

Abstract
Purpose of review
Chronic kidney disease (CKD) is a progressive disorder that is associated with development of elevated fibroblast growth factor 23 (FGF23) levels and anemia. Here, we review recent literature that extends our current knowledge on the interactions between FGF23 and anemia in CKD and the impact of anemia-targeting therapeutics on FGF23 elevation in CKD.
Recent findings
The anemia of CKD is primarily driven by a lack of erythropoietin (EPO) and iron deficiency. In addition to EPO and iron replacement, novel drug classes to treat anemia have been approved or are in clinical development. A recent observational study provides supportive evidence for the hypothesis that FGF23 elevation in CKD mediates adverse effects of iron deficiency on the cardiovascular system in patients with CKD. Preclinical and clinical studies revealed that ferric citrate (FC), and hypoxia induced factor-prolyl hydroxylase inhibitor (HIF-PHI) treatment may reduce elevated FGF23 levels in CKD, suggesting that correcting anemia in CKD could potentially lower FGF23 levels. However, as we describe, HIF-PHI have context-dependent effects. Moreover, whether a reduction in FGF23 will improve patient outcomes in patients with CKD remains to be determined.
Summary
With the emergence of novel therapeutics to treat oxygen and iron utilization deficits in CKD, studies have investigated the impact of these new drugs on FGF23. Several of these drugs, including FC and HIF-PHIs, alleviate iron homeostasis alterations in CKD and are associated with FGF23 reduction. Herein, we review the relationships between oxygen/iron sensing and FGF23 in CKD, recent findings which link FGF23 with cardiac dysfunction, as well as future translational and clinical avenues.
Keywords: FGF23, anemia, CKD, Iron, EPO
Introduction
Chronic kidney disease (CKD) is a global health concern, affecting between 7-12% of the world’s population [1, 2], and key pathology-driven mechanisms in CKD are the decrease of oxygen supply and iron delivery [3]. Previous clinical studies in humans [4] and translational experiments in mice [5], as well as in vitro studies on isolated osteoblast/osteocyte cells [6**], have shown that FGF23 is induced by anemia/hypoxia [5, 7], thus overlapping mechanisms of phosphate and iron handling exist. The anemia-targeting drug erythropoietin (EPO) is an FGF23 inducer, as transgenic mice overexpressing EPO, as well as humans and mice injected with EPO, showed elevated FGF23 [8, 9]. As will be discussed, these effects are context-dependent as the iron status of pre-clinical models likely affects responses to iron- and oxygen-controlling therapeutics. Poor iron utilization has been associated with poor clinical outcomes [10], and recent studies suggest that adverse effects of iron deficiency on the cardiovascular system in patients with CKD may be mediated by elevated FGF23 [11**]. Finally, increased circulating FGF23 is an independent risk factor for mortality in patients undergoing dialysis [12]. Emerging data suggest that therapies that improve iron handling and oxygenation in CKD may also regulate FGF23. Whether by reducing FGF23, these therapies lead to beneficial changes in patient outcomes should be further explored.
Regulation of FGF23 by anemia in rare diseases and in CKD
FGF23 is a hormone produced principally in bone osteoblasts/osteocytes, and controlled by multiple systemic factors, including elevated phosphate and 1,25(OH)2 vitamin D (1,25D) [13]. Circulating FGF23 binds to an FGF receptor (FGFR) and its co-receptor alpha-Klotho (KL), which is primarily expressed in the kidney and parathyroid glands. In renal cells, FGF23 acts as a counter-regulatory hormone to elevated 1,25D via suppressing the vitamin D 1α-hydroxylase (Cyp27b1), an anabolic enzyme for 1,25D, and in parallel increasing expression of the vitamin D 24-hydroxylase (Cyp24a1), a catabolic enzyme [13]. These parallel actions decrease 1,25-mediated intestinal phosphate absorption. FGF23 is also critical for coordinating renal phosphate handling by inhibiting kidney phosphate reabsorption via post-translational control of proximal tubule sodium phosphate cotransporters NPT2a and NPT2c [14–17]. During CKD, FGF23 gradually increases, and the mechanisms for this increase are under active investigation, including the idea that end-organ resistance due to reduction of kidney KL expression could lead to compensatory FGF23 increases [18]. At early CKD stages, FGF23 can be a sensitive marker of disease, and during later stages, FGF23 can reach markedly elevated concentrations in the ng/mL ranges (typically pg/mL) [19]. During studies defining the underlying pathogenic mechanisms of autosomal dominant hypophosphatemic rickets (ADHR) [5], an anemia-FGF23 relationship was discovered through the study of ADHR mice expressing a human activating point mutation in the Fgf23 gene which inhibits intracellular endoplasmic reticulum-trans golgi network (ER-TGN) proteolytic processing [5]. In ADHR mice fed a low iron diet to induce iron deficiency anemia (IDA), hypophosphatemia was observed [5]. It was found that the IDA was associated with significantly increased bone Fgf23 mRNA [5], resulting in elevated serum intact FGF23. Of note, recent clinical trials involving ADHR patients demonstrated that low dose iron administration reduced FGF23 and re-balanced phosphate handling [20**], galvanizing the connection between anemia and FGF23.
During CKD, patients lose the ability to generate erythropoietin (EPO). EPO is necessary for red blood cell production, and thus proper tissue oxygen and iron delivery [21, 22]. In addition to loss of EPO, CKD patients develop anemia due to EPO resistance, inadequate diet and compromised iron absorption, as well as bleeding and chronic inflammation [23]. The prevalence of anemia is raised with the progression of CKD: 8.4% at stage 1 to 53.4% by stage 5 [24, 25]. FGF23 also progressively increases as glomerular filtration rate falls, with the highest levels observed among individuals with end-stage renal disease (ESRD) [26]. In patients with ESKD, FGF23 is primarily produced as the intact form [27], thus the anemia of CKD may be a strong causative factor of chronically elevated bioactive FGF23, leading to downstream pathogenic manifestations.
Association of iron deficiency and FGF23 with cardiovascular outcomes
Cardiovascular disease is a severe and multifactorial manifestation of CKD. Elevated FGF23 has been directly linked to the pathogenesis of left ventricular hypertrophy (LVH) [28]. Evidence supports that these actions are KL-independent in cardiac tissue and likely occur through FGF23-FGFR4 off target binding. Indeed, conditional deletion of FGFR4 in cardiomyocytes using flox-FGFR4 mice offered protective mechanisms against FGF23-mediated LVH phenotypes [29]. Until recently, however, there has been a gap in our knowledge on the intersection of iron deficiency and FGF23 and risk of adverse clinical outcomes in patients with CKD. Using the Chronic Renal Insufficiency Cohort (CRIC) Study, Mehta et al assigned patients with CKD into subgroups based on iron parameters. In multivariable-adjusted models, iron deficiency independently associated with mortality and heart failure. In adjusted analysis, plasma FGF23 concentrations were inversely associated with iron stores, (highest in those with iron deficiency and lowest in high iron group), with intermediate iron groups having mid-range FGF23 concentrations. When FGF23 and the iron exposure groups were included together, the significant effects of FGF23 were unchanged, but the increased risks of mortality and heart failure in the iron deficiency group were fully mitigated (mortality: from 1.28 [95% CI] to 0.97 [95% CI]; heart failure: from 1.34 [95% CI] to 0.98 [95% CI]) [11**]. Mediation analyses showed that the effects of iron deficiency on mortality and heart failure were accounted by FGF23. Similar analyses of PTH and hemoglobin demonstrated that PTH did not influence outcomes for any group, whereas hemoglobin concentrations (reduced in CKD) were associated to a lesser degree than FGF23 with mortality and heart failure in iron deficiency. The findings from this study suggest that by reducing FGF23, iron repletion therapy in CKD may potentially improve clinical outcomes in patients with CKD. However, this hypothesis will need to be tested in future studies.
Ferric citrate and the effects on circulating FGF23 in clinical trials and pre-clinical models
Ferric citrate (FC) is an iron-based phosphate binder [30] designed to treat multiple CKD manifestations, including reducing serum phosphate concentrations through inhibiting phosphate absorption, and increasing iron in the bloodstream and tissues [30, 31]. FC is currently FDA approved in the United States: 1) in patients on dialysis, FC is used as a phosphate binder; and 2) to treat IDA in CKD patients who are not undergoing dialysis [30]. The proposed mechanism for FC’s effects on iron replacement is due to its iron-containing backbone. To this end, unbound FC (in the ferrous state) is absorbed in the small intestine, making iron bioavailable for erythropoiesis [30]. Translational and clinical studies [32–34] have associated decreases in plasma FGF23 with FC treatment [35]. In this regard, 5% dietary FC was administered to an Alport’s syndrome mouse model that develops CKD [35]. The FC treatment was associated with reductions in FGF23 and serum phosphate, as well as increased serum iron [35]. Interestingly, early FC intervention in the mice with CKD also reduced renal fibrosis and proteinuria, improved kidney function, and prolonged life span. It will be important in translational models to continue test these effects using a dosing regimen closer to human CKD patients, however this treatment improved kidney fibrosis and ameliorated adverse cardiac outcomes, providing a strong rationale for longer-term clinical trials.
In a short-term clinical trial, Block and colleagues [36] compared iron levels in patients receiving FC to those on placebo over a 12-week period. After the treatment period, patients provided with FC had an increase in ferritin and TSAT whereas in patients that received the placebo these values remained unchanged [36]. The placebo treatment group showed reduced hemoglobin whereas the FC-treated group’s hemoglobin levels increased. The study authors noted that there was a larger increase in the FC group’s hemoglobin levels than observed with other oral iron preparations [36]. During this study, patient’s FGF23 levels were also compared. Whereas both groups showed decreased FGF23 during the time course, in the FC-treated group FGF23 concentrations were reduced significantly more [36]. In a clinical trial performed by the same investigators, FGF23 levels were compared between a standard of care group (control) and an FC treatment group. By the end of the observational time point (9 months), the FGF23 levels in the control group increased over the study time course, however the FC group had stable FGF23 levels and with a significantly lower FGF23 when compared to controls at the endpoint [37]. Both IDA and high levels of phosphate are drivers of FGF23, thus it is possible that correcting these manifestations were the source of FC’s suppressive effect on FGF23 [36]. It has been established that high FGF23 was associated with increased all-cause mortality and poor cardiovascular outcomes [38], thus a reduction in FGF23 may benefit patients. The elevation of FGF23 in CKD patients and pre-clinical models treated with ferric citrate is however still markedly above normal ranges. Therefore, long-term clinical trials which could assess the effects of ferric citrate therapeutics on improving iron bioavailability associated with reduced circulating FGF23 are needed. Further, work in mice found that an isolated C-terminal tail of FGF23 [39] improved tissue iron mobilization to the circulation in CKD [40, 41]. Therefore, translational studies could consider combining FC treatments with other FGF23-related therapy designs.
A marked reduction of bioactive FGF23 using an anti-FGF23 antibody in rats [42] as well as deletion of FGF23 in mice leads to hyperphosphatemia and early death [43, 44]. Therefore, any treatment regimen should consider improvement of oxygen/iron utilization in CKD with a balance of maintaining FGF23 concentrations high enough that further hyperphosphatemia does not develop. Finally, emerging mechanistic data supports that therapeutic preparations that increase systemic oxygen delivery and improve iron handling may directly influence HIF1α activity and FGF23 synthesis in osteocytes (Fig. 1).
Figure 1.
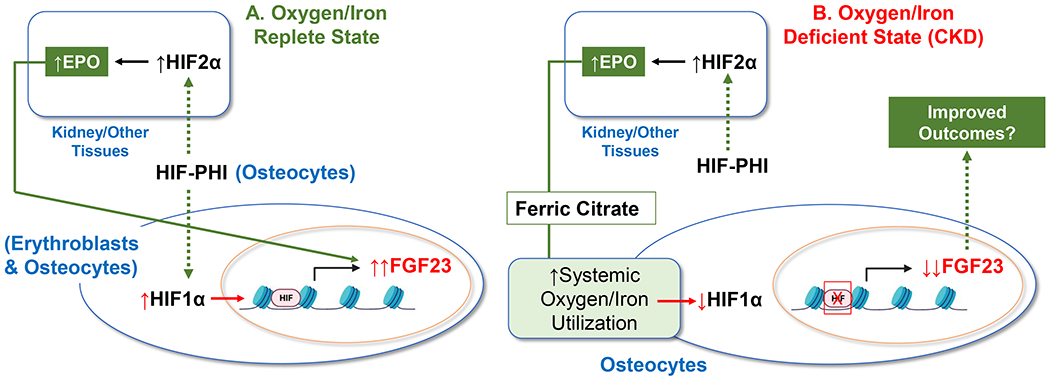
A. In a normal state of oxygen/iron utilization, as a molecular tool to mimic hypoxia, HIF-PHIs can activate HIF1α in osteocytes to increase FGF23 expression. HIF-PHI also stimulate HIF2α in kidney and other tissues leading to increased EPO synthesis. In both erythroblasts and osteocytes, EPO can stimulate FGF23. B. Under states of oxygen/iron deficiency, HIF-PHIs stimulate EPO, and the correction in systemic oxygen/iron utilization may suppress HIF1α activity and thus partially reduce FGF23 production in osteocytes. Similarly, ferric citrate may provide iron to directly act on osteocytes and inhibit FGF23 via a similar mechanism.
The impact of EPO therapeutics on FGF23 in CKD
Therapeutic regimen consisting of administrating EPO to overcome its deficiency in CKD patients have been developed and optimized for decades. Although these treatments are widely used, recent findings raised several concerns regarding the use of high EPO doses because of its association with increased cardiovascular disease risks and mortality [45, 46**]. Clinical and translational evidence supports a direct effect of EPO on FGF23 induction. In humans, healthy mice, or a mouse model of juvenile cystic kidney disease, EPO treatment increased FGF23 production and parallel proteolytic cleavage [9, 47]. Injection of rhEPO primarily increased circulating “total” FGF23 (proteolytic fragments plus intact bioactive hormone) in humans and rodents rather than primarily stimulating the intact, bioactive form of FGF23 [8, 9]. Further, these pharmacological-driven findings were confirmed in genetic mouse models as transgenic mice overexpressing EPO had increased FGF23. However, in a mouse model of “advanced” CKD disease, EPO treatment lowered FGF23 over 50% [48*], suggesting that the impact of EPO on FGF23 synthesis is context-dependent and may rely on the disease severity and health status of the models used (Figure 1). These findings suggest that the correction of systemic iron/oxygen handling during CKD using targeting therapeutics has a stronger influence on suppressing FGF23 than the stimulatory effects of some of these agents in isolation on osteocytic FGF23 synthesis.
In response to EPO, FGF23 is induced in bone and bone marrow [9]. Emerging data suggest that the presence of EPO in the marrow compartment drives FGF23 production and may influence marrow progenitor cells. Ishii et al [49], demonstrated in mice that in response to Granulocyte colony stimulating factor (G-CSF), erythroblasts produce FGF23 potentially through hypoxia. Interestingly, the authors found that deleting Fgf23 from hematopoietic cells blunted Hematopoietic stem and progenitor cell (HSPC) mobilization in response to G-CSF. Using in vitro evidence, it was suggested that FGF23-induced HSPC mobilization was a KL-dependent mechanism which impaired CXCL12/CXCR4 interactions [49]. It is not clear whether FGF23 production by erythroblasts could modulate osteocytic FGF23 production, and the mechanisms linking the activation of FGF23 production through G-CSF treatment remain to be elucidated.
HIF-PHI therapeutics in clinical trials for the anemia of CKD
A new class of orally administrated drugs, Hypoxia-inducible factor–prolyl hydroxylase domain inhibitors (HIF-PHIs) are in late-stage global clinical trials, and approved in some countries for the treatment of CKD anemia. In clinical studies, HIF-PHIs show potent stimulation of erythropoiesis in patients with anemia of non–dialysis-dependent and dialysis-dependent CKD (reviewed in [50]). Mechanistically, under normoxia the prolyl hydroxylase domain dioxygenases (PHDs) utilize oxygen and 2-oxoglutarate as substrates to regulate the stability of the HIF transcription factors by post-translational hydroxylation and subsequent degradation [51, 52]. The binding of HIF-PHIs to PHDs is therapeutically designed to stabilize HIF protein in tissues, particularly HIF2α in kidney and other tissues, to drive endogenous EPO production in addition to improving iron metabolism [53**].
In healthy mice, HIF-PHI treatment increases plasma FGF23 levels [9]. A pretreatment of mice with a neutralizing anti-EPO antibody abrogated FGF23 induction by HIF-PHI, highlighting an EPO-dependent mechanism [54]. In contrast to the increased FGF23 responses observed in healthy mice in response to HIF-PHIs, in mice with CKD the inhibition of HIF-prolyl hydroxylases markedly decreased both total and intact FGF23 [6**, 55**]. Thus, similar to parenteral EPO administration, HIF-PHIs may restore oxygen/iron utilization in models of CKD and thus suppress osteocyte FGF23 production (Figure 1). This is further supported by findings that osteocytes may directly respond to systemic iron changes to control FGF23 transcription via interactions with holo-transferrin [6**]. Thus, similar to iron provision via ferric citrate, the long-term effects of HIF-PHIs should be studied for impact on circulating FGF23 in CKD patients at different stages of disease.
Conclusions
In summary, recently approved and emerging therapeutics could provide benefit for treating the often severe manifestations of the anemia of CKD, which are associated with mortality. The use of these agents in pre-clinical models and as molecular tools in normal mice has provided critical insight into the interactions between oxygen/iron handling and FGF23, and their use in vivo are likely iron-status dependent. Further, human studies have shown that adverse effects of iron deficiency in patients with CKD are mediated by elevated FGF23 levels. Whether some individual or combined therapeutics that might lower FGF23 through their effects on oxygen/iron handling could provide beneficial outcomes to patients with CKD in the long term remains to be studied.
Key points.
CKD-related anemia develops as a consequence of EPO deficiency, iron deficiency and alterations in oxygen/iron utilization, and is a critical driver of excessive FGF23 synthesis in CKD.
Prospective observational studies show that iron deficiency is associated with adverse cardiovascular risks in patients with CKD, and although further study is required these findings suggest that therapies targeting anemia such as ferric citrate (FC) could potentially modulate the adverse outcomes through reducing FGF23.
HIF-prolyl hydroxylase inhibitors (HIF-PHIs) are emerging drugs that lower FGF23 in CKD preclinical models, and the effects of these agents are context dependent; when used as molecular tools in healthy mice to activate HIF transcription factors, HIF-PHIs induce FGF23 synthesis.
Financial support and sponsorship:
The authors would like to acknowledge NIH grants R21-AR059278, R01-DK112958, and R01-HL145528 (KEW); and The David Weaver Professorship (KEW).
Conflicts of interest:
KEW receives royalties for licensing the FGF23 gene to Kyowa Hakko Kirin, Ltd.; received previous funding from Akebia Therapeutics, Inc.; and current funding from Calico Labs. RA has no conflicts of interest.
References and recommended reading
Papers of particular interest, published within the annual period of review, have been highlighted as:
* of special interest
** of outstanding interest
- 1.Romagnani P, Remuzzi G, Glassock R, et al. , Chronic kidney disease. Nat Rev Dis Primers, 2017. 3: p. 17088. [DOI] [PubMed] [Google Scholar]
- 2.Hill NR, Fatoba ST, Oke JL, et al. , Global Prevalence of Chronic Kidney Disease - A Systematic Review and Meta-Analysis. PLoS One, 2016. 11(7): p. e0158765. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Hirakawa Y, Tanaka T, and Nangaku M, Renal Hypoxia in CKD; Pathophysiology and Detecting Methods. Front Physiol, 2017. 8: p. 99. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Wolf M, Koch TA, and Bregman DB, Effects of iron deficiency anemia and its treatment on fibroblast growth factor 23 and phosphate homeostasis in women. J Bone Miner Res, 2013. 28(8): p. 1793–803. [DOI] [PubMed] [Google Scholar]
- 5.Farrow EG, Yu X, Summers LJ, et al. , Iron deficiency drives an autosomal dominant hypophosphatemic rickets (ADHR) phenotype in fibroblast growth factor-23 (Fgf23) knock-in mice. 2011. 108(46): p. E1146–E1155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.**.Noonan ML, Ni P, Agoro R, et al. , The HIF-PHI BAY 85–3934 (Molidustat) Improves Anemia and Is Associated With Reduced Levels of Circulating FGF23 in a CKD Mouse Model. J Bone Miner Res, 2021. 36(6): p. 1117–1130. [DOI] [PMC free article] [PubMed] [Google Scholar]; This paper demonstrated that HIF-PHI reduce FGF23 during modeled CKD in mice and that iron may directly influence FGF23 production at the level of the osteocyte.
- 7.Clinkenbeard EL, Noonan ML, Thomas JC, et al. , Increased FGF23 protects against detrimental cardio-renal consequences during elevated blood phosphate in CKD. JCI Insight, 2019. 4(4). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Daryadel A, Bettoni C, Haider T, et al. , Erythropoietin stimulates fibroblast growth factor 23 (FGF23) in mice and men. Pflugers Arch, 2018. 470(10): p. 1569–1582. [DOI] [PubMed] [Google Scholar]
- 9.Clinkenbeard EL, Hanudel MR, Stayrook KR, et al. , Erythropoietin stimulates murine and human fibroblast growth factor-23, revealing novel roles for bone and bone marrow. Haematologica, 2017. 102(11): p. e427–e430. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Wheeler JA and Clinkenbeard EL, Regulation of Fibroblast Growth Factor 23 by Iron, EPO, and HIF. Current Molecular Biology Reports, 2019. 5(1): p. 8–17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.**.Mehta RC, Cho ME, Cai X, et al. , Iron status, fibroblast growth factor 23 and cardiovascular and kidney outcomes in chronic kidney disease. Kidney Int, 2021. 100(6): p. 1292–1302. [DOI] [PMC free article] [PubMed] [Google Scholar]; This report showed that anemia-driven FGF23 was associated with detrimental cardiac outcomes in patients with CKD.
- 12.Isakova T, Xie H, Yang W, et al. , Fibroblast growth factor 23 and risks of mortality and end-stage renal disease in patients with chronic kidney disease. JAMA, 2011. 305(23): p. 2432–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Agoro R, Ni P, Noonan ML, et al. , Osteocytic FGF23 and Its Kidney Function. Front Endocrinol (Lausanne), 2020. 11: p. 592. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Martin A, David V, and Quarles LD, Regulation and Function of the FGF23/Klotho Endocrine Pathways. Physiological Reviews, 2012. 92(1): p. 131–155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Clinkenbeard EL, Cass TA, Ni P, et al. , Conditional Deletion of MurineFgf23: Interruption of the Normal Skeletal Responses to Phosphate Challenge and Rescue of Genetic Hypophosphatemia. Journal of Bone and Mineral Research, 2016. 31(6): p. 1247–1257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Farrow EG, Summers LJ, Schiavi SC, et al. , Altered renal FGF23-mediated activity involving MAPK and Wnt: effects of the Hyp mutation. J Endocrinol, 2010. 207(1): p. 67–75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Larsson T, Marsell R, Schipani E, et al. , Transgenic mice expressing fibroblast growth factor 23 under the control of the alpha1(I) collagen promoter exhibit growth retardation, osteomalacia, and disturbed phosphate homeostasis. Endocrinology, 2004. 145(7): p. 3087–94. [DOI] [PubMed] [Google Scholar]
- 18.Hu MC, Shi M, Zhang J, et al. , Klotho deficiency causes vascular calcification in chronic kidney disease. J Am Soc Nephrol, 2011. 22(1): p. 124–36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Isakova T, Xie H, Barchi-Chung A, et al. , Fibroblast growth factor 23 in patients undergoing peritoneal dialysis. Clin J Am Soc Nephrol, 2011. 6(11): p. 2688–95. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.**.Imel EA, Liu Z, Coffman M, et al. , Oral Iron Replacement Normalizes Fibroblast Growth Factor 23 in Iron-Deficient Patients With Autosomal Dominant Hypophosphatemic Rickets. J Bone Miner Res, 2020. 35(2): p. 231–238. [DOI] [PMC free article] [PubMed] [Google Scholar]; This clinical investigation demonstrated that low dose iron cured ADHR patients by reducing elevated FGF23, solidifying the relationship between anemia and FGF23 production.
- 21.Shih HM, Wu CJ, and Lin SL, Physiology and pathophysiology of renal erythropoietin-producing cells. J Formos Med Assoc, 2018. 117(11): p. 955–963. [DOI] [PubMed] [Google Scholar]
- 22.Olmos G, Munoz-Felix JM, Mora I, et al. , Impaired erythropoietin synthesis in chronic kidney disease is caused by alterations in extracellular matrix composition. J Cell Mol Med, 2018. 22(1): p. 302–314. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Stauffer ME and Fan T, Prevalence of anemia in chronic kidney disease in the United States. PLoS One, 2014. 9(1): p. e84943. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.St Peter WL, Guo H, Kabadi S, et al. , Prevalence, treatment patterns, and healthcare resource utilization in Medicare and commercially insured non-dialysis-dependent chronic kidney disease patients with and without anemia in the United States. BMC Nephrol, 2018. 19(1): p. 67. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Portoles J, Martin L, Broseta JJ, et al. , Anemia in Chronic Kidney Disease: From Pathophysiology and Current Treatments, to Future Agents. Front Med (Lausanne), 2021. 8: p. 642296. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Kendrick J, Cheung AK, Kaufman JS, et al. , FGF-23 associates with death, cardiovascular events, and initiation of chronic dialysis. J Am Soc Nephrol, 2011. 22(10): p. 1913–22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Shimada T, Urakawa I, Isakova T, et al. , Circulating fibroblast growth factor 23 in patients with end-stage renal disease treated by peritoneal dialysis is intact and biologically active. J Clin Endocrinol Metab, 2010. 95(2): p. 578–85. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Faul C, Amaral AP, Oskouei B, et al. , FGF23 induces left ventricular hypertrophy. J Clin Invest, 2011. 121(11): p. 4393–408. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Han X, Cai C, Xiao Z, et al. , FGF23 induced left ventricular hypertrophy mediated by FGFR4 signaling in the myocardium is attenuated by soluble Klotho in mice. J Mol Cell Cardiol, 2020. 138: p. 66–74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Ganz T, Bino A, and Salusky IB, Mechanism of Action and Clinical Attributes of Auryxia® (Ferric Citrate). Drugs, 2019. 79(9): p. 957–968. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Chertow GM, Block GA, Neylan JF, et al. , Safety and efficacy of ferric citrate in patients with nondialysis-dependent chronic kidney disease. PLOS ONE, 2017. 12(11): p. e0188712. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Block GA, Block MS, Smits G, et al. , A Pilot Randomized Trial of Ferric Citrate Coordination Complex for the Treatment of Advanced CKD. J Am Soc Nephrol, 2019. 30(8): p. 1495–1504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Yokoyama K, Fukagawa M, Akiba T, et al. , Randomised clinical trial of ferric citrate hydrate on anaemia management in haemodialysis patients with hyperphosphataemia: ASTRIO study. Sci Rep, 2019. 9(1): p. 8877. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Maruyama N, Otsuki T, Yoshida Y, et al. , Ferric Citrate Decreases Fibroblast Growth Factor 23 and Improves Erythropoietin Responsiveness in Hemodialysis Patients. Am J Nephrol, 2018. 47(6): p. 406–414. [DOI] [PubMed] [Google Scholar]
- 35.Francis C, Courbon G, Gerber C, et al. , Ferric citrate reduces fibroblast growth factor 23 levels and improves renal and cardiac function in a mouse model of chronic kidney disease. Kidney Int, 2019. 96(6): p. 1346–1358. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Block GA, Fishbane S, Rodriguez M, et al. , A 12-Week, Double-Blind, Placebo-Controlled Trial of Ferric Citrate for the Treatment of Iron Deficiency Anemia and Reduction of Serum Phosphate in Patients With CKD Stages 3–5. American Journal of Kidney Diseases, 2015. 65(5): p. 728–736. [DOI] [PubMed] [Google Scholar]
- 37.Block GA, Block MS, Smits G, et al. , A Pilot Randomized Trial of Ferric Citrate Coordination Complex for the Treatment of Advanced CKD. J Am Soc Nephrol, 2019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Faul C, Amaral AP, Oskouei B, et al. , FGF23 induces left ventricular hypertrophy. Journal of Clinical Investigation, 2011. 121(11): p. 4393–4408. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Goetz R, Nakada Y, Hu MC, et al. , Isolated C-terminal tail of FGF23 alleviates hypophosphatemia by inhibiting FGF23-FGFR-Klotho complex formation. Proc Natl Acad Sci U S A, 2010. 107(1): p. 407–12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Agoro R, Montagna A, Goetz R, et al. , Inhibition of fibroblast growth factor 23 (FGF23) signaling rescues renal anemia. FASEB J, 2018. 32(7): p. 3752–3764. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Agoro R, Park MY, Le Henaff C, et al. , C-FGF23 peptide alleviates hypoferremia during acute inflammation. Haematologica, 2020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Shalhoub V, Shatzen EM, Ward SC, et al. , FGF23 neutralization improves chronic kidney disease-associated hyperparathyroidism yet increases mortality. J Clin Invest, 2012. 122(7): p. 2543–53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Sitara D, Razzaque MS, Hesse M, et al. , Homozygous ablation of fibroblast growth factor-23 results in hyperphosphatemia and impaired skeletogenesis, and reverses hypophosphatemia in Phex-deficient mice. Matrix Biol, 2004. 23(7): p. 421–32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Shalhoub V, Shatzen EM, Ward SC, et al. , FGF23 neutralization improves chronic kidney disease–associated hyperparathyroidism yet increases mortality. Journal of Clinical Investigation, 2012. 122(7): p. 2543–2553. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.McCullough PA, Barnhart HX, Inrig JK, et al. , Cardiovascular toxicity of epoetin-alfa in patients with chronic kidney disease. Am J Nephrol, 2013. 37(6): p. 549–58. [DOI] [PubMed] [Google Scholar]
- 46.Jackevicius CA, Fan CS, and Warner A, Clinical outcomes of erythropoietin use in heart failure patients with anemia of chronic kidney disease. J Card Fail, 2014. 20(5): p. 327–33. [DOI] [PubMed] [Google Scholar]
- 47.Sabbagh Y, Graciolli FG, O’Brien S, et al. , Repression of osteocyte Wnt/beta-catenin signaling is an early event in the progression of renal osteodystrophy. J Bone Miner Res, 2012. 27(8): p. 1757–72. [DOI] [PubMed] [Google Scholar]
- 48.*.Noonan ML, Clinkenbeard EL, Ni P, et al. , Erythropoietin and a hypoxia-inducible factor prolyl hydroxylase inhibitor (HIF-PHDi) lowers FGF23 in a model of chronic kidney disease (CKD). Physiol Rep, 2020. 8(11): p. e14434. [DOI] [PMC free article] [PubMed] [Google Scholar]; In a study of mice with CKD, both EPO and HIF-PHI reduced circulating FGF23, most likely through correcting the prevailing iron utilization defects.
- 49.Ishii S, Suzuki T, Wakahashi K, et al. , FGF-23 from erythroblasts promotes hematopoietic progenitor mobilization. Blood, 2021. 137(11): p. 1457–1467. [DOI] [PubMed] [Google Scholar]
- 50.Haase VH, Hypoxia-inducible factor-prolyl hydroxylase inhibitors in the treatment of anemia of chronic kidney disease. Kidney Int Suppl (2011), 2021. 11(1): p. 8–25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Huang J, Zhao Q, Mooney SM, et al. , Sequence determinants in hypoxia-inducible factor-1alpha for hydroxylation by the prolyl hydroxylases PHD1, PHD2, and PHD3. J Biol Chem, 2002. 277(42): p. 39792–800. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Epstein AC, Gleadle JM, McNeill LA, et al. , C. elegans EGL-9 and mammalian homologs define a family of dioxygenases that regulate HIF by prolyl hydroxylation. Cell, 2001. 107(1): p. 43–54. [DOI] [PubMed] [Google Scholar]
- 53.Sanghani NS and Haase VH, Hypoxia-Inducible Factor Activators in Renal Anemia: Current Clinical Experience. Adv Chronic Kidney Dis, 2019. 26(4): p. 253–266. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Flamme I, Ellinghaus P, Urrego D, et al. , FGF23 expression in rodents is directly induced via erythropoietin after inhibition of hypoxia inducible factor proline hydroxylase. PLoS One, 2017. 12(10): p. e0186979. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.**.Hanudel MR, Wong S, Jung G, et al. , Amelioration of chronic kidney disease-associated anemia by vadadustat in mice is not dependent on erythroferrone. Kidney Int, 2021. 100(1): p. 79–89. [DOI] [PubMed] [Google Scholar]; In this report, a HIF-PHI corrected anemia in a mouse model of CKD, and using the erythroferrone (Erfe)-KO mice, demonstrated that this effect was Erfe-independent.