectivity. Therefore, we replaced the kbt warhead with the larger substituted kbt-[V]-amide and the smaller ketothiazole (kt), however this modification when applied to compound 18 had no effect on HGFA potency (IC50 = 5378 nM) (compound 20) and the ketothiazole derivative showed no evidence of HGFA inhibition at concentrations tested (IC50 > 20 μM) (compound 21) and decreased hepsin and matriptase activity. Furthermore, when tested against the undesired trypsin-like serine proteases like Factor Xa and thrombin, we found that compound 20 was much less selective for HGFA, matriptase and hepsin; Factor Xa (IC50 = 118 nM) and thrombin (IC50 = 34 nM). In general, all compounds tested either lacked inhibition against thrombin up to 20 μM or showed only weak inhibition but alternately several compounds displayed moderate inhibition of Factor Xa.
In another focused SAR library based on compound 18, we kept the kbt warhead constant and varied the central P3 residue with several different amino acid residues (Table 2) with diverse functional groups. Surprisingly, we found that the identity of the P3 residue had only limited, if any effect on the activity of either protease, HGFA, matriptase, hepsin, factor Xa or thrombin. For example, when we change P3 from Leu to Met (28) and Phe (29) HGFA activity slightly improved (IC50 = 3.2 μM) vs (IC50 = 8.6 μM) (18). Interestingly, when the P3 Phe is replaced with Tyr (30) a loss of HGFA activity takes place with a corresponding 5-fold decrease in hepsin (IC50 = 44 nM) and matriptase (IC50 = 6.1 nM) potency in addition to a significant decrease in both factor Xa and thrombin inhibition.
4.2. Metabolic stability and pharmacokinetics of lead compounds
We tested all compounds for their in vitro stability in both mouse and human blood plasma (Table 3). The P4-Lys-P2-Asp/Glu linker cycloamides 15, 16 and 17 showed good stability in mouse and human plasma (Table 3) but when 16 was tested in mice (Figure 4), it displayed poor pharmacokinetic (PK) properties when dosed IP with a half-life of only 0.71 h and no detectable compound in plasma after 8 h. All other compounds with the P4-Asp/Glu-P2-Lys cycloamide linker (18-30) showed excellent stability in both mouse and human plasma with >289 min half-life except 18 and 19 which had a half-life 222 and 157 min in mouse plasma, respectively. Compound 23 had a half-life of 261.2 min in mouse plasma and 216.1 min in human plasma, 27 had 192.6 min in mouse plasma and 248.5 min in human plasma and 30 had half-life 276.8 min in mouse plasma. Based on potency, selectivity, in vitro properties we selected 3 additional compounds for in vivo pharmacokinetics (PK) in mice. Shown in Figure 4, compound 18 has an outstanding half-life of 8.7 h in mice with a high AUC and exposure in plasma beyond 24 h. Compound 19 had the worst PK with a half-life of less than 30 minutes and no compound exposure after 8 h but compound 29 had moderately good PK with a respectable half-life of 1.61 h and an AUC comparable to 18.
Table 3.
Plasma stability of selected compounds in both mouse and human plasma.
| Compound | Mouse Plasma Stability t1/2 (min) |
Human Plasma Stability t1/2 (min) |
Mouse PPB (% free) |
|---|---|---|---|
| 15 | 178 | >289 | 85.3 |
| 16 | 274 | >289 | 18.4 |
| 17 | 156 | 151 | 25.9 |
| 18 (VD2173) | 222 | >289 | 28.6 |
| 19 | 157 | >289 | 6.0 |
| 20 | >289.1 | >289.1 | ND |
| 21 | >289.1 | >289.1 | ND |
| 22 | >289.1 | >289.1 | |
| 23 | 261.2 | 216.1 | |
| 24 | >289.1 | >289.1 | |
| 25 | >289.1 | >289.1 | |
| 26 | >289.1 | >289.1 | |
| 27 | 192.6 | 248.5 | |
| 28 | >289.1 | >289.1 | |
| 29 | >289.1 | >289.1 | |
| 30 | 276.8 | >289.1 | |
| 31 (ZFH7116) | 184 | 140 | 16.7 |
| 32 | >289 | >289 |
Figure 4. Structures and mouse pharmacokinetics of 16, 18 (VD2173), 19, 29, 31 (ZFH7116) and 32.

Compounds were each dosed IP @20 mg/kg to 3 mice and compound blood plasma levels (ng/mL) were monitored at several timepoints for 24 h.
In summary, it is apparent that while we have solved the metabolic stability issue with the acyclic inhibitors, the PK properties are still variable in some cases with good half-life and compound exposure but in other cases with no improvement noted relative to the acyclic counterparts. This is likely due to rapid renal clearance in the kidney which is known to occur for peptides and related to their highly polar character, which is not a property that cyclic peptides can solve effectively. However, compound 18 with a long half-life and excellent compound exposure out to 24 h displays stellar PK even when compared to typical non-peptidyl small molecule drugs. In an attempt to increase HGFA activity of 18, we synthesized the N-Me amide linker version. This was accomplished via use of N-Me Lys in the P2 position (supplementary data). Shown in Figure 4, this analog (32) had similar HGFA activity but also less desirable PK with no compound exposure detected after 8 h. Taken together, compound 18 (VD2173) was selected as a lead candidate that we evaluated further in advanced biology studies for blocking the tumor-promoting activity of HGF in lung cancer as discussed in the next section.
4.3. VD2173 (18) and ZFH7116 (31) prevent pro-HGF processing and HGF activation
As discussed above and reported in previous publications, we have developed peptidomimetic inhibitors of matriptase, hepsin, and HGFA rationally designed based on the endogenous substrate pro-HGF 64-66. In this paper we discovered a new series of cyclopeptides and identified lead VD2173 (18). ZFH7116 (31) is a potent triplex inhibitor of matriptase (IC50 = 21 nM), hepsin (IC50 = 0.38 nM), and HGFA (IC50 = 26 nM), with good selectivity over thrombin (IC50 = 9000 nM) and factor Xa (IC50 = 770 nM) (Figure 4 & Table 4). The synthesis and initial in vitro biological evaluation have been reported previously 67.
Table 4.
Selectivity and biochemical characteristics of VD2173 and ZFH7116.
| VD2173 (18) | ZFH7116 (31) | |
|---|---|---|
| Matriptase IC50 (nM) | 2.2 | 21 |
| Hepsin IC50 (nM) | 11 | 0.38 |
| HGFA IC50 (nM) | 8610 | 26 |
| Thrombin IC50 (nM) | 8140 | 9000 |
| Factor Xa IC50 (nM) | 2048 | 770 |
| Solubility pH = 7.4 (μM) | 11 | 200 |
| Mouse [Human] Plasma Stability (t1/2, min) | 222 [>289] | 184 [140] |
| Mouse [Human] Microsomal Stability (t1/2, min) | >145 [>145] | >145 |
| Mouse PPB (% Free) | 28.6 | 16.7 |
Both ZFH7116 and VD2173 have good aqueous solubility, are stable in mouse and human microsomes and have good half-lives in both mouse and human plasma (Table 4). Both compounds also have low plasma protein binding and thus a high free fraction in mouse plasma. IP dosing in mice (20 mg/kg) demonstrated excellent pharmacokinetic properties for VD2173, (half-life of 8.7 h) and ZFH7116 (half-life of 2h). This demonstrates extended compound exposures above their IC50s (24 h for VD2173 and 8 h for ZFH7116).
To confirm that ZFH7116 and VD2173 inhibit the processing of pro-HGF into its active form, recombinant pro-HGF was incubated with matriptase, hepsin, or HGFA in the absence or presence of ZFH7116 or VD2173 (Figure 5). Consistent with data shown in Table 4, ZFH7116 potently inhibited proteolytic activation of pro-HGF by all three proteases. VD2173 potently inhibited pro-HGF activation by matriptase and hepsin but did not inhibit HGFA-induced pro-HGF activation as expected. Thus, while ZFH7116 is a triplex inhibitor of the three major HGF activating proteases, matriptase, hepsin, and HGFA, VD2173 is a dual inhibitor of matriptase and hepsin. In this study we compared how ZFH7116 and VD2173 interfered with pro-HGF-induced therapeutic resistance and HGF-mediated tumor progression using in vitro and in vivo models of lung cancer.
Figure 5. VD2173 and ZFH7116 prevent proteolytic processing of pro-HGF by matriptase, hepsin, or HGFA.

Recombinant pro-HGF (10 ng) was incubated for one hour with matriptase (1 nM), hepsin (10 nM), or HGFA (5 nM) in the absence or presence of VD2173 or ZFH7116 as indicated. Immunoblotting was performed using a polyclonal antibody that recognizes pro-HGF as well as the alpha and beta chains of active HGF. Active HGF was run as a positive control.
4.4. VD2173 and ZFH7116 inhibit HGF-mediated wound healing
HGF has been shown to induce scattering and migration of cancer cells 51. HCC827 cells were grown to confluence before a scratch (wound) was made. Indeed, HGF or pro-HGF promoted the migration of HCC827 cells. We next tested whether ZFH7116 and VD2173 could prevent HGF-mediated wound healing. We confirmed that both ZFH7116 and VD2173 significantly inhibited pro-HGF-mediated wound closure (Figure 6A-B).
Figure 6. VD2173 and ZFH7116 inhibit HGF-mediated wound healing.

HCC827 cells were grown to confluence before cells were serum-starved overnight. A scratch was made, and cells were incubated with pre-incubated with inhibitors (ZFH7116; 10μM, VD2173; 10μM) for 30 minutes before cells were stimulated with HGF or pro-HGF for 24h at which time the plate was stained with crystal violet. (a) Representative wound-healing images. (b) Average data of n=3. Statistical analysis was performed using one-way ANOVA and statistical differences indicated are compared to cells stimulated with pro-HGF only.
4.5. VD2173 and ZFH7116 overcome pro-HGF-mediated resistance to EGFR and MET targeting agents in lung cancer
HGF has been shown to induce resistance to EGFR inhibition in vitro 35, 37, 68. We first tested whether pro-HGF blocks the response to EGFR inhibitor gefitinib in the HCC827 lung cancer cell line, which carries a mutation in the EGFR tyrosine kinase domain (E746 - A750 deletion). We treated cells with gefitinib in the absence or the presence of pro-HGF and assessed cell viability. We confirmed that pro-HGF significantly lowers the activity of gefitinib. Treatment of HCC827 cells with ZFH7116 or VD2173 alone did not impact their growth or viability. However, both VD2173 and ZFH7116 blocked the pro-survival effects of pro-HGF in HCC827 cells (Figure 7A).
Figure 7. VD2173 and ZFH7116 overcome HGF-dependent resistance to anti-EGFR and anti-MET inhibition.

(a) HCC827 lung cancer cells were treated with gefitinib (20 nM) in the absence or presence of pro-HGF (100 ng/ml) and VD2713 or ZFH7116 (0.1-1μM) as indicated, and cell viability was determined after 72hrs by crystal violet staining. Data represent the means±S.E.M. for n=3-5. Statistical analysis was performed using one-way analysis of variance (ANOVA) with Dunnett’s multiple comparison test and statistical significance is indicated compared to HCC827 cells treated with gefitinib and pro-HGF. ***P<0.001 and ****P<0.0001. (b) EBC-1 lung cancer cells were pre-incubated with pro-HGF (100 ng/ml) in the absence or presence of VD2173 or ZFH7116 (1μM) for 24 hours before treatment with MET kinase inhibitor JNJ-38877605 (JNJ; 25nM). Cell viability was determined after 48 hrs by CellTiterGlo. Data represent the means±S.E.M. for n=3. Statistical analysis was performed using one-way analysis of variance (ANOVA) with Dunnett’s multiple comparison test and statistical significance is indicated compared to EBC-1 cells treated with JNJ and pro-HGF. **P<0.01.
MET amplification is a common cause of resistance to EGFR-targeting therapy. MET-amplified lung cancer cells do not respond to EGFR inhibition but are addicted to MET signaling and their growth is strongly inhibited by MET kinase inhibitors 15, 42. MET-amplified EBC1 lung cancer cells were treated with MET-specific inhibitor JNJ-38877605 in the absence or presence of pro-HGF. Pro-HGF significantly inhibited the activity of JNJ-38877605 and VD2173 and ZFH7116 blocked the pro-survival activity of pro-HGF (Figure 7B). This is consistent with our previous finding that HGF promotes survival of MET-amplified cancer cells upon pharmacological inhibition of MET 15, 42 and that the HGF-mediated survival is inhibited by SRI31215, one of our first sHAI 42. Together, these data suggest that MET or EGFR targeting agents should be combined with inhibitors of HGF for optimal therapeutic efficacy. Combined with the findings in the wound healing assay, these results also suggest that HGFA may not play a major role in the processing of pro-HGF in lung cancer cells, as both VD2173 and ZFH7116 displayed comparable activity inhibiting HGF-mediated wound healing and resistance to anti-EGFR and anti-MET inhibition.
4.6.. VD2173 and ZFH7116 inhibit HGF-driven growth of lung cancer cells
Having shown that VD2173 and ZFH7116 inhibit activation of pro-HGF and prevent HGF-mediated resistance against EGFR and MET kinase inhibitors in vitro, we tested whether they block HGF-dependent growth of lung cancer cells in vivo. We showed that mice tolerated VD2173 up to 20mg/kg daily (IP) for 7 days and ZFH7116 up to 100 mg/kg daily (IP) for 7 days (Supplementary Figures 4 and 5). Furthermore, no weight loss or indications of toxicity were observed upon prolonged treatment at these concentrations in experiments described below.
H596 lung cancer cell line harbors a MET exon 14 deletion and PIK3C mutation and does not respond to MET-targeted therapy 69-71. The exon 14 encodes the intracellular c-Met juxta-membrane domain which contains critical regulatory elements, such as tyrosine 1003, the direct binding site for Cbl, an E3 ubiquitin ligase, which promotes MET protein degradation. Our data show that a MET exon 14-deleted NSCLC cell line H596 shows HGF-dependent signaling (and is responsive to MET inhibition) in in vitro and in vivo models, is consistent with published literature72. We showed that human HGF (hHGF), but not mouse HGF (mHGF), stimulates the growth of H596 cells in vitro. Additionally, a conditioned medium (CM) from HGF-producing human fibroblasts also induced H596 cell growth (Figure 8A). We demonstrated that MET kinase inhibitor JNJ-38877605 and a non-specific serine protease inhibitor, Nafamostat, inhibited the pro-HGF-mediated growth of H596 cells (Figure 8B). Similarly, ZFH7116 blocked the growth-promoting activity of pro-HGF (Figure 8B).
Figure 8. H596 cell and tumor growth is dependent on hHGF, but not mHGF.

(a) H596 lung cancer cells were incubated with recombinant hHGF (50 ng/ml), mHGF (50ng/ml), or fibroblast conditioned medium (CM). Cell viability was determined after 72h by CellTiterGlo. Statistical significance was calculated using one-way ANOVA and is shown compared to CTRL. (b) H596 lung cancer cells were incubated with recombinant pro-HGF (50 ng/ml) in the presence or absence of JNJ-38877605 (JNJ; 500 nM), nafamostat (NAF; 33.3 μM), or ZFH7116 (10 μM). Statistical significance was calculated using one-way ANOVA and is shown compared to cells stimulated with pro-HGF. (c) The serum concentration of hHGF in male and female NSG and hHGFki-NSG mice was assessed by hHGF ELISA. n=3 for NSG males and females and n=6 for hHGF NSG males and females. Statistical significance was calculated using 2way ANOVA and is shown compared to NSG animals. (d) Comparison of H596 lung cancer cell growth in NSG and hHGFki-NSG mice. Mice were injected subcutaneously with 0.5 million H596 cells. n=3 for each group. Statistical significance was calculated using 2way ANOVA. (a-d) *P<0.05, **P<0.01, ***P<0.001, ****P<0.0001.
It has been shown that H596 cells rapidly form tumors in a mouse model where mouse HGF has been replaced by human HGF (hHGFki), but not in the parental mice 73. We used this model to test the activity of our compounds. We first compared serum levels of human HGF (hHGF) in male and female littermates of NSG and hHGFki-NSG mice (aged 8-11 weeks). We found that female and male hHGFki-NSG mice expressed similar levels of hHGF, whereas no hHGF was detected in control NSG mice (Figure 8C). We confirmed that the growth of H596 tumors in vivo was driven by hHGF (Figure 8D), as tumors rapidly developed in hHGFki-NSG mice, whereas only very small tumors had developed in NSG mice weeks after cancer cell inoculation.
We next tested whether ZFH7116 and VD2173 inhibit the growth of H596 cells in vivo. We first tested the ability of ZFH7116 to prevent tumor growth in a prevention model. hHGFki mice were injected with H596 cells, and daily treatment of animals with ZFH7116 (50 mg/kg; IP) or JNJ-38877605 (40 mg/kg; PO) was initiated immediately for 25 days. As shown in Figure 9A, ZFH7116 reduced H596 tumor growth with similar efficacy as JNJ-38877605, a specific MET kinase inhibitor. No drug toxicity was observed in animals after prolonged treatment with ZFH7116.
Figure 9. VD2173 and ZFH7116 block HGF-dependent tumor progression.


(a) H596 tumor growth in hHGF-KI NSG female mice injected with ten million H596 cells and treated with ZFH7116 (50 m/kg daily; IP) or JNJ-38877605 (40 mg/kg daily; PO) for 25 days. The treatment started immediately after tumor cell injection. n=3-5 for each group. (b) hHGF-KI NSG female mice were injected with 2 million H596 cells. When tumor volume reached ~ 40mm3, animals were divided into three groups and treated daily with control (IP and PO), VD2173 (20 mg/kg; IP), or JNJ-38877605 (40 mg/kg; PO) for 16 days. n=4-6 for each group. (c) hHGF-KI NSG male mice were injected with 0.5 million H596 cells. When tumor volume reached ~350mm3, animals were divided into three groups, and treated with VD2173 (20 mg/kg daily; IP) or JNJ-38877605 (40 mg/kg daily; PO) for 14 days. n=3-4 for each group. (d) Tumors in (c) were collected and weighed at the end of the experiment. n=3-4 for each group. Statistical analysis was performed using two- and one-way analysis of variance (ANOVA) with Dunnett’s multiple comparison test and statistical significance is indicated for sHAI or JNJ-treated mice compared to control-treated mice (a-c) *P<0.05, **P<0.01
The efficacy of VD2173 was tested in a therapeutic model. In the first study, treatment started when tumors were still relatively small (~40 mm3) (Fig. 9B), and in a second study when the average tumor volume reached ~350mm3 (Figure 9C). We demonstrated that JNJ-38877605 and VD2173 were equally potent at reducing the growth of established tumors (Figure 9B and 9C). Accordingly, the tumor weights were significantly reduced in treated animals (Figure 9D). No drug toxicity was observed after prolonged treatment with VD2173. Combined, these studies demonstrate that small-molecule inhibitors of HGF activation such as ZFH7116 and VD2173 inhibit HGF-driven tumor growth in vivo.
CONCLUSIONS
We have developed novel cyclic peptide-based small-molecule inhibitors of the HGF-activating proteases, matriptase, hepsin, and HGFA. The compounds were constructed using innovative methodology with the key step involving cyclization/cycloamidation of the peptides on resin. These compounds are potent inhibitors of matriptase and hepsin serine proteases which however are only weakly active against HGFA in contrast to our previously reported acyclic peptide counterparts which are triplex inhibitors of all three proteases. We surmise the reason for the lack of activity in this series of compounds is related to its lack of P2 residue binding which we know is highly preferred to be the large amino acid sidechain of Leu in the case of HGFA whereas hepsin and matriptase are relatively promiscuous in their specificity for P2 residues. In the computational model shown in Figure 3 it is evident that the P2 pocket of HGFA is not filled by the linker and in fact is directed away from the protein at that position. Our current efforts are exploring different linkers which do indeed fill that P2 space in HGFA and have now identified several subseries of cyclopeptides which have potent activity towards HGFA as well as matriptase and hepsin.
These unique and selective inhibitors of HGFA, matriptase and hepsin closely mimic the activity of their endogenous protease inhibitors, HAI-1 and 2, and efficiently prevent the activation of single chain pro-HGF into its active two-chain HGF form. Here we presented advanced biological data on two compounds, ZFH7116 (31), a triplex acyclic peptidomimetic inhibitor of the three HGF-activating proteases, and the new cyclic peptide inhibitor VD2173 (18), a duplex inhibitor of matriptase and hepsin (weak inhibitor of HGFA). Both compounds have good pharmacokinetic properties. We demonstrated that both ZFH7116 and VD2173 inhibited HGF-mediated wound healing and overcame resistance to EGFR and MET targeted therapy in in vitro lung cancer models of therapeutic resistance. Furthermore, both sHAIs inhibited HGF-mediated growth of lung cancer cells in vivo. While the treatments significantly reduced the growth of H596 tumors, none of the treatments blocked the tumor growth completely. The activity of ZFH7116 and VD2173 was comparable to the activity of a specific MET kinase inhibitor. Both compounds display similar activity in vitro and in vivo, despite VD2173 only inhibiting matriptase and hepsin. This is in accordance with gene expression for matriptase being the highest compared to hepsin and HGFA in all three cell lines tested here: HCC827, EBC-1, and H596 (data obtained from the Cancer Cell Line Encyclopedia (CCLE) distributed by the Broad Institute of MIT and Harvard) 74. This suggests that HGFA does not play a major role in lung cancer.
HGF has recently also been shown to regulate responses to immunotherapy, such as anti-PD1/PDL1. HGF expression and MET activation or amplification have been associated with PD-L1 expression in NSCLC, which was inhibited by MET-targeting drugs 75-78. Although the effect of HGF/MET signaling on the response to immunotherapeutic agents remains to be further investigated, this highlights another potential pathway by which inhibitors of HGF activation could improve outcomes for lung cancer patients.
Although the efficacy of ZFH7116 and VD2173 remains to be tested in animal models of HGF-mediated therapeutic resistance, our results highlight the clinical potential of sHAIs to prevent HGF-mediated tumor progression and resistance to targeted therapy in lung cancer patients as well as patients with other cancers that display HGF-mediated growth and therapeutic resistance. We have now developed novel cyclic peptide inhibitors which encompass good PK as well as potent triplex activity of all three HGF-activating proteases, HGFA, hepsin and matriptase. These new advanced lead compounds are currently being evaluated for their biological properties in vitro and in vivo and will be reported in a future manuscript.
Experimental
1. General synthesis, purification, and analytical chemistry procedures.
Starting materials, reagents, and solvents were purchased from commercial vendors unless otherwise noted. 1H NMR spectra were measured on a Varian 400 MHz NMR instrument. The chemical shifts were reported as δ ppm relative to TMS using residual solvent peak as the reference unless otherwise noted. The following abbreviations were used to express the multiplicities: s = singlet; d = doublet; t = triplet; q = quartet; m = multiplet; br = broad. High-performed liquid chromatography (HPLC) was carried out on GILSON GX-281 using Waters C18 5μM, 4.6*50mm and Waters Prep C18 5μM, 19*150mm reverse phase columns, eluted with a gradient system of 5:95 to 95:5 acetonitrile:water with a buffer consisting of 0.05% TFA. Mass spectra (MS) were performed on HPLC/MSD using electrospray ionization (ESI) for detection. All reactions were monitored by thin layer chromatography (TLC) carried out on Merck silica gel plates (0.25 mm thick, 60F254), visualized by using UV (254 nm) or dyes such as KMnO4, p-Anisaldehyde and CAM (Cerium Ammonium Molybdate or Hanessian's Stain). Silica gel chromatography was carried out on a Teledyne ISCO CombiFlash purification system using pre-packed silica gel columns (12g~330g sizes). All compounds used for biological assays are greater than 95% purity based on NMR and HPLC by absorbance at 220 nm and 254 nm wavelengths.
2. General procedures for the synthesis of macrocyclic peptides using Solid Phase Peptide Synthesis (SPPS).
Peptide coupling and deprotection steps of the Fmoc group:
Into a reaction vessel (with a fritted glass filter) containing Fmoc-L-Lys 79-Wang resin (5 g, 1.7 mmol), DCM (30 mL) was added under nitrogen atmosphere. The mixture was shaken at RT for 15 min and then filtered. The resin was washed with DMF (30 mL) 2 times. To the resulting resin, piperidine/DMF (20% v/v, 30 mL) was added and the mixture was shaken for 1-2 h at RT, then filtered. The resin was washed with DCM (2 x 25 mL), DMF (2 x 25 mL). Fmoc-AA-OH (1.8 g, 5.1 mmol), HBTU (2.26 g, 5.95 mmol), iPr2NEt (10.2 mmol), and DMF (40 mL) were added to the vessel and the resultant heterogeneous mixture was shaken at RT 2-3 h and then filtered. The resin was washed with DMF (4 x 25 mL), dried and then piperidine/DMF (20% v/v, 30 mL) was added and the mixture was shaken for 1-2 h at RT, then filtered. Fmoc-AA-OH (2.1 g, 5.1 mmol), HBTU (2.26 g, 5.95 mmol), iPr2NEt (10.2 mmol), and DMF (40 mL) were added to the vessel and the resultant heterogeneous mixture was shaken at RT 2-3 h and then filtered.
Acetyl capping of the peptides.
The peptide containing resin was suspended in 40 mL of DMF, (1 mL; 10.2 mmol) Ac2O, and 2.72 mmol iPr2Net and the mixture was shaken at RT for 1 h. The reaction was filtered and the resin was washed with DMF (2 x 20 mL) followed by DCM (2 x 20 mL) and dried.
Cleavage of Boc- and t-Bu groups of peptides.
To the resin in the vessel 40 mL of dry 4M HCl in 1, 4-dioxane was added and shaken at RT for 30-40 min., immediately filtered and washed the resin with DMF (4 x 20 mL). Note: Do not prolong the reaction it might cleave wang-resin in acidic condition.
Cyclization of peptide in presence of resin.
EDCI (0.98 g, 5.1 mmol), HOBt (0.78 g, 5.1 mmol), iPr2NEt (8.5 mmol), and DMF (50 mL) were added to the resin in the reaction vial and the resulting mixture was shaken for overnight at RT. The mixture was filtered, and the resin was washed with DMF (2 x 20 mL) followed by DCM (2 x 20 mL).
Cleavage of macrocyclic resin.
Acetyl capped macrocyclic resin was suspended in TFA (2 x 30 mL) and shaken for 30 min. The mixture was filtered, and the resin was washed with DCM (2 x 40 mL). The filtrate was concentrated, and the cold ether was added to the residue yielding the crude product as a precipitate, which was purified by flash chromatography yielded an off-white solid (400 mg).
Macrocyclic keto-benzothiazoles or keto-thiazoles.
The macrocyclic tripeptide acid (400 mg, 1.0 mmol) was dissolved in dry DMF (10 mL) under a nitrogen atmosphere at 0 °C and HATU (456 mg, 1.2 mmol) was added followed by stirring for 15 min, and then added the Arg(Pbf)-kbt. HCl (638 mg, 1.10 mmol) and iPr2NEt (0.87 mL, 5.00 mmol) at 0 °C. The reaction was allowed to reach RT and then stirred for 2-3 h. The DMF was removed under reduced pressure and water (250 mL) was added to the resulting residue. The precipitate formed was filtered and washed with water (2 x 50 mL) and dried. To this precipitate 10 mL of TFA/thioanisole/water (95:2.5:2.5 v/v/v) was added and the mixture was stirred for 2 h at RT. The solvent was removed, and then cold ether (100 mL) was added. The crude product was collected by centrifugation. The crude product was purified by HPLC (C18, 15 x 150 mm column; eluent: acetonitrile/water (0.05% TFA) to give the title compound as a white solid.
Ac-cyclo(KLD)-R-ketobenzothiazole (15):
Ac-cyclo(KLD)-OH (40 mg, 0.0969 mmol) and HATU (44 mg, 0.116 mmol) was taken in dry DMF under nitrogen atmosphere and the reaction mixture was cooled to 0 °C. N,N-diisopropylethylamine (0.08mL, 0.485 mmol) was then added drop wise to the reaction mixture and the reaction mixture was allowed to stir for 15 minutes followed by addition of HCl.Arg(Pbf)-kbt (56 mg, .097 mmol). The reaction mixture was stirred for 3 hours at 25 °C under nitrogen atmosphere and the completion of the reaction was confirmed by LC-MS monitoring. On completion, solvent was removed under reduced pressure. Water (100 mL) was added to the residue, product was precipitated out, which is filtered and dried. The crude product was directly taken to the next step without further purification.
Ac-cyclo(KLD)-R(Pbf)-kbt (90 mg, crude product from previous step) was taken in 5 mL TFA:thioanisole:H2O (95:2.5:2.5) and the reaction mixture was stirred for 3 hours at 25°C. The completion of the reaction was confirmed by LC-MS monitoring. On completion, the reaction mixture was concentrated under reduced pressure and triturated with diethyl ether to obtain the crude product as brown solid. The crude product was then subjected to reverse phase semi-preparative HPLC (Stationary phase: C18 column, mobile phase: H2O-Acetonitrile with 0.05% TFA in each, 10-55% Acetonitrile in H2O gradient for 20 minutes) to obtain the pure title product.
Yield (13 mg, 23%). 1H NMR (400MHz, DMSO-d6) δ = 8.55 (d, J = 6.7 Hz, 1 H), 8.28 (dt, J = 8.2, 13.5 Hz, 1 H), 7.83 - 7.76 (m, 2 H), 7.73 - 7.64 (m, 2 H), 7.47 (d, J = 6.7 Hz, 2 H), 6.53 (br. s, 1 H), 5.44 (br. s, 1 H), 4.46 - 4.41 (m, 1 H), 3.14 (d, J = 6.7 Hz, 7 H), 2.67 (br. s, 1 H), 2.54 (br. s, 1 H), 2.42 - 2.34 (m, 8 H), 1.79 (s, 3 H), 1.61 (br. s, 1 H), 1.51 - 1.44 (m, 3H), 1.33 - 1.22 (m, 2 H), 0.82 (dd, J = 6.1, 19.4 Hz, 7 H). ESI-MS [M+H]+ calcd for C31H46N9O6S+ 672.33, found 672.5.
Ac-cyclo(KLE)-R-ketobenzothiazole (16):
This titled compound was synthesized using same procedure as Ac-cyclo(KLD)-R-ketobenzothiazole (15). Compound was isolated as a white solid. Overall yield (10 mg, 20%). 1H NMR (400MHz, CD3OD) = 8.63 (d, J = 7.8 Hz, 1 H), 8.55 (d, J = 7.0 Hz, 1 H), 8.43 (d, J = 7.8 Hz, 1 H), 8.23 (d, J = 7.8 Hz, 1 H), 8.13 (d, J = 7.4 Hz, 1 H), 7.76 - 7.58 (m, 2 H), 5.75 (br. s., 1 H), 4.51 - 4.34 (m, 1 H), 3.58 (d, J = 4.7 Hz, 1 H), 2.99 - 2.80 (m, 2 H), 2.54 - 2.11 (m, 4 H), 1.76 - 1.53 (m,4 H), 2.07 - 1.51 (m, 6 H), 1.54 - 1.35 (m, 2 H), 1.27 (d, J = 6.7 Hz, 2 H), 0.95 (dd, J = 6.3, 18.8 Hz, 6 H). ESI-MS [M+H]+ calcd for C32H48N9O6S+ 686.34, found 686.6.
Ac-cyclo(KRE)-R- ketobenzothiazole (17):
This titled compound was synthesized using same procedure as Ac-cyclo(KLD)-R-ketobenzothiazole (15). Compound was isolated as a white solid. Overall yield (41 mg, 33%). 1H NMR (400MHz, DMSO-d6) δ = d = 8.50 - 8.40 (m, 1 H), 8.33 - 8.22 (m, 2 H), 8.15 - 8.04 (m, 1 H), 7.94 - 7.83 (m, 1 H), 7.69 (d, J = 3.1 Hz, 1 H), 7.58 - 7.43 (m, 1 H), 5.46 (br. s., 1 H), 4.42 - 4.28 (m, 2 H), 4.19 (d, J = 5.8 Hz, 1 H), 4.05 (d, J = 6.2 Hz, 1 H), 3.60 (br. s., 1 H), 3.39 (s, 2 H), 3.20 (s, 2H), 3.18 - 3.04 (m, 7H), 2.82 (d, J = 9.3 Hz, 1H), 2.34 - 2.14 (m, 2H), 2.08 - 1.89 (m, 2 H), 1.81 (s, 3 H), 1.69 - 1.36 (m, 9 H), 1.23 (br. s., 1 H). ESI-MS [M+H]+ calcd for C32H49N12O6S+ 729.36, found 729.6.
Ac-cyclo(DLK)-R- ketobenzothiazole (18):
This titled compound was synthesized using same procedure as Ac-cyclo(KLD)-R-ketobenzothiazole (15). Compound was isolated as a white solid. Overall yield (120 mg, 22%). 1H NMR (400MHz, DMSO-d6) δ = 8.51 (d, J = 6.7 Hz, 1 H), 8.26 (dd, J = 8.0, 15.1 Hz, 1 H), 7.98 (d, J = 7.4 Hz, 1 H), 7.93 - 7.83 (m, 2 H), 7.73 - 7.63 (m, 2 H), 7.53 (br. s., 1 H), 5.44 - 5.33 (m, 1 H), 4.60 - 4.48 (m, 1 H), 4.29 - 4.18 (m, 1 H), 3.42 (br. s., 4 H), 3.19 - 3.06 (m, 3 H), 2.96 (br. s., 1 H), 1.84 (s, 3 H), 1.78 - 1.69 (m, 1 H), 1.65 - 1.33 (m, 8 H), 1.23 - 1.07 (m, 2 H), 0.89 - 0.74 (m, 7 H). ESI-MS [M+H]+ calcd for C31H46N9O6S+ 672.33, found 672.5.
Ac-cyclo(ELK)-R- ketobenzothiazole (19):
This titled compound was synthesized using same procedure as Ac-cyclo(KLD)-R-ketobenzothiazole (15). Compound was isolated as a white solid. Overall yield (35 mg, 17%). 1H NMR (400MHz, DMSO-d6) δ = 8.60 (d, J = 6.3 Hz, 1 H), 8.34 - 8.19 (m, 1 H), 8.06 - 7.84 (m, 1 H), 7.76 - 7.63 (m, 1 H), 7.55 (br. s., 1 H), 7.40 - 7.27 (m, 1 H), 5.49 - 5.36 (m, 1 H), 5.00 (s, 1 H), 4.35 (d, J = 5.9 Hz, 2 H), 3.15 (d, J = 6.3 Hz, 2 H), 2.75 (br. s., 2 H), 2.19 - 2.09 (m, 4 H), 1.85 (br. s., 3 H), 1.81 (s, 3 H), 1.72 (br. s., 2 H), 1.65 - 1.39 (m, 11 H), 1.18 (br. s., 3 H), 0.82 (dd, J = 6.3, 14.1 Hz, 15 H). ESI-MS [M+H]+ calcd for C32H48N9O6S+ 686.34, found 686.5.
Ac-cyclo(DLK)-R- ketobenzothiazole-V-amide (20):
This titled compound was synthesized using same procedure as Ac-cyclo(KLD)-R-ketobenzothiazole (15). Compound was isolated as a white solid. Overall yield (16 mg, 20%). 1H NMR (400 MHz, DMSO-d6) δ ppm = 0.76 - 0.88 (m, 6 H), 0.95 (d, J=6.65 Hz, 6 H), 1.22 - 1.25 (m, 1 H), 1.46 - 1.61 (m, 1 H), 1.84 (s, 3 H), 2.09 - 2.17 (m, 1 H), 3.15 (d, J=6.26 Hz, 2 H), 4.20 - 4.36 (m, 2 H), 4.49 - 4.58 (m, 1 H), 5.34 - 5.42 (m, 1 H), 7.11 (br. s., 1 H), 7.52 (br. s., 1 H), 7.83 - 8.02 (m, 1 H), 8.14 (s, 1 H), 8.27 (s, 1 H), 8.38 - 8.43 (m, 1 H), 8.52 - 8.58 (m, 1 H), 8.81 (s, 1 H). ESI-MS [M+H]+ calcd for C37H56N11O8S+ 814.40, found 814.6.
Ac-cyclo(DLK)-R- ketothiazole (21):
This titled compound was synthesized using same procedure as Ac-cyclo(KLD)-R-ketobenzothiazole (15). Compound was isolated as a white solid. Overall yield (30 mg, 21%). 1H NMR (400MHz, CD3OD) δ ppm = 8.56 - 8.55 (m, 1 H), 8.55 (d, J = 7.8 Hz, 1 H), 8.58 - 8.52 (m, 1 H), 8.34 - 8.30 (m, 1 H), 8.32 (d, J = 7.8 Hz, 1 H), 8.15 (d, J = 7.8 Hz, 1 H), 8.18 - 8.13 (m, 1 H), 8.11 (s, 1 H), 8.13 - 8.10 (m, 1 H), 8.05 (d, J = 2.7 Hz, 1 H), 7.96 (t, J = 5.9 Hz, 1 H), 7.88 (d, J = 6.7 Hz, 1 H), 5.60 (br. s., 1 H), 4.73 - 4.66 (m, 1 H), 4.33 (br. s., 2 H), 3.26 (dd, J = 6.5, 11.5 Hz, 3 H), 2.84 - 2.64 (m, 3 H), 2.14 (t, J = 9.2 Hz, 1 H), 2.00 (s, 3 H), 1.88 - 1.50 (m, 4 H), 1.45 - 1.22 (m, 4 H), 0.95 (d, J = 5.9 Hz, 6 H), 0.89 (d, J = 5.9 Hz, 1 H). ESI-MS [M+H]+ calcd for C27H44N9O6S+ 622.31, found 622.5.
Ac-cyclo(DGK)-R- ketobenzothiazole (22):
This titled compound was synthesized using same procedure as Ac-cyclo(KLD)-R-ketobenzothiazole (15). Compound was isolated as a white solid. Overall yield (51 mg, 50%). 1H NMR (400MHz, DMSO-d6) δ ppm = 8.26 (br. s., 1 H), 7.77 (br. s., 1 H), 7.67 (br. s., 2 H), 7.49 (br. s., 2 H), 5.47 - 5.39 (m, 1 H), 4.54 - 4.43 (m, 1 H), 4.15 - 4.05 (m, 1 H), 3.69 (br. s., 3 H), 3.23 - 2.84 (m, 6 H), 2.54 (s, 3 H), 1.99 - 1.91 (s, 3 H), 1.84 (d, J = 3.1 Hz, 7 H), 1.60 (br. s., 3 H), 1.37 - 1.18 (m, 1 H). ESI-MS [M+H]+ calcd for C27H37N9O6S+ 616.27, found 616.40.
Ac-cyclo(DAK)-R- ketobenzothiazole (23):
This titled compound was synthesized using same procedure as Ac-cyclo(KLD)-R-ketobenzothiazole (15). Compound was isolated as a white solid. Overall yield (78 mg, 60%). 1H NMR (400MHz, DMSO-d6) δ ppm = 8.58 (br. s., 1 H), 8.25 (br. s., 2 H), 8.15 - 8.02 (m, 1 H), 7.66 (br. s., 2 H), 7.51 (br. s., 2 H), 5.56 - 5.37 (m, 1 H), 4.53 (br. s., 3 H), 4.21 (d, J = 5.9 Hz, 2 H), 3.13 (br. s., 2 H), 2.95 (br. s., 1 H), 2.01 (s, 3 H), 1.85 - 1.75 (m, 4 H), 1.59 (br. s., 9 H), 1.38 - 1.09 (m, 5 H). ESI-MS [M+H]+ calcd for C28H40N9O6S+ 630.28, found 630.4.
Ac-cyclo(DVK)-R- ketobenzothiazole (24):
This titled compound was synthesized using same procedure as Ac-cyclo(KLD)-R-ketobenzothiazole (15). Compound was isolated as a white solid. Overall yield (78 mg, 54%). 1H NMR (400MHz, DMSO-d6) δ = 8.27 - 8.12 (m, 4 H), 7.62 (br. s., 1 H), 7.45 (br. s., 1 H), 5.55 - 5.31 (m, 1 H), 4.55 (m, 1 H), 4.31 - 3.97 (m, 2 H), 3.09 (br. s., 4 H), 2.91 (br. s., 6 H), 2.31 (d, J = 12.9 Hz, 4 H), 1.91 (s, 3 H), 1.79 (br. s., 5 H), 1.55 (br. s., 4 H), 1.36 - 1.04 (m, 3 H), 0.90 - 0.60 (m, 4 H). ESI-MS [M+H]+ calcd for C30H44N9O6S+ 658.32, found 658.50.
Ac-cyclo(DNK)-R- ketobenzothiazole (25):
This titled compound was synthesized using same procedure as Ac-cyclo(KLD)-R-ketobenzothiazole (15). Compound was isolated as a white solid. Overall yield (57 mg, 31%). 1H NMR (400MHz, DMSO-d6) δ = 8.48 (br. s., 1 H), 8.26 (d, J = 5.9 Hz, 2 H), 7.97 (br. s., 1 H), 7.89 (br. s., 1 H), 7.67 (br. s., 2 H), 7.46 (br. s., 1 H), 7.36 (d, J = 14.5 Hz, 1 H), 6.81 (br. s., 1 H), 5.41 (br. s., 2 H), 4.56 - 4.42 (m, 1 H), 4.24 - 4.18 (m, 1 H), 3.15 (br. s., 6 H), 3.07 - 3.00 (m, 2 H), 2.68 - 2.61 (m, 5 H), 2.33 (br. s., 1 H), 1.85 (br. s., 11 H), 1.60 (br. s., 2 H). ESI-MS [M+H]+ calcd for C29H41N10O7S+ 673.29, found 673.50.
Ac-cyclo(DQK)-R- ketobenzothiazole (26):
This titled compound was synthesized using same procedure as Ac-cyclo(KLD)-R-ketobenzothiazole (15). Compound was isolated as a white solid. Overall yield (20 mg, 22%). 1H NMR (400MHz, DMSO-d6) δ = 9.19 (s, 1 H), 8.98 - 8.81 (m, 2 H), 8.78 - 8.67 (m, 1 H), 8.60 - 8.50 (m, 1 H), 8.38 - 8.24 (m, 1 H), 8.24 - 8.13 (m, 1 H), 7.89 - 7.81 (m, 1 H), 7.41 - 7.32 (m, 1 H), 7.29 - 7.11 (m, 1 H), 6.21 - 5.98 (m, 2 H), 5.25 - 5.08 (m, 1 H), 4.94 - 4.82 (m, 1 H), 4.05 (br. s., 2 H), 3.79 (t, J = 6.1 Hz, 6 H), 3.15 – 3.04 (m, 6 H), 2.69 - 2.56 (m, 3 H), 2.44 - 2.32 (m, 3 H), 2.04 (s, 3 H), 2.25 (br. s., 5 H). ESI-MS [M+H]+ calcd for C30H43N10O7S+ 687.30, found 687.50.
Ac-cyclo(DTK)-R- ketobenzothiazole (27):
This titled compound was synthesized using same procedure as Ac-cyclo(KLD)-R-ketobenzothiazole (15). Compound was isolated as a white solid. Overall yield (69 mg, 63%). 1H NMR (400MHz, DMSO-d6) δ = 8.25 (br. s., 2 H), 7.66 (br. s., 2 H), 7.59 - 7.47 (m, 2 H), 5.56 - 5.36 (m, 2 H), 3.13 (br. s., 7 H), 3.00 - 2.86 (m, 5 H), 2.03 (s, 3 H), 2.00 - 1.87 (m, 3 H), 1.89 - 1.73 (m, 6 H), 1.59 (br. s., 6 H), 1.01 (d, J = 6.7 Hz, 3 H). ESI-MS [M+H]+ calcd for C29H42N9O7S+ 660.29, found 660.50.
Ac-cyclo(DMK)-R- ketobenzothiazole (28):
This titled compound was synthesized using same procedure as Ac-cyclo(KLD)-R-ketobenzothiazole (15). Compound was isolated as a white solid. Overall yield (40 mg, 42%). 1H NMR (400MHz, DMSO-d6) δ = 8.54 (d, J = 6.3 Hz, 1 H), 8.31 - 8.07 (m, 2 H), 7.93 (d, J = 8.2 Hz, 1 H), 7.68 (br. s., 1 H), 7.49 (br. s., 1 H), 5.40 (br. s., 1 H), 4.51 (br. s., 1 H), 4.22 (br. s., 1 H), 3.15 (br. s., 6 H), 2.00 (d, J = 1.2 Hz, 4 H), 1.88 - 1.77 (s, 3 H), 1.58 (br. s., 3 H), 1.39 - 1.03 (m, 16 H). ESI-MS [M+H]+ calcd for C30H43N9O6S2+ 690.28, found 690.40.
Ac-cyclo(DFK)-R- ketobenzothiazole (29):
This titled compound was synthesized using same procedure as Ac-cyclo(KLD)-R-ketobenzothiazole (15). Compound was isolated as a white solid. Overall yield (33 mg, 48%). 1H NMR (400MHz, DMSO-d6) δ = 8.55 (d, J = 6.3 Hz, 2 H), 8.25 (d, J = 8.2 Hz, 2 H), 8.08 (s, 2 H), 7.80 (d, J = 7.8 Hz, 2 H), 7.71 - 7.51 (m, 2 H), 7.28 - 7.11 (m, 5 H), 5.56 - 5.35 (m, 1 H), 4.60 - 4.40 (m, 1 H), 4.31 - 4.23 (m, 1 H), 3.20 - 2.91 (m, 13 H), 2.01 - 1.90 (m, 1 H), 1.78 (d, J = 2.0 Hz, 3 H), 1.58 (br. s., 8 H). ESI-MS [M+H]+ calcd for C34H44N9O6S+ 706.31, found 706.50.
Ac-cyclo(DYK)-R- ketobenzothiazole (30):
This titled compound was synthesized using same procedure as Ac-cyclo(KLD)-R-ketobenzothiazole (15). Compound was isolated as a white solid. Overall yield (40mg, 36%). 1H NMR (400MHz, DMSO-d6) δ = 8.54 (d, J = 6.3 Hz, 2 H), 8.34 - 8.18 (m, 2 H), 7.91 - 7.77 (m, 2 H), 7.67 (br. s., 2 H), 7.55 - 7.41 (m, 2 H), 7.03 - 6.89 (m, 2 H), 6.59 (d, J = 6.7 Hz, 2 H), 4.56 (s, 1 H), 4.27 (d, J = 5.9 Hz, 1 H), 3.23 - 2.88 (m, 19 H), 2.01 (s, 3 H), 1.79 (s, 3 H), 1.58 (br. s., 2 H). ESI-MS [M+H]+ calcd for C34H44N9O7S+ 722.31, found 722.50.
Ac-cyclo(DYK(N-Me))-R- ketobenzothiazole (32):
This titled compound was synthesized using same procedure as Ac-cyclo(KLD)-R-ketobenzothiazole (15). Compound was isolated as a white solid. Overall yield (16 mg, 45%). 1H NMR (400MHz, DMSO-d 6) δ = 8.48 (d, J = 7.8 Hz, 2 H), 8.41 - 8.35 (m, 1 H), 8.31 - 8.22 (m, 6 H), 7.73 - 7.65 (m, 6 H), 7.50 (br. s., 2 H), 5.39 (br. s., 2 H), 5.20 (br. s., 1 H), 4.57 (br. s., 1 H), 4.39 - 4.23 (m, 2 H), 4.09 (br. s., 1 H), 3.15 (d, J = 4.7 Hz, 5 H), 3.06 (d, J = 6.3 Hz, 4 H), 2.88 (s, 3 H), 2.23 (d, J = 14.9 Hz, 1 H), 1.97 (br. s., 2 H), 1.85 (s, 2 H), 1.83 - 1.77 (m, 4 H), 1.77 - 1.52 (m, 10 H), 1.43 - 1.32 (m, 1 H), 0.93 - 0.75 (m, 15 H). ESI-MS [M+H]+ calcd for C32H47N9O6S+ 686.34, found 686.50.
3. Biochemical Studies
Fluorescent Kinetic Enzyme Inhibitor Assays of HGFA, matriptase, hepsin and thrombin:
Inhibitors (11-pt serial dilutions, 0-20 μM final concentration in reaction) were serially diluted in DMSO (2% DMSO final concentration) and then mixed with either recombinant serine protease domain of HGFA80, matriptase (Chares Craik, UCSF) or hepsin* (#4776-SE-010, R&D Systems, Minneapolis, Minnesota) in black 384 well plates (Corning # 3575. Corning, NY). The final assay concentration for HGFA, matriptase and hepsin 7.5 nM, 0.2 nM, and 0.3 nM, respectively in TNC buffer (25 mM Tris, 150 mM NaCl, 5 mM CaCl2, 0.01% Triton X-100, pH 8). After thirty minutes incubation at room temperature, Boc-QLR-AMC substrate (Km = 37 μM) was added to the HGFA assays and Boc-QAR-AMC substrate was added to the matriptase (Km = 93 μM) and hepsin (Km = 156 μM) assays. The final substrate concentrations for all assays were at the Km for the respective enzymes. Changes in fluorescence (excitation at 380 nm and emission at 460 nm) were measured at room temperature over time in a Biotek Synergy 2 plate reader (Winnoski, VT). Using GraphPad Prism version 6.04 software program, (GraphPad Software, San Diego, CA, www.graphpad.com), a four parameter curve fit was used to determine the inhibitor IC50s from a plot of the mean reaction velocity versus the inhibitor concentration. The IC50 values represent the average of three separate experimental determinations.
*Hepsin Activation:
Recombinant Hepsin (10 μg, 0.44mg/mL as received from R&D Systems, Catalogue # 4776-SE) was diluted to 2.4 μM in TNC buffer (25 mM Tris, 150 mM NaCl, 5 mM CaCl2, 0.01% Triton X-100, pH 8) and incubated at 37°C. After twenty-four hours, the hepsin was diluted in glycerol to 50%. This stock hepsin (1.2 μM) was stored in a −20°C freezer and diluted in TNC buffer for use in assays.
Chromogenic Kinetic Enzyme Assay of Thrombin and Factor Xa:
Inhibitors (11-pt serial dilutions, 0-20 μM final concentration) were serially diluted in DMSO (2% DMSO final concentration) and then mixed with recombinant thrombin (0.15 nM final concentration) or Factor Xa (0.35 nM final concentration) in TNC buffer (25 mM Tris, 150 mM NaCl, 5 mM CaCl2, 0.01% Triton X-100, pH 8) using clear 384 well plates. After incubating for 30 minutes at 25° C, the chromogenic substrate (S2238; D-Phe-Pip-Arg-pNA) for thrombin (Km = 14.5 μM) (#1473-SE-010, R&D Systems, Minneapolis, Minnesota) or (S2222; Bz-Ile-Glu-Gly-Arg-pNA) for Factor Xa (Km = 200 μM) was added to a final concentration of Km (4 x Km (50 μM) for thrombin) in a final reaction volume of 40 microliters. Changes in absorbance at 405 nm were measured over time in a Biotek Synergy 2 plate (Winnoski, VT). Using GraphPad Prism version 6.04 software program, (GraphPad Software, San Diego, CA, www.graphpad.com), a four parameter curve fit was used to determine the inhibitor IC50s from a plot of the mean reaction velocity versus the inhibitor concentration.
Statistical Analysis:
Data analysis was performed using Prism software (version 6.04: Graphpad, La Jolla, CA, USA). p-values of less than 0.05 were considered statistically significant.
Supplementary Material
Acknowledgments:
This research was funded by Alvin J. Siteman Cancer Research Fund, #16-FY18-02, pre-R01 SCC P30CA091842, Barnes Jewish Hospital Foundation Grant, BJHF 4984, Washington University School of Medicine, and the National Institutes of Health, National Cancer Institute grant award numbers 1R43CA224832 and 1R43CA243941. The authors would also like to thank BioSTL (Biogenerator) for private funding towards the efficacy studies, and the Center for Drug Discovery (CDC), Washington University School of Medicine for funding to do intermediate synthesis for scale-up of ZFH7116 (31) and VD2173 (18).
Non-standard Abbreviations Used
- EGFR
epidermal growth factor receptor
- HAI
HGF activation inhibitor
- HGF
hepatocyte growth factor
- Kt
ketothiazole
- Kbt
ketobenzothiazole
- NSCLC
non-small-cell lung cancer
- PK
pharmacokinetics
- PPB
plasma protein binding
- SPPS
solid-phase peptide synthesis
- TKI
tyrosine kinase inhibitors
Footnotes
Patents: The following patents are related to this work: PCT WO2016144654, US20180066015, PCT WO2020264187.
Supplementary Information Availability: The following are available online at https://pubs.acs.org/: Details of all biological experimental materials and methods. Description of the synthesis and characterization of N-Me 18 (32). Compound characterization for synthetic intermediates. HPLC, MS and NMR spectra for all final compounds. Molecular formula strings.
Institutional Review Board Statement: The study was conducted according to the guidelines of the Declaration of Helsinki and approved by Washington University’s Institutional Animal Care and Use Committee (Protocol 20180223, Approved Oct 18, 2018).
Conflicts of Interest: JWJ and LK are board members and owners of ProteXase Therapeutics, Inc. company stock.
REFERENCES
- [1].Siegel RL, Miller KD, and Jemal A (2020) Cancer statistics, 2020, CA: A Cancer Journal for Clinicians 70, 7–30. [DOI] [PubMed] [Google Scholar]
- [2].Meador CB, and Hata AN (2020) Acquired resistance to targeted therapies in NSCLC: Updates and evolving insights, Pharmacol Ther, 107522. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [3].Liang H, and Wang M (2020) MET Oncogene in Non-Small Cell Lung Cancer: Mechanism of MET Dysregulation and Agents Targeting the HGF/c-Met Axis, Onco Targets Ther 13, 2491–2510. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [4].D'Angelo SP, Pietanza MC, Johnson ML, Riely GJ, Miller VA, Sima CS, Zakowski MF, Rusch VW, Ladanyi M, and Kris MG (2011) Incidence of EGFR exon 19 deletions and L858R in tumor specimens from men and cigarette smokers with lung adenocarcinomas, J Clin Oncol 29, 2066–2070. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [5].Mitsudomi T, and Yatabe Y (2007) Mutations of the epidermal growth factor receptor gene and related genes as determinants of epidermal growth factor receptor tyrosine kinase inhibitors sensitivity in lung cancer, Cancer Sci 98, 1817–1824. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [6].Camidge DR, Pao W, and Sequist LV (2014) Acquired resistance to TKIs in solid tumours: learning from lung cancer, Nat Rev Clin Oncol 11, 473–481. [DOI] [PubMed] [Google Scholar]
- [7].Sequist LV, Waltman BA, Dias-Santagata D, Digumarthy S, Turke AB, Fidias P, Bergethon K, Shaw AT, Gettinger S, Cosper AK, Akhavanfard S, Heist RS, Temel J, Christensen JG, Wain JC, Lynch TJ, Vernovsky K, Mark EJ, Lanuti M, Iafrate AJ, Mino-Kenudson M, and Engelman JA (2011) Genotypic and histological evolution of lung cancers acquiring resistance to EGFR inhibitors, Sci Transl Med 3, 75ra26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [8].Janne PA, Yang JC, Kim DW, Planchard D, Ohe Y, Ramalingam SS, Ahn MJ, Kim SW, Su WC, Horn L, Haggstrom D, Felip E, Kim JH, Frewer P, Cantarini M, Brown KH, Dickinson PA, Ghiorghiu S, and Ranson M (2015) AZD9291 in EGFR inhibitor-resistant non-small-cell lung cancer, N Engl J Med 372, 1689–1699. [DOI] [PubMed] [Google Scholar]
- [9].Nagano T, Tachihara M, and Nishimura Y (2018) Mechanism of Resistance to Epidermal Growth Factor Receptor-Tyrosine Kinase Inhibitors and a Potential Treatment Strategy, Cells 7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [10].Sun Y (2016) Tumor microenvironment and cancer therapy resistance, Cancer Lett 380, 205–215. [DOI] [PubMed] [Google Scholar]
- [11].Wu T, and Dai Y (2017) Tumor microenvironment and therapeutic response, Cancer Lett 387, 61–68. [DOI] [PubMed] [Google Scholar]
- [12].Owusu BY, Galemmo R, Janetka J, and Klampfer L (2017) Hepatocyte Growth Factor, a Key Tumor-Promoting Factor in the Tumor Microenvironment, Cancers (Basel) 9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [13].Wilson TR, Fridlyand J, Yan Y, Penuel E, Burton L, Chan E, Peng J, Lin E, Wang Y, Sosman J, Ribas A, Li J, Moffat J, Sutherlin DP, Koeppen H, Merchant M, Neve R, and Settleman J (2012) Widespread potential for growth-factor-driven resistance to anticancer kinase inhibitors, Nature 487, 505–509. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [14].Ko B, He T, Gadgeel S, and Halmos B (2017) MET/HGF pathway activation as a paradigm of resistance to targeted therapies, Ann Transl Med 5, 4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [15].Pennacchietti S, Cazzanti M, Bertotti A, Rideout WM 3rd, Han M, Gyuris J, Perera T, Comoglio PM, Trusolino L, and Michieli P (2014) Microenvironment-derived HGF overcomes genetically determined sensitivity to anti-MET drugs, Cancer Res 74, 6598–6609. [DOI] [PubMed] [Google Scholar]
- [16].Zhang Y, Xia M, Jin K, Wang S, Wei H, Fan C, Wu Y, Li X, Li X, Li G, Zeng Z, and Xiong W (2018) Function of the c-Met receptor tyrosine kinase in carcinogenesis and associated therapeutic opportunities, Mol Cancer 17, 45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [17].Kuang WB, Deng QC, Deng CT, Li WS, Shu SW, and Zhou MR (2017) Hepatocyte growth factor induces breast cancer cell invasion via the PI3K/Akt and p38 MAPK signaling pathways to up-regulate the expression of COX2, Am J Transl Res 9, 3816–3826. [PMC free article] [PubMed] [Google Scholar]
- [18].Leung E, Xue A, Wang Y, Rougerie P, Sharma VP, Eddy R, Cox D, and Condeelis J (2017) Blood vessel endothelium-directed tumor cell streaming in breast tumors requires the HGF/C-Met signaling pathway, Oncogene 36, 2680–2692. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [19].Grotegut S, von Schweinitz D, Christofori G, and Lehembre F (2006) Hepatocyte growth factor induces cell scattering through MAPK/Egr-1-mediated upregulation of Snail, EMBO J 25, 3534–3545. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [20].Ogunwobi OO, and Liu C (2011) Hepatocyte growth factor upregulation promotes carcinogenesis and epithelial-mesenchymal transition in hepatocellular carcinoma via Akt and COX-2 pathways, Clin Exp Metastasis 28, 721–731. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [21].Zhang YW, Su YL, Volpert OV, and Van de Woude GF (2003) Hepatocyte growth factor/scatter factor mediates angiogenesis through positive VEGF and negative thrombospondin 1 regulation, P Natl Acad Sci USA 100, 12718–12723. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [22].Vermeulen L, De Sousa E Melo F, van der Heijden M, Cameron K, de Jong JH, Borovski T, Tuynman JB, Todaro M, Merz C, Rodermond H, Sprick MR, Kemper K, Richel DJ, Stassi G, and Medema JP (2010) Wnt activity defines colon cancer stem cells and is regulated by the microenvironment, Nature Cell Biology 12, 468–476. [DOI] [PubMed] [Google Scholar]
- [23].Donev IS, Wang W, Yamada T, Li Q, Takeuchi S, Matsumoto K, Yamori T, Nishioka Y, Sone S, and Yano S (2011) Transient PI3K inhibition induces apoptosis and overcomes HGF-mediated resistance to EGFR-TKIs in EGFR mutant lung cancer, Clin Cancer Res 17, 2260–2269. [DOI] [PubMed] [Google Scholar]
- [24].Nakamura T, Matsumoto K, Kiritoshi A, Tano Y, and Nakamura T (1997) Induction of hepatocyte growth factor in fibroblasts by tumor-derived factors affects invasive growth of tumor cells: in vitro analysis of tumor-stromal interactions, Cancer Res 57, 3305–3313. [PubMed] [Google Scholar]
- [25].Kawaguchi M, and Kataoka H (2014) Mechanisms of hepatocyte growth factor activation in cancer tissues, Cancers (Basel) 6, 1890–1904. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [26].Harvey P, Warn A, Newman P, Perry LJ, Ball RY, and Warn RM (1996) Immunoreactivity for hepatocyte growth factor/scatter factor and its receptor, met, in human lung carcinomas and malignant mesotheliomas, J Pathol 180, 389–394. [DOI] [PubMed] [Google Scholar]
- [27].Frampton GM, Ali SM, Rosenzweig M, Chmielecki J, Lu X, Bauer TM, Akimov M, Bufill JA, Lee C, Jentz D, Hoover R, Ou SH, Salgia R, Brennan T, Chalmers ZR, Jaeger S, Huang A, Elvin JA, Erlich R, Fichtenholtz A, Gowen KA, Greenbowe J, Johnson A, Khaira D, McMahon C, Sanford EM, Roels S, White J, Greshock J, Schlegel R, Lipson D, Yelensky R, Morosini D, Ross JS, Collisson E, Peters M, Stephens PJ, and Miller VA (2015) Activation of MET via diverse exon 14 splicing alterations occurs in multiple tumor types and confers clinical sensitivity to MET inhibitors, Cancer Discov 5, 850–859. [DOI] [PubMed] [Google Scholar]
- [28].Arrieta O, Cruz-Rico G, Soto-Perez-de-Celis E, Ramirez-Tirado LA, Caballe-Perez E, Martinez-Hernandez JN, Martinez-Alvarez I, Soca-Chafre G, Macedo-Perez EO, and Astudillo-de la Vega H (2016) Reduction in Hepatocyte Growth Factor Serum Levels is Associated with Improved Prognosis in Advanced Lung Adenocarcinoma Patients Treated with Afatinib: a Phase II Trial, Target Oncol 11, 619–629. [DOI] [PubMed] [Google Scholar]
- [29].Kasahara K, Arao T, Sakai K, Matsumoto K, Sakai A, Kimura H, Sone T, Horiike A, Nishio M, Ohira T, Ikeda N, Yamanaka T, Saijo N, and Nishio K (2010) Impact of serum hepatocyte growth factor on treatment response to epidermal growth factor receptor tyrosine kinase inhibitors in patients with non-small cell lung adenocarcinoma, Clin Cancer Res 16, 4616–4624. [DOI] [PubMed] [Google Scholar]
- [30].Arriola E, Canadas I, Arumi-Uria M, Domine M, Lopez-Vilarino JA, Arpi O, Salido M, Menendez S, Grande E, Hirsch FR, Serrano S, Bellosillo B, Rojo F, Rovira A, and Albanell J (2011) MET phosphorylation predicts poor outcome in small cell lung carcinoma and its inhibition blocks HGF-induced effects in MET mutant cell lines, Br J Cancer 105, 814–823. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [31].Chen JT, Lin TS, Chow KC, Huang HH, Chiou SH, Chiang SF, Chen HC, Chuang TL, Lin TY, and Chen CY (2006) Cigarette smoking induces overexpression of hepatocyte growth factor in type II pneumocytes and lung cancer cells, Am J Respir Cell Mol Biol 34, 264–273. [DOI] [PubMed] [Google Scholar]
- [32].Titmarsh HF, O'Connor R, Dhaliwal K, and Akram AR (2020) The Emerging Role of the c-MET-HGF Axis in Non-small Lung Cancer Tumor Immunology and Immunotherapy, Front Oncol 10, 54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [33].Masuya D, Huang C, Liu D, Nakashima T, Kameyama K, Haba R, Ueno M, and Yokomise H (2004) The tumour-stromal interaction between intratumoral c-Met and stromal hepatocyte growth factor associated with tumour growth and prognosis in non-small-cell lung cancer patients, Br J Cancer 90, 1555–1562. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [34].Yu HA, Arcila ME, Rekhtman N, Sima CS, Zakowski MF, Pao W, Kris MG, Miller VA, Ladanyi M, and Riely GJ (2013) Analysis of tumor specimens at the time of acquired resistance to EGFR-TKI therapy in 155 patients with EGFR-mutant lung cancers, Clin Cancer Res 19, 2240–2247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [35].Engelman JA, Zejnullahu K, Mitsudomi T, Song Y, Hyland C, Park JO, Lindeman N, Gale CM, Zhao X, Christensen J, Kosaka T, Holmes AJ, Rogers AM, Cappuzzo F, Mok T, Lee C, Johnson BE, Cantley LC, and Janne PA (2007) MET amplification leads to gefitinib resistance in lung cancer by activating ERBB3 signaling, Science 316, 1039–1043. [DOI] [PubMed] [Google Scholar]
- [36].Yano S, Yamada T, Takeuchi S, Tachibana K, Minami Y, Yatabe Y, Mitsudomi T, Tanaka H, Kimura T, Kudoh S, Nokihara H, Ohe Y, Yokota J, Uramoto H, Yasumoto K, Kiura K, Higashiyama M, Oda M, Saito H, Yoshida J, Kondoh K, and Noguchi M (2011) Hepatocyte growth factor expression in EGFR mutant lung cancer with intrinsic and acquired resistance to tyrosine kinase inhibitors in a Japanese cohort, J Thorac Oncol 6, 2011–2017. [DOI] [PubMed] [Google Scholar]
- [37].Yano S, Wang W, Li Q, Matsumoto K, Sakurama H, Nakamura T, Ogino H, Kakiuchi S, Hanibuchi M, Nishioka Y, Uehara H, Mitsudomi T, Yatabe Y, Nakamura T, and Sone S (2008) Hepatocyte growth factor induces gefitinib resistance of lung adenocarcinoma with epidermal growth factor receptor-activating mutations, Cancer Res 68, 9479–9487. [DOI] [PubMed] [Google Scholar]
- [38].Raghav K, Bailey AM, Loree JM, Kopetz S, Holla V, Yap TA, Wang F, Chen K, Salgia R, and Hong D (2018) Untying the gordion knot of targeting MET in cancer, Cancer Treat Rev 66, 95–103. [DOI] [PubMed] [Google Scholar]
- [39].Dhillon S (2020) Capmatinib: First Approval, Drugs 80, 1125–1131. [DOI] [PubMed] [Google Scholar]
- [40].Lokker NA, Mark MR, Luis EA, Bennett GL, Robbins KA, Baker JB, and Godowski PJ (1992) Structure-function analysis of hepatocyte growth factor: identification of variants that lack mitogenic activity yet retain high affinity receptor binding, EMBO J 11, 2503–2510. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [41].Organ SL, and Tsao MS (2011) An overview of the c-MET signaling pathway, Ther Adv Med Oncol 3, S7–S19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [42].Owusu BY, Thomas S, Venukadasula P, Han Z, Janetka JW, Galemmo RA Jr., and Klampfer L (2017) Targeting the tumor-promoting microenvironment in MET-amplified NSCLC cells with a novel inhibitor of pro-HGF activation, Oncotarget 8, 63014–63025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [43].Oberst MD, Johnson MD, Dickson RB, Lin CY, Singh B, Stewart M, Williams A, al-Nafussi A, Smyth JF, Gabra H, and Sellar GC (2002) Expression of the serine protease matriptase and its inhibitor HAI-1 in epithelial ovarian cancer: Correlation with clinical outcome and tumor clinicopathological parameters, Clinical Cancer Research 8, 1101–1107. [PubMed] [Google Scholar]
- [44].Yamauchi M, Kataoka H, Itoh H, Seguchi T, Hasui Y, and Osada Y (2004) Hepatocyte growth factor activator inhibitor types 1 and 2 are expressed by tubular epithelium in kidney and down-regulated in renal cell carcinoma, J Urol 171, 890–896. [DOI] [PubMed] [Google Scholar]
- [45].Nakamura K, Abarzua F, Kodama J, Hongo A, Nasu Y, Kumon H, and Hiramatsu Y (2009) Expression of hepatocyte growth factor activator inhibitors (HAI-1 and HAI-2) in ovarian cancer, Int J Oncol 34, 345–353. [PubMed] [Google Scholar]
- [46].Damalanka VC, Wildman SA, and Janetka JW (2019) Piperidine carbamate peptidomimetic inhibitors of the serine proteases HGFA, matriptase and hepsin, Medchemcomm 10, 1646–1655. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [47].Franco FM, Jones DE, Harris PK, Han Z, Wildman SA, Jarvis CM, and Janetka JW (2015) Structure-based discovery of small molecule hepsin and HGFA protease inhibitors: Evaluation of potency and selectivity derived from distinct binding pockets, Bioorg. Med. Chem 23, 2328–2343. [DOI] [PubMed] [Google Scholar]
- [48].Han Z, Harris PK, Karmakar P, Kim T, Owusu BY, Wildman SA, Klampfer L, and Janetka JW (2016) α-Ketobenzothiazole Serine Protease Inhibitors of Aberrant HGF/c-MET and MSP/RON Kinase Pathway Signaling in Cancer, ChemMedChem 11, 585–599. [DOI] [PubMed] [Google Scholar]
- [49].Damalanka VC, Han Z, Karmakar P, O'Donoghue AJ, La Greca F, Kim T, Pant SM, Helander J, Klefström J, Craik CS, and Janetka JW (2019) Discovery of Selective Matriptase and Hepsin Serine Protease Inhibitors: Useful Chemical Tools for Cancer Cell Biology, J. Med. Chem 62, 480–490. [DOI] [PubMed] [Google Scholar]
- [50].Han Z, Harris PK, Jones DE, Chugani R, Kim T, Agarwal M, Shen W, Wildman SA, and Janetka JW (2014) Inhibitors of HGFA, Matriptase, and Hepsin Serine Proteases: A Nonkinase Strategy to Block Cell Signaling in Cancer, ACS Med Chem Lett 5, 1219–1224. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [51].Owusu BY, Bansal N, Venukadasula PK, Ross LJ, Messick TE, Goel S, Galemmo RA, and Klampfer L (2016) Inhibition of pro-HGF activation by SRI31215, a novel approach to block oncogenic HGF/MET signaling, Oncotarget 7, 29492–29506. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [52].Damalanka VC, Kim Y, Alliston KR, Weerawarna PM, Galasiti Kankanamalage AC, Lushington GH, Mehzabeen N, Battaile KP, Lovell S, Chang K-O, and Groutas WC (2016) Oxadiazole-Based Cell Permeable Macrocyclic Transition State Inhibitors of Norovirus 3CL Protease, J. Med. Chem 59, 1899–1913. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [53].Lin JH (2009) Pharmacokinetics of biotech drugs: peptides, proteins and monoclonal antibodies, Curr Drug Metab 10, 661–691. [DOI] [PubMed] [Google Scholar]
- [54].Otvos L, and Wade JD (2014) Current challenges in peptide-based drug discovery, Frontiers in Chemistry 2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [55].Rezai T, Yu B, Millhauser GL, Jacobson MP, and Lokey RS (2006) Testing the conformational hypothesis of passive membrane permeability using synthetic cyclic peptide diastereomers, J. Am. Chem. Soc 128, 2510–2511. [DOI] [PubMed] [Google Scholar]
- [56].Buckton LK, Rahimi MN, and McAlpine SR (2021) Cyclic Peptides as Drugs for Intracellular Targets: The Next Frontier in Peptide Therapeutic Development, Chemistry – A European Journal 27, 1487–1513. [DOI] [PubMed] [Google Scholar]
- [57].Malde AK, Hill TA, Iyer A, and Fairlie DP (2019) Crystal Structures of Protein-Bound Cyclic Peptides, Chem. Rev 119, 9861–9914. [DOI] [PubMed] [Google Scholar]
- [58].Mallinson J, and Collins I (2012) Macrocycles in new drug discovery, Future Med Chem 4, 1409–1438. [DOI] [PubMed] [Google Scholar]
- [59].Alex A, Millan DS, Perez M, Wakenhut F, and Whitlock GA (2011) Intramolecular hydrogen bonding to improve membrane permeability and absorption in beyond rule of five chemical space, MedChemComm 2, 669–674. [Google Scholar]
- [60].Marsault E, and Peterson ML (2011) Macrocycles Are Great Cycles: Applications, Opportunities, and Challenges of Synthetic Macrocycles in Drug Discovery, J. Med. Chem 54, 1961–2004. [DOI] [PubMed] [Google Scholar]
- [61].White CJ, and Yudin AK (2011) Contemporary strategies for peptide macrocyclization, Nature Chemistry 3, 509–524. [DOI] [PubMed] [Google Scholar]
- [62].Giordanetto F, and Kihlberg J (2014) Macrocyclic Drugs and Clinical Candidates: What Can Medicinal Chemists Learn from Their Properties?, Journal of Medicinal Chemistry 57, 278–295. [DOI] [PubMed] [Google Scholar]
- [63].Mathiowetz AM, Leung SSF, and Jacobson MP (2015) Optimizing the permeability and oral bioavailability of macrocycles, In RSC Drug Discovery Series, pp 367–397. [Google Scholar]
- [64].Steinmetzer T, Schweinitz A, Sturzebecher A, Donnecke D, Uhland K, Schuster O, Steinmetzer P, Muller F, Friedrich R, Than ME, Bode W, and Sturzebecher J (2006) Secondary amides of sulfonylated 3-amidinophenylalanine. New potent and selective inhibitors of matriptase, J Med Chem 49, 4116–4126. [DOI] [PubMed] [Google Scholar]
- [65].Herter S, Piper DE, Aaron W, Gabriele T, Cutler G, Cao P, Bhatt AS, Choe Y, Craik CS, Walker N, Meininger D, Hoey T, and Austin RJ (2005) Hepatocyte growth factor is a preferred in vitro substrate for human hepsin, a membrane-anchored serine protease implicated in prostate and ovarian cancers, Biochem J 390, 125–136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [66].Ganesan R, Eigenbrot C, Wu Y, Liang WC, Shia S, Lipari MT, and Kirchhofer D (2009) Unraveling the allosteric mechanism of serine protease inhibition by an antibody, Structure 17, 1614–1624. [DOI] [PubMed] [Google Scholar]
- [67].Han Z, Harris PK, Karmakar P, Kim T, Owusu BY, Wildman SA, Klampfer L, and Janetka JW (2016) alpha-Ketobenzothiazole Serine Protease Inhibitors of Aberrant HGF/c-MET and MSP/RON Kinase Pathway Signaling in Cancer, ChemMedChem 11, 585–599. [DOI] [PubMed] [Google Scholar]
- [68].Kobayashi S, Boggon TJ, Dayaram T, Janne PA, Kocher O, Meyerson M, Johnson BE, Eck MJ, Tenen DG, and Halmos B (2005) EGFR mutation and resistance of non-small-cell lung cancer to gefitinib, N Engl J Med 352, 786–792. [DOI] [PubMed] [Google Scholar]
- [69].Jorge SE, Schulman S, Freed JA, VanderLaan PA, Rangachari D, Kobayashi SS, Huberman MS, and Costa DB (2015) Responses to the multitargeted MET/ALK/ROS1 inhibitor crizotinib and co-occurring mutations in lung adenocarcinomas with MET amplification or MET exon 14 skipping mutation, Lung Cancer 90, 369–374. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [70].Liu X, Jia Y, Stoopler MB, Shen Y, Cheng H, Chen J, Mansukhani M, Koul S, Halmos B, and Borczuk AC (2016) Next-Generation Sequencing of Pulmonary Sarcomatoid Carcinoma Reveals High Frequency of Actionable MET Gene Mutations, J Clin Oncol 34, 794–802. [DOI] [PubMed] [Google Scholar]
- [71].Cortot AB, Kherrouche Z, Descarpentries C, Wislez M, Baldacci S, Furlan A, and Tulasne D (2017) Exon 14 Deleted MET Receptor as a New Biomarker and Target in Cancers, J Natl Cancer Inst 109. [DOI] [PubMed] [Google Scholar]
- [72].Kong-Beltran M, Seshagiri S, Zha J, Zhu W, Bhawe K, Mendoza N, Holcomb T, Pujara K, Stinson J, Fu L, Severin C, Rangell L, Schwall R, Amler L, Wickramasinghe D, and Yauch R (2006) Somatic mutations lead to an oncogenic deletion of met in lung cancer, Cancer Res 66, 283–289. [DOI] [PubMed] [Google Scholar]
- [73].Zhang YW, Staal B, Essenburg C, Su Y, Kang L, West R, Kaufman D, Dekoning T, Eagleson B, Buchanan SG, and Vande Woude GF (2010) MET kinase inhibitor SGX523 synergizes with epidermal growth factor receptor inhibitor erlotinib in a hepatocyte growth factor-dependent fashion to suppress carcinoma growth, Cancer Res 70, 6880–6890. [DOI] [PubMed] [Google Scholar]
- [74].Ghandi M, Huang FW, Jane-Valbuena J, Kryukov GV, Lo CC, McDonald ER 3rd, Barretina J, Gelfand ET, Bielski CM, Li H, Hu K, Andreev-Drakhlin AY, Kim J, Hess JM, Haas BJ, Aguet F, Weir BA, Rothberg MV, Paolella BR, Lawrence MS, Akbani R, Lu Y, Tiv HL, Gokhale PC, de Weck A, Mansour AA, Oh C, Shih J, Hadi K, Rosen Y, Bistline J, Venkatesan K, Reddy A, Sonkin D, Liu M, Lehar J, Korn JM, Porter DA, Jones MD, Golji J, Caponigro G, Taylor JE, Dunning CM, Creech AL, Warren AC, McFarland JM, Zamanighomi M, Kauffmann A, Stransky N, Imielinski M, Maruvka YE, Cherniack AD, Tsherniak A, Vazquez F, Jaffe JD, Lane AA, Weinstock DM, Johannessen CM, Morrissey MP, Stegmeier F, Schlegel R, Hahn WC, Getz G, Mills GB, Boehm JS, Golub TR, Garraway LA, and Sellers WR (2019) Next-generation characterization of the Cancer Cell Line Encyclopedia, Nature 569, 503–508. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [75].Albitar M, Sudarsanam S, Ma W, Jiang S, Chen W, Funari V, Blocker F, and Agersborg S (2018) Correlation of MET gene amplification and TP53 mutation with PD-L1 expression in non-small cell lung cancer, Oncotarget 9, 13682–13693. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [76].Peng S, Wang R, Zhang X, Ma Y, Zhong L, Li K, Nishiyama A, Arai S, Yano S, and Wang W (2019) EGFR-TKI resistance promotes immune escape in lung cancer via increased PD-L1 expression, Mol Cancer 18, 165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [77].Demuth C, Andersen MN, Jakobsen KR, Madsen AT, and Sorensen BS (2017) Increased PD-L1 expression in erlotinib-resistant NSCLC cells with MET gene amplification is reversed upon MET-TKI treatment, Oncotarget 8, 68221–68229. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [78].Saigi M, Alburquerque-Bejar JJ, Mc Leer-Florin A, Pereira C, Pros E, Romero OA, Baixeras N, Esteve-Codina A, Nadal E, Brambilla E, and Sanchez-Cespedes M (2018) MET-Oncogenic and JAK2-Inactivating Alterations Are Independent Factors That Affect Regulation of PD-L1 Expression in Lung Cancer, Clin Cancer Res 24, 4579–4587. [DOI] [PubMed] [Google Scholar]
- [79].Bockus AT, Lexa KW, Pye CR, Kalgutkar AS, Gardner JW, Hund KC, Hewitt WM, Schwochert JA, Glassey E, Price DA, Mathiowetz AM, Liras S, Jacobson MP, and Lokey RS (2015) Probing the Physicochemical Boundaries of Cell Permeability and Oral Bioavailability in Lipophilic Macrocycles Inspired by Natural Products, J Med Chem 58, 4581–4589. [DOI] [PubMed] [Google Scholar]
- [80].Han Z, Harris PK, Jones DE, Chugani R, Kim T, Agarwal M, Shen W, Wildman SA, and Janetka JW (2014) Inhibitors of HGFA, Matriptase, and Hepsin Serine Proteases: A Nonkinase Strategy to Block Cell Signaling in Cancer, Acs Med Chem Lett 5, 1219–1224. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.