

Summary
The loss of protein homeostasis results in cytotoxic protein aggregates, a common hallmark of aging and neurological diseases. Here, we present an adjusted filter-trapping assay protocol to detect global aggregated proteins in human cell lines, via a high-sensitive protein staining method. This protocol also details an alternative approach to monitor specific protein aggregates trapped in the filter membrane, by subsequent immunoblotting of ectopically expressed and endogenous proteins.
For complete details on the use and execution of this protocol, please refer to Chhipi-Shrestha et al. (2022).
Subject areas: Molecular Biology, Protein Biochemistry, Protein expression and purification
Graphical abstract
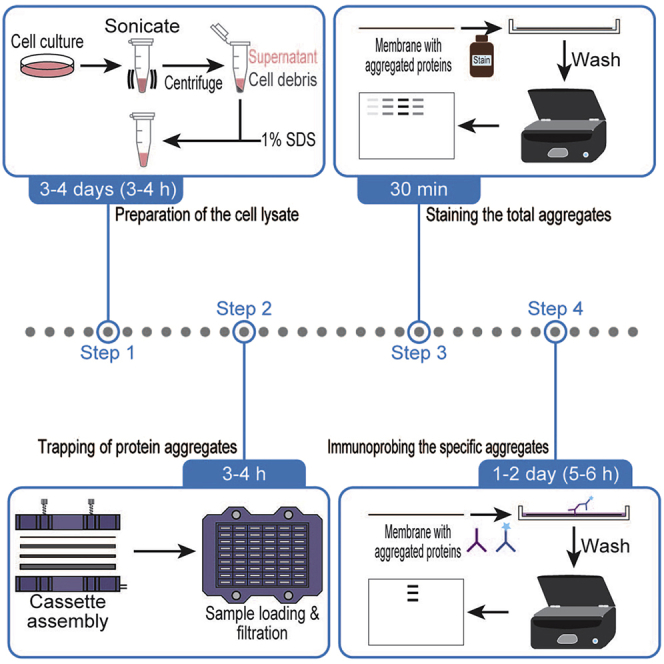
Highlights
-
•
Trapping SDS-resistant protein aggregates on a cellulose acetate membrane
-
•
Total protein staining on the filter membrane with high sensitivity
-
•
Immunoprobing specific aggregated proteins on the filter membrane
Publisher’s note: Undertaking any experimental protocol requires adherence to local institutional guidelines for laboratory safety and ethics.
The loss of protein homeostasis results in cytotoxic protein aggregates, a common hallmark of aging and neurological diseases. Here, we present an adjusted filter-trapping assay protocol to detect global aggregated proteins in human cell lines, via a high-sensitive protein staining method. This protocol also details an alternative approach to monitor specific protein aggregates trapped in the filter membrane, by subsequent immunoblotting of ectopically expressed and endogenous proteins.
Before you begin
Studies of protein aggregation under different cellular stresses have two main objectives: 1) investigation of alterations in global aggregating proteins and 2) investigation of alterations in specific aggregating protein targets. This protocol below describes the steps for both of them.
Here, we treated cells with compounds such as MG132 (dissolved in DMSO) and spliceostatin A (SSA) (dissolved in MeOH) (Kaida et al., 2007) to induce proteotoxicity. We used these conditions to accumulate system-wide protein aggregations and detected them by the application of Revert 700 Total Protein Stain (LI-COR Biosciences) with the infrared 700 channel with an Odyssey CLx Infrared Imaging System (LI-COR Biosciences).
Moreover, this protocol also covers the immunoprobing-based detection of specific aggregated target proteins trapped in the filter membrane. Cells can be transfected with different epitope-tagged plasmid constructs according to the topic of the study (Chhipi-Shrestha et al., 2022; Farrawell et al., 2015; Tsuboyama et al., 2020). Here, we used FLAG-tagged constructs to study the aggregation potential of specific ectopically expressed proteins. A similar technique should be applied to endogenous proteins if appropriate antibodies are available. We used secondary antibodies labeled with infrared fluorophores and collected images with an Odyssey CLx Infrared Imaging System (LI-COR Biosciences). Other immunoblot systems (such as chemiluminescence) can be employed with optimization.
The preparation of cell lysates and filter trap assays are common for both methods. The experiments were performed in 6-well plates; however, different scales of cell culture and downstream analysis should be usable if needed. Cells can be treated with drugs, gene knockdown/knockout/overexpression, environmental changes, etc., according to the goal of the aggregation study.
Key resources table
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| Mouse monoclonal anti-FLAG (M2) antibody (1:1,000) | Sigma-Aldrich | Cat#F1804; RRID: AB_262044 |
| Rabbit monoclonal anti-GFP (D5.1) antibody (1:1,000) | Cell Signaling Technology | Cat#2956, RRID: AB_1196615 |
| Goat polyclonal anti-mouse IgG antibody conjugated to IRDye 800CW (1:10,000) | LI-COR Biosciences | Cat#926-32210; RRID: AB_621842 |
| Goat polyclonal anti-rabbit IgG antibody conjugated to IRDye 800CW (1:10,000) | LI-COR Biosciences | Cat#926-32211; RRID: AB_621843 |
| Mouse monoclonal anti-ubiquitin (P4D1) antibody (1:500) | Santa Cruz Biotechnology | Cat#SC-8017; RRID: AB_628423 |
| Chemicals, peptides, and recombinant proteins | ||
| SSA (spliceostatin A) | (Kaida et al., 2007) | N/A |
| MG132 (Z-Leu-Leu-Leu-al) | FUJIFILM Wako Pure Chemical | Cat#135-18453 |
| Critical commercial assays | ||
| Tris-HCl | Nacalai Tesque | Cat#35433-15 |
| Tris-Base | Nacalai Tesque | Cat#35406-91 |
| NaCl | Nacalai Tesque | Cat#31320-05 |
| Tween-20 | Nacalai Tesque | Cat#35624-15 |
| DMEM, high glucose | FUJIFILM Wako Pure Chemical | Cat#044-29765 |
| Fetal bovine serum (FBS) | Thermo Fisher Scientific | Cat#10270106 |
| Phosphate-buffered saline, PBS (1×) | Nacalai Tesque | Cat #14249-95 |
| 0.5w/v% Trypsin-EDTA (10×) | FUJIFILM Wako Pure Chemical | Cat#208-17251 |
| Methanol (MeOH) | FUJIFILM Wako Pure Chemical | Cat#138-01836 |
| Dimethyl sulfoxide (DMSO) | FUJIFILM Wako Pure Chemical | Cat#047-29353 |
| Protease inhibitor cocktail | Sigma-Aldrich | Cat#P8340 |
| Pierce 660-nm Protein Assay Kit | Thermo Fisher Scientific | Cat#22662 |
| 10% SDS Solution | Nacalai Tesque | Cat#30562-04 |
| Revert 700 Total Protein Stain and Wash Solution Kit | LI-COR Biosciences | Cat#926-11015 |
| Opti-MEM I Reduced Serum Medium | Thermo Fisher Scientific | Cat#31985062 |
| FuGENE HD | Promega | Cat#E2311 |
| Odyssey Blocking Buffer (TBS) | LI-COR Biosciences | Cat#927-50000 |
| Deposited data | ||
| Original images used for the figures | This study | Mendeley Data: https://doi.org/10.17632/hb8mx8xt2n.1 |
| Experimental models: Cell lines | ||
| Human: Cell line HeLa S3: female | RIKEN BioResource Research Center | RCB1525 |
| Recombinant DNA | ||
| pCDNA5/FRT/TO | Thermo Fisher Scientific | Cat#V652020 |
| pCDNA5/FRT/TO-FTH1 | (Chhipi-Shrestha et al., 2022) | N/A |
| pCDNA5/FRT/TO-FTH1∗ | (Chhipi-Shrestha et al., 2022) | N/A |
| pCDNA5/FRT/TO-p27 | (Chhipi-Shrestha et al., 2022) | N/A |
| pCDNA5/FRT/TO-p27∗ | (Chhipi-Shrestha et al., 2022) | N/A |
| pCAGEN-FLAG-GFP | (Tsuboyama et al., 2020) | N/A |
| pCAGEN-GFP–TDP-43ΔNLS | (Tsuboyama et al., 2020) | N/A |
| Software and algorithms | ||
| Image Studio version 5.2 | LI-COR Biosciences | https://www.licor.com/bio/image-studio/ |
| Other | ||
| CO2 incubator | ESPEC | BNA-111 |
| Nunc Cell-Culture Treated Multidishes, 6-well | Thermo Fisher Scientific | Cat#140675 |
| DNA LoBind Tube 1.5 mL | Eppendorf | Cat#022431021 |
| Refrigerated microcentrifuge | TOMY | Cat#MX-307 |
| Sonicator | Cosmo Bio CO., LTD. | Bioruptor UCD-250 |
| Microplate reader | Molecular Devices | SpectraMax M2e |
| Bio-Dot SF Filter Paper | Bio-Rad | Cat#1620161 |
| 0.2-μm cellulose acetate membrane filter | Cytiva | Cat#10404180 |
| Bio-Dot SF Apparatus | Bio-Rad | Cat#1706542 |
| Square petri dish | Greiner Bio-One | Cat#07-000-330 |
| Seesaw shaker | Biocraft | BC-700 |
| Odyssey CLx Infrared Imaging System | LI-COR Biosciences | N/A |
Materials and equipment
20× Tris-buffered saline (TBS)
| Reagent | Final concentration | Amount |
|---|---|---|
| Tris-HCl | 0.3 M | 48 g |
| Tris-Base | 0.1 M | 11.2 g |
| NaCl | 3 M | 176 g |
| ddH2O | n/a | Add to 1 L |
| Total | n/a | 1 L |
Note: 20× TBS should be sterilized by autoclaving and can be store at 25°C for 1 month.
TBS with 0.1% Tween-20 (TBS-T)
| Reagent | Final concentration | Amount |
|---|---|---|
| 20× TBS | 1× | 250 mL |
| Tween-20 | 0.1% | 5 mL |
| ddH2O | n/a | 4745 mL |
| Total | n/a | 5 L |
Note: TBS-T can be store at 25°C for 1 month.
Note: Prepare 20% [v/v] Tween-20 as a working solution since 100% Tween-20 is viscous and difficult to pipet when a small volume is desired.
0.1× anti-mouse/anti-rabbit IgG antibody conjugated to IRDye 800CW
| Reagent | Final concentration | Amount |
|---|---|---|
| Anti-mouse IgG antibody conjugated to IRDye 800CW or anti-rabbit IgG antibody conjugated to IRDye 800CW | 1:10 | 10 μL |
| Odyssey Blocking Buffer (TBS) | n/a | 90 μL |
| Total | n/a | 100 μL |
Note: 0.1× anti-mouse/anti-rabbit IgG antibody conjugated to IRDye 800CW can be store at 4°C for 1 month.
Note: The Revert 700 Total Protein Stain and Wash Solution Kit requires methanol for preparation of the stain reagent. Follow the manufacturer’s instructions.
CRITICAL: Wear personal protective equipment such as gloves and safety glasses during experiments.
Alternatives: The Pierce 660-nm Protein Assay Kit can be replaced with other protein quantification kits as desired.
Alternatives: Instead of the Bio-Dot SF Apparatus (Bio-Rad), a similar setup, such as a slot blot microfiltration apparatus (48 wells) from Sanplatec, which was originally used in the previous work (Chhipi-Shrestha et al., 2022), should work successfully in a similar manner.
Alternatives: Instead of the Revert 700 Total Protein Stain and Wash Solution Kit, other total protein stains such as ponceau might be used with the corresponding optimum concentrations and incubation times. We mainly used the Revert 700 Total Protein Stain because of its high sensitivity and low background.
Alternatives: Instead of a goat polyclonal anti-mouse/rabbit IgG antibody conjugated to IRDye 800CW, equivalent antibodies, such as goat polyclonal anti-mouse/rabbit IgG antibody conjugated to IRDye 680RD (both LI-COR Biosciences), could be used, depending upon the source of the primary antibody.
Alternatives: Instead of FuGENE HD, other transfection reagents, such as X-tremeGENE 9 DNA transfection reagent (Sigma-Aldrich), could be used for plasmid transfection.
Alternatives: Stably expressing cells may be used instead of cells with transient expression induced by plasmid transfection.
Step-by-step method details
Detection of system-wide aggregated proteins
Cell culture and induction of proteotoxic stress
Timing: 1 h on day 1
This section describes the steps of cell growth and drug treatment to induce proteotoxic stress.
Day 1.
-
1.
Seed 3 × 105 HeLa S3 cells into each well of a 6-well plate using 2 mL of growth medium (DMEM supplemented with 10% FBS).
-
2.
Incubate the cells for 48 h at 37°C in a CO2 incubator.
Note: Prepare an appropriate number of replicates. Normally, triplicates are recommended.
Note: Since drug treatment (see below) may reduce cell growth, adjustment of the seeded cell number may be needed. We typically set up the cell number to yield ∼80% confluence at the time of cell lysis.
Day 3–4.
-
3.Treat the cells with the compounds and the same volume of solvent for the control.
-
a.For MG132:
-
i.Treat the cells with the compound at 5 μM or the solvent DMSO on day 4.
-
ii.Incubate them for 10 h.
-
i.
-
b.For SSA:
-
i.Treat the cells with the compound at 100 ng/mL or the solvent MeOH on day 3.
-
ii.Incubate them for 24 h.
-
i.
-
a.
Cell lysis
Day 4.
This section describes the method of cell harvesting and the preparation of cell lysate.
-
4.
Aspirate the medium from the wells of a 6-well plate.
-
5.
Rinse the wells gently with 1 mL of PBS and then aspirate the PBS.
-
6.
Add 150 μL of PBS and 15 μL of 10× Trypsin-EDTA, incubate for 5 min at 37°C in the CO₂ incubator, and then add 165 μL of warm medium. Tilt the dish and pipet the buffer several times from the pool back toward the top of the dish, scrape down the slope of the dish to ensure that all the cells are detached, and collect them in 1.5-mL microfuge tubes by pipetting.
-
7.
Centrifuge the cell solution at 800 × g for 4 min at 4°C.
-
8.
Remove the supernatant to collect the cell pellet.
-
9.
Rinse the cell pellet by 400 μL of ice-cold PBS and spin down the cells at 800 × g for 4 min at 4°C.
Note: From here, keep the sample on ice as much as possible until the protein samples are mixed with 1% SDS solution (step 16).
-
10.
Remove the supernatant and suspend the cells in ice-cold 125 μL of PBS containing protease inhibitor cocktail (1:100).
-
11.
Lyse the cells with the sonicator in “high” intensity mode for 6 min (On: 30 s and Off: 60 s) in a sonication bath containing water with crushed ice.
Note: Although our setup, including the sonicator (Bioruptor) and buffer (PBS), typically does not form bubbles, foaming generally affects the cell lysis efficacy and should be avoided.
-
12.
Centrifuge the cell lysate at 400 × g for 5 min at 4°C to remove cellular debris.
-
13.
Collect the supernatant by pipetting into a new 1.5-mL tube.
-
14.
Proceed to the filter trap assay immediately or flash-freeze the lysate with liquid nitrogen and store at –80°C.
Pause point: The lysate can be stored at –80°C.
Filter trap assay
This section describes the method to collect aggregated proteins in the lysate on a filter membrane.
-
15.
Measure the protein concentration in the lysate by Pierce 660-nm Protein Assay Kit with a plate reader according to the manufacturer’s instructions.
Note: The lysate typically contains 1–3 μg/μL of proteins.
-
16.
Prepare the sample mix in a new 1.5-mL tube to load onto a slot-blot filtration apparatus as follows:
| Reagent | Final concentration | Amount |
|---|---|---|
| Total protein in lysate (20 μg) | 0.17 μg/μL | X μL |
| 1× PBS | 0.33× (including PBS from the lysate) | 40 - X μL |
| 1% SDS | 0.67% | 80 μL |
| Total | n/a | 120 μL |
-
17.
Incubate the samples at 25°C for 10 min.
-
18.
Cut the cellulose acetate membrane filter to the same size as the Bio-Dot SF filter paper (11.3 × 7.7 cm).
-
19.
Soak three Bio-Dot SF filter papers and a cut cellulose acetate membrane filter in ∼50 mL of 1% SDS solution for 10 min.
-
20.
Thoroughly clean and dry the slot-blot filtration apparatus (Figure 1).
-
21.
Place the presoaked Bio-Dot SF filter papers on the bottom part of the cassette of the slot-blot apparatus and the presoaked cellulose acetate membrane on top (Figure 1).
Note: Avoid trapping air bubbles between filter paper and the membrane. Use a roller to ensure that there are no trapped bubbles.
-
22.
Assemble the cassette, putting the top part in place, and tighten all the screws.
Note: Screws are tightened in a diagonal manner to equalize the pressure.
-
23.
Apply vacuum and further tighten the screws. Turn off the vacuum.
-
24.
Load 100 μL of 1% SDS buffer into each of the wells and apply vacuum suction until the buffer passes through the filter. Wait until draining is complete.
Note: The well should not be dried before loading the sample. A dried membrane may lead to a nonuniform sample distribution, causing halos during vacuum filtration.
-
25.
Immediately, load 120 μL of the samples containing 20 μg of protein (prepared on step 16) for comparison in each sample slot.
Note: Excessive sample loading may lead to halos around the well and inefficient vacuum suction; hence, the optimum concentration of samples should be used according to the well size of the apparatus. In our setup, we often have good results with 5–60 μg of total proteins.
Note: Load 1% SDS in the empty wells. This is important to ensure uniform vacuum intensity in all the wells.
-
26.
Apply vacuum to drain the lysate to through the membrane filter. Wait until draining is complete and then turn off the vacuum immediately.
Note: Usually, the suction of lysate with 20 μg of protein takes a total of 0.5–2 min. We used relatively mild vacuum conditions.
-
27.
Load 100 μL of 1% SDS solution into the wells and apply vacuum to drain the solution. Wait until the solution is completely drained and then turn off the vacuum immediately to avoid overdrying.
Optional: This wash step may be repeated as necessary when the background signal is too high (see also the troubleshooting section).
-
28.
Remove all the screws of the cassette of the slot-blot apparatus under vacuum. Then, stop applying vacuum. Carefully disassemble the apparatus.
-
29.
Place the membrane on a square petri dish and rinse the membrane with Milli-Q water.
Note: The apparatus should be cleaned properly with detergent and Milli-Q water for the next use.
-
30.
Proceed immediately to the next step as described below.
Figure 1.

Assembly of the filter trapping apparatus
Schematic of the assembly of a cellulose acetate membrane and supporting filter papers in a slot blot cassette. Pre-soak both of the membrane and filter papers with 1% SDS solution. Place the cellulose acetate membrane on top of the filter papers. (Left, side view) Arrange this assembly above the bottom part of the slot-blot cassette (Gasket support plate) of the filter trapping apparatus and clamp this setup to the top part of cassette (sample template) with screws and tighten them well under vacuum connected to the vacuum. (Right, top view) Schematic view of the tightened cassette ready for sample loading into its well for the filter trapping experiment.
Detection of total filter-trapped proteins
This step describes the process for detecting the total proteins trapped on the cellulose acetate filter membrane.
-
31.
Aspirate the Milli-Q water from the dish.
-
32.
Pour 6 mL of Revert 700 Total Protein Stain onto the membrane.
-
33.
Incubate at 20°C–25°C with gentle shaking for 5 min.
-
34.
Aspirate the solution.
-
35.
Pour 6 mL of Revert Wash Solution onto the membrane.
-
36.
Incubate at 20°C–25°C with gentle shaking for 2 min.
-
37.
Repeat steps 34–36.
Note: Wash twice with Revert Wash Solution in total.
-
38.
Aspirate the solution.
-
39.
Pour 20 mL of Milli-Q water onto the membrane.
-
40.
Incubate at 20°C–25°C with gentle shaking for 2 min.
-
41.
Collect the images using an Odyssey CLx Infrared Imaging System with the infrared (IR) 680 channel.
-
42.
Quantify the Revert 700-stained signal in each sample lane using Image Studio software. Figure 2 shows a representative result.
Figure 2.

Representative results for the staining of the total proteins on the filter membrane
(A and B) Staining of the total proteins on the filter membrane. The lysates of HeLa S3 cells obtained after proteasome inhibition by MG132 (5 μM) for 10 h (A) and splicing modulation by SSA (100 ng/mL) for 24 h (B) were used. Titrated amounts of proteins (5, 10, and 20 μg) were tested. Proteins on the membrane were detected by Revert 700 staining. Total protein aggregation at 20 μg protein loading was quantified (right). The average signals detected in the control reagent treatment (DMSO for MG132 and MeOH for SSA) were set to 1. Data from three replicates (points) and the mean (bar) with s.d. (error bar) are shown.
Detection of specific accumulation of aggregated proteins
Cell culture and ectopic expression of epitope-tagged aggregated proteins
This section describes the steps of cell growth and plasmid transfection.
Day 1.
-
43.
Seed 3 × 105 HeLa S3 cells into each well of a 6-well plate using normal growth medium (DMEM supplemented with 10% FBS).
-
44.
Incubate the cells for 24 h at 37°C in a CO2 incubator.
Day 2.
-
45.
Mix the transfection reagents as follows:
| Reagent | Final concentration | Amount |
|---|---|---|
| Opti-MEM | n/a | 96-X μL |
| FuGENE HD | n/a | 4 μL |
| Plasmid: pCDNA5/FRT/TO, pCDNA5/FRT/TO-FTH1, pCDNA5/FRT/TO-FTH1∗, pCDNA5/FRT/TO-p27, pCDNA5/FRT/TO-p27∗, pCAGEN-FLAG-GFP, or pCAGEN-GFP–TDP-43ΔNLS | 0.01 μg/μL | X μL (1 μg) |
| Total | n/a | 100 μL |
Note: Bring the Opti-MEM and FuGENE HD to 20°C–25°C before mixing.
Note: The transfection condition (e.g., the type of transfection reagents, the ratio between the amounts of transfection reagent and plasmid DNA, incubation time, etc.) should be optimized for different cell types, according to the manufacturers’ instructions.
-
46.
Incubate the mixture at 20°C–25°C for 20 min.
-
47.
Add to the cell culture in a dropwise manner.
-
48.
Incubate cells for 48 h at 37°C in a CO2 incubator.
Cell lysis
Day 4.
This section describes the method of cell harvesting and the preparation of cell lysate.
-
49.
Proceed to cell harvesting from step 4 to step 14.
Filter trap assay
This section describes the method to collect aggregated proteins in the lysate on a filter membrane.
-
50.
Proceed to filter trap assay from step 15 to step 30.
Immunodetection of specific filter-trapped proteins
This step describes the process for detecting the specific proteins trapped on the cellulose acetate filter membrane by immunoprobing.
-
51.
Aspirate the Milli-Q water from the dish.
-
52.
Pour 6 mL of Odyssey Blocking Buffer (TBS) onto the membrane in a square petri dish.
-
53.
Incubate at 20°C–25°C with gentle shaking for 1 h.
Note: The membrane stained with Revert 700 Total Protein Stain (from step 31 to step 42) can be used for this assay. Destain the membrane in enough water for 30 min before blocking with Odyssey Blocking Buffer.
Note: The blocking buffer and the concentration of primary and secondary antibodies (see below) should be optimized depending upon epitopes, the imaging system, etc.
Optional: Blocking and antibody incubation could be conducted within plastic hybridization bags.
-
54.
Place 6 μL of primary antibody (anti-FLAG or anti-GFP) and 60 μL of 20% Tween-20 on the membrane in the blocking solution.
-
55.
Incubate at 20°C–25°C with gentle shaking for 1 h.
Optional: The incubation can be performed for 16–20 h at 4°C with gentle shaking.
-
56.
Place the membrane in a new square petri dish.
-
57.
Wash three times with 10 mL of TBS-T for 5 min each.
-
58.
Prepare the secondary antibody solution as follows:
| Reagent | Final concentration | Amount |
|---|---|---|
| 0.1× anti-mouse IgG antibody conjugated to IRDye 800CW or 0.1× anti-rabbit IgG antibody conjugated to IRDye 800CW | 1:10000 | 4 μL |
| Odyssey Blocking Buffer (TBS) | n/a | 4 mL |
| 20% Tween-20 | 0.2% | 40 μL |
| Total | n/a | 4044 μL |
-
59.
Place the membrane to a new square petri dish with 4 mL of the secondary antibody solution.
-
60.
Incubate at 20°C–25°C with gentle shaking for 1 h.
-
61.
Place the membrane to a new square petri dish.
-
62.
Wash three times with 10 mL of TBS-T for 5 min each.
-
63.
Collect images by using an Odyssey CLx Infrared Imaging System with the IR800 channel.
-
64.
Quantify each well of the total aggregated protein samples by using Image Studio software. Figure 3 shows a representative result.
Figure 3.

Representative results for specific aggregated proteins by immunoprobing
(A) Filter trap assay of the indicated proteins expressed in HeLa S3 cells. The tagged proteins were detected by an anti-FLAG antibody.
(B) Standard Western blot showing the levels of FLAG-tagged FTH1 and FTH1∗ proteins. Whole cell lysates were subfractionated by centrifugation into the supernatant and the pellet.
(C) Filter trap assay of the indicated proteins expressed in HeLa S3 cells. The tagged proteins were detected with an anti-GFP antibody.
(D) Filter trap assay of ubiquitinated proteins accumulated in HeLa S3 cells treated with MG132. The ubiquitinated proteins were detected using an anti-ubiquitin antibody.
Expected outcomes
The assessment of aggregated proteins is important for understanding cellular responses to different stresses and physiological conditions. Hence, this protocol provides a method for monitoring the aggregation of either total protein or specific proteins. Because the SDS that is present in sample and on filter membrane may dissolve typical proteins, they should pass through the membrane. Thus, only the aggregation-prone proteins should remain on the membrane.
A disturbance in protein homeostasis is the source of misfolded aggregation-prone proteins in cells, resulting in proteotoxic stress (Maghames et al., 2018). Consistently, we observed global aggregation of proteins upon inhibition of the proteasome by MG132 (Figure 2A) (Wilde et al., 2011). Moreover, as we recently reported, SSA led to the system-wide accumulation of misfolded protein (Figure 2B) because of the translation of exon–intron chimera mRNA generated by splicing modulation (Chhipi-Shrestha et al., 2022). A successful filter trap assay showed clear and distinct signal intensity on the filter membrane for the aggregated proteins. The signal intensity on the membrane was directly correlated with the amount of total protein loaded (5, 10, and 20 μg) and was dependent on the chemical compounds (Figure 2). Total protein loading (5–20 μg) was good for signal quantitation. Notably, this experiment may have a basal level of background signal originating from the natural cellular aggregates and/or other cell debris.
Additionally, this protocol is feasible to investigate individual aggregation-prone proteins, presenting important cellular and pathological implications. For illustration, we investigated aggregation-prone exon–intron chimeric proteins; the chimeric protein of ferritin heavy chain 1 (FTH1∗) led to condensate formation, which was probed using an antibody against the FLAG epitope fused to the protein (Figure 3A) (Chhipi-Shrestha et al., 2022). In contrast, the chimeric-intron protein p27 (or CDKN1B)∗ and full-length FTH1 and p27 did not show this behavior (Figure 3A) (Chhipi-Shrestha et al., 2022). The aggregation-prone nature of FTH1∗ was also validated by its precipitation by centrifugation and the subsequent standard Western blot analysis (Figure 3B) [see a detailed description of the method in (Chhipi-Shrestha et al., 2022)]. This analysis further ensured that the signal on the filter trap membrane originates from the FLAG-tagged FTH1∗ protein, but not from a non-specific signal attributed to the antibody.
Similarly, pathological protein aggregation trapped on the filter was monitored by immunoprobing. Aggregation of Trans-activation response element (TAR) DNA-binding protein of 43 kDa (TDP-43 or TARDBP) often occurs in individuals with amyotrophic lateral sclerosis (ALS), a neurodegenerative disease (Neumann et al., 2006). Its forced localization to the cytoplasm by depletion of nuclear localization signal (NLS) leads to ectopic aggregation. Our experimental setup also detected the aggregation of this pathological protein (GFP–TDP-43ΔNLS) (Figure 3C) (Tsuboyama et al., 2020).
In addition to ectopically expressed proteins, immunoprobing facilitates the detection of endogenous aggregated proteins. As the proteasome inhibition promotes the accumulation of polyubiquitinated proteins, those proteins were trapped on the filter (Figure 3D). This method could be conducted as described in steps 51–64 with 12 μL of an anti-ubiquitin antibody (1:500) at step 54 to detect MG132-treated samples prepared using the procedures described in steps 1–14.
Limitations
Higher protein loading (> 100 μg) negatively impacts the efficiency of the filtration process. Therefore, a limited amount of protein should be used. If loading of a higher protein amount is desired for detection, this method may not be suitable for the study. The limit of loading also varies with cell type, as it depends on total aggregated protein rather than total protein loading. Notably, the maximum limit of loading may vary with the well size of the filter trapping apparatus from different manufacturers. Preliminary optimization with different dilutions of the lysate materials can provide the maximum loading limit for high sensitivity and appropriate technical efficiency.
The sonication equipment (Bioruptor) used for cell lysis may be critical for reproducible experiments. We tested detergent (Triton X-100)-based cell lysis (and the combination with sonication) for the filter trap assay (Figure 4A); however, the tolerance to detergent may depend on the types of protein aggregates. SSA-induced filter-trapped proteins were lost when Triton X-100 was used (Figure 4A, left panel), while pathogenic TDP-43ΔNLS aggregates tolerated the detergent (Figure 4A, right panel). We recommend the use of sonication and avoiding the use of detergent during cell lysis to cover the maximum spectrum of protein aggregates.
Figure 4.

Limitation of the protocol for cell lysis and freeze–thaw cycles
(A) Different cell lysis methods were compared for the filter trap assay. Left panel: Staining of the total proteins on filter membrane. The lysate of HeLa S3 cells obtained after splicing modulation by SSA (100 ng/mL) for 24 h was used. Proteins on the membrane were detected by performing Revert 700 staining. Right panel: Filter trap assay of the indicated proteins expressed in HeLa S3 cells. The tagged proteins were detected using an anti-GFP antibody.
(B) The effect of repeated freeze–thaw cycles was tested. The experiments were conducted as described in A, but the lysate were subjected to freeze–thaw cycles before samples were loaded onto the membrane.
We typically freeze the sample with liquid nitrogen and store the sample at −80°C until use. We tested the effects of freeze–thaw cycles on the experiments (Figure 4B). In our hands, repeated cycles up to 2 may not significantly change the results for SSA-induced total filter-trapped proteins (Figure 4B, left panel) and ectopically expressed GFP–TDP-43ΔNLS (Figure 4B, right panel). However, because the tolerance to freeze–thaw cycles may depend on the type of protein aggregate, the number of repeated cycles should be kept to a minimum.
Troubleshooting
Problem 1
Inefficient filtration of lysate through the filter membrane (in step 26).
Potential solution
The presence of air bubbles may affect the filtration efficiency. Make sure to prevent bubble formation. Additionally, loading more sample than the optimum relative to the well size could be another possible reason. Make sure to use only the optimum sample amount. No leakage should be detected in the vacuum system. Wells without samples should be filled with 1% SDS buffer.
Problem 2
Halo formation or cross-well contamination (visible in steps 42 and 64). See Figure 5 for an example.
Figure 5.
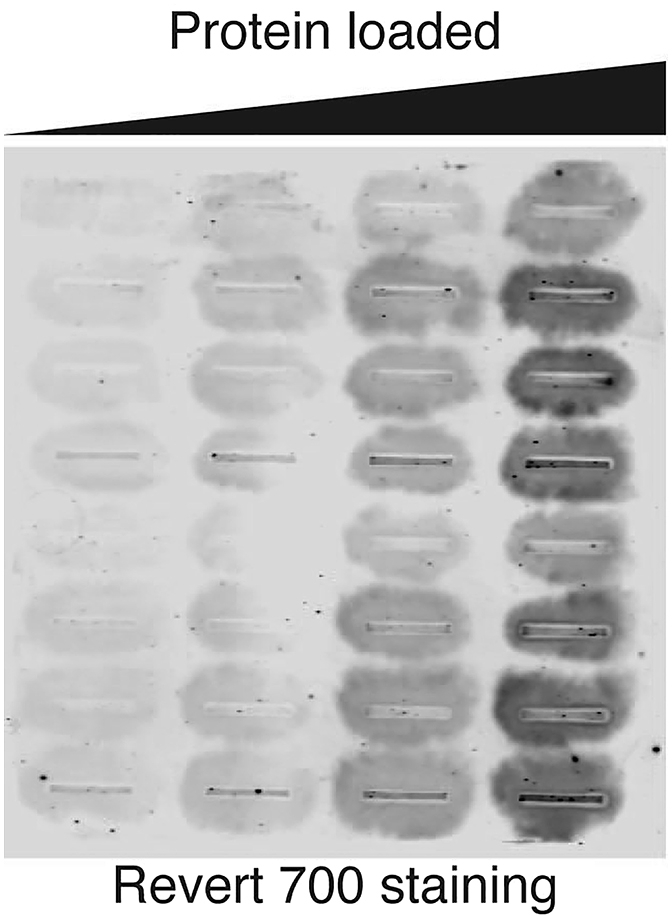
Representative image of Revert 700 staining with halo formation during the filter trap assay
Potential solution
Consider rehydration of the membrane before sample loading. Dried membranes may cause these problems. Turn off the vacuum as soon as all the liquids are drained to prevent overdrying. Alternatively, ensure proper assembly of the cassette. Retightening the screw of the cassette under vacuum should minimize this problem. Moreover, consider optimizing the optimum number and thickness of supporting filter papers required in the cassette.
Problem 3
Weak signal intensity (visible in steps 42 and 64).
Potential solution
Insufficient aggregated proteins or target protein expression will provide a weak signal. Increase the loading of high-concentration protein material, but avoid overloading, as mentioned above.
Problem 4
High background signal (visible in steps 42 and 64).
Potential solution
Consider staining and destaining for proper times in steps 33 and 36. Additionally, the number of washing steps may be adjusted during filter trapping of proteins in step 27. When immunoprobing a specific protein, ensure proper blocking of filter membrane in step 53 and washing after primary and secondary antibody incubations. Conventional blocking reagents such as BSA or skim milk may result in a high background.
Problem 5
Variance in reproducibility (visible in steps 42 and 64).
Potential solution
Ensure proper mixing of the sample before loading to the well of the filter trap apparatus.
Resource availability
Lead contact
Further information and requests for resources and reagents should be directed to and will be fulfilled by the lead contact, Shintaro Iwasaki (shintaro.iwasaki@riken.jp).
Materials availability
This study did not generate new unique reagents.
Acknowledgments
We thank all the members of the Iwasaki and Yoshida laboratories for constructive discussions, technical help, and critical reading of the manuscript. This study used pCAGEN-FLAG-GFP and pCAGEN-GFP–TDP-43ΔNLS plasmids that were kindly provided by Kotaro Tsuboyama and Yukihide Tomari. M.Y. was supported in part by the Grant-in-Aid for Scientific Research (S) (JP19H05640) from the Japan Society for the Promotion of Science (JSPS) and the Grant-in-Aid for Scientific Research on Innovative Areas “Ubiquitin new frontier driven by Chemo-technologies” (JP18H05503) from the Ministry of Education, Culture, Sports, Science and Technology (MEXT). S.I. was supported by the Grant-in-Aid for Transformative Research Areas (B) “Parametric Translation” (JP20H05784) from MEXT, AMED-CREST (JP21gm1410001) from the Japan Agency for Medical Research and Development (AMED), and RIKEN (the Pioneering Project “Biology of Intracellular Environments”). J.K.C.S. was supported by the Grant-in-Aid for Early-Career Scientists (JP20K15420 and JP22K14801) from JSPS.
Author contributions
Conceptualization, J.K.C.S., S.I., and M.Y.; methodology, J.K.C.S, S.I., and M.Y.; formal analysis, J.K.C.S.; investigation, J.K.C.S.; writing – original draft, J.K.C.S. and S.I.; writing – review & editing, J.K.C.S., S.I., and M.Y.; visualization, J.K.C.S.; supervision, S.I. and M.Y.; project administration, S.I.; funding acquisition, J.K.C.S., S.I., and M.Y.
Declaration of interests
The authors declare no competing interests.
Contributor Information
Minoru Yoshida, Email: yoshidam@riken.jp.
Shintaro Iwasaki, Email: shintaro.iwasaki@riken.jp.
Data and code availability
Original images used for the figures are deposited in the Mendeley database (https://doi.org/10.17632/hb8mx8xt2n.1).
References
- Chhipi-Shrestha J.K., Schneider-Poetsch T., Suzuki T., Mito M., Khan K., Dohmae N., Iwasaki S., Yoshida M. Splicing modulators elicit global translational repression by condensate-prone proteins translated from introns. Cell Chem. Biol. 2022;29:259–275.e10. doi: 10.1016/j.chembiol.2021.07.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Farrawell N.E., Lambert-Smith I.A., Warraich S.T., Blair I.P., Saunders D.N., Hatters D.M., Yerbury J.J. Distinct partitioning of ALS associated TDP-43, FUS and SOD1 mutants into cellular inclusions. Sci. Rep. 2015;5:13416. doi: 10.1038/srep13416. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kaida D., Motoyoshi H., Tashiro E., Nojima T., Hagiwara M., Ishigami K., Watanabe H., Kitahara T., Yoshida T., Nakajima H., et al. Spliceostatin A targets SF3b and inhibits both splicing and nuclear retention of pre-mRNA. Nat. Chem. Biol. 2007;3:576–583. doi: 10.1038/nchembio.2007.18. [DOI] [PubMed] [Google Scholar]
- Maghames C.M., Lobato-Gil S., Perrin A., Trauchessec H., Rodriguez M.S., Urbach S., Marin P., Xirodimas D.P. NEDDylation promotes nuclear protein aggregation and protects the Ubiquitin Proteasome System upon proteotoxic stress. Nat. Commun. 2018;9:4376. doi: 10.1038/s41467-018-06365-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Neumann M., Sampathu D.M., Kwong L.K., Truax A.C., Micsenyi M.C., Chou T.T., Bruce J., Schuck T., Grossman M., Clark C.M., et al. Ubiquitinated TDP-43 in frontotemporal lobar degeneration and amyotrophic lateral sclerosis. Science. 2006;314:130–133. doi: 10.1126/science.1134108. [DOI] [PubMed] [Google Scholar]
- Sin O., Mata-Cabana A., Seinstra R.I., Nollen E.A.A. Filter retardation assay for detecting and quantifying polyglutamine aggregates using Caenorhabditis elegans lysates. Bio Protoc. 2018;8:e3042. doi: 10.21769/bioprotoc.3042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tsuboyama K., Osaki T., Matsuura-Suzuki E., Kozuka-Hata H., Okada Y., Oyama M., Ikeuchi Y., Iwasaki S., Tomari Y. A widespread family of heat-resistant obscure (Hero) proteins protect against protein instability and aggregation. PLoS Biol. 2020;18:e3000632. doi: 10.1371/journal.pbio.3000632. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wilde I.B., Brack M., Winget J.M., Mayor T. Proteomic characterization of aggregating proteins after the inhibition of the ubiquitin proteasome system. J. Proteome Res. 2011;10:1062–1072. doi: 10.1021/pr1008543. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
Original images used for the figures are deposited in the Mendeley database (https://doi.org/10.17632/hb8mx8xt2n.1).