

Abstract
Hypothalamic hamartoma with gelastic seizures is a well-established cause of drug-resistant epilepsy in early life. The development of novel surgical techniques has permitted the genomic interrogation of hypothalamic hamartoma tissue. This has revealed causative mosaic variants within GLI3, OFD1 and other key regulators of the sonic-hedgehog pathway in a minority of cases. Sonic-hedgehog signalling proteins localize to the cellular organelle primary cilia. We therefore explored the hypothesis that cilia gene variants may underlie hitherto unsolved cases of sporadic hypothalamic hamartoma. We performed high-depth exome sequencing and chromosomal microarray on surgically resected hypothalamic hamartoma tissue and paired leukocyte-derived DNA from 27 patients. We searched for both germline and somatic variants under both dominant and bi-allelic genetic models. In hamartoma-derived DNA of seven patients we identified bi-allelic (one germline, one somatic) variants within one of four cilia genes—DYNC2I1, DYNC2H1, IFT140 or SMO. In eight patients, we identified single somatic variants in the previously established hypothalamic hamartoma disease genes GLI3 or OFD1. Overall, we established a plausible molecular cause for 15/27 (56%) patients. Here, we expand the genetic architecture beyond single variants within dominant disease genes that cause sporadic hypothalamic hamartoma to bi-allelic (one germline/one somatic) variants, implicate three novel cilia genes and reconceptualize the disorder as a ciliopathy.
Introduction
Hypothalamic hamartoma is a benign congenital lesion of the hypothalamus associated with a well-recognized developmental and epileptic encephalopathy defined by gelastic (laughing) seizures in infancy, which evolve into a drug-resistant epilepsy. Novel surgical techniques have allowed access to these small deep-seated lesions, permitting tissue analysis. Following the discovery that syndromic hypothalamic hamartoma in Pallister Hall syndrome is predominantly caused by germline or de novo variants in GLI3, a transcriptional regulator of the sonic hedgehog (SHH) pathway (1,2), we and others have demonstrated that sporadic hypothalamic hamartoma can be caused by single somatic variants of GLI3, OFD1 and other genes within the SHH pathway (3–6).
It has also been shown that the SHH pathway proteins localize to the ubiquitous cell organelle, primary cilia, to facilitate cell-to-cell interactions via signal transduction pathways during development (7–12). The importance of this relationship is evidenced by functional studies showing that disruption of cilia genes leads to altered SHH signalling responses during development (8,9,11). Recently, the cilia gene DYNC2H1 [MIM:603297] was implicated in sporadic hypothalamic hamartoma when two Japanese patients with bi-allelic (germline and somatic) variants in hamartoma tissue were described (13).
Here, we systematically analysed a cohort of 27 sporadic patients with hypothalamic hamartoma (Fig. 1) to establish the contribution of cilia gene variants and increase diagnostic yield.
Figure 1.
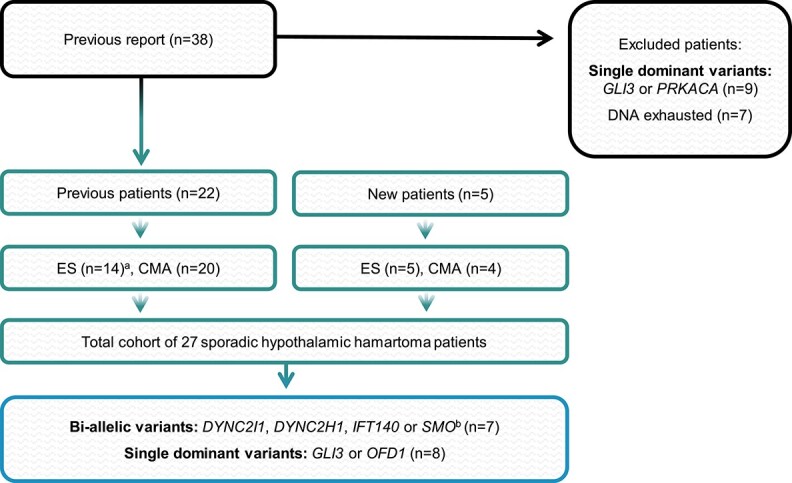
Sporadic hypothalamic hamartoma cohort and genetic analyses. The 27 sporadic hypothalamic hamartoma patients derived from our previously published report (4) and new cases that have not previously been investigated. ES; exome sequencing, CMA; chromosomal microarray. Patients from our previous report with molecular diagnoses (n = 9) or insufficient DNA remaining (n = 7) for analysis were excluded. aNine patients had exome data re-analysed from our previous study (4). The remaining five underwent exome analysis as part of this study. bThree cases had previously identified CNVs that overlap with candidate SHH pathway genes.
Results
Genetic analyses
Initial analysis of known hypothalamic hamartoma disease genes revealed eight patients with a single somatic variant (Table 1). Four of these patients were found to harbour frameshift variants within the SHH transcription factor GLI3 [MIM:165240], while the other four (all male) had somatic frameshift variants in the X-linked cilia gene OFD1 [MIM:300170], including a recurrent variant in two of them. Consistent with these findings, there was a predominance of males (74%) in our cohort (Table 1), as has been observed in other independent hypothalamic hamartoma case series (61%, (14) 64% (15) and 65% (16)), indicating potential under-recognition of OFD1 or other X-linked genes in the aetiology of hypothalamic hamartoma.
Table 1.
Cases with genetic findings
| Somatic VAF a | ||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Case | Sex | ES | CMA | Germline/Somatic | Gene | RefSeq | Variant/s | AC | SIFT(19) | PP2(20) | HH | Blood |
| T322 | F | + (P) | + | Somatic | GLI3 | NM_000168.6 | c.3098del (p. Pro1033ArgfsTer46) | 0 | N/A | N/A | 4/10 (40%) | 0/27 (0%) |
| T1165 | M | + (P) | − | Somatic | GLI3 | c.3313C > T (p.Gln1105Ter) | 0 | N/A | N/A | 196/522 (38%) | 0/208 (0%) | |
| T25069 | F | + (P) | + | Somatic | GLI3 | c.2977C > T (p.Gln993Ter) | 0 | N/A | N/A | 23/158 (15%) | 0/67 (0%) | |
| T1276 c | M | + (P) | − | Somatic | GLI3 | c.3189_3219del (p.Cys1065TrpfsTer4) | 0 | N/A | N/A | 25/139 (18%) | 0/48 (0%) | |
| T929 c | M | + (P) | + | Somatic | OFD1 d | NM_003611.3 | c.702dup (p.Tyr238ValfsTer2) | 1 | N/A | N/A | 80/143 (56%) | 0/53 (0%) |
| T25052 | M | + (P) | + | Somatic | OFD1 d | c.1193_1196del (p.Gln398LeufsTer2) | 0 | N/A | N/A | 5/15 (33%) | 0/30 (0%) | |
| T25056 | M | + (P) | + | Somatic | OFD1 d | c.702del (p.Lys237SerfsTer6) | 30 | N/A | N/A | 22/30 (73%) | 0/50 (0%) | |
| T25073 | M | + (P) | + | Somatic | OFD1 d | c.702del (p.Lys237SerfsTer6) | 30 | N/A | N/A | 45/80 (56%) | 0/41 (0%) | |
| T1198 e | F | + (P) | + | Germline & Somatic | DYNC2I1/chr7q11.21-q36.3 | NM_018051.5 | c.1703-1G > A, chr7q LOH (chr7:58814064– 159 138 663) |
0, N/A | N/A | N/A | N/A | N/A |
| T25059 | F | + (P) | + | Germline & Somatic | DYNC2I1 | c.1083_1083del (p.Asp361ValfsTer2), c.673_676del (p.Lys226ThrfsTer91) | 0, 0 | N/A | N/A | 52/99 (53%) | 0/42(0%) | |
| T25063 e | F | − | + | Germline & Somatic | DYNC2I1/chr7p22.1–q36.3 | c.2190_2191del (p. Arg731AlafsTer73), chr7p22.1–q36.3 CNG, CNL (chr7:986211–60 069 242, 58 814 064–159 138 663) | 0, N/A | N/A | N/A | N/A | N/A | |
| T25079 | M | + (HH) | + | Germline & Somatic | DYNC2H1 | NM_001080463.2 | c.11186C > A (p.Pro3729His), c.448C > T (p.Arg150Ter) | 6, 1 | D, N/A | PrD, N/A | 15/36 (42%) | N/A |
| T25080 | M | + (P) | + | Germline & Somatic | DYNC2H1 | c.8145_8146delinsAT(p.TyrGln2715Ter), c.11437C > T (p.Arg3813Cys) | 0, 4 | N/A, D | N/A, PrD | 89/347 (26%) | 0/79 (0%) | |
| T735 e | F | + (P) | + | Germline & Somatic | IFT140/chr16p11.2-p13.3 | NM_014714.4 | c.1901del (p.Lys634ArgfsTer10), chr16p LOH (chr16:0–31 543 619) | 0, N/A | N/A | N/A | N/A | N/A |
| T1094 b | M | + (P) | − | Germline & Somatic | SMO | NM_005631.5 | c.1453C > G (p.Arg485Gly), c.1274_1275del (p.Leu426ValfsTer13) | 0, 1 | D, N/A | D, N/A | 135/400 (34%) | 0/87 (0%) |
ES; exome sequencing, CMA; chromosomal microarray, AC; gnomAD allele count, PP2; PolyPhen2, (HH); hypothalamic hamartoma only, (P); paired blood and hypothalamic hamartoma tissue, LOH; loss of heterozygosity, CNG; copy number gain, CNL; copy number loss, N/A; not applicable, D; damaging, PrD; probably damaging
Variant allele fraction; Variant reads/Total reads
Confirmed bi-allelic variants
Not validated with Sanger sequencing or ddPCR due to insufficient DNA
X-linked gene, hence mosaic VAF can be >50% in hemizygous males
CNV Identified in previous analyses (4)
For gene discovery, we then interrogated cilia genes in the remaining 19 unsolved cases and revealed seven cases with confirmed or putative bi-allelic variants, each comprised of a germline and a somatic variant in one of four genes: DYNC2I1 [MIM:615462], DYNC2H1, IFT140 [MIM:614620] and SMO [MIM:601500]. In four cases the variants were confirmed as bi-allelic, whereas in three cases this remained putative (Table 1, Fig. 2).
Figure 2.

The exome and Sanger sequencing data supporting a bi-allelic allelic phase in the three cases T1198, T25063 and T735. In all three cases, the corresponding germline variant is the most prevalent allele in hypothalamic hamartoma tissue indicating the somatic CNVs detected in these cases are on the alternate allele. (A) Integrative genomics viewer (IGV) shot of paired exome sequencing of T1198 showing the DYNC2I1 c.1703-1G > A variant, top panel within IGV is hypothalamic hamartoma and bottom panel is blood. (B) IGV shot of paired exome sequencing of T735 showing the IFT140 c.1901del; p.Lys634ArgfsTer10 variant, top panel within IGV is hypothalamic hamartoma and bottom panel is blood. (C) Sanger sequencing of DYNC2I1 c.2190_2191delAA; p. Arg731AlafsTer73 variant within T25063.
Three of the seven bi-allelic cases (T1198, T25059, T25063) had distinct germline and somatic variants in DYNC2I1 that encodes the intermediate chain of the ciliary intraflagellar transport (IFT) dynein-2 complex (Table 1) (17). Two of these cases (T1198, T25063) harboured large somatic copy number variants (CNVs) that encompass DYNC2I1 on one allele with a novel germline DYNC2I1 sequence variant on the other; we confirmed these were bi-allelic in hamartoma tissue using both exome and Sanger sequencing (Fig. 2). The third case (T25059) carried two frameshift deletions that were presumed to be bi-allelic, but this could not be confirmed due to the large distance (~22 kb) between the variants precluding allelic discrimination given hamartoma tissue RNA was not available from the patient. Three of the four sequence variants found in DYNC2I1 were frameshifts resulting in premature stop codons, two of which occur early in the transcript meaning the respective transcripts are likely subject to nonsense-mediated decay, although RNA was unavailable to confirm this. The fourth DYNC2I1 variant detected was a splice site variant predicted by SpliceAI (18) to alter splicing through disruption of the splice acceptor site at the 5′ end of exon 14 (Δ score = 0.92); tissue RNA was also unavailable from this case.
Two cases (T25079, T25080) were found to have germline and somatic variants in DYNC2H1 that encodes the ciliary heavy chain IFT motor protein dynein-2 (Table 1). In these two cases, we identified protein-truncating variants (PTVs) (nonsense or frameshift) in conjunction with missense variants that are predicted damaging by in silico tools (19,20). The large genomic distances (189 and 108 kb) between these respective sequence variants precluded phasing in the absence of hamartoma tissue RNA.
We also identified one patient (T735) with bi-allelic variants in the IFT140 gene encoding one of six subunits of the IFT-A protein complex (Table 1) (12). This patient harboured a large somatic CNV encompassing IFT140 in combination with a novel germline frameshift variant that introduces a premature stop codon. As this variant occurs in the last nucleotide of exon 16 it is predicted to result in loss of the consensus donor splice site by SpliceAI (Δ score = 1.00). A trans configuration in hamartoma tissue for these variants was confirmed on sequencing (Fig. 2).
The seventh case (T1094) had bi-allelic variants in the cilia gene SMO that encodes a frizzled class receptor within the SHH pathway known to be trafficked and signal within the primary cilium (Table 1) (7). In this patient, we identified a novel missense germline variant in combination with a somatic frameshift variant introducing a premature stop codon that may also lead to nonsense-mediated decay. Due to the close genomic proximity between these two variants, we were able to perform allelic discrimination by restriction digest, which confirmed these two variants were in a trans configuration (Supplementary Material, Fig. S1).
Gene enrichment analysis
Given the above discoveries, and in the absence of further analysable tissue samples, we sought alternate indirect evidence for a possible bi-allelic mechanism involving cilia genes. We posited there may be an excess of potentially damaging rare germline variants in cilia genes (detectable in blood) within sporadic hypothalamic hamartoma cases. We therefore sought to quantify the presence of rare germline qualifying variants with a population-specific minor allele frequency <0.1% in sporadic hypothalamic hamartoma patients compared with 2413 controls across four different gene-sets (Supplementary Material). In order to assess these germline contributions, we also included an additional 25 sporadic hypothalamic hamartoma cases on which we had access to exome data generated from blood-derived DNA.
Given the recessive inheritance of most ciliopathies (21), we reasoned that the germline variants would also be present in the general population. PTVs in known cilia genes which showed a trend towards enrichment were further divided into ultra-rare (absent in gnomAD) and rare (present in gnomAD but population-specific allele frequency <0.1%) with ion channel genes as a control (Fig. 3). Among PTVs in known cilia genes, PTVs were primarily rare (OR = 2.5, CI = [0.89–6.2], p = 0.039, adj.p = 0.83) and not ultra-rare (OR = 0.5, CI = [0.0–3.0], p = 0.72, adj.p = 1.0), which is consistent with our hypothesis (Fig. 3). Despite no statistically significant association being found, our data revealed a trend that could be strengthened in future by increasing the size of our cohort (Supplementary Material, Fig. S6). Additionally, as ciliopathies are generally recessively inherited, our findings may reflect the weak selection pressure on deleterious heterozygous germline variants as has been demonstrated with disease genes for disorders with autosomal recessive inheritance (21,22).
Figure 3.
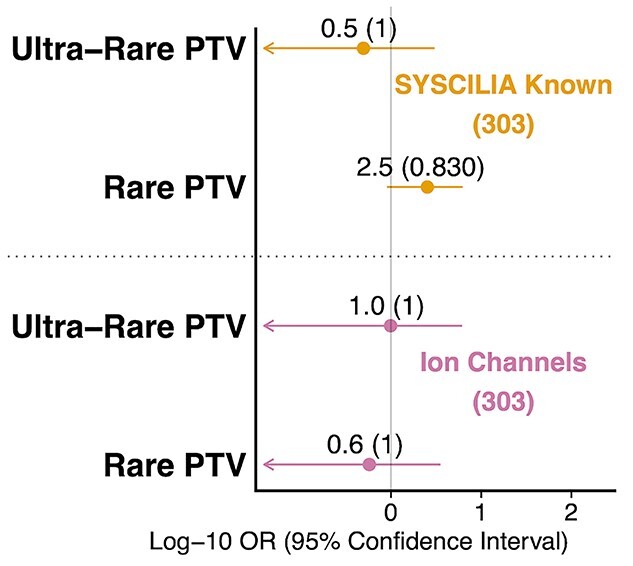
Exploring burden of protein truncating variants in hypothalamic hamartoma patients in two gene sets separated by minor allele frequency. The two gene sets listed are known cilia genes and ion channel genes. ‘Ultra-rare’ refers to variants absent in gnomAD, ‘Rare’ refers to variants present in gnomAD but with a population-specific minor allele frequency less than 0.1%. ‘PTV’ indicates protein truncating variant. Number of genes in set in parenthesis. Odds ratio, confidence intervals and FDR corrected p-value were generated from the Fisher’s exact test. Odds ratio is displayed for each test with FDR-adjusted p-values in parentheses. X-axis displays the log of the odds ratio and confidence intervals. Arrows indicate lower bound of confidence interval has been truncated.
Discussion
We have expanded the molecular architecture of sporadic hypothalamic hamartoma by implicating three new cilia genes (DYNC2I1, IFT140, SMO) involved in intraflagellar transport (IFT), and replicated previous findings (13) of bi-allelic DYNC2H1 variants. Our findings of confirmed or putative bi-allelic variants within a single gene comprising of a germline and somatic variant are analogous to other reported brain lesions that can cause epilepsy including subependymal giant cell astrocytomas, cortical tubers in tuberous sclerosis complex and focal cortical dysplasias (23–25).
Primary cilia comprise a single non-motile axoneme that is constructed and maintained by protein complexes known as the IFT machinery (12). The IFT machinery consists of two protein complexes, IFT-A and IFT-B, which are required for retrograde and anterograde transport, respectively (12). It has been demonstrated in numerous studies that SHH pathway constituents localize to primary cilia and are dependent upon IFT machinery for signal transduction (7–10,26). DYNC2H1 and DYNC2I1 encode subunits of the IFT dynein-2 molecular motor together with DYNLT2B [MIM:617353], DYNC2LI1 [MIM:617083] and DYNC2I2 [MIM:613363] (12). This dynein-2 motor drives the IFT-A protein complex, of which IFT140 is a subunit, to facilitate retrograde transport (Fig. 4) (12).
Figure 4.

The sonic hedgehog pathway and primary cilia. In response to the hedgehog ligand binding to PTCH1, SMO localizes to the axoneme of primary cilia (7). The GLI protein complexes including GLI3 localize to the tip of primary cilia (8) and are processed into the activated form, GLIA. IFT mediated retrograde transport and specifically the IFT dynein-2 and IFT-A complexes facilitate the movement of the GLIA complex to regulate downstream targets of the pathway. Red text; the genes in which bi-allelic variants have been identified within our sporadic hypothalamic hamartoma cohort. Blue text; the genes in which dominant somatic variants have previously been identified in sporadic hypothalamic hamartoma. OFD1 encodes a protein that localizes to the basal body of primary cilia and is required for ciliogenesis (12). Single somatic PRKACA variants have previously been identified in sporadic hypothalamic hamartoma (4) which encodes one of two catalytic subunits of the PKA protein complex that localizes to primary cilia (42). Figure adapted from (12).
‘Ciliopathies’ is a term applied to a heterogeneous group of disorders, typically recessively inherited with germline variants in cilia genes causing disruption of ciliogenesis or IFT (21). Interestingly, bi-allelic germline variants in the DYNC2H1, DYNC2I1 and IFT140 genes are associated with the related skeletal ciliopathies Jeune Asphyxiating Thoracic Dystrophy (JATD [MIM: 208500]), Short Rib Polydactyly (SRPS [MIM: 615503]) and Mainzer–Saldino (MSS [MIM 266920]) syndromes (27–30). Functional interrogation of bi-allelic germline variants within DYNC2H1, DYNC2I1 or IFT140 has demonstrated disruption of retrograde trafficking and ciliogenesis within primary cilia (27,29,30). The lack of skeletal features in our cases is presumably explained by the brain-specific somatic second hit. Notably, hypothalamic hamartoma has not been reported in these ciliopathies with two germline variants. This may represent disease-specific genotype–phenotype associations, similar to those well established for Pallister–Hall and Greig Cephalopolysyndactyly syndromes due to pathogenic GLI3 variants (2). We observe that bi-allelic variants identified in patients with hypothalamic hamartoma may have more severe impact. For example, patients we report with DYNC2I1, IFT140 and SMO bi-allelic variants have one PTV in combination with either a second PTV or loss of heterozygosity (LOH) copy number variant (CNV) which may severely disrupt the protein (Table 1). In contrast, germline bi-allelic variants in DYNC2I1 and IFT140 associated with ciliopathies tend to include a combination of a potentially less damaging missense variant and a PTV (28–31). The recent identification of germline SMO variants has demonstrated that patients with hypothalamic hamartoma have at least one PTV as opposed to patients without hypothalamic hamartoma carrying only missense variants (32,33). However, these observations are based on only small numbers and further cases and functional analyses of variants is required to establish any mutational spectrum and genotype–phenotype associations.
Our data implicate genes involved in IFT-mediated retrograde trafficking in primary cilia in the molecular pathogenesis of sporadic hypothalamic hamartoma. Notably, the pathological mechanisms underlying formation of hypothalamic hamartomas remain unclear even for the established GLI3 variants (5). For example, the GLI3 mouse model of Pallister–Hall syndrome does not develop hypothalamic hamartomas (34). Disruption of SHH pathway signalling appears to be central to the pathogenesis. With respect to this, the cilia genes (OFD1, DYNC2I1, DYNC2H1, IFT140 or SMO) identified in this study have all been shown to disrupt SHH pathway proteins and lead to aberrant SHH signalling in prior studies (5,30,33,35,36). We propose that dysfunction of specific components of the IFT machinery localized to hamartoma cells interferes with normal SHH signalling, a mechanism which will require functional interrogation in future studies. Consistent with this, we implicated a third cilia gene in sporadic hypothalamic hamartoma, Smoothened (SMO), which is also a regulator of the SHH pathway. It encodes a seven-transmembrane protein that localizes to primary cilia and depends on IFT to regulate SHH signalling during development (Fig. 4) (7,26). Moreover, germline SMO bi-allelic variants have recently been reported in five individuals with syndromic hypothalamic hamartoma and other developmental anomalies including post-axial polydactyly, microcephaly and skeletal abnormalities that often present in the aforementioned skeletal ciliopathies (32,33,37).
In summary, we have identified plausible molecular causes on a research basis in 15/27 (56%) cases in our sporadic cohort including confirmed or putative single somatic or bi-allelic variants. Taken together, our results suggest that disrupted trafficking and SHH signalling in the primary cilium underlie the pathogenesis of sporadic hypothalamic hamartoma through two distinct genetic mechanisms. Firstly, through single somatic variants in disease genes such as GLI3 and OFD1 that function within primary cilia during development, a mechanism that we and others previously established (3–6). Secondly, through bi-allelic variants in cilia genes disrupting IFT and SHH signalling, suggesting we can reconceptualize sporadic hypothalamic hamartoma as a ciliopathy.
Materials and Methods
Patient cohort and sample collection
We studied resected hypothalamic hamartoma tissue from 22 previously reported unsolved (4) and five newly recruited patients with sporadic disease (Fig. 1), comprising 20 (74%) males and seven (26%) females. Epilepsy commenced in the first year of life for 22/27 of these patients, and all had gelastic seizures. Additional features included intellectual disability (n = 20) and central precocious puberty (n = 11) (Supplementary Material, Table S1). None had additional syndromic features of digital, oro-facial abnormalities or visceral malformations, and none had a family history of hypothalamic hamartoma. An additional 25 sporadic hypothalamic hamartoma cases with only blood-derived DNA available were studied, exclusively in our gene enrichment analysis.
DNA was extracted from fresh frozen or formalin-fixed paraffin-embedded hamartoma tissue, obtained during surgical treatment of hamartomas, and from whole-blood using Qiagen All Prep DNA/RNA, FFPE Tissue and QIAamp DNA Blood Maxi Kits (Hilden, Germany), respectively, and previously reported standard protocols (4). The Human Research Ethics Committees of Austin Health and The Royal Children’s Hospital in Melbourne and the Institutional Review Board of St. Joseph’s Hospital and Medical Center in Phoenix approved this study. Informed consent was obtained from affected individuals or their parents or legal guardians in the case of minors, those with intellectual disability or deceased individuals.
Exome sequencing
There was insufficient hamartoma-derived DNA to perform exome sequencing and chromosomal microarray (CMA) analysis on all cases. Exome sequencing was possible on 19/27 patients including 18 with paired hamartoma/blood-derived DNA and one with only hamartoma-derived DNA (Fig. 1). Exome sequencing was performed using the Agilent SureSelect DNA Human All Exon V6, 96RXN kit (Agilent Technologies, Santa Clara, CA) and the Illumina NovaSeq 6000 System (Illumina, San Diego, CA, USA) or the Illumina TruSeq Exome Enrichment kit and HiSeq 2000 System (Illumina, San Diego, CA, USA). Briefly, reads were aligned to the hg19 reference genome with BWA-MEM v0.7.17-r1188, then duplicate marking and base quality score recalibration performed with the Genome Analysis Toolkit (GATK). Germline variant calling was performed with GATK HaplotypeCaller and somatic variant calling with GATK Mutect2 v4.0.1.2, VarScan v2.4.3 and Strelka v2.9.10. Variants were annotated using vcfanno and ANNOVAR.
We performed a targeted search for germline and somatic variants in a curated list of known or candidate cilia and SHH pathway genes compiled from the SYSCILIA Gold Standard (38) and the Kyoto Encyclopedia of Genes and Genomes (KEGG) (39) catalogues. Variants were filtered according to the following criteria: located in a coding or splice site region, frequency of less than or equal to 0.001 in the Genome Aggregation Database (gnomAD v2.1.1) (40), and variant type—missense, nonsense, coding indel or splice site.
CMA analysis
CMA data were generated from 24 cases, 20/24 of these as part of our previous study (Fig. 1) (4). For the four new cases, the Illumina Global Diversity array platform (GDAv1.0 San Diego, CA) was used to perform genome-wide CNVs and LOH screening at a resolution of 200 Kb and 2 Mb, respectively, on hamartoma and blood-derived DNA. The data were analysed using NxClinicalv6.0 (BioDicovery, CA) using genome reference sequence NCBI37/hg19.
PCR and Sanger sequencing
Amplification was performed using gene-specific primers (available on request) designed to reference human gene transcripts using NCBI Gene (https://www.ncbi.nlm.nih.gov/gene) and Primer3 v4.1.0 (http://bioinfo.ut.ee/primer3/). Amplification reactions were cycled using a standard protocol on a Veriti Thermal Cycler (Applied Biosystems, Carlsbad, CA) and sequencing performed with a BigDyeTM v3.1 Terminator Cycle Sequencing Kit (Applied Biosystems). Sequencing products were resolved using a 3730 XL DNA Analyzer (Applied Biosystems). All nucleotide changes were called using Codon Code Aligner software (CodonCode Corporation, Dedham, MA).
Droplet digital PCR
Custom probes and primers were ordered from Integrated DNA Technologies, Inc. (Iowa, USA) and were utilized to validate and quantify somatic variants. Droplet generation, PCR cycling and droplet reading were performed according to the manufacturer’s recommendations (Bio-Rad, Hercules, CA). Briefly, probes and primers were mixed with 2x ddPCR Supermix (No dUTPs) for probe (Bio-Rad) at 250 and 900 nm final concentrations for each probe and each of the primers, respectively, and mixed with 10 ng of DNA sample to a final volume of 23 μL. Twenty microliters of reactions were loaded in an eight-channel droplet generator cartridge (Bio-Rad) and droplets were generated with 70 μL of droplet generation oil (Bio-Rad) using the manual QX200 Droplet Generator. Following droplet generation, samples were manually transferred to a 96-well PCR plate, heat-sealed and amplified on a C1000 Touch thermal cycler using the following cycling conditions: 95°C for 10 min for one cycle, followed by 40 cycles at 94°C for 30 s and 55°C for 60 s, one cycle at 98°C for 10 min and 4°C hold. Post-PCR products were read on the QX200 droplet reader (Bio-Rad) and analysed using the QuantaSoft software.
Allelic discrimination by restriction digestion
Two restriction enzymes, FastDigest PfoI and TaaI (Thermo Scientific™, Waltham, MA), were utilized according to the manufacturer’s recommendations in a 30 μL reaction volume to sequentially differentiate variant alleles. Restriction digest reactions were resolved with standard agarose gels and visualized under UV light with a UVP GelDoc-It310 imaging system. Digested fragments were gel extracted using the Qiagen QIAquick Gel Extraction Kit (Hilden, Germany) as per the manufacturer’s instructions (Supplementary Material).
Gene enrichment analysis
Gene enrichment analysis was performed as previously described (41) by quantifying ultra-rare germline variants in sporadic patients with hypothalamic hamartoma and 2413 ethnically matched population controls using four different gene-sets: SHH pathway genes (49 genes) from the KEGG database, and known cilia genes (303 genes) and potential cilia genes (410 genes) from SYSCILIA and 303 randomly selected ion channel genes from the HGNC database given the lack of known overlap between these proteins and hypothalamic hamartoma aetiology (Supplementary Material) (38,39,42).
Supplementary Material
Acknowledgements
We thank the families for their participation in this study, and we value the advocacy from the Hope for Hypothalamic Hamartoma Foundation (http://hopeforhh.org). Joshua Reid (Epilepsy Research Centre) performed genomic DNA extractions from tissues. Brett Copeland, Joshua Bridgers and Sitharthan Kamalakaran (Institute for Genomic Medicine) provided bioinformatics support. Greta Gillies and Kate Pope (Murdoch Childrens Research Institute) assisted with collection of tissue and Jeffrey Rosenfeld and Wirginia Maixner (Royal Children’s Hospital, Melbourne) performed surgeries. Figure 4 was created with BioRender.com.
Conflict of Interest statement. The authors have no conflicts of interest to declare.
Contributor Information
Timothy E Green, Epilepsy Research Centre, Department of Medicine, The University of Melbourne, Austin Health, Heidelberg, VIC 3084, Australia.
Joshua E Motelow, Institute for Genomic Medicine, Columbia University, New York, NY 10032, USA.
Mark F Bennett, Epilepsy Research Centre, Department of Medicine, The University of Melbourne, Austin Health, Heidelberg, VIC 3084, Australia; Population Health and Immunity Division, The Walter and Eliza Hall Institute of Medical Research, Melbourne, Victoria 3052, Australia; Department of Medical Biology, University of Melbourne, Melbourne, Victoria 3052, Australia.
Zimeng Ye, Epilepsy Research Centre, Department of Medicine, The University of Melbourne, Austin Health, Heidelberg, VIC 3084, Australia.
Caitlin A Bennett, Epilepsy Research Centre, Department of Medicine, The University of Melbourne, Austin Health, Heidelberg, VIC 3084, Australia.
Nicole G Griffin, Institute for Genomic Medicine, Columbia University, New York, NY 10032, USA.
John A Damiano, Epilepsy Research Centre, Department of Medicine, The University of Melbourne, Austin Health, Heidelberg, VIC 3084, Australia.
Richard J Leventer, Department of Neurology, The Royal Children’s Hospital, Parkville, VIC 3052, Australia; Department of Paediatrics, University of Melbourne, Royal Children's Hospital, Parkville, VIC 3052, Australia; Murdoch Children’s Research Institute, The Royal Children’s Hospital, Parkville, VIC 3052, Australia.
Jeremy L Freeman, Department of Neurology, The Royal Children’s Hospital, Parkville, VIC 3052, Australia; Murdoch Children’s Research Institute, The Royal Children’s Hospital, Parkville, VIC 3052, Australia.
A Simon Harvey, Department of Neurology, The Royal Children’s Hospital, Parkville, VIC 3052, Australia; Department of Paediatrics, University of Melbourne, Royal Children's Hospital, Parkville, VIC 3052, Australia; Murdoch Children’s Research Institute, The Royal Children’s Hospital, Parkville, VIC 3052, Australia.
Paul J Lockhart, Department of Paediatrics, University of Melbourne, Royal Children's Hospital, Parkville, VIC 3052, Australia; Murdoch Children’s Research Institute, The Royal Children’s Hospital, Parkville, VIC 3052, Australia.
Lynette G Sadleir, Department of Paediatrics and Child Health, University of Otago, Wellington 6242, New Zealand.
Amber Boys, Victorian Clinical Genetics Services, Parkville, VIC 3052, Australia.
Ingrid E Scheffer, Epilepsy Research Centre, Department of Medicine, The University of Melbourne, Austin Health, Heidelberg, VIC 3084, Australia; Department of Neurology, The Royal Children’s Hospital, Parkville, VIC 3052, Australia; Murdoch Children’s Research Institute, The Royal Children’s Hospital, Parkville, VIC 3052, Australia; The Florey Institute of Neuroscience and Mental Health, Parkville, VIC 3052, Australia.
Heather Major, Department of Pediatrics, The University of Iowa, Iowa City, IA 52246, USA.
Benjamin W Darbro, Department of Pediatrics, The University of Iowa, Iowa City, IA 52246, USA.
Melanie Bahlo, Population Health and Immunity Division, The Walter and Eliza Hall Institute of Medical Research, Melbourne, Victoria 3052, Australia; Department of Medical Biology, University of Melbourne, Melbourne, Victoria 3052, Australia.
David B Goldstein, Institute for Genomic Medicine, Columbia University, New York, NY 10032, USA; Department of Genetics and Development, Columbia University, New York, NY 10032, USA.
John F Kerrigan, Division of Pediatric Neurology, Barrow Neurological Institute, Phoenix Children's Hospital, Phoenix, AZ 85013, USA.
Erin L Heinzen, Eshelman School of Pharmacy, Division of Pharmacotherapy and Experimental Therapeutics, and Department of Genetics, University of North Carolina, Chapel Hill, NC 27599, USA.
Samuel F Berkovic, Epilepsy Research Centre, Department of Medicine, The University of Melbourne, Austin Health, Heidelberg, VIC 3084, Australia.
Michael S Hildebrand, Epilepsy Research Centre, Department of Medicine, The University of Melbourne, Austin Health, Heidelberg, VIC 3084, Australia; Murdoch Children’s Research Institute, The Royal Children’s Hospital, Parkville, VIC 3052, Australia.
Funding
National Health and Medical Research Council Program Grant (1091593) to I.E.S. and S.F.B., a Project Grant (1129054) to S.F.B., a Project Grant (1079058) to M.S.H., a Practitioner Fellowship (1006110) to I.E.S., a Senior Research Fellowship (1102971) to M.B., and a R.D Wright Career Development Fellowship (1063799) to M.S.H. R.J.L. is supported by a Melbourne Children’s Clinician Scientist Fellowship and P.J.L. is supported by the Vincent Ciodo Foundation. Z.Y. is supported by a University of Melbourne Australian Postgraduate Award International Graduate Research Training Scholarship, and a private scholarship from the Tang Lixin Education Development Fund. I.E.S. was also funded by grants from the National Institutes of Health, Australian Research Council, Health Research Council of New Zealand, CURE, American Epilepsy Society, US Department of Defense Autism Spectrum Disorder Research Program, March of Dimes and Perpetual Charitable Trustees. E.L.H. was supported by a National Institutes of Health grant (R21-NS078657). J.E.M. is supported by the National Institutes of Health (TL1TR001875). This work was made possible through Victorian State Government Operational Infrastructure Support and Australian Government NHMRC IRIISS. This study was supported by a Cure Kids (3576) 15/070 project grant to L.G.S. M.F.B was supported by a Taking Flight Award from CURE Epilepsy.
Author Contribution
M.S.H. and S.F.B developed the idea and directed the project. T.E.G., Z.Y., N.G.G., J.a.d., H.M., B.W.D., E.L.H. and M.S.H. performed molecular genetics experiments. N.G.G., J.M., M.F.B., M.B., A.B and E.L.H. performed bioinformatics analysis. E.J.C., R.B., R.J.L. A.S.H., J.L.F., P.J.L., L.G.S., I.E.S., J.F.K. and S.F.B. conducted clinical phenotyping or specimen collection. M.S.H., R.J.L., P.J.L., D.B.G., E.L.H. and S.F.B. provided equipment and reagents. T.E.G., S.F.B. and M.S.H. drafted the paper. All authors discussed the results and commented on the final manuscript.
References
- 1. Kang, S., Graham, J.M., Olney, A.H. and Biesecker, L.G. (1997) GLI3 frameshift mutations cause autosomal dominant Pallister-hall syndrome. Nat. Genet., 15, 266–268. [DOI] [PubMed] [Google Scholar]
- 2. Johnston, J.J., Olivos-Glander, I., Killoran, C., Elson, E., Turner, J.T., Peters, K.F., Abbott, M.H., Aughton, D.J., Aylsworth, A.S., Bamshad, M.J.et al. (2005) Molecular and clinical analyses of Greig Cephalopolysyndactyly and Pallister-hall syndromes: robust phenotype prediction from the type and position of GLI3 mutations. Am. J. Hum. Genet., 76, 609–622. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Craig, D.W., Itty, A., Panganiban, C., Szelinger, S., Kruer, M.C., Sekar, A., Reiman, D., Narayanan, V., Stephan, D.A. and Kerrigan, J.F. (2008) Identification of somatic chromosomal abnormalities in hypothalamic hamartoma tissue at the GLI3 locus. Am. J. Hum. Genet., 82, 366–374. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Hildebrand, M.S., Griffin, N.G., Damiano, J.A., Cops, E.J., Burgess, R., Ozturk, E., Jones, N.C., Leventer, R.J., Freeman, J.L., Harvey, A.S.et al. (2016) Mutations of the sonic hedgehog pathway underlie hypothalamic hamartoma with gelastic epilepsy. Am. J. Hum. Genet., 99, 423–429. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Saitsu, H., Sonoda, M., Higashijima, T., Shirozu, H., Masuda, H., Tohyama, J., Kato, M., Nakashima, M., Tsurusaki, Y., Mizuguchi, T.et al. (2016) Somatic mutations in GLI3 and OFD1 involved in sonic hedgehog signaling cause hypothalamic hamartoma. Ann. Clin. Transl. Neurol., 3, 356–365. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Wallace, R.H., Freeman, J.L., Shouri, M.R., Izzillo, P.A., Rosenfeld, J.V., Mulley, J.C., Harvey, A.S. and Berkovic, S.F. (2008) Somatic mutations in GLI3 can cause hypothalamic hamartoma and gelastic seizures. Neurology, 70, 653–655. [DOI] [PubMed] [Google Scholar]
- 7. Corbit, K.C., Aanstad, P., Singla, V., Norman, A.R., Stainier, D.Y.R. and Reiter, J.F. (2005) Vertebrate smoothened functions at the primary cilium. Nature, 437, 1018–1021. [DOI] [PubMed] [Google Scholar]
- 8. Haycraft, C.J., Banizs, B., Aydin-Son, Y., Zhang, Q., Michaud, E.J. and Yoder, B.K. (2005) Gli2 and Gli3 localize to cilia and require the intraflagellar transport protein polaris for processing and function. PLoS Genet., 1, 53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Huangfu, D. and Anderson, K.V. (2005) Cilia and hedgehog responsiveness in the mouse. Proc. Natl. Acad. Sci. U. S. A., 102, 11325–11330. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Rajat, R., Ljiljana, M. and Matthew, P.S. (2007) Patched1 regulates hedgehog signaling at the primary cilium. Science, 317, 372–376. [DOI] [PubMed] [Google Scholar]
- 11. Ocbina, P.J.R. and Anderson, K.V. (2008) Intraflagellar transport, cilia, and mammalian hedgehog signaling: analysis in mouse embryonic fibroblasts. Dev. Dyn., 237, 2030–2038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Goetz, S.C. and Anderson, K.V. (2010) The primary cilium: a signalling Centre during vertebrate development. Nat. Rev. Genet., 11, 331–344. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Fujita, A., Higashijima, T., Shirozu, H., Masuda, H., Sonoda, M., Tohyama, J., Kato, M., Nakashima, M., Tsurusaki, Y., Mitsuhashi, S.et al. (2019) Pathogenic variants of DYNC2H1, KIAA0556, and PTPN11 associated with hypothalamic hamartoma. Neurology, 93, e237–e251. [DOI] [PubMed] [Google Scholar]
- 14. Hiroshi, S., Hiroshi, M. and Shigeki, K. (2020) Repeat stereotactic radiofrequency thermocoagulation in patients with hypothalamic hamartoma and seizure recurrence. Epilepsia Open., 5, 107–120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Ferrand-Sorbets, S., Fohlen, M., Delalande, O., Zuber, K., Bulteau, C., Levy, M., Chamard, P., Taussig, D., Dorison, N., Bekaert, O.et al. (2020) Seizure outcome and prognostic factors for surgical management of hypothalamic hamartomas in children. Seizure, 75, 28–33. [DOI] [PubMed] [Google Scholar]
- 16. Curry, D.J., Raskin, J., Ali, I. and Wilfong, A.A. (2018) MR-guided laser ablation for the treatment of hypothalamic hamartomas. Epilepsy Res., 142, 131–134. [DOI] [PubMed] [Google Scholar]
- 17. Asante, D., Stevenson, N.L. and Stephens, D.J. (2014) Subunit composition of the human cytoplasmic dynein-2 complex. J. Cell Sci., 127, 4774–4787. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Jaganathan, K., Kyriazopoulou Panagiotopoulou, S., McRae, J.F., Darbandi, S.F., Knowles, D., Li, Y.I., Kosmicki, J.A., Arbelaez, J., Cui, W., Schwartz, G.B.et al. (2019) Predicting splicing from primary sequence with deep learning. Cell, 176, 535–548. [DOI] [PubMed] [Google Scholar]
- 19. Sim, N.-L., Kumar, P., Hu, J., Henikoff, S., Schneider, G. and Ng, P.C. (2012) SIFT web server: predicting effects of amino acid substitutions on proteins. Nucleic Acids Res., 40, W452–W457. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Adzhubei, I.A., Schmidt, S., Peshkin, L., Ramensky, V.E., Gerasimova, A., Bork, P., Kondrashov, A.S. and Sunyaev, S.R. (2010) A method and server for predicting damaging missense mutations. Nat. Methods, 7, 248–249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Braun, D.A. and Hildebrandt, F. (2017) Ciliopathies. Cold Spring Harb. Perspect. Biol., 9, a028191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Cassa, C.A., Weghorn, D., Balick, D.J., Jordan, D.M., Nusinow, D., Samocha, K.E., O'Donnell-Luria, A., MacArthur, D.G., Daly, M.J. and Beier, D.R. (2017) Estimating the selective effects of heterozygous protein-truncating variants from human exome data. Nat. Genet., 49, 806–810. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Martin, K.R., Zhou, W., Bowman, M.J., Shih, J., Au, K.S., Dittenhafer-Reed, K.E., Sisson, K.A., Koeman, J., Weisenberger, D.J., Cottingham, S.L.et al. (2017) The genomic landscape of tuberous sclerosis complex. Nat. Commun., 8, 15816. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Bongaarts, A., Giannikou, K., Reinten, R.J., Anink, J.J., Mills, J.D., Jansen, F.E., Spliet, W.G.M., denDunnen, W.F.A., Coras, R., Bluemcke, I.et al. (2017) Subependymal giant cell astrocytomas in tuberous sclerosis complex have consistent TSC1/TSC2 biallelic inactivation, and no BRAF mutations. Oncotarget, 8, 95516–95529. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Ribierre, T., Deleuze, C., Bacq, A., Baldassari, S., Marsan, E., Chipaux, M., Muraca, G., Roussel, D., Navarro, V., Leguern, E.et al. (2018) Second-hit mosaic mutation in mTORC1 repressor DEPDC5 causes focal cortical dysplasia-associated epilepsy. J. Clin. Invest., 128, 2452–2458. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. May, S.R., Ashique, A.M., Karlen, M., Wang, B., Shen, Y., Zarbalis, K., Reiter, J., Ericson, J. and Peterson, A.S. (2005) Loss of the retrograde motor for IFT disrupts localization of Smo to cilia and prevents the expression of both activator and repressor functions of Gli. Dev. Biol., 287, 378–389. [DOI] [PubMed] [Google Scholar]
- 27. Schmidts, M., Arts, H.H., Bongers, E.M.H.F., Yap, Z., Oud, M.M., Antony, D., Duijkers, L., Emes, R.D., Stalker, J. and Yntema, J.B.L. (2013) Exome sequencing identifies DYNC2H1 mutations as a common cause of asphyxiating thoracic dystrophy (Jeune syndrome) without major polydactyly, renal or retinal involvement. J. Med. Genet., 50, 309–323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Cossu, C., Incani, F., Serra, M.L., Coiana, A., Crisponi, G., Boccone, L. and Rosatelli, M.C. (2016) New mutations in DYNC2H1 and WDR60 genes revealed by whole-exome sequencing in two unrelated Sardinian families with Jeune asphyxiating thoracic dystrophy. Clin. Chim. Acta, 455, 172–180. [DOI] [PubMed] [Google Scholar]
- 29. Perrault, I., Saunier, S., Hanein, S., Filhol, E., Bizet, A.A., Collins, F., Salih, M.A., Gerber, S., Delphin, N., Bigot, K.et al. (2012) Mainzer-Saldino syndrome is a ciliopathy caused by IFT140 mutations. Am. J. Hum. Genet., 90, 864–870. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. McInerney-Leo, A.M., Schmidts, M., Corte’s, C.R., Leo, P.J., Gener, B., Courtney, A.D., Gardiner, B., Harris, J.A., Lu, Y., Marshall, M.et al. (2013) Short-rib polydactyly and Jeune syndromes are caused by mutations in WDR60. Am. J. Hum. Genet., 93, 515–523. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Schmidts, M., Frank, V., Eisenberger, T., alTurki, S., Bizet, A.A., Antony, D., Rix, S., Decker, C., Bachmann, N., Bald, M.et al. (2013) Combined NGS approaches identify mutations in the Intraflagellar transport gene IFT140 in skeletal ciliopathies with early progressive kidney disease. Hum. Mutat., 34, 714–724. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Rubino, S., Qian, J., Pinheiro-Neto, C.D., Kenning, T.J. and Adamo, M.A. (2019) A familial syndrome of hypothalamic hamartomas, polydactyly, and SMO mutations: a clinical report of 2 cases. J. Neurosurg. Pediatr., 23, 98–103. [DOI] [PubMed] [Google Scholar]
- 33. Le, T.-L., Sribudiani, Y., Dong, X., Huber, C., Kois, C., Baujat, G., Gordon, C.T., Mayne, V., Galmiche, L., Serre, V.et al. (2020) Bi-allelic variations of SMO in humans cause a broad Spectrum of developmental anomalies due to abnormal hedgehog signaling. Am. J. Hum. Genet., 106, 779–792. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Bose, J., Grotewold, L. and Ruther, U. (2002) Pallister-hall syndrome phenotype in mice mutant for Gli3. Hum. Mol. Genet., 11, 1129–1135. [DOI] [PubMed] [Google Scholar]
- 35. Ocbina, P.J.R., Eggenschwiler, J.T., Moskowitz, I. and Anderson, K.V. (2011) Complex interactions between genes controlling trafficking in primary cilia. Nat. Genet., 43, 547–553. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Miller, K.A., Ah-Cann, C.J., Welfare, M.F., Tan, T.Y., Pope, K., Caruana, G., Freckmann, M.-L., Savarirayan, R., Bertram, J.F., Dobbie, M.S.et al. (2013) Cauli: a mouse strain with an Ift140 mutation that results in a skeletal ciliopathy modelling Jeune syndrome. PLoS Genet., 9, e1003746–e1003746. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Green TE, Schimmel M, Schubert S, Lemke J, Bennett MF, Hildebrand MS and Berkovic SF. (2021) Bi-allelic SMO variants in Hypothalamic Hamartoma: a recessive cause of Pallister-Hall syndrome. Eur J Hum Genet.. Accepted 6th December. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Van Dam, T.J., Wheway, G., Slaats, G.G., Huynen, M.A. and Giles, R.H. (2013) The SYSCILIA gold standard (SCGSv1) of known ciliary components and its applications within a systems biology consortium. Cilia, 2, 1–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Kanehisa, M. and Goto, S. (2000) KEGG: Kyoto encyclopedia of genes and genomes. Nucleic Acids Res., 28, 27–30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Karczewski, K.J., Francioli, L.C., Tiao, G., Cummings, B.B., Alföldi, J., Wang, Q., Collins, R.L., Laricchia, K.M., Ganna, A., Birnbaum, D.P.et al. (2020) The mutational constraint spectrum quantified from variation in 141,456 humans. Nature, 581, 434–443. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Motelow, J.E., Povysil, G., Dhindsa, R.S., Stanley, K.E., Allen, A.S., Feng, Y.-C.A., Howrigan, D.P., Abbott, L.E., Tashman, K., Cerrato, F.et al. (2021) Sub-genic intolerance, ClinVar, and the epilepsies: a whole-exome sequencing study of 29,165 individuals. Am. J. Hum. Genet., 108, 965–982. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Tweedie, S., Braschi, B., Gray, K., Jones, T.E.M., Seal, R.L., Yates, B. and Bruford, E.A. (2021) Genenames.Org: the HGNC and VGNC resources in 2021. Nucleic Acids Res., 49, D939–D946. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.