

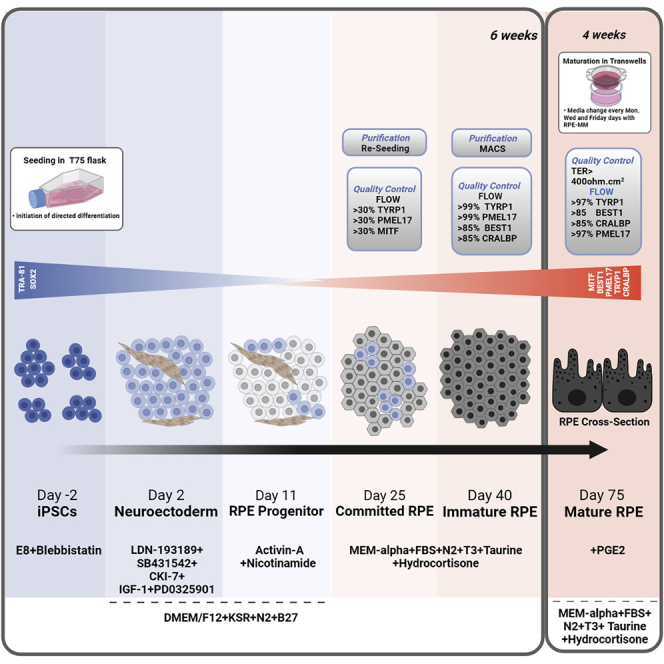
Summary
RPE tissues are derived from induced pluripotent stem cells (iPSCs) to model retinal diseases and as a replacement therapy for macular degeneration. Here, we developed a robust and efficient directed differentiation protocol to generate pure RPE cells that form a polarized monolayer. This protocol describes how to set up RPE differentiation and to obtain a pure population that expresses mature RPE markers and forms strong tight junctions.
For complete details on the use and execution of this protocol, please refer to Sharma et al., 2019, Sharma et al., 2021 and Miyagishima et al. (2021).
Subject areas: Cell Biology, Cell culture, Cell isolation, Flow Cytometry/Mass Cytometry, Health Sciences, Stem Cells, Cell Differentiation
Graphical abstract
Highlights
-
•
The protocol follows developmentally guided cues to generate RPE from iPSC
-
•
Magnetic cell sorting produces a high yield of pure RPE cells
-
•
Flow cytometry confirms RPE cell purity and expressions of maturation markers
-
•
Mature cells form strong tight junctions as measured by trans-epithelial resistance
Publisher’s note: Undertaking any experimental protocol requires adherence to local institutional guidelines for laboratory safety and ethics.
RPE tissues are derived from induced pluripotent stem cells (iPSCs) to model retinal diseases and as a replacement therapy for macular degeneration. Here, we developed a robust and efficient directed differentiation protocol to generate pure RPE cells that form a polarized monolayer. This protocol describes how to set up RPE differentiation and to obtain a pure population that expresses mature RPE markers and forms strong tight junctions.
Before you begin
Follow sterile cell culture practices for growing cells and cleaning the bio safety cabinet (BSC). Our protocol has been used to differentiate over 50 iPSC lines derived from patients with different retinal degenerative diseases and from unaffected controls confirming its reproducibility, efficiency, and ability to generate pure RPE cells in high numbers. The protocol has been used in clinical manufacturing of iPSC-RPE as autologous transplants for macular degeneration patients. For clinical manufacturing iPSC-RPE are differentiated using the same timeline and process but replacing research-grade reagents with xenofree and cell therapy-grade reagents.
Institutional permissions
These iPSCs were derived under a NIH IRB approved protocol #11-E1-0245.
Prepare reagents for iPSC expansion
Timing: 1–2 days
This section will cover the details on reagents preparation for iPSC culture, which includes Vitronectin coating and making complete E8 media.
-
1.2.5 μg/mL Vitronectin solution (VTN).
-
a.Thaw the VTN-N vial at room temperature (RT) and dilute it by mixing it in 200 mL of DPBS (without Ca2+/Mg2).
-
b.Filter it through a 0.22 μm filter unit.
-
c.Add diluted vitronectin (2.5 μg/mL) to the plastic surface of the culture dish as per the volume mentioned in coating volume table.
Surface type Volume VTN concentration Vol/surface area 6-well plate 2 mL/well 2.5 μg/mL 200 μL/cm2 T-75 flask 20 mL 2.5 μg/mL T-150 flask 30 mL 2.5 μg/mL 12 well trans well plate 0.5 mL on apical side 2.5 μg/mL -
d.Leave the plates at RT for an hour.
-
e.Aspirate the vitronectin solution from wells (or flasks) prior to seeding cells. Ensure that the wells (flasks) do not dry out prior to seeding.
-
f.If plates/flasks are not used immediately wrap coated plates/flasks in aluminum foil and store them at 4°C for maximum of two weeks.
-
g.Before using stored plates, bring them to RT for at least an hour.
 CRITICAL: Avoid more than one freeze-thaw cycle of VTN. Don't place the unused coated plates back in the fridge once they are stored at 4°C and taken out at RT for use.
CRITICAL: Avoid more than one freeze-thaw cycle of VTN. Don't place the unused coated plates back in the fridge once they are stored at 4°C and taken out at RT for use.
-
a.
-
2.Prepare E8 complete media.
-
a.Prepare E8 complete media by thawing 10 mL E8 supplement bottle and mix in 500 mL of E8 basal media.
-
a.
-
3.Prepare 1000× (10 mM) rock inhibitor (Y27632).
-
a.Stock concentration: 10 mM; working concentration: 10 μM.
-
b.Dissolve 10 mg of rock inhibitor in required volume of cell culture grade sterile water and mix the vial well on vortex. Pool all the vials in one 50 mL tube and mix it well by placing the tube on a vortex again.
-
c.Prepare 100 μL aliquots, report volume, concentration, and date of preparation on the label and store in −80°C.
-
d.The stock solution at −80°C freezer is suitable to use within six months.
-
a.
-
4.Prepare E8 thawing media.
-
a.Add rock Inhibitor aliquot to E8 complete media at the dilution of 1:1000 to make a working concentration of 10 μM.
-
b.Mix it well in a 50 mL conical tube and filter it through a 0.22 μm filter unit.
-
c.Media is good to use within 24 h of preparation.
-
a.
Prepare reagents and media for iPSC to RPE differentiation
Reagents and media preparation is as critical and essential as the starting iPSC culture. This section will focus on preparing the stock and working aliquots of the growth factors, small molecules, compounds, and recombinant proteins needed for RPE differentiation.
-
5.Prepare 10,000× (25 mM) blebbistatin stock solution.
-
a.Stock concentration: 25 mM; working concentration: 2.5 μM.
-
b.Reconstitute 5 mg of blebbistatin powder in 0.384 mL (384 μL) of DMSO and collect in a sterile microcentrifuge tube.
-
c.Rinse the stock bottle containing the powder with 0.3 mL (300 μL) of DMSO and collect in the same 1.5 mL microcentrifuge tube.
-
d.Dispense the above stock solution into 14 μL aliquots in fresh 1.5 mL microcentrifuge tubes.
-
e.Label the tubes including information on volume, concentration, and date of preparation and store them at −80°C freezer.
-
f.The stock solution at −80°C freezer is suitable to use within six months.
-
a.
-
6.Prepare 1,000× (200 mM) ascorbic acid stock solution.
-
a.Stock concentration: 200 mM; working concentration: 200 μM.
-
b.For 50 mL of ascorbic solution, weigh 1.76 g of ascorbic acid and dissolve in 25 mL of cell culture grade sterile water. Vortex the tube for 3–5 min for complete dissolution and make up the volume to 50 mL.
-
c.Filter the solution through 0.22 μm filter unit.
-
d.Make aliquots of 550 μL aliquots each to be added to 500 mL of differentiation media.
-
e.Label the tubes including information on volume, concentration, and date of preparation and store in −80°C freezer.
-
f.The stock solution at −80°C freezer is suitable to use within six months.
-
a.
-
7.Prepare 100,000× (10 mM) LDN-193189 stock solution.
-
a.Stock concentration: 10 mM; working concentration: 100 nM (for RDM), 10 nM (for RIM).
-
b.Thaw the stock vial at RT and make 7 μL aliquots for 500 mL of RDM.
-
c.Dilute 10 mM LDN-193189 1:1000 in DMEM/F12 to make a stock concentration of 10 μM for RIM and make the aliquots of 550 μL for 500 mL of RIM.
-
d.Label the tubes including information on volume, concentration, and date of preparation and store them at −80°C freezer.
-
e.The stock solution at −80°C freezer is suitable to use within six months.
-
a.
-
8.Prepare 4000× (40 mM) SB431542 stock solution.
-
a.Stock concentration: 40 mM; working concentration:10 μM.
-
b.Dissolve 10 mg of SB431542 in the appropriate volume of DMSO.
-
c.Let the vial sit inside the BSC for 2–3 min. Ensure that the powder is well suspended.
-
d.Invert the vial upside down to make sure that the powder stuck to the cap is resuspended in the DMSO.
-
e.Leave the vial again for a few minutes in the BSC. Gently tap vials to collect all liquid at the bottom of each vial. Pool solutions from all the vials in one 50 mL tube and mix it well again.
-
f.Make aliquots of 130 μL for 500 mL of RDM and 15 μL for 500 mL of RIM.
-
g.Label the tubes including information on volume, concentration, and date of preparation and freeze at −80°C.
-
h.The stock solution at −80°C freezer is suitable to use within six months.
-
a.
-
9.Prepare 1000× (5 mM) CKI-7 stock solution.
-
a.Stock concentration: 5 mM; working concentration: 5 μM (RDM), 0.5 μM (RIM).
-
b.Dissolved in cell culture grade sterile water.
-
c.Dissolve 5 mg in 2.8 mL for a final concentration of 5 mM.
-
d.Mix the vial on vortex, sometimes incubation in dry bath is also needed.
-
e.Pool all the vials in one 50 mL tube and mix it well by placing the tube on a vortex again. Make aliquots of 520 μL for RDM and 60 μL for RIM.
-
f.Label the tubes including information on volume, concentration, and date of preparation and freeze at −80°C.
-
g.The stock solution at −80°C freezer is suitable to use within six months.
-
a.
-
10.Prepare 10,000 × (100 μg/mL) AF-IGF-1 stock solution.
-
a.Stock concentration: 100 μg/mL; working concentration: 10 ng/mL (RDM), 1 ng/mL (RIM).
-
b.Dissolve 1 mg of IGF-1 in 10 mL of DPBS.
-
c.Let the vial mix well on vortex and place it in a 50 mL conical tube.
-
d.Make aliquots of 55 μL for 500 mL of RDM and 7 μL for 500 mL of RIM.
-
e.Label the tubes including information on volume, concentration, and date of preparation and store in −80°C freezer.
-
f.The stock solution at −80°C freezer is suitable to use within six months.
-
a.
-
11.Prepare (50 μg/mL) Activin-A stock solution.
-
a.Stock concentration: 50 μg/mL; working concentration: 50 ng/mL.
-
b.The stock bottle of Activin-A has 1–2 mg/mL in a volume of 1–3 mL of 30% (v/v) Acetonitrile and 0.1% (v/v) TFA.
-
c.Prepare aliquots of 50 μg in 50–100 μL volume for 500 mL of media.
-
d.The stock solution at −80°C freezer is suitable to use within six months.
-
a.
-
12.Prepare 1000× (1 M) nicotinamide stock solution.
-
a.Stock concentration:1 M; working concentration: 1 mM.
-
b.Weight 12.12 grams of nicotinamide and dissolve in 50 mL of cell culture grade sterile water.
-
c.Stir the solution until dissolved and make up the volume to 100 mL.
-
d.Filter the solution through 0.22 μm filter unit.
-
e.Make aliquots of 6 mL in sterile 15 mL tubes for 500 mL medium.
-
f.Label the tubes including information on volume, concentration, and date of preparation and store in −80°C freezer.
-
g.The stock solution at −80°C freezer is suitable to use within six months.
-
a.
-
13.Prepare 200× (50 mg/mL) taurine stock solution.
-
a.Stock concentration: 50 mg/mL; working concentration:250 μg/mL.
-
b.Dissolve in DPBS. Take 10 g of taurine, dissolve in 200 mL of DPBS. Keep it in dry bath and vortex the vial for few mins, if needed.
-
c.Filter it through 0.2 μM PES filter unit.
-
d.Make 3 mL aliquots for 500 mL.
-
e.Label the tubes including information on volume, concentration, and date of preparation store in −80°C freezer.
-
f.The stock solution at −80°C freezer is suitable to use within six months.
-
a.
-
14.Prepare 905× (50 μM) Hydrocortisone stock solution.
-
a.Stock concentration: 50 μM; working concentration: 18 nM.
-
b.Compound comes in a solution at concentration of 50 μM. Make aliquots of 600 μL for 500 mL medium.
-
c.Label the tubes including information on volume, concentration, and date of preparation store in −80°C freezer.
-
d.The stock solution at −80°C freezer is suitable to use within six months.
-
a.
-
15.Prepare 1538,461× (20 mg /mL) (T3) 3,3′,5-Triiodo-L-thyronine stock solution.
-
a.Stock concentration:20 mg/mL; working concentration:0.013 μg/mL.
-
b.Take 200 μL of 10 M NaOH and dilute it in 1.8 mL of cell culture grade sterile water to make a 1 M NaOH.
-
c.To make T3 stock- add 1 mL of 1 M NaOH, 1 mg of 3,3′,5-Triiodo-L-thyronine in 49 mL of MEM Alpha.
-
d.Make aliquots of 200 μL medium, label the tubes with the information citing volume, concentration, and date of preparation and store at −80°C.
-
e.The stock solution at −80°C freezer is suitable to use within six months.
-
f.For making 500 mL media - take 1 μL from the 200 μL aliquot, add 1 mL of MEM alpha to it, mix well and take 330 μL from this diluted stock and add to the 500 mL media.
-
a.
-
16.Prepare E8 plating media.
-
a.Thaw an aliquot of blebbistatin and add it as 1:10,000 dilution to E8 complete media.
-
b.Mix the media well by inverting the tube 2–3 times before filtering it through 0.22 μm filter unit.
-
c.Media is suitable to use within 48 h at 4°C.
-
a.
| Reagent | Stock concentration | Final concentration | Volume |
|---|---|---|---|
| E8 complete media | 1× | n/a | 500 mL |
| Blebbistatin | 25 mM | 2.5 μM | 50 μL |
-
17.Prepare Retinal Induction Media (RIM) for the induction of neuroectoderm- day 0 - day 1 of culture.
-
a.Thaw N2, B27 and KSR at 4°C overnight.
-
b.Bring the reagent aliquots from the −80°C freezer and media components to the BSC.
-
c.Follow the media recipe from the table below.
-
d.Make sure the aliquots are made appropriately and media is mixed well before filtration using a 0.22 μm filter unit.
-
e.Media is suitable to use within 2 weeks at 4°C.
-
a.
| RIM | Stock concentration | Final concentration | Volume |
|---|---|---|---|
| DMEM/F12 | 1× | N/A | 500 mL |
| CTS™ KnockOut™ SR XenoFree Or Knockout KSR |
1× | 1.5% | 7.5 mL |
| MEM non-essential AA | 10 mM | 0.1 mM | 5 mL |
| Sodium pyruvate | 100 mM | 1.0 mM | 5 mL |
| CTS™ N-2 supplement or N-2 supplement | 100× | 1.0% | 5 mL |
| B-27® supplement (+VitA) | 50× | 2.0% | 10 mL |
| Ascorbic acid | 200 mM | 200 μM | 500 μL |
| LDN-193189 | 10 mM | 10 nM | 500 μL |
| SB 431542 | 40 mM | 1.0 μM | 12.5 μL |
| CKI-7 Dihydrochloride | 5 mM | 0.5 μM | 50 μL |
| AF-IGF-1 | 100 μg/mL | 1 ng/mL | 5 μL |
-
18.Prepare Retinal Differentiation Media (RDM) for the induction of RPE fate day 2 - day 10 of culture.
-
a.Thaw N2, B27 and KSR at 4°C overnight.
-
b.Bring the reagent aliquots from the −80°C freezer and media components to the BSC.
-
c.Follow the media recipe from the table below.
-
d.Make sure the aliquots are made appropriately and media is mixed well before filtration using a 0.22 μm filter unit.
-
e.Media is suitable to use within 2 weeks at 4°C.
-
a.
| RDM | Stock concentration | Final concentration | Volume |
|---|---|---|---|
| DMEM/F12 | 1× | N/A | 500 mL |
| CTS™ KnockOut™ SR XenoFree Or Knockout KSR | 1× | 1.5% | 7.5 mL |
| MEM non-essential AA | 10 mM | 0.1 mM | 5 mL |
| Sodium pyruvate | 100 mM | 1.0 mM | 5 mL |
| CTS™ N-2 supplement or N-2 supplement | 100× | 1% | 5 mL |
| B-27® supplement (VitA) | 50× | 2% | 10 mL |
| Ascorbic acid | 200 mM | 200 μM | 500 μL |
| LDN-193189 | 10 mM | 100 nM | 5 μL |
| SB 431542 | 40 mM | 10 μM | 125 μL |
| CKI-7 Dihydrochloride | 5 mM | 5 μM | 500 μL |
| AF-IGF-1 | 100 μg/mL | 10 ng/mL | 50 μL |
| PD0325901 | 10 mM | 1 μM | 50 μL |
-
19.Prepare Retinal Media with Nicotinamide and Activin-A (RMNA) media for the commitment of RPE fate day 11 – day 20 of culture.
-
a.Thaw N2, B27 and KSR at 4°C overnight.
-
b.Bring the reagent aliquots from the −80°C freezer and media components to the BSC.
-
c.Follow the media recipe from the table below.
-
d.Make sure the aliquots are made appropriately and media is mixed well before filtration using a 0.22 μm filter unit.
-
e.Media is suitable to use within 2 weeks at 4°C.
-
a.
| RM-NA media | Stock concentration | Final concentration | Volume |
|---|---|---|---|
| DMEM/F12 | 1× | N/A | 500 mL |
| CTS™ KnockOut™ SR XenoFree or Knockout KSR | 1× | 1.5% | 7.5 mL |
| MEM non-essential AA | 10 mM | 0.1 mM | 5 mL |
| Sodium pyruvate | 100 mM | 1.0 mM | 5 mL |
| CTS™ N-2 supplement or N-2 supplement | 100× | 1% | 5 mL |
| B-27® supplement (+VitA) | 50× | 2% | 10 mL |
| Ascorbic acid | 200 mM | 200 μM | 500 μL |
| Activin-A | 100 μg/mL | 100 ng/mL | n/a |
| Nicotinamide | 1 M | 10 mM | 5 mL |
-
20.Prepare RPE-MM media for the RPE progenitors’ fate day 21 – day 40 of culture.
-
a.Thaw N2, B27 and KSR at 4°C overnight.
-
b.Bring the reagent aliquots from the −80°C freezer and media components to the BSC.
-
c.Follow the media recipe from the table below.
-
d.Make sure the aliquots are made appropriately and media is mixed well before filtration using a 0.22 μm filter unit.
-
e.Media is suitable to use within 2 weeks at 4°C.
-
a.
| RPE-MM | Stock concentration | Final concentration | Volume |
|---|---|---|---|
| MEM alpha | 1× | N/A | 500 mL |
| Heat inactivated fetal bovine serum | 1× | 5% | 25 mL |
| N-2 supplement | 100× | 1% | 5 mL |
| MEM non-essential amino acids | 10 mM | 0.1 mM | 5 mL |
| Sodium pyruvate | 100 mM | 1 mM | 5 mL |
| Penicillin-Strep | 1× | 5 mL | |
| Taurine | 50 mg/mL | 250 μg/mL | 2.5 mL |
| Hydrocortisone | 18.1 mg/L | 20 μg/L | 553 μL |
| T3 | 20 mg/mL | 0.013 μg/L | 330 μL |
-
21.Prepare MACS buffer for the enrichment of mature RPE.
-
a.Thaw heat inactivated FBS overnight in 4°C.
-
b.Follow the recipe given in the table below.
-
c.Buffer is suitable to use within 2 weeks at 4°C.
-
a.
| MACS buffer | Stock concentration | Final concentration | Volume |
|---|---|---|---|
| DPBS (without Ca2+ and Mg2+) | 1× | N/A | 1000 mL |
| Heat inactivated fetal bovine serum | 1× | 2% | 20 mL |
| Ultra-Pure EDTA solution | 0.5 M | 2 mM | 4 mL |
Prepare reagents for day 40 and day 75 purity analysis by flow cytometry
This section describes the buffers, antibodies, and fixative recipes for the FLOW purity assays.
-
22.Fixation Buffer: 4%PFA in DPBS without Ca2+ /Mg2+.
-
a.Follow the recipe given in the table below.
-
b.Buffer is suitable to use within 2 weeks at 4°C.
-
a.
| Fixation buffer | Final concentration | Volume |
|---|---|---|
| DPBS (without Ca2+ and Mg2+) | N/A | 49 mL |
| 36.5% Formaldehyde | 4% | 1 mL |
-
23.Permeabilization Buffer: 2%FBS in DPBS without Ca2+ /Mg2+ + 0.2%Triton-X-100.
-
a.Thaw heat inactivated FBS overnight in 4°C.
-
b.Follow the recipe given in the table below.
-
c.Buffer is suitable to use within 2 weeks at 4°C.
-
a.
| Permeabilization buffer | Final concentration | Volume |
|---|---|---|
| DPBS (without Ca2+ and Mg2+) | 98% | 49 mL |
| Heat inactivated fetal bovine serum | 2% | 1 mL |
| Triton-X-100 | 0.2% | 0.1 mL |
Prepare reagents and media for RPE maturation
This section focuses on preparing reagent and RPE maturation media.
-
24.Prepare 1000× (50 μM) PGE2.
-
a.Stock concentration: 50 mM; working concentration: 50 μM.
-
b.Dissolve 10 mg in appropriate volume of DMSO.
-
a.
-
25.Prepare RPE-MM with PGE2 for day 50 – day 75 of culture.
-
a.Make sure the aliquots are made appropriately and media is mixed well before filtration using a 0.22 μm filter unit.
-
b.Follow the recipe given in the table below.
-
c.Media is suitable to use within 2 weeks at 4°C.
-
a.
| RPE-MM | Final concentration | Volume |
|---|---|---|
| 5%RPE media | N/A | 500 mL |
| PGE2 | 50 μM | 500 μL |
Establish iPSC culture for setting up differentiation
The state of iPSC culture is one of the critical factors determining the efficiency of RPE differentiation. Therefore, we will cover all the steps that might affect the iPSC culture’s health, ranging from thawing to passaging and media change.
Pick the iPSC line that is characterized for pluripotency markers, normal karyotype, and stable genotype.
-
26.Thaw iPSC line(s):
-
a.Prepare E8 thawing media as above and let E8 thawing media equilibrate at RT for 20 min before using it.
-
b.Take out an iPSC vial from the liquid nitrogen tank and thaw the vial either in a water bath, ThawSTAR, or dry beads.
-
c.For thawing in ThawSTAR, transfer the vial, push it in the socket, wait until it beeps. Take the vial out of ThawSTAR, spray it with 70% ethanol, wipe it, before bringing it to the BSC.
-
d.For dry beads thawing, place the vial in 37°C dry beads. Thaw until last the ice crystal is visible, which takes between 3–4 min.
-
e.Aliquot 10 mL of thawing media in 50 mL conical tube.
-
f.Bring the thawed vial to the BSC. With serological pipette, aspirate cell clumps from the vial and start adding them dropwise to the 50 mL conical tube containing the thawing media with constant shaking of tube.
-
g.Wash the vial with 1 mL of thawing media.
-
h.Centrifuge the tube @ 400 g for 5 min.
-
i.Aspirate the supernatant and add 6 mL of thawing media to the 50 mL tube using a 5 mL serological pipette.
-
j.Break the cell pellet gently by pipetting with a 5 mL serological pipette 1–2 times and mix it well by inverting the tube.
-
k.Aspirate VTN from the 2 wells of 6 well plates prepared in step 1. Add 3 mL of the cell suspension to each VTN coated well.
-
l.Switch to complete E8 media 24 h post thawing.
-
m.Feed the cells every day with 3 mL/well (300 μL/cm2) of E8 complete media.
-
a.
-
27.Passaging the iPSC lines:
-
a.Spilt iPSCs using Versene solution when culture reaches 70%–80% confluent.
-
b.Wash the well with 2 mL of DPBS, aspirate DPBS and add 1 mL of Versene solution per well.
-
c.After 5 min at RT or 3 min at 37°C, look under the microscope to identify rounding of cells.
-
d.Aspirate the Versene solution when you see small holes in an intact colony or cells losing contact from neighboring cells and add 3 mL of complete E8 media in that well. Versene is usually on for 7–8 min.
-
e.With 5 mL serological pipette to collect cells from the well. In 2–3 rounds of pipetting up and down the E8 medium, the cells should come off. Collect them in a 50 mL tube containing a volume of 10 mL of E8 media.
-
f.Aspirate Vitronectin from the plate to be used for seeding.
-
g.After thaw, the first split should be at a 1:3 ratio, whereas subsequent splits can be at a 1:6–1:12 ratio to get the confluency of 70%–80% by day 4–5.
-
h.Add 3 mL of cell suspension per well of a 6 well plate and distribute it evenly across the entire well of 6-well plate and place the culture plate back in the incubator.
-
a.
Key resources table
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| PAX6 (1:1500) | Covance | PRB-278P |
| MITF (1:200) | Exalpha | X1405M |
| TYRP1 (1:250) | Novus Biologicals | NBP2-32901 |
| CD24 (1:500) | BD Biosciences | 655154 |
| CD56 (1:500) | BD Biosciences | 340723 |
| Anti-FITC MicroBeads (1:10) | Miltenyi | 130-048-701 |
| Anti-mouse Alexa-Flour-488 (1:50) | Life Technologies | A21202 |
| Chemicals, peptides, and recombinant proteins | ||
| Activin-A | R&D Systems | 338-AC/CF |
| IGF-1 | R&D Systems | AFL291 |
| PD0325901 | R&D Systems | 4192 |
| Nicotinamide | Sigma | N0636 |
| LDN-193189 | Stemgent | 04-0074 |
| SB 431542 | R&D Systems | 1614/10 |
| CKI-7 Dihydrochloride | Sigma | C0742 |
| Blebbistatin | Sigma | B0560 |
| Ascorbic Acid | Sigma | A4544 |
| Rock-Inhibitor | R&D Systems | 1254 |
| 3,3′,5-Triiodo-L-thyronine | Sigma | T5516 |
| Taurine | Sigma | T0625 |
| Hydrocortisone | Sigma | H6909 |
| Water for Injection (WFI) | Thermo Fisher Scientific | A12873-01 |
| 10 M NaOH | Sigma | 72068 |
| Vitronectin | Life Technologies | A14701SA |
| DPBS | Life Technologies | 14190-144 |
| CryoStor® CS10 | STEMCELL Technologies |
07930 |
| FBS | Hyclone | SH30071.03 |
| KnockOut™ SR XenoFree | Life Technologies | 10828028 |
| KnockOut™ SR | Life Technologies | 10828010 |
| MEM Alpha Thermo Fisher | Life Technologies | 12571-063 |
| DMEM/F12 | Life Technologies | 11330-032 |
| MEM non-essential AA | Life Technologies | 11140 |
| E8 media | Life Technologies | A1517001 |
| Penicillin-Strep | Life Technologies | 15140-148 |
| Sodium Pyruvate | Life Technologies | 11360-070 |
| CTS N-2 Supplement | Life Technologies | A13707-01 |
| N-2 Supplement | Life Technologies | 17502048 |
| B-27® Supplement (+VitA) | Life Technologies | 17504-044 |
| 0.5 M EDTA | Life Technologies | 15575-020 |
| DMSO | Sigma | D2650 |
| 16% Formaldehyde | Electron Microscopy Sciences | 50-980-487 |
| Prostaglandin E2 | R&D Systems | 2296 |
| Versene | Life Technologies | 15040066 |
| TrypLE Express | Life Technologies | 12605028 |
| Experimental models: Cell lines | ||
| Human: iPSC, Female, 83 years, healthy subject | in house generated | N/A |
| Other | ||
| 6-well plates | Nunc | 140675 |
| T-75 flasks | Nunc | 156499 |
| T-150 flasks | TPP | 90151 |
| 96 well plates- v bottom for FLOW assay | Thermo Fisher Scientific | 249570 |
| 20 μm sterifilp | Sigma | SCNY00020 |
| 0.22 μm filter units-50 mL | Sigma | SCGP00525 |
| 0.22 μm filter units-250 mL | Corning | 431096 |
| 0.22 μm filter units-500 mL | Corning | 431118 |
| LD Columns | Miltenyi | 130-042-901 |
| MidiMACS or QuadroMACS separator | Miltenyi | 130-042-302 /130-090-976 |
| ThawSTAR CFT | BioLife | BCS601 |
Materials and equipment
Retinal Induction Media (RIM)
| Reagent | Final concentration | Amount |
|---|---|---|
| DMEM/F12 | N/A | 500 mL |
| CTS™ KnockOut™ SR XenoFree Or Knockout KSR |
1.5% | 7.5 mL |
| MEM non-essential AA | 0.1 mM | 5 mL |
| Sodium pyruvate | 1.0 mM | 5 mL |
| CTS™ N-2 supplement or N-2 supplement | 1.0% | 5 mL |
| B-27® supplement (+VitA) | 2.0% | 10 mL |
| Ascorbic acid | 200 μM | 500 μL |
| LDN-193189 | 10 nM | 500 μL |
| SB 431542 | 1.0 μM | 12.5 μL |
| CKI-7 Dihydrochloride | 0.5 μM | 50 μL |
| AF-IGF-1 | 1 ng/mL | 5 μL |
| Total | NA | 500 mL |
Retinal Differentiation Media (RDM)
| Reagent | Final concentration | Amount |
|---|---|---|
| DMEM/F12 | N/A | 500 mL |
| CTS™ KnockOut™ SR XenoFree Or Knockout KSR | 1.5% | 7.5 mL |
| MEM non-essential AA | 0.1 mM | 5 mL |
| Sodium pyruvate | 1.0 mM | 5 mL |
| CTS™ N-2 supplement or N-2 supplement | 1% | 5 mL |
| B-27® supplement (VitA) | 2% | 10 mL |
| Ascorbic acid | 200 μM | 500 μL |
| LDN-193189 | 100 nM | 5 μL |
| SB 431542 | 10 μM | 125 μL |
| CKI-7 Dihydrochloride | 5 μM | 500 μL |
| AF-IGF-1 | 10 ng/mL | 50 μL |
| PD0325901 | 1 μM | 50 μL |
| Total | NA | 500 mL |
Retinal Media with Nicotinamide and Activin-A (RM-NA)
| Reagent | Final concentration | Amount |
|---|---|---|
| DMEM/F12 | N/A | 500 mL |
| CTS™ KnockOut™ SR XenoFree or Knockout KSR | 1.5% | 7.5 mL |
| MEM non-essential AA | 0.1 mM | 5 mL |
| Sodium pyruvate | 1.0 mM | 5 mL |
| CTS™ N-2 supplement or N-2 supplement | 1% | 5 mL |
| B-27® supplement (+VitA) | 2% | 10 mL |
| Ascorbic acid | 200 μM | 500 μL |
| Activin-A | 100 ng/mL | n/a |
| Nicotinamide | 10 mM | 5 mL |
| Total | NA | 500 mL |
RPE Maturation Media (RPE-MM)
| Reagent | Final concentration | Amount |
|---|---|---|
| MEM alpha | N/A | 500 mL |
| Heat inactivated fetal bovine serum | 5% | 25 mL |
| N-2 supplement | 1% | 5 mL |
| MEM non-essential amino acids | 0.1 mM | 5 mL |
| Sodium pyruvate | 1 mM | 5 mL |
| Penicillin-Strep | 1× | 5 mL |
| Taurine | 250 μg/mL | 2.5 mL |
| Hydrocortisone | 20 μg/L | 553 μL |
| T3 | 0.013 μg/L | 330 μL |
| Total | NA | 500 mL |
MACS buffer
| Reagent | Final concentration | Amount |
|---|---|---|
| DPBS (without Ca2+ and Mg2+) | N/A | 1000 mL |
| Heat inactivated fetal bovine serum | 2% | 20 mL |
| Ultra-Pure EDTA solution | 2 mM | 4 mL |
| Total | NA | 1000 mL |
Permeabilization buffer
| Reagent | Final concentration | Amount |
|---|---|---|
| DPBS (without Ca2+ and Mg2+) | 98% | 49 mL |
| Heat inactivated fetal bovine serum | 2% | 1 mL |
| Triton-X-100 | 0.2% | 0.1 mL |
| Total | NA | 50 mL |
Step-by-step method details
Setting up differentiation for RPE cells
This section will cover the steps involved in generating RPE progenitors (day25) and immature RPE (day40) from iPSC.
Note: Before setting up differentiation, prepare E8 plating media, RIM media, RDM media, RMNA media, RPE-MM media, coated plates and flasks, and have the iPSC line ready for differentiation. Keep VTN coated 6-well/T-75 or T150 flasks ready.
-
1.Set up differentiation:
-
a.The day of seeding is termed as Day-2 (day minus 2).
-
b.Use iPSC culture with 60%–70% confluency. Troubleshooting 1.
-
c.Aspirate E8 media, wash cells with 3 mL of DPBS.
-
d.Add 1 mL of TrypLE to each well of 6-well plate.
-
e.Incubate the plate in 37°C incubator for 5–6 min.
-
f.Check the plate after 2–3 min, and as soon as the cells in the iPSC colonies start rounding off (become brighter), start collecting cells in TrypLE using a 5 mL serological pipette.
-
g.Wash the plate with E8 plating media, and collect the cell suspension in a 50 mL conical tube.
-
h.Centrifuge at 400 g for 5 min.CRITICAL: Avoid using 1 mL pipette (always use 5 mL serological pipette) to collect the cell suspension as it affects the viability and hence negatively affects the differentiation.
-
i.Aspirate the supernatant and resuspend the cell pellet in 5 mL (do not use I mL pipette) of E8 plating media.
-
j.Count cells using ViCell automated cell counter at proper cell counting settings. Use the viable cell count for seeding cells for differentiation.CRITICAL: This protocol is optimized on cell count from ViCell instrument specifically. There could be variation in cell counts when compared to other cell counters. However, this protocol works well on a range of cell densities.
-
k.Start with 200K/well seeding density but the protocol is optimized for a range of 200–400K cells/well of a 6 well plate.
-
l.Add 3 mL of E8 plating media with cell suspension and bring the plates to the incubator.
Culture surface Seeding numbers Media volume Number of plates/flasks 6 well plates 200 K–400 K/well 3 mL per well 3- 6-well plates T75 1.5–3.0 million 15 mL 2 T75 T150 3.0–5.0 million 30 mL 1 T150 -
m.Make sure to rock the plate in orthogonal directions to ensure even distribution of cells in every well.CRITICAL: Homogenous distribution of cells is a necessary event to have a good efficiency in differentiation process.
-
n.The health of iPSC culture - dead cells, spontaneously differentiating cells, homogenous size of colonies, all affect the efficiency of differentiation. Figure 1.CRITICAL: When working with a new iPSC line, set up three different seeding densities (200 K, 300 K and 400 K cells/well of a 6 well plate) for the first round of differentiation.
-
a.
-
2.
Day -1: Change the media with E8 complete media.
Note: Make sure to aspirate the media from the corner of the well to avoid damaging cells or aspirating them off. Dispense media slowly from the edge of the well.
-
3.
Day 0 and Day 1: Prepare RIM media. Change E8 media with 3 mL of RIM media/well of 6-well plate for two days. Figure 2.
-
4.
Day 2–10 RDM: From day 2 to day 10, feed RDM media every day. Add 3 mL of RDM medium to each well. Be careful while changing the media, make sure you are not aspirating off or damaging the cells. Troubleshooting 2.
Note: The last couple of days of RDM may show some cell death in culture plates. This is part of the differentiation process, and it indicates an enrichment process towards RPE cells. Figure 3.
Figure1.

Morphology of iPSC colonies for setting up RPE differentiation
(A and B) iPSC colonies ready to be used for differentiation. The culture should be more homogeneous with minimum dead cell floating and compact colonies.
(C and D) iPSC colonies that should not be used for RPE differentiation. Heterogenous colony size (blue arrowheads), high cell death, and spiky cells (white arrowheads) lead to poor differentiation.
Scale bar, 200 μm.
Figure 2.

Morphology of iPSC cells transition to RIM phase: Day 2 to day 1
(A and B) Differentiating cells at day-1 (A) and day 0 (B). The cells at day-1 are spiky because of the blebbistatin in culture media. The images represent good density and morphology of differentiating cells.
(C and D) Differentiating cells at day 0. The arrowheads represent the abnormal morphology that should be avoided in day 0 cells. The probable reason for such morphologies is the excessive use of TrypLE and mechanical force during passaging.
Scale bar, 200 μm.
Figure 3.

Morphology of differentiating cells in RDM phase: Day 2 to day 10
(A and B) Differentiating cells at day 5 (A) and day 10 (B). At day 10, expect lot of cell death indicating the priming to RPE fate. The differentiation runs lacking the cell death at the end of RDM phase often brings poor efficiency.
Scale bar, 200 μm.
-
5.
Day 11–20 RMNA: From day 11 to day 20, feed RMNA media every day. Add 3 mL of RMNA medium to each well. Be careful while changing the media, make sure you are not aspirating off or damaging the cells. At the end of RMNA, neuronal clusters will start to appear in wells which is a positive sign indicating that cells differentiated into RPE. Figure 4.
-
6.
Day 21–25 5% RPE: From day 21, feed the media every other day. Add 3 mL of 5% RPE medium to each well. At this stage you will start to see flat areas with RPE progenitor cells with islands of neuronal clusters. Pigment also starts to appear in these 4 days of media change. The non-RPE cells will die off in this phase. In extreme cases of failed differentiation, the whole well peels off as a layer. In case of mixed cultures of RPE and non-RPE cells, there will be some signs of peeling; if that happens its recommended to collect cells at day 23 or day 24. At this stage cells can be either frozen down or plated for MACS purification and enrichment. Figure 5. Troubleshooting 3 and Troubleshooting 4.
Figure 4.

Morphology of differentiating cells in RMNA phase: Day 11 to day 20
(A and B) Differentiating cells at day 16 (A) and day 20 (B). At day 20, appearance of little bumps of neuronal clusters (arrowhead marked) indicates successful run. The flat land around bumps RPE progenitor cells.
(C and D) Images of bad cultures at day 16 (A) and day 20 (B). The opaque cultures lacking any islands of neuronal islands end up dying at the end of day 20, i.e., RMNA phase.
Scale bar, 20 μm.
Figure 5.

Morphology of differentiating cells in RPE phase from committed to immature stage
(A) RPE progenitor cells at day 25.
(B) Reseeded cells at day 27.
(C) Immature RPE cells before MACS purification. The images in this panel represent the good differentiation run. Scale bar, 200 μm.
(D) RPE progenitor cells at day 25.
(E) Reseeded cells at day 27.
(F) Immature RPE cells before MACS purification. The images in this panel represent the bad differentiation run with no appearance of neuronal clusters at day 25 (D) and only neurons at day 27.
Scale bar, 200 μm.
Peeling off cells during RIM and RDM stage could happen because of several reasons: Nunc plates were not used; vitronectin coating was not done correctly; iPSC cells were not maintained well or medium change was not done carefully. Be careful to set the dispense speed on pipetman at 2 or 3 before adding the medium to the wells. Always add or aspirate the medium from the edge of the well.
Peeling off cells could also happen during the last phase of 5% RPE, be careful while changing media.
Some morphological signs about the differentiation: noticeable cell death at the end of RDM and the beginning of the RMNA phase. That is a good sign. It tells that non-RPE primed cells are dying off.
Big holes in the differentiation plate during the 5% RPE medium step and complete peeling off signal that the culture was full of neurons. Figure 6.
Figure 6.
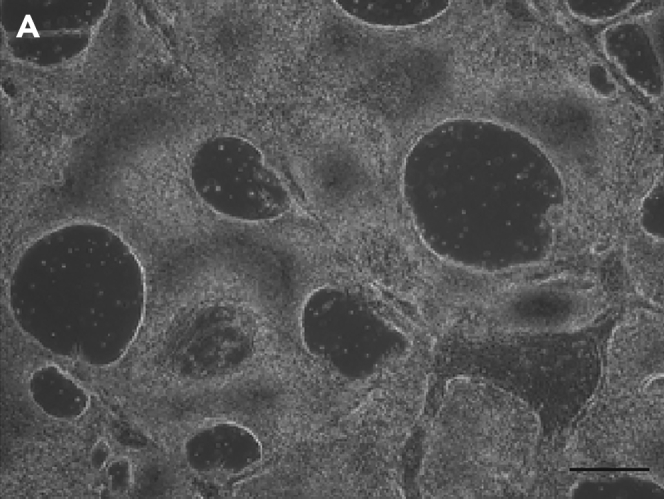
Morphology of differentiating cells at day 25
(A) Differentiating cells at day 25, the big holes in the. monolayer of differentiating cells indicates the failed run and that the culture is full of neurons. Scale bar, 200 μm.
The color of the day 25 pellet will give an indication of the efficiency of differentiation. Figure 7. Off-white pellet suggests cultures cull of neurons (BAD differentiation), brown to black pellet suggests differentiating RPE cells (GOOD differentiation).
Figure 7.

RPE pigmentation chart
RPE enrichment step or purifying mature RPE cells
Preparing immature RPE cells for purification through magnetic associated cell sorting will be described in this section. The steps will cover preparing LD columns and cells for staining.
The quality of the differentiation run is critical to ensure the successful Day25 (RPE progenitors) replating. The replating process gets rid of neuronal clusters. Prepare MACS Buffer, RPE plating media, permeabilization buffer, fixation buffer and FACS buffer.
-
7.Day 25 replating for MACS purification.
-
a.Prepare RPE plating media and vitronectin coated flasks.
-
b.Aspirate the 5% RPE medium and wash the well twice with DPBS - 2 mL/well of a 6-well plate.
-
c.Aspirate DPBS and add 1 mL of TrypLE to each well of 6-well plate and incubate at 37°C for 5 min. This will remove neuronal clusters. Take the plate out from incubator and start tapping gently to get rid of neuronal clusters. Aspirate TrypLE and perform two DPBS washes.
-
d.Add 1 mL of TrypLE and leave in incubator for 15 more minutes. Collect the cells with 5 mL serological pipette and pool cells in a 50 mL conical tube. Wash the wells with RPE plating media, pool all media and centrifuge the conical tube at 400 g for 5 min.Note: If the quality of differentiation run is poor, all cells start to detach as a monolayer, and the process will not need the second step of TrypLE. On the other hand, in case of good quality differentiation runs, the RPE progenitors come off after the 15 min TrypLE incubation.
-
e.Aspirate the supernatant and resuspend cells in 5–10 mL of plating media.
-
f.Pass the cell suspension through 20 μm steriflip cell strainer. Count the cells using ViCell counter (expect 5–6 million cells/mL). Seed cells at 24–30 million in T75 flask in 20 mL of RPE plating media. The yield of RPE progenitors varies from 100–120 million cells/75cm2. Keep the flask in incubator.
-
g.For replated cultures, 48 h after seeding, change medium using room temperature equilibrated 5% RPE media.
-
h.Flasks are fed every other day.
-
a.
-
8.Freezing Day 25/RPE progenitors:
-
a.Aspirate the media and wash cells twice with DPBS (without calcium and magnesium) with 2 mL/well of 6-well plate.
-
b.Aspirate DPBS and add TrypLE (1 mL/well of a 6 well-plate).
-
c.Incubate at 37°C for ∼5 min. Tap the vessel gently to detach neuronal clusters.
-
d.Aspirate TrypLE and wash twice with DPBS (2 mL/well of a 6 well-plate).
-
e.Aspirate the DPBS and add TrypLE (1 mL/well of a 6 well-plate).
-
f.Incubate at 37°C for an additional ∼15 min.
-
g.After incubation, collect the cells using a serological pipette. Collect the cell suspension in a 50 mL conical tube. Wash each well with 2–3 mL of RPE plating media.
-
h.Aspirate the supernatant and resuspend cells in 5–10 mL of RPE plating media (depending on the size of the pellet).
-
i.Pass the cell suspension through a 20 μm steriflip cell strainer. Count the cells using ViCell counter. Use the viable cell number as the cell concentration.
-
j.Centrifuge the cells again at 400 g for 5 min.
-
k.Aspirate the supernatant and gently resuspend with 5 mL serological pipette (pipet 3–5 times) the cells in cold CS10 freezing media roughly 30 million cells/1–1.5 mL of CS10 per vial.
-
l.Aliquot the cell suspension into pre-labeled cryovials with cell line name, number of cells frozen and date of freezing.
-
m.Place the cryovials in a cold (∼4°C) freezing container.
-
n.Transfer the freezing container with a freezing speed of 1 ºC/min to a −80°C freezer.
-
o.Transfer the vials to the vapor phase of liquid nitrogen after 24 h.
-
a.
-
9.MACS purification step: Day 40 Replating with MACS based enrichment.
-
a.On day 40, aspirate the media from T75 flask and wash twice with 30 mL of DPBS (without Ca2+/Mg2+).
-
b.Aspirate the DPBS and add 15 mL of TrypLE.
-
c.Incubate at 37°C for 40–50 min.Note: Check the flask under the microscope after 30 min, cells derived from some iPSC lines come off easier than others. Longer TrypLE incubation reduces the viability and hence will negatively affect the yield.
-
d.After incubation, collect cells using a 5 mL serological pipet. Collect the cell suspension in a 50 mL conical tube. Wash each well with enough room temperature equilibrated 5% RPE Plating Medium. Centrifuge the cells at 400 g for 5 min.
-
e.Aspirate the supernatant and resuspend the cells in 10 mL of room temperature 5% RPE Plating Medium.
-
f.Filter the cell suspension using a 20 μm steriflip cell strainer.
-
g.Count number of cells using ViCell. Use the total cell number as the cell concentration. The expected cell count ranges from 9–10 million/mL.
-
h.Add 20 mL MACS Buffer to the cell suspension and mix using a serological pipet. Centrifuge the cells at 400 g for 5 min.
-
i.Aspirate the supernatant and resuspend the cells in MACS Buffer at 1 × 107 cells/mL.
-
j.Label cells with primary antibodies.Add FITC anti-CD24 at 1:500 dilution (2 μL antibody per mL cell suspension).Add FITC anti-CD56 at 1:500 dilution (2 μL antibody per mL cell suspension).Note: As an alternative, it is acceptable to pre-dilute the antibodies in MACS Buffer and resuspend the cells at 1 × 107 cells/mL in the antibody solution.
-
k.Mix the cell suspension by pipetting or swirling and incubate at 4°C for 20 min.
-
l.After incubation, add 20 mL MACS Buffer. Mix by pipetting or swirling and centrifuge at 400 g for 5 min.
-
m.Aspirate the supernatant and resuspend the cell pellet in 20 mL MACS Buffer using a serological pipet. Centrifuge at 400 g for 5 min.
-
n.Aspirate the supernatant and resuspend the cell pellet in MACS Buffer at 1.11 × 108 cells/mL (90 μL/1 × 107 cells).
-
o.Add Anti-FITC Microbeads at a 1:10 dilution.Note: As an alternative, it is acceptable to pre-dilute the Microbeads in MACS Buffer and resuspend the cells at 1.11 × 108 cells/mL in the bead solution.
-
p.Mix by vigorously swirling the cell suspension and incubate at 4°C for 20 min. During incubation, prepare the magnet, magnet stand and LD columns for use. Wet each LD column by adding 2 mL MACS Buffer. Discard the flow through.
-
q.After incubation, add 20 mL MACS Buffer to the cell suspension. Mix by vigorously swirling and centrifuge at 400 g for 5 min.
-
r.Aspirate the supernatant and resuspend the cell pellet in 20 mL MACS Buffer using a serological pipet. Centrifuge at 400 g for 5 min.
-
s.Aspirate the supernatant and resuspend the cell pellet (up to 1.25 × 108 cells) in 500 μL MACS Buffer using a 1000 μL pipettor. For higher cell numbers, scale accordingly.
-
t.Add the cell suspension to a LD column and collect flow through in a tube.
-
u.Wash the LD column by adding 2 mL MACS buffer to the column and collect flows through in the same tube used in step ‘t’.
-
v.Repeat the wash step by adding 2 mL MACS buffer to the column and collect the flow through in the same tube used in step ‘t’.
-
w.After the flow from the column has stopped, centrifuge the collected cell suspension at 400 g for 5 min.
-
x.Aspirate the supernatant and resuspend the cell pellet in RPE-MM Plating Medium. Count the cell suspension using the ViCell. Use the total cell number as the cell concentration.
-
y.Remove 2 × 106 cells for post-sort purity FLOW analysis.
-
z.Seed remaining cells at the density of 24–30 million in a T75 flask.
-
aa.Two days post seeding freeze day42 cells. Follow instructions given in point 6 Freezing Day 42 immature cells.
-
a.
-
10.FLOW assay for purity.
-
a.Collect cells after splitting with TrypLE.
-
b.Wash the pellet with FACS Wash Buffer and cell count needed for FLOW assay is 200K cells. (You can fix 1–2 million cells and save them for a backup if something goes wrong in the assay.).
-
c.Aspirate the Wash Buffer and add the 1 mL of fixation buffer and fix the cells for 15 min at RT.
-
d.Add 5 mL of FACS Wash Buffer and spin the cells down at 400 g for 4 min.
-
e.Decant the FACS Wash Buffer. Add 2 mL of Wash Buffer and resuspend the pellet by using 1 mL pipette.
-
f.Add the cells in 96 well plate, so that each well has minimum 200K cells. Make sure you have the 96 well plate map ready with you.Note: Always use conical bottom plates.
-
g.Add 100 μL of Wash Buffer and spin down the 96 well plate @400 g for 5 min.
-
h.Decant the plate carefully and then tap the plate carefully on paper tissues to get rid of left-over buffer from the well. Make sure you don’t use excessive force on the plates.
-
i.Add 50 μL of permeabilization buffer in each well containing cells. Add the primary antibodies according to the dilution table. Make sure you mix the cells well, before putting the plate for incubation.
Antibodies First dilution Volume (μL) of Ab needed in 50 μL of permeabilization buffer Second
DilutionVolume of Ab needed in 50 μL of permeabilization buffer Anti-PMEL17 1:25 2 μL 1:10 5 μL Anti-TYRP1 1:25 2 μL 1:10 5 μL IgG1 (Isotype control for PMEL-17, TYRP1) 1:25 2 μL 1:25 2 μL IgG Alexa Fluor-488 1:10 5 μL 1:5 10 μL Anti-PAX6 1:1500 Anti-MITF 1:200 -
j.Incubate the plate covered in foil for overnight at 4°C on rocking shaker.
-
k.After incubation is over, add 100 μL of FACS Wash Buffer and spin down the plate@400 g for 5 min.
-
l.Again, be careful in decanting the plate. Add 50 μL of permeabilization buffer and add secondary antibody as per the dilution table. Incubate the plate at RT for 30 min.
-
m.Add 100 μL of FACS Wash Buffer and spin down the plate 400 g for 5 min.
-
n.Again, be careful in decanting the plate. Add 150 μL of Wash Buffer and the plate is ready for running on FLOW machine. Troubleshooting 5.
-
a.
-
11.Freezing day 42 immature cells.
-
a.On day 42, aspirate the media and wash the cells twice with 20 mL of DPBS.
-
b.Aspirate DPBS and add 15 mL of TrypLE to T75 flask and incubate at 37°C for ∼15 min.
-
c.Pipet off the cells after 15 min of incubation using a 5 mL serological pipet.
-
d.Collect the cell suspension in a 50 mL conical tube.
-
e.Wash each well or flask with 15–20 mL of room temperature 5% RPE media.
-
f.Centrifuge the cells at 400 g for 5 min.
-
g.Aspirate the supernatant and resuspend the cells in an appropriate volume of room temperature 5% RPE media.
-
h.Filter the cell suspension using a 20 μm steriflip cell strainer.
-
i.Count the cell suspension using the ViCell. Use the total cell number as the cell concentration.
-
j.For cryopreservation, aspirate the supernatant and gently resuspend the cells at an appropriate density in cold CryoStor CS10. Freeze the cells as per the need- 6 million to 12 million in 1–1.5 mL of freezing media.
-
k.Aliquot the cell suspension into pre-labeled cryovials.
-
l.Place the cryovials in a cold (∼4°C) freezing container.
-
m.Transfer the freezing container to a −80°C freezer for 12–24 h before moving the frozen vials to liquid nitrogen tank for long term storage.
-
a.
-
12.Seeding on transwells: thawing and culture day 42 RPE cells.Note: Before starting to thaw the cells, prepare the vitronectin coated transwells or any other plastic surface which is planned to use. Take the plates out on the day of seeding the cells and bring the plates to room temperature by at least keeping them for an hour.
-
a.Transfer cells from liquid nitrogen to cell culture room using ThawSTAR cell transporter or dry ice.
-
b.Place the vial in a ThawSTAR automated thawing system to thaw. In the absence of ThawSTAR use dry bead bath at 37°C for 3–5 min.
-
c.Remove the vials from the ThawSTAR or dry bead bath, spray with 70% ethanol, wipe dry and place in a biosafety cabinet.
-
d.Transfer the content of the vial (1 mL) to 50 mL conical tube using P1000 pipette. Avoid repeated pipetting of the thawed cell suspension.
-
e.Add 1 mL of room temperature 5% RPE plating medium to cryovial to recover any remaining cells, and transfer to 50 mL tube.
-
f.Using 5 mL serological pipette, slowly add 8 mL of room temperature 5% RPE plating dropwise to the 50 mL conical tube.Note: Dropwise addition of medium to thawed cell suspension is critical to minimize osmotic shock to ensure maximum viability of the cells.
-
g.Mix the cell suspension by swirling. Avoid vigorous shaking or vortexing of the cell suspension.
-
h.Centrifuge the cell suspension at 400 g for 5 min.
-
i.Aspirate the supernatant. Resuspend the pellet in 5 mL 5% RPE plating media. Count the cells using ViCell cell counter.
-
j.Based on the cell count seed the cells in a desired culture plates/scaffold plates at the following cell concentration:
-
k.For 12-well Transwell plates, add to each basal compartment (well) 1.5 mL of 5% RPE plating media.
-
l.Into each apical compartment chamber, seed 275K cells in 0.5 mL of the same media.
-
m.After seeding the plates, wait for 10 min before putting them in the incubator. Do the first media change after 24–48 h with 5% RPE maturation media, and at 10 days post- seeding, change the 5% RPE maturation media to 5% RPE media + PGE2 after 10 days of post seeding and continue for 5 weeks with PGE2-supplemented media.
-
n.Return cultures to the incubator at 37°C; 5% CO2.
-
o.The regular media change days for D40 or D42 immature RPE cells seeded are Mondays, Wednesday, and Fridays.
-
a.
-
13.Trans-epithelial resistance measurement.Note: Trans-epithelial resistance (TER) is a measure of intactness of tight junctions between neighboring cells and reflects mature monolayer tissue. TER across the iRPE monolayers is measured using commercially available Epithelial Volt/Ohm Meter (EVOM2, WPI).
-
a.EVOM2: The electrodes in the form of chopsticks (STX2, WPI) were applied on the apical and basal sides of iRPE, the current was passed, and resistance values (Ohms) were noted down for each condition. Actual resistance (Ohms.cm2) is calculated by multiplying TER by area of measurement (12 mm diameter transwell in this case).
-
a.
Expected outcomes
The protocol is well optimized and established for healthy or patient derived iPSC lines from diverse genetic backgrounds. The expected yield of RPE progenitors (day25) is 110–150 million cells when differentiation is started in one T75 flask or in three 6-well plates. The average efficiency of the RPE purification step, i.e., immature stage (day40), is between 30%–70%. Overall, amplification of cells is between 40 and 100.
Limitations
This protocol has been successfully used to produce RPE cells from over 65 iPSC lines but has failed to work on 5% of the lines. It is not clear how those 5% iPSC lines are different from the lines that have worked but it suggests that our protocol may not work on certain stages of pluripotency of iPSC lines. Another limitation is that this protocol has not yet been tested on rodent, avian, porcine, or monkey iPSCs or embryonic stem cells (ESCs). One may need to adjust the timing of different phases of media change to reflect species’ s developmental stages. The protocol can be expensive because of all the growth factors and reagents; purchasing reagents in bulk allows reducing reagent cost.
Troubleshooting
Problem 1
Poor iPSC cultures with heterogenous size of colonies and high cell death- “setting up differentiation for RPE cells” section, step 1.
Potential solution
iPSC cultures are sensitive to the handling and culture conditions; if not handled properly, they tend to spontaneously differentiate or show signs of cell death. To avoid that, make sure the VTN coated plates are not older than 2 weeks, while passaging avoids breaking colonies into single cells; iPSC colonies prefer small clumps. Different iPSC lines have variable growth rates but passage them no later than 7 days. The colony size and the confluency of the culture determine the efficiency of differentiation. 70%–80% of culture confluency is suitable to set up differentiation.
Problem 2
Peeling at the stage of early RDM- “setting up differentiation for RPE cells” section, step 2.
Potential solution
If the iPSC culture, after thawing, hasn’t been well adapted to culture before using in differentiation, it will end up peeling during the early RDM stages. Therefore, passage a given cultures at least 3 times before setting up differentiation.
Problem 3
Failed differentiation runs- “setting up differentiation for RPE cells” section, step 6.
Potential solution
Failed differentiation runs become evident near the end of the RM-NA stage when the culture appears dense, and there are no neuronal clusters or flat RPE land. There could be several possible reasons for it – inappropriately aliquoted SB431542, LDN-193189, and Activin-A stocks. So, it is imperative to prepare reagents very accurately. The plastic surface matters in RPE differentiation. The recommended plastic surface for the differentiation is Nunc.
In our experience, the protocol is very robust, well-optimized, and works over 90% of the time with minimal donor variability.
Problem 4
Failed differentiation and maturation- “setting up differentiation for RPE cells” section, step 6.
Potential solution
Make sure the days of feeding, media preparation is well followed. Below is the breakdown and easy version of a schematic for users to follow.
Problem 5
Failed FLOW purity assays- “RPE enrichment step or purifying mature RPE cells” section, step 10n.
Potential solution
Check for the antibodies, aliquots, and buffers used in the assay. Make sure that buffers are prepared correctly. Check Figure 8 for the breakdown steps.
Figure 8.

Breakdown culture schematic detailing all the steps
Resource availability
Lead contact
Further information and requests for resources and reagents should be directed to and will be fulfilled by the lead contact Kapil Bharti (Kapil.bharti@nih.gov).
Materials availability
This study didn’t generate new unique reagents.
Acknowledgments
The authors thank Helena Herzog and Dr. Jason Miller for making schematics used in graphical abstracts and troubleshooting sections. The graphical abstract and schematic were created with BioRender.com. This work was supported by funds from the NEI Intramural Research Program to K.B.
Author contributions
Methodology: K.B. and R.S. Validation: D.B., J.M., D.O., and R.S. Resources: K.B. Writing—original draft: R.S.; Writing—review & editing: K.B., D.O., and J.M.
Declaration of interests
We, the authors, have a patent related to this work.
Contributor Information
Ruchi Sharma, Email: fnu.ruchi2@nih.gov.
Kapil Bharti, Email: kapil.bharti@nih.gov.
Data and code availability
This study did not generate/analyze datasets/code.
References
- Miyagishima K.J., Sharma R., Nimmagadda M., Clore-Gronenborn K., Qureshy Z., Ortolan D., Bose D., Farnoodian M., Zhang C., Fausey A., et al. AMPK modulation ameliorates dominant disease phenotypes of CTRP5 variant in retinal degeneration. Commun. Biol. 2021;4:1360. doi: 10.1038/S42003-021-02872-X. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sharma R., Khristov V., Rising A., Jha B.S., Dejene R., Hotaling N., Li Y., Stoddard J., Stankewicz C., Wan Q., et al. Clinical-grade stem cell-derived retinal pigment epithelium patch rescues retinal degeneration in rodents and pigs. Sci. Transl. Med. 2019;11:eaat5580. doi: 10.1126/SCITRANSLMED.AAT5580. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sharma R., George A., Nimmagadda M., Ortolan D., Karla B.S., Qureshy Z., Bose D., Dejene R., Liang G., Wan Q., et al. Epithelial phenotype restoring drugs suppress macular degeneration phenotypes in an iPSC model. Nat. Commun. 2021;12:7293. doi: 10.1038/S41467-021-27488-X. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
This study did not generate/analyze datasets/code.