

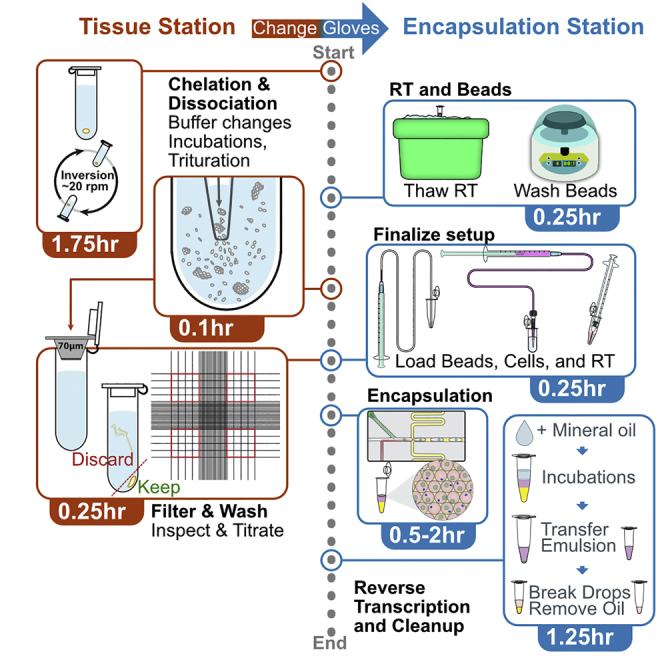
Summary
In droplet-based single-cell RNA-sequencing (scRNA-seq) experiments, cells, along with some of their surrounding buffer and ambient material, are encapsulated into droplets for mRNA capture and barcoding. This protocol details the steps for human gut tissue dissociation using cold active protease, and subsequent isolation of single epithelial cells, with enrichment of viability through washes. Next, the steps for encapsulation on the inDrops scRNA-seq platform are described. This procedure has been demonstrated to be applicable to polyps, cancers, and inflamed tissues.
For complete details on the use and execution of this protocol, please refer to Chen et al. (2021).
Subject areas: Single Cell, RNAseq
Graphical abstract
Highlights
-
•
Dissociation of gut tissues using cold-active protease
-
•
Enrichment for viable single cells
-
•
Setup of inDrops reagents and machine
-
•
Encapsulation of cells, reverse transcription, and cleanup
Publisher’s note: Undertaking any experimental protocol requires adherence to local institutional guidelines for laboratory safety and ethics.
In droplet-based single-cell RNA-sequencing (scRNA-seq) experiments, cells, along with some of their surrounding buffer and ambient material, are encapsulated into droplets for mRNA capture and barcoding. This protocol details the steps for human gut tissue dissociation using cold active protease, and subsequent isolation of single epithelial cells, with enrichment of viability through washes. Next, the steps for encapsulation on the inDrops scRNA-seq platform are described. This procedure has been demonstrated to be applicable to polyps, cancers, and inflamed tissues.
Before you begin
While many clinical research endeavors are conveniently decoupled from the clinic through fixation, freezing, or other forms of preservation, little is known of the effects of variable ischemic storage time on scRNAseq data across large human cohorts, and thus it is generally advisable to minimize and record any variations in tissue storage and processing times.
Institutional permissions
All participants contributing specimens were provided written informed consent approved by the Vanderbilt University Medical Center Institutional Review Board. The user should abide by their institution’s policies with regards to working with human subjects prior to engaging in human studies.
Preparation for tissue dissociation
Timing: Days to hours before experiment
Preparation and storage of dissociation reagents should be kept separate from encapsulation (distinct work areas and pipettes, gloves changed) to prevent carryover of DNase into encapsulation reagents.
-
1.
Ensure that DNase and protease have been hydrated and frozen in aliquots of the proper size for use (see table).
-
2.Reagents should be on-hand, with DPBS stored at 4°C.
-
a.Chelation Buffer should be made day of procedure and kept on ice until tissue arrives.
-
a.
-
3.
Prepare tubes and ice buckets.
Preparation for encapsulation: Early machine setup
This section describes the setup of the first 3 syringe pumps, which can be completed well before the procedure begins, and may be used for multiple experiments with occasional replacement of reagents or syringes as needed.
-
4.Bead oil pump (pushes beads into chip).
-
a.Load a 1 mL Normject syringe with HFE 7500 using a p1000 pipette.
-
i.Using pipettor, fill tip with 1 mL HFE.
-
ii.Dock syringe (open end up) with luer fitting to end of pipette tip (pointing down into syringe).
-
iii.Pull syringe plunger to fill syringe with HFE.
-
i.
-
b.Attach a 27 gauge needle to the syringe luer.
-
c.Attach ∼5 cm of tubing to the needle and, with needle pointing up, prime air out of syringe until HFE enters tubing.
-
d.Invert syringe and ensure that no bubbles are present in the oil within syringe.
-
e.Carefully load syringe onto pump and ensure mechanical fittings around plungers are closed and tightened.
-
f.Engage pump at 2000 μL/h until oil moves in tubing.
-
i.This is to remove any mechanical lag and bubbles from the syringe assembly and will save time later.
-
i.
-
a.
-
5.Droplet making oil pump.
-
a.Load a 1 mL Normject syringe with Droplet stabilizing oil (DSO)using a p1000 pipette.
-
i.Using pipettor, fill tip with 1 mL DSO.
-
ii.Dock syringe (open end up) with luer fitting to end of tip (pointing down into syringe).
-
iii.Pull syringe plunger to fill syringe with DSO.
-
i.
-
b.Attach a 27 gauge needle to the syringe luer.
-
c.Attach ∼20 cm of tubing to the needle and, with needle pointing up, prime air out of syringe until DSO enters tubing.
-
d.Invert syringe and ensure that no bubbles are present.
-
e.Carefully load syringe onto pump and ensure mechanical fittings around plungers are closed and tightened.
-
a.
-
6.Cell Oil pump (pushes cells into chip).
-
a.Load a 1 mL Normject syringe with red mineral oil (RMO) using a p1000 pipette.
-
i.Using pipettor, fill tip with 1 mL RMO.
-
ii.Dock syringe (open end up) with luer fitting to end of tip (pointing down into syringe).
-
iii.Pull syringe plunger to fill syringe with RMO.
-
i.
-
b.Attach luer-to-tip adapter to syringe luer.
-
c.Attach a cell loading tip to the end of the luer-to-tip adapter.
-
d.Prime assembly with mineral oil, ensuring that no air bubbles remain in line.
-
e.Carefully load syringe onto pump and ensure mechanical fittings around plungers are closed and tightened.
-
f.Use a 27ga needle or other sterile luer fitting to cover the end of the luer-to-tip adapter until ready to load cells.
-
a.
Pause point: Machine is ready for run. Reagents currently loaded on machine are typically stable for at least 2 weeks but users should watch for the formation of air bubbles in syringes as they may need to be primed out periodically.
Key resources table
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Chemicals, peptides, and recombinant proteins | ||
| DPBS (no calcium or magnesium) | Corning | 21-031-CV |
| EDTA 0.5 M | Corning | 46-034-CI |
| DTT 1 M | Teknova | D9750 |
| DNASE | Sigma-Aldrich | DN25-100MG |
| Protease from Bacillus licheniformis (Cold Protease) | Sigma-Aldrich | P5380-250MG |
| 10% Bovine Serum Albumin | Sigma-Aldrich | 126615-25ml |
| Optiprep Density Gradient Medium | Sigma-Aldrich | D1556 |
| Ultrapure Distilled Water (DNase, RNase, Free) | Invitrogen | 10977-015 |
| Mineral Oil | Sigma-Aldrich | M5310 |
| Igepal CA-630 | Alfa Aesar | J61055 |
| DNTPs | New England Biolabs | N0447L |
| Maxima H Minus Reverse transcriptase | Thermo Scientific | EP0753 |
| 5× RT buffer (Included with Reverse Transcriptase) | Thermo Scientific | EP0753 |
| Potassium Chloride (KCL) 1 M | Alfa Aesar | J6422 |
| Oil Red O | Alfa Aesar | A12989 |
| Droplet Stabilization Oil | Droplet Genomics | DG-DSO-15 |
| HFE 7500 | Novec | 7500 |
1H, 1H, 2H, 2H-Perfluoro-1-octanol (PFO) 
|
Sigma-Aldrich | 370533-5G |
| Critical commercial assays | ||
| InDrop Photocleavable beads | RAN Biotechnologies | FGB |
| Other | ||
| Tube Revolver Rotator | Thermo Scientific | 88881001 |
| Disposable Hemocytometers | BullDog Bio | DHC-N420 |
| 70 μm PluriStrainer Mini Cell Strainers | PluriSelect | 43-10070-40 |
| 70 μm Flowmi Filters | Bel-Art | H136800070 |
| 100 μL Disposable tip adapter | Hamilton | 31330 |
| Male-to-Male Luer adapter | Qosina | 12090 |
| 27G blunt dispensing needles | CML Supply | 901-27-100L |
| 1 mL 27G Insulin Syringes | Becton Dickinson | 329412 |
| 1 mL Luer syringes | Norm-Ject | NJ-9166017-02 |
| 27G Precision Glide Needles | Becton Dickinson | 305109 |
| 100ft Micro medical Tubing .015″I.D. × 0.043″O.D. | Scientific Commodities | BB31695-PE/2 |
| Cell Barcoding Chip | Droplet Genomics | DG-CBC2-80 |
| Variable Speed Mini Centrifuge | G-Biosciences | BT604 |
| Curved Forceps | FisherBrand | 16-100-110 |
| Razor blades | FisherBrand | 12-640 |
| 2 mL Round bottom tubes | FisherBrand | 14-666-315 |
| 1.5 mL DNA LoBind Tubes | Eppendorf | 022431021 |
| 0.5 mL DNA LoBind Tubes | Eppendorf | 022431005 |
| Cool rack | Corning | 432038 |
| UV Barcode Cleaving Device | Droplet Genomics | DG-BRD |
| Mini Dry Bath | FisherBrand | 14-955-219 |
Materials and equipment
The equipment needed for this dissociation workflow is a tube rotator in a refrigerated environment, an ice bucket to hold samples between incubations, and a dedicated p1000 pipette for buffer changes and trituration. inDrops (Klein et al., 2015) encapsulation can be performed on a microfluidic setup similar to that described in (Zilionis et al., 2017). There are also many off-the-shelf systems available from companies such as Dolomite Microfluidics or Droplet Genomics. Any system that allows a user to independently control the flow of 4 solutions at rates between 10 and 2000 μL/h while visualizing the encapsulation on an inverted microscope using a high-speed camera capable of 10× magnification is sufficient.
Luer-to-tip adapter
This allows the operator to load cells into a 200 μL pipette tip and then push them into the chip using mineral oil. Loading cells in this way can be beneficial when working with cells which are in low numbers or cannot withstand transit in small-bore tubing. The luer-to-tip adapter consists of a section of small bore tubing with a female luer adapter on each end, connected to a 200 μL pipette tip (non-filter) using an adapter. This can be achieved using the standard tubing connected to 27ga needles (Figure 1), or using barbed luer adapters with slightly larger tubing. Tips should be changed between samples, and no more than 150 μL of cells loaded in order to prevent cells from entering the adapter.
Figure 1.
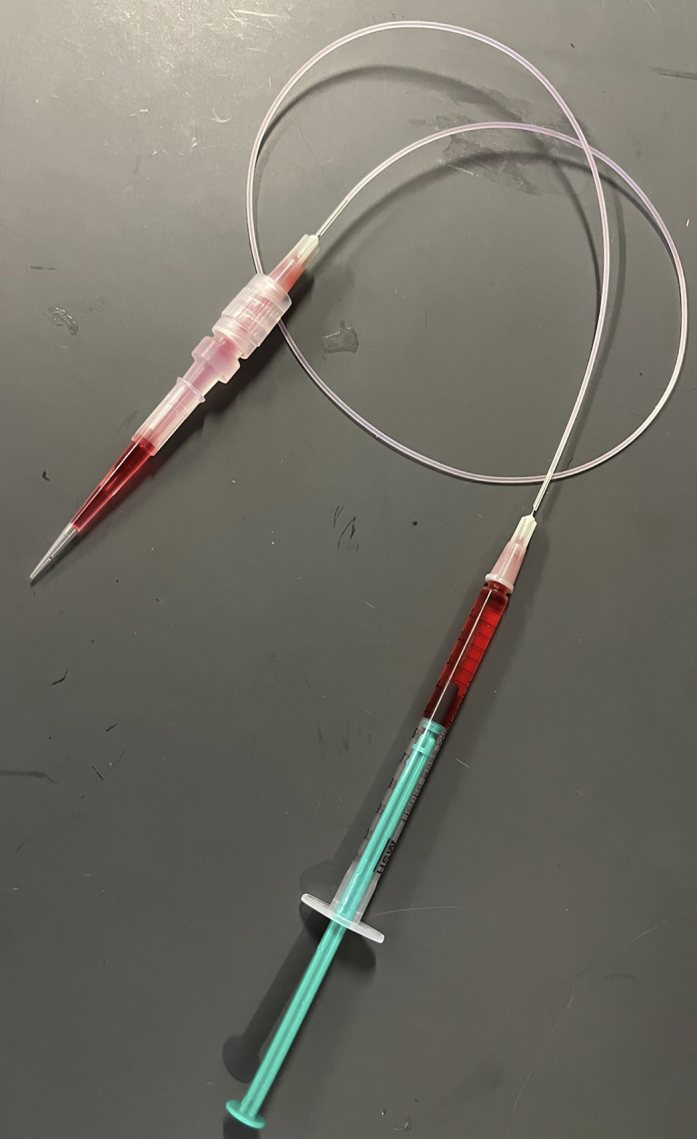
Example luer-to-tip adapter construction
Dissociation reagents
Chelation Buffer
| Reagent | Final concentration | Amount |
|---|---|---|
| 1× DPBS (no ca/mg) | ∼1× | 24.75 mL |
| EDTA (500 mM) | 4 mM | 250 μL |
| DTT (100 mM) | 0.5 mM | 125 μL |
| Total | n/a | 25 mL |
DPBS/EDTA can be mixed up to 6 weeks in advance, but DTT should be added day of use.
2× DNase
| Reagent | Final concentration | Amount |
|---|---|---|
| 1× DPBS (no ca/mg) | – | 20 mL |
| DNase | 5 mg/mL | 100 mg |
| Total | n/a | 20 mL |
Store 1 mL aliquots in 2 mL tubes at −20°C. Good for at least 6 months frozen.
20× Cold Protease
| Reagent | Final concentration | Amount |
|---|---|---|
| Molecular Bio Grade H2O | – | 2.5 mL |
| Cold-Active Protease | 100 mg/mL | 250 mg |
| Total | n/a | 2.5 mL |
Store 100 μL aliquots in 2 mL tubes at −20°C. Good for at least 6 months frozen.
Dissociation Buffer
| Reagent | Final concentration | Amount |
|---|---|---|
| 1× DPBS (no ca/mg) | ∼1× | 0.9 mL |
| DNase (5 mg/mL in DPBS) | 2.5 mg/mL | 1 mL |
| 20× Cold protease | 5 mg/mL | 100 μL |
| Total | n/a | 2 mL |
DNase and Cold Protease are stored at −20°C in aliquots sized for 1 sample each.
Encapsulation reagents
15% Optiprep
| Reagent | Final concentration | Amount |
|---|---|---|
| Molecular bio Grade H2O | – | 7.5 mL |
| 10× DPBS (no ca/mg) | 1× | 1 mL |
| Optiprep | 15% | 1.5 mL |
| Total | n/a | – |
Avoid prolonged light exposure, store at 4°C for up to 2 wks.
RT Premix
| Reagent | Final concentration | Amount |
|---|---|---|
| 5× RT | 1× | 1 mL |
| Molecular Bio Grade H2O | – | 600 μL |
| 10% Igepal | 0.75% | 150 μL |
| (10 mM ea.) DNTP mix | 1.25 mM ea | 250 μL |
| Total | n/a | 2 mL |
Divide into 200 μL aliquots (for ∼45 min runs) in 1.5 mL tubes and store at −20°C until use.
Bead Wash Buffer
| Reagent | Final concentration | Amount |
|---|---|---|
| Molecular Bio Grade H2O | – | 489.9 mL |
| 1 M Tris pH 8 | 10 mM | 5 mL |
| 10% Tween | 0.1% | 5 mL |
| 0.5 M EDTA | 0.1 mM | 100 μL |
| Total | n/a | 500 mL |
Store stock at 4°C and keep working aliquot of 50 mL at 22°C.
2× Gel Concentrating Buffer
| Reagent | Final concentration | Amount |
|---|---|---|
| Molecular Bio Grade H2O | – | 36.2 mL |
| 1 M KCl | 150 mM | 7.5 mL |
| 1 M Tris HCl pH 8 | 100 mM | 5 mL |
| 10% Igepal | 0.2% | 1 mL |
| 1 M MgCl | 6 mM | 300 μL |
| Total | n/a | 50 mL |
Divide into 10 mL aliquots. Keep one at 4°C and store the rest at −20°C.
TET Bead Storage Buffer
| Reagent | Final concentration | Amount |
|---|---|---|
| Molecular Bio Grade H2O | – | 48 mL |
| 0.5 M EDTA | 10 mM | 1 mL |
| 1 M Tris HCl pH 8 | 10 mM | 500 μL |
| 10% Tween | 0.1% | 500 μL |
| Total | n/a | 50 mL |
Store at 4°C. Good for up to 6 months.
Barcoded Gel Bead aliquots
| Reagent | Final concentration | Amount |
|---|---|---|
| Barcoded Gel beads | 20%–40% | 100–200 μL |
| TET Buffer | 1× | 500 μL |
| Total | n/a | ∼700 μL |
Store aliquots at 4°C in a box, protecting from excess light exposure.
20% PFO
| Reagent | Final concentration | Amount |
|---|---|---|
| HFE 7500 | 80% v/v | 8 mL |
| PFO | 20% v/v | 2 mL |
| Total | n/a | 10 mL |
Make 1 mL aliquots and store wrapped in parafilm, good for at least 3 months at 22°C. A working aliquot can be kept on the bench for multiple uses until spent.
CRITICAL: PFO should be made in a chemical or biosafety cabinet to ensure proper ventilation. Wear adequate PPE. Consult SDS for hazards.
Red Mineral oil (use only for cell syringe)
| Reagent | Final concentration | Amount |
|---|---|---|
| Mineral Oil | >99% | 50 mL |
| Oil Red | ∼0.01 mg/mL | ∼0.5 mg |
| Total | n/a | 45 mL |
Color is to allow for visual contrast to cell buffer and should be pink (not quite red).
Store at 22°C, keeping at least three 2 mL working aliquots on hand. Good for at least 6 months.
Step-by-step method details
Tissue: Assessment
This procedure begins with the receipt of tissues, which can vary in size from a biopsy millimeters in length to a resection several centimeters in length. Specimen should be collected immediately into a physiological buffer (typically RPMI) and stored/transported on ice or at 4°C until processed. While we have isolated cells from large tissues stored on RPMI for up to 3 days, it is best practice to minimize the time between collection and processing. Likewise, the timing for each step (including collection, storage, and all processing steps) should be recorded, as timing and other observations gathered during this and subsequent processes can be important pre-analytical factors in accounting for anomalies or bias in the downstream data.
-
1.
Record details about the tissue, such as accession number, time received, size, number of fragments, appearance, storage buffer, and any other sample-specific information which may be written on the tube it is coming from.
Note: It is ideal to standardize the collection of this metadata into a form which is generated for each sample as a physical record of its processing (Figure 2).
Figure 2.

Example dissociation workflow form
Tissue: Transfer/downsizing
This method has been developed for biopsy-sized samples (fragments of a polyp or 1–2 biopsy bites of mucosal surface, no more than 2 mm2 each). If larger tissues are to be processed, then tissues will need to be scaled down to the dissociation. If tissue is in biopsy form, no further action will be needed, other than transferring to a 2 mL tube (step 4).
-
2.Pour off excess storage/transport buffer.
-
a.Upon removing the lid, check for additional tissue in the threads.
-
b.Extra buffer may need to be retained if unused excess tissue is to be stored.
-
a.
-
3.If tissue pieces are larger than 2 mm2 transfer to a shallow weigh boat or dish (with 1–3 mL of buffer for wetting) to cut.
-
a.If total tissue is roughly 4 mm2 or less, cut into pieces ∼2 mm2 or less by bisecting 1–2 times with a razor blade or scalpel.
-
b.If tissue is larger than 4 mm2 then it will need to be scaled down.
-
i.Select 2–3 representative regions from which to cut 1–2 2 mm2 sized pieces.
-
ii.Avoid edges or excessively damaged regions.
-
iii.If a tissue presents with multiple morphologically distinct regions, consider creating individual samples from each region, taking notes on the descriptions for each.
-
i.
-
a.
-
4.
Transfer tissue to a 2 mL tube using forceps, pipette, or careful pouring.
-
5.
Remove any remaining storage buffer, including any debris resulting from cutting tissue, and resuspend in DPBS.
-
6.
Gently remove and replace DPBS if needed to further wash excess storage buffer, blood, mucus, or other material from specimens.
Tissue: Chelation
This step serves to loosen the epithelium by removing calcium from the proteins connecting epithelial cells to ECM and one another. While chelation is often used for separating the epithelium and the stroma into different fractions, biopsy-sized samples provide too little material for reductive processing steps, and so total material is carried forward.
-
7.
Gently remove DPBS and add 1.9 mL of chelation buffer.
-
8.
Incubate at 4°C with rotation/ inversion for 1 h and 15 min.
Figure 3.

Tissue in Chelation buffer
Encapsulation: RT and bead prep
It is crucial to separate this setup process from cell dissociation to prevent contamination of encapsulation reagents. However, timing will dictate that some encapsulation setup will need to take place during tissue prep. For this reason, we make distinctions between tissue/cell prep and encapsulation work areas and their reagents. Gloves should be changed when moving from dissociation to encapsulation work areas, and separate dedicated pipettors used for the respective procedures. With practice, an individual can alternate between both processes, setting up the encapsulation during tissue processing incubations, and have cells proceed immediately from washes to encapsulation.
-
9.
Bring out 200 μL RT Premix and leave on ice to thaw.
-
10.Prepare Beads.
-
a.Spin a Hydrogel bead aliquot at 1000 rcf for 1 min.Note: Beads will be difficult to see, and must be pelleted to gauge their volume. A 100 μL pellet of beads will be adequate for approximately 45 min of encapsulation (Figure 4)
-
b.Remove supernatant down to 500 μL.
-
i.Careful not to disturb or aspirate beads, bead volume should be no higher than 300 μL (1–2 mm below 0.5 mL meniscus).
-
i.
-
c.Add 500 μL of bead wash buffer and briefly vortex.
-
d.Repeat wash twice, leaving beads in 500 μL of wash buffer.Note: Beads will expand in the wash buffer, increasing the size of the pellet.
-
e.Add 500 μL of 2× GCB (beads should now be in 1 mL total) and briefly vortex.
-
f.Place beads on a tube rack in the dark (a drawer).Note: Beads will shrink in the GCB buffer, returning to their original size.Pause point: Beads and RT will be finalized immediately before encapsulation.
-
a.
Figure 4.

Bead pellet in TET buffer
Tissue: Dissociation
During this step the tissue is digested in a cold-active protease (Adam et al., 2017) to release individual cells with gentle pipetting. DNase aliquot should be thawed just before going into this step.
-
11.
Move sample to ice or a cooled rack, and allow tissue to settle.
-
12.
Gently remove chelation buffer and resuspend tissue in DPBS.
-
13.Prepare Dissociation buffer.
-
a.Add 0.9 mL of DPBS to 2× DNase aliquot and mix by pipetting.
-
i.Ensure that DNase thaws before or during this step.
-
i.
-
b.Add 1.9 mL of thawed 1× DNase to 100 μL frozen cold protease aliquot and pipette to thaw and mix.
-
a.
-
14.Gently remove DPBS from tissue and resuspend in Dissociation buffer.
-
a.Remove excess buffer if needed to ensure agitating bubble is present.
-
b.Incubate at 4°C on the rotator for 25 min.
-
a.
-
15.Trituration - Using a p1000 pipette, pipette tissue pieces 10–20 times, as needed to release and disaggregate the epithelium from the tissue (Figure 5).
-
a.The tissue should become softer and release material into the buffer.
-
i.Pipette the tissue to release material.
-
ii.Pipette material in the buffer to disaggregate cells.
-
i.
-
b.If tissue does not immediately enter the tip, it (please specify what “it” is, tissue or tip) can be gently pressed to the bottom of the tube to assist.
-
c.The pipette tip may need to be trimmed to initially allow the tissue to pass – however, in the interest of optimization, it is best to strive for an initial tissue size that permits it to barely fit through the tip at this stage (cut the tissue into smaller pieces next time).
-
i.If a trimmed tip is used, a normal p1000 will still need to be used in order to disaggregate clusters.
-
i.
-
a.
-
16.Place a 70 μm Pluriselect strainer on a clean 2 mL tube, label with sample identifier, and transfer disaggregated cell suspension into filter (Figure 6).
-
a.Transfer ∼500 μL at a time and ensure it passes to prevent overflow.
-
a.
Figure 5.

Tissue after trituration
Figure 6.
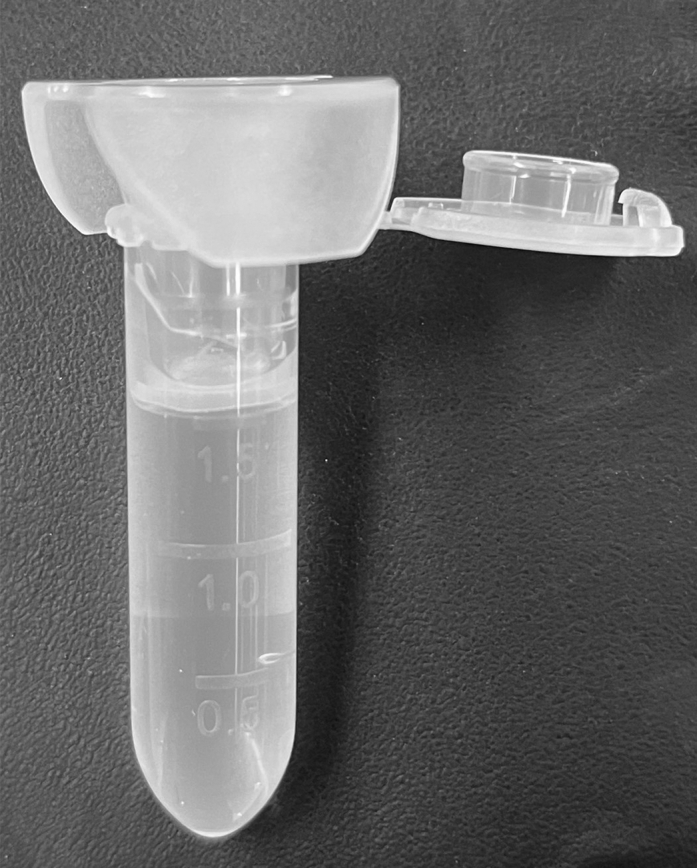
70 μm filtration setup
Tissue: Cell washes
Having achieved a suspension of mostly-viable cells, further success is dependent entirely on the effective washing of cells. The goal is to remove dead cells, debris, and free DNA/RNA, leaving cells in clean DPBS for encapsulation. However, depending on the composition of the suspension, different strategies may be necessary to achieve this.
-
17.
Pellet cells at 600 × g for 5 min at 4°C–6°C.
-
18.Inspect pellet (Figure 7).
-
a.While it is not uncommon for some material to remain suspended in the supernatant, there should always be a visible pellet.
-
b.If no pellet forms, see troubleshooting.
-
a.
-
19.Aspirate supernatant.
-
a.Pellet may be tethered to material in supernatant and dislodge early, leave 100–200 μL of supernatant if necessary to prevent loss of pellet.
-
a.
-
20.Resuspend in DPBS to half previous volume (1 mL for first wash).
-
a.Use tube graduations to estimate this volume (this is faster and easier than volumetric pipetting).
-
b.Gently resuspend cells by pipetting, then mount 5 μL on a disposable hemocytometer.
-
i.We recommend using a p1000 or large bore pipette tip to avoid damaging cells in the loading process.
-
i.
-
a.
-
21.Inspect cells using phase contrast at 10–20× to discern viability (ideally >70% and improving through washes).
-
a.Under phase contrast, viable cells will have a whiter appearance, with a faint “halo” around them.
-
b.Dying cells may still have a halo, but their contents will appear darker and more granular.
-
c.Dead cells will appear dark and may have a bit of chromatin extruding from their membrane, or shrink into a bit of debris (Figure 8).
-
d.Alternative methods for assessing cell quality include trypan blue and automated cell counters that implore fluorescent viability dyes, but visual inspection is preferred as it allows the user to account for debris and quickly move forward through washes.
-
a.
-
22.Count and titrate cells for next wash.
-
a.If (roughly) >25 cells/square (>250,000 cells/ mL) then double the volume of DPBS (same as previous wash).
-
b.If <25 cells/square, keep wash in current volume (reducing wash volume).
-
a.
-
23.
Proceed to wash 2, repeating steps 17–22.
-
24.
Proceed to wash 3, repeating steps 12–19.
-
25.
Resuspend cells in 15% optiprep at ½ previous volume.
-
26.Inspect and count cells.
-
a.If cell concentration is >200,000 cells/mL and cells are in >200 μL then filter using a 70 μm Flowmi:
-
i.Pull at least 100 μL of cells into a p1000 tip.
-
ii.Attach a flowmi filter to the end.
-
iii.Dispense cells through filter into a clean new 2 mL tube.
-
iv.Load and inspect cells on hemocytometer.
-
i.
-
b.Dilute with more 15% optiprep as needed to reach a concentration of ∼100,000 cells/mL.
-
i.If too many cells are in the tube to sufficiently dilute within 2 mL, a portion can be moved to a new tube for further dilution.
-
i.
-
a.
Figure 7.

Example of a large cell pellet
Figure 8.

Good (left) and poor (right) quality cells under phase contrast microscopy
Encapsulation: Encapsulation of cells
The second leg of this procedure will be encapsulation using microfluidics (Figure 9), which immediately follows cell washes. Cold cell isolation procedures can help preserve cell viability throughout dissociation, but many cells may be prone to death thereafter. Any delay in loading cells may decrease data quality. The aliquot and working volumes described in this protocol will allow for ∼45 min of encapsulation time, or 2–4 samples. This working time can be scaled up or down with changes to reagent volumes. However, the RT may lose activity when running for longer than 2 h. This can be mitigated by keeping the solution cool on the machine with a small ice pack that contacts the syringe without obstructing the pump. For each sample that is ran, the operator will need to use a fresh microfluidic device and p200 for cell loading. Once sample and reagents are properly flowing into the device, observations will need to be recorded in order to calculate the duration of collection required to collect 3,000–4,000 cells. To better organize these details, it is recommended to record encapsulation details in a form that standardizes the process (Figure 10).
-
27.
Place cell tube in chilled cool rack on the inDrops station’s microscope stage.
-
28.Begin Cell loading:
-
a.Remove primed cell loading tip from mineral oil.
-
b.Wipe the tip of excess oil with lens paper.
-
c.Ensure that no air bubbles are in the tip.
-
d.Place primed cell loading tip into cell tube, ensuring that tip is submerged and contacting the bottom of the tube.
-
e.Begin aspiration for the cell syringe at 3000 μL/h.CRITICAL: Do not allow tip to invert going forward, such that mineral oil will remain above the more dense cell solution.
-
f.Monitor cell loading (no longer than 6 min/200 μL) – this can be done while skipping ahead and finishing RT and Beads.
-
g.When 100–200 μL of cells have been loaded, reverse the pump and allow 20–30 s for the pump to catch up and cells to begin injecting.
-
h.Stop the cell pump. Cells are primed and ready. The cell loading tip can remain in cell tube until encapsulation begins.
-
a.
-
29.Load BHM.
-
a.Spin BHM at 1K × g for 1 min and remove most of the excess buffer using a p1000.
-
i.Leave 2–5 mm above pellet to avoid aspirating beads.
-
i.
-
b.Spin BHM at 1K × g for 1 min again and remove excess buffer using a p200.
-
c.Cut a 40 cm length of tubing and attach to the end of an insulin syringe.
-
d.Carefully hold the other end of the tubing at the bottom of the bead tube and slowly pull the syringe plunger to load beads into tubing (Figure 11).
-
i.The goal is to load 25–35 cm of beads into tubing with no bubbles.
-
ii.Monitor the bead’s position in the tubing, to ensure that beads do not overfill tubing and enter syringe.
-
iii.Monitor the tubing’s position in the bead tube, ensuring that bubbles do not enter the tubing.
-
i.
-
e.When beads have sufficiently filled tubing (or there is no more to load without bubbles) remove the tubing from bead tube and the needle and dispose of the syringe.Note: It is okay and in fact helpful to have some bubbles (no longer than ∼5 mm) at the very end of the tubing from which beads are loaded. This will serve as a visual indicator of the last beads which will eventually load into the chip.
-
f.Remove the tubing attached to the Bead oil syringe on pump.
-
g.Attach the end of the bead tubing that was in the bead tube to the needle/syringe on pump.
-
h.Trim the other (potentially damaged) end of the bead tubing that was on the needle used to load beads.
-
i.If beads are not fully in the end of the tubing, they can be primed using the pump at 500 μL/h, or the tubing can be trimmed back (as long as it is long enough to reach the chip with 2–3 extra cm for stage movement).
-
j.Any beads which were not loaded into tubing can be resuspended in TET buffer and returned to 4°C storage for later use.
-
k.Beads should now be primed and ready.
-
a.
-
30.Finalize and load RT:
-
a.To RT solution, add 25 μL of RNaseout! and mix by pipetting.
-
b.To RT solution, add 25 μL of Maxima Hminus RTase and mix by pipetting.CRITICAL: Be careful not to introduce bubbles while pipetting. If significant bubbles are formed, RT solution can be briefly centrifuged to remove them.
-
c.Load RT solution into chilled insulin syringe with a single pull of the plunger, careful not to pull in excess air.CRITICAL: Moving the plunger back and forth can slough syringe lubricant into the solution, so excessive aspiration should be avoided.
-
d.Attach 20 cm of tubing to the needle and aspirate any air bubbles followed by 1–10 cm of RT solution into tubing.
-
e.Invert syringe and attach to pump along with cooling tube.
-
f.Tube can be trimmed as needed to minimize excess length, just make sure to allow a few extra cm for stage movement.
-
g.Using pump control software, prime RT at 2000 μL/h until it reaches the end of tube and stop. RT is now primed and ready.
-
a.
-
31.
Ensure that the Droplet oil is primed to the end of its tubing by running the pump at 2000 μL/h. All reagents should now be ready for encapsulation.
-
32.
Place a cell barcoding chip on the stage and peel away the covering for a device.
-
33.
Carefully insert the cell loading tip into the Cell port.
-
34.
Using curved forceps, insert tubes for RT, Beads, and Oil into their respective ports.
-
35.Make sure that pumps are set for aspiration at the following rates:
-
a.Beads at 100 μL/h.
-
b.Droplet Oil at 400 μL/h.
-
c.Cells at 200 μL/h.
-
d.RT at 200 μL/h.
-
a.
-
36.Position microscope camera field of view over the droplet formation channel and start pumps.
-
a.Solutions should begin to enter chip, ideally oil first.
-
a.
-
37.Cut a 7 cm length of tubing and insert into the droplet outlet of the device. Direct the other end of the tube into a clean tube on the cold rack.
-
a.This tube will be for collecting emulsion prior to optimization of droplets, and thus will not be good for data generation. It can, however, be used for practice for later steps.
-
a.
-
38.Continue monitoring the solutions as they enter the chip, and displace air.
-
a.Ensure that all solutions move unobstructed in the chip.
-
b.Air will form bubbles with wide, dark borders in the oil.
-
c.As aqueous solutions begin replacing air, the droplets will have a finer interface with the oil (Figure 12A).
-
d.Watch for beads entering the chip and appearing in droplets (Figure 12B).
-
a.
-
39.As beads begin loading into droplets, monitor their entrance and decrease the flow rate as they become more compact (suggest but not require adding images or words to define compact. For example, is Figure 12B just right, too compact, too spaced out, etc?).
-
a.If >20% of the bead inlet still contains air, remove the bubble by pressing gently above it.CRITICAL: Remove pressure very slowly so as not to pull cells and RT into the bead channel.
-
b.Lower flow rate to 50 μL/h when beads have filled the inlet.
-
a.
-
40.Monitor Droplet size and shape.
-
a.Ensure that droplets are symmetrical and consistent.
-
b.Increase oil in increments of 10 μL/h if droplet length is greater than twice their width.
-
c.Increase oil if droplets elongate or drag to one side of the chip.
-
a.
-
41.Monitor bead loading.
-
a.Using the camera set to “strobe” watch droplets for doublets and estimate the fraction of droplets containing beads (# of total droplets / droplets with a bead).
-
b.Increase or decrease Bead flow rate in increments of 5 μL/h to adjust bead loading.
-
c.This will need to be monitored and may need to be adjusted throughout run.
-
a.
-
42.Check cell loading.
-
a.Using the camera set to “high speed” watch cells entering the chip.
-
b.Record a 10 s video of cells entering the chip.
-
a.
-
43.If all solutions are entering chip, and droplet formation and bead loading are adequate (No fluctuation in drop size and no doublets, >50% of droplets contain beads) for at least 1 min then collection can begin.
-
a.Route output tube into a labeled collection tube.
-
b.Start a count-up timer and record start time.
-
a.
-
44.If everything continues to remain stable, play cell video and count cells entering the chip.
-
a.Lower frame rate to make this easier if needed.
-
b.If counting takes too long, video may need to be paused to check on encapsulation and make sure droplets are still forming adequately.
-
c.(Cells counted for 10 s video / 10) = cells per sec.
-
a.
-
45.Calculate the collection duration needed to capture the desired number of cells. We typically aim for 3500 cells as described below.
-
a.Watch the droplet channel in slow motion, and/or pausing to estimate the average fraction of droplets containing beads.
-
i.i.e., If 8 drops are visible and 6 contain beads, then 6/8=0.75.
-
i.
-
b.3500 cells / (Bead loading ∗ Cells/second∗ 60 s/min).
-
c.Ideal fraction collection time is 5–10 min. Above or below this and the library volume may be more difficult and less efficient to process.
-
a.
-
46.Determine how many fractions to collect.
-
a.Consider how many other samples need to be ran, if a fraction takes longer than 10 min it may be best to collect a single fraction and move on.
-
a.
-
47.Continue monitoring the encapsulation, maintaining consistent bead loading and droplet formation until a 3500 cell fraction is collected.
-
a.If another fraction is to be collected, route the end of the tubing into a new tube.
-
b.If collection for the sample is complete, stop all pumps and disconnect tubes from chip.
-
i.Discard cell tip with remaining cells and mineral oil.
-
ii.Cover end of cell/mineral oil tubing with a capped needle if done, or another cell tip if loading additional samples.
-
i.
-
c.Move the collected emulsion tube to ice for up to ten minutes (Sample should proceed to UV cleaving and RT start as soon as timing permits).
-
a.
Figure 9.

Schematic of an example encapsulation microfluidic chip
Figure 10.

Example Encapsulation workflow form
Figure 11.
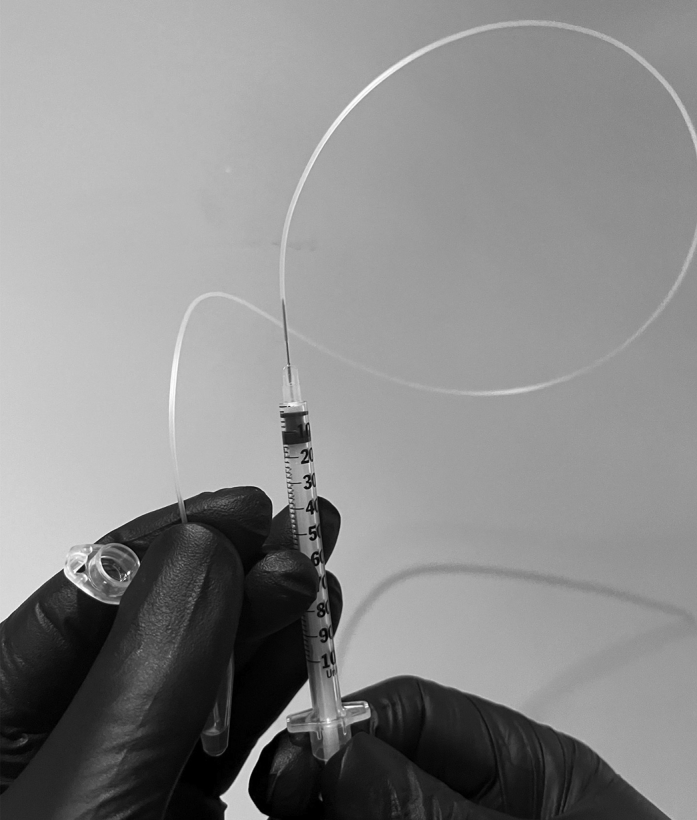
Bead loading procedure
Figure 12.

Droplet channel
(A and B) Examples of air bubbles (A) and aqueous drops loaded with beads (B).
Encapsulation: RT and cleanup
-
48.Collected emulsions can sit on ice a few minutes if additional samples need to be loaded, etc. but ideally should move on to UV cleavage and reverse transcription within 10 min of collection.
-
a.Move collected emulsion to UV barcode cleaver and activate.
-
b.Carefully pipette ∼500 μL of mineral oil (clear) around the sides of the tube, such that the mineral oil covers the emulsion without significantly disturbing it (Figure 13A).
-
i.This is crucial to prevent excessive evaporation of material in droplets.
-
i.
-
c.Move collected emulsion to UV cleaver and activate for a second exposure.
-
a.
-
49.
Move the emulsion to the 50°C heat block and incubate for 1 h.
-
50.
After 1 h, move the tube to 85°C for 5 min to inactivate enzymes.
-
51.
Move tube to ice and allow to cool for 5 min before starting emulsion processing.
-
52.
Label a 0.5 mL LoBind tube for each sample.
-
53.When a sample is cooled, begin by removing and discarding mineral oil from above the emulsion (Figure 13B).
-
a.Take care not to aspirate the emulsion.
-
b.Start with a p1000 to remove most, then follow up with a p200 to remove more.
-
c.Removing 100% of mineral oil is next to impossible, try to remove enough that the layer breaks and the emulsion and HFE oil can be accessed.
-
a.
-
54.Using a clean new tip, slowly remove excess HFE oil from below the emulsion, then remove any additional mineral oil from above the emulsion (Figure 13C).
-
a.As the emulsion collects at the bottom of the tube, remaining mineral oil may become more apparent.
-
b.Rotate the tube to find “pockets” of mineral oil above emulsion and remove them if possible.
-
c.Gel loading tips may assist in this process.
-
a.
-
55.Using a clean new p200 tip, carefully transfer emulsion to a clean, labeled 0.5 mL tube (Figure 13D).
-
a.Take care to leave remaining mineral oil behind.
-
a.
-
56.Add 40 μL of 20% PFO to the emulsion to break droplets.
-
a.Emulsion will become clear within 30 s as aqueous phase consolidates above the HFE oil.
-
b.Emulsions collected longer than 10 min may require a second 40 μL to resolve.
-
a.
-
57.Spin briefly (∼10 s) on minicentrifuge (with 0.5 mL adapter).
-
a.Solution should have 2 distinct phases with no emulsion remaining at interface (Figure 13E).
-
i.Beads may be visible at interface.
-
i.
-
a.
-
58.
Using a p200, carefully remove and discard the HFE from beneath the aqueous phase.
-
59.
Move tube containing aqueous phase (barcoded cDNA) to −80°C for storage up to 6 months (Figure 13F).
Figure 13.

Emulsion cleanup
(A–E) Progress of the emulsion cleanup from (A) to (E).
Expected outcomes
For an average sized biopsy sample (roughly 2 mm2), we typically get between 20,000–200,000 viable cells after washes. Depending on the sample, these cells may remain stable on ice for hours, or die within minutes. While cell death during encapsulation is normal, too much can quickly ruin a run by clogging the cell channel, releasing debris resulting in diminished wetting/droplet integrity, or by simply contributing too much ambient RNA contaminant to all droplets that results in excessive background noise in data.
Limitations
As previously mentioned, this dissociation protocol is most useful when applied to biopsy-sized pieces of tissue, as it is intended to minimize death in samples which are already limiting in material. Too much tissue will result in insufficient dissociation, saturation of filters, and difficulty pelleting. Because the cold protease enzyme primarily aids in the separation of epithelial cells from one another, the protocol does not yield high numbers of non-epithelial cells.
Troubleshooting
Most issues with tissue dissociation will occur (or become apparent) during the cell washes. As epithelial cells die, they tend to release chromatin that can entangle other cells and leave them floating in large precipitations. Checking cells on a hemocytometer between washes is necessary to anticipate and troubleshoot such issues.
In an ideal encapsulation, solutions will enter the chip and form consistent droplets, with drop size and bead loading reliably controlled by adjusting bead and oil flow rates. In reality, a number of small variables can contribute to significant fluctuations in consistency. As every second counts, the ability to quickly diagnose and resolve issues is crucial to a successful encapsulation. Keep in mind that any time droplets are not of the desired composition, quality or consistency, the collection tubing should be routed into a waste tube to prevent the collection of stochastic droplets until issues are resolved.
Problem 1
Tissue does not dissociate sufficiently during step 15 (solution does not become cloudy and/or contains less than 100,000 cells total yield) after 25 min incubation and up to 2 min of trituration.
Potential solution
Assess the appearance of the tissue: Has it changed?
If the tissue is still relatively intact, continue incubation up to 10 additional minutes and try again.
If the tissue is stringy and has come apart (but the supernatant is clear of chunks and doesn’t contain many cells) then carefully remove the tissue remnant and proceed directly to pelleting (do not filter) and perform subsequent resuspension in 500 μL or less. Adding BSA at 0.05% into DPBS washes may be necessary to retain low numbers of cells.
Problem 2
No cell pellet forms during cell washes after centrifugation, but cell number is high >100K/mL, or pellet forms but does not stay on bottom of tube during washes.
Potential solution
Total material (cells + debris) may be too high, ensure that cells are resuspended and washed at no higher than ∼500,000 cells per mL. Moving half of the cells to a new tube and adding more PBS can be an effective means of dilution, as it leaves half of the cells as a backup. Also make sure that optiprep is not used during washes (only for final resuspension) as it adjusts the density of the buffer and will make cells more difficult to effectively pellet.
Problem 3
No cell pellet forms during cell washes after centrifugation, cell number is low (<100,000 cells/mL).
Potential solution
Low numbers of cells may be more difficult to pellet in DPBS as the protein level diminishes. Clean (DNase/RNase free) BSA can be spiked directly into a suspension of cells and used in subsequent DPBS washes at 0.05% final to assist with pelleting.
Problem 4
Debris or fibers in chip during encapsulation.
Potential solution
It is not uncommon for tiny fibers to end up within the chip during encapsulation, typically in the bead channel. While these are often inconsequential, they can be problematic if they obstruct the bead or droplet formation juncture. Taking measures to prevent the introduction of dust and fibers are key to avoiding these issues. Ensure that ends of tubing are free of dust when plugged into chip. Keep workstation clean and free of dust. Avoid wearing garments that shed fibers (especially at the sleeves). When opening syringes and needles, do not pop them through the paper, as this can introduce microscopic fibers; peel them open.
Problem 5
Clog in bead channel during encapsulation.
Potential solution
Sometimes material may lodge in the bead loading inlet. If bead loading is not significantly impaired it may be okay to continue, but if beads are blocked or loading becomes stochastic (drops containing either several beads or none at all) then intervention may be necessary. Try gently pressing on the chip just above the obstruction, while routing the collection tube into waste. If the clog is not resolved, stop all flow and start over with a new chip.
Problem 6
Clog in cell channel during encapsulation.
Potential solution
Clogs in the cell channel are usually due to large debris, cell clusters, or the accumulation of dead cells in the inlet. The first two of these issues can and should be resolved through optimization of the washes and filtration prior to loading. Dead cells and other debris accumulating in the inlet is generally permissible if it does not completely clog the channel, however, if the cell channel becomes completely clogged, droplet size will shrink and at the cell/RT juncture it will be apparent that the cell solution is less than 50% (Figure 14). The collection tubing outlet should be routed to waste, and gentle pressure above the cell inlet can help facilitate clearing. Allow time for the clog to pass into the waste and droplets to stabilize before resuming collection. In general, the rapid accumulation of dead cells (before collection of a single fraction) does not bode well for data quality.
Figure 14.

Visual inspection of increasing dead cell accumulation in chip from left to right
Problem 7
Large droplets or elongation of droplets upon formation during encapsulation.
Potential solution
Droplet size is mainly influenced by the amount of oil relative to aqueous solution, and the degree of wetting (hydrophilicity) inside the droplet channel. As aqueous solutions interact with the droplet channel, protein and other material can cause the inner PDMS walls to become more hydrophilic, resulting in asymmetrical droplets that drag against the walls of the channel. Most issues with droplet size or stability can be resolved by increasing the droplet oil flow rate, which immediately provides more oil relative to aqueous solution and decreases the degree of aqueous interaction with the droplet channel surfaces. Occasionally, downstream resistance will contribute to a delay in droplet shrinkage even with an increase in the oil flow rate. In this case, gently moving or trimming the outlet tubing can help spur droplets to become smaller. Recurring wetting issues that don’t resolve when droplet oil flow is increased may be attributable to excess debris or protein in cell solution, loss of hydrophobic coating in chip, or reduced surfactant in oil, each of which can be isolated and identified through the use of new reagents or a cleaner cell suspension.
Problem 8
Fluctuations or irregularity during encapsulation in droplet formation, droplet size, or bead loading with no visible clog.
Potential solution
Fluctuations in the flow rate of any solution can lend to stochastic droplets or bead loading, and the first step to solving this is to identify the solution that is not flowing consistently. Examine each solution as it interfaces with others. Aberrations in cell and bead loading are easy to identify as brief pauses or lurches in loading. RT and cell fluctuations can be spotted at the RT/Cell interface, which should visibly remain at 1:1 (Figure 12). Oil fluctuations will appear as variations in the droplet formation interface. Make sure that tubes are well connected into the chip and no solutions are leaking out. Fluctuations not caused by objects or air in the chip are typically due to an air bubble upstream in one of the syringes. Occasionally, multiple bubbles in separate syringes can even culminate to create these issues. While some air in the syringe is permissible, it should be kept to a minimum to prevent or mitigate potential stochasticity.
Resource availability
Lead contact
Further information and requests for resources and reagents should be directed to and will be fulfilled by the lead contact, Ken S. Lau (ken.s.lau@vanderbilt.edu).
Materials availability
This study did not generate new unique reagents.
Acknowledgments
This work is funded by R01DK103831 (NIDDK), P50CA236733 (NCI), and U2CCA233291 (NCI). The funders played no role in the research conducted. The authors wish to thank other contributing investigators, including Austin Southard-Smith, Yanwen Xu, Cherie’ Scurrah, Thomas Wise, Qi Liu, Robert Coffey, Martha Shrubsole, Michael Brenan, Colin Brenan, and the rest of the Vanderbilt COLON MAP team. We would also like to acknowledge the VANTAGE Core at Vanderbilt for sequencing services.
Author contributions
A.J.S. developed the protocol. K.S.L. supervised the research and obtained funding. Both authors contributed to writing the manuscript.
Declaration of interests
The authors declare no competing interests.
Contributor Information
Alan J. Simmons, Email: alan.j.simmons@vanderbilt.edu.
Ken S. Lau, Email: ken.s.lau@vanderbilt.edu.
Data and code availability
This study did not generate datasets or code.
References
- Adam M., Potter A.S., Potter S.S. Psychrophilic proteases dramatically reduce single-cell RNA-seq artifacts: a molecular atlas of kidney development. Development. 2017;144:3625–3632. doi: 10.1242/dev.151142. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen B., Scurrah C.R., McKinley E.T., Simmons A.J., Ramirez-Solano M.A., Zhu X., Markham N.O., Heiser C.N., Vega P.N., Rolong A., et al. Differential pre-malignant programs and microenvironment chart distinct paths to malignancy in human colorectal polyps. Cell. 2021;184:6262–6280.e26. doi: 10.1016/j.cell.2021.11.031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Klein A.M., Mazutis L., Akartuna I., Tallapragada N., Veres A., Li V., Peshkin L., Weitz D., Kirschner M., Zilionis R., et al. Droplet barcoding for single-cell transcriptomics applied to embryonic stem cells. Cell. 2015;161:1187–1201. doi: 10.1016/j.cell.2015.04.044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Southard-Smith A.N., Simmons A.J., Chen B., Jones A.L., Ramirez Solano M.A., Vega P.N., Scurrah C.R., Zhao Y., Brenan M.J., Xuan J., et al. Dual indexed library design enables compatibility of in-Drop single-cell RNA-sequencing with exAMP chemistry sequencing platforms. BMC Genom. 2020;21:456. doi: 10.1186/s12864-020-06843-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zilionis R., Nainys J., Veres A., Savova V., Zemmour D., Klein A.M., Mazutis L. Single-cell barcoding and sequencing using droplet microfluidics. Nat. Protoc. 2017;12:44–73. doi: 10.1038/nprot.2016.154. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
This study did not generate datasets or code.