

Abstract
Transfer RNA (tRNA)-derived fragment (tRDF) is a novel small non-coding RNA that presents in different types of cancer. The comprehensive understanding of tRDFs in non-small cell lung cancer remains largely unknown. In this study, 1,550 patient samples of non-small cell lung cancer (NSCLC) were included, and 52 tRDFs with four subtypes were identified. Six tRDFs were picked as diagnostic signatures based on the tRDFs expression patterns, and area under the curve (AUC) in independent validations is up to 0.90. Two signatures were validated successfully in plasma samples, and six signatures confirmed the consistency of distinguished expression in NSCLC cell lines. Ten tRDFs along with independent risk scores can be used to predict survival outcomes by stages; 5a_tRF-Ile-AAT/GAT can be a prognosis biomarker for early stage. Association analysis of tRDFs-signatures-correlated mRNAs and microRNA (miRNA) were targeted to the cell cycle and oocyte meiosis signaling pathways. Five tRDFs were assessed to associate with PD-L1 immune checkpoint and correlated with the genes that target in PD-L1 checkpoint signaling pathway. Our study is the first to provide a comprehensive analysis of tRDFs in lung cancer, including four subtypes of tRDFs, investigating the diagnostic and prognostic values, and demonstrated their biological function and transcriptional role as well as potential immune therapeutic value.
Keywords: tRNA-derived fragments, lung cancer, diagnosis, survival outcome, tumor immune microenvironment
Graphical abstract
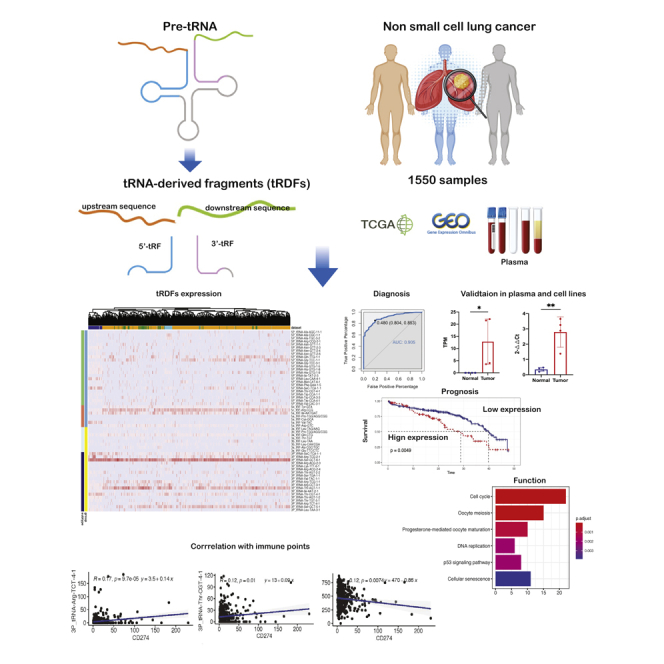
Identified six tRDFs with excellent diagnosis ability. tRDFs were confirmed in plasma data and NSCLC cell lines. Ten tRDFs associate with survival outcomes by stages. 5a_tRF-Ile-AAT/GAT is early-stage biomarker. tRDFs were related to the cell cycle and oocyte meiosis. Five tRDFs associate with PD-L1 immune checkpoint.
Introduction
Non-coding RNAs, including long non-coding RNA and small non-coding RNA, have been elucidated over the past few decades on the complex mechanism and crucial roles in the development of cancers.1, 2, 3 Transfer RNA (tRNA), as one type of non-coding RNA, is traditionally considered an adapter molecule in protein transition.4, 5, 6 In recent years, a new class of small non-coding RNA has been discovered as tRNA-derived fragments (tRDFs) in next-generation sequencing dataset, which was derived from tRNA precursor or mature sequences.7, 8, 9 Instead of random degradation from tRNA, the biogenesis of these fragments is the products that have precise site cutting under specific tRNA modification and result in lengths of 14–50 nt.10,11
In general, based on the length and cleavage sites of tRNAs, tRDFs from mature or primary tRNA can be classified into two main groups, including tRNA-derived tRNA halves (tiRNAs) and small RNA fragments (tRFs).4 tiRNAs are also called tRNA-derived stress-induced RNAs and are generated by the cleavage around the anticodon loop of tRNA by angiogenin (ANG).4,12 Recent discoveries have classified them into two subtypes: 5ʹ-tiRNAs and 3ʹ-tiRNAs.13,14 Depending on the original location from tRNA, tRFs are placed into four major categories: internally derived tRFs (i-tRFs), mainly derived from the internal region of mature tRNA; 5ʹ-derived tRFs (5ʹ-tRFs), which are cleaved from the 5ʹ end in the D-loop by Dicer;15,16 3ʹ-derived tRFs (3ʹ-tRFs), which are cleaved from the 3ʹ end in the T-loop by Dicer, ANG, or other members of the ribonuclease A superfamily; and 1ʹ-tRFs, which are derived from 3ʹ trailer of primary tRNA and formed from the maturation process of the tRNA precursor sequence by RNase Z.4,17
Due to the development of sequencing technologies, increasing evidence has shown that tRNA-derived fragments are involved in gene regulation at transcriptional and post-transcriptional levels.4,18, 19, 20 They participate in various biological processes, including microRNA (miRNA)-mediated silencing,21, 22, 23 mRNA stabilization,24 translation regulation,25 epigenetic regulation,26 and cell differentiation.27 Moreover, tRDFs have been gradually discovered as aberrantly expressed in major diseases, such as cancer,24,28, 29, 30, 31, 32 viral infectious disease,33, 34, 35, 36, 37 and neurodegenerative disease,7,38, 39, 40 which is expected with novel biomarkers on the identification of organ damage.41
A first-time report on the relation of tRDFs with cancer by Lee et al.16 found the expression of tRF-1001 is tightly correlated with cell proliferation, and it is expressed highly in a wide range of prostate cancer cell lines. In recently published studies, more evidence has shown that tRDFs are associated with cancer progression through cell proliferation and with a confirmed presence of high-abundant tRDFs in different types of human cell lines, tissues, or extracellular body fluids.42, 43, 44, 45, 46 Balatti et al.28 discovered four tRNA-derived small RNAs (tsRNAs) that were generated from pre-tRNA 3ʹ end cleavage and were downregulated in chronic lymphocytic leukemia (CLL) and lung cancer. tRF-Leu-CAG was strongly expressed in non-small cell lung cancer (NSCLC) tissues compared with normal tissues.47 5ʹ-tRF-Gly-GCC was dramatically increased in plasma of colorectal cancer patients in comparison with the healthy control.48 Four tRFs expressed significantly higher levels in plasma exosomes of liver cancer. Consistent evidence indicated the potential value as biomarker of tRDFs in cancer diagnosis.49
Currently, the diversity of tRNA fragments has been reported in many review articles as well as multiple online databases, such as tRFdb and MINTbase.8,32,50,51 tRDFs are usually classified into different subtypes based on its section and length; however, different databases of tRDFs would result in different names based on the barcodes the authors created. In some ways, it is hard to find the feature of each tRDF in common in terms of these assorted names, let alone if combining multiple databases all together for a comprehensive analysis of tRDFs. In this study, we assembled four tRDF resources that included four sections of tRNA fragments—upstream sequences, 5-tRF, 3-tRF, and downstream sequences (1ʹ-tRFs)—and consolidated them into a unified form according to tRDF’s definition and sources from tRNA.
Up until now, no tRDFs study has comprehensively investigated all types of tRDFs expression profile and its biological function in NSCLC. In the present study, we integrated four tRDFs data resources by combining different sections of tRNA fragments all together and examining the expression pattern of tRDFs in 1,550 samples from The Cancer Genome Atlas (TCGA), Gene Expression Omnibus (GEO), and plasma sequencing data, and we are the first to include four types of tRNA-derived fragment into analysis, especially the upstream sequences. We successfully identified 58 tRDFs and distinguished 11 tRDFs with significantly higher expression in NSCLC patients than in healthy controls and did validation in cell lines. We also highlighted groups of tRDFs associated with a prognosis value based on different cancer stages and cancer subtypes. We then examined the attributes of the mRNA and miRNA that correlated with tRDFs, and potential target genes were identified. Finally, we investigated the dependencies of tRDFs on the tumor microenvironment and hidden association with immune checkpoint and target signaling pathways.
Results
We mined all possible datasets from TCGA-lung adenocarcinoma (LUAD) and GEO repositories; only four cohorts conducting miRNA sequencing with pairwise samples were available in this study. GEO: GSE83527 contains 34 adenocarcinoma patients and 34 healthy controls, GEO: GSE62182 included 94 adenocarcinoma patients and paired healthy samples, and GEO: GSE110907 had 48 adenocarcinoma patients and 48 healthy tissues. TCGA-LUAD cohort contained 519 patients and 47 solid healthy samples, and the TCGA-lung squamous cell carcinoma (LUSC) cohort contained 478 patients and 44 healthy samples. Patients’ information was summarized in Table S1.
Based on the reference variety of tRNA-derived fragments, we gathered all fragments from 5ʹ end, including “5P-tRNA” (upstream sequences) and 5ʹ-tRF (D-loop), to be categorized as 5ʹ-tRDF, and fragments from 3ʹ end, including 3ʹ-tRF (T-loop) and 3P-tRNA/1ʹ-tRF (downstream sequences), to be categorized as 3ʹ-tRDF (Figure 1A). Due to the specialty name of each tRDF from different databases potentially hiding some biological information, like its tRNA section and length, we also unified all tRDFs into the mutual way as “5P/3P/5-tRF/3tRF-tRNA”. Some specific tRDFs sequences from 5-tRF and 3-tRF that we collected from tRNAdb were identified to overlap in multiple tRNA; for example, tRFdb-5010a as a 5a-tRF could be found from tRNA-Ile-AAT-2-1/5–4/5–5/5–1/5–2/5–3/4–1/7–1/7–2/1–1/6–1/8–1/12–1 and so on. Therefore, 5a_tRF-Ile-AAT/GAT was used in short to present the fragment. Each tRNA name corresponding to the numbered tRDFs can be found in Tables S2 and S3.
Figure 1.

Expression pattern of two types of tRDFs in LUAD
(A) The structure of tRNA and each fragment derived from tRNA, including upstream sequence (orange), downstream sequence (green), 5ʹ-tRF (blue), and 3ʹ-tRF (purple). (B) Composition of each type of tRDFs in each dataset is shown. (C) Composition of tRDFs from different tRNA sections in each subtype among all datasets is shown. (D) Venn plot shows 152 tRDFs were identified as common in three GEO adenocarcinoma cohorts and TCGA-LUAD cohort. (E) Clustering analysis from t-SNE of overall cohorts is shown. The cluster of GEO: GSE110907 was separated from the rest of cohorts shown in orange color and named as cluster A; cluster B is shown in blue. (F) Heatmap and hierarchical clustering shows the landscape of each type of tRDF expression in each group; tRDFs were classified into four subtypes as 5P/upstream sequences, 5ʹ-tRF, 3ʹ-tRF, and 3P/downstream sequences.
Through the overview of tRDFs composition in each dataset, 5P (upstream sequences) dominated the five datasets (62.6% in GEO: GSE83527, 63.3% in GEO: GSE62182, 71.3% in GEO: GSE110907, 67.4% in TCGA-LUAD, and 63.9% in TCGA-LUSC), while 3P was merely one-third of the 5P (Figure 1B). In the distribution of different subtypes of tRDFs from each tRNA, 5P and 3P tRNA that derived from up- and downstream sequences were identified more than the 5ʹ- and 3ʹ-tRF from D-loop and T-loop. 5P tRNA can be frequently found in Ala-, Asn-, Gly-, His-, and Trp-tRNA, while 3P can be found more in Arg-, Ser-, and Thr-tRNA (Figure 1C).
After filtering out tRDFs with zero expression, we have 362 tRDFs from five datasets. The average length for all types of tRDFs is 22 nt; the longest is 3P_tRNA-SeC-TCA-1-1 with a length of 47 nt and the shortest tRDFs is 5a_tRF-Val-TAC, 5a_tRF_Ser-AGA/TGA, and 5a_tRNA-Leu-TAA with the length of 14 nt. The prevalent length of 5P is 20 nt, while the length of 3P averaged in 27 nt with outliers such as 3P_tRNA-Leu-TAA-3-1 (15 nt) and 3P_tRNA-Thr-CGT-4-1 (40 nt) (Figure S1A; Table S4).
Expression pattern of two types of tRDFs in LUAD
The annotation results of tRDFs in each adenocarcinoma dataset showed different compositions; 152 tRDFs were identified in common within three GEO adenocarcinoma cohorts and the TCGA-LUAD cohort (Figure 1D). Figure S1B presents the length distribution of 152 tRDFs common in four datasets, in comparison with the 362 tRDFs length distribution (Figures S1A and S1B) that presents that more 5P were removed but still dominated the most. Clustering by t-distributed stochastic neighbor embedding (t-SNE) combined with Gaussian mixture models (GMMs) was applied to 152 tRDFs in 695 tumor samples from TCGA-LUAD cohort and three GEO cohorts. Two clusters of tRDFs showed significantly different expression profiles among four groups of cohorts (Figure 1E). Samples from GEO: GSE110907 were identified as one independent cluster A, while the rest of GEO and TCGA-LUAD cohorts were classified under cluster B. After filtering out low-expression tRDFs among four cohorts, 58 common tRDFs remain (sequence information could be found in Table S5).
The heatmap visualized the 58 tRDFs expression pattern of four subclasses of tRDFs among the four datasets in tumor samples only. The unsupervised clustering showed the two different clustering modes that separated the GEO: GSE110907 from the rest of the cohorts successfully. 58 tRDFs contain 24 5P-tRNAs and 19 3P-tRNAs. 5a_tRF-Asp-GTC, 5a_tRF-Ile-AAT/GAT, 5c_tRF-Arg-CCG, 5b_tRF-Tyr-GTA, 5P_tRNA-Gly-TCC-1-1, and 5P_tRNA-Gln-TGG-1-1 were significantly expressed tRDFs in 5ʹ-tRDFs. 3P_tRNA-Ser-GCT-5-1, 3P_tRNA-Thr-CGT-4-1, 3P_tRNA-Ser-GCT-6-1, 3P_tRNA-SeC-TCA-1-1, 3P_tRNA-Thr-AGT-1-1, 3P_tRNA-Arg-CCT-3-1, 3P_tRNA-Arg-TCG-2-1, 3P_tRNA-Thr-AGT-2-2, and 3a_tRF-Gln-CTG were distinguished in 3ʹ-tRDFs. The landscape of 58 tRDFs expression pattern is shown in Figure 1F.
Moreover, we investigated the certain tRDFs that were derived from the same tRNA but were cleaved from different loops, 17 in total that were presented in Figure S1C and were defined as paired tRDFs. The Pearson correlation analysis showed less significant or low association of each paired tRDF (Table S6). Heatmap as Figure S1D also proved that no cluster of tRDFs derived from the same tRNA.
DE tRDFs present a diagnostic value associated with tumor and normal samples differentiation
Based on clustering results, we investigated the differentially expressed (DE) tRDFs in cluster B to gain a comprehensive understanding of the expression pattern of the tRDFs between tumor and normal samples. Due to the different data platforms, GEO and TCGA were separately calculated. We used Deseq2 and t test in two GEO datasets (GEO: GSE83527 and GEO: GSE62812) and TCGA-LUAD cohort based on both raw read counts and normalized expression profile transcripts per million mapped reads (TPM), respectively. Both tumor and normal samples were included, and the DE tRDFs were chosen with the selection standard adjusted p value (Padj)/false discovery rate (FDR) < 0.05 and |log2fold change| > 0.58.
In the GEO: GSE83527, 32 DE tRDFs were identified, including 18 5ʹ-tRDFs and 14 3ʹ-tRDFs (Tables S1–S7). In the GEO: GSE628182, 16 DE tRDFs in total were found as significant DE tRDFs, including eight 5ʹ-tRDFs (5P_tRNA-Gly-TCC-1-1, 5P_tRNA-Asn-GTT-2-3, 5b_tRF-Tyr-GTA, 5c_tRF-Arg-CCG-2-1, 5P_tRNA-Gly-TCC-3-1, 5P_tRNA-Gln-TCC-1-1, 5a_tRF-Asp-GTC, and 5a_tRF-Ile-AAT/GAT) and eight 3ʹ-tRDFs (3P_tRNA-Val-TAC-1-1, 3P_tRNA-SeC-TCA-1-1, 3P_tRNA-Thr-CGT-4-1, 3P_tRNA-Arg-TCT-4-1, 3P_tRNA-Ser-GCT-6-1, 3a_tRF-Leu-TAG/AAG, 3P_tRNA-Arg-TCG-1-1, and 3b_tRF-Leu-CAA/CGA) (Table S7; Figures 2A and 2B).
Figure 2.

Differentially expressed (DE) tRDFs in tumor and normal samples
(A and B) Heatmap shows the DE tRDFs in two GEO datasets; samples were pairwise by tumor and normal. Up- and downregulation of DE tRDFs could be separated by unsupervised clustering. (A) DE tRDFs in GEO: GSE83527 are shown; (B) DE tRDFs in GEO: GSE62182 are shown. (C and D) Boxplot shows the 11 tRDFs that were identified as common DE in GEO: GSE83527 and GSE62182 datasets; p value of each group is less than 0.05. (E) Correlogram shows positive (blue) and negative (red) correlation among 11 DE tRDFs as determined by the Pearson correlation analysis. (F and G) AUCs show the independent validation results of GEO: GSE1109067 (AUC: 0.914) and TCGA-LUAD (AUC: 0.905) from random forest model. (H) Boxplot shows the expression level of six DE tRDFs in TCGA-LUAD cohort that were found in common from GEO DE tRDFs. (I) AUCs show the diagnostic effect of six tRDFs signatures from five independent validations. Each validation contains different groups of tRDFs from signatures; one tRDF would be removed from previous group in order as 5P_tRNA-Asn-GTT-2-3, 5a_tRF-Asp-GTC, 3P_tRNA-Arg-TCG-1-1, 3P_tRNA-Arg-TCT-4-1, 5a_tRF-Ile-AAT/GAT, and 5P_tRNA-Gly-TCC-1-1 and finally ends as last two tRDFs.
Finally, 11 mutual tRDFs in two GEO datasets were identified, including five upregulated tRDFs—3P_tRNA-Ser-GCT-6-1, 3P_tRNA-Arg-TCG-1-1, 5a_tRF-Asp-GTC, 3P_tRNA-Arg-TCT-4-1, and 5a_tRF-Ile-AAT/GAT—and six downregulated tRDFs—5b_tRF-Tyr-GTA, 5P_tRNA-Gly-TCC-1-1, 5P_tRNA-Gly-TCC-3-1, 5P_tRNA-Asn-GTT-2-3, 3P_tRNA-Val-TAC-1-1, and 3P_tRNA-SeC-TCA-1-1 (Figures 2C and 2D). The log2 fold changes of two datasets can be found in Figure S1E. We also included the genome tracks of 11 tRDFs in Figures S2 and S3. Besides DE tRDFs, three unchanged tRDFs, 3P_tRNA-Arg-CCT-3-1, 3P_tRNA-Arg-TCG-2-1, and 5P_tRNA-SeC-TCA-1-1, were found overlapped in GEO and TCGA datasets (Figure S4).
To explore the relationship within these candidate tRDFs, we calculated pairwise correlations among the expression of 11 tRDFs in GEO: GSE83527 and GEO: GSE62182 (Figure 2E). Notably, the expression of two downregulated tRDFs (5P_tRNA-Gly-TCC-1-1 and 5P_tRNA-Asn-GTT-2-3) was remarkably correlated with the rest of tRDFs (absolute value of correlation coefficients is between 0.18 and 0.47). We also noticed that the expression of 5ʹ-tRDFs was not only correlated in the same category but also significantly correlated with 3ʹ-tRDFs. 5P_tRNA-Gly-TCC-1-1 with 3P_tRNA-Val-TAC-1-1 and 5P_tRNA-Asn-GTT-2-3 was positively correlated, and the highest correlation coefficient between upregulated tRDFs 5a_tRF-Ile-AAT/GAT and 5a_tRF-Asp-GTC is 0.42. A negative correlation was found between upregulated and downregulated tRDFs, such as 5P_tRNA-Gly-TCC-1-1 and 3P_tRNA-Arg-TCG-1-1, with a coefficient of −0.44.
In terms of the GMM-t-SNE cluster result, we decided to take advantage of the machine learning technique, random forest, to investigate the diagnostic value of the 11 tRDFs on distinguishing the tumor and normal samples. Two GEO datasets (GEO: GSE83527 and GEO: GSE62182) that contain pairwise data (N: 128; T: 128) were merged and used as training datasets; we also included two independent validation groups from the TCGA-LUAD cohort (N: 46; T: 460) and GEO: GSE110907 (N: 48; T: 48). Eleven DE tRDFs were used as variables to build this classification model, with an out-of-bag (OOB) estimate of error rate at 8.09%. Independent validations on TCGA-LUAD and GEO: GSE110907 achieved excellent area under the curve (AUC) (GEO: GSE110907: 0.914; TCGA-LUAD: 0.905) (Figures 2F and 2G), and all sensitivity, specificity, and accuracy were above 0.80 (Table S7). The results suggest the performance of 11 DE tRDFs in the random forest model was significantly associated with adenocarcinoma and can be used for tumor and normal diagnostic determination.
In addition, the calculation results of TCGA-LUAD identified 21 DE tRDFs, including 12 5ʹ-tRDRs and 9 3ʹ-tRDFs (Tables S5–S7). When comparing the candidates with GEO, six tRDFs (5P_tRNA-Gly-TCC-1-1, 5a_tRF-Ile-AAT/GAT, 3P_tRNA-Arg-TCG-1-1, 3P_tRNA-Arg-TCT-4-1, 5P_tRNA-Asn-GTT-2-3, and 5a_tRF-Asp-GTC) in TCGA-LUAD were found overlapping with 11 DE tRDFs candidates from the independent validation by random forest models, which was referred to as a signature to be diagnostic biomarkers (Figure 2H).
We also tested the diagnostic ability of six tRDF signatures with the same training and validation datasets in five combinations in descending order from six signatures to two signatures (5a_tRF-Ile-AAT/GAT and 5P_tRNA-Gly-TCC-1-1); the results exhibited excellent diagnostic value of tRDFs that combined in groups as AUCs more than 0.87, and sensitivity and specificity are all above 0.77. The best performance of grouped tRDFs is the combination of 3P_tRNA-Arg-TCG-1-1, 3P_tRNA-Arg-TCT-4-1, 5a_tRF-Ile-AAT/GAT, and 5P_tRNA-Gly-TCC-1-1, presenting AUC as 0.91 (Figure 2I). We then tested the diagnostic value of individual signatures; however, the identification ability showed lower accuracy compared with grouped tRDFs. 5a_tRF-Ile-AAT/GAT exhibited best performance AUC is 0.786, while 5a_tRF-Asp-GTC has the worst accuracy among all signature with 0.587 AUC (Figure S1F).
We also evaluated the DE tRDFs between normal and tumor samples in TCGA-LUSC cohort; 35 DE tRDFs were identified, and six DE tRDFs were found to exhibit the diagnostic features (Tables S6 and S7).
Identification and validation of tRDFs in lung cancer patient plasma and NSCLC cell lines
To validate the diagnostic value of 11 tRDF candidates on LUAD patients, we collected blood samples from 50 patients and 60 healthy controls and performed small RNA sequencing. After the same upstream and downstream analysis, six tRDFs can be identified. We compared the tRDFs expression level between tumor and healthy samples. The results showed that two tRDFs, 3P_tRNA-Arg-TCG-1-1 and 5P_tRNA-Asn-GTT-2-3, had significant differences between two groups (Figures 3B and 3E).
Figure 3.

Plasma sample validation in NSCLC patients and healthy donors
TPM expression of six tRDFs 3P_tRNA-Ser-GCT-6-1 (A), 3P_tRNA-Arg-TCG-1-1 (B), 3P_tRNA-SeC-TCA-1-1 (C), 3P_tRNA-Val-TAC-1-1 (D), 5P_tRNA-Asn-GTT-2-3 (E), and 5b_tRF-Tyr-GTA (F) between tumor and healthy control. ∗p ≤ 0.05 and ∗∗p ≤ 0.01. ns, not significant.
No statistical differences can be identified among the rest of the four tRDFs. However, the expression pattern of tRDF can still be evaluated, as the trend of expression in two types of samples has the same performance in GEO and TCGA-LUAD. In the group of upregulated tRDFs, the 3P_tRNA-Ser-GCT-6-1 in tumor samples showed higher expression than the normal sample. On the other hand, three downregulated tRDFs (3P_tRNA-SeC-TCA-1-1, 3P_tRNA-Val-TAC-1-1, and 5b_tRF-Tyr-GTA) are all highly expressed in normal samples.
We also assessed 11 DE tRDFs expression in human lung cancer line ABC-1 and in a normal lung cell line MRC-9. Stem-loop primer design for tRDFs referred to the methods of Zhu et al.49 Analysis of tRDFs expression by real-time quantitative polymerase chain reaction (qPCR) showed eight of 11 tRDFs, including four upregulated tRDFs and four downregulated tRDFs, were significantly different between tumor and normal cell lines, which was consistent with the bioinformatics analyses (Figure 4). In this result, six tRDF signatures (5a_tRF-Ile-AAT/GAT, 5a_tRF-Asp-GTC, 3P_tRNA-Arg-TCG-1-, 3P_tRNA-Arg-TCT-4-1, 5P_tRNA-Gly-TCC-1-1, and 5P_tRNA-Asn-GTT-2-3) were all identified and validated successfully.
Figure 4.

Real-time qPCR validation in NSCLC cell lines and normal cell lines
Real-time qPCR analyses of 5b_tRF-Tyr-GTA (A), 5P_tRNA-Asn-GTT-2-3 (B), 3P_tRNA-Val-TAC-1-1 (C), 5P_tRNA-Gly-TCC-1-1 (D), 3P_tRNA-Arg_TCG-1-1 (E), 5a_tRF-Asp-GTC (F), 5a_tRF-Ile-AAT/GAT (G), and 3P_tRNA-Arg-TCT-4-1 (H). ∗∗p ≤ 0.01; ∗∗∗p ≤ 0.001; ∗∗∗∗ p ≤ 0.0001.
We then conducted TOPO TA cloning to validate the accuracy of the fragments that were amplified; 9 out of 11 (5a_tRF-Ile-AAT/GAT, 5a_tRF-Asp-GTC, 3P_tRNA-Ser-GCT-6-1, 3P_tRNA-Arg-TCG-1-1, 3P_tRNA-Arg-TCT-4-1, 5b_tRF-Tyr-GTA, 5P_tRNA-Gly-TCC-1-1, 5P_tRNA-Asn-GTT-2-3, and 3P_tRNA-Val-TAC-1-1) can be found by Sanger sequencing as well as one non-changed tRDF 3P_tRNA-Arg-CCT-3-1 also identified (Figure S5). Primer information can be found in Table S17.
The distinct prognostic pattern of tRDFs associated with cancer stages
To investigate the prognostic patterns of tRDFs, 506 patients in TCGA-LUAD and 470 patients in TCGA-LUSC were included in our calculation, and 52 tRDFs were put into analysis after filtering out low expression from TCGA cohorts. The clinical information of each cohort was shown in Table S1. We characterized data according to tumor stages by combining stages I and II into an early stage and merging stages III and IV as a later stage.
In the TCGA-LUAD cohort, univariate Cox regression showed that 3P_tRNA-Arg-CCT-3-1, 5P_tRNA-Ala-TGC-3-1, and 3P_tRNA-SeC-TCA-1-1 correlated with LUAD prognosis in all four stages (Figures S6A–S6C), which exhibited statistically significant differences. High expression of 3P_tRNA-SeC-TCA-1-1 was associated with longer survival time in LUAD patients, while shorter survival times were found in high expression of 3P_tRNA-Arg-CCT-3-1 and 5P_tRNA-Ala-TGC-3-1 in LUAD patients.
5P_tRNA-Ala-TGC-3-1, 3P_tRNA-Ser-TGA-1-1, and 3b_tRF-Leu-CCA/CGA were identified as significant tRDFs in early stages (Figures S7A–S7C), while 5P_tRNA-SeC-TCA-1-1, 5P_tRNA-Phe-GAA-1-5, 5P_tRNA-Arg-CCG-2-1, and 5c_tRF-Pro-AGG/TGG were the four tRDFs dramatically correlated with patient prognosis in later stages (Figures S7F–S7I).
In the TCGA-LUSC cohort, 5P_tRNA-Asn-GTT-2-3, 3b_tRF-Leu-CCA/CGA, 3P_tRNA-Ser-GCT-6-1, 3a_tRF-Ala-CGC/TGC, and 3P_tRNA-Arg-CCG-1-3 were found significantly correlated with patient survival across four stages (Figures S8A–S8E). In early stages, 5P_tRNA-Asn-GTT-2-3, 5P_tRNA-Gly-TCC-3-1, 3b_tRF-Leu-CCA/CGA, 3P_tRNA-Arg-CCG-1-3, 3P_tRNA-Ser-GCT-6-1, and 3a_tRF-Ala-CGC/TGC were found to affect the survival prognosis (Figures S9A–S9F). 5P_tRNA-Asn-GTT-1-1 and 5P_tRNA-SeC-TCA-1-1 are two significant tRDFs in later stages (Figures S9I and S9J).
The risk score model was constructed based on DE tRDFs to quantify the prognosis prediction effect in each group. To evaluate the clinical relevance of risk score, we divided the score into high- and low-risk group by a cutoff value that was decided by the Survminer package. Patients with low risk scores demonstrated a prominent survival benefit. To examine whether the risk score could serve as an independent prognostic factor, we performed multivariate Cox regression analysis, including patient age, smoking status, sex, and pathologic_t. We found that risk score was a robust and independent prognostic biomarker for predicting and evaluating patient clinical survival in NSCLC (Figures S6D, S6E, S7D, S7E, S7J, S7K, S8F, S9G, S9H, S9K, and S9L). Consistent with these findings, the multivariate Cox regression analysis showed that the low-risk score group had a better overall clinical outcome than the high-risk score group. These results imply that the risk score reflects the tRDF expression patterns and predicts the prognosis of NSCLC patients. All information about univariate Cox regression models can be found in Table S8.
Except for the survival analysis based on 52 tRDFs by stages and subtypes, we also did the analysis based on tRDFs signatures with specific endpoints. We categorized the survival time into several month periods, such as 6 months, 12 months, 24 months, 36 months, 48 months, and 60 months. 5a_tRF-Ile-AAT/GAT showed a comprehensive prognostic association on the endpoint of 48 months among patients from all stages and early stages (Figures 5A and 5C). Moreover, low expression of 5a_tRF-Ile-AAT/GAT had a significantly better prognostic value with survival time ending at 36 months and 24 months among early-stages patients (Figures 5D and 5E). High expression of 3P_tRNA-Arg-TCT-4-1 was identified to associate with unfavorable survival outcome within 12 months among all-stage patients as well as 12 and 6 months from early-stages patients (Figures 5B, 5G, and 5H). Among advanced-stages patients, 5a_tRF-Asp-GTC was associated with 48 months and 6 months survival time (Figures S10A and S10D). 5P_tRNA-Asn-GTT-2-3 and 3P_tRNA-Arg-TCG-1-1 had prognostic link with patients within 36 months and 12 months (Figures S10B and S10C).
Figure 5.

Prognostic value of tRDF signatures
(A and B) Kaplan-Meier curves show 48 months and 12 months survival between high (red) and low expression (blue) in 5a_tRF-Ile-AAT/GAT and 3P_tRNA-Arg-TCT-4-1 from the TCGA-LUAD cohort; all stages are included. The group of TCGA-LUAD samples is shown at the bottom of the chart. p < 0.05 in the two-sided log rank test was considered statistically significant. (C–H) Kaplan-Meier curve shows prognostic relation from early-stages patients in three signatures. 5a_tRF-Ile-AAT/GAT related survival in 48 months (C), 36 months (D), 24 months (E), 5a_tRF-Asp-GTC (F), and 3P_tRNA-Arg-TCT-4-1 (G and H) in 12 months and 6 months. p < 0.05 in the two-sided log rank test was considered statistically significant.
tRDFs involved in transcriptional and post-transcriptional regulation
To further investigate tRDFs in transcriptional and post-transcriptional events, we computed positive and negative tRDF-miRNA Pearson correlation coefficient using TCGA-LUAD tumor data and expression of 52 tRDFs received after removing near-zero variables; the sequence information of 52 tRDFs can be found in Table S9. We then separated 52 tRDFs into 5ʹ-tRDFs and 3ʹ-tRDFs for correlation analysis with the selection standard as |Pearson correlation| ≥ 0.2 and p < 0.05.
Our results showed 69 miRNAs correlated with 5ʹ-tRDFs, including 63 miRNAs that were positively correlated with 15 5ʹ-tRDFs and 23 miRNAs that were negatively correlated with six 5ʹ-tRDFs. has-let-7a-5p had the most correlation with 5ʹ-tRDFs, and has-miR-30c-5P had the greatest link with 3ʹ-tRDFs. 5a_tRF-Asp-GTC had the most correlation with miRNAs among 5ʹ-tRDFs. It also showed the most significant coefficient as 0.51 with hsa-miR-145-5p, and it was negatively correlated with hsa-let-7a-5p. 5b_tRF-Tyr-GTA exhibited the second highest correlation and negatively correlated with hsa-miR-26b-3p and hsa-miR-199b-5p. Eighty-eight miRNAs were positively correlated with 17 3ʹ-tRDFs, and 30 miRNAs were negatively correlated with six 3ʹ-tRDFs. 3P_tRNA-Arg-CCG-1-3 and 3P_tRNA-Thr-AGT-1-1 had the most positive correlation and negative correlation in 3ʹ-tRDFs, respectively (Table S10; Figures S11A and S11B).
Next, we performed the correlation analysis between miRNA and tRDF signatures (5P_tRNA-Gly-TCC-1-1, 5a_tRF-Ile-AAT/GAT, 3P_tRNA-Arg-TCG-1-1, 3P_tRNA-Arg-TCT-4-1, 5P_tRNA-Asn-GTT-2-3, and 5a_tRF-Asp-GTC). We had tRDF-miRNA pairs with the same selection standard, finding 60 miRNAs correlated with six target tRDFs. Fifty-nine out of sixty miRNAs were positively correlated with six target tRDFs, while 8 out of 60 miRNAs were negatively correlated. 5a_tRF-Asp-GTC dominated the most correlations with miRNAs (Figure S11C).
The miRNA-targeted genes were significantly correlated with phosphatidylinositol 3-kinase (PI3K)-Akt, mitogen-activated protein kinase (MAPK), endocytosis, Ras, and other signaling pathways (Figure S11D). Fifty-seven out of sixty miRNA target genes were enriched in PI3K-Akt, 56 out of 60 miRNA target genes were enriched in MAPK and endocytosis, and 55 out of 60 miRNA target genes were enriched in Ras signaling pathway.
The molecular function from Gene Ontology (GO) enrichment indicated miRNA target genes were mostly shown in small protein serine and threonine kinase activity, guanosine triphosphatase (GTPase) binding, Ras GTPase binding, and transcription coregulator activity (Figure S11E). The biological processed enrichment showed a high gene ratio in axonogenesis and cell morphogenesis and positive regulation of neurogenesis (Figure S11F).
Functional analysis of tRDFs
To investigate the implicational biological function of tRDFs in LUAD, we examined the correlation between tRDFs and mRNA based on the TPM expression profile in the TCGA-LUAD cohort. After filtering out low counts exclusive in TCGA-LUAD, 52 tRDFs remain. tRDF-mRNA pairs were selected with |Pearson correlation| ≥ 0.2 and FDR < 0.05. 3ʹ-tRDF showed more frequent correlations than 5ʹ-tRDFs in a comparison of 6,219 genes versus 1,607 genes.
3P_tRNA-Thr-AGT-1-1 and 3a_tRF-Thr-TGT are two 3ʹ-tRDFs that correlated genes the most as 1,259 and 1,385 genes, respectively. 3P_tRNA-Arg-TCT-4-1 and 3P_tRNA-Ser-GCT-6-1, two of the six tRDFs signatures, were the second most downstream sequences that linked with over 600 genes. Among downstream sequences, 3P_tRNA-Arg-ACG-2-3 performed remarkable correlation with several genes, such as OR10G1P (R = 0.700), RAPGEF4-AS1 (R = 0.697), and C3P1(R = 0.620). 3a_tRF-Leu-TAG/AAG was identified as the most positive correlation with FAM50B (R = 0.700), MIR4503 (R = 0.626), and SLC10A6 (R = 0.467) among all 3ʹ-tRFs (Table S11).
5P_tRNA-Thr-CGT-4-1 and 5c_tRF-Pro-AGG/TGG were identified to be correlated with most genes among tRDFs in 5P and 5-tRF. 5P_tRNA-Thr-CGT-4-1 also showed highly positive correlation with genes like TNMD (R = 0.580), OR5BN2P (R = 0.569), and RNU7-164P (R = 0.569). 5c_tRF-Pro-AGG/TGG has significant association with histone coding genes, such as H3C12 (R = 0.423), H3C13 (R = 0.410), and H4C13 (R = 0.378) (Table S11).
We calculated the fold changes of 5,815 genes that were identified to be correlated with tRDFs in TCGA-LUAD cohort, including 497 tumor samples and 56 healthy samples. Our selection standard of DE genes was FDR < 0.05 and abs log2 fold change > 1. Among all genes that correlated with tRDFs, 1,806 genes were statistically significantincluding 1560 upregulated genes, and 246 downregulated genes (Figure 6A).
Figure 6.

tRDFs-correlated genes enrichment analysis
(A) Volcano plot showed 5,782 genes that correlated with 52 tRDFs; FDR < 0.05 and log2 fold change > 1 on the two sides were considered as statistically significant. (B) Thirteen KEGG pathways from GSEA that were identified common in the signaling pathways that miRNA target genes enriched in are shown. (C) The mutation frequency of top 25 genes from 653 DE-correlated candidate genes in 464 LUAD patients from the TCGA cohort; each column represents individual patients. The number on the right indicates the mutation frequency in each correlated gene. The right bar shows the proportion of each variant type, and the stacked bar graph below exhibited the fraction of conversions in each patient. (D) The six signaling pathways enriched from 657 genes that were correlated with six tRDF signatures are shown. (E) The network presents 31 gene targets in six signaling pathways with mutation frequency that correlated with three tRDF signatures.
We performed gene set enrichment analysis (GSEA) and found that tRDF-correlated genes are mainly enriched in regulation of gene silencing, regulation of post-transcriptional gene silencing, and negative regulation of gene expression in epigenetic. Protein-macromolecule adaptor activity, oxygen carrier activity, and globin binding are several probable molecular functions of correlated genes. And these genes were mostly identified in mitochondrial protein complex, nuclear nucleosome, hemoglobin complex, and the other cellular component (Figures S12A–S12C).
After comparing the Kyoto Encyclopedia of Genes and Genomes (KEGG) pathway analysis results from tRDFs-correated miRNA target genes, 13 signaling pathways were found in common from the enriched genes that correlated with tRDFs, and proteoglycans in cancer, endocytosis, T cell receptor signaling pathway, and transcriptional mis-regulation in cancer and cell cycle are the top five pathways (Figure 6B). To further assess the functional role of tRDF signatures (5P_tRNA-Gly-TCC-1-1, 5a_tRF-Ile-AAT/GAT, 3P_tRNA-Arg-TCG-1-1, 3P_tRNA-Arg-TCT-4-1, 5P_tRNA-Asn-GTT-2-3, and 5a_tRF-Asp-GTC), we expanded the selection criteria as |Pearson correlation| ≥ 0.15. After filtering out the genes that were not DE, correlation results in six tRDF signatures exhibited that upregulated tRDFs being associated with abundant positive correlations involving 630 DE genes (Figure S12D), an obvious comparison of connection from two downregulated tRDFs that merely correlated with 50 genes (Figure S12E); finally, 657 genes in total correlated with six tRDF signatures (Table S12). In terms of GO enrichment analysis of correlated genes, upregulated tRDF signatures were mostly found in the biological processes, such as mitotic nuclear division, chromosome segregation, and nuclear division. Downregulated tRDF signatures were identified to be correlated in positive regulation of endothelial and epithelial cell migration and synaptic vesicle recycling (Figures S12F and S12G).
To determine the genetic alterations among these correlated genes, we assessed the prevalence of somatic mutations among 657 genes. Out of 464 LUAD samples, 344 (74.14%) had mutations of genes that correlated with six tRDF signatures (Figure 6C). Out of 303 genes were identified with mutations, ASPM has the highest rate (15%), followed by MYH8 and MAGEC1 (Table S13). KEGG enrichment analysis of 303 genes are significantly enriched in the cell cycle, oocyte meiosis, progesterone-mediated oocyte maturation, DNA replication, p53 signaling pathway, and cellular senescence (Figure 6D). Thirty-one genes with mutations were found among six signaling pathways (Figure 6E). These genes are correlating with 3P_tRNA-Arg-TCT-4-1, 3P_tRNA-Arg-TCG-1-1, and 5a_tRF-Ile-AAT/GAT. Copy number variation (CNV) results were based on 25 genes with top mutation frequency. CDKN2A with highest mutation frequency also showed higher CNV loss and was identified in cell cycle, p53, and cellular senescence signaling pathway, which correlated with 3P_tRNA-Arg-TCT-4-1. TTK is the second gene with higher mutation frequency that correlated with 3P_tRNA-Arg-TCT-4-1 and 5a_tRF-Ile-AAT/GAT in cell cycle (Table S14; Figure S12H).
tRDFs expression pattern associated with immune infiltration and tumor microenvironment
Currently, no studies have conducted association between tRDFs and TME-infiltrating immune cells; therefore, we used CIBSORT methods to further understand the functional role of tRDFs in TME-infiltrating immune cells. Thirty-five tRDFs were associated with tumor immune microenvironment (TIME) cell infiltration, with correlation coefficient over 0.1 or less than −0.1. For example, 5P_tRNA-Thr-CGT-4-1 correlated with dendritic cells activated (R = 0.29), 3b_tRF-Leu-CCA/CGA correlated with T cells follicular helper (R = 0.24), and 5b_tRF-Tyr-GTA (R = 0.20) linked with T cells CD4 memory activated. T cells CD4 memory activated had the most link with tRDFs (Table S15).
Differences in TIME cell infiltration between two types of tRDFs were also analyzed; 14 immune cells were found in common between two types of tRDFs. We noticed 5ʹ-tRDFs were strongly positively correlated with T cells CD8 and T cells CD4 memory activated. Both 5ʹ-tRDFs and 3ʹ-tRDFs had a positive link with dendritic cells resting (Figure 7A). Three 5ʹ-tRDFs had negative association with neutrophils, while 3P_tRNA-Arg-CCG-1-3 had positive correlation. Four 3ʹ-tRDFs were positively correlated with macrophages M0. In order to explore any prognostic value of tRDFs that combined with immune infiltration, we conducted survival analysis based on prognostic related tRDFs we analyzed by all stages, early stages, and advanced stages from TCGA-LUAD cohorts (3P_tRNA-Arg-CCT-3-1, 3P_tRNA-SeC-TCA-1-1, 5P_tRNA-Ala-TGC-3-1, 3b_tRF-Leu-CCA/CGA, 3P_tRNA-Ser-TGA-1-1, 5P_tRNA-Phe-GAA-1-5, 5c_tRF-Pro-AGG/TGG, and 5P_tRNA-Arg-CCG-2-1). Thirteen types of immune cells were found to be correlated with all tRDFs except 5c_tRF-Pro-AGG/TGG (Figure S13A). Survival analysis was based on all-stage patients among 13 immune cells, and only the plasma cell that correlated with 3P_tRNA-Arg-CCT-3-1 (R = −0.1) was identified to be significantly related to overall survival (Figure 7B).
Figure 7.

Immune infiltration and tumor microenvironment correlated with tRDFs
(A) Bar plot shows the correlation between tRDFs and TIME cell infiltration evaluated by Pearson correlation. The length of column indicates the correlation; immune cells were selected from the mutual correlation with 5ʹ-tRDFs and 3ʹ-tRDFs. (B) Kaplan-Meier curves show overall survival of plasma cells that correlated with prognostic tRDFs. Red color indicates the high abundance of plasma cells, and blue color indicates the low abundance of plasma cells. (C) Network shows the tRDF-targeted immune-related signaling pathways in TCGA-LUAD cohort, including primary immunodeficiency, viral protein interaction with cytokine and cytokine receptor, cytokine-cytokine receptor interaction, hematopoietic cell lineage, T cell receptor signaling pathway, and the other signaling pathway. IgA, immunoglobulin A. (D) The correlation between tRDF with immune checkpoint CD274 (PD-L1), CTLA4, CD80, and CD86.
We also characterized the functional role of tRDFs that highly correlated with immune-infiltration-related genes. We identified 100 immune-related genes from LM22 that were correlated with 30 tRDFs (Figure S13B). The enrichment analysis results presented that these genes were enriched in biological processes, particularly those related to B cell proliferation, activating cell surface receptor signaling pathway, and activating signal transduction (Figure S13C). Immune receptor activity, cytokine activity, and signaling receptor activator activity are specific immune-related genes that enriched in molecular function (Figure S13D).
According to the results of KEGG pathway enrichment, there are 20 tRDF-correlated genes identified to be enriched in signaling pathways, such as cytokine-cytokine receptor interaction, chemokine signaling pathway, and T cell receptor signaling pathway (Figure 7C). Only one tRDF signature was detected among 20 tRDFs: 3P_tRNA-Arg-TCT-4-1 is identified as targeting in cytokine-cytokine receptor interaction, viral protein interaction with cytokine-cytokine receptor, and chemokine signaling pathway. Besides the signature, we also noticed three tRDFs, tRNA-Ala-TGC-3-1, tRNA-Arg-CCG-2-1, and 3b_tRF-Leu-CCA/CGA, that demonstrated association with prognosis were enriched in some immune-related signaling pathways. For example, tRNA-Ala-TGC-3-1, identified as correlated with all-stage and early-stage patient survival outcome in LUAD cohorts, was correlated in T cell receptor signaling pathway, which may indicate some links of tRDFs prognosis with possible immune therapy.
We finally conducted correlation analysis based on four immune checkpoints: CD274 (PD-L1), CD80, CD86, and CTLA4. The correlation with 10 tRDFs was identified (Figure 7D). 5P_tRNA-Gln-TTG-1-1 had the most negative correlation with three checkpoints, including CTLA4, CD80, and CD86. 5a_tRF-Cys-GCA, 3P_tRNA-Ser-GCT-6-1, 3P_tRNA-Thr-CGT-4-1, 3P_tRNA-Arg-TCT-4-1, and 5P_tRNA-Trp-CCA-3-3 were five tRDFs that correlated with CD274.
Figure S13E showed the correlation network of tRDFs and correlated gene in the signaling pathway of PD-L1 expression and PD-1 checkpoint pathways in cancer that was enriched by GSEA. Three tRDFs, including 3P_tRNA-Ser-GCT-6-1, 3P_tRNA-Thr-CGT-4-1, and 3P_tRNA-Arg-TCT-4-1, were correlated with genes that target in this pathway (R = 0.200), which were also commonly associated with PD-L1 checkpoint.
Discussion
tRDFs are generally named in terms of the cleavage positions on the pre- and mature tRNAs and can be roughly classified into four categories. We have tRNA fragments from both D-loop and T-loop as well as up- and downstream sequences and classified them based on sequence location from 5ʹ or 3ʹ. In terms of some publications, tRNA fragments derived from D-loop can be further classified into three subtypes based on their incision loci and lengths—tRDF-5a (14–16 nt), tRDFs-5b (22–24 nt), and tRDFs-5c (28–30 nt)7,52,53—but there has been no identification about the fragment from upstream sequences, and we are the first to include type of fragment into a comprehensive landscape of NSCLC study.
Due to the abundant types of 5P tRNA and prominent expression level, it is necessary to emphasize the importance of this study. We are also the first to bring all tRNA-derived fragments together in coupled names, eight pairs of tRDF we identified to share the same biogenesis. For example, tRFdb-5010a and ts-67 are 5a-tRF and downstream sequences named in two tRDFs databases; however, they are both derived from tRNA 5a-tRF-Ile-AAT-2-1 but different sections. Unified names like 5a-tRF-Ile-AAT and 3P_tRNA-Ile-AAT-2-1 made it much more convenient to identify each tRDF, not only about its section but also length. Unfortunately, no strong correlation was detected between these pairs, which indicates the cleavage by different enzymes resulted into independent biological processes.8
In this study, 11 tRDFs were screened out from two GEO datasets, and two independent validations confirmed the capacity of 11 tRDFs to be used as a diagnostic biomarker in terms of high accuracy and AUC. In the comparison of DE tRDFs we obtained from a different data platform, six tRDFs named as signatures were found in overlap with 11 tRDFs from TCGA-LUAD; the disparities may be due to the bias from study design, sample, sequencing, and different platforms. Also, the independent validation of tRDF signatures showed high accuracy in diagnosis prediction, but not as well as individual tRDF prediction, which indicates the necessity of combination of tRDF signatures in terms of better diagnosis results.
Sequencing results of plasma validated the potential diagnostic value of 11 tRDF candidates and two tRDFs (3P_tRNA-Arg-TCG-1-1 and 5P_tRNA-Asn-GTT-2-3) that were shown statistically different between normal and tumor samples. The reasoning may stem from quality disparities of blood samples compared with tissue samples from TCGA and GEO. However, the trending of fold changes in the rest of tRDFs proved the consistency with expression pattern we found in our analysis. Currently, most samples in study came from human tissue in which tRDFs can be much easier detected. Increasing numbers of abnormal tRDF expressions have been discovered in bodily fluids in cancers,46,54 and clinical attention of non-invasive, biofluid-based markers for cancer is emerging. The limitation of clinical and pathological methods makes it essential to find accurate diagnosis for early-stage patients.8 Also, sample type variety can strengthen and solidify the conclusion, and combination of plasma and tissue can contribute to better understanding of tRDFs in cancer research.
We also identified the 11 tRDFs candidates in NSCLC cell lines through real-time qPCR validation combined with TOPO TA cloning experiment. The real-time qPCR results confirmed the consistency of the diagnosis value of tRDFs we got from bioinformatic analysis. Four upregulated and two downregulated tRDF signatures were found to be significantly expressed between NSCLC cell lines and normal cell lines. In order to validate the fragments we amplified through qPCR, we used the TOPO TA cloning to assist the Sanger sequencing of tRDFs due to the short length of tRDFs. Finally, 9 out of 11 amplified tRDFs can be detected by Sanger sequencing as well as one unchanged tRDF. The validation experiment provided solid and robust evidence of the existence and accuracy of the tRDFs we analyzed through bioinformatics analyses.
Survival-related tRDFs play an indispensable role in clinical outcome prediction in NSCLC, and the 5-year survival rates of early-stage NSCLC showed demonstrably better outcomes than advanced stage. Here, we revealed a systematic survival analysis by focusing on stages as well as subtypes. Different groups of tRDFs were identified and validated as independent factors related with survival time, suggesting the tRDFs are associated with development of cancers. 3P_tRNA-SeC-TCA-1-1 shown with diagnostic value is also identified as an independent prognostic biomarker when predicting patient’s clinical outcome of all stages and advanced stages in LUAD cohort. In comparison of LUAD cohort, LUSC cohort showed different prognostic patterns with different significant tRDFs associated with stages. We also included a survival analysis based on the certain endpoints of follow-up time in 6–60 months; 5a_tRF-Ile-AAT/GAT exhibited excellent prognostic value in both early stages and all stages. 3P_tRNA-Arg-TCT-4-1 was identified to be sensitive in early following days, which was within 1 year.
We identified the correlated miRNA and the target genes enriched in signaling pathways, such as PI3K-Akt, MAPK, endocytosis, and Ras. In comparison with the correlation analysis between tRDFs-mRNA and tRDFs-miRNA, endocytosis signaling pathway is enriched in both association analyses. hsa-miR-145-5p has been reported many times as a suppressor that targets various tumor-specific genes and proteins in different cancers and was found strongly correlated with one of our signatures, 5a_tRF-Asp-GTC. Moreover, significant tRDFs were identified as affecting axonogenesis, cell morphogenesis, and positive regulation of neurogenesis by mediating the differential expression of miRNA. Nerves were not considered an important factor for tumor progression while emerging evidence of underlying axonogenesis was simulated by the release of neurotrophic growth factors from cancer cells.55,56 In aggressive tumors, axonogenesis is identified as a characteristic showing that nerve-growth-factor-induced cholinergic innervation may potentially simulate colorectal cancer. For example, cholinergic signaling can induce nerve growth factor (NGF) secretion to drive tumor axonogenesis in gastric cancer.56 As for the significantly activated biological processes enriched in tRDF-correlated miRNAs, the link between the aggressive tumor and neurogenetic gene expression can be further clarified in future studies.
Our study identified differences in functional enrichment of 52 tRDFs and six signature tRDFs. Both GSEA and GO enrichment suggested that tRDF-correlated mRNAs were identified to affect cell cycle, oocyte meiosis, and other signaling pathways. 3P_tRNA-Arg-TCT-4-1 performed with the most correlation tRDFs in cell-cycle-signaling pathway both among 52 tRDFs and tRDF signatures. Biological function, such as RNA silencing, translation regulation, and epigenetic regulation, has been addressed many times in publications. Cell cycle and oocyte meiosis are two pathways that could be identified from mRNA/miRNA/tRDF signatures. 5a_tRF-Ile-AAT/GAT was identified as having high expression in tumor tissue and was one of the tRDFs in signatures. The study from Sun et al.57 has proven the abundance of 5a_tRF-Ile-AAT/GAT in vitro in lung cancer and can regulate the cell cycle. The oocyte meiosis associated with tRDFs may result in some embryo-specific defects.
In addition, we found in both correlation analyses with miRNA and mRNA that 3ʹ-tRDFs were more strongly correlated with miRNA or mRNA than 5ʹ-tRDFs. There are 13 signaling pathways in common from miRNA and mRNA KEGG results, such as cell cycle and transcriptional mis-regulation of cancer, which indicated the potential regulation roles of tRDFs that participate in tumorigenesis. Mutation analysis based on each correlated gene also revealed several potential tRDF targets in six signaling pathways. We identified 31 genes with mutations that correlated with the six tRDF signatures; CDKN2A with mutation, which is the most correlated gene with tRDF signature 3P_tRNA-Arg-TCT-4-1, has been already most identified in LUAD among all types of cancers.58 It can be involved in the inactivation mechanism in NSCLC;59 another publication revealed the loss of function of CDKN2A also negatively impacts clinical outcome in advanced NSCLC treated with immune checkpoint blockade.60 The further mechanisms in which CDKN2A participated that are correlated with tRDFs need to be investigated.
Immune regulation of tRDFs has been an emerging target in recent years, and tRDFs can be found both in hematopoietic and lymphoid tissue as well as the blood circulation system.61 Previous studies found that a rapid increase of tRDFs during the acute inflammation stage probably involves immune responses.62 Given the current knowledge regarding tRDFs and tumor-infiltrating immune cells, we established a framework to identify potential tRDFs infiltrating immune cells in TIMEs, which provide a new perspective on cancer immunity. Based on the integrative analysis of non-coding transcriptome and immunogenomics profile, both types of tRDF were found to be strongly correlated with the infiltrating levels of 14 immune cell types. Enrichment analysis results showed 30 tRDFs that correlated with immune-related genes and also correlated with T cell CD4 memory activated, resting, CD8, etc.; combined with T-cell-receptor-signaling-pathway-enriched result, tRDFs do perform as a potential role in immune modulation. Several tRDFs were also assessed as potential immune checkpoint targets to help the immune therapy. Overall, we identified the impact of tRDF expression on immune-related biological processes and signaling pathway in the TIME and provided a potential target and therapeutic value for immune treatment by tRDF-mediated cancer immunity in NSCLC.
We also have some limitations in this study. Even though we included five datasets, only TCGA-LUAD/LUSC contain follow-up time, so we just did the prognosis analysis based on these two cohorts. Also, we merely analyzed the mRNA expression from TCGA-LUAD, as no more mRNA expression profiles available associated with small RNA sequencing (RNA-seq) from GEO. We also did not include 5ʹ-tiRNA and 3ʹ-tiRNA due to the length differences from the other four subtypes.
Conclusion
Our systematically integrated analysis of four types of tRDFs revealed novel expression patterns in NSCLC, as well as their diagnostic value for cancer patients. We also comprehensively investigated their relationship with prognosis by stages and subtypes, and several tRDFs candidates have been identified and had a risk score established as independent risk factor to predict survival outcome. Functional analysis also exhibited tRDF-target genes and signaling pathways that show how the biological role tRDFs regulate in lung cancer. tRDFs were also found to participate in transcriptional and post-transcriptional events that related to cancerous pathways. tRDFs also take a role in immune infiltration and affect TIME. This work highlights the crucial clinical implications of tRDFs and helps provide new perspectives on therapeutic strategies for NSCLC patients.
Materials and methods
Data collection and upstream analysis
Public gene expression data and complete clinical annotation from the same sequencing platform were retrieved in the TCGA database and GEO. mRNA expression (FPKM), isoacceptor (read per million mapped reads [RPM]) expression, and clinical data, including tumor stage, pathologic stage, histology subtype, sex, age, smoking history, treatment, and follow-up days, were obtained from the TCGA database, which can be used for further analysis. The fastq raw data from small RNA-seq, including three GEO non-small cell adenocarcinoma cancer cohorts (GEO: GSE83527, GSE62182, and GSE110907) and the TCGA-LUAD/LUSC cohort were downloaded by fastq-dump. The data information is summarized in Table S1.
All data were processed by trim galore and fastp for adapters trimming. Customized annotation tRDF GTF files were referred from trfexplorer (https://trfexplorer.cloud/)51 and complied with the reference human genome hg38. This contains over 1,500 tRDFs, and all tRFs can be classified into four subtypes: upstream sequences, 5ʹ-tRF, 3ʹ-tRF, and downstream sequence/1ʹ-tRF. The tRDFs come from GtRNAdb (http://gtrnadb.ucsc.edu/genomes/eukaryota/Hsapi38/Hsapi38-gene-list.html),63 tRFdb (http://genome.bioch.virginia.edu/trfdb/index.php),50 and precursor fragment from 3ʹ of tRNA.31 We used Tophat2 to take control of the annotation file to label tRDFs on hg38 and used bowtie2 to perform the alignment. HTSeq was used to get the read counts quantification of tRDFs, and raw counts were normalized into two different normalization methods: TPM and RPM. FPKM was transformed into TPM. All these procedures were done in the Linux environment.
Removing batch effect and cohort clustering
The “ComBat” algorithm of sva package and “limma” package were used to correct the batch effect caused by non-biotechnological bias.64,65 t-SNE was used to perform dimensionality reduction and embedding the data into a low-dimensional space. Standard clustering algorithm k-means from Gaussian mixture models was used on TPM data from four cohorts on this embedding to get clusters. Different clusters were set into training and validation group for further analysis. tRDFs were filtered out through identification of near zero variance predictors (nearZeroVar) in R package “carat.”
DE tRDFs identification and machine-learning selection
To identify sample-type-related DE tRDFs, the read counts and TPM data were calculated by the Deseq266 and “limma” packages to analyze the DE tRDFs between normal and tumor samples. DE tRDFs were chosen by FDR < 0.05 and |log2fold change| > 0.58. Machine learning methods by random forest were used as a diagnostic model in training and independent validation group with various feature selections, including “tumor,” “normal,” “mean decrease Gini,” “mean decrease accuracy,” and “node size.” Receiver operating characteristic (ROC) analysis was performed to determine AUC from the independent validation group.
Survival analysis
Follow-up time in TCGA LUAD/LUSC cohorts ranged from days to death and days to last follow-up. Tumor stage, pathologic stage, histology subtype, sex, age, smoking history, treatment, and vital status were included. TPM normalized data were used for analysis. After removing tRDFs with low expression, we applied univariate Cox regression analysis to identify the tRDFs that significantly correlated with patient survival and considered p < 0.05 as statistical significance. We also divided patients into three different groups based on tumor stages: all stages, early stages (stage I and stage II), and later stages (stage III and stage IV). Kaplan-Meier curve and log rank test were used to determine the significance of the difference.
The construction of scoring system was based on the prognostic value of each tRDF signature score, as we applied risk score calculation based on univariate Cox model Progscore = (beta × Exp). We then combined age, smoking, sex, pathologic_t, and risk score as variables to perform multivariate Cox regression model analysis. All statistical analysis was two sided, and a p < 0.05 was considered as statistically significant.
Correlations and functional enrichment
We computed positive and negative tRDF-mRNA and tRDF-isoacceptors Pearson correlation coefficient and Pearson correlation coefficient using only tumor datasets in TCGA for TCGA-LUAD cohort. Pearson correlation > |0.2| or Pearson correlation > |0.15| and FDR < 0.05 were selection standards. Correlated genes log2 fold changes were calculated. The GSEA and DOSE and clusterProfiler R package were used for enrichment analysis and functional annotation of highly correlated mRNAs and miRNAs.67, 68, 69 “org.hs.eg.db” was used as annotation to carry out enrichment analysis of GO and KEGG in gene set. Mutation frequency was obtained from R package maftools. Network visualizations were performed by Cytoscape.
Calculation of TIME cell invasion abundance
We used CIBERSORT algorithm (https://cibersort.stanford.edu/) to quantify the relative abundance of 22 types of immune cells in TCGA-LUAD tRDF-mRNA correlation coefficient results with the following parameters: the input mixture matrix is our gene expression matrix, the input of gene signature reference for 22 immune cell types from Newman et al.,70 1,000 times for permutation test, and RNA-seq data with quantile normalization. Pearson correlations were used to compute tRDFs with TIME cell invasion abundance results, Pearson correlation coefficient > |0.1|, and p < 0.05 as selection standard. We screened out all the immune cells that correlated with tRDFs, which showed prognostic value among all stages and conducted survival analysis. All downstream analyses were performed in R studio 4.0.0.
Plasma sample collection
Plasma was obtained from the Rush University and the Lung Cancer Biospecimen Resource Network (LCBRN). All blood samples were collected using EDTA-blood tubes. Plasma was purified by centrifuging at 1,500g for 15 min. Adenocarcinoma subjects were grouped into five pools to make all pools statistically identical. Each pool involved 10 white subjects with early-stage cancers ranging from 1a to 2a, including two males and eight females. The average age of subjects in each group was 70.2 years old, and the average tumor size was 19.4 mm. Kruskal-Wallis test did not show any statistical differences between pools. We prepared five pooled plasma samples for LUAD and six pooled plasma samples for healthy controls. This study involved in human blood samples was approved by the Institutional Review Boards (IRBs) of University of Hawaii at Manoa, protocol number 2018–00636.
RNA extraction from plasma
We used miRNeasy Serum/Plasma Kit (QIAGEN) for RNA extraction from plasma following the manufacturer’s protocol. Plasma was mixed with acid-phenol/guanidine-based lysis buffer to denature protein complexes. After adding chloroform, total RNAs were purified by centrifuging. The aqueous-phase-contained total RNAs were applied to the RNeasy MinElute spin column to wash away phenol and other contaminants. High-quality RNAs were then eluted by RNase-free water. We took 50 μL plasma from each subject, resulting in 500 μL from 10 subjects per pool. Due to the capacity of the kit, we treated 250 μL plasma from one pool at a time and combined two products at the step of MinElute spin column. After eluting with 14 μL RNase-free water, we added 1 μL of RNase inhibitor.
Small RNA-seq
Library prep and small RNA-seq were performed by the Genomics and Bioinformatics Shared Resources (GBSR) at the University of Hawaii Cancer Center. QIAseq miRNA Library Kit and QIAseq miRNA NGS 12 Index IL from QIAGEN were used for making the library, and NextSeq 500 from Illumina was used for sequencing to obtain 10M reads/sample. Raw data were processed by the upstream analysis pipeline, and raw counts were normalized into TPM. DE tRDFs were computed by DEseq2 and Limma t test.
Cell culture
ABC-1 and MRC9 cell lines were cultured in Dulbecco’s modified Eagle’s medium (DMEM) supplemented with 10% fetal bovine serum (FBS) and 1% penicillin-streptomycin at 37°C in 5% CO2 incubator.
RNA extraction from cell lines
We used miRNeasy Serum/Plasma Kit (QIAGEN) for RNA extraction from cell lines following the manufacturer’s protocol.
Reverse transcription and real-time qPCR
Total RNAs was subjected to cDNA synthesis by TAKARA PrimeScript RT Reagent Kit (Perfect Real Time), and qPCR was processed with Quanta bio PerfeCta SYBR Green FastMix. miR-16 was chosen as internal control for tRDFs quantification in cell lines.49 The relative expression levels were calculated via the 2-ΔΔCt method. The primers for RT and qPCR are listed in Table S17.
TOPO TA cloning and Sanger sequencing
cDNA products were amplified by Accuris Taq, and PCR products were purified by Zymoresearch PCR purification kit. The ligation reaction of PCR products was performed with pGEM-T easy vectors and 2× Rapid Ligation Buffer. JM109 competent cells were used for transformation to the ligation reaction. Transformation culture was incubated onto duplicate LB/ampicillin/IPTG/X-Gal plates, and white colonies were selected. Bacterial colonies were screened with PCR, and target bands were detected by agarose gel electrophoresis. Sanger sequencing was performed by University of Hawaii at Manoa ASGPB.
Availability of data and materials
The small RNA-seq or mRNA sequencing data are available on GEO: GSE83527, GSE62182, and GSE110907 and TCGA-LUAD/LUSC project.
Acknowledgments
This study involved in human blood samples was approved by the Institutional Review Boards (IRBs) of University of Hawaii at Manoa, protocol number 2018–00636. We are very grateful to the Gene Expression Omnibus (GEO) and The Cancer Genome Atlas (TCGA) database for providing the transcriptome and clinical information. Y.D. is supported by the National Institutes of Health (NIH) grants 1R01CA223490, 5P30GM114737, 5P20GM103466, 5U54MD007601, 5P30CA071789, 1R01CA230514, and P20GM139753. G.H. is supported by National Natural Science Foundation of China grant nos. 82127807 and 81830052, National Key Research and Development Program of China no. 2020YFA0909000, and Shanghai Key Laboratory of Molecular Imaging 18DZ2260400. Z.G. is supported by the grants from National Institutes of Health R25CA244073. We also appreciated the technique support from H. Guo, J. Wu, J. Song, and M. Ge.
Author contributions
Z.G. and Y.D. conceived and collected the data; Z.G., S.C., and Y.F. integrated and analyzed all the data. Z.G. drafted the manuscript, Z.G. and J.X. did data visualization, M.J. and M.N. conducted plasma experiment, and H.B., T.G., and Y.C. revised the article. G.H. and X.H. provided the data support.
Declaration of interests
The authors declare no competing interests.
Footnotes
Supplemental information can be found online at https://doi.org/10.1016/j.omto.2022.07.002.
Contributor Information
Xiamin Hu, Email: huaximing@163.com.
Gang Huang, Email: huangg@sumhs.edu.cn.
Youping Deng, Email: dengy@hawaii.edu.
Supplemental information
References
- 1.Taft R.J., Pang K.C., Mercer T.R., Dinger M., Mattick J.S. Non-coding RNAs: regulators of disease. J. Pathol. 2010;220:126–139. doi: 10.1002/path.2638. [DOI] [PubMed] [Google Scholar]
- 2.Esteller M. Non-coding RNAs in human disease. Nat. Rev. Genet. 2011;12:861–874. doi: 10.1038/nrg3074. [DOI] [PubMed] [Google Scholar]
- 3.Slaby O., Laga R., Sedlacek O. Therapeutic targeting of non-coding RNAs in cancer. Biochem. J. 2017;474:4219–4251. doi: 10.1042/BCJ20170079. [DOI] [PubMed] [Google Scholar]
- 4.Sun C., Fu Z., Wang S., Li J., Li Y., Zhang Y., Yang F., Chu J., Wu H., Huang X., et al. Roles of tRNA-derived fragments in human cancers. Cancer Lett. 2018;414:16–25. doi: 10.1016/j.canlet.2017.10.031. [DOI] [PubMed] [Google Scholar]
- 5.Kirchner S., Ignatova Z. Emerging roles of tRNA in adaptive translation, signalling dynamics and disease. Nat. Rev. Genet. 2015;16:98–112. doi: 10.1038/nrg3861. [DOI] [PubMed] [Google Scholar]
- 6.Schimmel P. The emerging complexity of the tRNA world: mammalian tRNAs beyond protein synthesis. Nat. Rev. Mol. Cell Biol. 2018;19:45–58. doi: 10.1038/nrm.2017.77. [DOI] [PubMed] [Google Scholar]
- 7.Li S., Xu Z., Sheng J. tRNA-derived small RNA: a novel regulatory small non-coding RNA. Genes (Basel) 2018;9:E246. doi: 10.3390/genes9050246. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Yu M., Lu B., Zhang J., Ding J., Liu P., Lu Y. tRNA-derived RNA fragments in cancer: current status and future perspectives. J. Hematol. Oncol. 2020;13:121. doi: 10.1186/s13045-020-00955-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Tosar J.P., Cayota A. Extracellular tRNAs and tRNA-derived fragments. RNA Biol. 2020;17:1149–1167. doi: 10.1080/15476286.2020.1729584. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Zhang Y., Qian H., He J., Gao W. Mechanisms of tRNA-derived fragments and tRNA halves in cancer treatment resistance. Biomark. Res. 2020;8:52. doi: 10.1186/s40364-020-00233-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Zhu L., Ge J., Li T., Shen Y., Guo J. tRNA-derived fragments and tRNA halves: the new players in cancers. Cancer Lett. 2019;452:31–37. doi: 10.1016/j.canlet.2019.03.012. [DOI] [PubMed] [Google Scholar]
- 12.Fu H., Feng J., Liu Q., Sun F., Tie Y., Zhu J., Xing R., Sun Z., Zheng X. Stress induces tRNA cleavage by angiogenin in mammalian cells. FEBS Lett. 2009;583:437–442. doi: 10.1016/j.febslet.2008.12.043. [DOI] [PubMed] [Google Scholar]
- 13.Tao E.W., Cheng W.Y., Li W.L., Yu J., Gao Q.Y. tiRNAs: a novel class of small noncoding RNAs that helps cells respond to stressors and plays roles in cancer progression. J. Cell. Physiol. 2020;235:683–690. doi: 10.1002/jcp.29057. [DOI] [PubMed] [Google Scholar]
- 14.Jiang P., Yan F. tiRNAs & tRFs biogenesis and regulation of diseases: a review. Curr. Med. Chem. 2019;26:5849–5861. doi: 10.2174/0929867326666190124123831. [DOI] [PubMed] [Google Scholar]
- 15.Kumar P., Mudunuri S.B., Anaya J., Dutta A. tRFdb: a database for transfer RNA fragments. Nucleic Acids Res. 2015;43:D141–D145. doi: 10.1093/nar/gku1138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Lee Y.S., Shibata Y., Malhotra A., Dutta A. A novel class of small RNAs: tRNA-derived RNA fragments (tRFs) Genes Dev. 2009;23:2639–2649. doi: 10.1101/gad.1837609. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Guzzi N., Bellodi C. Novel insights into the emerging roles of tRNA-derived fragments in mammalian development. RNA Biol. 2020;17:1214–1222. doi: 10.1080/15476286.2020.1732694. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Maute R.L., Schneider C., Sumazin P., Holmes A., Califano A., Basso K., Dalla-Favera R. tRNA-derived microRNA modulates proliferation and the DNA damage response and is down-regulated in B cell lymphoma. Proc. Natl. Acad. Sci. USA. 2013;110:1404–1409. doi: 10.1073/pnas.1206761110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Kumar P., Kuscu C., Dutta A. Biogenesis and function of transfer RNA-related fragments (tRFs) Trends Biochem. Sci. 2016;41:679–689. doi: 10.1016/j.tibs.2016.05.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Zhu L., Liu X., Pu W., Peng Y. tRNA-derived small non-coding RNAs in human disease. Cancer Lett. 2018;419:1–7. doi: 10.1016/j.canlet.2018.01.015. [DOI] [PubMed] [Google Scholar]
- 21.Haussecker D., Huang Y., Lau A., Parameswaran P., Fire A.Z., Kay M.A. Human tRNA-derived small RNAs in the global regulation of RNA silencing. RNA. 2010;16:673–695. doi: 10.1261/rna.2000810. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Schorn A.J., Gutbrod M.J., LeBlanc C., Martienssen R. LTR-retrotransposon control by tRNA-derived small RNAs. Cell. 2017;170:61–71.e11. doi: 10.1016/j.cell.2017.06.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Kumar P., Anaya J., Mudunuri S.B., Dutta A. Meta-analysis of tRNA derived RNA fragments reveals that they are evolutionarily conserved and associate with AGO proteins to recognize specific RNA targets. BMC Biol. 2014;12:78. doi: 10.1186/s12915-014-0078-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Goodarzi H., Liu X., Nguyen H.C.B., Zhang S., Fish L., Tavazoie S.F. Endogenous tRNA-derived fragments suppress breast cancer progression via YBX1 displacement. Cell. 2015;161:790–802. doi: 10.1016/j.cell.2015.02.053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Sobala A., Hutvagner G. Small RNAs derived from the 5' end of tRNA can inhibit protein translation in human cells. RNA Biol. 2013;10:553–563. doi: 10.4161/rna.24285. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Shen L., Tan Z., Gan M., Li Q., Chen L., Niu L., Jiang D., Zhao Y., Wang J., Li X., et al. tRNA-derived small non-coding RNAs as novel epigenetic molecules regulating adipogenesis. Biomolecules. 2019;9:E274. doi: 10.3390/biom9070274. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Goncalves K.A., Silberstein L., Li S., Severe N., Hu M.G., Yang H., Scadden D.T., Hu G.F. Angiogenin promotes hematopoietic regeneration by dichotomously regulating quiescence of stem and progenitor cells. Cell. 2016;166:894–906. doi: 10.1016/j.cell.2016.06.042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Balatti V., Nigita G., Veneziano D., Drusco A., Stein G.S., Messier T.L., Farina N.H., Lian J.B., Tomasello L., Liu C.G., et al. tsRNA signatures in cancer. Proc. Natl. Acad. Sci. USA. 2017;114:8071–8076. doi: 10.1073/pnas.1706908114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Dhahbi J.M., Spindler S.R., Atamna H., Boffelli D., Martin D.I. Deep sequencing of serum small RNAs identifies patterns of 5' tRNA half and YRNA fragment expression associated with breast cancer. Biomark. Cancer. 2014;6:37–47. doi: 10.4137/BIC.S20764. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Zheng L.L., Xu W.L., Liu S., Sun W.J., Li J.H., Wu J., Yang J.H., Qu L.H. tRF2Cancer: a web server to detect tRNA-derived small RNA fragments (tRFs) and their expression in multiple cancers. Nucleic Acids Res. 2016;44:W185–W193. doi: 10.1093/nar/gkw414. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Pekarsky Y., Balatti V., Palamarchuk A., Rizzotto L., Veneziano D., Nigita G., Rassenti L.Z., Pass H.I., Kipps T.J., Liu C.G., Croce C.M. Dysregulation of a family of short noncoding RNAs, tsRNAs, in human cancer. Proc. Natl. Acad. Sci. USA. 2016;113:5071–5076. doi: 10.1073/pnas.1604266113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Pliatsika V., Loher P., Magee R., Telonis A.G., Londin E., Shigematsu M., Kirino Y., Rigoutsos I. MINTbase v2.0: a comprehensive database for tRNA-derived fragments that includes nuclear and mitochondrial fragments from all the Cancer Genome Atlas projects. Nucleic Acids Res. 2018;46:D152–D159. doi: 10.1093/nar/gkx1075. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Cho H., Lee W., Kim G.W., Lee S.H., Moon J.S., Kim M., Kim H.S., Oh J.W. Regulation of La/SSB-dependent viral gene expression by pre-tRNA 3' trailer-derived tRNA fragments. Nucleic Acids Res. 2019;47:9888–9901. doi: 10.1093/nar/gkz732. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Ruggero K., Guffanti A., Corradin A., Sharma V.K., De Bellis G., Corti G., Grassi A., Zanovello P., Bronte V., Ciminale V., D'Agostino D.M. Small noncoding RNAs in cells transformed by human T-cell leukemia virus type 1: a role for a tRNA fragment as a primer for reverse transcriptase. J. Virol. 2014;88:3612–3622. doi: 10.1128/JVI.02823-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Deng J., Ptashkin R.N., Chen Y., Cheng Z., Liu G., Phan T., Deng X., Zhou J., Lee I., Lee Y.S., Bao X. Respiratory syncytial virus utilizes a tRNA fragment to suppress antiviral responses through a novel targeting mechanism. Mol. Ther. 2015;23:1622–1629. doi: 10.1038/mt.2015.124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Wang Q., Lee I., Ren J., Ajay S.S., Lee Y.S., Bao X. Identification and functional characterization of tRNA-derived RNA fragments (tRFs) in respiratory syncytial virus infection. Mol. Ther. 2013;21:368–379. doi: 10.1038/mt.2012.237. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Zhou J., Liu S., Chen Y., Fu Y., Silver A.J., Hill M.S., Lee I., Lee Y.S., Bao X. Identification of two novel functional tRNA-derived fragments induced in response to respiratory syncytial virus infection. J. Gen. Virol. 2017;98:1600–1610. doi: 10.1099/jgv.0.000852. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Blanco S., Dietmann S., Flores J.V., Hussain S., Kutter C., Humphreys P., Lukk M., Lombard P., Treps L., Popis M., et al. Aberrant methylation of tRNAs links cellular stress to neuro-developmental disorders. EMBO J. 2014;33:2020–2039. doi: 10.15252/embj.201489282. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Ivanov P., O'Day E., Emara M.M., Wagner G., Lieberman J., Anderson P. G-quadruplex structures contribute to the neuroprotective effects of angiogenin-induced tRNA fragments. Proc. Natl. Acad. Sci. USA. 2014;111:18201–18206. doi: 10.1073/pnas.1407361111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Schaffer A.E., Eggens V.R.C., Caglayan A.O., Reuter M.S., Scott E., Coufal N.G., Silhavy J.L., Xue Y., Kayserili H., Yasuno K., et al. CLP1 founder mutation links tRNA splicing and maturation to cerebellar development and neurodegeneration. Cell. 2014;157:651–663. doi: 10.1016/j.cell.2014.03.049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Mishima E., Inoue C., Saigusa D., Inoue R., Ito K., Suzuki Y., Jinno D., Tsukui Y., Akamatsu Y., Araki M., et al. Conformational change in transfer RNA is an early indicator of acute cellular damage. J. Am. Soc. Nephrol. 2014;25:2316–2326. doi: 10.1681/ASN.2013091001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Honda S., Loher P., Shigematsu M., Palazzo J.P., Suzuki R., Imoto I., Rigoutsos I., Kirino Y. Sex hormone-dependent tRNA halves enhance cell proliferation in breast and prostate cancers. Proc. Natl. Acad. Sci. USA. 2015;112:E3816–E3825. doi: 10.1073/pnas.1510077112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Farina N.H., Scalia S., Adams C.E., Hong D., Fritz A.J., Messier T.L., Balatti V., Veneziano D., Lian J.B., Croce C.M., et al. Identification of tRNA-derived small RNA (tsRNA) responsive to the tumor suppressor, RUNX1, in breast cancer. J. Cell. Physiol. 2020;235:5318–5327. doi: 10.1002/jcp.29419. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Zhang M., Li F., Wang J., He W., Li Y., Li H., Wei Z., Cao Y. tRNA-derived fragment tRF-03357 promotes cell proliferation, migration and invasion in high-grade serous ovarian cancer. Onco. Targets Ther. 2019;12:6371–6383. doi: 10.2147/OTT.S206861. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Dhahbi J.M., Atamna H., Selth L.A. Data mining of small RNA-seq suggests an association between prostate cancer and altered abundance of 5' transfer RNA halves in seminal fluid and prostatic tissues. Biomark. Cancer. 2018;10 doi: 10.1177/1179299X18759545. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Wang J., Ma G., Ge H., Han X., Mao X., Wang X., Veeramootoo J.S., Xia T., Liu X., Wang S. Circulating tRNA-derived small RNAs (tsRNAs) signature for the diagnosis and prognosis of breast cancer. NPJ Breast Cancer. 2021;7:4. doi: 10.1038/s41523-020-00211-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Shao Y., Sun Q., Liu X., Wang P., Wu R., Ma Z. tRF-Leu-CAG promotes cell proliferation and cell cycle in non-small cell lung cancer. Chem. Biol. Drug Des. 2017;90:730–738. doi: 10.1111/cbdd.12994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Wu Y., Yang X., Jiang G., Zhang H., Ge L., Chen F., Li J., Liu H., Wang H. 5'-tRF-GlyGCC: a tRNA-derived small RNA as a novel biomarker for colorectal cancer diagnosis. Genome Med. 2021;13:20. doi: 10.1186/s13073-021-00833-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Zhu L., Li J., Gong Y., Wu Q., Tan S., Sun D., Xu X., Zuo Y., Zhao Y., Wei Y.Q., et al. Exosomal tRNA-derived small RNA as a promising biomarker for cancer diagnosis. Mol. Cancer. 2019;18:74. doi: 10.1186/s12943-019-1000-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Kumar P., Mudunuri S.P., Anaya J., Dutta A. tRFdb: a relational database of tranfer RNA related Fragments. Nucleic Acids Res. 2012;43:D141–D145. doi: 10.1093/nar/gku1138. http://genome.bioch.virginia.edu/trfdb/index.php [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.The tRF Explorer. 2019. https://trfexplorer.cloud/
- 52.Yu X., Xie Y., Zhang S., Song X., Xiao B., Yan Z. tRNA-derived fragments: mechanisms underlying their regulation of gene expression and potential applications as therapeutic targets in cancers and virus infections. Theranostics. 2021;11:461–469. doi: 10.7150/thno.51963. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Zeng T., Hua Y., Sun C., Zhang Y., Yang F., Yang M., Yang Y., Li J., Huang X., Wu H., et al. Relationship between tRNA-derived fragments and human cancers. Int. J. Cancer. 2020;147:3007–3018. doi: 10.1002/ijc.33107. [DOI] [PubMed] [Google Scholar]
- 54.Gu W., Shi J., Liu H., Zhang X., Zhou J.J., Li M., Zhou D., Li R., Lv J., Wen G., et al. Peripheral blood non-canonical small non-coding RNAs as novel biomarkers in lung cancer. Mol. Cancer. 2020;19:159. doi: 10.1186/s12943-020-01280-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Faulkner S., Jobling P., March B., Jiang C.C., Hondermarck H. Tumor neurobiology and the war of nerves in cancer. Cancer Discov. 2019;9:702–710. doi: 10.1158/2159-8290.CD-18-1398. [DOI] [PubMed] [Google Scholar]
- 56.Hayakawa Y., Sakitani K., Konishi M., Asfaha S., Niikura R., Tomita H., Renz B.W., Tailor Y., Macchini M., Middelhoff M., et al. Nerve growth factor promotes gastric tumorigenesis through aberrant cholinergic signaling. Cancer Cell. 2017;31:21–34. doi: 10.1016/j.ccell.2016.11.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Sun X., Yang J., Yu M., Yao D., Zhou L., Li X., Qiu Q., Lin W., Lu B., Chen E., et al. Global identification and characterization of tRNA-derived RNA fragment landscapes across human cancers. NAR Cancer. 2020;2:zcaa031. doi: 10.1093/narcan/zcaa031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.AACR Project GENIE Consortium AACR project GENIE: powering precision medicine through an international consortium. Cancer Discov. 2017;7:818–831. doi: 10.1158/2159-8290.CD-17-0151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Tam K.W., Zhang W., Soh J., Stastny V., Chen M., Sun H., Thu K., Rios J.J., Yang C., Marconett C.N., et al. CDKN2A/p16 inactivation mechanisms and their relationship to smoke exposure and molecular features in non-small-cell lung cancer. J. Thorac. Oncol. 2013;8:1378–1388. doi: 10.1097/JTO.0b013e3182a46c0c. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Gutiontov S.I., Turchan W.T., Spurr L.F., Rouhani S.J., Chervin C.S., Steinhardt G., Lager A.M., Wanjari P., Malik R., Connell P.P., et al. CDKN2A loss-of-function predicts immunotherapy resistance in non-small cell lung cancer. Sci. Rep. 2021;11:20059. doi: 10.1038/s41598-021-99524-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Dhahbi J.M., Spindler S.R., Atamna H., Yamakawa A., Boffelli D., Mote P., Martin D.I.K. 5' tRNA halves are present as abundant complexes in serum, concentrated in blood cells, and modulated by aging and calorie restriction. BMC Genomics. 2013;14:298. doi: 10.1186/1471-2164-14-298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Zhang Y., Zhang Y., Shi J., Zhang H., Cao Z., Gao X., Ren W., Ning Y., Ning L., Cao Y., et al. Identification and characterization of an ancient class of small RNAs enriched in serum associating with active infection. J. Mol. Cell Biol. 2014;6:172–174. doi: 10.1093/jmcb/mjt052. [DOI] [PubMed] [Google Scholar]
- 63.GtRNAdb: tRNAscane-SE analysis of complete genomes. Nucleic Acids Res. 2021;37:D93–D97. http://gtrnadb.ucsc.edu/ [Google Scholar]
- 64.Leek J.T., Johnson W.E., Parker H.S., Jaffe A.E., Storey J.D. The sva package for removing batch effects and other unwanted variation in high-throughput experiments. Bioinformatics. 2012;28:882–883. doi: 10.1093/bioinformatics/bts034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Ritchie M.E., Phipson B., Wu D., Hu Y., Law C.W., Shi W., Smyth G.K. Limma powers differential expression analyses for RNA-sequencing and microarray studies. Nucleic Acids Res. 2015;43:e47. doi: 10.1093/nar/gkv007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Love M.I., Huber W., Anders S. Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol. 2014;15:550. doi: 10.1186/s13059-014-0550-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Wu T., Hu E., Xu S., Chen M., Guo P., Dai Z., Feng T., Zhou L., Tang W., Zhan L., et al. clusterProfiler 4.0: a universal enrichment tool for interpreting omics data. Innovation. 2021;2:100141. doi: 10.1016/j.xinn.2021.100141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Yu G., Wang L.G., Han Y., He Q.Y. clusterProfiler: an R package for comparing biological themes among gene clusters. OMICS. 2012;16:284–287. doi: 10.1089/omi.2011.0118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Yu G., Wang L.G., Yan G.R., He Q.Y. DOSE: an R/Bioconductor package for disease ontology semantic and enrichment analysis. Bioinformatics. 2015;31:608–609. doi: 10.1093/bioinformatics/btu684. [DOI] [PubMed] [Google Scholar]
- 70.Newman A.M., Liu C.L., Green M.R., Gentles A.J., Feng W., Xu Y., Hoang C.D., Diehn M., Alizadeh A.A. Robust enumeration of cell subsets from tissue expression profiles. Nat. Methods. 2015;12:453–457. doi: 10.1038/nmeth.3337. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The small RNA-seq or mRNA sequencing data are available on GEO: GSE83527, GSE62182, and GSE110907 and TCGA-LUAD/LUSC project.