

Abstract
Non-syndromic cleft lip with or without cleft palate (NSCL/P) is a common, severe craniofacial malformation that imposes significant medical, psychosocial and financial burdens. NSCL/P is a multifactorial disorder with genetic and environmental factors playing etiologic roles. Currently, only 25% of the genetic variation underlying NSCL/P has been identified by linkage, candidate gene and genome-wide association studies. In this study, whole-genome sequencing and genome-wide genotyping followed by polygenic risk score (PRS) and linkage analyses were used to identify the genetic etiology of NSCL/P in a large three-generation family. We identified a rare missense variant in PDGFRA (c.C2740T; p.R914W) as potentially etiologic in a gene-based association test using pVAAST (P = 1.78 × 10−4) and showed decreased penetrance. PRS analysis suggested that variant penetrance was likely modified by common NSCL/P risk variants, with lower scores found among unaffected carriers. Linkage analysis provided additional support for PRS-modified penetrance, with a 7.4-fold increase in likelihood after conditioning on PRS. Functional characterization experiments showed that the putatively causal variant was null for signaling activity in vitro; further, perturbation of pdgfra in zebrafish embryos resulted in unilateral orofacial clefting. Our findings show that a rare PDGFRA variant, modified by additional common NSCL/P risk variants, have a profound effect on NSCL/P risk. These data provide compelling evidence for multifactorial inheritance long postulated to underlie NSCL/P and may explain some unusual familial patterns.
Introduction
Non-syndromic cleft lip with or without cleft palate (NSCL/P) is a common craniofacial malformation affecting 1/700 live births and 135 000 newborns worldwide each year. NSCL/P requires lifelong medical, dental, speech and psychosocial interventions thus imposing familial and public health burdens. A multifactorial etiology has long been hypothesized with genetic and environmental factors and their interactions playing a role (1,2). Family studies provide strong evidence for a genetic component; however, a specific Mendelian pattern of inheritance is rarely identified. Linkage, mutation screening, candidate gene, genome-wide association studies (GWAS) and recent whole-exome sequencing (WES) studies have identified ~40 NSCL/P genes/loci (1,3). However, most of the identified NSCL/P risk variants have modest effects that collectively account for a small fraction (~25%) of variance in NSCL/P risk (4–12). More recently, a combination of rare and common variants has been proposed to explain the etiology of complex heterogeneous conditions, such as NSCL/P (13,14). These findings underlie the challenges in identifying all of genetic variation contributing to NSCL/P and impact accurate risk assessment and genetic counseling.
In this study, we performed whole-genome sequencing (WGS) and genome-wide genotyping followed by polygenic risk score (PRS) analysis to identify the genetic etiology of NSCL/P in a large multigenerational family. In vitro functional characterization experiments and zebrafish mutants were used to evaluate the effects of the putatively causal variant.
Results
Platelet-derived growth factor receptor alpha (PDGFRA) was identified as the second-highest ranking gene (P = 1.78 × 10−4) in our whole exome gene-based pVAAST analysis (Supplementary Material, Table S1). The observed signal at PDGFRA was driven by a rare variant in exon 20 (Chr4: 55155031; c.C2740T; p.R914W) with a VAAST score of 19.07 and a logarithm of the odds (LOD) score of 1.2. This variant had not previously been reported among 141 456 individuals in the gnomAD database and is located in a highly conserved region (Supplementary Material, Fig. S1). Sanger sequencing was used for variant segregation analysis on 15 individuals with available DNA. For individual I-1 whose DNA was not available, genotypes were inferred based on the haplotype inheritance pattern to confirm paternal inheritance of the c.C2740T variant by the affected individuals in the second generation. The variant was found in all five affected individuals (II-2, II-5, II-7, III-1 and III-2) and in three (I-1, III-5 and III-6) of the 11 unaffected individuals (Fig. 1A).
Figure 1.
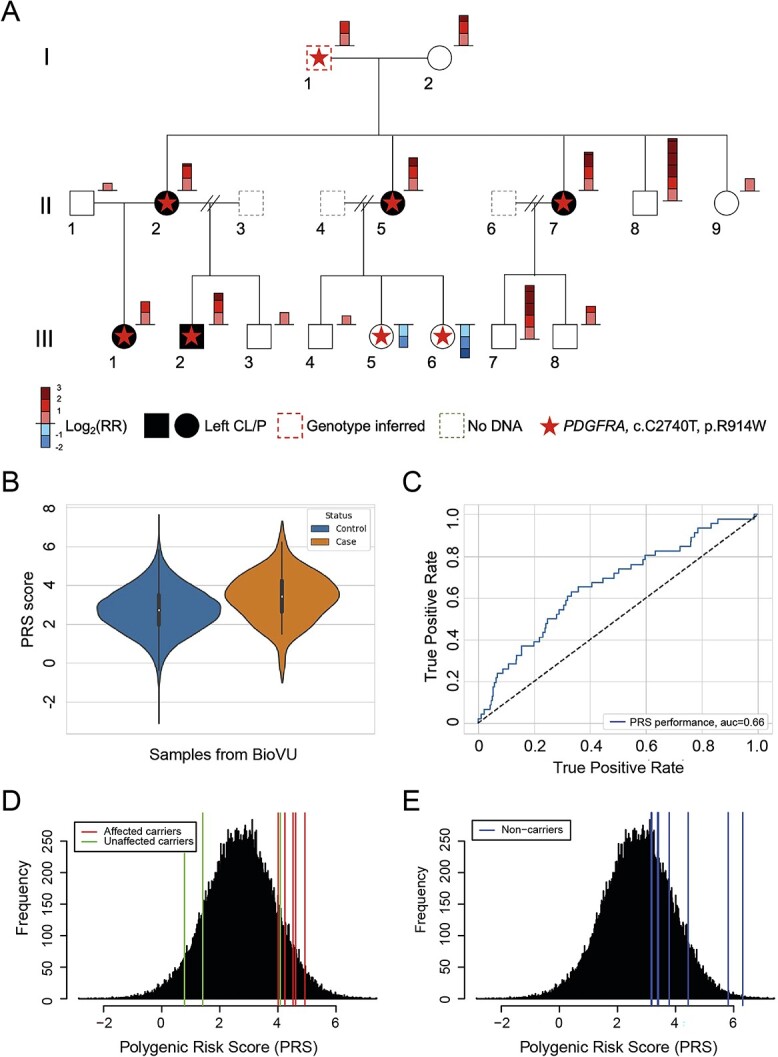
The penetrance of PDGFRA variant is modified by PRS from known NSCL/P risk loci in a multigenerational family. (A) WGS and PRS results for a three-generation NSCL/P family. Individuals with NSCL/P are depicted by solid black rectangles and circles. The red dashed rectangle indicates the individual with imputed genotypes. Gray dashed rectangles indicate individuals without available DNA or genotype information. The colored bar to the right of each individual shows the PRS-derived relative risk (RR) of NSCL/P on a log2 scale. RR is calculated as the ratio of estimated PRS of each individual versus the average PRS of 72 496 non-Hispanic European individuals from BioVU. The red star indicates carrier status for PDGFRA c.C2740T. Individuals I-2, II-1, II-5, II-7, III-1 and III-2 were submitted for WGS. (B) Violin plots of the distributions of PRS in 46 cases and 72 496 controls from BioVU. (C) Receiver operating characteristic (ROC) curve for PRS for the diagnosis of NSCL/P in BioVU samples. ROC was performed for PRS using NSCL/P patients and controls from BioVU. (D–E) The comparison of PRS of the NSCL/P family and the background distribution of controls from BioVU. Each vertical line represents one individual in the family: red lines indicate affected individuals with PDGFRA c.C2740T variant, green lines indicate unaffected individuals with PDGFRA c.C2740T variant, and blue lines indicate unaffected individuals without PDGFRA c.C2740T variant.
Although the segregation pattern of PDGFRA c.C2740T was consistent with autosomal dominant inheritance, the inheritance pattern observed in the siblings from generation III (III-5 and III-6) suggested the introduction of protective variants in modifier genes from the marry-in parent in generation II (II-4). This pattern was hypothesized to occur by genetic interaction(s) between PDGFRA c.C2740T and common variants known to influence NSCL/P risk. To test the hypothesis of polygenic contribution to NSCL/P, a population-specific polygenic risk score (PRS) analysis for NSCL/P was performed using 29 risk NSCL/P variants identified in previous GWAS (9,10,15,16) with allele frequencies of the affective allele ranging from 0.014 to 0.567 and odds ratios (ORs) ranging from 0.23 to 2.26 (Supplementary Material, Table S2). The predictive performance of this PRS was validated in an independent set of 46 cases and 72 496 controls obtained from the BioVU repository (17). Our results showed significantly higher PRSs among affected individuals (P = 1.19 × 10−4) with an area under curve (AUC) of 0.66 (Fig. 1B and C). PRSs were then calculated for 16 individuals in the pedigree (Supplementary Material, Tables S3 and S4) and the scores compared with controls in BioVU (Fig. 1D and E). All affected individuals exhibited a PRS substantially above the population mean, with scores ranging from 4.0 to 4.8, corresponding to a 3.7- to 9.1-fold increased risk of NSCL/P (relative to the mean PRS in BioVU). Among the three unaffected individuals with the c.C2740T variant, I-1 had an inferred PRS of 4.1, corresponding to a 3.9-fold increased risk. Two unaffected individuals (III-5 and III-6) had PRSs of 1.41, and 0.88, corresponding to 3.7- and 7.1-fold decreased risk, respectively.
The PRS results support a multifactorial disease model (2) of NSCL/P in this family. To quantify the evidence supporting this model, linkage analysis was conducted with and without conditioning on the PRS. Under the assumption that the penetrance of the causal variant is fixed regardless of genetic background, the estimated penetrance was 0.62 with a LOD score of 1.3 at c.C2740T. Alternatively, under the assumption that PRS and a rare, high-penetrance variant are independent risk factors for NSCL/P in a logistic regression model, the estimated penetrance of c.C2740T for the five affected individuals ranged from 0.86 to 0.94, whereas the estimated penetrance for the two unaffected individuals was 0.31 and 0.19 (Supplementary Material, Table S5). The LOD score in the PRS model was 2.2, indicating that the alternative model is 7.4-fold more likely under the assumption that the PRS and c.C2740T independently influence NSCL/P risk. In the genome-wide linkage analysis conditioned on PRS, a region with a higher LOD score than c.C2740T was observed at chr14:33M-51M (LOD = 2.4, Supplementary Material, Fig. S2). However, WGS identified no rare variants in this region with predicted function to explain the phenotype (Supplementary Material, Table S6 and Supplementary Material, Fig. S3). In contrast, individually, the NSCL/P PRS variants confer modest changes in risk but collectively they result in up to a 5-fold difference in the penetrance of c.C2740T; p.R914W.
To determine the effects of the PDGFRA c.C2740T variant on gene/protein function, in silico and in vitro functional characterization experiments were performed, and the pdgfra locus was genetically altered in zebrafish embryos. A published structure (PDB5K5X) shows that the wild-type (R914) forms direct hydrogen bonds with I909, Y913 and W933 plus three additional amino acids (I909, G912 and S935) via bridged interactions [Fig. 2A; (18)]. Alphafold2 predicts that the R914W substitution eliminates all seven hydrogen bonds bridging two regions of the C-terminal kinase domain and that 914W is rotated out of the groove causing a hydrophobic bump and subtly alters the surface charge of that domain [(19); Supplementary Material, Fig. S4].
Figure 2.

The R914W mutation is a functional null mutation. (A) Image showing the hydrogen-binding contacts (dashed lines) of R914 in the intracellular domain of PDGFRA. R914 and all interacting amino acids are labeled. Individual red circles are water molecules from the solvent. Image from the RCSB PDB (rcsb.org) of PDB 5K5X (18). (B) Schematic showing the domain structure of PDGFRA. Key domains and tested mutations are shown. IgL, immunoglobulin-like domains; JM, juxta-membrane domain; TK #1, tyrosine kinase domain 1; TK #2, tyrosine kinase domain 2. (C) Summary of flow cytometry data measuring the responsiveness (total EGFP signal) of the receptors to PDGF ligands. The horizontal dashed line indicates the baseline signal (WT receptor, no ligand). Data shown are the means ± SD of three independent biological replicate experiments. The results were evaluated using two-way ANOVA (alpha = 0.05) and all tested variables show a P-value < 0.0001 (Supplementary Material, Tables S9 and S10).
PDGFRA point mutations were generated to assay the functional consequence of R914W (Fig. 2B). All mutants, including R914W, decreased cell-surface PDGFRA protein compared with wild-type (Supplementary Material, Figs S5A and S6). PDGF ligands were assayed for their ability to activate an EGFP reporter in the context of wild-type or mutant PDGFRA. As expected, PDGFRA cell-surface levels dipped with ligand addition and receptor internalization (20). Confirming previous reports, K627R and D842V behaved as dominant-negative and constitutively active mutations, respectively [Fig. 2C; (18)]. Strikingly, R914W eliminated the ability of PDGFRA to respond to its ligands (Fig. 2C); adding R914W to the constitutively active D842V background nearly eliminated signaling from the double mutant. When controlled for cell-surface receptor levels, the R914W variant overrides the activating effect of D842V (Supplementary Material, Figs S5B and S7). Overall, these results show that the R914W variant has a dominant-negative effect and is a functional null mutation with respect to PDGF signaling.
Genetic alteration of the pdgfra locus using CRISPR/Cas9 in transgenic zebrafish embryos with the surface epithelium (or periderm) fluorescently labeled revealed a vertical groove in the upper lip in 65% compared with 0% in uninjected (negative) or tyrosinase-injected (positive) controls (Fig. 3, Supplementary Material, Fig. S8). Pharmacological inhibition of pdgfr generated a similar phenotype to the pdgfra F0 CRISPR-injected embryos (93% in inhibitor versus 0% controls had vertical groove) (Supplementary Material, Fig. S9), demonstrating the influence of pdgfra signaling on the formation of a vertical groove in the upper lip. The observed ventral groove formation in the upper lip of the F0 zebrafish embryos was consistent with previous studies of pdgfra in zebrafish (21) that mainly focused on the palatal abnormalities.
Figure 3.
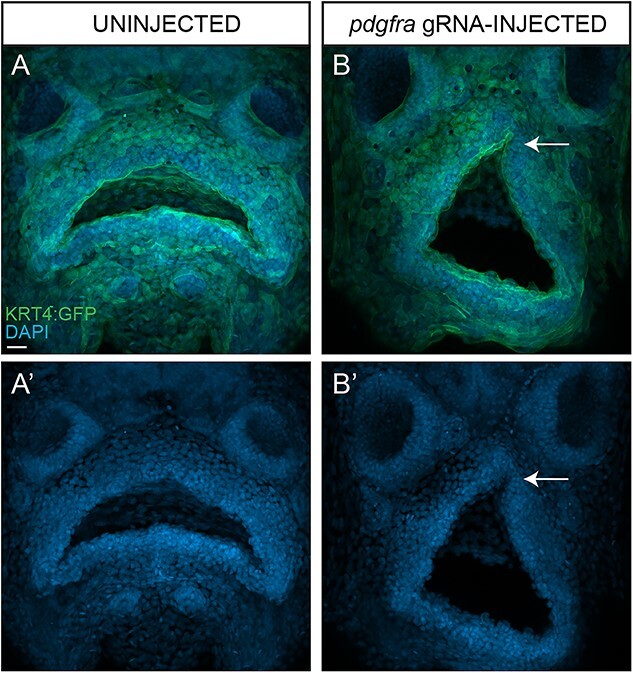
Perturbation of pdgfra in developing zebrafish embryos is sufficient to promote formation of a hypoplastic lip with a ventral groove. Maximum intensity projections of confocal images of uninjected (A) and pdfrga gRNA-injected (B) Tg(Krt4:GFP) transgenic zebrafish embryos counterstained with DAPI (A′–B′) to label nuclei. Arrow denotes hypoplastic upper lip with a ventral groove (B′). Scale bar size = 20 μm.
Discussion
Recent evidence suggests that multiple rare and/or common variants (each with a modest marginal effect) together explain a large proportion of the genetic basis of NSCL/P and other common complex disorders (1,22). Here, we identified a rare missense variant in PDGFRA (c.C2740T; p.R914W), exhibiting decreased penetrance, as potentially etiologic. PRS analysis suggested that variant penetrance was modified by common NSCL/P risk variants, with lower scores found among unaffected carriers.
Various signaling pathways regulate the development of craniofacial structures across vertebrates, including the platelet-derived growth factor (PDGF) signaling pathway. PDGFRA is a cell-surface receptor tyrosine kinase for platelet-derived growth factors that plays an important role in craniofacial and neural crest development and palatogenesis (23,24). Expression of PDGFRA in a temporal and spatial manner is required for proper neural crest cell migration and murine embryonic development (21,25). Further, variation in this gene has been associated with various congenital developmental abnormalities including neural tube closure defects and orofacial clefts (26,27).
Although the PDGFRA variant was found in all affected individuals suggesting autosomal dominant inheritance, it was also found in three unaffected individuals suggesting that this variant alone could not account for the NSCL/P segregation pattern in this family and that additional genes/variants might be influencing NSCL/P risk. To identify additional variants contributing to NSCL/P, we developed a PRS for a population of non-Hispanic European ancestry (NHEA) considering 29 NSCL/P risk variants from previous GWAS (6,9,10,12,15,16,28). The predictive performance of this PRS was validated using samples from the BioVU repository. Although all individuals in the study carried multiple NSCL/P risk alleles, individuals with NSCL/P in the BioVU repository had significantly higher PRSs than controls (P = 1.19 × 10−4). Polygenic risk scores have provided evidence that disease risk may be the result of the effects of common variants in an individual’s genome. Furthermore, it provides an estimate of the genetic propensity to a trait at the individual level (14). Our results showed significantly higher PRSs among affected individuals in the studied family (all whom had the c.C2740T variant), which were also markedly above the population mean for predicted NSCL/P risk. Interestingly, for those unaffected individuals who also had the c.C2740T variant, PRSs were significantly lower and associated with decreased risk of NSCL/P.
Our PRS model adopted the standard assumption that common risk variants independently contribute to NSCL/P risk under an additive model. In addition, our linkage model assumed independent contributions to disease risk from PRS and c.C2740T. Alternatively, and as observed in other diseases (29), it is possible that only a few of the 29 PRS variants act as strong modifiers of c.C2740T. These observations could potentially explain the unaffected status of the grandfather in generation I (I-1), who had the c.C2740T variant and a PRS of 4.1, corresponding to a 3.9-fold increase in risk. However, among the 29 common NSCL/P risk variants identified from GWAS, there were no unique genotypes shared by the grandfather (I-1) and the two unaffected individuals with the c.C2740T variant in generation III (III-5 and III-6). We further explored whether PDGFRA shares one or more pathways with a subset of the genes implicated in GWAS, observing a modest enrichment in the MAPK signaling pathway involving PGFRA, MKNK2 and GADD45G, although the result was not significant (P = 0.06, Supplementary Material, Table S7) and the risk variants in MKNK2 and GADD45G did not segregate with affected status (Supplementary Material, Tables S2 and S4). Somatic/germline mosaicism in the grandfather is another possible explanation for his unaffected status, but all offspring in generation II with the c.C2740T haplotype were carriers of c.C2740T (Supplementary Material, Fig. S10), providing no indication that c.C2740T was mosaic in the grandfather. Unfortunately, this mechanism cannot be explored further due to the lack of available biospecimens from the grandfather. Assuming independent risks, we estimate that the penetrance of c.C2740T after adjusting for the grandfather’s PRS is 0.87 (Supplementary Material, Fig. S10), and thus the probability that the grandfather would be unaffected is 0.13. With the available evidence, we conclude that the incomplete penetrance with independent contributions to risk from PRS and c.C2740T is the most likely explanation for the observed inheritance pattern.
To better understand the impact of mutant PDGFRA on PDGF signaling, we tested the effects of single- and double-point mutations on PDGFRA activation. The results suggest three possible mechanisms to explain how R914W eliminates PDGF signaling. First, with seven hydrogen bonds serving as a bridge between two regions of PDGFRA, R914 is likely a lynchpin for the local structure of the protein. Two other crystal structures show that drugs (crenolanib and sunitinib, 6JOK and 6JOJ, respectively) that bind PDGFRA some distance away can disrupt some (but not all) of these interactions (30,31). The R914W variant would eliminate most or all of those bonds, possibly destabilizing vital protein:protein interactions required to maintain the activity of PDGFRA’s C-terminal tyrosine kinase domain. Another possibility is that R914W would disrupt hydrogen-bonding interactions with W933 and S935, which likely help maintain the proper orientation of the internalization sequence predicted to span amino acids 944–958. Indeed, the increase in cell-surface PDGFRA in the D842V + R914W mutant is consistent with this hypothesis. Second, interference with the kinase activity of the receptor, either dependent upon- or independent from- receptor dimerization, is another possibility. Finally, as PDGFRA is glycosylated, the mutation may somehow impair the glycosylation of the receptor during or after processing (32).
Lastly, our in vivo analysis showed that genetic perturbation of pdgfra in developing zebrafish embryos resulted in the formation of a hypoplastic lip with a ventral groove, consistent with previous studies (21). Taken together, our study suggests that disruption of PDGF signaling is sufficient to promote clefting and generation of a hypoplastic lip, supporting our observations in this three-generation NSCL/P family.
Our findings have significant implications for understanding the genetic etiology of NSCL/P and other complex birth defects. Here, we show that the penetrance of a rare PDGFRA variant is modified by common NSCL/P risk variants further supporting the multifactorial model of NSCL/P. This finding provides a paradigm for identifying the missing genetic liability of NSCL/P and an explanation for multiplex families segregating NSCL/P in a non-Mendelian pattern. Importantly, these results have the potential to improve recurrence risk estimates for NSCL/P.
Materials and Methods
Study subjects
The UTHealth Committee for the Protection of Human Subjects approved this study (HSC-MS-03-090). DNA (saliva or blood) was collected from 15 individuals (5 affected with NSCL/P and 10 unaffected) of self-reported NHEA (Fig. 1) as part of our ongoing NSCL/P study that has previously been described (5,8,33,34). The family members were completely phenotyped and no other anomalies were identified. I-1 was deceased but reportedly had no orofacial anomalies. Family members II-2 and III-2, III-1 had complete left NSCL/P and II-5 and II-7 had left CL only.
Data generation and acquisition
WGS was performed on DNA samples of four affected (II-5, II-7, III-1 and III-2) and two unaffected (I-2 and II-1) individuals using the Illumina HiSeq, Illumina Inc., San Diego, CA, platform by Hudson Alpha Institute for Biotechnology (Huntsville, AL). Genome-wide genotyping was conducted on all samples using the Illumina Expanded Multi-Ethnic Genotyping Array (MEGAEX) at Vanderbilt University Medical Center. Controls consisted of WES data from 3764 individuals of European ancestry from the National Database for Autism Research (NDAR). The controls from NDAR were unaffected parents of offspring with Autism Spectrum Disorder (ASD). Other than ASD status, NSCL/P phenotype information was unavailable for the controls and thus a small proportion may have been misclassified, although the potential misclassification bias would have only a modest effect on statistical power.
Sequencing data process
Genome alignment was conducted using GRCh37 as the reference genome for the WGS data of the NSCL/P family and for the WES data of the NDAR controls (35,36). Genome Analysis Toolkit (GATK) Best Practices workflow (37) was then used to conduct joint genotype calling and variant quality recalibration (VQSR). XPAT (cross-platform association toolkit) (38) was employed to conduct quality control for cross-platform data and to identify variants potentially influenced by cross-platform biases. VQSR tranche score thresholds were set as 99.9 for single-nucleotide variants (SNVs) and 98.0 for insertions and deletions (INDELs). XPAT was used to conduct an external principal component analysis (PCA) to project individuals onto a reference panel of 1000 genome project samples, verifying the European ethnicity of NSCL/P family and selecting 3764 European controls among NDAR samples. After that, XPAT was used to conduct an internal PCA for cases and controls only excluding the reference panel to characterize the population stratification of samples involved in this study.
Gene-based association analyses
pVAAST (the Pedigree Variant Annotation, Analysis, and Search Tool) (39), a unified test that combines linkage, case–control association and a priori functional variant prioritization, was used to perform gene-based analyses. All rare protein-coding variants that passed QC standards and with a minor allele frequency (MAF) < 0.005 among populations of non-Finnish Europeans in gnomAD (40) were evaluated. The first five PCs from the internal PCA in the previous step were incorporated as covariates in the association analysis to control population and technical stratification. Both LOD score and CLRT score calculated by pVAAST were used to select informative sites. pVAAST simulated genotype error in the data with an error rate of 10−5. The maximum prevalence was set to be 0.001. pVAAST scores only genes with a positive LOD score. See the Supplementary Material, Table S1 for the list of nominal significant genes (P < 0.05) in pVAAST analysis.
Calculation of polygenic risk scores
To develop an NHEA-specific polygenic risk scores (PRS) for NSCL/P, 29 variants were identified from prior GWAS (6,7,9,10,12,15,16,28) that met the following criteria: (i) the association was genome-wide significant (P < 5 × 10−8) in one or more NHEA GWAS, or (ii) the association was genome-wide significant in a multi-ethnic GWAS and was significant after Bonferroni correction (adjusted P < 0.05) in an NHEA GWAS (Supplementary Material, Table S2). If multiple variants at a given 1 Mb locus met these criteria, the variant with the lowest P-value at the locus was included. The potential for multiple independent signals at each locus was evaluated, but for each locus, all other significant variants were in linkage disequilibrium (r2 < 0.1) with the lead variant (calculated using LDlink, https://ldlink.nci.nih.gov).
The MEGAEX included six of the 29 NSCL/P variants included in the PRS. The remaining 23 SNPs were imputed using the Michigan Imputation Server (https://imputationserver.sph.umich.edu) with the European ancestry Haplotype Reference Consortium (HRC) reference panel (r1.1 2016; Supplementary Material, Table S3). For three variants with imputation scores < 0.7, we verified the genotypes based on the haplotype inheritance pattern observed in individuals with WGS data based on the phased data from the Michigan Imputation Server (Supplementary Material, Table S4).
Because DNA was not available for individual I-1, the genotypes were inferred based on the haplotype inheritance pattern of generations I and II (Fig. 1). For each variant, the haplotypes in 500K region of the target variant in samples I-2, II-5 and II-7 were collected. Each of the two haplotypes from offspring II-5 and II-7 were compared with their maternal haplotypes (I-2) and the inherited haplotype was determined according to the proportion of consistent genotypes, which was >99.9%. The remaining haplotypes (not inherited from sample I-2) were then compared with determine our ability to infer one or two alleles of sample I-1. If a genotype could not be reliably imputed using either the Michigan Imputation Server or haplotype inheritance patterns, the alleles were assigned dosages based on the allele frequency of non-Finnish Europeans from gnomAD.
Individualized PRSs were then calculated as the sum of the natural logarithm of the per allele OR multiplied by the number of alleles. To reduce the winner’s curse bias, the OR for each variant was obtained from the most recent NHEA GWAS. The PRSs were tested with individuals from the BioVU repository (https://www.vumc.org/dbmi/biovu), including 46 cases diagnosed with cleft lip and palate or cleft lip, unspecified cleft type and 72 496 controls.
Penetrance estimate
To conduct linkage analysis conditioned on PRS, individual penetrance for all samples with known genotypes were estimated (41). To establish individual penetrance estimates that account for background PRS in each individual, the PRS estimates were centered around the population mean PRS (2.72). Thus, individuals with below average PRS estimates had reduced estimated penetrance, whereas individuals with above average PRS estimates had increased estimated penetrance. Taking as the vector of observed sample phenotypes and as the vector of observed risk allele carriers, penetrance can then be estimated for each sample as, where is the estimated coefficient (log-odds) for the risk allele in a logistic regression model with intercept fixed to the population incidence, and PRS is the vector of centered PRS estimates.
Linkage analysis
MEGAEX data for all genotyped individuals was cleaned to remove variants with call rate < 95% and monomorphic variants, followed by confirmation of pedigree structure using PRIMUS (42). Variants with Mendelian errors were identified using PedCheck (43) and removed. Multipoint parametric linkage was performed using ALLEGRO (44) with individual-specific penetrance estimates based on the PRS for each individual (Supplementary Material, Table S5). Variant frequency in ALLEGRO input was specified as the frequency in the PRIMUS-generated maximally unrelated subset (< 0.09) of the HapMap 3 CEU population.
Two-point linkage using the genotypes at the PDGFRA variant, c.C2740T, including the imputed genotype for individual I-1, was also conducted using ALLEGRO. Analyses were performed with family-wide penetrance estimates of f0,1,2 = 0.0014, 0.625, 0.625 and with individual PRS-based penetrance estimates.
Rare variant identification in chr14:33M-51M
In the GWAS analysis of this NSCL/P family, the region of chr14:33M-51M was reported to have a strong association with the phenotype. All rare variants from the six samples with WGS data were identified in order to investigate potential disease susceptibility variants or haplotype(s) in this region. To identify rare variants, variants with reported allele frequencies > 0.1% in genomAD database (v3.1.1) that contains 76 156 whole genomes were excluded. CNVnator (45) was used for CNV detection for samples with WGS data using default parameters. Abnormal read depth distribution was observed for sample I-2 (likely due to the quality of the original blood specimen) and was excluded from CNV analysis. Only fully segregating variants among four affected and two unaffected individuals in the pedigree were considered. To reduce low-quality variants, variants in repeats and low complexity regions were removed as well as variants with alternative allele of A (T, G or C) and next to poly-A (poly-C, poly-G and poly-T) region (> 8 nt).
Variant functional annotation was conducted using ANNOVAR (46). TraP score (47) for non-intergenic variants and pre-computed combined annotation dependent depletion (CADD)-based scores [C-scores; (48)] were obtained for all variants (Supplementary Material, Table S6). AliBaba 2.1 (49), Patch (http://gene-regulation.com/pub/programs.html#patch) and PROMO (50) were used to predict transcription factor binding sites in the promoter region induced by the rare variants identified in the upstream region of a gene.
Structural modeling
The crystal structure of the intracellular domain of PDGFRA (PDB 5K5X) was previously described (18), accessed, and rendered using the RCSB protein data bank https://www.rcsb.org/3d-view/5K5X [Fig. 2A; (51)]. The effects of the R914W mutation were modeled using Alphafold2 https://colab.research.google.com/github/sokrypton/ColabFold/blob/main/AlphaFold2.ipynb#scrollTo=kOblAo-xetgx. Specifically, the salient portion of the intracellular domain of PDGFRA (amino acids 541–1020) with and without the R914W substitution was used as the input amino acid sequence for Alphafold2 (19). Five models were generated for each sequence using the MMseqs2 (UniRef+Environmental) setting and the 3D structure was generated with the ‘show_sidechains’ setting selected. Alphafold2’s structure for the wild-type (WT) sequence recapitulated the observed crystal structure. Further analyses were performed and graphics generated with UCSF ChimeraX (52). Briefly, Alphafold2-generated models were uploaded into ChimeraX and aligned via chimera’s matchmaker command using default parameters. Hydrogen bonds for residue 914 were analyzed and visualized using ChimeraX’s H-Bonds tool (default parameters except for retaining pre-existing H-Bonds). Hydrophobicity and electrostatic potential maps were also generated using Chimera’s tools with default parameters.
Molecular cloning
A pCEP4-PDGFRA-reporter plasmid containing WT human PDGFRA (with an extracellular Myc-tag) and a PEST-tagged EGFP reporter was obtained from Addgene [#136 456; (53)]. Single and double PDGFRA point mutations (K627R, D842V, R914W, K627R + R914W, D842V + R914W) were constructed using the QuikChange Lightning Site-Directed Mutagenesis Kit (Agilent) and all point mutations were confirmed by Sanger sequencing (see https://doi.org/10.6084/m9.figshare.15077172). Oligonucleotide sequences and plasmids used are listed in Supplementary Material, Table S8. All plasmids used in this study are available via Addgene (ID#: 136 456, 172 596–172 600).
Cell culture, transfection and receptor activation
Expi293F suspension cells (Gibco, Waltham, Massachussetts) were cultured in Expi293 Expression Medium (Gibco) at 37°C in 8% CO2 and under constant rotation (135RPM) as previously described (53). Approximately 2 × 106 cells were transfected with either wild-type or mutant PDGFRA + Reporter (500 ng DNA per ml of culture) plasmids using Expifectamine (Gibco). Cells were stimulated by adding either 7.5 μl (PDGF-BB) or 15 μl (PDGF-AA, AB and CC) of recombinant PDGF ligands (2 μm, R&D Systems, Minneapolis, MN) or water (mock) during transfection into 6-well dishes with 1 ml of media. Cells were collected 24 h post-transfection and split into two pools for use in staining and flow cytometry, and other assays.
Flow cytometry
Cell staining and flow cytometry were performed as previously described (53) with slight modifications. Alexa 647-conjugated anti-Myc antibody (1:50 dilution, Invitrogen, Carlsbad, CA) was used to measure Myc-tagged surface receptor (APC-A channel) and functional GFP-PEST expression (FITC-A channel) in a BD FACS Fortessa flow cytometer (BD Biosciences, Franklin Lakes, NJ). At least 20 000 cells were counted for each replicate and three independent biological replicate transfections were assayed for each condition. Gating parameters for EGFP and Myc signals were determined by independent pilot experiments and remained uniform for all replicates. The numerical values of counted cells were entered into Graphpad software (San Diego, CA) and the resulting values were tested using 2-way ANOVA (Supplementary Material, Tables S9 and S10). All replicate and numerical data for flow cytometry experiments are available via Figshare.
Zebrafish studies
CRISPR/Cas9 genome editing was used to perturb the pdgfra locus in developing zebrafish embryos (Supplementary Material, Table S11). Sixty-two embryos were injected with 5 um of guide RNA and 2 ug/ml of Cas9 enzyme (51 survived, 37 were imaged and 24 had a vertical upper lip groove compared with 0/24 control embryos). DNA was extracted from individual CRISPR-injected animals and subsequently used for genotyping. Targeted bar-coded deep sequencing (54) detected deletions in exon 6 ranging from 3 to 24 bps, confirming alteration of the pdgfra locus (Supplementary Material, Table S11). A three primer PCR strategy as described by (54) was used to add a unique 6 bp barcode to each individual embryo. Then, 6 μl from each of the uniquely bar-coded embryos was pooled and the pooled sample run through a MinElute Reaction Cleanup Kit (cat. no. 28 204, Qiagen, Hilden, Germany). The resulting solution was subjected to deep sequencing on a MiSeq instrument using a V2, 500 cycle kit at the University of Texas MD Anderson Advanced Technology Genomics Core Facility (Houston, TX). The raw reads obtained from the deep sequencing were de-multiplexed using the FASTQ/A Barcode Splitter tool from the FASTX-Toolkit (http://hannonlab.cshl.edu/fastx_toolkit/index.html). To estimate the amount of CRISPR-mediated gene editing observed in each embryo, fastq files for individual animals were run though the command line version of the CRISPResso2 software pipeline (55) in the amplicon mode.
To determine the impact of pharmacological inhibition of PDGFRA signaling, Tg(Krt4:GFP) transgenic embryos were immersed in 0.6 um Pdgfr inhibitor V (Calbiochem, San Diego, CA) in E3 from 10 to 30 h post-fertilization (hpf). Embryos were fixed at 5 days post-fertilization (dpf), stained with DAPI and mounted rostrally for imaging. The resulting phenotype was dose-dependent, with no effect at 0.125 um and gross severe abnormalities at 2 um. At 0.6 um, 37/40 had a vertical groove in the upper lip compared with 0/14 controls.
Supplementary Material
Acknowledgements
We thank Maria E. Serna and Rosa Martinez for technical assistance. The author(s) acknowledge the support of Dr David Haviland of the Houston Methodist Flow Cytometry Core and the High-Performance Computing for research facility at the University of Texas MD Anderson Cancer Center for providing computational resources that have contributed to these research results.
Conflict of Interest statement. The authors declare no conflict of interests.
Contributor Information
Yao Yu, Department of Epidemiology, University of Texas MD Anderson Cancer Center, Houston, TX 77030, USA.
Rolando Alvarado, Center for RNA Therapeutics, Department of Cardiovascular Sciences, Houston Methodist Research Institute, Houston, TX 77030, USA; Department of Cardiovascular Sciences, Houston Methodist Research Institute, Houston, TX 77030, USA.
Lauren E Petty, Vanderbilt Genetics Institute, Vanderbilt University Medical Center, Nashville, TN 37232, USA.
Ryan J Bohlender, Department of Epidemiology, University of Texas MD Anderson Cancer Center, Houston, TX 77030, USA.
Douglas M Shaw, Vanderbilt Genetics Institute, Vanderbilt University Medical Center, Nashville, TN 37232, USA.
Jennifer E Below, Vanderbilt Genetics Institute, Vanderbilt University Medical Center, Nashville, TN 37232, USA.
Nada Bejar, Center for RNA Therapeutics, Department of Cardiovascular Sciences, Houston Methodist Research Institute, Houston, TX 77030, USA; Department of Cardiovascular Sciences, Houston Methodist Research Institute, Houston, TX 77030, USA.
Oscar E Ruiz, Department of Genetics, University of Texas MD Anderson Cancer Center, Houston, TX 77030, USA.
Bhavna Tandon, Department of Pediatrics and Pediatric Research Center, UTHealth McGovern Medical School, Houston, TX 77030, USA.
George T Eisenhoffer, Department of Genetics, University of Texas MD Anderson Cancer Center, Houston, TX 77030, USA.
Daniel L Kiss, Center for RNA Therapeutics, Department of Cardiovascular Sciences, Houston Methodist Research Institute, Houston, TX 77030, USA; Department of Cardiovascular Sciences, Houston Methodist Research Institute, Houston, TX 77030, USA.
Chad D Huff, Department of Epidemiology, University of Texas MD Anderson Cancer Center, Houston, TX 77030, USA.
Ariadne Letra, Department of Diagnostic and Biomedical Sciences, UTHealth School of Dentistry at Houston, Houston, TX 77054, USA; Center for Craniofacial Research, UTHealth School of Dentistry at Houston, Houston 77054, TX, USA.
Jacqueline T Hecht, Department of Pediatrics and Pediatric Research Center, UTHealth McGovern Medical School, Houston, TX 77030, USA; Center for Craniofacial Research, UTHealth School of Dentistry at Houston, Houston 77054, TX, USA.
Funding
National Institutes of Health (R01-DE011931 to J.T.H.; R35GM137819 to D.L.K.; R01GM124043 to G.T.E.; R01GM133169 to C.D.H. and J.E.B.); HMRI Career Cornerstone (to D.L.K.); Gulf Coast Consortium (to G.C.C.); John S. Dunn Collaborative Research Award (to J.T.H. and G.T.E.); Cancer Prevention Research Institute of Texas (RR140077) and Mark and Linda Quick Basic Science Award (to G.T.E.).
References
- 1. Dixon, M.J., Marazita, M.L., Beaty, T.H. and Murray, J.C. (2011) Cleft lip and palate: understanding genetic and environmental influences. Nat. Rev. Genet., 12, 167–178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Carter, C.O. (1969) Polygenic inheritance and common diseases. Lancet, 1, 1252–1256. [DOI] [PubMed] [Google Scholar]
- 3. Leslie, E.J. and Marazita, M.L. (2013) Genetics of cleft lip and cleft palate. Am. J. Med. Genet. C Semin. Med. Genet., 163C, 246–258. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Letra, A., Zhao, M., Silva, R.M., Vieira, A.R. and Hecht, J.T. (2014) Functional significance of MMP3 and TIMP2 polymorphisms in cleft lip/palate. J. Dent. Res., 93, 651–656. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Cvjetkovic, N., Maili, L., Weymouth, K.S., Hashmi, S.S., Mulliken, J.B., Topczewski, J., Letra, A., Yuan, Q., Blanton, S.H., Swindell, E.C.et al. (2015) Regulatory variant in FZD6 gene contributes to nonsyndromic cleft lip and palate in an African-American family. Mol. Genet. Genomic Med., 3, 440–451. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Ludwig, K.U., Mangold, E., Herms, S., Nowak, S., Reutter, H., Paul, A., Becker, J., Herberz, R., AlChawa, T., Nasser, E.et al. (2012) Genome-wide meta-analyses of nonsyndromic cleft lip with or without cleft palate identify six new risk loci. Nat. Genet., 44, 968–971. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Ludwig, K.U., Ahmed, S.T., Bohmer, A.C., Sangani, N.B., Varghese, S., Klamt, J., Schuenke, H., Gultepe, P., Hofmann, A., Rubini, M.et al. (2016) Meta-analysis reveals genome-wide significance at 15q13 for nonsyndromic clefting of both the lip and the palate, and functional analyses implicate GREM1 as a plausible causative gene. PLoS Genet., 12, e1005914. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Chiquet, B.T., Henry, R., Burt, A., Mulliken, J.B., Stal, S., Blanton, S.H. and Hecht, J.T. (2011) Nonsyndromic cleft lip and palate: CRISPLD genes and the folate gene pathway connection. Birth Defects Res. A Clin. Mol. Teratol., 91, 44–49. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Leslie, E.J., Carlson, J.C., Shaffer, J.R., Butali, A., Buxo, C.J., Castilla, E.E., Christensen, K., Deleyiannis, F.W., Leigh Field, L., Hecht, J.T.et al. (2017) Genome-wide meta-analyses of nonsyndromic orofacial clefts identify novel associations between FOXE1 and all orofacial clefts, and TP63 and cleft lip with or without cleft palate. Hum. Genet., 136, 275–286. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Leslie, E.J., Carlson, J.C., Shaffer, J.R., Feingold, E., Wehby, G., Laurie, C.A., Jain, D., Laurie, C.C., Doheny, K.F., McHenry, T.et al. (2016) A multi-ethnic genome-wide association study identifies novel loci for non-syndromic cleft lip with or without cleft palate on 2p24.2, 17q23 and 19q13. Hum. Mol. Genet., 25, 2862–2872. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Leslie, E.J., Liu, H., Carlson, J.C., Shaffer, J.R., Feingold, E., Wehby, G., Laurie, C.A., Jain, D., Laurie, C.C., Doheny, K.F.et al. (2016) A genome-wide association study of nonsyndromic cleft palate identifies an etiologic missense variant in GRHL3. Am. J. Hum. Genet., 98, 744–754. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Mangold, E., Ludwig, K.U., Birnbaum, S., Baluardo, C., Ferrian, M., Herms, S., Reutter, H., deAssis, N.A., Chawa, T.A., Mattheisen, M.et al. (2010) Genome-wide association study identifies two susceptibility loci for nonsyndromic cleft lip with or without cleft palate. Nat. Genet., 42, 24–26. [DOI] [PubMed] [Google Scholar]
- 13. Dickson, S.P., Wang, K., Krantz, I., Hakonarson, H. and Goldstein, D.B. (2010) Rare variants create synthetic genome-wide associations. PLoS Biol., 8, e1000294. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Crouch, D.J.M. and Bodmer, W.F. (2020) Polygenic inheritance, GWAS, polygenic risk scores, and the search for functional variants. Proc. Natl. Acad. Sci. U. S. A., 117, 18924–18933. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Carlson, J.C., Anand, D., Butali, A., Buxo, C.J., Christensen, K., Deleyiannis, F., Hecht, J.T., Moreno, L.M., Orioli, I.M., Padilla, C.et al. (2019) A systematic genetic analysis and visualization of phenotypic heterogeneity among orofacial cleft GWAS signals. Genet. Epidemiol., 43, 704–716. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. van Rooij, I.A., Ludwig, K.U., Welzenbach, J., Ishorst, N., Thonissen, M., Galesloot, T.E., Ongkosuwito, E., Berge, S.J., Aldhorae, K., Rojas-Martinez, A.et al. (2019) Non-syndromic cleft lip with or without cleft palate: genome-wide association study in europeans identifies a suggestive risk locus at 16p12.1 and supports SH3PXD2A as a clefting susceptibility gene. Genes, 10, 1023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. McGregor, T.L., Van Driest, S.L., Brothers, K.B., Bowton, E.A., Muglia, L.J. and Roden, D.M. (2013) Inclusion of pediatric samples in an opt-out biorepository linking DNA to de-identified medical records: pediatric BioVU. Clin. Pharmacol. Ther., 93, 204–211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Liang, L., Yan, X.E., Yin, Y. and Yun, C.H. (2016) Structural and biochemical studies of the PDGFRA kinase domain. Biochem. Biophys. Res. Commun., 477, 667–672. [DOI] [PubMed] [Google Scholar]
- 19. Jumper, J., Evans, R., Pritzel, A., Green, T., Figurnov, M., Ronneberger, O., Tunyasuvunakool, K., Bates, R., Zidek, A., Potapenko, A.et al. (2021) Highly accurate protein structure prediction with AlphaFold. Nature, 596, 583–589. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Heldin, C.H. and Lennartsson, J. (2013) Structural and functional properties of platelet-derived growth factor and stem cell factor receptors. Cold Spring Harb. Perspect. Biol., 5, a009100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Eberhart, J.K., He, X., Swartz, M.E., Yan, Y.L., Song, H., Boling, T.C., Kunerth, A.K., Walker, M.B., Kimmel, C.B. and Postlethwait, J.H. (2008) MicroRNA Mirn140 modulates Pdgf signaling during palatogenesis. Nat. Genet., 40, 290–298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Leslie, E.J. and Murray, J.C. (2013) Evaluating rare coding variants as contributing causes to non-syndromic cleft lip and palate. Clin. Genet., 84, 496–500. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Tallquist, M.D. and Soriano, P. (2003) Cell autonomous requirement for PDGFRalpha in populations of cranial and cardiac neural crest cells. Development, 130, 507–518. [DOI] [PubMed] [Google Scholar]
- 24. Xu, X., Bringas, P., Jr., Soriano, P. and Chai, Y. (2005) PDGFR-alpha signaling is critical for tooth cusp and palate morphogenesis. Dev. Dyn., 232, 75–84. [DOI] [PubMed] [Google Scholar]
- 25. Qian, C., Wong, C.W.Y., Wu, Z., He, Q., Xia, H., Tam, P.K.H., Wong, K.K.Y. and Lui, V.C.H. (2017) Stage specific requirement of platelet-derived growth factor receptor-alpha in embryonic development. PLoS One, 12, e0184473. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Rattanasopha, S., Tongkobpetch, S., Srichomthong, C., Siriwan, P., Suphapeetiporn, K. and Shotelersuk, V. (2012) PDGFRa mutations in humans with isolated cleft palate. Eur. J. Hum. Genet., 20, 1058–1062. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Joosten, P.H., Toepoel, M., Mariman, E.C. and Van Zoelen, E.J. (2001) Promoter haplotype combinations of the platelet-derived growth factor alpha-receptor gene predispose to human neural tube defects. Nat. Genet., 27, 215–217. [DOI] [PubMed] [Google Scholar]
- 28. Ludwig, K.U., Bohmer, A.C., Bowes, J., Nikolic, M., Ishorst, N., Wyatt, N., Hammond, N.L., Golz, L., Thieme, F., Barth, S.et al. (2017) Imputation of orofacial clefting data identifies novel risk loci and sheds light on the genetic background of cleft lip +/− cleft palate and cleft palate only. Hum. Mol. Genet., 26, 829–842. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Shawky, R.M. (2014) Reduced penetrance in human inherited disease. Egypt. J. Med. Hum. Genet., 15, 103–111. [Google Scholar]
- 30. Liang, L., Yan, X.E. and Yun, C.H. (2020) Crystal structure of PDGFRA T674I in complex with crenolanib by soaking. Protein Data Bank. 10.2210/pdb6joj/pdb. [DOI] [Google Scholar]
- 31. Liang, L., Yan, X.E. and Yun, C.H. (2020) Crystal structure of PDGFRA in complex with sunitinib by soaking. Protein Data Bank. 10.2210/pdb6jok/pdb. [DOI] [Google Scholar]
- 32. Ip, C.K.M., Ng, P.K.S., Jeong, K.J., Shao, S.H., Ju, Z., Leonard, P.G., Hua, X., Vellano, C.P., Woessner, R., Sahni, N.et al. (2018) Neomorphic PDGFRA extracellular domain driver mutations are resistant to PDGFRA targeted therapies. Nat. Commun., 9, 4583. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Hashmi, S.S., Waller, D.K., Langlois, P., Canfield, M. and Hecht, J.T. (2005) Prevalence of nonsyndromic oral clefts in Texas: 1995-1999. Am. J. Med. Genet. A, 134, 368–372. [DOI] [PubMed] [Google Scholar]
- 34. Letra, A., Silva, R.M., Motta, L.G., Blanton, S.H., Hecht, J.T., Granjeirol, J.M. and Vieira, A.R. (2012) Association of MMP3 and TIMP2 promoter polymorphisms with nonsyndromic oral clefts. Birth Defects Res. A Clin. Mol. Teratol., 94, 540–548. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Fischbach, G.D. and Lord, C. (2010) The Simons Simplex Collection: a resource for identification of autism genetic risk factors. Neuron, 68, 192–195. [DOI] [PubMed] [Google Scholar]
- 36. Iossifov, I., O'Roak, B.J., Sanders, S.J., Ronemus, M., Krumm, N., Levy, D., Stessman, H.A., Witherspoon, K.T., Vives, L., Patterson, K.E.et al. (2014) The contribution of de novo coding mutations to autism spectrum disorder. Nature, 515, 216–221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. DePristo, M.A., Banks, E., Poplin, R., Garimella, K.V., Maguire, J.R., Hartl, C., Philippakis, A.A., delAngel, G., Rivas, M.A., Hanna, M.et al. (2011) A framework for variation discovery and genotyping using next-generation DNA sequencing data. Nat. Genet., 43, 491–498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Yu, Y., Hu, H., Bohlender, R.J., Hu, F., Chen, J.S., Holt, C., Fowler, J., Guthery, S.L., Scheet, P., Hildebrandt, M.A.T.et al. (2018) XPAT: a toolkit to conduct cross-platform association studies with heterogeneous sequencing datasets. Nucleic Acids Res., 46, e32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Hu, H., Roach, J.C., Coon, H., Guthery, S.L., Voelkerding, K.V., Margraf, R.L., Durtschi, J.D., Tavtigian, S.V., Shankaracharya, W., W.et al. (2014) A unified test of linkage analysis and rare-variant association for analysis of pedigree sequence data. Nat. Biotechnol., 32, 663–669. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Karczewski, K.J., Francioli, L.C., Tiao, G., Cummings, B.B., Alfoldi, J., Wang, Q., Collins, R.L., Laricchia, K.M., Ganna, A., Birnbaum, D.P.et al. (2020) The mutational constraint spectrum quantified from variation in 141,456 humans. Nature, 581, 434–443. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Shete, S., Amos, C.I., Hwang, S.J. and Strong, L.C. (2002) Individual-specific liability groups in genetic linkage, with applications to kindreds with Li-Fraumeni syndrome. Am. J. Hum. Genet., 70, 813–817. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Staples, J., Qiao, D., Cho, M.H., Silverman, E.K., University of Washington Center for Mendelian, G, Nickerson, D.A. and Below, J.E (2014) PRIMUS: rapid reconstruction of pedigrees from genome-wide estimates of identity by descent. Am. J. Hum. Genet., 95, 553–564. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. O'Connell, J.R. and Weeks, D.E. (1998) PedCheck: a program for identification of genotype incompatibilities in linkage analysis. Am. J. Hum. Genet., 63, 259–266. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Gudbjartsson, D.F., Jonasson, K., Frigge, M.L. and Kong, A. (2000) Allegro, a new computer program for multipoint linkage analysis. Nat. Genet., 25, 12–13. [DOI] [PubMed] [Google Scholar]
- 45. Abyzov, A., Urban, A.E., Snyder, M. and Gerstein, M. (2011) CNVnator: an approach to discover, genotype, and characterize typical and atypical CNVs from family and population genome sequencing. Genome Res., 21, 974–984. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Wang, K., Li, M. and Hakonarson, H. (2010) ANNOVAR: functional annotation of genetic variants from high-throughput sequencing data. Nucleic Acids Res., 38, e164. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Gelfman, S., Wang, Q., McSweeney, K.M., Ren, Z., La Carpia, F., Halvorsen, M., Schoch, K., Ratzon, F., Heinzen, E.L., Boland, M.J.et al. (2017) Annotating pathogenic non-coding variants in genic regions. Nat. Commun., 8, 236. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Rentzsch, P., Witten, D., Cooper, G.M., Shendure, J. and Kircher, M. (2019) CADD: predicting the deleteriousness of variants throughout the human genome. Nucleic Acids Res., 47, D886–D894. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Grabe, N. (2002) AliBaba2: context specific identification of transcription factor binding sites. In Silico Biol., 2, S1–S15. [PubMed] [Google Scholar]
- 50. Messeguer, X., Escudero, R., Farre, D., Nunez, O., Martinez, J. and Alba, M.M. (2002) PROMO: detection of known transcription regulatory elements using species-tailored searches. Bioinformatics, 18, 333–334. [DOI] [PubMed] [Google Scholar]
- 51. Berman, H.M., Westbrook, J., Feng, Z., Gilliland, G., Bhat, T.N., Weissig, H., Shindyalov, I.N. and Bourne, P.E. (2000) The Protein Data Bank. Nucleic Acids Res., 28, 235–242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Pettersen, E.F., Goddard, T.D., Huang, C.C., Meng, E.C., Couch, G.S., Croll, T.I., Morris, J.H. and Ferrin, T.E. (2021) UCSF ChimeraX: structure visualization for researchers, educators, and developers. Protein Sci., 30, 70–82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Park, J., Gill, K.S., Aghajani, A.A., Heredia, J.D., Choi, H., Oberstein, A. and Procko, E. (2020) Engineered receptors for human cytomegalovirus that are orthogonal to normal human biology. PLoS Pathog., 16, e1008647. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Varshney, G.K., Pei, W., LaFave, M.C., Idol, J., Xu, L., Gallardo, V., Carrington, B., Bishop, K., Jones, M., Li, M.et al. (2015) High-throughput gene targeting and phenotyping in zebrafish using CRISPR/Cas9. Genome Res., 25, 1030–1042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Clement, K., Rees, H., Canver, M.C., Gehrke, J.M., Farouni, R., Hsu, J.Y., Cole, M.A., Liu, D.R., Joung, J.K., Bauer, D.E.et al. (2019) CRISPResso2 provides accurate and rapid genome editing sequence analysis. Nat. Biotechnol., 37, 224–226. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.