

Abstract
INTRODUCTION:
Human T cell responses to antigen stimulation, including the production of cytokines, are critical for healthy immune function and can be dysregulated in autoimmunity, immunodeficiencies, and cancer. A systematic understanding of the regulators that orchestrate T cell activation with gain-of-function and loss-of-function gene perturbations would offer additional insights into disease pathways and further opportunities to engineer next-generation immunotherapies.
RATIONALE:
Although CRISPR activation (CRISPRa) and CRISPR interference (CRISPRi) screens are powerful tools for gain-of-function and loss-of-function studies in immortalized cell lines, deploying them at scale in primary cell types has been challenging. Here, we developed a CRISPRa and CRISPRi discovery platform in primary human T cells and performed genome-wide screens for functional regulators of cytokine production in response to stimulation.
RESULTS:
We optimized lentiviral methods to enable efficient and scalable delivery of the CRISPRa machinery into primary human T cells. This platform allowed us to perform genome-wide pooled CRISPRa screens to discover regulators of cytokine production. Pools of CRISPRa-perturbed cells were isolated by fluorescence-activated cell sorting into high and low bins based on levels of endogenous Interleukin-2 (IL-2) production in CD4+ T cells or interferon-γ (IFN-γ) production in CD8+ T cells. Hits included proximal T cell receptor (TCR) signaling pathway genes, indicating that overexpression of these components could overcome signaling “bottlenecks” and tune stimulation and cytokine production.
Reciprocal genome-wide loss-of-function screens with CRISPRi detected hits with critical regulatory functions, including some missed by CRISPRa. By contrast, CRISPRa also identified hits that may not be required and in some cases were expressed at only low levels under the conditions of the screen. This was strongly exemplified by regulation of IFN-γ production by the nuclear factor κ B (NF-κB) signaling pathway, in which CRISPRi identified a required TCR–NF-κB signaling circuit (including MALT1 and BCL10). CRISPRa selectively detected a set of tumor necrosis factor superfamily receptors that also signal through NF-kB, including 4–1BB, CD27, CD40, and OX40. These receptors were not individually required for signaling in our experimental conditions but could promote IFN-γ when overexpressed. Thus, CRISPRa and CRISPRi complement each other for the comprehensive discovery of functional cytokine regulators.
Arrayed CRISPRa perturbation validated the effects of key hits in CD4+ and CD8+ T cells. We also assessed how individual CRISPRa perturbations more broadly reprogram cytokine production beyond IL-2 and IFN-γ by measuring a panel of secreted cytokines and chemokines.
Finally, we developed a platform for pooled CRISPRa perturbations coupled with single-cell RNA-sequencing (scRNA-seq) readout (CRISPRa Perturb-seq) in primary human T cells. We used CRISPRa Perturb-seq for deep molecular characterization of single-cell states caused by 70 genome-wide screen hits and controls to reveal how regulators of cytokine production both tune T cell activation and program cells into different stimulation-responsive states.
CONCLUSIONS:
Our study demonstrates a robust platform for large-scale pooled CRISPRa and CRISPRi in primary human T cells. Paired CRISPRa and CRISPRi screens enabled comprehensive functional mapping of gene networks that can modulate cytokine production. Follow-up of CRISPRa hits with arrayed phenotypic analyses and with pooled scRNA-seq approaches enabled precise functional characterization of key screen hits, revealing how key perturbations may tune T cells to therapeutically relevant states. Future CRISPRa and CRISPRi screens in primary cells could identify targets for improved next-generation cellular therapies.
Regulation of cytokine production in stimulated T cells can be disrupted in autoimmunity, immunodeficiencies, and cancer. Systematic discovery of stimulation-dependent cytokine regulators requires both loss-of-function and gain-of-function studies, which have been challenging in primary human cells. We now report genome-wide CRISPR activation (CRISPRa) and interference (CRISPRi) screens in primary human T ceils to identify gene networks controlling interleukin-2 (IL-2) and interferon-γ (IFN-γ) production. Arrayed CRISPRa confirmed key hits and enabled multiplexed secretome characterization, revealing reshaped cytokine responses. Coupling CRISPRa screening with single-cell RNA sequencing enabled deep molecular characterization of screen hits, revealing how perturbations tuned T cell activation and promoted cell states characterized by distinct cytokine expression profiles. These screens reveal genes that reprogram critical immune cell functions, which could inform the design of immunotherapies.
Graphical Abstract
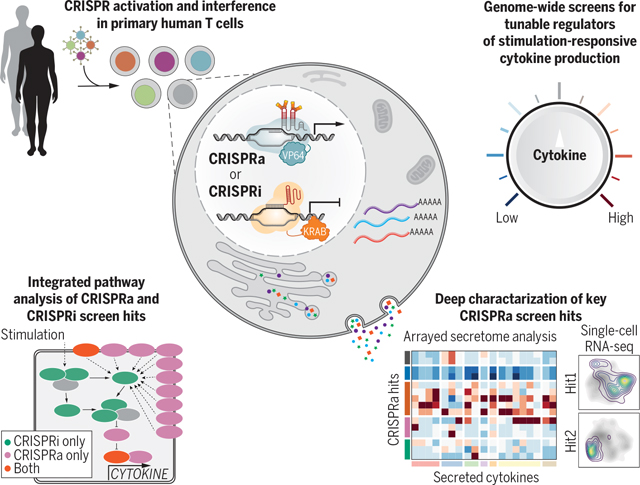
Genome-wide CRISPRa/i screens discover tunable regulators of stimulation-responsive cytokine production in primary human T cells. Genome-wide CRISPRa/i gain-of-function and loss-of-function screens in human T cells allowed for systematic identification of regulators of cytokine production. Follow-up on key CRISPRa screen hits with secretome and scRNA-seq analysis helped to decode how these regulators tune T cell activation and program cells into different stimulation-responsive states.
Regulated T cell cytokine production in response to stimulation is critical for balanced immune responses. Cytokine dysregulation can lead to autoimmunity, immunodeficiency, and immune evasion in cancer (1–4). Interleukin-2 (IL-2), which is secreted predominantly by CD4+ T cells, drives T cell expansion (5) and is therapeutically applied in autoimmunity and cancer at different doses (6). Interferon-γ (IFN-γ) is a cytokine secreted by both CD4+ and CD8+ T cells that promotes a type I immune response against intracellular pathogens, including viruses (4), and is correlated with positive cancer immunotherapy responses (7–9). Much of our current understanding of the pathways leading to cytokine production in humans originates from studies in transformed T cell lines, which often are not representative of primary human cell biology (10–12). Comprehensive understanding of pathways that control cytokine production in primary human T cells would facilitate the development of next-generation immunotherapies.
Unbiased forward genetic approaches can uncover the components of regulatory networks systematically, but challenges with efficient Cas9 delivery have limited their application in primary cells. Genome-wide CRISPR knockout screens have been completed using primary mouse immune cells from Cas9-expressing transgenic mice (13–15), including a screen for regulators of innate cytokine production in dendritic cells (13). Genome-scale CRISPR studies in human primary cells have recently been accomplished using transient Cas9 electroporation to introduce gene knockouts (16, 17). However, comprehensive discovery of regulators requires both gain-of-function and loss-of-function studies. For example, CRISPR activation (CRISPRa) gain-of-function screens can discover genes that may not normally be active in the tested conditions but can promote phenotypes of interest (18, 19). In contrast to a CRISPR knockout, CRISPRa or CRISPR interference (CRISPRi) require the sustained expression of an endonuclease-dead Cas9 (dCas9) and, because of poor lentiviral delivery, has been limited to small-scale experiments in primary cells (20, 21). Here, we developed a CRISPRa and CRISPRi screening platform in primary human T cells, which allowed for the systematic discovery of genes and pathways that can be perturbed to tune stimulation-dependent cytokine responses.
Genome-wide CRISPRa screens identify regulators of IL-2 and IFN-γ production in T cells
To enable scalable CRISPRa in primary human T cells, we developed an optimized high-titer lentiviral production protocol with a minimal dCas9-VP64 vector (pZR112), allowing for transduction efficiencies up to 80% (fig. S1). A second-generation CRISPRa synergistic activation mediator (SAM) system (22, 23) induced robust increases in target expression of established surface markers (fig. S2). Next, we scaled up our platform to perform pooled genome-wide CRISPRa screens targeting >18,800 protein-coding genes with >112,000 single-guide RNAs (sgRNAs) (22). We used fluorescence-activated cell sorting (FACS) to separate IL-2–producing CD4+ T cells and IFN-γ–producing CD8+ T cells into high and low bins (Fig. 1A and fig. S3A to D). Subsequent sgRNA quantification confirmed that sgRNAs targeting IL-2 (IL2) and IFN-γ (IFNG) were strongly enriched in the respective cytokine high populations, and nontargeting control sgRNAs were not enriched in either bin (Fig. 1B). Both CRISPRa screens were highly reproducible in two different human blood donors (Fig. 1, C and D, and fig. S3, E and F). Gene-level statistical analysis of the IL-2 and IFN-γ CRISPRa screens revealed 444 and 471 hits, respectively, including 171 shared hits (Fig. 1E; fig. S3, G and H; and tables S1 and S2). Thus, CRISPRa screens provide a robust platform to discover gain-of-function regulators of stimulation-dependent responses in primary cells.
Fig. 1. Genome-wide CRISPRa screens for cytokine production in stimulated primary human T cells.

(A) Schematic of CRISPRa screens. (B) sgRNA log2-fold changes for genes of interest in IL-2 (left) and IFN-γ (right) screens. Bars represent the mean log2-fold change for each sgRNA across two human blood donors. Density plots above represent the distribution of all sgRNAs. (C and D) Scatter plots of median sgRNA log2-fold change (high/low sorting bins) for each gene, comparing screens in two donors, for IL-2 (C) and IFN-γ (D) screens. (E) Comparison of gene log2-fold change (median sgRNA, mean of two donors) in IL-2 and IFN-γ screens.
CRISPRa hits included components of the T cell receptor (TCR) signaling pathway and T cell transcription factors. Activation of TBX21 (encoding T-bet), which promotes both memory CD8+ T cell and CD4+ T helper cell 1 (TH1) differentiation (24–26), selectively enhanced the signature type I cytokine IFN-γ (Fig. 1E). By contrast, sgRNAs activating GATA3, which promotes type II differentiation by antagonizing T-bet (25, 27), had the opposite effects (Fig. 1E). Overexpression of members of the proximal TCR signaling complex, such as VAV1, CD28, LCP2 (encoding SLP-76), and LAT (28, 29) reinforced T cell activation and were enriched in both cytokine-high bins. Conversely, the negative TCR signaling regulators MAP4K1 and SLA2 were depleted in these bins (Fig. 1, B and E) (30, 31). Thus, CRISPRa identifies critical “bottlenecks” in signals leading to cytokine production.
Complementary CRISPRa and CRISPRi screens comprehensively reveal circuits of cytokine production in T cells
CRISPRa screens were effective in identifying limiting factors in cytokine production but they could miss necessary components that would only be identified through loss-of-function studies. We therefore performed reciprocal genome-wide CRISPRi screens, adapting our optimized lentiviral protocols (Fig. 2, A and B; fig. S4; and tables S1 and 2). Dropout of gold standard essential genes (32) and reproducibility across two human donors confirmed the screen quality (fig. S5). The CRISPRi IL-2 and IFN-γ screens identified 226 and 203 gene hits, respectively, including 92 shared hits (Fig. 2, A and B). As expected, the CRISPRi hits were biased toward genes with high mRNA expression, including members of the CD3 complex, whereas CRISPRa additionally identified regulators that were expressed either at low levels or not at all in T cells under the screened conditions (Fig. 2, C and D, and fig. S6). For example, PIK3AP1 and IL1R1 were expressed at low levels under the screened conditions (fig. S7A). They are potentially inducible in some T cell contexts (fig. S7, B to D); however, they were detected as hits by CRISPRa but not CRISPRi.
Fig. 2. Integrated CRISPRa and CRISPRi screens mapping the genetic circuits underlying T cell cytokine response in high resolution.

(A and B) Median sgRNA log2-fold change (high/low sorting bins) for each gene, comparing CRISPRi screens in two donors, for IL-2 (A) and IFN-γ (B) screens. (C) Distributions of gene mRNA expression for CRISPRa and CRISPRi cytokine screen hits in resting CD4+ T cells (this study). (D) Comparison of IL-2 CRISPRi and CRISPRa screens with genes belonging to the TCR signaling pathway (KEGG pathways) indicated in colors other than gray. (E) Comparison of IFN-γ CRISPRi and CRISPRa screens with manually selected NF-κB pathway regulators labeled. All other genes are shown in gray. (F) Map of NF-κB pathway regulators labeled in (D). (G) Map of screen hits with previous evidence of defined function in T cell stimulation and costimulation signal transduction pathways. Genes shown are significant hits in at least one screen and were selected based on review of the literature and pathway databases (e.g., KEGG and Reactome). Tiles represent proteins encoded by indicated genes with the caveat that, because of space constraints, subcellular localization is inaccurate because many of the components shown in the cytoplasm occur at the plasma membrane. Tiles are colored according to log2-fold change Z score, as shown in the subpanel, with examples of different hits. Large arrows at the top represent stimulation/costimulation sources. (H) Select screen hits with less well-described functions in T cells in the same format as (G). For (H), only significant hits from the top 20 positive and negative ranked genes by log2-fold change for each screen were candidates for inclusion.
The power of coupling activation and interference screening was exemplified further by the identification of two IFN-γ–regulating circuits. CRISPRi screens identified key components of the nuclear factor κ B (NF-κB) pathway that are required for IFN-γ production (and, to a lesser extent, IL-2 production). CRISPRi detected a circuit of T cell stimulation signaling through MALT1, BCL10, TRAF6, and TAK1 (encoded by MAP3K7) to the inhibitor of the NF-κB complex (IκB complex, encoded by CHUK, IKBKB, and IKBKG) that promotes IFN-γ production (Fig. 2, E and F, and fig. S8A). By contrast, CRISPRa revealed a set of positive IFN-γ regulators that included members of the tumor necrosis factor receptor superfamily (TNFRSF) and IL1R1. These regulators also signal through NF-κB even though they are not individually required and therefore not detected by CRISPRi (Fig. 2, E and F). Thus, CRISPRa and CRISPRi complement each other for the comprehensive discovery of functional cytokine regulators.
To gain insights into functional pathways enriched across CRISPRi and CRISPRa screens, we completed gene set enrichment analysis (GSEA) of Kyoto Encyclopedia of Genes and Genomes (KEGG) pathways, identifying multiple immune-related pathways as being enriched across screens (fig. S8B). Furthermore, we analyzed data from numerous genome-wide association studies (GWAS) to determine whether the heritability of complex immune traits was enriched in genomic regions harboring our screen hits by stratified linkage disequilibrium score regression (s-LDSC). Both CRISPRi and CRISPRa regulators of IFN-γ and CRISPRa regulators of IL-2 were in regions enriched for immune trait heritability compared with nonimmune traits or an expression-matched background set (fig. S8C). Thus, these forward genetic screens may serve as a resource to help prioritize candidate functional genes in genomic regions associated with complex immune diseases.
We next completed integrative analyses of gene hits across CRISPRa and CRISPRi screens for both cytokines. We found that a few genes were identified across all screens (e.g., ZAP70 as a positive regulator and CBLB as a negative regulator), representing core regulators of stimulation-responsive cytokine production in T cells. Most hits, however, were either cytokine-specific (IL-2 in CD4+ T cells or IFN-γ in CD8+ T cells) or perturbation-specific (activation or interference) (fig. S8D). For a few target genes, including PTPRC (CD45), CRISRPa and CRISPRi both influenced cytokine production in the same direction, suggesting that for some genes, activation and interference both impair optimal levels (fig. S8E). The marked overlap in regulators between IL-2 in CD4+ T cells and IFN-γ in CD8+ T cells led us to perform additional genome-wide CRISPRa screens for IL-2, IFN-γ, and TNF-α in CD4+ T cells, allowing for direct comparisons of type 1 cytokine regulators in CD4+ T cells (fig. S9). Many of the strongest positive (e.g., VAV1, CD28, and LCP2) and negative hits (e.g., MAP4K1, LAT2, and GRAP) overlapped across all CRISPRa screens, likely representing core regulators of type 1 cytokine production in response to stimulation and costimulation. Additionally, these screens identified hits that could potentially increase or decrease individual cytokines selectively. Thus, CRISPRi and CRISPRa hits reveal both core and context-specific regulators of cytokine production.
We used our integrated dataset combined with literature review to build a high-resolution map of tunable regulators of signal transduction pathways leading to cytokine production (Fig. 2G). This included calcium pathway signaling genes (e.g., PLCG1, PLCG2, PRKCB, PRKD2, and NFATC2), and cytokine signaling genes (e.g., STAT3, JAK1, JAK3, and SOCS3), the latter suggesting feedback circuits among cytokine signals. In particular, CRISPRa identified regulators absent from previous literature (e.g., APOBEC3A/D/C, FOXQ1, and EMP1) (Fig. 2H), underscoring the need for gain-of-function screens for comprehensive discovery. Thus, CRISPRa and CRISPRi screens complement one another to map the tunable genetic circuits controlling T cell stimulation–responsive cytokine production.
Arrayed characterization of selected CRISPRa screen hits
We next performed arrayed CRISPRa experiments for deeper phenotypic characterization of screen hits (Fig. 3A). We selected 14 screen hits (from different screen categories) (Fig. 3B) including the established regulators VAV1 and MAP4K1 and the positive controls IL2 and IFNG. Notably, we included genes with relatively low expression in T cells under our experimental conditions, FOXQ1, IL1R1, LHX6, and PIK3AP1 (fig. S7). First, we validated that selected sgRNAs increased the expression of target gene mRNA (fig. S10). Next, we assessed IL-2, IFN-γ, and TNF-α by intracellular staining in both CD4+ and CD8+ T cells. Thirteen of 14 target genes caused significant (q < 0.05) changes in the proportion of cells positive for the relevant cytokine(s), with at least one sgRNA (Fig. 3, C and D, and fig. S11). Furthermore, we observed effects on both IL-2 and IFN-γ double- and single-positive populations (fig. S12, A to C). With the exception of TNFRSF1A (and IL2 or IFNG), positive regulators did not cause spontaneous cytokine production without stimulation (Fig. 3D and fig. S11B). Although IL-2 was screened in CD4+ T cells and IFN-γ in CD8+ T cells, CRISPRa sgRNA effects were highly correlated across both lineages (Fig. 3E). We also assessed T cell differentiation and observed that FOXQ1 and TNFRSF1A significantly decreased the percentage of CD62L+ cells, indicating a shift toward effector T cell states as a potential mechanism (fig. S12D). Thus, these studies validate the pooled CRISPRa screens and begin to characterize cytokine production and cell differentiation states promoted by activation of key target genes.
Fig. 3. Characterization of CRISPRa screen hits by arrayed profiling.

(A) Schematic of arrayed experiments. (B) Comparison of IL-2 (in CD4+ T cells) and IFN-γ (in CD8+ T cells) CRISPRa screens, with genes targeted by the arrayed sgRNA panel indicated, as well as their screen hit categorization. Paralogs of arrayed panel genes that were also highly ranked hits are additionally indicated. (C) Representative intracellular cytokine staining flow cytometry for indicated cytokines in control (NO-TARGET_1 sgRNA) or VAV1 (VAV1_1 sgRNA) CRISPRa T cells after 10 hours of stimulation. (D) Intracellular cytokine staining of full arrayed sgRNA panel, showing the percentage of cells that gated positive for the indicated cytokines in CD4+ or CD8+ T cells. Points represent the mean value of four donors, with and without stimulation. Dashed vertical lines represent the mean no-target control sgRNA control value with stimulation. *q < 0.05, **q <0.01, Mann–Whitney U test, followed by q value multiple-comparisons correction. Full data are provided in fig. S11B. The medium stimulation dose is shown for IL-2 and IFN-γ, and low-dose stimulation is shown for TNF-α. (E) Scatter plot comparison of log2-fold changes in the percentage of cytokine-positive cells for arrayed panel sgRNAs versus the mean of no-target control sgRNAs in stimulated CD4+ and CD8+ cells using the same data from (D). (F) Secreted cytokine staining arrayed panel grouped by indicated gene categories, with sgRNAs targeting the IL2 and IFNG genes removed. Points represent a single gene and donor measurement. *P < 0.05, **P < 0.01, ***P < 0.001, Mann–Whitney U test. (G) Principal component analysis of secreted cytokine measurements resulting from the indicated CRISPRa sgRNAs. (H) Heatmap of selected secreted cytokine measurements grouped by indicated biological category. Values represent the median of four donors, followed by Z-score scaling for each cytokine.
We next tested whether genes identified by CRISPRa could also regulate cytokines when overexpressed as cDNA transgenes, because continuous expression of CRISPRa would present challenges in cell therapies caused by Cas9 immunogenicity (33) (fig. S13A). cDNA transgene overexpression of CRISPRa hits affected cytokine production in T cells stimulated with antibodies or antigen-positive cancer cells (fig. S13, B to D). Thus, this strategy could potentially be used to implement CRISPRa discoveries in engineered T cell therapies.
We next assessed how individual CRISPRa perturbations reprogram cytokine production by measuring a broad panel of 48 secreted cytokines and chemokines, 32 of which were detected in control samples (fig. S14A and table S6). After confirming that the effects on IL-2, IFN-γ, and TNF-α measurements were generally consistent with intracellular staining (Fig. 3F and fig. S14B), we performed principal component analysis and hierarchical clustering on all cytokines. We observed sgRNA categorical grouping consistent with that observed in the screens, with sgRNAs targeting genes identified as regulators of both cytokines, causing broad increases or decreases in cytokine concentration (Fig. 3G and fig. S14C). There were distinct patterns in the classes of cytokines increased by different regulators (Fig. 3H). VAV1 and FOXQ1 (a transcription factor that has not been well characterized in T cells) led to preferential increases in type 1 signature cytokines and dampened type 2 cytokines. Unexpectedly, OTUD7B, a positive regulator of proximal TCR signaling (34), had a distinct effect and increased type 2 cytokines (fig. S14D). We next investigated whether modulations in the secretome correlated with transcriptional control of the corresponding genes. Taking FOXQ1 as an example, we performed bulk RNA sequencing (RNA-seq) on FOXQ1 and control sgRNA CD4+ T cells and found that it correlated strongly with the secretome effects (fig. S15). Thus, the identified regulators may not only modulate TCR stimulation and signaling but also tune the T cell secretome toward specific signatures.
CRISPRa Perturb-seq characterizes the molecular phenotypes of cytokine regulators
To assess the global molecular signatures resulting from each CRISPRa gene induction, we developed a platform to couple pooled CRISPRa perturbations with barcoded single-cell RNA-seq (scRNA-seq) readouts (CRISPRa Perturb-seq) (Fig. 4A). Because similar CRISPRa Perturb-seq approaches have been powerful in cell lines and animal models (35–37), we incorporated a direct-capture sequence into the CRISPRa-SAM modified sgRNA scaffold to enable compatibility with droplet-based scRNA-seq methods (fig. S16).
Fig. 4. CRISPRa Perturb-seq captures diverse T cell states driven by genome-wide cytokine screen hits.

(A) Schematic of CRISPRa Perturb-seq experiment. (B) Categorical breakdown of genes targeted by the sgRNA library comprising hits from our primary genome-wide CRISPRa cytokine screens as indicated. Genes with a summed log2-fold change less than zero across both screens (diagonal line) are categorized as negative regulators. (C) UMAP projection of post–quality control filtered restimulated T cells, colored by blood donor. (D) Distribution of CD4+ and CD8+ T cells across restimulated T cell UMAP projection. Each bin is colored by the average log2(CD4/CD8) transcript levels of cells in that bin. (E) Restimulated T cell UMAP colored by average cell activation score in each bin. (F) Boxplots of restimulated T cells’ activation scores grouped by sgRNA target genes. Dashed line represents the median activation score of no-target control cells. *P < 0.05, **P < 0.01, ***P < 0.001, Mann–Whitney U test with Bonferroni correction. (G) Restimulated T cell UMAP with cells colored by cluster. (H) Heatmap of differentially expressed marker genes in each cluster. The top 50 statistically significant (FDR < 0.05) differentially up-regulated genes for each cluster are shown, with genes that are up-regulated in multiple clusters being given priority to the cluster with the higher log2-fold change for the given gene. To the right of the heatmap are (left to right), the top marker genes by log2-fold change in each clusters’ section, the top overrepresented sgRNAs in each cluster by odds ratio (full data are provided in fig. S20G), and the top differentially up-regulated cytokine genes in each cluster. Mean cell log2(CD4/CD8) cell transcript values in each cluster are shown on the far right. (I) Restimulated T cell UMAP with the expression of indicated genes shown. (J) Contour density plots of restimulated cells assigned to indicated sgRNA targets in UMAP space. The no-target control contour is shown in grayscale underneath. “Perturbed cells” represents all cells assigned a single sgRNA other than no-target control sgRNAs.
We performed CRISPRa Perturb-seq characterization of regulators of stimulation responses in ~56,000 primary human T cells, targeting 70 hits and controls from our genome-wide CRISPRa cytokine screens (Fig. 4, A and B, and fig. S17, A to C). First, we confirmed that sgRNAs led to significant increases in the expression of their target genes (fig. S17D). Next, uniform manifold approximation and projection (UMAP) dimensionality reduction revealed discrete separation of the resting and restimulated cells (fig. S17E) and showed relatively even distribution of cells from two donors (Fig. 4C and fig. S17F). Gene signatures allowed us to resolve most T cells as either CD4+ or CD8+ (Fig. 4D and fig. S17, G and H). Thus, we generated a high-quality CRISPRa Perturb-seq dataset.
Cytokine production can be tuned by reinforced TCR signaling. To identify CRISPRa gene perturbations that tune the general strength of stimulation-responsive genes, we calculated a scRNA-seq “activation” score based on a gene signature that we derived by comparing resting and restimulated cells within the nontargeting control sgRNA group (fig. S18). Projecting activation scores on the stimulated cell UMAP revealed discrete regions of higher and lower activation scores among the restimulated cells (Fig. 4E). We next examined activation scores across CRISPRa perturbations (Fig. 4F). Negative regulators except IKZF3 (encoding the transcription factor Aiolos) decreased activation scores, suggesting that they act to broadly dampen stimulation strength. By contrast, IKZF3 reduced IFNG expression without reducing the overall activation score (Fig. 4F and fig. S19A), indicative of a possible distinct mechanism of cytokine gene regulation. Many of the positive regulators significantly increased activation score, with VAV1 causing the strongest activation potentiation (Fig. 4F). Thus, many, but not all, hits act by tuning overall T cell activation to varying degrees.
We next investigated how different perturbations affected the expression of cytokine and other effector genes in stimulated cells. We analyzed pseudobulk differential gene expression under restimulated conditions for each sgRNA target cell group compared with no-target control cells (fig. S19, A and B). IFNG was differentially expressed in 29 different sgRNA targets, with only sgRNAs targeting negative regulators causing decreased expression. IL2, however, was barely detectable by scRNA-seq (fig. S19C). Only IL2 and VAV1 sgRNAs caused its increased expression, consistent with our observations that VAV1 activation caused the greatest level of IL-2 release (Fig. 3H). Many of the negative regulators drove a stereotyped pattern of differential cytokine gene expression, whereas positive regulators generally promoted more diverse cytokine expression patterns than negative regulators (fig. S19A). TBX21 (T-bet) modulated the expression of most detectable cytokine genes. Furthermore, unlike most perturbations, it altered cytokine expression independently of stimulation (fig. S19D).
We next used clustering analysis to characterize CRISPRa-driven cell states in restimulated and resting T cells (Fig. 4G and fig. S20). For each cluster, we identified the top up-regulated gene expression markers and cytokine genes, contributions of CD4+/CD8+ T cells, and overrepresented sgRNAs revealing a diverse landscape of T cell states promoted by CRISPRa (Fig. 4, H to J, and fig. S20, D to G). Negative cytokine regulators (e.g., MAP4K1) were highly enriched in cluster 2, marked by LTB expression and low activation score. Only GATA3 promoted a T helper 2 (Th2) phenotype (cluster 3), suggesting that altered Th differentiation was not a common mechanism among negative IFNG regulators. Thus, Perturb-seq reveals cell states promoted by the overexpression of different key regulators.
We identified two IL2-expressing clusters, despite poor capture of the transcript, with both clusters consisting primarily of CD4+ T cells. Cluster 13 had the higher IL2 expression of the two and was promoted by VAV1 and OTUD7B sgRNAs. VAV1 sgRNAs were strongly enriched in both IFNG- and IL2-expressing clusters, suggesting that VAV1-mediated potentiation of T cell stimulation may drive differentiation toward multiple distinct cytokine-producing populations.
We also identified two distinct clusters of cells expressing IFNG (clusters 1 and 12) and containing both CD4+ and CD8+ T cells. Cluster 1 was marked by high expression of CCL3 and CCL4 and was enriched for sgRNAs with strong activation score potentiation such as VAV1, CD28, and FOXQ1. By contrast, cluster 12 was enriched for sgRNAs known to activate the NF-κB pathway, such as IL1R1, TRAF3IP2, TNFRSF1A, and TNFRSF1B. These observations suggest that potentiated stimulation/costimulation may drive T cells to an activated IFNG-expressing state distinct from more specific signaling through the NF-κB pathway. Activation of a subset of TNFRSF receptor genes (TNFRSF1A, TNFRSF1B, LTBR, and CD27) also promoted cell states (clusters 5 and 6) marked by the high expression of cell cycle genes. LTBR and CD27 sgRNAs were almost exclusively found in cells of this cluster, whereas TNFRSF1A/B sgRNAs appeared to push cells to both proliferative and IFNG-expressing states. Thus, CRISPRa Perturb-seq reveals how regulators of cytokine production both tune T cell activation and program cells into different stimulation-responsive states.
Discussion
Paired CRISPRa and CRISPRi screens complement one another to decode the genetic programs regulating stimulation-responsive cytokine production in primary human T cells. CRISPRi identified required cytokine regulators, whereas CRISPRa uncovered key signaling bottlenecks in pathway function as well as regulators that are not necessarily active in ex vivo–cultured T cells. Future screens performed in various other experimental conditions will have the potential to identify additional regulators of T cell states and functions.
The technologies developed in this study will enable screening approaches in primary human T cells and potentially other primary cell types, such as screens for functional noncoding regions of the human genome (18, 38, 39). Furthermore, this screening framework should be adaptable to other nonheritable editing applications of the CRISPR toolkit (40), continuing to expand opportunities to investigate complex biological questions in primary cells, especially when CRISPR perturbations are coupled with single-cell analyses.
Major efforts are underway to discover gene modifications that enhance the efficacy of adoptive T cell therapies. Although we do not expect all perturbations that lead to increased cytokine production to translate to enhanced in vivo antitumor efficacy, we are encouraged by the identification of genes in various stages of therapeutic development, including CD5 (41), TNFRSF9 (encoding 4–1BB), CD27, CD40, and TNFRSF4 (encoding OX40). Recent preclinical work (42) highlights c-JUN overexpression to limit T cell exhaustion and further enhance cell therapies. Thus, loss- and gain-of-function discovery platforms can guide efforts to engineer T cells for different clinical indications. Future CRISPRa and CRISPRi screens in human T cells will continue to nominate targets for improved next-generation cellular therapies.
Materials and Methods
Isolation and culture of human T cells
Human T cells were sourced from PBMC-enriched leukapheresis products (Leukopaks, STEMCELL Technologies, catalog no. 70500.2) from healthy donors, after institutional review board–approved informed written consent (STEMCELL Technologies). Bulk T cells were isolated from Leukopaks using EasySep magnetic selection following the manufacturers’ recommended protocol (STEMCELL Technologies, catalog no. 17951). Unless stated otherwise, bulk T cells were frozen in Bambanker Cell Freezing Medium at 5 × 107 cells/ml (Bulldog Bio, catalog no. BB01) and kept at −80°C for short-term storage or in liquid nitrogen for long-term storage immediately after isolation. Unless otherwise noted, thawed T cells were cultured in X-VIVO 15 (Lonza Bioscience, catalog no. 04–418Q) supplemented with 5% fetal calf serum (FCS), 55 mM 2-mercaptoethanol, 4 mM N-acetyl L-cysteine, and 500 IU/ml of recombinant human IL-2 (Amerisource Bergen, catalog no. 10101641). Primary T cells were activated using anti-human CD3/CD28 CTS Dynabeads (Fisher Scientific, catalog no. 40203D) at a 1:1 cell:bead ratio at 106 cells/ml.
Cell line maintenance
Lenti-X HEK293T cells (Takara Bio, catalog no. 632180) were maintained in high-glucose Dulbecco’s modified Eagle’s medium with GlutaMAX (Fisher Scientific, catalog no. 10566024), supplemented with 10% FCS, 100 U/ml of penicillin/streptomycin (PenStrep; Fisher Scientific, catalog no. 15140122), 1 mM sodium pyruvate (Fisher Scientific, catalog no. 11360070), 1× minimal essential medium (MEM) nonessential amino acids (Fisher Scientific, catalog no. 11140050), and 10 mM HEPES solution (Sigma-Aldrich, catalog no. H0887–100ML). Cells were passaged every 2 days using Tryple Express (Fisher Scientific, catalog no. 12604013) for dissociation and maintained at <60% confluency.
NALM6 cells were engineered to express NY-ESO-1 peptide in an HLA-A0201 background, recognizable with the 1G4 TCR by the Eyquem laboratory at University of California San Francisco (UCSF) and provided for TCR stimulation coculture experiments. For simplicity, these cells are referred to as NALM6. NALM6 cells were cultured in RPMI (Invitrogen, catalog no. 21870092) supplemented with 10% FCS, 100 U/ml PenStrep (Fisher Scientific, catalog no. 15140122), 1 mM sodium pyruvate (Fisher Scientific, catalog no. 11360070), and 1X MEM nonessential amino acids (Fisher Scientific, catalog no. 11140050), 10 mM HEPES solution (Sigma-Aldrich, catalog no. H0887–100ML), and 2 mM L-glutamine (Lonza Bioscience, catalog no. 17–605E).
Plasmids
dCas9-VP64 originated from lentiSAMv2 (Addgene, catalog no. 75112) and cloned into the lentiCRISPRv2-dCas9 backbone (Addgene, catalog no. 112233) with Gibson Assembly. The promoter was switched to SFFV and mCherry was introduced upstream of dCas9-VP64, separated by a P2A sequence resulting in the pZR112 plasmid. The LTR-LTR range was minimized to enhance lentiviral titer. For CRISPRi, BFP in pHR-SFFV-dCas9-BFP-KRAB (Addgene, catalog no. 46911) was switched to mCherry with Gibson Assembly, resulting in pZR071.
Single sgRNAs for arrayed experiments have been introduced by Golden Gate Cloning as described previously (22). Briefly, DNA oligomers with Golden Gate overhangs were annealed and subsequently cloned into the nondigested target plasmid using the Golden Gate Assembly Kit (BsmBI-v2, New England Biolabs, catalog no. E1602L). sgRNAs have been cloned into pXPR_502 (Addgene, catalog no. 96923) for CRISPRa and into CROPseq-Guide-Puro (43) (Addgene, catalog no. 86708) for CRISPRi. All single sgRNAs used in this study can be found in table S3.
The genome-wide CRISPRa (Calabrese A, catalog no. 92379 and Calabrese B, catalog no. 92380) and CRISPRi libraries (Dolcetto A, catalog no. 92385 and Dolcetto B, catalog no. 92386) (22) were obtained from Addgene. Forty nanograms of each library were transformed into Endura ElectroCompetent Cells (Lucigen, catalog no. 60242–2) following the manufacturer’s instructions. After transformation, Endura cells were grown in a shaking incubator for 16 hours at 30°C in the presence of ampicillin. Library plasmid has been isolated using the Plasmid Plus MaxiKit (Qiagen, catalog no. 12963) and sequenced for sgRNA representation as described under the section titled “Genome-wide CRISPRa and CRISPRi screens.”
For cDNA-mediated target overexpression, the lentiCRISPRv2 (Addgene, catalog no. 75112) backbone was rebuilt to a lentiviral cDNA cloning plasmid with an SFFV promoter followed by BsmBI restriction sites and P2A-Puro. Transgene cDNAs were purchased from Genscript, choosing the canonical (longest) isoform for each gene, and BsmBI restriction sites were introduced by polymerase chain reaction (PCR). The final lentiviral transfer plasmids were assembled using the Golden Gate Assembly Kit (BsmBI-v2, New England Biolabs, catalog no. E1602L).
To clone direct-capture compatible CRISPRa-SAM plasmids for Perturb-seq, different sgRNA designs were synthesized as G-Blocks (Integrated DNA Technologies) and cloned into pXPR_502 (Addgene, catalog no. 96923) by Gibson assembly, replacing its sgRNA cassette.
Lentivirus production
Unless otherwise stated, human embryonic kidney (HEK) 293T cells were seeded in Opti-MEM I Reduced Serum Medium (OPTI-MEM) with GlutaMAX Supplement (Invitrogen, catalog no. 31985088) supplemented with 5% FCS, 1 mM sodium pyruvate (Fisher Scientific), and 1× MEM nonessential amino acids (Fisher Scientific) (cOPTI-MEM) at 3.6 × 107 cells per T225 flask in 45 ml of medium overnight to achieve confluency between 85 and 95% at the time point of transfection. The following morning, HEK293Ts cells were transfected with second-generation lentiviral packaging plasmids and transfer plasmid using Lipofectamine 3000 transfection reagent (Fisher Scientific, catalog no. L3000075). Briefly, 165 μl of Lipofectamine 3000 reagent was added to 5 ml of room-temperature OPTI-MEM without supplements. Forty-two micrograms of Cas9 transfer plasmid, 30 mg of psPAX2 (Addgene 12260), 13 μg of pMD2.G (Addgene 12259), and 145 μl of p3000 reagent were added to 5 ml of room-temperature OPTI-MEM without supplements and mixed by gentle inversion. The plasmid and Lipofectamine 3000 mixtures were combined, mixed by gentle inversion, and incubated for 15 min at room temperature. After incubation, 20 ml of medium was removed from the T225 flask and the 10-ml transfection mixture was carefully added without detaching HEK293T cells. After 6 hours, the transfection medium was replaced with 45 ml of cOPTI-MEM supplemented with 1× ViralBoost (Alstem Bio, catalog no. VB100). Lentiviral supernatant was harvested 24 hours after transfection (first harvest) and replaced with 45 ml of fresh cOPTI-MEM. A second harvest was performed 48 hours after transfection. Immediately after collection, the medium was centrifuged at 500g for 5 min at 4°C to clear cellular debris. Unless otherwise noted, Lenti-X-Concentrator (Takara Bio, catalog no. 631232) was added to the collected supernatant, and lentivirus was concentrated following the manufacturer’s instructions and resuspended in OPTI-MEM in 1% of the original culture volume without supplements. Lentiviral particles were subsequently aliquoted and frozen at −80°C.
Flow cytometry
Aria 2, Aria 3, and Aria Fusion cell sorters (BD Biosciences) at the UCSF Parnassus Flow Core and the Gladstone Institute Flow Core were used for sorting. The Attune NxT (Thermo Fisher Scientific) and LSRFortessa X-20 (BD Biosciences) flow cytometers were used for flow cytometry. Antibodies used for flow cytometric analyses and sorting are summarized in table S4.
Intracellular cytokine staining
Unless indicated otherwise, T cells were stimulated with ImmunoCult Human CD3/CD28/CD2 T Cell Activator (STEMCELL Technologies, catalog no. 10990) with 6.25 μl/ml of culture medium at 2 × 106 cells/ml. One hour after restimulation, Golgi Plug protein transport inhibitor (BD Biosciences, catalog no. 555029) was added at a 1/1000 dilution. Nine hours after the addition of Golgi Plug, T cells were stained for surface antigens before fixation and subsequently processed for intracellular cytokine staining using the BD Cytofix/Cytoperm kit instructions (BD Biosciences, catalog no. 554714).
Genome-wide CRISPRa and CRISPRi screens
One day after activation, T cells from two human blood donors were infected with 2% v/v concentrated dCas9-VP64 lentivirus. Two days after activation, T cells were split into two populations and infected with 1% v/v [multiplicity of infection (MOI) ~ 0.5] Calabrese Set A (Addgene, catalog no. 92379) or 0.8% v/v (MOI ~0.5) Calabrese Set B (Addgene, catalog no. 92380) lentivirus. These two sets were independently cultured and processed in parallel until analysis. Three days after activation, fresh medium with IL-2 (final concentration 500 IU/ml) and puromycin (final concentration 2 μg/ml) was added to bring cells to 3 × 105 cells/ml. Cells were split 2 days later and fresh medium with IL-2 was added to bring cells to 3 × 105 cells/ml. Two days later, fresh medium without IL-2 was added to bring the concentration to 106/ml. Eight days after initial activation, cells were harvested, centrifuged at 500g for 5 min, and resuspended at 2 × 106 cells/ml X-VIVO 15 without supplements. The following day, cells were restimulated and stained for FACS as described under the “Intracellular cytokine staining” section. Over the subsequent 2 days, cells were sorted at the Parnassus Flow Cytometry Core (PFCC) facility into IL-2lo and IL-2hi CD4+ and IFN-γlo and IFN-γhi CD4− T cell populations (see fig. S3C for gating strategy). Sorted cells were stored in EasySep Buffer (phosphate-buffered saline with 2% FCS and 1 mM EDTA) overnight until genomic DNA isolation.
The same experimental procedure using T cells from the same donors was followed for the CRISPRi screens. T cells were infected with dCas9-mCherry-KRAB at 2% v/v and Dolcetto A (Addgene, catalog no. 92385) and B (Addgene, catalog no. 92386) sgRNA libraries at 10% v/v or 25% v/v unconcentrated virus, respectively (~0.5 MOI).
Genomic DNA was extracted from fixed cells as described previously (44). Integrated sgRNA sequences were amplified as described previously (22), and sequencing libraries were subsequently agarose gel purified using NucleoSpin Gel and PCR Clean-up Mini kit (Machery-Nagel, catalog no. 740609.50). Libraries were sequenced on a NextSeq500 instrument to a targeted depth of 100-fold coverage.
For the supplementary CD4+ T cell set of genome-wide CRISPRa screens, CD4+ T cells were isolated from Leukopaks using magnetic negative selection (STEMCELL Technologies, catalog no. 17952) and subsequently stimulated as described in the section entitled “Isolation and culture of human T cells.” T cells were then cultured and infected with lentivirus as described for the primary CRISPRa screens above. For library lentivirus production, Calabrese Set A and Set B plasmid were mixed at equimolar ratios before transfection, and the pooled lentiviral particles from both sets was used for transduction. CD4 flow cytometry staining on day 7 after T cell activation confirmed >98% purity. T cells were further processed and restimulated as described above. T cells were separately stained for IL-2, IFN-γ, or TNF-α for FACS. After our initial analysis, it appeared that the IFN-γ screen was potentially undersampled because of lower hit resolution than the other screens. To address this, additional fixed cells from the same experiment were stained and sorted as an additional technical replicate and then computationally merged (described below).
CRISPR screen analysis
Reads were aligned to the appropriate reference library using MAGeCK version 0.5.9.2 (45) using the –trim-5 22,23,24,25,26,28,29,30 argument to remove the staggered 5’ adapter. Next, raw read counts across both library sets were normalized to the total read count in each sample, and each of the matching samples across two sets were merged to generate a single normalized read count table. Normalized read counts in high versus low bins were compared using mageck test with –norm-method none, –paired, and –control-sgrna options, pairing samples by donor and using nontargeting sgRNAs as controls, respectively. Gene hits were classified as having a median absolute log2-fold change >0.5 and a false discovery rate (FDR) <0.05. For supplemental CD4+ screens (fig. S9), reads were aligned to the full Calabrese A and B library in a single reference file. For the supplemental CD4+ IFN-γ screen, which was sorted and sequenced as two technical replicates, normalized counts were averaged across technical replicates before analysis with mageck test.
Gene-set enrichment analysis
Gene-set enrichment analysis (GSEA) was completed with the fgsea Bioconductor R package using the default settings (46). KEGG pathways version 7.4 were obtained from GSEA mSigDB http://www.gsea-msigdb.org/gsea/downloads.jsp. The KEGG NF-κB signaling pathway (entry hsa04064) was missing from this dataset and added manually from https://www.genome.jp/entry/pathway+hsa04064.
Stratified linkage disequilibrium score analysis
GWAS summary statistics were downloaded from the Price laboratory website (https://alkesgroup.broadinstitute.org/sumstats_formatted/ and https://alkesgroup.broadinstitute.org/UKBB/). Linkage disequilibrium (LD) scores were created for each screen [corresponding to a set of single-nucleotide polymorphisms (SNPs) within 100 kb of genes identified as significant hits in each screen or their corresponding matched background sets] using the 1000G Phase 3 population reference. Each annotation’s heritability enrichment for a given trait was computed by adding the annotation to the baselineLD model and regressing against trait chi-squared statistics using HapMap3 SNPs with the stratified LD score regression package (47). Heritability enrichments were then meta-analyzed across immune or nonimmune traits using inverse variance weighting. The sets of background genes were sampled from the set of all genes that were expressed in the control sgRNA, stimulated bulk RNA-Seq data. For each screen, the background genes were sampled to match the significant screen hits in number and based on deciles of gene expression. Immune traits used for analysis were: “Eosinophil Count,” “Lymphocyte Count,” “Monocyte Count,” “White Count,” “Autoimmune Disease All,” “Allergy Eczema Diagnosed,” “Asthma Diagnosed,” “Celiac,” “Crohn’s Disease,” “Inflammatory Bowel Disease,” “Lupus,” “Multiple Sclerosis,” “Primary Biliary Cirrhosis,” “Rheumatoid Arthritis,” “Type 1 Diabetes,” “Ulcerative Colitis.” Nonimmune traits used were: “Heel Tscore,” “Balding1,” “Balding4,” “Bmi,” “Height,” “Type 2 Diabetes,” “Neuroticism,” “Anorexia,” “Autism,” “Bipolar Disorder,” “Depressive Symptoms,” “Fasting Glucose,” “Hdl,” “Ldl,” “Triglycerides,” and “Fasting Glucose.”
Arrayed CRISPRa experiments
For each gene chosen to target in follow-up experiments, one sgRNA was chosen from the Calabrese library used in screens. The first sgRNAs (“_1”) were manually chosen for consistent log2-fold change observed in both donors. The second sgRNA (“_2”) was picked from the hCRISPRa-v2 genome-wide library (48), choosing the top-ranked sgRNA not present in Calabrese libraries for each gene. sgRNAs were cloned into the pXPR_502 vector as described in the plasmid section.
Primary human T cells were transduced with 2% v/v mCherry-2A-dCas9-VP64 lentivirus (pZR112) 1 day after activation. The following day (day 2), the dCas9-VP64–transduced cells were split into 96-well flat-bottom plates, avoiding edge wells, and transduced with a different sgRNA lentivirus in each well (5% v/v). One day after sgRNA transduction, fresh medium was added with IL-2 (500 IU/ml) and 2 μg/ml puromycin (final culture concentrations). Cells were passaged 2 days later, adding fresh medium with 500 IU/ml of IL-2 and maintaining a concentration of 3 × 105 to 1 × 106 cells/ml, with 96-well plates copied as needed to maintain this concentration. On day 8, cells from copied plates were pooled and samples were counted. Cells were pelleted and resuspended at a concentration of 2 × 106 cells/ml in fresh X-VIVO-15 without additives. On day 9, cells were restimulated with anti–CD3/CD28/CD2 ImmunoCult T Cell Activator (as described in the “Intracellular cytokine staining” section) or left resting.
RT-qPCR
T cells were prepared as described under the “Arrayed CRISPRa experiments” section. Seven days after sgRNA transduction, 100,000 T cells per well were pelleted at 500g for 5 min at 4°C. Cells were lysed and RNA was extracted using the Quick-RNA 96 kit (Zymo Research) following the manufacturer’s protocol but skipping the option of in-well DNase treatment. DNase treatment and cDNA synthesis were subsequently completed with Maxima First Strand cDNA Synthesis Kit for reverse transcription quantitative PCR (RT-qPCR) with double-stranded DNase (Thermo Fisher Scientific). qPCR was performed with the PrimeTime PCR Master Mix (Integrated DNA Technologies) and PrimeTime qPCR probe assays (Integrated DNA Technologies; a list of probes used is provided in table S5) on an Applied Biosystems Quantstudio 5 real-time PCR system. Data were analyzed using the ΔΔCt method. The mean Ct values of two housekeeping genes, PPIA and GUSB, to calculate the ΔCt, and the mean ΔCt of nontargeting controls to calculate ΔΔCt.
cDNA experiments
See fig. S13A for an experimental overview. One day after activation, T cells were transduced with the 1G4 TCR lentivirus recognizing the NY-ESO-1 antigen or nontransduced for immunocult assay. One day later, cells were transduced with the transgenes in cDNA format. Three days after initial activation, puromycin was added to obtain a final concentration of 2 μg/ml, along with fresh X-VIVO 15 medium with 500 IU/ml of IL-2, and further cultured and expanded analogous to the genome-wide CRISPR screens. Nine days after initial activation, T cells were centrifuged and resuspended at 2 × 106 cells/ml in X-Vivo 15 without supplements. On the same day, 1G4 TCR expression was assessed by flow cytometry after dextramer staining (Immudex, catalog no. WB3247-PE) to ensure even expression across different cDNA constructs. The following day, T cells were restimulated with either 6.25 μl/ml of Immunocult or NALM6 cells at an effector: target ratio of 1:2 for 1G4 TCR-transduced cells. Cells were further processed as described under the “Intracellular cytokine staining” section. CD22 was used as a marker for NALM6 cells to discriminate them from T cells in the coculture. Overexpression of OTUD7B cDNA together with the 1G4 TCR (but not alone) caused toxicity and was therefore excluded from analyses. Two donors were excluded from the 1G4 TCR assay because of poor TCR transduction.
Cytokine Luminex assay
T cells were prepared as explained under the “Arrayed CRISPRa experiments” section. On day 9 after activation, T cells at a concentration of 2 × 106 cells/ml were restimulated with ImmunoCult Human CD3/CD28/CD2 (STEMCELL Technologies, catalog no. 10970) at 6.25 μl/ml. Twenty-four hours after restimulation, supernatant was collected and frozen at −20°C. After a serial pilot titration, cytokine analyses were performed at a 1/200 dilution by Eve Technologies with the Luminex xMAP technology on the Luminex 200 system (Luminex). To remove very lowly expressed cytokines for downstream analysis, any group in which three of four donors had undetectable cytokines, the cytokine was removed. Additionally, the sgIL1R1–1 donor 4 measurement for IL-1α was removed manually because this was an extremely high outlier.
Bulk RNA-seq sample preparation
FOXQ1 and nontargeting sgRNA control primary human T cells from four donors were transduced and expanded as described in the “Arrayed CRISPRa experiments” section. On day 8, mCherry+CD4+ populations were sorted and resuspended in X-VIVO-15 without additives at 2 × 106 cells/ml. On day 9, cells were restimulated with 6.25 μl/ml of anti-CD3/CD28/CD2 ImmunoCult or left unperturbed for resting (nonstimulated) condition. Twenty-four hours later, cells were lysed for RNA.
RNA was purified using the Quick-RNA Microprep kit (Zymo Research) without the optional in-well DNase treatment step. Purified RNA was treated with TURBO DNase (Thermo Fisher Scientific) to remove potential contaminating DNA. RNA was subsequently purified using the RNA Clean & Concentrator-5 kit (Zymo Research). RNA quality control was performed using an RNA ScreenTape assay (Agilent Technologies), with all samples having an RNA integrity number >7. RNA-seq libraries were prepared using the Illumina Stranded mRNA Prep kit with 100 ng of input RNA. Libraries were sequenced using paired-end 72-bp reads on a NextSeq500 instrument to an average depth of 3.2 × 107 clusters per sample.
Bulk RNA-seq data analysis
Adapters were trimmed from fastq files using cutadapt version 2.10 (49) with default settings keeping a minimum read length of 20 bp. Reads were mapped to the human genome GRCh38 keeping only uniquely mapping reads using STAR version 2.7.5b (50) with the setting “–outFilterMultimapNmax 1.” Reads overlapping genes were then counted using featureCounts version 2.0.1 (51) with the setting “-s 2” and using the Gencode version 35 basic transcriptome annotation.
The count matrix was imported into R. Only genes with at least 1 count per million across at least four samples were kept. TMM normalized counts were used for heatmaps. Differentially expressed genes between FOXQ1 overexpression and control samples were then identified using limma version 3.44.3 (52) while controlling for any differences between donors. Significant differentially expressed genes were defined as having an FDR-adjusted P value <0.05.
Perturb-seq library design and cloning
The CRISPRa Perturb-seq target genes were selected from the primary IL-2 and IFN-γ CRISPRa screen results. First, genes that had a significant fitness defect were removed from the gene list (fig. S5). Next, genes were ranked by median sgRNA log2-fold change and the top ranked, not previously selected gene, was picked in the following order: (1) IL-2–positive hit, (2) IFN-γ–positive hit, (3) IL-2–positive hit, (4) IFN-γ–positive hit, and (5) IL-2– or IFN-γ–negative hit (alternating each round), such that positive hits outnumbered negative hits at a 4:1 ratio. Only hits that were significant (FDR < 0.05) were selected in each round. The one exception was TCF7, which was added manually because we considered it worthwhile to analyze due to its known effects on T cell function. To select sgRNAs, the top two enriched sgRNAs by log2-fold change in the screen for which the gene was selected were used. The library was ordered as pooled single-stranded oligos, PCR amplified, and cloned into the CRISPRa-SAM direct-capture design I cloning vector (pZR158).
Perturb-seq sample preparation and sequencing
Bulk CD3+ primary human T cells from two donors were transduced and cultured as described in the “Genome-wide CRISPRa and CRISPRi screens” section, except library transduction was completed at lower MOI of 0.3. Cells in the stimulated condition were stimulated with 6.25 μl/ml of anti–CD3/CD28/CD2 immunocult. Twenty-four hours later, cells from both the stimulated and nonstimulated condition were sorted for mCherry+ (marking dCas9-VP64). Sorted cells were processed to single-cell RNA-seq and sgRNA sequencing libraries by the Institute for Human Genetics (IHG) Genomics Core using Chromium Next GEM Single Cell 3’ Reagent Kit version 3.1 with feature barcoding technology for CRISPR screening, following the manufacturer’s protocol. Before loading the Chromium chip, sorted cells from two blood donors were normalized to 1000 cells/μl and mixed at a 1:1 ratio for each condition. Twenty microliters of cell suspension was loaded into four replicate wells per condition, for a total 80,000 cells loaded per condition. Final sgRNA sequencing libraries were further purified for the correct size fragment by 4% agarose E-Gel EX Gels (Thermo Fisher Scientific) and gel extracted. Libraries were sequenced over two NovaSeq S4 lanes (two stimulated wells and two nonstimulated wells per lane) at a 2:1 molar ratio of the gene expression libraries to sgRNA libraries.
Perturb-seq analysis
Alignments and count aggregation of gene expression and sgRNA reads were completed with Cell Ranger version 6.1.1. Gene expression and sgRNA reads were aligned using cellranger count, with default settings. Gene expression reads were aligned to the “refdata-gex-GRCh38-2020-A” human transcriptome reference downloaded from 10x Genomics. sgRNA reads were aligned to the Perturb-seq library using the pattern (BC)GTTTAAGAGCTATG. Counts were aggregated with cellranger aggr with default arguments. To assign sgRNAs to cells, cellranger count output files “protospacer_calls_per_cell.csv” were used, filtering out droplets with >1 sgRNA called, returning a median of 133 sgRNA UMIs in sgRNA singlets. For increased stringency, only droplets with ≥5 sgRNA UMIs were used in further analysis.
Cell donors were genetically demultiplexed using Souporcell (53) (https://github.com/wheaton5/souporcell). The input for each run was the bam file and barcodes.tsv file from the cellranger count output and the reference fasta. Donor calls across wells were harmonized using the vcf file outputs from Souporcell using a publicly available python script (https://github.com/hyunminkang/apigenome/blob/master/scripts/vcf-match-sample-ids).
Gene expression data were imported and analyzed in R with the Seurat version 4.0.3 Read10X function (54). Cells were initially quality filtered for percentage of mitochondrial reads <25% and number of detected RNA features >400 and <6000, removing 4% of cells. After filtering, a median of 401 cells per sgRNA target gene per condition (median of 127 sgRNA unique molecular indices (UMIs) per singlet) were recovered, along with ~2000 cells with no-target control guides per condition. Four sgRNA targets, HELZ2, TCF7, PRDM1, and IRX4, were removed from downstream analysis because of low cell counts (<100).
Gene-expression counts were normalized and transformed using the Seurat SCTransform function (55), with the following variables regressed: percentage mitochondrial reads, S-phase score, and G2/M-phase score, performing the regression as described on the Satija laboratory website (https://satijalab.org/seurat/articles/cell_cycle_vignette.html). Normalized and transformed counts were used for all downstream analysis. To call CD4+ and CD8+ T cells, a CD4/CD8 score for each cell using following formula was used: log2[CD4/mean(CD8A, CD8B)], with a score <−0.9 called as a CD8+ cell and a score >1.4 called a CD4+ cell (fig. S17G).
For both restimulated and resting conditions, UMAP reduction was performed with dimensions 1 to 20, and otherwise default settings of the RunUMAP Seurat function. For clustering, FindClusters was run using algorithm 3, resolution 0.4 for the restimulated condition and resolution 0.5 for the resting condition. Two clusters in the restimulated condition were manually merged to form “Cluster 2: Negative Regulators.” The merged clusters showed highly similar gene expression patterns, with one cluster containing the bulk of cells containing negative regulator sgRNAs and the other containing sgRNAs targeting the negative regulator MUC1. Cluster trees shown were generated using the Seurat BuildClusterTree function with default arguments. For pseudobulk differential expression analyses, the Seurat FindMarkers function was used with the default method, Wilcoxon rank sum test.
To generate the T cell activation score, pseudobulk differential expression analysis was first performed on restimulated versus resting no-target control sgRNAs, and log2-fold change outputs were used as gene weights. Only genes that had an absolute log2-fold change >0.25 and were detected in 10% of restimulated or resting cells were used for gene weights. For a given cell, the activation score was calculated as sum(GE × GW/GM), where GE is a gene’s normalized/transformed expression count, GW is the gene’s weight, and GM is the gene’s mean expression in no-target control cells (to correct for differential levels of baseline expression).
Statistical analysis
All statistical analyses were performed in R version 4.0.2 unless otherwise noted. To address ties in nonparametric tests, Mann–Whitney U tests were performed using the wilcox_test function of the Coin R package (version 1.4–1), with default arguments. For q-value–based multiple-comparisons correction, the R qvalue package (version 2.20.0) was used, with default arguments.
Supplementary Material
ACKNOWLEDGMENTS
We thank E. Shifrut, J. Carnevale, and V. Tobin for help related to the development of CRISPRa technologies in T cells; J. Eyquem for providing NY-ESO-1–expressing NALM6 cells; D. Goodman, D. Lee, and B. Hwang for advice related to development of the Perturb-seq platform and analysis; all members of the Marson laboratory for critical insight and discussion over the course of this study; and the staff of the PFCC for support with sorting. Some of the figures were generated using BioRender (https://biorender.com/).
Funding:
This work was supported by the National Institute of Diabetes and Digestive and Kidney Diseases (grant DP3DK111914-01 to A.M.); Simons Foundation (A.M.); Burroughs Wellcome Fund, Career Award for Medical Scientists (A.M.); The Cancer Research Institute Lloyd J. Old STAR grant (A.M.); Parker Institute for Cancer Immunotherapy (A.M.); Innovative Genomics Institute (A.M.); National Institutes of Health grant P30 DK063720 to the Parnassus Flow Cytometry Core and A.M.; National Institutes of Health grant S10 1S10OD021822-01 to the PFCC and A.M.; gifts from B. Byers, B. Bakar, K. Jordan, and E. Radutzky (A.M.); Parker Institute for Cancer Immunology scholarship (Z.S.); Austrian Exchange Service and Austrian Society of Laboratory Medicine Fellowships (R.S.); Max Kade Foundation (R.S.); Care-for-Rare Foundation and German Research Foundation Fellowships (F.B.); National Institutes of Health grant R01HG008140 (J.W.F.); and National Institutes of Health grant S10 RR028962 and James B. Pendleton Charitable Trust to the Gladstone Institutes Flow Cytometry Core Facility. A.M. and C.J.Y. are Chan Zuckerberg Biohub investigators.
Footnotes
Competing interests: A.M. is a compensated cofounder, member of the boards of directors, and member of the scientific advisory boards of Spotlight Therapeutics and Arsenal Biosciences. A.M. and C.J.Y. are cofounders, members of the boards of directors, and members of the scientific advisory board of Survey Genomics. A.M. is a compensated member of the scientific advisory board of NewLimit. A.M. was a compensated member of the scientific advisory board at PACT Pharma and was a compensated adviser to Juno Therapeutics. A.M. owns stock in Arsenal Biosciences, Spotlight Therapeutics, NewLimit, Survey Genomics, PACT Pharma, and Merck. A.M. has received fees from Vertex, Merck, Amgen, Trizell, Genentech, AlphaSights, Rupert Case Management and Bernstein and is an investor in and informal adviser to Offline Ventures and a client of EPIQ. The Marson laboratory has received research support from Juno Therapeutics, Epinomics, Sanofi, GlaxoSmithKline, Gilead, and Anthem. C.J.Y. is a Scientific Advisory Board member for and holds equity in Related Sciences and ImmunAI, a consultant for and holds equity in Maze Therapeutics, and a consultant for TReX Bio. C.J.Y. has received research support from Chan Zuckerberg Initiative and Genentech. J.W.F. is a consultant for NewLimit. R.S., Z.S., and A.M. are listed as inventors on a patent application related to this work. The remaining authors declare no competing interests.
Data and materials availability: All raw sequencing data critical to the findings of this study, including pooled CRISPR screens and RNA-seq, are deposited at the National Center for Biotechnology Information (NCBI) Gene Expression Omnibus under accession numbers GSE174292, GSE190604, and GSE190846. Custom code critical to reproducing the findings of this study, in addition to analyzed Perturb-seq data, are archived at Zenodo (56). All other data are available in the main text or the supplementary materials.
REFERENCES AND NOTES
- 1.Abbas AK, Trotta E, R Simeonov D, Marson A, Bluestone JA, Revisiting IL-2: Biology and therapeutic prospects. Sci. Immunol. 3, eaat1482 (2018). doi: 10.1126/sciimmunol.aat1482 [DOI] [PubMed] [Google Scholar]
- 2.Ni L, Lu J, Interferon gamma in cancer immunotherapy. Cancer Med. 7, 4509–4516 (2018). doi: 10.1002/cam4.1700 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Castro F, Cardoso AP, Gonçalves RM, Serre K, Oliveira MJ, Interferon-gamma at the crossroads of tumor immune surveillance or evasion. Front. Immunol. 9, 847 (2018). doi: 10.3389/fimmu.2018.00847 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Ivashkiv LB, IFNγ: Signalling, epigenetics and roles in immunity, metabolism, disease and cancer immunotherapy. Nat. Rev. Immunol. 18, 545–558 (2018). doi: 10.1038/s41577-018-0029-z [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Malek TR, The biology of interleukin-2. Annu. Rev. Immunol. 26, 453–479 (2008). doi: 10.1146/annurev.immunol.26.021607.090357 [DOI] [PubMed] [Google Scholar]
- 6.Boardman DA, Levings MK, Cancer immunotherapies repurposed for use in autoimmunity. Nat. Biomed. Eng. 3, 259–263 (2019). doi: 10.1038/s41551-019-0359-6 [DOI] [PubMed] [Google Scholar]
- 7.Gao J et al. , Loss of IFN-γ pathway genes in tumor cells as a mechanism of resistance to anti-CTLA-4 therapy. Cell 167, 397–404.e9 (2016). doi: 10.1016/j.cell.2016.08.069 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Zaretsky JM et al. , Mutations associated with acquired resistance to PD-1 blockade in melanoma. N. Engl. J. Med. 375, 819–829 (2016). doi: 10.1056/NEJMoa1604958 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Ayers M et al. , IFN-γ-related mRNA profile predicts clinical response to PD-1 blockade. J. Clin. Invest. 127, 2930–2940 (2017). doi: 10.1172/JCI91190 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Bartelt RR, Cruz-Orcutt N, Collins M, Houtman JCD, Comparison of T cell receptor-induced proximal signaling and downstream functions in immortalized and primary T cells. PLOS ONE 4, e5430 (2009). doi: 10.1371/journal.pone.0005430 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Colin-York H, Kumari S, Barbieri L, Cords L, Fritzsche M, Distinct actin cytoskeleton behaviour in primary and immortalised T-cells. J. Cell Sci. 133, jcs.232322 (2019). doi: 10.1242/jcs.232322 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Astoul E, Edmunds C, Cantrell DA, Ward SG, PI 3-K and T-cell activation: Limitations of T-leukemic cell lines as signaling models. Trends Immunol. 22, 490–496 (2001). doi: 10.1016/S1471-4906(01)01973-1 [DOI] [PubMed] [Google Scholar]
- 13.Parnas O et al. , A genome-wide CRISPR screen in primary immune cells to dissect regulatory networks. Cell 162, 675–686 (2015). doi: 10.1016/j.cell.2015.06.059 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Dong MB et al. , Systematic immunotherapy target discovery using genome-scale in vivo CRISPR screens in CD8 T cells. Cell 178, 1189–1204.e23 (2019). doi: 10.1016/j.cell.2019.07.044 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Henriksson J et al. , Genome-wide CRISPR screens in T helper cells reveal pervasive crosstalk between activation and differentiation. Cell 176, 882–896.e18 (2019). doi: 10.1016/j.cell.2018.11.044 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Shifrut E et al. , Genome-wide CRISPR screens in primary human T cells reveal key regulators of immune function. Cell 175, 1958–1971.e15 (2018). doi: 10.1016/j.cell.2018.10.024 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Ting PY et al. , Guide Swap enables genome-scale pooled CRISPR-Cas9 screening in human primary cells. Nat. Methods 15, 941–946 (2018). doi: 10.1038/s41592-018-0149-1 [DOI] [PubMed] [Google Scholar]
- 18.Simeonov DR et al. , Discovery of stimulation-responsive immune enhancers with CRISPR activation. Nature 549, 111–115 (2017). doi: 10.1038/nature23875 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Liu Y et al. , CRISPR activation screens systematically identify factors that drive neuronal fate and reprogramming. Cell Stem Cell 23, 758–771.e8 (2018). doi: 10.1016/j.stem.2018.09.003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Chen X et al. , Functional interrogation of primary human T cells via CRISPR genetic editing. J. Immunol. 201, 1586–1598 (2018). doi: 10.4049/jimmunol.1701616 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Nasrallah R et al. , A distal enhancer at risk locus 11q13.5 promotes suppression of colitis by Treg cells. Nature 583, 447–452 (2020). doi: 10.1038/s41586-020-2296-7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Sanson KR et al. , Optimized libraries for CRISPR-Cas9 genetic screens with multiple modalities. Nat. Commun. 9, 5416 (2018). doi: 10.1038/s41467-018-07901-8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Konermann S et al. , Genome-scale transcriptional activation by an engineered CRISPR-Cas9 complex. Nature 517, 583–588 (2015). doi: 10.1038/nature14136 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Kaech SM, Cui W, Transcriptional control of effector and memory CD8+ T cell differentiation. Nat. Rev. Immunol. 12, 749–761 (2012). doi: 10.1038/nri3307 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Zhu J, Yamane H, Paul WE, Differentiation of effector CD4 T cell populations (*). Annu. Rev. Immunol. 28, 445–489 (2010). doi: 10.1146/annurev-immunol-030409-101212 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Lazarevic V, Glimcher LH, Lord GM, T-bet: A bridge between innate and adaptive immunity. Nat. Rev. Immunol. 13, 777–789 (2013). doi: 10.1038/nri3536 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Evans CM, Jenner RG, Transcription factor interplay in T helper cell differentiation. Brief. Funct. Genomics 12, 499–511 (2013). doi: 10.1093/bfgp/elt025 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Courtney AH, Lo W-L, Weiss A, TCR signaling: Mechanisms of initiation and propagation. Trends Biochem. Sci. 43, 108–123 (2018). doi: 10.1016/j.tibs.2017.11.008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Gaud G, Lesourne R, Love PE, Regulatory mechanisms in T cell receptor signalling. Nat. Rev. Immunol. 18, 485–497 (2018). doi: 10.1038/s41577-018-0020-8 [DOI] [PubMed] [Google Scholar]
- 30.Holland SJ et al. , Functional cloning of Src-like adapter protein-2 (SLAP-2), a novel inhibitor of antigen receptor signaling. J. Exp. Med. 194, 1263–1276 (2001). doi: 10.1084/jem.194.9.1263 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Shui J-W et al. , Hematopoietic progenitor kinase 1 negatively regulates T cell receptor signaling and T cell-mediated immune responses. Nat. Immunol. 8, 84–91 (2007). doi: 10.1038/ni1416 [DOI] [PubMed] [Google Scholar]
- 32.Hart T et al. , Evaluation and design of genome-wide CRISPR/ SpCas9 knockout screens. G3 7, 2719–2727 (2017). doi: 10.1534/g3.117.041277 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Ferdosi SR et al. , Multifunctional CRISPR-Cas9 with engineered immunosilenced human T cell epitopes. Nat. Commun. 10, 1842 (2019). doi: 10.1038/s41467-019-09693-x [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Hu H et al. , Otud7b facilitates T cell activation and inflammatory responses by regulating Zap70 ubiquitination. J. Exp. Med. 213, 399–414 (2016). doi: 10.1084/jem.20151426 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Mimitou EP et al. , Multiplexed detection of proteins, transcriptomes, clonotypes and CRISPR perturbations in single cells. Nat. Methods 16, 409–412 (2019). doi: 10.1038/s41592-019-0392-0 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Alda-Catalinas C et al. , A single-cell transcriptomics CRISPR-activation screen identifies epigenetic regulators of the zygotic genome activation program. Cell Syst. 11, 25–41.e9 (2020). doi: 10.1016/j.cels.2020.06.004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Replogle JM et al. , Combinatorial single-cell CRISPR screens by direct guide RNA capture and targeted sequencing. Nat. Biotechnol. 38, 954–961 (2020). doi: 10.1038/s41587-020-0470-y [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Thakore PI et al. , Highly specific epigenome editing by CRISPR-Cas9 repressors for silencing of distal regulatory elements. Nat. Methods 12, 1143–1149 (2015). doi: 10.1038/nmeth.3630 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Fulco CP et al. , Systematic mapping of functional enhancerpromoter connections with CRISPR interference. Science 354, 769–773 (2016). doi: 10.1126/science.aag2445 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Xu X, Qi LS, A CRISPR-dCas toolbox for genetic engineering and synthetic biology. J. Mol. Biol. 431, 34–47 (2019). doi: 10.1016/j.jmb.2018.06.037 [DOI] [PubMed] [Google Scholar]
- 41.Chun I et al. , CRISPR-Cas9 knock out of CD5 enhances the anti-tumor activity of chimeric Antigen Receptor T cells. Blood 136 (Supplement 1), 51–52 (2020). doi: 10.1182/blood-2020-136860 [DOI] [Google Scholar]
- 42.Lynn RC et al. , c-Jun overexpression in CAR T cells induces exhaustion resistance. Nature 576, 293–300 (2019). doi: 10.1038/s41586-019-1805-z [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Datlinger P et al. , Pooled CRISPR screening with single-cell transcriptome readout. Nat. Methods 14, 297–301 (2017). doi: 10.1038/nmeth.4177 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Freimer JW et al. , Systematic discovery and perturbation of regulatory genes in human T cells reveals the architecture of immune networks. bioRxiv 2021.04.18.440363 (2021); https://www.biorxiv.org/content/10.1101/2021.04.18.440363v1. [DOI] [PMC free article] [PubMed]
- 45.Li W et al. , MAGeCK enables robust identification of essential genes from genome-scale CRISPR/Cas9 knockout screens. Genome Biol. 15, 554 (2014). doi: 10.1186/s13059-014-0554-4 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Korotkevich G, Sukhov V, Budin N, Shpak B, Artyomov MN, Sergushichev A, Fast gene set enrichment analysis. bioRxiv 060012 (2016); https://www.biorxiv.org/content/10.1101/060012v3. doi: 10.1101/060012 [DOI]
- 47.Finucane HK et al. , Partitioning heritability by functional annotation using genome-wide association summary statistics. Nat. Genet. 47, 1228–1235 (2015). doi: 10.1038/ng.3404 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Horlbeck MA et al. , Compact and highly active next-generation libraries for CRISPR-mediated gene repression and activation. eLife 5, e19760 (2016). doi: 10.7554/eLife.19760 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Martin M, Cutadapt removes adapter sequences from highthroughput sequencing reads. EMBnet. J. 17, 10–12 (2011). doi: 10.14806/ej.17.1.200 [DOI] [Google Scholar]
- 50.Dobin A et al. , STAR: Ultrafast universal RNA-seq aligner. Bioinformatics 29, 15–21 (2013). doi: 10.1093/bioinformatics/bts635 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Liao Y, Smyth GK, Shi W, featureCounts: An efficient general purpose program for assigning sequence reads to genomic features. Bioinformatics 30, 923–930 (2014). doi: 10.1093/bioinformatics/btt656 [DOI] [PubMed] [Google Scholar]
- 52.Ritchie ME et al. , limma powers differential expression analyses for RNA-sequencing and microarray studies. Nucleic Acids Res. 43, e47 (2015). doi: 10.1093/nar/gkv007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Heaton H et al. , Souporcell: Robust clustering of single-cell RNA-seq data by genotype without reference genotypes. Nat. Methods 17, 615–620 (2020). doi: 10.1038/s41592-020-0820-1 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Satija R, Farrell JA, Gennert D, Schier AF, Regev A, Spatial reconstruction of single-cell gene expression data. Nat. Biotechnol. 33, 495–502 (2015). doi: 10.1038/nbt.3192 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Hafemeister C, Satija R, Normalization and variance stabilization of single-cell RNA-seq data using regularized negative binomial regression. Genome Biol. 20, 296 (2019). doi: 10.1186/s13059-019-1874-1 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Steinhart Z, Code repository for: “CRISPR activation and interference screens decode stimulation responses in primary human T cells,” Zenodo (2022); doi: 10.5281/zenodo.578465 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.