

Abstract
Memory CD8+ T cells are characterized by their ability to persist long after the initial antigen encounter and their capacity to generate a rapid recall response. Recent studies have identified a role for metabolic reprogramming and mitochondrial function in promoting the longevity of memory T cells. However, detailed mechanisms involved in promoting their rapid recall response are incompletely understood. Here, we identify a role for the initial and continued activation of the trifunctional rate-limiting enzyme of the de novo pyrimidine synthesis pathway CAD (carbamoyl-phosphate synthetase 2, aspartate transcarbamylase, and dihydroorotase) as critical in promoting the rapid recall response of previously-activated CD8+ T cells. We found that CAD was rapidly phosphorylated upon naïve T cell activation in an mTORC1-dependent manner, yet remained phosphorylated long after initial activation. Previously-activated CD8+ T cells displayed continued de novo pyrimidine synthesis in the absence of mitogenic signals, and interfering with this pathway diminished the speed and magnitude of cytokine production upon rechallenge. Inhibition of CAD did not affect cytokine transcript levels, but diminished available pre-rRNA, the polycistronic rRNA precursor whose synthesis is the rate-limiting step in ribosomal biogenesis. CAD inhibition additionally decreased levels of detectable ribosomal proteins in previously-activated CD8+ T cells. Conversely, overexpression of CAD improved both the cytokine response and proliferation of memory T cells. Overall, our studies reveal a critical role for CAD-induced pyrimidine synthesis and ribosomal biogenesis in promoting the rapid recall response characteristic of memory T cells.
One Sentence Summary:
Pyrimidine synthesis fuels ribosomal biogenesis to facilitate rapid recall responses in CD8+ T cells.
Introduction
Memory CD8+ T cells are characterized by two important functional properties: they persist at relatively increased frequencies long after their initial antigen encounter and, upon rechallenge, exhibit both a rapid and robust recall memory T cell response (1,2). While previous studies have identified metabolic reprogramming as promoting persistence (3), less is known about the biochemical properties that endow memory CD8+ cells with superior recall capacity. The kinase mTOR, acting through its two canonical signaling complexes mTORC1 and mTORC2, integrates environmental cues to direct a myriad of transcriptional and translational programs downstream of T cell activation (4,5). Specifically, mTORC1 promotes CD8+ effector generation and mTORC2 inhibits the generation of memory CD8+ T cells, although the downstream targets of mTOR giving rise to these different T cell fates have not been fully elucidated (6). To this end, asymmetric partitioning of mTOR activity promotes in part the simultaneous generation of effector and memory T cells during an immune response (7).
Naïve T cells maintain low baseline levels of mTOR activity and divide infrequently without TCR engagement during homeostatic proliferation. Upon T cell activation, the PI3K/Akt/mTOR signaling cascade results in robust mTORC1 activity that is detectable within minutes and returns to baseline by 48–72 hours (9). This mTORC1 activity, in concert with other signaling pathways, dramatically alters the physiology and metabolism of lymphocytes to promote glycolytic metabolism that is permissive for the additional synthesis of necessary metabolites (5,6). Recently, mTORC1 acting via the downstream kinase S6K1 was shown to phosphorylate and activate the rate-limiting trifunctional enzyme of the de novo pyrimidine synthesis pathway, CAD (carbamoyl-phosphate synthetase 2, aspartate transcarbamylase, and dihydroorotase) in HEK-293 and HeLa cell lines (10). In the absence of mitogenic signals, T cells do not require de novo pyrimidine synthesis for cytidine and thymidine nucleosides, instead relying on the pyrimidine salvage pathway (14,15). The rapid clonal expansion of activated T cells is supported in part by rapid de novo synthesis of nucleic acids, consistent with multiple rounds of cell division each requiring replication of genomic DNA. The same radiolabeling studies that helped describe this process over fifty years ago also noted the continued accumulation of tritiated nucleotides into RNA in previously-activated lymphocytes nine days after stimulation, well after incorporation of new nucleotides into DNA had diminished (16), although an explanation for this phenomenon was not explored at the time.
Given that full T cell activation leading to effector cell generation results in mTORC1 activation and CAD-induced pyrimidine synthesis is mTORC1-dependent, we hypothesized that the ability of mTORC1 to promote effector cell generation was due in part to the acute activation of CAD. Surprisingly, while we did indeed find that TCR engagement leads to CAD phosphorylation, we also observed that phosphorylated CAD remains detectable in resting cells long after T cell activation at a time when mTOR activity had returned to baseline. This prompted us to explore the role of persistent CAD activation in previously activated/memory T cells. Our studies demonstrate that persistent CAD-induced pyrimidine synthesis contributes to the rapid recall response of memory T cells. Mechanistically, the contribution of CAD is not mediated through increased cytokine transcription, but rather promotion of ribosomal biogenesis secondary to CAD-mediated pre-rRNA synthesis.
Results
CAD undergoes TORC1-mediated phosphorylation upon CD8+ T cell activation and remains phosphorylated in resting cells
The trifunctional CAD protein catalyzes the first three steps in de novo pyrimidine synthesis (Figure 1A). Regulation of CAD activity by phosphorylation has been described at multiple residues by a diverse array of kinases (8), including an activating phosphorylation at Ser1859 by mTORC1 acting via the downstream kinase S6K1 (10). As mTOR signaling is instrumental in CD8+ T cell activation and differentiation (5,6), we interrogated C57BL/6J (referred to as WT) CD8+ T cells for CAD phosphorylation via western blot at this site following T cell activation. CAD is phosphorylated at Ser1859 in a mTORC1-dependent manner acutely following T cell activation, as T cells activated in the presence of the mTORC1 inhibitor rapamycin showed no increase in pCAD or mTOR activity as measured by pS6K1 (Figure 1B). This phosphorylation was further confirmed to be mTORC1-dependent by utilizing CD8+ cells isolated from T cell-specific Rptor or Rictor knockout mice with T cells deficient in mTORC1 or mTORC2 signaling, respectively (Figure 1C). In these experiments, the elimination of mTORC1 signaling abrogated CAD phosphorylation while genetic inhibition of mTORC2, as expected, had no effect. After validating the use of our commercially available pCAD antibody for flow cytometry applications via shRNA knockdown of CAD and secondary antibody isotype staining (Figure S1), we confirmed mTORC1-dependent phosphorylation of CAD upon CD8+ T cell activation utilizing flow cytometry (Figure 1D). Thus, TCR engagement leads to CAD phosphorylation in an mTORC1-dependent fashion.
Figure 1: mTORC1-dependent phosphorylation of CAD at S1859 persists beyond T cell activation.
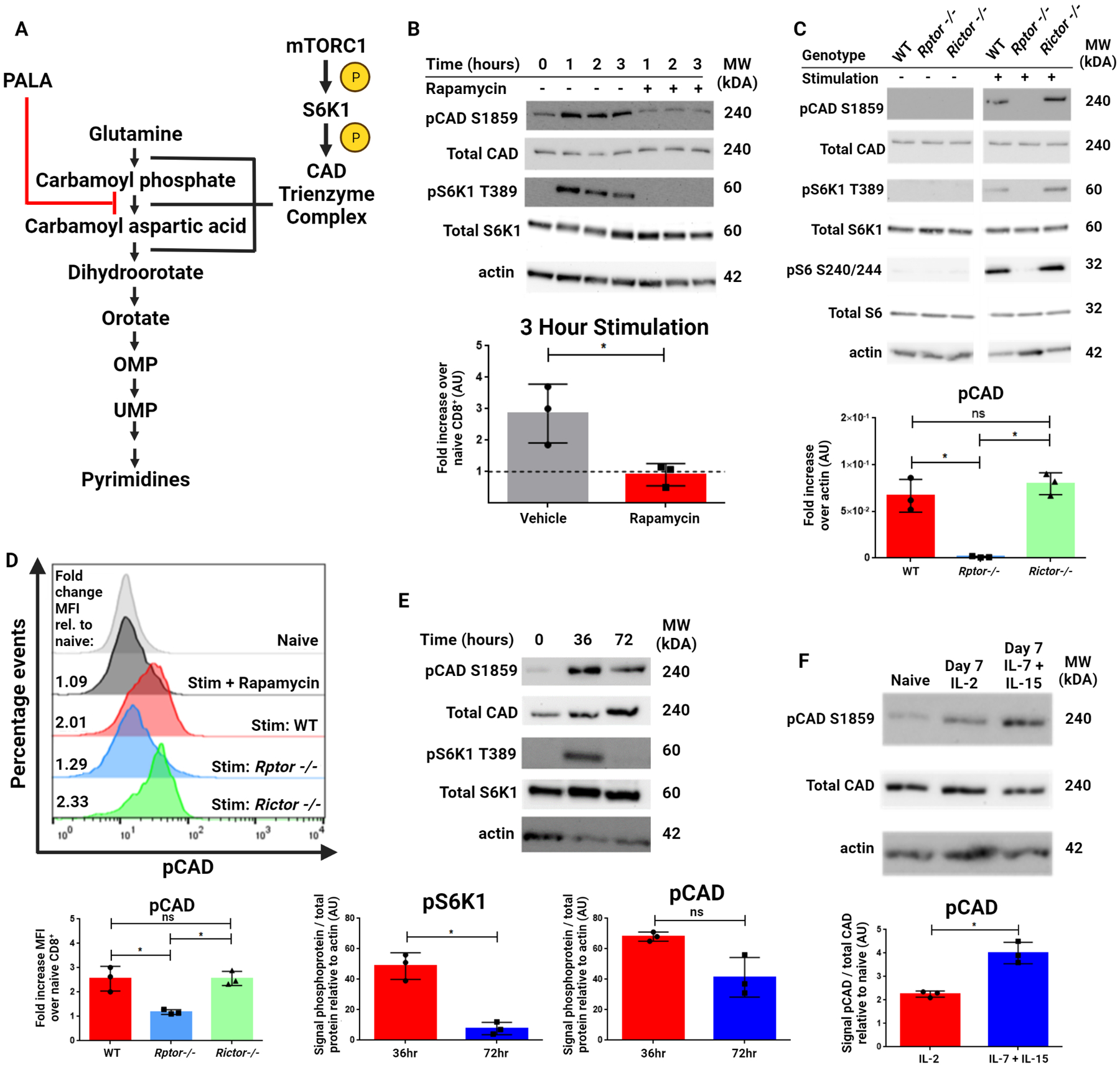
(A) The de novo pyrimidine synthesis pathway. The rate-limiting trifunctional enzyme CAD is phosphorylated and activated by S6K1, which is in turn phosphorylated and activated by mTORC1. The CAD protein catalyzes the first three steps of this pathway and is inhibited by the small molecule PALA (shown in red). (B) Western blot analysis of CD8+ T cells isolated from spleens of naïve WT mice and stimulated with anti-CD3/28 antibodies for indicated time periods with or without rapamycin (100 nM). (C) Western blot analysis of CD8+ T cells isolated from spleens of naïve WT, Rptor−/− and Rictor−/− mice stimulated with anti-CD3/28 antibodies for indicated three hours. (D) Flow cytometric analysis of WT, Rptor−/− and Rictor−/− mice for pCAD. CD8+ T cells from WT mice were analyzed as either unstimulated (grey) or stimulated using anti-CD3/28 antibodies for three hours with 100 nM rapamycin (black) or without rapamycin (red). Rptor−/− (blue) and Rictor−/− (green) CD8+ T cells were also stimulated for three hours. (E) Western blot analysis of CD8+ T cells isolated from spleens of naïve WT mice and stimulated with anti-CD3/28 antibodies for 36 and 72 hours. (F) Western blot analysis of CD8+ T cells isolated from spleens of naïve C57BL/6J mice and stimulated with anti-CD3/28 antibodies for 48 hours, then expanded in media containing IL-2 or IL-7 + IL-15 for an additional five days. Data in (B) includes representative image and densitometry of fold increase in signal relative to actin across three experiments. Data in (C) includes representative image and densitometry of signal relative to actin across three experiments. Data in (D) included representative image and depicts fold change in MFI over naïve CD8+cells across three experiments. Data in (E) and (F) include representative image and densitometry of signal ratio of phosphoprotein to total protein relative to actin across three experiments. Data in (B), (E), and (F) analyzed by two-tailed t test. Data in (C) and (D) analyzed by one-way ANOVA followed by Tukey’s HSD test. *P <0.05, ns = not significant.
Next, we cultured CD8+ T cells for up to 72 hours after activation to further interrogate the kinetics of CAD phosphorylation. At 36 hours after activation, both pCAD and pS6K1 are robustly elevated. However, by 72 hours of stimulation, mTORC1 activity begins to decrease while pCAD signal remains elevated when compared to naïve cells (Figure 1E). That is, we observed prolonged phosphorylation of CAD in previously-activated T cells long after mTORC1 activity diminished. This continuation of pCAD signal led us to hypothesize that elevated pCAD might not just be important for acute activation of T cells, but also may play a role in the identity and function of long-lived memory CD8+ T cells. Expansion of activated CD8+ T cells with exogenous supplementation of the cytokines IL-7 and IL-15 induces a memory-like phenotype in vitro when compared to expansion with supplemented IL-2, which induces an effector-like phenotype (11). Indeed, CD8+ T cells cultured for seven days following activation have elevated pCAD compared to naïve cells, with activated cells cultured in IL-7 and IL-15 being further enriched in pCAD than cells cultured in IL-2 (Figure 1F). Taken together, these findings identify Ser1859 of CAD as a downstream target of mTORC1 in CD8+ T cells. Upon T cell activation, this site is phosphorylated in an mTORC1-dependent manner yet remains phosphorylated in vitro even after mTORC1 activity decreases.
CD8+ T cells maintain an activation-induced increase in de novo pyrimidine synthesis without further stimulation
To confirm whether the elevated pCAD seen in previously-activated CD8+ T cells reflected an increase in de novo pyrimidine synthesis, we utilized a mass spectrometry approach to quantify metabolites in this pathway. Consistent with previous findings, naïve T cells generated limited amounts of carbamoyl aspartic acid, orotate, and uridine monophosphate (UMP), three intermediates of de novo pyrimidine synthesis (Figure 2A). Activation of naïve T cells for 24 hours led to an increase in these metabolites consistent with prior studies (17). To further interrogate the fates of metabolic intermediates in the de novo pyrimidine synthesis pathway, we quantified metabolic flux utilizing amide-labeled 15-N glutamine (10) (Figure 2B). Of note, as the amide nitrogen of one molecule of glutamine is condensed with CO2 in the first step of this pathway, all downstream metabolites except for CTP will be reflected in the M+1 isotopic peak (Figure 2C). Naïve CD8+ T cells exhibited limited flux into de novo pyrimidine synthesis which increased upon TCR stimulation. Surprisingly, previously-activated cells maintained a high baseline level of flux into this pathway even in the absence of further stimulation (Figure 2D). These findings demonstrate that the increased pCAD seen in previously-activated CD8+ T cells in vitro indeed correlates with an increase in both detectable metabolites in the de novo pyrimidine synthesis pathway and flux into this pathway via amide-labeled nitrogen tracing.
Figure 2: CD8+ T cells continue to conduct de novo pyrimidine synthesis in vitro beyond T cell activation.
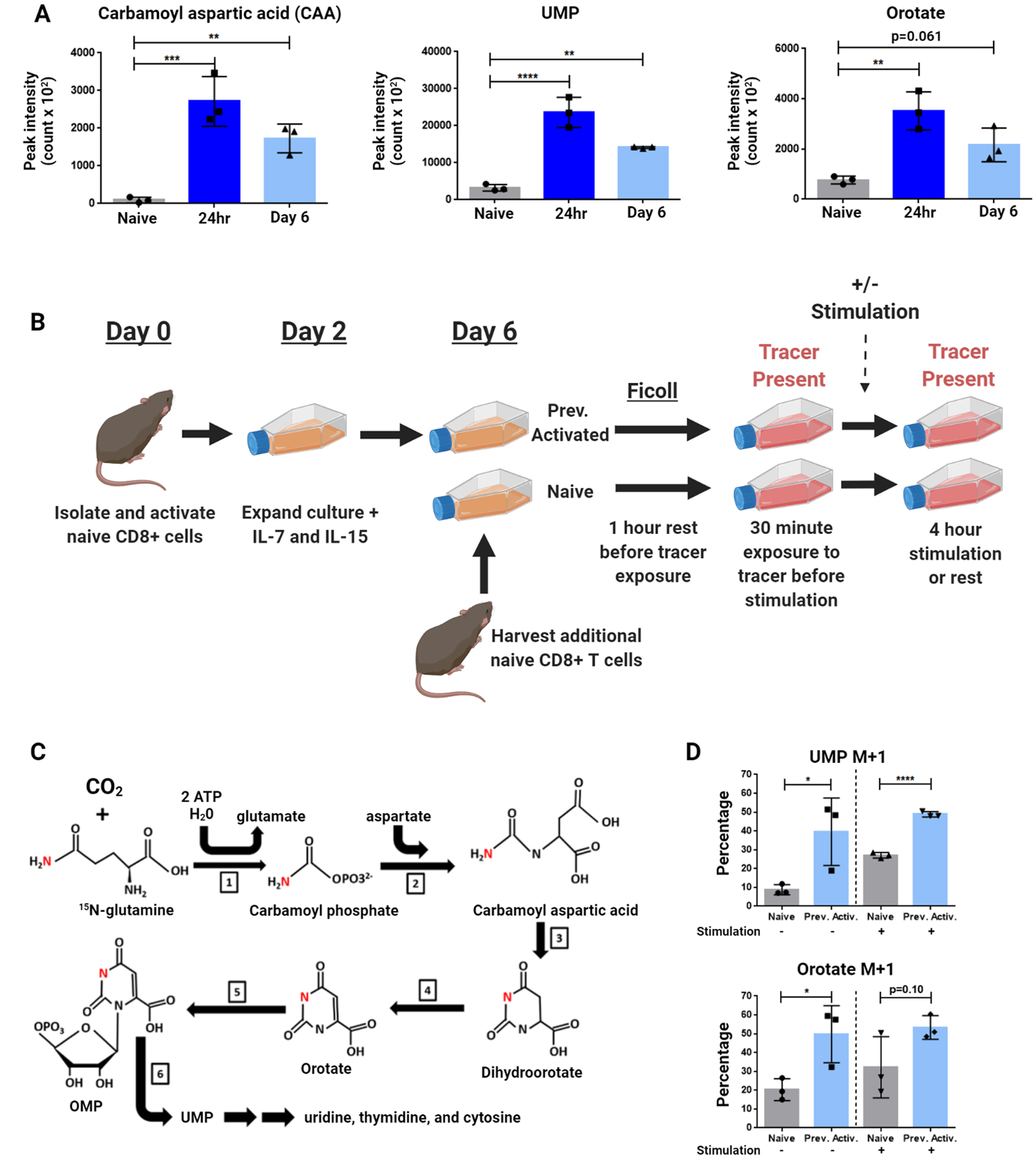
(A) Peak area for de novo pyrimidine synthesis metabolites measured by LC/MS. CD8+ T cells isolated from spleens of naïve C57BL6/J (WT) mice were stimulated with anti-CD3/28 antibodies. A portion of cells was collected and frozen in liquid nitrogen 24 hours after activation. Remaining cells were expanded at 48 hours into culture with IL-7 + IL-15, then rested four more days to generate the Day 6 condition. Additional naïve CD8+ T cells were isolated before analysis (Naïve condition). (B-C) Schematic of amide-labeled 15-N glutamine tracing. Labeled nitrogen depicted in red. Previously activated cells were generated by stimulating isolated CD8+ cells from naïve WT mice with anti-CD3/28 antibodies for 48 hours before expanding media with IL-7 + IL-15 and rested until Day 6 of culture, when an additional naïve WT mouse was harvested and CD8+ splenocytes isolated. Naïve and previously activated cells were then rested before transfer to glutamine-free media supplemented with 4mM amide-labeled 15-N glutamine, and a portion of each condition was stimulated with anti-CD3/28 antibodies. Labeled reactions consist of: [1] Carbamoyl phosphate synthetase II – [2] Aspartate transcarbamoylase – [3] Dihydroorotase – [4] Dihydroorotate dehydrogenase – [5] Orotate phosphoribosyl transferase – [6] Orotidine 5’phosphate decarboxylase. (D) Percentage M+1 peak data for de novo pyrimidine synthesis metabolites measured by LC/MS. Data are representative of triplicate samples analyzed from two independent experiments (A and D). Data are mean ± SD analyzed by one-way ANOVA followed by Tukey’s HSD test (A) and two-tailed t test (D). *P <0.05, **P<0.01, ***P<0.001, ****P<0.0001, ns = not significant.
Phosphorylated CAD is enriched in murine central memory CD8+ cells only with intact mTORC1 signaling at activation
To investigate the phosphorylation status of CAD in vivo, we first isolated spleens from nine-week-old uninfected mice for analysis by flow cytometry (Figure 3A). In the absence of any experimental infection, pCAD was enriched in central memory-phenotype CD8+ cells (CD44+ CD62L+) over naïve CD8+ cells within the same host (CD44−CD62L+). To quantify pCAD levels in an antigen-specific setting, we isolated CD8+ cells from transgenic OT-I mice overexpressing the TCR specific for OVA257–264 (SIINFEKL) epitope. These cells also expressed the congenic marker Thy1.1, allowing us to adoptively transfer them into Thy1.2+ recipients and infect them with Vaccinia virus expressing OVA (Vac-OVA) to expand a population of antigen-specific cells. Antigen-specific central memory CD8+ cells (CD44+CD62L+Thy1.1+) at Day 45 post-infection were enriched in pCAD relative to naïve CD8+ T cells in each mouse (CD44−CD62L+Thy1.1−) (Figure 3B). To investigate enrichment of pCAD in relation to mTOR activity, we repeated this experiment but harvested splenocytes both at the peak of Vac-OVA infection (Day 8) and during the contraction phase (Day 15). Both mTOR activity (as measured by pS6) and pCAD were elevated in antigen-responding cells compared to endogenous naïve cells at the peak of infection (Figure 3C). During the contraction phase, however, pS6 returned to the baseline level seen in naïve cells or lower while pCAD remained enriched in OVA-specific T cells in all mice across all experiments. These findings demonstrate that the persistence of pCAD in the absence of continued mTORC1 activity as seen in previously-activated CD8+ T cells in vitro extends to central memory CD8+ T cells in vivo.
Figure 3: pCAD is detectable in central memory CD8+ cells in vivo when mTORC1 is intact at T cell activation.
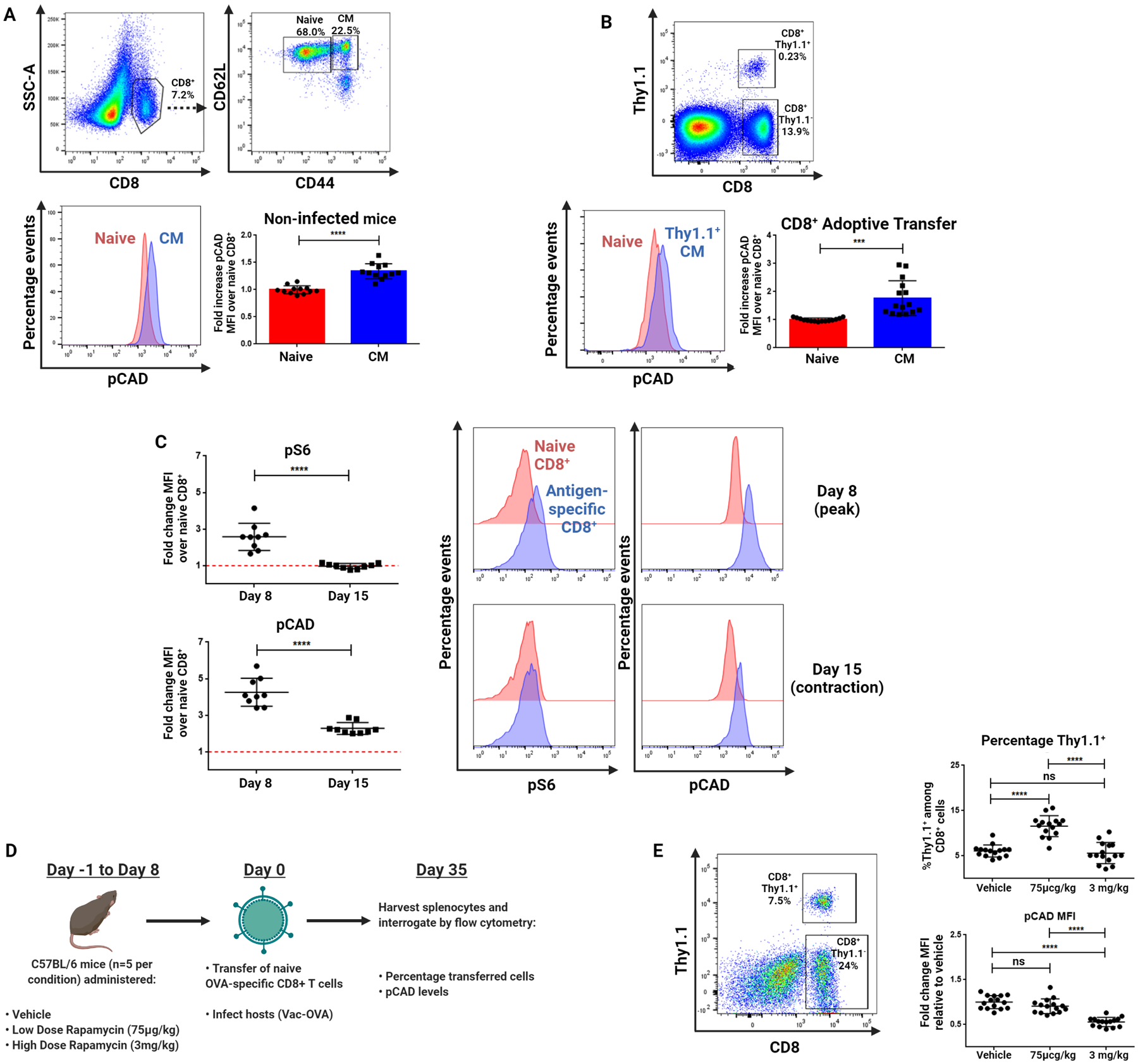
(A) Flow cytometric analysis of pCAD signal in naïve cells (CD8+CD44−CD62L+) and CM cells (CD8+CD44+CD62L+) isolated from the spleens of uninfected WT mice. (B) Flow cytometric analysis of pCAD in naïve and antigen-specific CM CD8+ cells. CD8+ cells were isolated from spleens of naïve OT-I mice and adoptively transferred into WT hosts which were immediately infected with Vac-OVA with splenocytes harvested forty five days later. (C) Flow cytometric analysis of pCAD and pS6 during viral infection. Experiment was performed as in (B) with splenocytes harvested eight days and fifteen days post-infection. (D) Schematic of in vivo rapamycin administration. Mice were administered Vehicle, Low Dose (75 μg/kg) or High Dose (3 mg/kg) rapamycin for Day −1 to Day 8 of Listeria-OVA infection with adoptive transfer of cells freshly isolated from naïve OT-I mice on Day 0. Splenocytes were harvested on Day 35 of infection for analysis by flow cytometry. (E) Summary pCAD intensity and Thy1.1+ data from splenocytes in experiments described in (D) across three replicates. Data in (A) and (B) depict representative gating and fluorescent staining alongside summary MFI values ± SD across three independent experiments normalized to naïve CD8+ cell values within each experiment analyzed by paired two-tailed t-test (n=5 mice per experiment). Data in (C) show mean fluorescence intensity values across three independent experiments normalized to naïve values within each experiment analyzed by paired two-tailed t-test (n=3 mice per experiment). Data in (E) depict representative gating and percentage of Thy1.1+ cells among CD8+ cells and pCAD MFI values ± SD across three independent experiments normalized to vehicle treated CD8+ cell values within each experiment analyzed by one-way ANOVA followed by Tukey’s HSD test. *P <0.05, **P<0.01, ***P<0.001, ****P<0.0001, ns = not significant.
Our data thus far demonstrate that upon initial activation, TCR-induced mTORC1 activation leads to phosphorylation of CAD, which in turn remains phosphorylated in memory CD8+ T cells. At first glance, these data appeared to contradict previous studies demonstrating increased memory T cell generation in the presence of rapamycin (12). To further clarify the role of mTORC1 signaling in CAD phosphorylation, we modified the experimental design utilized by Araki et al. to evaluate the effect of mTOR inhibition during the naïve-to-effector transition of CD8+ cells responding to infection (12). We adoptively transferred OVA-specific Thy1.1+CD8+ T cells into wild-type recipients and infected them with Vac-OVA (Figure 3D). The mice were administered low-dose rapamycin (75 μg/kg), high-dose rapamycin (3 mg/kg), or vehicle from day −1 to day 8 of infection and the spleens were harvested on day 35. Consistent with previous findings (12), low-dose rapamycin administered during the naïve-to-effector transition increased the frequency of memory cells. However, this low dose did not inhibit the phosphorylation of CAD when compared to vehicle controls (Figure 3E). Thus, our findings concerning the role of pCAD in promoting memory T cell function are compatible with the previously established model in which low-dose rapamycin promotes memory T cell formation (13). Alternatively, administration of high-dose rapamycin during this time decreased detectable pCAD. Together, these data suggest pCAD is enriched in previously-activated cells in vivo and in vitro and this enrichment is abrogated in situations where mTORC1 is inhibited at T cell activation.
Reduction of CAD activity before rechallenge decreases the speed and magnitude of CD8+ recall responses
Blasting CD8+ cells are known to require nucleotides produced by de novo pyrimidine synthesis (15,17). Thus, we hypothesized that the CAD-induced increase in de novo pyrimidine synthesis promoted the rapid recall response of previously activated CD8+ cells. To this end, we sought to inhibit CAD activity during the resting phase prior to rechallenge. We activated and expanded antigen-specific CD8+ cells and treated them with the CAD inhibitor N-phosphonacetyl-L-aspartate (PALA), a highly selective competitive inhibitor of CAD (18). To confirm PALA’s activity as a CAD inhibitor, we treated previously-activated cells with PALA for the final 24 hours of their six-day culture period, washed out any remaining drug, then exposed them to amide-labeled 15-N glutamine-containing media alongside naïve cells with and without TCR stimulation. PALA pretreatment decreased flux into de novo pyrimidine synthesis in both resting cells and rechallenged cells in the absence of continued drug exposure (Figure 4A). PALA did not cause global metabolic inhibition, as PALA treatment modestly increased flux into unrelated pathways such as the de novo purine synthesis pathway in cells at rest and at rechallenge (Figure S2). These results confirm PALA as a specific inhibitor of de novo pyrimidine synthesis for use in our studies.
Figure 4: CAD inhibition in resting cells impairs their response at rechallenge.
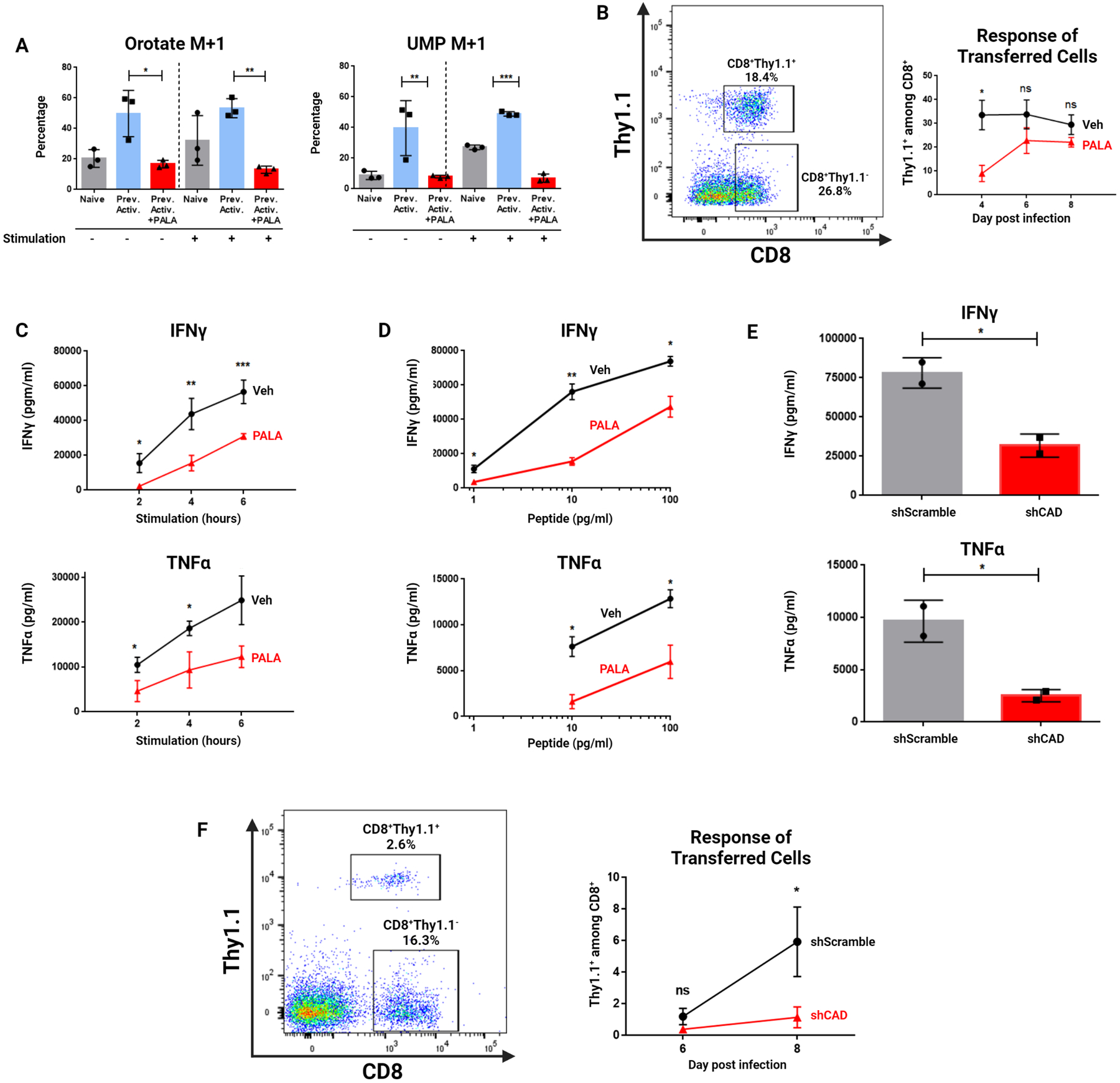
(A) Percentage M+1 peak data for de novo pyrimidine synthesis metabolites measured by LC/MS. Previously activated cells were exposed to amide-labeled 15-N glutamine as in Figure 2C, with the addition of Vehicle or 1 mM PALA for the final 24 hours of culture. Naïve and previously activated cells analyzed are the same triplicate samples from two independent experiments utilized in Figure 2C. (B) Response of transferred previously activated P14 cells to LCMV-Armstrong infection with and without PALA pretreatment. Mice were bled on indicated days with percentage of Thy1.1+ cells among CD8+ cells in blood quantified by flow cytometry. (C) Cytokine response of previously activated cells over a time course of restimulation as measured by ELISA. Vehicle and PALA-treated cells were generated as in (A). On Day 6 of the experiment, naïve OT-I splenocytes were harvested, irradiated, and incubated with OVA257–264 (SIINFEKL) peptide before plating with cultured T cells. (D) Cytokine response of previously activated cells to decreasing peptide doses as measured by ELISA. Vehicle and PALA-treated cells were generated as in (A) from OT-I mice. On Day 6 of the experiment, naïve OT-I splenocytes were harvested, irradiated, and incubated with indicated dose of OVA257–264 (SIINFEKL) peptide before plating with cultured T cells with supernatant analysis after five hours. TNFα was not detectable at 1 pg/ml peptide concentration. (E) Cytokine production of previously activated cells following lentiviral transduction with Cad-targeted (shCAD) or scramble (shScramble) viral constructs. (F) Response of transferred previously activated P14 cells to LCMV-Armstrong infection with and without shRNA knockdown of CAD. Mice were bled on indicated days for analysis of Thy1.1+ cells. Data in (A) representative of two independent experiments analyzed by two-way ANOVA followed by Tukey’s HSD test. Data in (B) depicts representative gating of three independent experiments analyzed by two-tailed t test at each time point with n=5 mice per condition. Data in (C) and (D) depict mean concentration ± SD of two to three dilutions of supernatant each from three independent experiments analyzed by two-tailed t test. Data in (E) representative of two independent experiments analyzed by two-tailed t test. Data in (F) depict representative gating and Thy1.1 positivity from three independent experiments of n=5 mice per condition analyzed by two-tailed t test. *P <0.05, **P<0.01, ***P<0.001, ns = not significant.
To explore the effects of CAD inhibition in the context of antigenic challenge, we generated previously-activated CD8+ T cells from P14 transgenic mice and pretreated them with PALA before adoptively transferring them into mice which were then infected with LCMV-Armstrong. The percentage of Thy1.1+ cells in the blood of these animals were monitored on days 4, 6, and 8 post-infection. PALA pretreatment decreased the speed with which the previously-activated cells expanded in response to antigen encounter (Figure 4B). Thus, CAD inhibition at the time of rechallenge impaired the recall response of CD8+ T cells in vivo.
We next sought to determine the functional consequences of CAD inhibition. Previously-activated OT-I cells were generated as described, and PALA was added for 24 hours on the sixth day of culture. Next the PALA was washed out and viable cells were enriched by Ficoll gradient prior to rechallenge utilizing irradiated murine antigen-presenting cells pre-loaded with OVA257-264 (SIINFEKL) peptide. PALA pretreatment decreased the speed with which previously-activated cells were able to produce both IFNγ and TNFα (Figure 4C). The defect in cytokine production resulting from PALA exposure was partially reversible with the supplementation of exogenous uridine, suggesting a role for nucleotide synthesis restriction in promoting a decrease in recall response (Figure S3). Of note, PALA is not present during rechallenge, and thus these observations reflect inhibition of CAD activity during the baseline rest period.
In addition to a more rapid response to antigen, memory T cells can also respond robustly to lower doses of antigen (19). Thus, we hypothesized that inhibition of CAD in memory cells would lead to a decrease in the peptide dose-response curve of these cells. To address this, we generated previously-activated cells pretreated with PALA (as in Figure 4A) and exposed them to irradiated antigen-presenting cells loaded with different doses of OVA257-264 (SIINFEKL) peptide, with supernatants harvested at 5 hours. PALA pretreatment decreased the ability of previously-activated cells to respond at rechallenge to a wide range of peptide concentrations (Figure 4D). Overall, our results suggest that inhibition of CAD following initial T cell expansion – but prior to rechallenge – impairs both the speed and magnitude of the CD8+ recall response. Thus, in both in vitro and in vivo settings, inhibition of de novo pyrimidine synthesis in previously-activated CD8+ cells impaired their recall response.
As further validation of these findings, we utilized the shRNAs (described in Figure S1) to generate CAD-deficient previously-activated CD8+ T cells which were then subject to both in vitro and in vivo rechallenge. Similar to PALA pretreatment, introduction of shRNA against the Cad gene following T cell activation but before rechallenge impaired cytokine production upon rechallenge (Figure 4E). Adoptive transfer of CAD-deficient antigen-specific CD8+ T cells into mice which were then challenged with infection led to the impaired expansion of those cells (Figure 4F). Taken together, these findings demonstrate that genetic ablation or pharmacologic inhibition of CAD in previously-activated CD8+ T cells impairs their ability to respond rapidly and robustly to antigen encounter, both in vitro and in vivo.
Overexpressing CAD improves CD8+ recall responses in vitro and in vivo
Thus far, our studies demonstrate that either pharmacologic inhibition of CAD or its genetic deletion leads to decreased recall responses. As such, we next sought to increase CAD activity in previously-activated CD8+ T cells by retroviral overexpression of CAD. We cloned the wild-type Cad gene sequence into the GFP-containing MigR1 retroviral backbone to generate CAD-MigR1, allowing us to visualize cell populations with increased CAD expression. To confirm overexpression, previously-activated CD8+ T cells were generated and transduced with CAD-MigR1 or EV-MigR1, with CAD overexpression increasing both total CAD protein levels and pCAD levels at Ser1859 (Figure 5A). To confirm that this increase in active CAD also led to increased flux into de novo pyrimidine synthesis, we again employed amide-labeled 15-N glutamine tracing. Previously-activated CAD overexpressing CD8+ T cells demonstrated increased flux into de novo pyrimidine synthesis both at rest and upon rechallenge (Figure 5B). When these cells were rechallenged and analyzed for cytokine production in vitro, CAD overexpressing cells were more polyfunctional when assayed for IFNγ and TNFα production than empty vector controls (Figure 5C).
Figure 5: CAD overexpression improves recall response in CD8+ T cells.
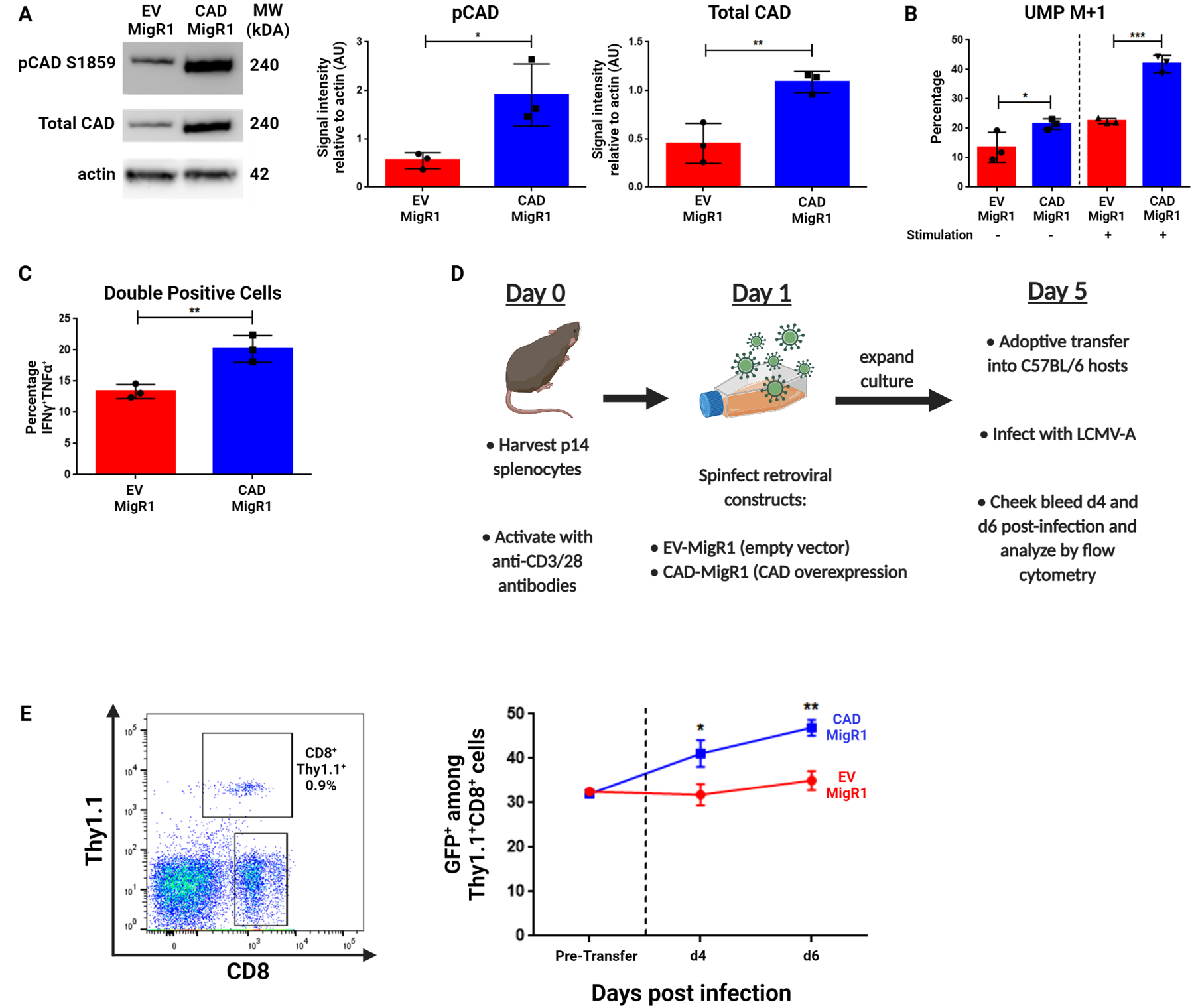
(A-C) CD8+ cells isolated from WT mice were stimulated with anti-CD3/28 antibodies for 24 hours before introduction of indicated retroviral constructs with cells analyzed on Day 6 of culture. (A) Western blot analysis of pCAD (S1859) and total CAD levels in CD8+ cells following retroviral transduction with EV-MigR1 or CAD-MigR1 constructs. (B) Percentage M+1 peak data for de novo pyrimidine synthesis metabolites measured by LC/MS between EV-MigR1 and CAD-MigR1 cells. On Day 6 of culture, freshly-isolated naïve, EV-MigR1 and CAD-MigR1-transduced cells were rested in culture media before transfer to glutamine-free media supplemented with 4 mM amide-labeled 15-N glutamine, with a portion of each condition stimulated with anti-CD3/28 antibodies. (C) Summary data of cytokine response of retrovirally-transduced cells across three experiments generated as in (B) following 4 hours stimulation with PMA/ionomycin. Data gated on live CD8+GFP+ cells with percentage IFNγ+TNFα+ cells reported. (D) Schematic of adoptive transfer experiments with previously activated P14 retrovirally-transduced cells. (E) Percentage GFP+ before transfer and at Day 4 and Day 6 following adoptive transfer and infection with LCMV-Armstrong. Data in (A) depict representative western blot images and densitometry quantification of indicated protein relative to actin from three independent experiments analyzed by two-tailed t test. Data in (B) are mean ± SD of triplicate samples representative of two independent experiments analyzed by two-way ANOVA followed by Tukey’s HSD test. Data in (C) depicts percentage double positive cells across three independent experiments analyzed by two-tailed t test. Data in (E) depict representative gating strategies and GFP+ ± SD analyzed with two-tailed t test at each indicated time point (n=5 mice per condition). *P <0.05, **P<0.01, ***P<0.001.
To explore the effects of CAD overexpression in an in vivo setting, we employed CAD-MigR1 or EV-MigR1 to generate populations of previously-activated P14 CD8+ T cells with equal percentages of GFP-positivity. These cells were then adoptively transferred into hosts immediately infected with LCMV-Armstrong (Figure 5D). In response to antigenic challenge, the frequency of GFP+ cells in the CAD-overexpression condition increased, while the frequency of GFP+ cells remained largely unchanged in empty vector controls (Figure 5E). This preferential expansion of CAD-overexpressing cells provides evidence in vivo of a superior recall response when the ability of CD8+ T cells to conduct de novo pyrimidine synthesis is enhanced.
Modulating CAD activity does not alter mRNA transcript levels but affects production of ribosomal RNA precursors
To further elucidate mechanisms for the defect in cytokine production observed in PALA-pretreated cells, we generated previously-activated cells and rechallenged them as before, isolating RNA for qPCR analysis of cytokine transcripts. Intriguingly, CAD inhibition did not impair the magnitude of CD8+ recall response at the level of Ifng transcription (Figure 6A). As CAD inhibition consistently impaired cytokine protein production (Figure 4), we hypothesized that the defect in pyrimidine synthesis caused a post-transcriptional inhibition of protein synthesis in these cells. Nucleotide availability has been demonstrated to preferentially affect the production of ribosomal RNA (rRNA) over mRNA in cancer cells (20), leading us to hypothesize a preferential role for rRNA synthesis in resting previously-activated and memory CD8+ T cells. Mammalian RNA polymerase I (RNA pol I) transcribes a 13kb pre-rRNA, a polycistronic precursor later processed into the mature 18S, 28S, and 5.8S ribosomal RNA species (Figure 6B). As the precursor rRNA molecule is relatively short-lived and rapidly undergoes post-transcriptional processing and maturation, the detection of regions undergoing rapid processing such as the 5’ externally transcribed spacer region (5’ETS) can be used to assess active pre-rRNA synthesis (21,22). PALA pretreatment decreased pre-rRNA as measured by qPCR using primers for 5’ETS (Figure 6C). These observations suggest that the role of CAD-induced de novo pyrimidine synthesis in previously-activated and memory T cells is not to enhance cytokine transcription, but rather to enhance the synthesis of rRNA.
Figure 6: Inhibition of de novo pyrimidine synthesis restricts rRNA synthesis without affecting mRNA production.
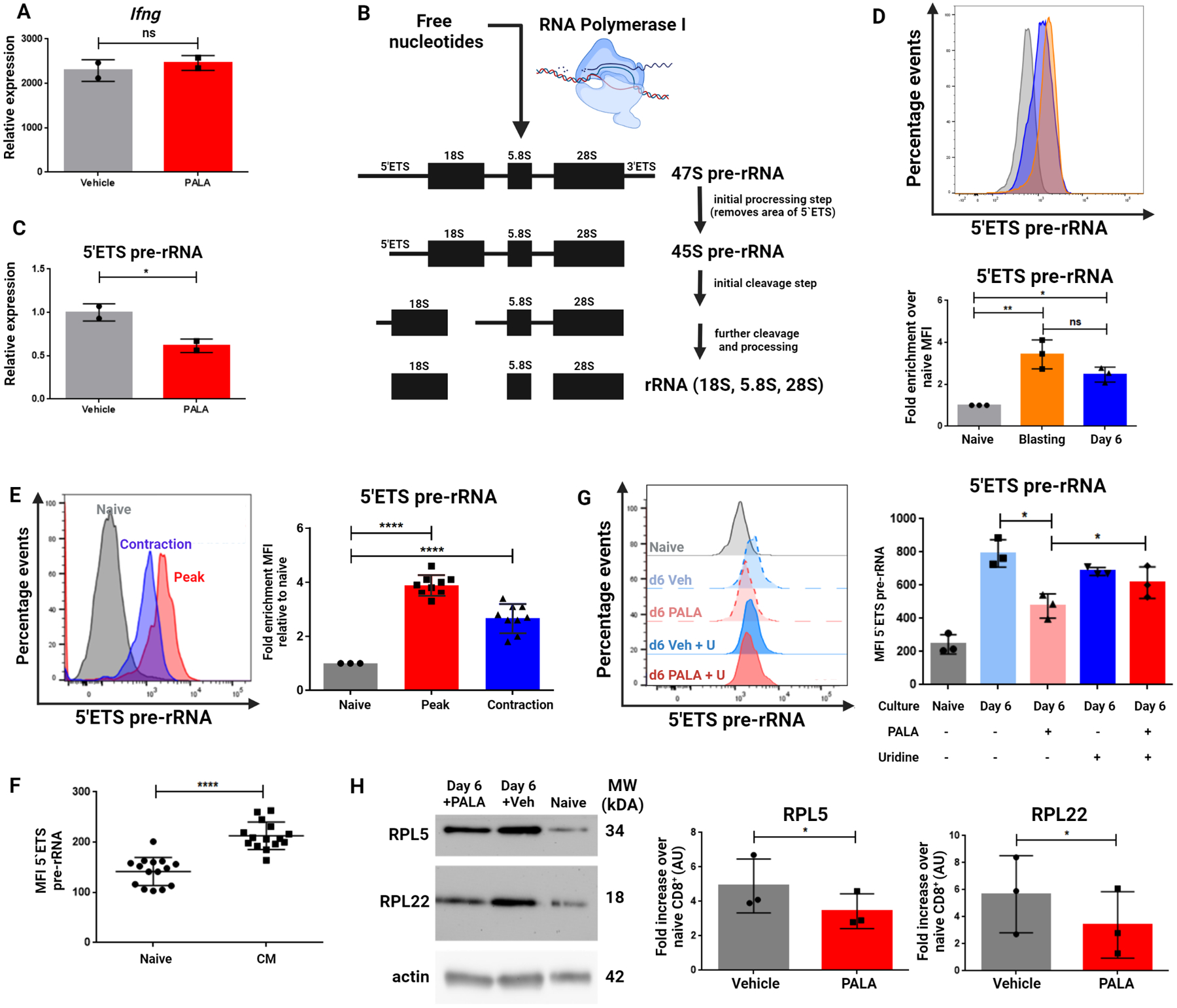
(A) Ifng transcript levels in previously activated CD8+ cells generated as in Figure 4A and stimulated with anti-CD3/28 antibodies on Day 6 of culture relative to resting vehicle-treated cells. (B) Diagram of the 45S pre-rRNA molecule transcribed by RNA Polymerase I before processing into 18S, 28S, and 5.8S ribosomal RNA molecules. (C) 5’ETS pre-rRNA expression levels in previously activated CD8+ cells treated with vehicle or PALA generated with expression relative to vehicle-treated cells. (D) 5’ETS pre-rRNA MFI during CD8+ cell culture. Naïve CD8+ cells were isolated and fixed immediately (Naïve) or stimulated with anti-CD3/28 for 48 hours (Blasting) with the remainder expanded in media with IL-7+15 for an additional four days (Day 6). (E) PrimeFlow® analysis of 5’ETS pre-rRNA signal intensity in CD8+ cells responding to infection. Naïve OT-I cells were isolated with a portion fixed and the remainder transferred into WT hosts infected with Vac-OVA. Splenocytes were harvested at Day 8 (Peak) and Day 15 (Contraction). (F) 5’ETS pre-rRNA MFI in naïve and central memory cells of uninfected mice. (G) 5’ETS pre-rRNA MFI plotted from previously activated vehicle or PALA treated cells generated as in Figure 4A. (H) Western blots of previously-activated CD8+ T cells generated as in Figure 4A. Data in (A) and (C) depict representative mean ± SD of technical duplicates from three independent experiments analyzed by two-tailed t test. Data in (D) depict representative MFI plots ± SD and summary data from three independent experiments generated by normalizing MFI to signal intensity in the naïve condition in each experiment and analyzing via one-way ANOVA followed by Tukey’s HSD test. Data in (E) depict representative MFI plots ± SD and summary data from three independent experiments generated by normalizing MFI to signal intensity in the naïve condition in each experiment and analyzing via one-way ANOVA followed by Tukey’s HSD test (n=3 mice per condition). Data in (F) show MFI ± SD indicated cell populations analyzed by two-tailed t-test. Data in (G) depicts MFI summary data ± SD from three independent experiments analyzed by two-tailed t test (n=5 mice per experiment). Data in (H) depict representative image and summary densitometry analysis from three independent experiments and normalization of each signal intensity to the naïve condition in each experiment ± SD and analyzing with paired two-tailed t test. *P <0.05, **P <0.01, ****P<0.0001, ns = not significant.
Assessment of mRNA and pre-rRNA on a per-cell basis is not possible with PCR-based techniques. The PrimeFlow® RNA Assay, an in-situ hybridization assay utilizing branched-DNA technology which is compatible with flow cytometry, has been demonstrated to specifically and robustly amplify signals from mRNA and miRNA species (23,24). Expanding the potential of this technology, we designed PrimeFlow® probes to target the 5’ETS of pre-rRNA to quantify the rates of rRNA synthesis in different cell populations on a per-cell basis. Radiolabeling experiments from the 1960s demonstrated that naïve lymphocytes synthesize limited amounts of ribosomal RNA at rest but rapidly induce synthesis during the first few days after activation (16). Consistent with these findings, PrimeFlow® analysis of pre-rRNA showed low levels of rRNA in naïve CD8+ T cells which were elevated in blasting cells at 48 hours post-activation in vitro and persisted at six days of resting in culture (Figure 6D). We next utilized this flow cytometry-based assay to determine if we could detect persistence of pre-rRNA synthesis in vivo. Indeed, antigen-specific CD8+ cells analyzed during the contraction phase of response to an infection retained elevated pre-rRNA levels compared to naïve cells, although the signal was lower than that observed at the peak of infection (Figure 6E). In a separate series of experiments (similar to those described in Figure 3A), pre-rRNA was enriched in central memory-phenotype CD8+ T cells (CD44+CD62L+) over naïve CD8+ T cells (CD44−CD62L+) in uninfected mice (Figure 6F). These findings suggest a continuation of pre-rRNA synthesis in memory CD8+ cells beyond the initial burst of rRNA synthesis that is known to accompany the activation of naïve T cells.
Lastly, we tested the effects of CAD inhibition on pre-rRNA production and ribosomal biogenesis. PALA pretreatment of previously-activated cells decreased pre-rRNA detected by PrimeFlow® relative to vehicle treatment, in alignment with our qPCR data (Figure 6G). Notably, this deficit in pre-rRNA production could be rescued via supplementation of exogenous uridine to the cell culture media, again confirming a specific role for pyrimidine nucleotide abundance in regulating the ability of CD8+ T cells to synthesize pre-rRNA. The synthesis of pre-rRNA is the rate-limiting step in ribosomal biogenesis, with individual ribosomal proteins undergoing proteolytic degradation in the absence of assembly with processed pre-rRNA precursors in the nucleolus (36). PALA pretreatment of previously-activated cells also decreased detectable ribosomal proteins, consistent with the finding that inhibition of pre-rRNA synthesis leads to reduced ribosomal protein content (Figure 6H). Taken together, these findings demonstrate a role for de novo pyrimidine synthesis and active CAD for the generation of rRNA and ribosomal biogenesis. These pathways play a role not only at initial activation, but also continuously in previously activated/memory T cells, facilitating the robust recall response upon rechallenge.
Human memory CD8+ cells are enriched in phosphorylated CAD and modulating CAD activity affects their recall response
Finally, we sought to determine whether the ability of CAD to promote ribosomal biogenesis in memory T cells was also critical to human T cell responses. To this end, we analyzed human PBMCs. pCAD was enriched in the CD45RA−CR45RO+CCR7+ central memory CD8+ population compared to the CD45RA+CD45RO− naïve CD8+ populations in healthy donors (Figure 7A). Utilizing pcDNA3 mammalian expression vectors, we next transiently transfected human CD8+ T cells with CAD (also referred to as pcDNA3-CAD) or empty vector (pcDNA3-EV) using electroporation. The presence of an HA tag in each construct allowed for gating exclusively on transfected cells. pcDNA3-CAD human CD8+ T cells demonstrated higher basal levels of pCAD than those expressing empty vector (Figure 7B). To investigate whether functional differences would arise in human CD8+ T cells with differing levels of CAD activity, we rechallenged pcDNA3-CAD or pcDNA3-EV cells and analyzed cytokine production (Figure 7C). CAD overexpressing CD8+ T cells produced more cytokine than empty vector controls upon rechallenge (Figure 7D). This increase in cytokine production was abrogated by pretreatment of these cells after transfection with PALA or a human RNA pol I-specific inhibitor BMH-21. BMH-21 has previously been shown to specifically arrest RNA pol I transcriptional activity without eliciting DNA damage and with negligible effect on RNA pol II (34,35). This supported that the increase in functionality seen in CAD overexpressing cells was contingent upon their ability to conduct de novo pyrimidine synthesis and the intact ability of RNA pol I to conduct rRNA synthesis.
Figure 7: CAD activity in human lymphocytes influences recall response and modulation of pyrimidine synthesis affects ribosomal protein content in human CD8+ T cells.
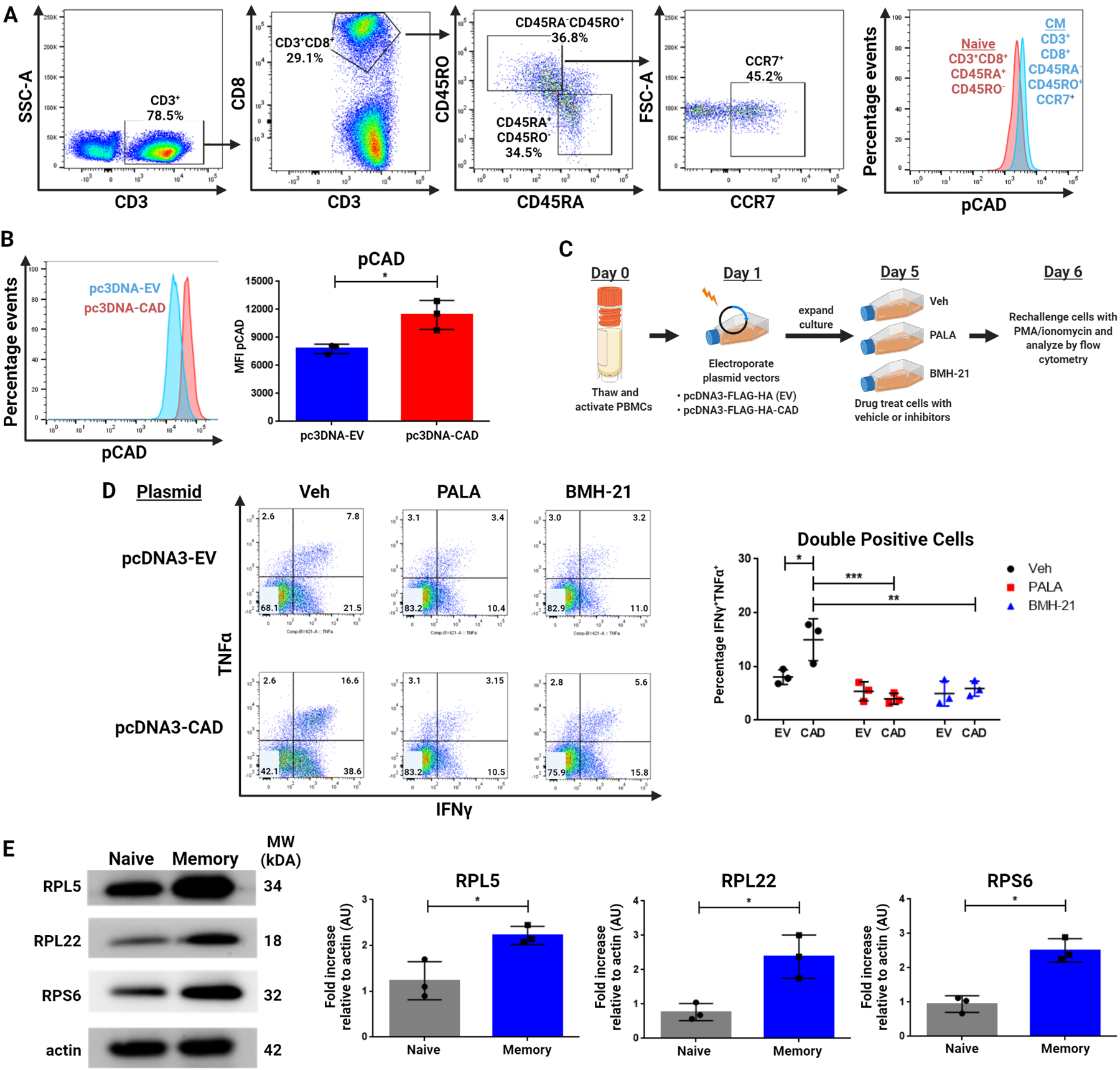
(A) Flow cytometric analysis of pCAD in naïve (CD45RA+CD45RO−) and central memory (CD45RA−CD45RO+CCR7+) human CD3+CD8+ cells. PBMCs were freshly isolated from healthy donor blood before fixation and analysis. (B) Flow cytometric analysis of pCAD levels in Cad-overexpressing (pcDNA3-CAD) or empty vector-expressing human (pcDNA3-EV) cells. Data shown are gated on live CD3+CD8+HA+ cells. (C) Schematic for overexpression of HA-tagged CAD construct in cultured human CD8+ T cells. (D) Flow cytometric analysis of IFNγ and TNFα production by CD8+ cells with and without Cad overexpression following indicated drug treatment. Cells were exposed to drug for 24 hours before drug removal and four hours of restimulation with PMA/ionomycin. Data shown are gated on CD3+CD8+HA+ cells. (E) Western blot of ribosomal proteins in naïve and memory CD8+ T cells. Human PBMCs were thawed and magnetically sorted into naive (CD8+CD45RO−) and memory (CD8+CD45RO+) populations for lysis and analysis. Data in (A) representative of three healthy donors and is depicted alongside representative gating. Data in (B), (D), and (E) depict representative images as well as summary data across three independent experiments with three frozen PBMC aliquots from three separate healthy donors with data in (B) and (E) analyzed by two-tailed t test and data in (D) analyzed by two-way ANOVA followed by Tukey’s HSD test. *P <0.05, **P<0.01, ***P<0.001.
As the product of RNA pol I activity is ultimately the generation of the ribosomes themselves, we sought to analyze the ribosomal protein content of memory cells in comparison to naïve cells. Ribosomal proteins were enriched in isolated memory cells compared to naïve cells (Figure 7E). Taken together, these findings suggest a requirement in human CD8+ T cells for active CAD and that modulation of nucleotide availability or direct RNA pol I inhibition in previously activated cells prior to rechallenge can modulate the recall response.
Discussion
The heightened response of memory CD8+ T cells is one of the hallmarks of adaptive immunity and requires relatively quiescent cells to rapidly reacquire effector function (1,2,25). Memory CD8+ T cells emerge from quiescence so quickly that they can divide in as little as two hours after encountering their cognate antigen in vivo (26). In this study, we propose that active CAD in resting memory CD8+ T cells provides a mechanism to support this superior recall response by fueling ribosomal biogenesis. Naïve T cells have been shown to contain low levels of ribosomal proteins and 45S rRNA which increase in an ERK-MAPK-dependent manner following TCR stimulation (27). The demands for protein synthesis placed on a newly-activated T cell are immense, and it is not unreasonable to expect a concomitant increase in ribosomal biogenesis to provide the necessary protein translation machinery. The role of this process as it pertains to previously-activated T cells is not well-studied, although an upregulation of genes involved in ribosomal biogenesis such as those encoding ribosomal subunits and small nucleolar RNAs (snoRNAs) has been reported in memory T cells (28). The current study both extends the investigation of ribosomal biogenesis beyond the effector phase of a T cell response and provides a mechanism for the superior recall response of memory CD8+ T cells through active CAD and continued pre-rRNA generation.
Although we identify CAD as a downstream phosphorylation target of mTORC1 in CD8+ T cell activation, the maintenance of pCAD in resting cells is mTOR-independent. The precise mechanisms which promote the continued phosphorylation of CAD remain to be determined. Previous studies have identified CAD as a binding partner for 14-3-3ζ (37). As members of the 14-3-3 family of proteins are known to sequester targets by binding phosphorylated residues, it is possible that this interaction could protect or maintain a pool of pCAD in CD8+ T cells. Likewise, it is possible that other kinases or inhibition of certain phosphoprotein phosphatases may be involved. We can, however, conclude that the mTORC1-dependent reprogramming that takes place during the initial cell activation is integral to the generation of phosphorylated CAD in previously-activated cells. Along these lines, our findings support the notion that while rapamycin and mTORC1 inhibition can promote memory T cell formation, this is subsequent to the activation mTORC1 during the initial antigen recognition. To this end, our findings are consistent with the role of low dose rapamycin in promoting memory while high dose rapamycin can promote T cell anergy (13). Our group has previously demonstrated that the specific inhibition of mTORC2 leads to metabolic reprogramming via enhancement of FOXO1 activity that greatly enhances T cell persistence and hence T cell memory without inhibiting effector function (6). Thus, our data support the development of mTORC2-specific inhibitors in order to enhance immunologic processes requiring the generation of long-lived memory cells, such as in optimizing the efficacy of vaccines. In this way, we envision that unbridled mTORC1 activity can promote CAD activation and enhance the speed and robustness of the recall response, with inhibition of mTORC2 leading to enhanced FOXO1 activity and improved memory generation.
Given that CAD promotes de novo pyrimidine synthesis, we initially hypothesized that inhibition of CAD activity would lead to inhibition of cytokine production upon rechallenge at the transcriptional level. To our surprise, inhibition of CAD prior to rechallenge led to decreased cytokine protein without inhibiting mRNA levels. Pursuant to these findings, we consequently identified a role of CAD in promoting rRNA synthesis. Ribosomal biogenesis is among the most energy-intensive cellular processes in eukaryotes, requiring dozens of ribosomal subunit proteins, over 200 different assembly factors and the synthetic activity of all three RNA polymerases to assemble functional ribosomes and export them from the nucleolus (29). RNA polymerase I, whose sole transcriptional product is the polycistronic pre-rRNA molecule, accounts for over half of all cellular transcription (30). The synthesis of ribosomal RNA is the rate-limiting step in ribosomal biogenesis and ribosomal proteins are proteolytically degraded in the absence of appropriate ribosomal RNA molecules (36). Indeed, our studies suggest that the rapid and robust response of memory T cells is facilitated in part by the continuous generation of ribosomes, such that upon rechallenge, these cells are poised to rapidly respond. As such, our data identifies a new line of inquiry for studying defective T cell responses, such as T cell exhaustion. Overexpression of CAD and increased flux into de novo pyrimidine synthesis facilitated expansion of previously-activated antigen-specific CD8+ T cells in our murine models and we propose that a continuation of pyrimidine synthesis is a defining property of memory cells.
As cellular therapies with more memory-like transferred cell populations survive longer and control tumors to a greater degree than effector-like populations (31,32), increasing CAD activity could have implications in adoptive cell therapy (ACT) or CAR T-cell therapy in clinical settings for treatment of malignancy. Alternatively, inhibition of ribosomal biogenesis could be utilized in the treatment of autoimmune conditions in which the response of memory T cells is known to contribute heavily to pathology, such as multiple sclerosis (33). Identification of pre-rRNA synthesis and its relation to ribosomal biogenesis as a potential regulator of T cell memory function therefore invites opportunities to modulate this process in diverse therapeutic settings.
Materials and Methods
Study Design
These experiments began as an investigation of CAD phosphorylation downstream of T cell activation-induced mTORC1 activity. The observation of persistent CAD phosphorylation after activation then led us to explore a role in memory. We utilized flow cytometry, western blot analysis, and ELISA to demonstrate a functional role for CAD activity in T cell responses. The hypothesis regarding utilization of nucleotides for rRNA was generated later after qPCR data revealed no difference in cytokine transcript levels under our experimental nucleotide restriction conditions. This approach necessitated the development and use of a pre-rRNA-targeted PRIME Flow® assay. Age-matched mice were randomized to each experimental condition for all murine experiments. Unless otherwise stated in figure legends, laboratory experiments were conducted at least three separate times with representative data and/or summary data shown.
Mice
Male or female mice between six and nine weeks of age with appropriate age- and sex-matching were used for each experiment. Following matching, mice were randomized to all described experimental conditions. All mouse experiments and procedures were approved by the Johns Hopkins University Institutional Animal Care and Use Committee and conducted under protocol #MO16M103. C57BL/6J, Cd4-Cre, Rptor-loxP, OT-I, and Cd90.1 mice were obtained from The Jackson Laboratory. P14 TCR transgenic mice were kindly provided by Dr. David A. Hildeman (Cincinnati Children’s Medical Center). Rictor-loxP mice were kindly provided from Dr. Mark Magnuson (Vanderbilt University Medical Center).
Drugs
Rapamycin was purchased from LC Laboratories. PALA (N-phosphonacetyl-L-aspartate) was provided courtesy of the Developmental Therapeutics Program (DTP) at the National Cancer Institute. BMH-21 was provided courtesy of Dr. Marikki Laiho (Johns Hopkins University).
Antibodies and Reagents
Primary antibodies included CD8α (53–6.7), CD44 (IM7), IL-2 (JES6-5H4), TNF-α (MP6-XT22), IFN-γ (XMG1.2), CD45RO (UCHL1), CD45RA (HI100), CD3 (UCHT1), IFN-γ (B27), and TNF-α ((RUO(GMP)) from BD Biosciences, CD62L (MEL-14) from BioLegend, anti-CAD (11933S), anti pCAD (S1859, 12662S), anti-S6 (2217), anti-pS6 (S240/244, D68F8), anti S6K (9202), anti-pS6K (T389, 9205S), anti-RPL5 (14568S), and anti-β-Actin (D6A8, 8457) from Cell Signaling, Fixable Viability Dye eFluor 780 (65-0865-14) from eBioscience, anti-HA Alexa Fluor 488 conjugated (F-7, sc-7392) from Santa Cruz Biotechnology, and anti-RPL22 (PA5-97192) from ThermoFisher. Secondary antibodies included anti-rabbit HRP (7074) from Cell Signaling and Alexa Fluor 647 donkey anti-rabbit (A-31571) from ThermoFisher. Ficoll Paque (17-1440-02) was purchased from GE Healthcare. IFNγ and TNFα capture and detection antibodies, Avidin HRP (18-4100-51) and TMB Substrate (N301) were purchased from ThermoFisher. PRIME Flow® #CPF-204 Assay ID VP47VTP “5 Prime ETS” was purchased from ThermoFisher. Stimulatory anti-CD3 (2C11) and anti-CD28 (37.51) were purified in-house from hybridoma supernatants.
Flow Cytometry
Flow cytometry experiments were performed using a FACS Celesta (BD Biosciences) and analyzed using FlowJo software (v.10.3). All samples from all experiments are gated on viable cells utilizing Fixable Viability Dye eFluor 780 (65-0865-14, eBioscience). Intracellular staining for cytokines, pCAD, and pS6 was performed with BD Fixation/Permeabilization solution (BD Biosciences, 554714). For PRIME Flow® studies, samples were prepared in accordance with the provided protocol (ThermoFisher). Data are reported as mean fluorescence intensity and displayed as a percentage of events recorded relative to the mode values for each data set collected.
qPCR
Total RNA from cells was extracted with TRIzol Reagent (Life Technologies, 15596018). RNA was converted to cDNA using the ProtoScript II Reverse Transcriptase (New England BioLabs, M0368). qRT-PCR was performed with PowerSYBR® Green PCR Master Mix (ThermoFisher). 5’ ETS pre-rRNA, Ifng, and Actb primers were ordered from IDT:
5’ ETS: F 5’-TTTTGGGGAGGTGGAGAGTC-3’ | R 5’-AGAGAACTCCGGAGCACCAC-3’. Ifng: F 5’-CGGCACAGTCATTGAAAGCCTA-3’ | R 5’-GTTGCTGATGGCCTGATTGTC-3’. Actb: F 5’-CATTGCTGACAGGATGCAGAAGG-3’ | R 5’- TGCTGGAAGGTGGACAGTGAGG-3’. Reactions were run using an Applied Biosystems StepOnePlus 96-well Real-Time PCR. ΔΔCt values were normalized to β-actin and a control group of interest.
Cell Cultures
T cells were stimulated with soluble anti-CD3 (5 μg/ml) and anti-CD28 (2 μg/ml) for 48 hours, followed by a 10-fold media expansion with IL-2 (1 ng/ml) or IL-7 (10 ng/ml) and IL-15 (20 ng/ml) for an additional 4 days. For viral transduction with MigR1 vectors, cells were spinfected with 5μg/ml polybrene and viral constructs at 2500 rpm for 90 minutes at 24 hours post-activation. Media was changed 24 hours post-transduction to remove polybrene. At the end of the culture, live cells were separated by Ficoll density gradient and then restimulated by incubation with irradiated APCs from WT mice that had been pre-loaded with indicated concentrations of SIINFEKL peptide for 5 hours with subsequent harvesting of supernatant or by PMA (10 ng/ml) and ionomycin (1 μg/ml) in the presence of GolgiPlug (BD Biosciences) for indicated time periods.
ELISA
Supernatants were obtained as described in the Cell Cultures section and incubated overnight in pre-coated 96-well plates with IFN gamma or TNF alpha capture antibodies described in the Antibodies and Reagents section. Plates were then washed and incubated with IFN gamma and TNF alpha detection antibodies for 2 hours at concentrations recommended by the manufacturer. Plates were then washed and incubated with Avidin HRP substrate for 1 hour. Plates were then washed and incubated with TMB Substrate for 15 minutes before addition of 1 M phosphoric acid as stop solution. Plates were read on a SpectraMax M3 Series Multi-mode Microplate Reader.
Human T cell experiments
Frozen human PBMCs were thawed and plated at 5×106/ml and activated with 1ug/ml each of anti-CD3/28 antibodies. Twenty-four hours into culture, 1×106 million cells were suspended in 30μl of EP buffer (Lonza) with 3μg of either pcDNA3-EV or pcDNA3-CAD (provided by Dr. Brendan Manning, Harvard University). Cells were nucleofected with Lonza Amaxa 4-D Nucleofactor per protocol and replated at 1×106/ml in fresh media containing IL-2 for an additional four days before analysis. CD45RO+ humam memory CD8+ T cells were isolated with subsequent CD8 and CD45RO MicroBeads (Miltenyi) before analysis.
Cad Overexpression Experiments
The CAD-MigR1 retroviral construct was prepared by digesting 2 μg MigR1 plasmid with EcoRI and XhoI (NEB) at 37 degrees for one hour. HA-FLAG-CAD (provided by Dr. Brendan Manning, Harvard University) was digested with amplified with primers to introduce recognition sites for the EcoRI and XhoI cuts made in MigR1. The resulting product was transformed into DH5α (ThermoFisher), allowed to grow overnight at 37 °C, then isolated using a QIAprep Spin Miniprep Kit (Qiagen, 27104). The isolated products were then sequenced to ensure proper addition of the CAD gene sequence to the MigR1 backbone. For isolation of virus, Phoenix-ECO cells (ATCC, CRL#3214) were plated on 6 well plates at 1.2×106 cells per well and allowed to grow in antibiotic-free DMEM for 24 hours before exposure to plasmid and Lipofectamine® 3000 per protocol (ThermoFisher). Media was changed to DMEM with antibiotics 24 hours after transfection with resulting supernatant harvested and filtered through a 0.45-micron filter and incubated with Retro-X® Concentrator (Takara, 631456) for viral isolation. The viral products were then introduced to T cells 24 hours post-activation (see Cell Cultures section) by spinfection at 2500 rpm for 2 hours at 37 °C in the presence of 5 μg/ml polybrene. Media was changed to remove polybrene 24 hours after spinfection.
Cad Knockdown Experiments
Naïve CD8+ T cells were stimulated with soluble anti-CD3 (5 μg/ml) and anti-CD28 (2 μg/ml) for 48 hours before introduction of 1.6 μg empty vector or Cad-targeted shRNA (TRCN0000032550, Sigma). Cells were cultured for an additional 24 hours, then washed and plated in fresh media for an additional 3 days of culture before analysis.
Western Blotting
Cell were lysed at indicated time points in whole cell lysis buffer (RIPA). Protein content of lysates was quantified by Bradford assay using a SpectraMax M3 Series Multi-mode Microplate Reader (Molecular Devices). Lysates were run on NuPAGE 4–12% Bis-Tris Gels (ThermoFisher) and transferred to PVDF membranes (Biorad). Membranes were incubated with indicated primary antibodies overnight in 4% BSA in TBST, washed in TBST 0.1% Tween, and incubated with anti-rabbit HRP-linked secondary antibody for 2 hours (Cell Signaling Technologies). Membranes were washed in TBST 0.1% Tween and developed with SuperSignal West Pico PLUS Chemiluminescent Substrate (Thermo Fisher Scientific, 34578). The images were captured using an UVP BioSpectrum 500 Imaging System and analyzed with the accompanying VisionWorks Pro software.
Infectious Models
In adoptive transfer (AT) experiments with naïve cells, C57BL/6J Thy1.2+ host mice were injected intraperitoneally (i.p.) with 106 PFU Vaccinia-OVA (Vac-OVA) or 106 PFU LCMV-Armstrong and retro-orbitally administered 5×105 naïve CD8+Thy1.1+ OT-I or P14 T cells, respectively. Detection of adoptively transferred cells based on CD8+Thy1.1+ staining. For AT experiments with previously-activated cells, C57BL/6J Thy1.2+ host mice were injected i.p. with 106 PFU Vac-OVA or 106 PFU LCMV-Armstrong and administered 5×105 CD8+Thy1.1+ OT-I or P14 T cells cultured as described in the Cell Cultures section. These cells were expanded with the indicated cytokines and either pre-treated with drug, exposed to shRNA, or transfected with MigR1 retroviral construct per respective sections of the Methods section.
Metabolic Flux Tracing
Previously Activated cells were generated by stimulating isolated CD8+ cells from naïve C57BL/6J mice with anti-CD3/28 antibodies for 48 hours before expanding media with IL-7 and IL-15, with 1 mM PALA added for the final 24 hours of culture in the Previously Activated + PALA condition. Culture contents were then subjected to a Ficoll gradient with additional naïve WT CD8+ cells isolated on Day 6 of the experiment. Targeted metabolite analysis was performed with liquid-chromatography tandem mass spectrometry (LC-MS/MS). Metabolites from cells were extracted with 80% (v/v) methanol solution equilibrated at −80 °C, and the metabolite-containing supernatants were dried under nitrogen gas. Dried samples were re-suspended in 50% (v/v) acetonitrile solution and 4 ml of each sample were injected and analyzed on a 5500 QTRAP mass spectrometer (AB Sciex) coupled to a Prominence ultra-fast liquid chromatography (UFLC) system (Shimadzu). The instrument was operated in selected reaction monitoring (SRM) with positive and negative ion-switching mode as described. The optimized MS parameters were: ESI voltage was +5,000V in positive ion mode and −4,500V in negative ion mode; dwell time was 3ms per SRM transition and the total cycle time was 1.57 seconds. Hydrophilic interaction chromatography (HILIC) separations were performed on a Shimadzu UFLC system using an amide column (Waters XBridge BEH Amide, 2.1 × 150 mm, 2.5 μm). The LC parameters were as follows: column temperature, 40 °C; flow rate, 0.30 ml/min. Solvent A, Water with 0.1% formic acid; Solvent B, Acetonitrile with 0.1% formic acid; A non-linear gradient from 99% B to 45% B in 25 minutes with 5min of post-run time. Peak integration for each targeted metabolite in SRM transition was processed with MultiQuant software (v2.1, AB Sciex). The preprocessed data with integrated peak areas were exported from MultiQuant and re-imported into Metaboanalyst software for further data analysis (e.g. statistical analysis, fold change, etc.).
Statistical analysis
All statistics were completed using GraphPad Prism Version 7 software. Statistical tests were used for comparison of data points and generation of P values as described at the end of each figure legend. For comparison of two conditions, paired and unpaired two-tailed t tests were utilized. For comparison of three or more groups with one experimental variable, one-way ANOVA with Tukey’s HSD was utilized. For comparison of three or more groups with two experimental variables, two-way ANOVA with Tukey’s HSD was utilized. Unless otherwise stated in the corresponding figure legend, all data is presented as mean ±SD. Significance was defined as *P <0.05, **P<0.01, ***P<0.001, ****P<0.0001.
Supplementary Material
Acknowledgements:
We would like to acknowledge Dr. Brendan Manning of the Harvard T.H. Chan School of Public Health for providing the wild-type CAD plasmid. We thank Im-Hong Sun and Chirag Patel of the Bloomberg Kimmel Institute for Cancer Immunotherapy at the Johns Hopkins University School of Medicine for consulting about experimental design and review of the manuscript. Diagrams were licensed and created with BioRender.com.
Funding:
We acknowledge our funding sources including NIH Grant #5R01AI077610-08 (J.D.P.) and NIGMS R01GM121404, NCI P30 CA006973 to M.L.
Footnotes
Competing Interests: The authors declare that they have no competing interests.
Data and materials availability:
All data needed to support the conclusions of the paper are present in the paper or the Supplementary Materials.
References and Notes
- 1.Barber DL, Wherry EJ, Ahmed R. Cutting edge: rapid in vivo killing by memory CD8 T cells. J Immunol. 2003. Jul 1;171(1):27–31. [DOI] [PubMed] [Google Scholar]
- 2.Lalvani A, Brookes R, Hambleton S, Britton WJ, Hill AV, McMichael AJ. Rapid effector function in CD8+ memory T cells. J Exp Med. 1997. Sep 15;186(6):859–65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.O’Sullivan D The metabolic spectrum of memory T cells. Immunol Cell Biol. 2019. Aug;97(7):636–646. [DOI] [PubMed] [Google Scholar]
- 4.Powell JD, Pollizzi KN, Heikamp EB & Horton MR. Regulation of Immune Responses by mTOR. Annu Rev Immunol. 2012;30:39–68. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Pollizzi KN, Powell JD. Regulation of T cells by mTOR: The known knowns and the known unknowns. Trends Immunol. 2015;36(1):13–20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Pollizzi KN, Patel CH, Sun IH, Oh MH, Waickman AT, Wen J, Delgoffe GM, Powell JD. mTORC1 and mTORC2 selectively regulate CD8+ T cell differentiation. J Clin Invest. 2015. May;125(5):2090–108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Pollizzi K, Sun IH, Patel C et al. Asymmetric inheritance of mTORC1 kinase activity during division dictates CD8+ T cell differentiation. Nat Immunol 17, 704–711 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Graves LM, Guy HI, Kozlowski P, Huang M, Lazarowski E, Pope RM, Collins MA, Dahlstrand EN, Earp HS 3rd, Evans DR. Regulation of carbamoyl phosphate synthetase by MAP kinase. Nature. 2000. Jan 20;403(6767):328–32. [DOI] [PubMed] [Google Scholar]
- 9.Rao RR, Li Q, Odunsi K & Shrikant PA. The mTOR kinase determines effector versus memory CD8+ T cell fate by regulating the expression of transcription factors T-bet and Eomesodermin. Immunity. 2010. Jan 29;32(1):67–78. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Ben-Sahra I, Howell JJ, Asara JM, Manning BD. Stimulation of de novo pyrimidine synthesis by growth signaling through mTOR and S6K1. Science. 2013. Mar 15;339(6125):1323–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Carrio R, Bathe OF, Malek TR. Initial antigen encounter programs CD8+ T cells competent to develop into memory cells that are activated in an antigen-free, IL-7- and IL-15-rich environment. J Immunol. 2004. Jun 15;172(12):7315–23. [DOI] [PubMed] [Google Scholar]
- 12.Araki K, Turner AP, Shaffer VO, Gangappa S, Keller SA, Bachmann MF, Larsen CP, Ahmed R. mTOR regulates memory CD8 T-cell differentiation. Nature. 2009. Jul 2;460(7251):108–12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Araki K, Youngblood B, Ahmed R. The role of mTOR in memory CD8 T-cell differentiation. Immunol Rev. 2010. May;235(1):234–43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Fairbanks LD, Bofill M, Ruckemann K, and Simmonds HA. Importance of ribonucleotide availability to proliferating T-lymphocytes from healthy humans: disproportionate expansion of pyrimidine pools and contrasting effects of de novo synthesis inhibitors. J. Biol. Chem 1995. 270:29682.20. [PubMed] [Google Scholar]
- 15.Ruckemann K, Fairbanks, Carrey E, Hawrylowicz C, Richards DF, Kirschbaum B, and Simmonds HA. Leflunomide inhibits pyrimidine de novo synthesis in mitogen-stimulated T-lymphocytes from healthy humans. J. Biol. Chem 1998. 273:21682. [DOI] [PubMed] [Google Scholar]
- 16.Cooper HL. Ribosomal ribonucleic acid wastage in resting and growing lymphocytes. J Biol Chem. 1969. Oct 25;244(20):5590–6. [PubMed] [Google Scholar]
- 17.Dimitrova P, Skapenko A, Herrmann ML, Schleyerbach R, Kalden JR, Schulze-Koops H. Restriction of de novo pyrimidine biosynthesis inhibits Th1 cell activation and promotes Th2 cell differentiation. J Immunol. 2002. Sep 15;169(6):3392–9. [DOI] [PubMed] [Google Scholar]
- 18.Collins KD, Stark GR. Aspartate transcarbamylase. Interaction with the transition state analogue N-(phosphonacetyl)-L-aspartate. J Biol Chem. 1971. Nov;246(21):6599–605. [PubMed] [Google Scholar]
- 19.Pihlgren M1, Dubois PM, Tomkowiak M, Sjögren T, Marvel J. Resting memory CD8+ T cells are hyperreactive to antigenic challenge in vitro. J Exp Med. 1996. Dec 1;184(6):2141–51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Huang F, Ni M, Chalishazar MD, Huffman KE, Kim J, Cai L, Shi X, Cai F, Zacharias LG, Ireland AS, Li K, Gu W, Kaushik AK, Liu X, Gazdar AF, Oliver TG, Minna JD, Hu Z, DeBerardinis RJ. Inosine Monophosphate Dehydrogenase Dependence in a Subset of Small Cell Lung Cancers. Cell Metab. 2018. Sep 4;28(3):369–382.e5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Lazdins IB1, Delannoy M, Sollner-Webb B. Analysis of nucleolar transcription and processing domains and pre-rRNA movements by in situ hybridization. Chromosoma. 1997. Jun;105(7–8):481–95. [DOI] [PubMed] [Google Scholar]
- 22.Mullineux ST1, Lafontaine DL. Mapping the cleavage sites on mammalian pre-rRNAs: where do we stand? Biochimie. 2012. Jul;94(7):1521–32. [DOI] [PubMed] [Google Scholar]
- 23.Angelini G, Flego D, Vinci R, Pedicino D, Trotta F, Ruggio A, Piemontese GP, Galante D, Ponzo M, Biasucci LM, Liuzzo G, Crea F. Matrix metalloproteinase-9 might affect adaptive immunity in non-ST segment elevation acute coronary syndromes by increasing CD31 cleavage on CD4+ T-cells. Eur Heart J. 2018. Apr 1;39(13):1089–1097. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Gustafson CE, Cavanagh MM, Jin J, Weyand CM, Goronzy JJ. Functional pathways regulated by microRNA networks in CD8 T-cell aging. Aging Cell. 2019. Feb;18(1):e12879. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Cho BK, Wang C, Sugawa S, Eisen HN, Chen J. Functional differences between memory and naive CD8 T cells. Proc Natl Acad Sci USA. 1999;96(6):2976–2981. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Yoon H, Kim TS, Braciale TJ. The cell cycle time of CD8+ T cells responding in vivo is controlled by the type of antigenic stimulus. PLoS One. 2010. Nov 8;5(11):e15423. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Asmal M, Colgan J, Naef F, Yu B, Lee Y, Magnasco M, Luban J. Production of ribosome components in effector CD4+ T cells is accelerated by TCR stimulation and coordinated by ERK-MAPK. Immunity. 2003. Oct;19(4):535–48. [DOI] [PubMed] [Google Scholar]
- 28.Best JA, Blair DA, Knell J, Yang E, Mayya V, Doedens A, Dustin ML, Goldrath AW. Immunological Genome Project Consortium. Transcriptional insights into the CD8(+) T cell response to infection and memory T cell formation. Nat Immunol. 2013. Apr;14(4):404–12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Awad D, Prattes M, Kofler L, et al. Inhibiting eukaryotic ribosome biogenesis. BMC Biol. 2019;17(1):46. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Russell J, Zomerdijk J. The RNA polymerase I transcription machinery. Biochemical Society Symposium. 2006. 73 (73): 203–16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Maus MV & June CH. Making Better Chimeric Antigen Receptors for Adoptive T-cell Therapy. Clin Cancer Res. 2016. Apr 15;22(8):1875–84. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Baitsch L et al. The three main stumbling blocks for anticancer T cells. Trends Immunol. 2012. Jul;33(7):364–72. [DOI] [PubMed] [Google Scholar]
- 33.Kivisäkk P, Mahad DJ, Callahan MK, Sikora K, Trebst C, Tucky B, Wujek J, Ravid R, Staugaitis SM, Lassmann H, Ransohoff RM. Expression of CCR7 in multiple sclerosis: implications for CNS immunity. Ann Neurol. 2004. May;55(5):627–38. [DOI] [PubMed] [Google Scholar]
- 34.Colis L, Peltonen K, Sirajuddin P, Liu H, Sanders S, Ernst G, Barrow JC, Laiho M. DNA intercalator BMH-21 inhibits RNA polymerase I independent of DNA damage response. Oncotarget. 2014. Jun 30;5(12):4361–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Peltonen K, Colis L, Liu H, Trivedi R, Moubarek MS, Moore HM, Bai B, Rudek MA, Bieberich CJ, Laiho M. A targeting modality for destruction of RNA polymerase I that possesses anticancer activity. Cancer Cell. 2014. Jan 13;25(1):77–90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Lam Y, Lamond A, Mann M, Andersen J. Analysis of Nucleolar Protein Dynamics Reveals the Nuclear Degradation of Ribosomal Proteins. Curr Biol. 2007. May 1; 17(9): 749–760. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Meek SE, Lane WS, Piwnica-Worms H. Comprehensive proteomic analysis of interphase and mitotic 14-3-3-binding proteins. J Biol Chem. 2004. Jul 30;279(31):32046–54. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
All data needed to support the conclusions of the paper are present in the paper or the Supplementary Materials.