

Abstract
Previous genome-wide association studies (GWAS) of hematological traits have identified over 10 000 distinct trait-specific risk loci. However, at these loci, the underlying causal mechanisms remain incompletely characterized. To elucidate novel biology and better understand causal mechanisms at known loci, we performed a transcriptome-wide association study (TWAS) of 29 hematological traits in 399 835 UK Biobank (UKB) participants of European ancestry using gene expression prediction models trained from whole blood RNA-seq data in 922 individuals. We discovered 557 gene-trait associations for hematological traits distinct from previously reported GWAS variants in European populations. Among the 557 associations, 301 were available for replication in a cohort of 141 286 participants of European ancestry from the Million Veteran Program. Of these 301 associations, 108 replicated at a strict Bonferroni adjusted threshold ( = 0.05/301). Using our TWAS results, we systematically assigned 4261 out of 16 900 previously identified hematological trait GWAS variants to putative target genes. Compared to coloc, our TWAS results show reduced specificity and increased sensitivity in external datasets to assign variants to target genes.
= 0.05/301). Using our TWAS results, we systematically assigned 4261 out of 16 900 previously identified hematological trait GWAS variants to putative target genes. Compared to coloc, our TWAS results show reduced specificity and increased sensitivity in external datasets to assign variants to target genes.
Introduction
The study of blood cells in humans is well motivated by the role of blood cells as both facilitators of physiological processes and endophenotypes for complex diseases. Blood cells facilitate key physiological processes in human health such as immunity, oxygen transport and clotting. Additionally, measures of blood cells in humans are endophenotypes for complex diseases including asthma, several autoimmune conditions and cardiovascular disease. Thousands of genetic loci associated with blood cell traits have been previously discovered in large genome-wide association studies (GWAS) in both European cohorts and multipopulation studies (1–4).
While GWAS provide general insights into the genetic architecture of blood cell traits, transcriptome-wide association studies (TWAS) are an alternative study design to both identify new genetic loci for complex traits and prioritize potential causal genes at known loci (5–8). A TWAS tests the association between a phenotype of interest and imputed gene expression from genotype-based prediction models trained in a reference dataset. TWAS can have increased statistical power to discover trait-associated genetic loci compared to single variant association tests when a trait association is driven by multiple variants mediated by the expression of a gene or genes. TWAS can gain power by aggregating these multiple mediated single variant signals into a combined test (9). Additionally, TWAS results can shed light on the functional mechanisms underlying variant-trait associations by linking variants to target genes through the gene expression prediction models. Designing appropriate functional experiments to interrogate biological mechanisms or to identify potential drug targets necessitates accurately assigning GWAS variants to target genes. Often, variants are linked to target genes using distance-based approaches, which can lead to inaccurate assignments (10,11). Colocalization-based methods evaluate the evidence that a GWAS variant coincides with an expression quantitative trait locus (eQTL) signal for a gene in a relevant cell type and if these signals are likely driven by the same biological process or the same set of variants. If there is evidence of colocalization, these methods can be used to assign the GWAS variant to a target gene via the eQTL signal. While useful, colocalization methods may be unreliable in situations where there are multiple variants which are associated both with a complex trait in GWAS and linked to the same target gene but with low or moderate effect size. By explicitly linking variants to target genes by including them in gene expression prediction models, TWAS results can provide similar target gene suggestions for GWAS-associated variants.
In this study, we conducted a large TWAS of 29 hematological traits by studying 399 835 participants of European ancestry from the UKB to discover novel loci and assign known GWAS variants to potential target genes (Fig. 1) (12). First, we trained gene expression prediction models using a reference dataset of 922 participants of European ancestry from the Depression Genes and Networks (DGN) cohort with both genotype and RNA-seq data from whole blood (13). Second, we applied the gene expression prediction models trained in DGN to our discovery UKB participants (n = 399 835) to obtain predicted gene expression levels and performed association testing between predicted gene expression values and blood cell phenotypes. Third, we attempted to replicate associations identified in UKB in 141 286 European ancestry participants from the Million Veteran Program (MVP) study (14). Fourth, we performed follow-up analyses including conditional association tests on known GWAS variants, fine-mapping of TWAS loci and pathway analysis in order to interpret TWAS loci. Finally, we systematically assigned the 16 900 conditionally distinct variant-trait associations identified by Vuckovic et al. to target genes and compared our TWAS-based assignments to those from coloc, a commonly used eQTL colocalization method (Fig. 2). By conducting a TWAS with thorough secondary analyses and systematic variant-to-gene assignments in UKB Europeans, our study reveals novel biology and increases functional understanding of genetic loci associated with blood cell traits. We compare the results of our study to a recent GWAS of blood cell traits in UKB Europeans to understand the advantages of TWAS compared to single-variant analyses (3).
Figure 1.

UKB TWAS of blood cell traits overview. (A) We trained gene expression prediction models using whole blood gene expression data from 922 Depression Genes and Networks (DGN) European ancestry participants by fitting an elastic net model on the cis-SNPs (±1 Mb) for each gene. Models with r2 > 0.05 are considered sufficiently predicted, and are subject to association testing in UKB. w represents the TWAS weights in the prediction model. (B) Using our DGN-trained models, we predicted gene expression in 399 835 UKB participants of European ancestry and performed association testing with 29 hematological traits. 11 759 gene-trait associations were significant at the Bonferroni adjusted threshold (out of 290116 tested). (C) TWAS results from UKB were replicated in 141 286 MVP participants of European ancestry for 15 hematological traits available in MVP. (D) We further conditioned our TWAS significant associations in UKB on GWAS signals reported from Vuckovic et al. to determine which TWAS gene-trait associations were driven by previously reported GWAS variants (TWAS CA for TWAS CA). (E) We used the TWAS associations and prediction models (blue stars) to assign GWAS signals from Vuckovic et al. (gold star) to plausible target genes (see Fig. 2), assessing correlation between each GWAS variant and predicted gene expression of each TWAS significant gene.
Figure 2.
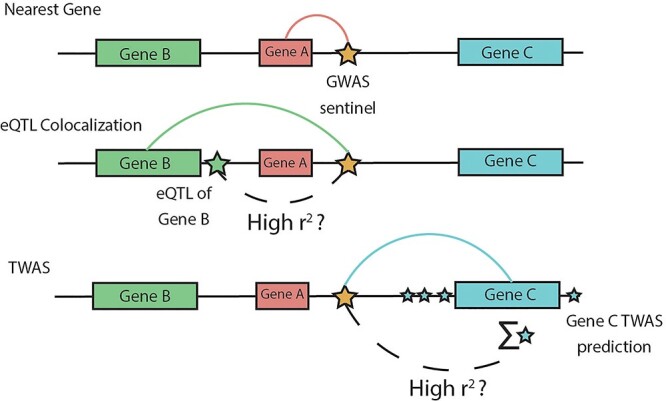
TWAS variant-to-gene approach. Comparison of our TWAS-based approach to variant-to-gene assignment with two commonly used approaches: distance-based and colocalization-based assignments. We consider the problem of assigning a GWAS variant (gold star) in a non-coding region to a target gene. The nearest gene approach assigns the variant to the closest gene at the locus (Gene A), but ignores epigenomic evidence at the locus. Colocalization-based approaches assign the variant to a target gene based on evidence that the GWAS signal is not distinct from an eQTL signal for a target gene (green star, Gene B). Our TWAS-based approach assesses the correlation between the GWAS variant and TWAS predicted gene expression which aggregates smaller effect cis-eQTLs for a gene (blue stars, Gene C). For presentation brevity, we use ‘high r2’ but the threshold to define high correlation can be lenient.
Results
Marginal TWAS results
Using an elastic net-based pipeline, we trained gene expression prediction models using imputed genotypes and whole blood RNA-seq data from 922 European ancestry participants from the DGN cohort (13). In total, we trained prediction models for 12 989 genes, 10 004 of which passed our quality control filter (model R2 > 0.05 and >1 variant selected in model) (Supplementary Material, Fig. S1).
We conducted a TWAS in 399 835 participants of European ancestry from the UKB for 29 blood cell phenotypes: 11 white blood cell indices, 4 platelet indices and 14 red blood cell indices (see Supplementary Material, Table S1). 11 759 gene-trait associations were transcriptome-wide significant at the Bonferroni adjusted threshold of 1.72 × 10−7. The 11 759 associations were grouped into 4835 trait-specific TWAS loci (see Methods) with the most significant gene at each TWAS locus assigned as the sentinel TWAS gene. This procedure resulted in 1792 unique sentinel genes. Among these 1792 sentinel genes, 1112 were sentinel genes for more than one trait (see Supplementary Material, Fig. S2). Of the 4835 TWAS loci, 2375 (49.1%) had multiple TWAS significant genes. We examined the utility of TWAS conditional analysis (CA) for fine-mapping loci with multiple TWAS significant genes and highlighted one such TWAS loci near the erythropoietin gene, EPO (Supplementary Material, Results).
We generated credible sets at all TWAS loci using FINEMAP (see Methods for details) (15). 8928 out of 11 759 (76%) marginal TWAS associations were included in the FINEMAP credible sets for their trait-specific loci. The average number of genes in each FINEMAP credible set was 3.97 (SD = 2.3) and the median was 4 (see Supplementary Material, Table S2). In 297 (6.1%) trait-specific loci, the sentinel TWAS gene was not included in the credible set.
Next, to explore potential biological pathways identified through our TWAS, we performed pathway analysis with clusterProfiler on genes in the FINEMAP credible sets for each phenotype to test for enrichment for gene ontology (GO) terms (see Methods). Thirteen out of the 29 gene sets were enriched for at least one GO term at FDR = 0.05 when compared to the set of all genes that passed QC for their gene expression prediction model (see Supplementary Material, Table S3). Several gene sets were enriched for biologically plausible GO terms: immune response was enriched in the lymphocyte count gene set, erythrocyte development was enriched in the red blood cell distribution width gene set and platelet degranulation was enriched in the platelet count gene set (Supplementary Material, Table S3). These results suggest that TWAS can identify biologically plausible genes associated with complex hematological traits.
In order to replicate significant results from our marginal TWAS analysis, we predicted gene expression values in 141 286 European ancestry participants from MVP using models trained in DGN (see Methods) (14). For the replication analysis, 15 out of the 29 UKB analyzed blood cell traits were available in MVP. 9492 out of the 10 004 (94.8%) gene expression prediction models were comprised of variants that overlapped completely with variants available in MVP. Replication was thus attempted in MVP for 5993 gene-trait associations with fully matching phenotype and gene expression prediction model variants. Among the attempted 5993 gene-trait associations marginally significant in UKB (marginal in contrast to conditional on nearby GWAS variants), 2357 (39.3%) replicated in MVP at the Bonferroni corrected threshold ( = 8.34 × 10−6) with the same direction of effect (Supplementary Material, Fig. S3).
= 8.34 × 10−6) with the same direction of effect (Supplementary Material, Fig. S3).
Conditional analyses adjusting for nearby GWAS variants
We then used CA to determine which of the 11 759 gene-trait associations in UKB represent novel findings beyond a recently published GWAS in UKB Europeans (see Methods for details) (3). Of the 11 759 marginal gene-trait associations, 557 were conditionally significant at the Bonferroni corrected threshold ( = 0.05/11 759 = 4.25 × 10−6, Fig. 3). These 557 associations represent 395 distinct genes in 463 trait-specific TWAS loci; 276 genes were conditionally significant for one trait and 119 for multiple traits (Supplementary Material, Fig. S4). Of the 557 conditionally significant associations, 256 associations could not be replicated in MVP. First, 222 associations did not have matching phenotypes available in MVP. An additional 34 associations did not have complete matching in MVP for variants in the TWAS gene expression prediction model. Thus, we tested 301 genes for replication in MVP. 108 associations (35.9%) replicate at a Bonferroni adjusted threshold (0.05/301 = 1.66 × 10−4) with matching direction of effect (Supplementary Material, Fig. S5).
= 0.05/11 759 = 4.25 × 10−6, Fig. 3). These 557 associations represent 395 distinct genes in 463 trait-specific TWAS loci; 276 genes were conditionally significant for one trait and 119 for multiple traits (Supplementary Material, Fig. S4). Of the 557 conditionally significant associations, 256 associations could not be replicated in MVP. First, 222 associations did not have matching phenotypes available in MVP. An additional 34 associations did not have complete matching in MVP for variants in the TWAS gene expression prediction model. Thus, we tested 301 genes for replication in MVP. 108 associations (35.9%) replicate at a Bonferroni adjusted threshold (0.05/301 = 1.66 × 10−4) with matching direction of effect (Supplementary Material, Fig. S5).
Figure 3.

Manhattan Plot of TWAS conditional analysis results. Figure shows the −log10(P-value) for TWAS gene-trait associations after conditioning on distinct GWAS variants from Vuckovic et al. for a given phenotype category. The red dashed line denotes the Bonferroni adjusted significance threshold (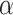 = 4.25 × 10−6). Named genes have −log10(P-value) > 12. The conditional TWAS analysis assesses whether a TWAS signal is driven primarily from signals at previously discovered GWAS loci, which is a crucial step for our analysis of well-studied hematological traits. The maximum −log10(P-value) for each gene is plotted and stratified by phenotype category.
= 4.25 × 10−6). Named genes have −log10(P-value) > 12. The conditional TWAS analysis assesses whether a TWAS signal is driven primarily from signals at previously discovered GWAS loci, which is a crucial step for our analysis of well-studied hematological traits. The maximum −log10(P-value) for each gene is plotted and stratified by phenotype category.
Below, we discuss two subsets of our TWAS CA results which demonstrate the advantages of TWAS over single-variant analyses in UKB and may reveal novel blood cell biology. First, 9 of the 557 conditionally significant gene-trait associations were not within 1 Mb of any distinct GWAS variants for any blood cell trait from Vuckovic et al. (Table 1). These nine TWAS associations are therefore considered loci discovered only by TWAS in UKB Europeans. The RBCK1, IRAK1BP1, SNHG5 and BNIP3 regions were recently reported as associated with their respective traits in large multipopulation GWAS meta-analyses (2,4) validating these TWAS only associations from UKB Europeans. Second, we identified 92 conditionally distinct associations grouped into 70 TWAS loci with no distinct GWAS variants for the corresponding phenotype category within 1 Mb of the gene. This second subset supports that the previously reported GWAS association at the locus is extended to a new class of blood phenotypes. For example, this second subset might include the extension of a locus already associated with red blood cell-related traits to platelet indices.
Table 1.
Blood cell trait genes discovered by TWAS
| Phenotype | Gene Name | Chr | TSS | Model R2 | TWAS Beta | Log10 REGENIE_p | CA Beta | Log10 Conditional_p | MVP Beta | Log10 MVP_p |
|---|---|---|---|---|---|---|---|---|---|---|
| Eosinophil count | RBCK1 | 20 | 407 498 | 0.48 | −0.03 (0.006) | 7.00 | −0.029 (−0.029) | 6.91 | −0.007 (0.006) | 0.57 |
| Mean platelet volume | MFAP3L | 4 | 170 033 031 | 0.07 | 0.034 (0.006) | 7.83 | 0.033 (0.033) | 7.53 | −0.006 (0.007) | 0.36 |
| Mean platelet volume | IRAK1BP1 | 6 | 78 867 472 | 0.23 | 0.03 (0.004) | 11.02 | 0.033 (0.033) | 13.27 | 0.025 (0.005) | 5.47 |
| Mean platelet volume | LPCAT4 | 15 | 34 367 278 | 0.10 | −0.025 (0.004) | 9.63 | −0.024 (−0.024) | 9.46 | −0.005 (0.006) | 0.43 |
| Neutrophil count | SNHG5 | 6 | 85 678 736 | 0.87 | −0.015 (0.003) | 7.88 | −0.013 (−0.013) | 6.18 | −0.009 (0.004) | 2.21 |
| Eosinophil percentage | RBCK1 | 20 | 407 498 | 0.48 | −0.033 (0.006) | 8.77 | −0.033 (−0.033) | 8.74 | NA | NA |
| Monocyte percentage | TMEM144 | 4 | 158 201 604 | 0.53 | 0.031 (0.006) | 6.88 | 0.03 (0.03) | 6.61 | NA | NA |
| Neutrophil count | BNIP3 | 10 | 131 982 013 | 0.08 | 0.022 (0.004) | 6.83 | 0.022 (0.022) | 6.86 | NA | NA |
| Platelet distribution width | GTF2H2 | 5 | 71 067 689 | 0.24 | −0.037 (0.005) | 12.93 | −0.036 (−0.036) | 12.46 | NA | NA |
Four genes could not be replicated in MVP: genes names followed by an asterisk did not have available matching phenotypes in MVP and gene names followed by a plus sign did not have complete variants for the respective gene expression prediction model. Abbreviations: transcription start site (TSS), conditional analysis (CA)
TWAS discovers loci missed by UKB European GWAS
We identified nine gene-trait associations that had no distinct GWAS variants within 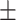 1 Mb of the locus for any blood cell trait in Vuckovic et al. Among the nine associations, three were unable to be assessed for replication in MVP due to phenotype unavailability and one was unable to be assessed due to missing variants in the gene expression prediction model. Three out of the remaining five associations replicated in MVP at a nominal significance threshold (
1 Mb of the locus for any blood cell trait in Vuckovic et al. Among the nine associations, three were unable to be assessed for replication in MVP due to phenotype unavailability and one was unable to be assessed due to missing variants in the gene expression prediction model. Three out of the remaining five associations replicated in MVP at a nominal significance threshold (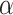 = 0.05) with the same direction of effect as in UKB, namely, interleukin 1 receptor-associated kinase 1 binding protein 1 (IRAK1BP1) for mean platelet volume (beta = 0.025, P = 3.4 × 10−6), and SNHG5 for neutrophil count (beta = −0.0146, P = 0.0061) and white blood cell count (beta = −0.0134, P = 0.013). Below, we highlight the biological implications of the IRAK1BP1 association with mean platelet volume. These results represent gene-trait associations identified by TWAS that were not discovered by single variant analyses in UKB Europeans (3).
= 0.05) with the same direction of effect as in UKB, namely, interleukin 1 receptor-associated kinase 1 binding protein 1 (IRAK1BP1) for mean platelet volume (beta = 0.025, P = 3.4 × 10−6), and SNHG5 for neutrophil count (beta = −0.0146, P = 0.0061) and white blood cell count (beta = −0.0134, P = 0.013). Below, we highlight the biological implications of the IRAK1BP1 association with mean platelet volume. These results represent gene-trait associations identified by TWAS that were not discovered by single variant analyses in UKB Europeans (3).
IRAK1BP1 (chr6:79577189–79656157)
In our TWAS, IRAK1BP1 demonstrated evidence of association with mean platelet volume despite no conditionally distinct GWAS variants within 1 Mb of the gene. The 1 Mb region around IRAK1BP1 contains several genome-wide significant variants in Vuckovic et al., with lead variant chr6:79617522_T_C (P = 6.4 × 10−13, all variants in this manuscript are formatted with the following fields: chromosome number, hg19 position, reference allele and alternate allele) (Fig. 4A). However, this region was grouped into a mean platelet volume locus over 8 Mb away via individual-level CA (sentinel variant chr6:71326034_G_A). Importantly, in the Vuckovic et al. results, no target gene was identified based on proximity for chr6:71326034_G_A via Ensembl Variant Effect Predictor (VEP) (16) limiting the biological interpretation of the findings at the GWAS locus. Our TWAS prediction model for IRAK1BP1 is primarily driven by variants in high LD with chr6:79617522; of the top 15 variants in terms of absolute value of the TWAS weights, 13/15 are in high LD (r2 > 0.8 in TOP-LD Europeans) with chr6:79617522_T_C (Fig. 4B).
Figure 4.

IRAK1BP1 locus EpiGenome browser. Figure demonstrates the cell type-specific epigenetic information linking IRAK1BP1 and mean platelet volume. Figure A shows that variants within IRAK1BP1 were identified as GWAS significant variants in Vuckovic et al. (3), but the signal at this locus was attenuated after conditioning on a locus 8 Mb away (sentinel variant chr6:71326034_G_A). (B) Several of these variants are included in the IRAK1BP1 TWAS prediction model. (C) Promoter-capture Hi-C data support that TWAS model variants for IRAK1BP1 form a loop with the promoter region of LCA5, a Mendelian disease gene for Leber congenital amaurosis. LCA5 was not available in our DGN expression dataset. (D) LCA5 is more strongly expressed in MK cell lines compared to IRAK1BP1. (E) TWAS model variants overlap with MK ATACseq peaks.
After conditioning on all distinct platelet-related variants on chromosome 6, including chr6:71326034_G_A, the marginal TWAS association for IRAK1BP1 and mean platelet volume (beta =0.030, P = 9.47 × 10−12) was not attenuated (beta = 0.033, P = 5.33 × 10−14), demonstrating that the IRAK1BP1 TWAS signal is distinct from previously reported GWAS variants. Furthermore, the association between IRAK1BP1 and mean platelet volume was replicated in MVP Europeans at the Bonferroni adjusted threshold (P = 3.4 × 10−6). Thus, with TWAS, we combined several trait-associated variants at the IRAK1BP1 locus into a stronger signal which demonstrated statistical independence from the previously reported chr6:71326034_G_A signal and all other distinct platelet variants on chromosome 6. Additionally, these results link the variants to putative target genes via our gene expression prediction models.
Figure 4 shows that there is cell type-specific epigenetic evidence that supports our findings. IRAK1BP1 is a component of the IRAK1-dependent TNFRSF1A signaling pathway, which can activate NF-kappa-B and regulate cellular apoptosis and inflammation (17). Variants in the gene expression prediction model for IRAK1BP1 in high LD with chr6:79617522_T_C overlapped with megakaryocyte ATACseq peaks from BLUEPRINT (Fig. 4E) (18). Additionally, we observed via megakaryocyte pcHi-C data that these same variants in the IRAK1BP1 prediction model interact with the promoter region for the nearby gene, lebercilin LCA5 (LCA5) (Fig. 4C). LCA5 plays roles in centrosomal functions in non-ciliary cells (19). While both IRAK1BP1 and LCA5 are expressed in megakaryocyte cells using expression data from BLUEPRINT, the expression level is higher for LCA5, suggesting a potential role for LCA5 in platelet trait variability, despite not being captured by TWAS (Fig. 4D). LCA5 is not present in the DGN reference panel, and thus, unavailable to fit a prediction model, likely because of low expression in whole blood (median transcripts per million 0.018 in Genotype-Tissue Expression (GTEx v8) (20). Integration of our TWAS results with expression and chromatin conformation data in platelet producing megakaryocyte cells revealed candidate genes at this genomic locus; it is possible that the variants in the IRAK1BP1 locus aggregated by the TWAS prediction model impact the expression of LCA5 through spatial proximity to the promoter region of the gene. The IRAK1BP1 locus shows the importance of full consideration of other potential target genes as well as complementary functional annotation resources in the biological interpretation of a TWAS identified signal.
TWAS implicates genes in novel phenotype categories
To further understand the biological significance of our TWAS results, we partitioned the distinct GWAS variants for 29 traits from Vuckovic et al. into three phenotype categories: red blood cell, white blood cell and platelet traits (3). A phenotype category represents a group of biologically related phenotypes. Our TWAS CA identified 92 conditionally significant associations grouped into 70 TWAS loci with no distinct GWAS variants for the corresponding phenotype category within 1 Mb of the gene. Our results support that the previously reported association at the locus is extended to a new class of correlated phenotypes. Of the 92 associations, 42 associations could not be replicated in MVP. First, 33 associations did not have matching phenotypes available in MVP. An additional nine associations did not have complete matching in MVP for variants in the TWAS gene expression prediction model. Thus, we tested 50 genes for replication in MVP. Seventeen out of 50 are replicated at the Bonferroni adjusted threshold for the total number of conditionally significant associations (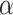 = 0.05/557 = 8.98 × 10−5). CD79B is highlighted as an example of the biological significance of these findings.
= 0.05/557 = 8.98 × 10−5). CD79B is highlighted as an example of the biological significance of these findings.
CD79B (chr17:62006100–62009714)
One such example is the 1 Mb region surrounding B-cell antigen receptor complex-associated protein beta chain (CD79B), which was associated with lymphocyte count (P = 9.81 × 10−10), hematocrit (P = 1.22 × 10−9), plateletcrit (P = 3.37 × 10−9), white blood cell count (P = 8.49 × 10−9) and hemoglobin percentage (P = 1.21 × 10−7) in our TWAS marginal analysis. Supporting the role of this gene in blood cell indices, an extremely rare mutation in CD79B, rs267606711, has been reported to cause agammaglobulinemia 6 [MIM: 612692], an immunodeficiency characterized by profoundly low or absent serum antibodies and low or absent circulating B cells due to an early block of B-cell development (21,22).
In Vuckovic et al., the region surrounding CD79B contained several borderline genome-wide significant variants for lymphocyte count, with lead variant 17:62008437_C_T (P = 2.3 × 10−9). However, in their CA, the region was clumped into nearby lymphocyte count GWAS signals, namely, 17:57929535_A_G (P = 1.16 × 10−25) with annotated target gene RNA, U6 small nuclear 450, pseudogene (RNU6-450P) and 17:65087308_G_C (P = 4.34 × 10−10) with target gene helicase with zinc finger (HELZ) (with both genes assigned based on distance). After conditioning on the set of 186 white blood cell count distinct variants identified by GWAS CA on chromosome 17, including 17:57929535_A_G and 17:65087308_G_C, CD79B continued to demonstrate evidence of association with lymphocyte count (P = 9.8 × 10−10) and white blood cell count (P = 8.5 × 10−9).
Further, there were six distinct GWAS variants from individual-level GWAS CA across both red blood cell and platelet traits within 1 Mb of CD79B. To control for confounding due to correlated hematological traits, we further conditioned on the six distinct variants for red blood cell and platelet traits in addition to the set of 186 white blood cell distinct variants. The association with lymphocyte count remained nominally significant (P = 3.03 × 10−4) and the white blood cell count association was attenuated (P = 0.16). CD79B demonstrated some evidence of association with lymphocyte count in MVP Europeans as well (P = 1.1 × 10−5) with matching direction of association, despite not achieving the Bonferroni adjusted threshold (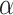 = 8.34 × 10−6). Our findings suggest the biologically plausible CD79B association with lymphocyte count was likely distinct from previously reported genetic loci in the neighborhood, supporting the increased power of TWAS above single variant TWAS.
= 8.34 × 10−6). Our findings suggest the biologically plausible CD79B association with lymphocyte count was likely distinct from previously reported genetic loci in the neighborhood, supporting the increased power of TWAS above single variant TWAS.
TWAS-based assignment of variants to target genes
In addition to identifying gene-trait associations, our study also aimed to assign blood cell trait-associated variants to target genes via TWAS. Our TWAS results allowed us to assign putative target genes to 10 239 variant-trait associations across 10 hematological traits from Vuckovic et al. These 10 239 variant-trait associations had previously been analyzed in an eQTL colocalization analysis with coloc (see Methods). In order to explore the properties of our TWAS-based assignments, we compared the TWAS assignments to those generated by coloc for the same set of GWAS variants.
In their analyses, coloc identified 427 out of 10 239 associations (4.2%) that colocalized with at least one eQTL (Fig. 5A). We assigned the eGene(s) corresponding to these eQTLs as the coloc target gene(s). Our TWAS-based approach assigned target genes to 1738 variant-trait associations, a 4-fold increase compared to coloc. Of the 269 associations assigned to at least one gene by both methods, 80% of the associations have at least one assigned gene in common, demonstrating that the two methods tend to assign variants to the same genes where they both assign a target gene. Of the 158 associations assigned to genes by coloc but not by our TWAS-based approach, 13 were assigned to genes with no expression data in our DGN reference dataset, 23 were assigned to genes with poor model predictive performance (model R2 ≤ 0.05), 51 variant-trait associations were not within ±1 MB of any TWAS loci, 49 were only nearby TWAS loci with a non-significant sentinel gene and 22 had low correlations between variant dosage for the lead GWAS variant and imputed TWAS gene expression (r2 < 0.2) (Supplementary Material, Fig. S6).
Figure 5.

TWAS and coloc variant-to-gene assignments. We compare our TWAS-based variant-to-gene assignments with assignments from coloc using a set of 10 239 variants associated with 10 hematological traits. (A) Coloc successfully assigns 427 variants to target causal genes, while our TWAS-based approach assigns 1738 to target genes. (B, C) We compare these assignments to several external datasets, using variant-to-gene assignments both considering phenotype-specific and phenotype-agnostic approaches. BLUEPRINT Any indicates the target gene is specifically expressed in any cell type in BLUEPRINT dataset, while BLUEPRINT Exact means that the phenotype matches the cell type in BLUEPRINT. OT Any corresponds to the target gene matching any gene indicated as a target gene, while OT Max indicates the target gene was the most likely target for OT. The TWAS-based approach has increased sensitivity to assign genes to potentially causal genes (B) and decreased specificity to coloc (C).
To illustrate one example where the two methods agree, Figure 6 highlights the concordant TWAS and coloc assignment of rs6062304 (chr20:62351539_A_T), a distinct variant for lymphocyte percentage, to Lck-interacting transmembrane adaptor 1 (LIME1), a gene with known involvement in T cell signaling (23,24). In Vuckovic et al., rs6062304 was assigned via VEP annotation to zinc finger CCCH-type and G-patch domain containing (ZGPAT), which has no clear link to blood cells. Figure 6A shows Vuckovic et al. GWAS results overlaid with the marginal TWAS results for lymphocyte percentage. Six TWAS gene-trait associations are significant, and a subset of three genes are included in the FINEMAP 95% credible set: LIME1, ZGPAT and regulator of telomere elongation helicase 1 (RTEL1). Figure 6A shows that LIME1 predicted expression is highly correlated (r2 = 0.905) with rs6062304, while ZGPAT is moderately correlated (r2 = 0.556). Coloc assigns LIME1 as an eGene because of the high LD (r2 = 0.916) between rs6062304 and an eQTL for LIME1, rs6062497 (Fig. 6B). Similarly, Fig. 6C demonstrates that variants with the largest weights in the LIME1 gene expression prediction model are in high LD with rs6062304. In contrast, Figure 6D reveals that variants in high LD with rs6062304 have smaller TWAS weights in the ZGPAT model, suggesting that the ZGPAT association with lymphocytes at this locus is not primarily due to rs6062304. While both LIME1 and ZGPAT correlations pass the r2 cutoff for the TWAS-based gene assignment (r2 > 0.2), LIME1 predicted expression is much more correlated with rs6062304, and is the most likely target gene at this locus according to the TWAS-based approach. This highlights the value of considering correlation of predicted gene expression with the lead GWAS variant in TWAS assignment of likely target genes, as done in our pipeline. Thus, using different approaches, TWAS-based and coloc-based variant-to-gene assignment methods assign rs6062304 to a biologically plausible target gene, improving upon distance-based approaches.
Figure 6.

TWAS and coloc variant-to-gene assignments agree at LIME1 locus. The LIME1-lymphocyte percentage associated locus illustrates one example where the TWAS- and coloc-based variant-to-gene assignments agree. (A) Genes in the FINEMAP credible set are colored by their correlation with rs6062304. The predicted gene expression with LIME1 is highly correlated with rs6062304, whereas neither ZGPAT nor RTEL1 are. (B) An eQTL for LIME1, rs6062497, is in high LD with rs6062304, and in turn coloc assigns LIME1 as an eGene. (C, D) Model variants for LIME1 and ZGPAT are colored by their LD with rs6062304, respectively. The variants with the largest effect sizes in the TWAS prediction model for LIME1 are in high LD with rs6062304, whereas those for ZGPAT are not.
As reported above, the TWAS based approach assigned four times as many variants to target genes. Figure 5A shows that there are 1469 variant-trait pairs which are assigned to a target gene via TWAS not assigned to a gene by coloc. One such example is the TWAS assignment of rs1985157 to leucine-rich repeat containing 25 (LRRC25) (chr19:18513594_T_C), a distinct variant for neutrophil count and neutrophil percentage. Neither VEP nor coloc assigned rs1985157 to a target gene. Our TWAS marginal analysis identified four significant genes for neutrophil count at this locus, LRRC25, elongation factor for RNA polymerase II (ELL), single-stranded DNA binding protein 4 (SSBP4) and inositol-3-phosphate synthase 1 (ISYNA1) (Fig. 7A). However, only LRRC25 predicted gene expression values have a strong correlation with rs1985157 (r2 = 0.863). Two other TWAS-assigned genes are moderately correlated with rs1985157 (ELL r2 = 0.46) and (SSBP4 r2 = 0.47). Figure 7C shows that variants in the LRRC25 prediction model that are in high LD with rs1985157 have the largest weights in absolute value. In contrast, Figure 7D shows that SSBP4 predicted expression is driven by variants in moderate LD with rs1985157. Several studies have suggested that LRRC25 plays a key role in innate immune response and autophagy (25,26). Further, cell type-specific gene expression data from BLUEPRINT suggest that LRRC25 is specifically expressed in neutrophils (18). Our results show that TWAS-based variant-to-gene assignment methods can identify biologically plausible target genes, even when coloc fails to do so.
Figure 7.

TWAS assigns rs1985157 to LRRC25. Figure illustrates how TWAS assigned rs1985157 to LRRC25 when coloc failed to do so with no individual significant eQTL in the region. (A) LRRC25 predicted gene expression was highly correlated with rs1985157 (r2 = 0.863), whereas the prediction from SSBP4 (r2 = 0.47) and ELL (r2 = 0.457) were not as highly correlated despite both genes being significant. (B) LD patterns for variants at the locus. (C, D) Only model variants for LRRC25 and SSBP4 are colored by their LD with rs1985157, respectively. Variants with the largest TWAS weights (in absolute values) for LRRC25 are in high LD with rs1985157, whereas those for SSBP4 are not.
Annotating target genes assigned by TWAS and coloc
In order to understand the differences in the TWAS and coloc gene assignments and to examine whether the additional variants assigned to genes by TWAS over coloc have relevant epigenetic evidence to the phenotype of interest, we compared the gene assignments of TWAS and coloc using BLUEPRINT cell type-specific expression data and Open Targets V2G scores (see Methods for details) (27). While the presence of evidence from BLUEPRINT cell type-specific expression analyses or Open Targets scores does not prove that an assigned gene is the true target gene, a similar preponderance of evidence in external datasets is often used in practice to select genes for functional validation experiments. Therefore, we are primarily interested in exploring the utility of TWAS and coloc to generate plausible hypotheses of target genes for GWAS variants.
We found that the TWAS-based approach assigned GWAS variants to genes identified by external datasets at a slightly lower rate than the coloc assignments, but identified target genes for more than double the number of variants (see Fig. 5B and C). Specifically, Figure 5C shows that 84% of the TWAS-based variant-to-gene assignments are supported by Open Targets (OT Any genes), and 64% of genes assigned by TWAS are the most likely target gene as identified by Open Targets (OT Max gene). In comparison, 88% of the coloc assigned genes are supported by OT Any genes and 78% as the OT Max gene. On the other hand, Figure 5C shows that 294 TWAS pairs are assigned to an OT Any gene and 226 pairs assigned to an OT Max gene, much larger number of supported assignments than the 85 and 76 coloc pairs, a 3.46- and 1.97-fold increase, respectively. The proportion of variants assigned to cell type specifically expressed genes in BLUEPRINT expression data is lower compared to the Open Targets assignments (Fig. 5C). However, the TWAS-based approach matches 3.12-fold more variants to specifically expressed genes in trait-relevant cell types and 3.73-fold more genes to specifically expressed genes for any blood cell compared to coloc. Therefore, our results suggest that TWAS, compared to coloc, is less specific but more sensitive when assigning variants to target genes supported by external annotations.
Since not all traits were considered for the previous eQTL colocalization analysis in UKB Europeans, we applied our TWAS-based variant-to-gene assignment to all 29 hematological traits considered in our UKB TWAS. We successfully assigned 4261 variant-trait associations to 1842 distinct potentially causal genes with an average of 1.45 (SD = 0.81) genes assigned per variant-trait association (see Supplementary Material, Table S4). Of the 4261 associations, 746 (17.5%) were assigned to specifically expressed genes in trait-relevant cell types, and 1982 (46.5%) were assigned to specifically expressed genes for any blood cell. Both rates were comparable to the performance of the TWAS variant-to-gene assignments in the phenotype restricted results above. For the 813 overlapping variant-to-gene assignments from the Open Targets datasets, the replication rates were similar to the phenotype restricted results for OT Any genes (78.2%), but the replication rate decreased for the OT Max gene (54.5%).
Discussion
Our TWAS of blood cell traits in UKB Europeans identified loci missed by a GWAS in UKB Europeans and extended known loci to additional phenotype categories, even in well-studied hematological traits for which over 10 000 loci have been reported by previous GWAS (2,3). We identified nine loci that were undiscovered by GWAS of UKB Europeans for blood cell traits. For example, the IRAK1BP1 locus was associated with mean platelet volume, and our secondary analyses suggest that both IRAK1BP1 and LCA5 may be plausible target genes for genetic variants at this locus. As noted above, five of the nine loci were reported in large multipopulation or cross-cohort meta-analyses (2,4), but had not been previously reported in the UKB GWAS, supporting the validity of the additional TWAS findings. These results demonstrate advantages of TWAS over single-variant analyses for novel locus identification within the same cohort.
Further, we extended 92 previously reported associations at genomic loci to a new class of correlated phenotypes. Due to the shared genetic architecture of blood cell traits which is mediated through the differentiation of common progenitor cells, variants which impact one class of blood cell traits may have an effect on other hematological traits. For example, the CD79B locus demonstrated a robust association with lymphocyte count despite conditioning on previously identified white blood cell, red blood cell and platelet distinct variants at the locus. This robust association confirms previously reported biological roles for CD79B with lymphocyte function, and establishes relevant variant-level candidates for functional validation through the TWAS prediction model (21,22). Our results suggest several insights into the genetic architecture of blood cell traits through TWAS loci discovery above single-variant studies and extension of known loci to new phenotypes.
We addressed challenges with interpreting TWAS loci at the scale of biobank-sized analyses through our adapted TWAS fine-mapping via FINEMAP (28). To our knowledge, all TWAS fine-mapping methods and software are currently designed for summary statistics-based TWAS approaches, including the recently published FOCUS method, with limited functionality to input user-generated TWAS statistics from individual-level data such as those generated by our REGENIE-based approach (29,30). To overcome this challenge, we substituted the variant-level LD matrix for the predicted expression correlation matrix in our UKB sample in the FINEMAP software to generate credible sets of genes. Using fine-mapped sets of TWAS results, we conducted pathway analysis, and the identified gene sets were enriched for trait-relevant GO terms (Supplementary Material, Table S3), highlighting the biological plausibility of our fine-mapped gene sets. One shortcoming of our approach is that we addressed correlation at the gene level via predicted expression values, and this substitution for TWAS fine-mapping may not be valid since FINEMAP was originally designed for GWAS fine-mapping. Our extension of FINEMAP is an ad hoc solution to the problem of TWAS loci fine-mapping, which is more complex than variant-level fine-mapping due to correlations at both the variant and gene levels (30,31). In addition to previous research, future methodological research and software development should be done to address this challenge (28,30,31).
Additionally, we performed systematic variant-to-gene assignment for distinct hematological trait GWAS signals using a TWAS-based approach, and demonstrated that many of our assignments are supported by external datasets. While the use of external datasets such as Open Targets or BLUEPRINT does not prove that the TWAS or coloc gene is the true causal gene, a preponderance of evidence from tissue-specific external datasets suggest that a gene could be a good candidate for follow-up with functional experiments. As identifying candidates for functional follow-up is often one goal of large association studies, we believe that this in silico external replication metric is reasonable for our TWAS and colocalization comparisons.
Our variant-to-gene assignment results support complementary roles of TWAS and colocalization approaches. The TWAS-based approach of assigning GWAS variants to target genes mapped more variants to target genes using biobank scale data compared to an eQTL colocalization approach. However, this increased number of variants assigned to target genes decreased sensitivity in external annotations. One possible explanation of this result is that in scenarios where eQTLs have not been identified in a target tissue of interest, likely due to small sample size for a given expression dataset, TWAS-based methods, which combine multiple potential eQTLs which may be in LD with a GWAS variant, are more likely to assign GWAS variants to target genes. To systematically assign GWAS variants to target genes, we propose first using colocalization to assign GWAS variants to target genes using available cell type-specific eQTL data relevant to the trait of interest, and then leveraging the additional assignments generated by TWAS for GWAS variants not assigned to a target gene.
There are still several future directions for the improvement of biobank-scale TWAS. First, increasing sample sizes in tissue-specific expression datasets will allow future TWAS studies to train gene expression prediction models in cell/tissue types which are directly relevant to traits of interest. Already, several TWAS methods have been developed to leverage multiple tissues to train better gene expression prediction models (7,8,32). For example, at the identified IRAK1BP1 locus, it would be useful to have larger megakaryocyte-specific gene expression datasets available for TWAS model training; similar cell type-specific panels would be useful for other hematological indices and for complex trait analysis more generally. Additionally, the TWAS variant-to-gene assignment approach would benefit from larger expression datasets to train cell/tissue type-specific gene expression prediction models to assess the correlation between predicted expression and a GWAS variant of interest across several relevant models. Such cell type-specific reference panels are becoming increasingly available, though not always in adequate sample sizes for TWAS and not always with publicly available individual-level data (33). Second, extending the variable selection procedure for prediction models past the 1 Mb cis-region surrounding genes either via trans-eQTL datasets or by selecting variants which are highly likely to be in interesting epigenomic regions will improve TWAS models (34,35).
In summary, we conducted a large-scale TWAS of well-studied hematological traits and identified loci undiscovered by GWAS in the same cohort. We showed that TWAS-based approaches for assigning variants to their target genes were comparable in specificity to co-localization-based approaches, but were able to assign many more variants (4.07-fold increase) to target genes. Our careful use of CA, TWAS-based fine-mapping and TWAS-based variant-to-gene assignments in the context of blood cell traits will be broadly useful to the practice of TWAS for other complex traits.
Materials and Methods
Included cohorts
Depression genes and networks (DGN)
The DGN study was designed to collect samples of individuals with and without major depressive disorder, ages 21–60, from a survey research panel broadly representative of the United States population (13). Genotyping and RNA-sequencing procedures have been described previously (13). For 922 European ancestry participants from the DGN study, we obtained both genotype data imputed to the Trans-Omics in Precision Medicine (TOPMed) Freeze 8 reference panel and RNA-seq data (36,37). Whole blood samples were from PAXgene tubes, which retain red blood cells unlike peripheral blood mononuclear cells. Therefore, we may be more likely to detect associations with blood cell traits in DGN versus peripheral blood mononuclear cell-based datasets.
Quality control in DGN
For training gene expression prediction models, we included bi-allelic variants that are common and well-imputed (MAF > 0.05, Rsq > 0.8) in both DGN and in the UKB. In all, 5 652 397 variants were included, here forward referred to as QC variants. DGN whole-blood RNA-seq data for both coding and non-coding genes was obtained for 922 European ancestry participants (13). As described previously, quantified gene expression values were normalized using the hidden covariates with the prior method (38), correcting for technical and biological factors, including blood cell type frequencies and the time of the blood draw (13).
UK Biobank (UKB) Europeans
UKB recruited 500 000 people aged between 40 and 69 years in 2006–2010, establishing a prospective biobank study to understand risk factors for common diseases such as cancer, heart disease, stroke, diabetes, and dementia (12). Participants are being followed-up through health records from the UK National Health Service. UKB has genotype data, imputed with UK10K as reference, on all enrolled participants, as well as extensive baseline questionnaires and physical measures and stored blood and urine samples. Hematological traits were assayed as previously described (1). Genotyping on custom Axiom arrays, subsequent quality control and imputation has been previously described (12).
For our TWAS, we analyzed UKB participants of European ancestry to match the genetic ancestry of DGN participants used for model training. Participants were included in our analysis if identified as European through a combination of self-reported ethnicity and k-means clustering of genetic principal components (PCs) in order to minimize genomic inflation due to population stratification, and for consistency with previously published blood cell trait GWAS in UKB (3). First, we calculated PCs and their loadings for all 488 377 genotyped UKB participants using LD pruned variants (pairwise r2 < 0.1) with MAF 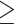 0.01 and missing rate
0.01 and missing rate 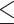 0.015 in the UKB data set that overlapped with the participants in the 1000G Phase 3 v5 (1KG) reference panel (12). Reference ancestries used included 504 European, 347 American, 661 African, 504 East Asian and 489 South Asian samples (overall 2504). We projected the 1KG reference panel dataset on the calculated PC loadings from UKB. We then used k-means clustering with four dimensions, defined by the first four PCs, to identify individuals that clustered with the majority of 1KG reference panels in each ancestry. We used self-reported ethnicity, in some circumstances, to adjust these groups. UKB participants defined as European ancestry include those that cluster with most 1KG Europeans by k-means clustering. We adjusted this group by removing those that self-reported as Indian, Pakistani, Bangladeshi, any other Asian background, Black or Black British, Caribbean, African, any other Black background or Chinese (n = 32). Additionally, we removed any individuals with self-reported mixed ancestry (n = 402). A total of 451 305 participants remained in the European ancestry group. Participants were also excluded based on factors likely to cause major perturbations in hematological indices including positive pregnancy status, drug treatments, cancer self-report, ICD9 and ICD10 disease codes (see Supplementary Material, Supplemental Text) and surgical procedures. Participants were included only if they had complete data for all covariates and phenotypes. In total, 399 835 samples were included in the analysis.
0.015 in the UKB data set that overlapped with the participants in the 1000G Phase 3 v5 (1KG) reference panel (12). Reference ancestries used included 504 European, 347 American, 661 African, 504 East Asian and 489 South Asian samples (overall 2504). We projected the 1KG reference panel dataset on the calculated PC loadings from UKB. We then used k-means clustering with four dimensions, defined by the first four PCs, to identify individuals that clustered with the majority of 1KG reference panels in each ancestry. We used self-reported ethnicity, in some circumstances, to adjust these groups. UKB participants defined as European ancestry include those that cluster with most 1KG Europeans by k-means clustering. We adjusted this group by removing those that self-reported as Indian, Pakistani, Bangladeshi, any other Asian background, Black or Black British, Caribbean, African, any other Black background or Chinese (n = 32). Additionally, we removed any individuals with self-reported mixed ancestry (n = 402). A total of 451 305 participants remained in the European ancestry group. Participants were also excluded based on factors likely to cause major perturbations in hematological indices including positive pregnancy status, drug treatments, cancer self-report, ICD9 and ICD10 disease codes (see Supplementary Material, Supplemental Text) and surgical procedures. Participants were included only if they had complete data for all covariates and phenotypes. In total, 399 835 samples were included in the analysis.
Quality control in UKB
As mentioned previously, we included only bi-allelic and well-imputed common variants (Rsq > 0.8, MAF > 0.05) in UKB. All 29 blood cell phenotypes were adjusted for age, age2, top 10 genotype PCs, center, genotyping array and sex. For white blood cell traits, phenotypes were log10(x + 1) transformed before regression. Residuals from these regression models were inverse normal transformed and serve as phenotypes.
Million veteran program (MVP) Europeans
The MVP is an observational cohort study and mega-biobank in the Department of Veteran Affairs healthcare system which began enrollment in 2011. As of Release 3, 318 725 individuals of European ancestry (as defined by HARE (39)) have available electronic health records, survey and genotype data. After quality control largely following the guidelines established in Marees et al. (40), 308 778 individuals of European ancestry remained.
Quality control in MVP
Only a subset of 15 hematological traits out of the 29 analyzed in UKB was available for replication in MVP. For our replication study, participants were limited to those with available data among these 15 traits (n = 141 286). Phenotypes were adjusted for covariates following the same procedure as in UKB.
Training of gene expression prediction models
We trained gene expression prediction models using an elastic net pipeline following the well-established PrediXcan methodology (9). We set 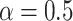 for all gene expression prediction models since the Elastic Net with
for all gene expression prediction models since the Elastic Net with 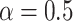 has been previously demonstrated to be a robust choice for modeling gene expression compared to LASSO or Ridge Regression (9,41).
has been previously demonstrated to be a robust choice for modeling gene expression compared to LASSO or Ridge Regression (9,41).
Our decision to use an in-house pipeline rather than the publicly available weights from PrediXcan was 2-fold. First, we performed TOPMed freeze 8-based imputation, enhancing genome coverage and imputation quality compared to the reference panel underlying the PrediXcan weights, the 1000G Phase 1 v3 ShapeIt2 (no singletons) panel. Second, by training our own prediction models, we ensured that every variant present in the prediction models was available in our UKB dataset.
For each gene, we included variants within a 1 Mb window of the gene start and end positions and excluded variants in high pairwise LD (r2 > 0.9) with other variants in the window. We tuned the elastic net penalty parameters using 5-fold cross-validation with the ‘glmnet’ function in R. We obtained 12 898 elastic net models where more than one variant was included in the prediction model. Models with a single variant were excluded from our TWAS. This modeling decision was made in order to differentiate our TWAS from previous single-variant analyses of hematological traits. Single-variant prediction models could still provide useful information linking a single-variant to a target gene, but are not considered here. We further excluded models with model R2 ≤ 0.05, leading to 10 004 models for subsequent analysis (Supplementary Material, Fig. S1).
Association testing with REGENIE
Using the 10 004 models trained in DGN, we predicted gene expression values in UKB European ancestry participants. We then performed association testing between predicted gene expression and covariate-adjusted blood cell phenotypes with REGENIE (42). We used an LD (linkage disequilibrium) pruned (plink—indep-pairwise 50 5 0.1) set of 174 957 variants with MAF > 0.01 in the genotype data available for UKB Europeans to fit the REGENIE null model accounting for cryptic relatedness. We analyze all 29 phenotypes simultaneously using the grouping option available in REGENIE and set the number of blocks to 1000.
To control Type I error at 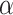 = 0.05, we considered a TWAS association significant if P < 0.05/(10 004 * 29) = 1.72*10−7. Note that this Bonferroni adjusted significance threshold is rather conservative due to correlations among the blood cell phenotypes and among predicted expressions of genes. Results from this TWAS association analysis are referred to throughout the manuscript as the marginal TWAS results.
= 0.05, we considered a TWAS association significant if P < 0.05/(10 004 * 29) = 1.72*10−7. Note that this Bonferroni adjusted significance threshold is rather conservative due to correlations among the blood cell phenotypes and among predicted expressions of genes. Results from this TWAS association analysis are referred to throughout the manuscript as the marginal TWAS results.
For each trait, we grouped multiple significant TWAS gene-trait associations within the same region into TWAS loci via the following procedure. First, we selected the most significant TWAS gene as the TWAS sentinel gene for the locus. Second, we assigned all genes within 1 Mb of the gene as a member of the locus defined by the TWAS sentinel gene. Third, we repeated this process only considering genes not yet assigned to a TWAS locus. The procedure is complete when all TWAS significant genes for a given trait are assigned a locus.
Conditional analysis
In order to assess which marginally significant TWAS gene-trait associations provide novel findings above and beyond the single variant discoveries in GWAS of blood cell traits in Europeans (3), we tested the association between predicted gene expression and phenotype while conditioning on reported blood cell trait GWAS variants. This methodology has been described in a previous TWAS of blood cell traits from our group (28). We partitioned the distinct GWAS variants from Vuckovic et al. into three phenotype categories: red blood cell, white blood cell and platelet traits (3). We considered all distinct GWAS variants as determined by CA on individual-level data, referred to as conditionally independent variants by Vuckovic et al. For a TWAS gene associated with one trait in the above categories, we conditioned on any distinct variant reported as associated with any trait within the corresponding phenotype category on the same chromosome.
Replication analysis in MVP
We conducted two replication analyses in MVP Europeans to follow-up on our results from the UKB TWAS: one for the marginal TWAS results and a second restricted to only conditionally significant genes. In both analyses, our DGN trained gene expression prediction models were used to impute gene expression values in MVP Europeans. Association testing was performed via boltLMM (43). The Bonferroni adjusted thresholds for replication were determined by the number of marginal or conditional associations in the UKB available for replication, respectively.
TWAS fine-mapping via FINEMAP
We modified the FINEMAP software to compute credible sets of genes from our marginal TWAS results (15). We substituted GWAS summary statistics for our TWAS summary statistics from the marginal TWAS analysis. In place of an LD matrix, we used a gene–gene correlation matrix computed on the predicted gene expression values in UKB Europeans. We compute the FINEMAP credible sets and posterior probabilities of inclusion for all TWAS loci with at least two genes.
Pathway analysis
We conducted a pathway analysis using the clusterProfiler R package to search for GO terms that were enriched among FINEMAP credible sets of TWAS significant genes (44). For each phenotype, we defined the gene set for each phenotype as the set of TWAS significant genes with FINEMAP posterior probability of inclusion in the credible set >0.5. The universe of genes was the set of 10 004 genes with gene expression prediction models that passed prediction QC. Multiple testing was addressed by setting the false discovery rate for each phenotype to 0.05. The minimum gene set size for genes annotated with a GO term was set to 10.
TWAS variant-to-gene assignments
We assigned the distinct GWAS variants from Vuckovic et al. to putative target genes using our TWAS results (3). For a GWAS variant-trait association, we considered all significant TWAS gene-trait associations for the matching trait in any TWAS locus within 1 Mb of the variant. We assigned the variant to a gene if the TWAS gene had both a FINEMAP posterior probability of inclusion greater than 0.5, and evidence of correlation (r2 > 0.2) between the variant genotype and predicted gene expression. We performed our TWAS assignments on 10 239 variant-trait associations across 10 hematological traits from Vuckovic et al. (3) In their original paper, these 10 traits were chosen by Vuckovic et al. based on data availability for eQTLs in relevant cell types including platelets, CD4+, CD8+, CD14+, CD15+ and CD19+ cells. In their work, they performed eQTL colocalization analyses using coloc. For a GWAS variant, we assigned the eGene(s) corresponding to any colocalizing eQTL as the target gene.
Open targets
Open Targets Genetics is an open-access integrative resource which aggregates human GWAS and functional genomics data including gene expression, protein abundance, chromatin interaction and conformation data in order to make robust connections between GWAS loci and potentially causal genes (27). In order to assign potentially causal genes to a given GWAS variant, Open Targets provides a disease-agnostic variant-to-gene (V2G) score which combines a single aggregated score for each GWAS variant-gene prediction. This analysis combines four different data types: eQTL and pQTL datasets, chromatin interaction and conformation datasets, VEP scores and distance from the canonical transcription start site for a target gene. We compare the TWAS and coloc variant-to-gene assignments to the sets of potentially causal genes identified by Open Targets. Performance is assessed by checking if any TWAS/coloc assigned gene for a given variant is either the most likely gene identified by Open Targets (OT Max) or any gene identified by Open Targets (OT Any).
BLUEPRINT specifically expressed genes
We also assessed the quality of the gene assignments for the TWAS and coloc-based methods by determining if the assigned gene is cell type specifically expressed in gene expression data from BLUEPRINT (18). We group available expression data into five cell type groups: erythrocytes, megakaryocytes, macrophages and monocytes, nCD4 cells and neutrophils. We classified genes as cell type group-specific or shared via Shannon entropy across the five cell type groups. We first exponentiated the BLUEPRINT MMSEQ expression quantifications, to be comparable to RPKM. Then, for each gene, we calculated the normalized gene expression by dividing gene expression in each cell type group by the sum across all five cell type groups. Next, we calculated Shannon entropy using the normalized gene expression values. We defined the shared genes across cell type groups as those with entropy <0.1 and the cell type-specific genes as those with entropy >0.5 and gene expression >1 in the respective cell type. Biologically plausible cell type groups selected for the 29 phenotypes analyzed are detailed in Supplementary Material, Table S1.
Supplementary Material
Acknowledgements
This research has been conducted using the UKB Resource under Application Number 25953. Support for title page creation and format was provided by AuthorArranger, a tool developed at the National Cancer Institute.
This research is based on data from the Million Veteran Program, Office of Research and Development, Veterans Health Administration and was supported by awards #MVP006 and #MVP001. This publication does not represent the views of the Department of Veteran Affairs or the United States Government.
The Genotype-Tissue Expression Project was supported by the Common Fund of the Office of the Director of the National Institutes of Health, and by National Cancer Institute, National Human Genome Research Institute, National Heart, Lung and Blood Institute (NHLBI), National Institute on Drug Abuse, National Institute of Mental Health and National Institute of Neurological Disorders and Stroke. The data used for the analyses described in this manuscript were obtained from: the GTEx Portal on 05/01/2021.
Trans-Omics in Precision Medicine program imputation panel (version TOPMed-r2) supported by the NHLBI; see www.nhlbiwgs.org. TOPMed study investigators contributed data to the reference panel, which can be accessed through the TOPMed Imputation Server. The panel was constructed and implemented by the TOPMed Informatics Research Center at the University of Michigan (3R01HL-117626-02S1; contract HHSN268201800002I). The TOPMed Data Coordinating Center (3R01HL-120393-02S1; contract HHSN268201800001I) provided additional data management, sample identity checks and overall program coordination and support. We gratefully acknowledge the studies and participants who provided biological samples and data for TOPMed.
Codes to analyze the data can be found at https://github.com/brycerowland/UKB_BCT_TWAS.git.
Conflict of Interest statement. The authors have no conflicts of interest.
Contributor Information
Bryce Rowland, Department of Biostatistics, University of North Carolina at Chapel Hill, Chapel Hill, NC 27599, USA.
Sanan Venkatesh, Department of Genetics and Genomic Sciences, Icahn School of Medicine at Mount Sinai, New York City, NY 10029, USA; Mental Illness Research, Education, and Clinical Center (VISN 2 South), James J. Peters VA Medical Center, Bronx, NY 10468, USA.
Manuel Tardaguila, Department of Human Genetics, Wellcome Sanger Institute, Hinxton CB10 1SA, UK.
Jia Wen, Department of Genetics, University of North Carolina at Chapel Hill, Chapel Hill, NC 27599, USA.
Jonathan D Rosen, Department of Biostatistics, University of North Carolina at Chapel Hill, Chapel Hill, NC 27599, USA.
Amanda L Tapia, Department of Biostatistics, University of North Carolina at Chapel Hill, Chapel Hill, NC 27599, USA.
Quan Sun, Department of Biostatistics, University of North Carolina at Chapel Hill, Chapel Hill, NC 27599, USA.
Mariaelisa Graff, Department of Epidemiology, University of North Carolina at Chapel Hill, Chapel Hill, NC 27599, USA.
Dragana Vuckovic, Department of Epidemiology and Biostatistics, School of Public Health, Faculty of Medicine, Imperial College London, London SW7 2AZ, UK.
Guillaume Lettre, Montreal Heart Institute, Université de Montréal, Montreal, Quebec, Canada.
Vijay G Sankaran, Division of Hematology/Oncology, Boston Children's Hospital, Boston, MA 02115, USA; Department of Pediatric Oncology, Dana-Farber Cancer Institute, Boston, MA 02115, USA; Broad Institute of MIT and Harvard, Cambridge, MA 02142, USA.
Georgios Voloudakis, Mental Illness Research, Education, and Clinical Center (VISN 2 South), James J. Peters VA Medical Center, Bronx, NY 10468, USA; Department of Genetics and Genomic Sciences, Icahn School of Medicine at Mount Sinai, New York City, NY 10029, USA; Department of Psychiatry, Icahn School of Medicine at Mount Sinai, New York City, NY 10029, USA.
Panos Roussos, Mental Illness Research, Education, and Clinical Center (VISN 2 South), James J. Peters VA Medical Center, Bronx, NY 10468, USA; Department of Genetics and Genomic Sciences, Icahn School of Medicine at Mount Sinai, New York City, NY 10029, USA; Department of Psychiatry, Icahn School of Medicine at Mount Sinai, New York City, NY 10029, USA.
Jennifer E Huffman, Center for Population Genomics, Massachusetts Veterans Epidemiology Research and Information Center (MAVERIC), VA Boston Healthcare System, Boston, MA 02130, USA.
Alexander P Reiner, Department of Epidemiology, University of Washington, Seattle, WA 98195, USA.
Nicole Soranzo, Department of Human Genetics, Wellcome Sanger Institute, Hinxton CB10 1SA, UK.
Laura M Raffield, Department of Genetics, University of North Carolina at Chapel Hill, Chapel Hill, NC 27599, USA.
Yun Li, Department of Biostatistics, University of North Carolina at Chapel Hill, Chapel Hill, NC 27599, USA; Department of Genetics, University of North Carolina at Chapel Hill, Chapel Hill, NC 27599, USA; Department of Computer Science, University of North Carolina at Chapel Hill, Chapel Hill, NC 27599, USA.
Funding
National Science Foundation (DGN-1650116 to B.R.); Brain and Behavior Research Foundation (29350 to G.V.); and the National Institutes of Health (K08MH122911 to G.V., R01DK103794 to V.S., KL2TR002490 to L.M.R., R01 HL129132 to Y.L., R01 HL146500 to A.P.R. R01 GM105785 to Y.L. U01HG011720 to Y.L.).
References
- 1. Astle, W.J., Elding, H., Jiang, T., Allen, D., Ruklisa, D., Mann, A.L., Mead, D., Bouman, H., Riveros-Mckay, F., Kostadima, M.A.et al. (2016) The allelic landscape of human blood cell trait variation and links to common complex disease. Cell, 167, 1415–1429.e19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Chen, M.-H., Raffield, L.M., Mousas, A., Sakaue, S., Huffman, J.E., Moscati, A., Trivedi, B., Jiang, T., Akbari, P., Vuckovic, D.et al. (2020) Trans-ethnic and ancestry-specific blood-cell genetics in 746,667 individuals from 5 global populations. Cell, 182, 1198–1213.e14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Vuckovic, D., Bao, E.L., Akbari, P., Lareau, C.A., Mousas, A., Jiang, T., Chen, M.H., Raffield, L.M., Tardaguila, M., Huffman, J.E.et al. (2020) The polygenic and monogenic basis of blood traits and diseases. Cell, 182, 1214–1231.e11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Sakaue, S., Kanai, M., Tanigawa, Y., Karjalainen, J., Kurki, M., Koshiba, S., Narita, A., Konuma, T., Yamamoto, K., Akiyama, M.et al. (2021) A cross-population atlas of genetic associations for 220 human phenotypes. Nat. Genet., 53, 1415–1424. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Gusev, A., Mancuso, N., Won, H., Kousi, M., Finucane, H.K., Reshef, Y., Song, L., Safi, A., McCarroll, S., Neale, B.M.et al. (2018) Transcriptome-wide association study of schizophrenia and chromatin activity yields mechanistic disease insights. Nat. Genet., 50, 538–548. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Bhattacharya, A., García-Closas, M., Olshan, A.F., Perou, C.M., Troester, M.A. and Love, M.I. (2020) A framework for transcriptome-wide association studies in breast cancer in diverse study populations. Genome Biol., 21, 42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Hu, Y., Li, M., Lu, Q., Weng, H., Wang, J., Zekavat, S.M., Yu, Z., Li, B., Gu, J., Muchnik, S.et al. (2019) A statistical framework for cross-tissue transcriptome-wide association analysis. Nat. Genet., 51, 568–576. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Zhou, D., Jiang, Y., Zhong, X., Cox, N.J., Liu, C. and Gamazon, E.R. (2020) A unified framework for joint-tissue transcriptome-wide association and Mendelian randomization analysis. Nat. Genet., 52, 1239–1246. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Gamazon, E.R., Wheeler, H.E., Shah, K.P., Mozaffari, S.V., Aquino-Michaels, K., Carroll, R.J., Eyler, A.E., Denny, J.C., Nicolae, D.L., Cox, N.J.et al. (2015) A gene-based association method for mapping traits using reference transcriptome data. Nat. Genet., 47, 1091–1098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Zhu, Z., Zhang, F., Hu, H., Bakshi, A., Robinson, M.R., Powell, J.E., Montgomery, G.W., Goddard, M.E., Wray, N.R., Visscher, P.M.et al. (2016) Integration of summary data from GWAS and eQTL studies predicts complex trait gene targets. Nat. Genet., 48, 481–487. [DOI] [PubMed] [Google Scholar]
- 11. Musunuru, K., Strong, A., Frank-Kamenetsky, M., Lee, N.E., Ahfeldt, T., Sachs, K.V., Li, X., Li, H., Kuperwasser, N., Ruda, V.M.et al. (2010) From noncoding variant to phenotype via SORT1 at the 1p13 cholesterol locus. Nature, 466, 714–719. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Bycroft, C., Freeman, C., Petkova, D., Band, G., Elliott, L.T., Sharp, K., Motyer, A., Vukcevic, D., Delaneau, O., O’Connell, J.et al. (2018) The UK biobank resource with deep phenotyping and genomic data. Nature, 562, 203–209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Battle, A., Mostafavi, S., Zhu, X., Potash, J.B., Weissman, M.M., McCormick, C., Haudenschild, C.D., Beckman, K.B., Shi, J., Mei, R.et al. (2014) Characterizing the genetic basis of transcriptome diversity through RNA-sequencing of 922 individuals. Genome Res., 24, 14–24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Gaziano, J.M., Concato, J., Brophy, M., Fiore, L., Pyarajan, S., Breeling, J., Whitbourne, S., Deen, J., Shannon, C., Humphries, D.et al. (2016) Million veteran program: a mega-biobank to study genetic influences on health and disease. J. Clin. Epidemiol., 70, 214–223. [DOI] [PubMed] [Google Scholar]
- 15. Benner, C., Spencer, C.C.A., Havulinna, A.S., Salomaa, V., Ripatti, S. and Pirinen, M. (2016) FINEMAP: efficient variable selection using summary data from genome-wide association studies. Bioinformatics, 32, 1493–1501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. McLaren, W., Gil, L., Hunt, S.E., Riat, H.S., Ritchie, G.R.S., Thormann, A., Flicek, P. and Cunningham, F. (2016) The Ensembl Variant Effect Predictor. Genome Biol., 17, 122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Conner, J.R., Smirnova, I.I., Moseman, A.P. and Poltorak, A. (2010) IRAK1BP1 inhibits inflammation by promoting nuclear translocation of NF-kappaB p50. Proc. Natl. Acad. Sci. U. S. A., 107, 11477–11482. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Martens, J.H.A. and Stunnenberg, H.G. (2013) BLUEPRINT: mapping human blood cell epigenomes. Haematologica, 98, 1487–1489. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. den Hollander, A.I., Koenekoop, R.K., Mohamed, M.D., Arts, H.H., Boldt, K., Towns, K.V., Sedmak, T., Beer, M., Nagel-Wolfrum, K., McKibbin, M.et al. (2007) Mutations in LCA5, encoding the ciliary protein lebercilin, cause Leber congenital amaurosis. Nat. Genet., 39, 889–895. [DOI] [PubMed] [Google Scholar]
- 20. GTEx Portal https://www.gtexportal.org/home/.
- 21. Dobbs, A.K., Yang, T., Farmer, D., Kager, L., Parolini, O. and Conley, M.E. (2007) Cutting edge: a hypomorphic mutation in Igbeta (CD79b) in a patient with immunodeficiency and a leaky defect in B cell development. J. Immunol., 179, 2055–2059. [DOI] [PubMed] [Google Scholar]
- 22. Ferrari, S., Lougaris, V., Caraffi, S., Zuntini, R., Yang, J., Soresina, A., Meini, A., Cazzola, G., Rossi, C., Reth, M.et al. (2007) Mutations of the Igbeta gene cause agammaglobulinemia in man. J. Exp. Med., 204, 2047–2051. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Brdicková, N., Brdicka, T., Angelisová, P., Horváth, O., Spicka, J., Hilgert, I., Paces, J., Simeoni, L., Kliche, S., Merten, C.et al. (2003) LIME: a new membrane raft-associated adaptor protein involved in CD4 and CD8 coreceptor signaling. J. Exp. Med., 198, 1453–1462. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Hur, E.M., Son, M., Lee, O.-H., Choi, Y.B., Park, C., Lee, H. and Yun, Y. (2003) LIME, a novel transmembrane adaptor protein, associates with p56lck and mediates T cell activation. J. Exp. Med., 198, 1463–1473. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Du, Y., Duan, T., Feng, Y., Liu, Q., Lin, M., Cui, J. and Wang, R.-F. (2018) LRRC25 inhibits type I IFN signaling by targeting ISG15-associated RIG-I for autophagic degradation. EMBO J., 37, 351–366. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Feng, Y., Duan, T., Du, Y., Jin, S., Wang, M., Cui, J. and Wang, R.-F. (2017) LRRC25 functions as an inhibitor of NF-κB Signaling pathway by promoting p65/RelA for autophagic degradation. Sci. Rep., 7, 13448. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Ghoussaini, M., Mountjoy, E., Carmona, M., Peat, G., Schmidt, E.M., Hercules, A., Fumis, L., Miranda, A., Carvalho-Silva, D., Buniello, A.et al. (2021) Open targets genetics: systematic identification of trait-associated genes using large-scale genetics and functional genomics. Nucleic Acids Res., 49, D1311–D1320. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Tapia, A.L., Rowland, B.T., Rosen, J.D., Preuss, M., Young, K., Graff, M., Choquet, H., Couper, D.J., Buyske, S., Bien, S.A.et al. (2021) Full title: a large-scale transcriptome-wide association study (TWAS) of 10 blood cell phenotypes reveals complexities of TWAS fine-mapping. Genet. Epidemiol., 46, 3–16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Wu, C. and Pan, W. (2020) A powerful fine-mapping method for transcriptome-wide association studies. Hum. Genet., 139, 199–213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Mancuso, N., Freund, M.K., Johnson, R., Shi, H., Kichaev, G., Gusev, A. and Pasaniuc, B. (2019) Probabilistic fine-mapping of transcriptome-wide association studies. Nat. Genet., 51, 675–682. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Wainberg, M., Sinnott-Armstrong, N., Mancuso, N., Barbeira, A.N., Knowles, D.A., Golan, D., Ermel, R., Ruusalepp, A., Quertermous, T., Hao, K.et al. (2019) Opportunities and challenges for transcriptome-wide association studies. Nat. Genet., 51, 592–599. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Barbeira, A.N., Pividori, M.D., Zheng, J., Wheeler, H.E., Nicolae, D.L. and Im, H.K. (2019) Integrating predicted transcriptome from multiple tissues improves association detection. PLoS Genet., 15, e1007889. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Kammers, K., Taub, M.A., Rodriguez, B., Yanek, L.R., Ruczinski, I., Martin, J., Kanchan, K., Battle, A., Cheng, L., Wang, Z.Z.et al. (2021) Transcriptional profile of platelets and iPSC-derived megakaryocytes from whole-genome and RNA sequencing. Blood, 137, 959–968. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Luningham, J.M., Chen, J., Tang, S., De Jager, P.L., Bennett, D.A., Buchman, A.S. and Yang, J. (2020) Bayesian genome-wide TWAS method to leverage both cis- and trans-eQTL information through summary statistics. Am. J. Hum. Genet., 107, 714–726. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Bhattacharya, A., Li, Y. and Love, M.I. (2021) MOSTWAS: multi-Omic strategies for transcriptome-wide association studies. PLoS Genet., 17, e1009398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Das, S., Forer, L., Schönherr, S., Sidore, C., Locke, A.E., Kwong, A., Vrieze, S.I., Chew, E.Y., Levy, S., McGue, M.et al. (2016) Next-generation genotype imputation service and methods. Nat. Genet., 48, 1284–1287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. TOPMed Imputation Server https://imputation.biodatacatalyst.nhlbi.nih.gov/#.
- 38. Mostafavi, S., Battle, A., Zhu, X., Urban, A.E., Levinson, D., Montgomery, S.B. and Koller, D. (2013) Normalizing RNA-sequencing data by modeling hidden covariates with prior knowledge. PLoS One, 8, e68141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Fang, H., Hui, Q., Lynch, J., Honerlaw, J., Assimes, T.L., Huang, J., Vujkovic, M., Damrauer, S.M., Pyarajan, S., Gaziano, J.M.et al. (2019) Harmonizing genetic ancestry and self-identified race/ethnicity in genome-wide association studies. Am. J. Hum. Genet., 105, 763–772. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Marees, A.T., deKluiver, H., Stringer, S., Vorspan, F., Curis, E., Marie-Claire, C. and Derks, E.M. (2018) A tutorial on conducting genome-wide association studies: quality control and statistical analysis. Int. J. Methods Psychiatr. Res., 27, e1608. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Mogil, L.S., Andaleon, A., Badalamenti, A., Dickinson, S.P., Guo, X., Rotter, J.I., Johnson, W.C., Im, H.K., Liu, Y. and Wheeler, H.E. (2018) Genetic architecture of gene expression traits across diverse populations. PLoS Genet., 14, e1007586. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Mbatchou, J., Barnard, L., Backman, J., Marcketta, A., Kosmicki, J.A., Ziyatdinov, A., Benner, C., O’Dushlaine, C., Barber, M., Boutkov, B.et al. (2021) Computationally efficient whole-genome regression for quantitative and binary traits. Nat. Genet., 53, 1097–1103. [DOI] [PubMed] [Google Scholar]
- 43. Loh, P.-R., Tucker, G., Bulik-Sullivan, B.K., Vilhjálmsson, B.J., Finucane, H.K., Salem, R.M., Chasman, D.I., Ridker, P.M., Neale, B.M., Berger, B.et al. (2015) Efficient Bayesian mixed-model analysis increases association power in large cohorts. Nat. Genet., 47, 284–290. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Wu, T., Hu, E., Xu, S., Chen, M., Guo, P., Dai, Z., Feng, T., Zhou, L., Tang, W., Zhan, L.et al. (2021) clusterProfiler 4.0: a universal enrichment tool for interpreting omics data. Innovation (N Y), 2, 100141. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.