

Abstract
Activation of inert molecules like CO2 is often mediated by cooperative chemistry between two reactive sites within a catalytic assembly, the most common form of which is Lewis acid/base bifunctionality observed in both natural metalloenzymes and synthetic systems. Here, we disclose a heterobinuclear complex with an Al-Fe bond that instead activates CO2 and other substrates through cooperative behavior of two radical intermediates. The complex Ldipp(Me)Al-Fp (2, Ldipp = HC{(CMe)(2,6-iPr2C6H3N)}2, Fp = FeCp(CO)2) was found to insert CO2 and cyclohexene oxide, producing LdippAl(Me)(μ:κ2-O2C)Fp (3) and LdippAl(Me)(μ-OC6H10)Fp (4), respectively. Detailed mechanistic studies indicate unusual pathways in which (i) the Al-Fe bond dissociates homolytically to generate formally AlII and FeI metalloradicals, then (ii) the metalloradicals add to substrate in a pairwise fashion initiated by O-coordination to Al. The accessibility of this unusual mechanism is aided, in part, by the redox non-innocent nature of Ldipp that stabilizes the formally AlII intermediates, instead giving them predominantly AlIII-like physical character. The redox non-innocent nature of the radical intermediates was elucidated through direct observation of LdippAl(Me)(OCPh2) (22), a metalloradical species generated by addition of benzophenone to 2. Complex 22 was characterized by X-band EPR, Q-band EPR, and ENDOR spectroscopies as well as computational modeling. The “radical pair” pathway represents an unprecedented mechanism for CO2 activation.
Graphical Abstract

INTRODUCTION
Identifying new reaction pathways for activation of inert substrates enables rational design of catalysts for challenging synthetic transformations. Although historically the activation of inert substrates by coordination complexes has focused on unsaturated mononuclear systems with single-site reactivity, recently there has been a resurgence in cooperative bond activation studies in which reactivity is delocalized over two or more reactive sites within a well-defined system. Examples include frustrated Lewis pairs (FLPs),1 metal-ligand bifunctional catalysts,2 homo- and heterobinuclear metal complexes,3 and multinuclear metal clusters.4 As a representative example, one can consider the coordination chemistry of CO2. While CO2 activation by mononuclear metal complexes has been mapped extensively,5 remaining challenges in catalytic CO2 fixation have motivated numerous studies on cooperative CO2 activation.6 The dominant paradigm is one of Lewis acid/base bifunctionality (Figure 1a): a Lewis basic reactive site adds a reactive electron pair to the carbon center of CO2, while the resultant buildup of negative charge on oxygen is simultaneously stabilized by a Lewis acidic reactive site.7 Since the seminal work of Floriani on cooperative CO2 activation by low-valent Co/M bifunctional systems (M = alkali metal),8 numerous examples of metal- and non-metal-based systems functioning by this paradigm have been identified.9 Moreover, Lewis acid/base bifunctionality is thought to be the operative paradigm in nature, stabilizing CO2-activated intermediates in both aerobic (Mo/Cu) and anaerobic (Ni/Fe) carbon monoxide dehydrogenase enzymes.10 Identifying reaction pathways beyond this prevailing CO2 activation manifold has the potential to open new catalyst design strategies.11
Figure 1.

Cooperative CO2 activation pathways and representative examples: (a) Lewis acid/base bifunctionality, (b) radical pair chemistry (this work).
In this report, we disclose a system that activates CO2 and other substrates by a radical pair mechanism, which is a novel mechanistic paradigm for cooperative CO2 reactivity (Figure 1b). In our discovered system, a heterobinuclear complex dissociates homolytically to generate two metalloradical intermediates, which then cooperatively activate CO2 as a radical pair that donates one electron per metal. In part due to the unfavorable one-electron reduction potential of CO2 (E° = −1.9 V at pH 7), activation of CO2 by metalloradical pairs is quite rare. In the cases that have been observed, generation of high-energy radical pairs required photochemical or electrochemical activation of a precursor complex.12a The concept of frustrated radical pairs has recently emerged in the field of FLP chemistry,12b–d but typically such systems are limited to activation of relatively weak bonds (e.g. H-SnR3, RO-OR) or substrates possessing radical character of their own (e.g. O2, TEMPO). On the contrary, here the heterobimetallic radical pair is capable of cooperatively activating not only CO2 but also the C-O bond of an epoxide under ambient conditions with no external stimulus.
As detailed below, our discovery emerged from studies of an Al-Fe heterobinuclear system. Despite extensive recent work on heterobimetallic chemistry,13 heterobimetallic complexes bearing aluminum (Earth’s most abundant metal) are not thoroughly studied and their reactivity remains underexplored,14 especially for heterobimetallic complexes in which aluminum is paired with another earth-abundant metal.15–20 Only a few examples of cooperative CO2 activation by Al/M heterobinuclear complexes have been reported, with their mechanistic pathways currently under debate in some cases. In 2019, Goicoechea and Aldridge reported CO2 insertion into an Al-Au bond (Figure 2a).21 A cooperative Lewis acid/base mechanism involving nucleophilic auride (i.e., AlIII-Au−I) was proposed initially, although recent theoretical analysis by Sorbelli et al. disfavors that proposal and instead supports diradical-like reactivity (i.e., AlII-Au0).22 In 2021, Frenking, Goicoechea, and Aldridge disclosed analogous reactivity of the Al-Cu and Al-Ag derivatives (Figure 2a), again proposing Lewis acid/base mechanisms.23 A closely related set of Al-Cu reactions with CO2 were reported by Hill and McMullin in 2021 (Figure 2b).18 In 2021, Goicoechea and Aldridge reported CO2 insertion into an Al-Zn bond (Figure 2c), although no mechanistic studies were reported.24 Finally, in 2021, Camp reported CO2 deoxygenation by a AlIII-IrIII complex that was proposed to go by a concerted transition state (Figure 2d).9e
Figure 2.
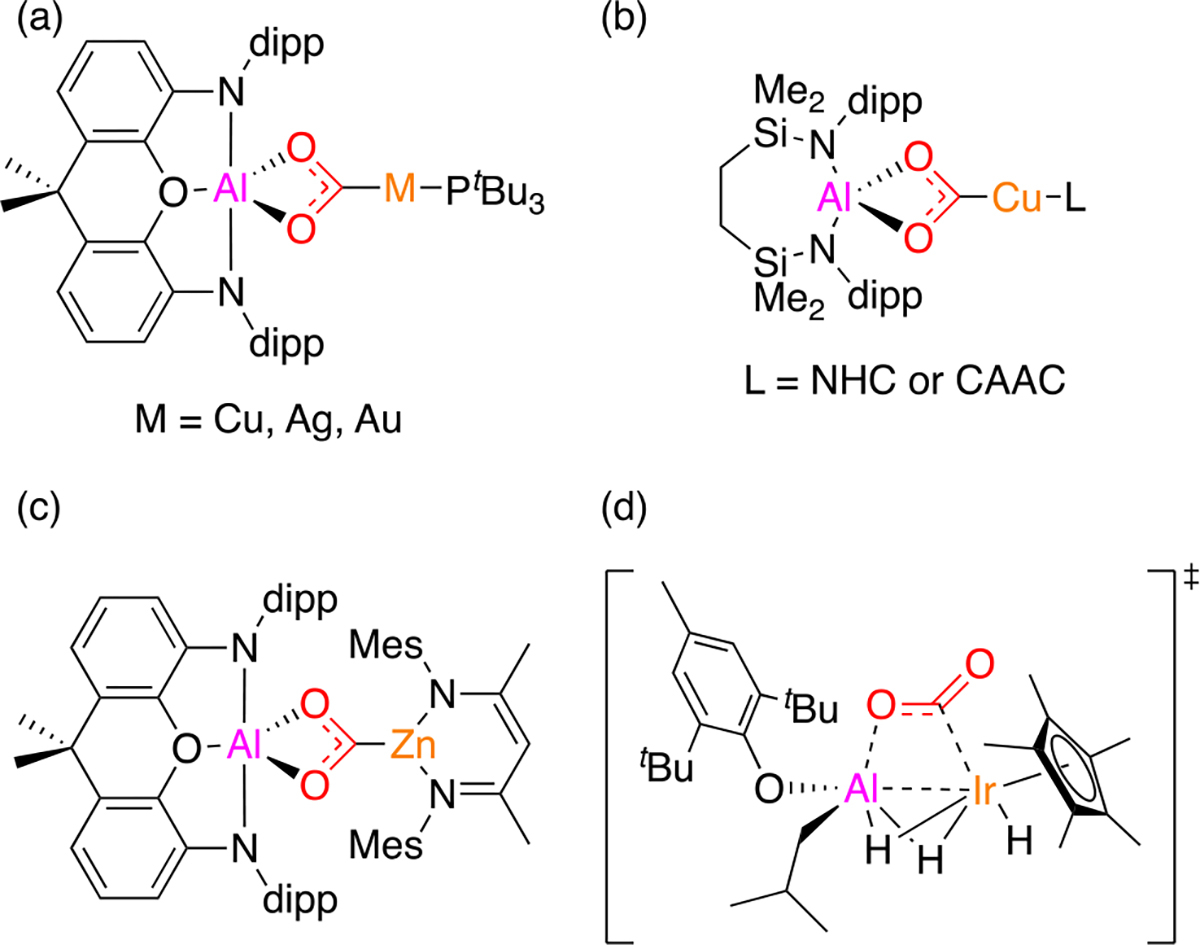
Previously reported examples of complexes derived from CO2 insertion into Al-M bonds.
We became interested in pursuing the chemistry of Al-Fe complexes, particularly those derived from AlIII rather than AlI precursors. Despite aluminum and iron being the two most abundant metals in earth’s crust (7.4% and 5%, respectively), only a few Al-Fe heterobimetallic complexes have been reported and their reactivity has never been studied.15,16,25 Herein we report that the β-diketiminate-supported Al-Fe heterobimetallic complex, Ldipp(Me)Al-FeCp(CO)2 (Ldipp = HC{(CMe)(2,6-i-Pr2C6H3N)}2), is able to insert CO2 and cyclohexene oxide. Using a combined experimental/computational approach, the reaction pathways for CO2 activation and epoxide ring opening were elucidated to involve (i) homolytic Al-Fe dissociation to metalloradical intermediates, then (ii) pairwise addition to substrate prior to solvent cage escape. This “radical pair” mechanism is a novel mode of cooperative CO2 activation, and its extension to epoxide ring-opening may open new avenues for C-O activation.
RESULTS & DISCUSSION
Synthesis and reactivity.
To synthesize Al-Fe heterobimetallic complex LdippAl(Me)Fp (2, Fp = FeCp(CO)2), reaction of LdippAl(Me)I (1) was carried out with KFp in a mixture of toluene and ether for 16 h (Scheme 1). Compound 2 was obtained as a colorless solid in moderate yield (66 %) and was found to be soluble in toluene and diethyl ether but to decompose in tetrahydrofuran, chloroform, and dichloromethane. Furthermore, solutions of 2 in toluene slowly decay to produce Fp2 over extended periods of time at room temperature.
Scheme 1.

Thermal and photochemical reactivity studies of an Al-Fe heterobinuclear system, including solid-state structures of complexes 2-5 determined by X-ray crystallography. For clarity, hydrogen atoms and co-crystallized solvent molecules are omitted from the crystal structures; ligand backbones (except nitrogen) are shown as wireframes, and all other atoms are shown as 50% probability ellipsoids.
Reactions of 2 with CO2 and epoxide were carried out since they are both useful feedstocks for preparation of cyclic or polycarbonate products, often with the involvement of Al catalysts.26 Accordingly, reaction of compound 2 with CO2 in toluene resulted in CO2-inserted product LdippAl(Me)(μ:κ2-O2C)Fp (3) as a colorless solid in 81% yield (Scheme 1). This reaction was found to occur with equal efficiency when shielded from light. Insertion of CO2 into Al-M bonds with κ2 binding to Al is rare, with previously reported examples involving low valent AlI rather than AlIII precursors.18,21,23,24 Compound 3 was stable to thermolysis and vacuum exposure, and we observed no evidence for conversion back to 2 under these conditions. In the 13C{1H} NMR spectrum of 3, the carbon atom of the [κ2-O2C] moiety resonates at 220.8 ppm, which is shifted downfield compared to the bridging CO2 unit in [Cp2Zr(Cl)(μ:κ2-O2C)FeCp(CO)2] (δ =212.6 ppm).27 Stoichiometric reaction of 2 with cyclohexene oxide was carried out at room temperature, resulting in ring-opened product LdippAl(Me)(μ-OC6H10)Fp (4, 57 %, Scheme 1). Ring opening of epoxides using Al/Co heterobimetallic catalysts has been proposed by the Coates group for several carbonylation reactions,28 but such an intermediate has never been isolated until now. Compound 4 is stable at −25 °C but decomposes slowly at room temperature inside the glove box. The solid-state IR spectrum of 2 has features at 1903 and 1963 cm−1 assigned to the CO ligands that are shifted to higher energy in 3 (1981, 2021 cm−1) and 4 (1935, 2002 cm−1), corresponding with a change in formal Fe oxidation state from Fe(0) to Fe(II).
The molecular structures of 2 and 4 feature four-coordinate aluminum centers with distorted tetrahedral geometries, while 3 has a penta-coordinate aluminum center with distorted square pyramidal geometry, as determined by X-ray crystallography. The Fe-Al bond length [2.478(9) Å] in 2 is comparable to the value of 2.480(1) Å reported for the related compound LdippAl(Cl)Fp* (Fp* = Cp*Fe(CO)2; Cp* = C5Me5)25c but longer than the other previously reported Fe-Al bonds (2.231(3)-2.349(9) Å).25d–25f The Al-O bond lengths in 3 [1.980(1) and 1.897(1) Å] are slightly longer to the corresponding bonds [1.880(5) and 1.861(4) Å] present in (NON)Al(κ2-O2C)AuPtBu3 (NON = 4,5-bis(2,6-diisopropylanilido)-2,7-di-tert-butyl-9,9-dimethylxanthene).21 The O-C bond lengths for the Al(κ2-O2C)Fe unit in 3 [1.271(2) and 1.298(2) Å] fall between the typical ranges for C-O single and double bonds. The Fe-C bond [1.933(1) Å] of the Al(κ2-O2C)Fe unit is comparable with the Fe-C bond [1.935(14) Å] seen in HC(SiMe2NC6H4F-2)3-Zr(κ2-S2C)Fe(CO)2Cp.29 The molecular structure of 4 reveals that LAl(Me)O and Fp moieties are in trans orientation about the cyclohexane unit. The Al-O bond length [1.728(2) Å] in 4 is much shorter than those in 3. The Fe-CCy bond length is 2.111(3) Å, which is in range of reported Fe-Calkyl bonds.30
Complex 3 is stable thermally (up to 60 °C) in C6D6 for at least 12 h but was found to react further upon irradiation with UV light. A C6D6 solution of 3 was irradiated at ambient temperature for 5 h. The 1H NMR spectrum reveals complete conversion of 3 to 5 (Scheme 1). In the 1H NMR spectrum of compound 3, the Al-CH3 protons appear as a singlet at −0.20 ppm but are shifted downfield to 1.71 ppm in 5, indicating methyl migration from the aluminum center to the ligand backbone. The crystal structure of 5 revealed that its aluminum center is tetra-coordinate with two nitrogen and two oxygen atoms in its immediate environment. It also confirmed that the methyl group migrated from aluminum to the β-diketiminate backbone as anticipated from 1H NMR spectroscopy. The methyl migration to the β-diketiminate ligand transforms it into a dianionic ligand. Although the M-C bond migration phenomenon has been observed in transition metal β-diketiminate chemistry, this is first report involving an aluminum β-diketiminate.31,32 The C3N2Al six membered ring is puckered with both Al-N bonds being almost equal in distance [1.774(3) Å and 1.808(3)]. The Al-O bond lengths [1.862(2) Å and 1.848(2)] are shorter than in 3, possibly due to change in coordination number at the aluminum center.
Computational analysis of atomic charges & bonding.
To better understand the origins of reactivity of our Al-Fe heterobimetallic complex LdippAl(Me)Fp (2), we decided to study the charge distribution using NBO33 (Figure S18) and QTAIM34 (Figure 3) analysis methods on pre-optimized geometries for complex 2 and, for modelling purposes, its simplified counterparts with methyl (LMeAl(Me)Fp, 6) and phenyl (LPhAl(Me)Fp, 7) substituents in place of 2,6-diisopropylphenyl (dipp) in 2. QTAIM charges, being origin-independent35 and more “chemically intuitive”, are consistent with representation of complexes 2, 6 and 7 as featuring strongly polarized covalent AlIII-Fe0 bonds. For comparison, we also calculated NBO and QTAIM charges for a well-studied heterobimetallic system,36 MeIMesCuFp 8 (MeIMesMes = 1,3-bis(2,4,6-trimethylphenyl)-4,5-dimethylimidazol-2-ylidene, Figures S18 and 3). QTAIM charges in this model system are much closer to traditional ideas of valence, with Cu being in the oxidation state of (almost) one and Fe being in the oxidation state of zero.
Figure 3.

Calculated QTAIM charges (red) for selected atoms in 2 and 6-8, with M-Fe Wiberg bond indices (black) indicated below the structures.
To further confirm the presence of the covalent bond between two metals, we calculated Wiberg bond indices (WBIs) in Löwdin orthogonalized basis37 (Figure 3 and Table S1). From the obtained data we can conclude that the Al-Fe bond in 2 is almost twice as covalent as the Cu-Fe bond in 8. Thus, some caution must be used in describing the oxidation levels of the Al and Fe centers in 2.
Although the used methods gave quite differing charge values, trends between structures in question are consistent. This allowed us to (initially) simplify our model and study Me-substituted complex 6 in lieu of complex 2 to study its reactivity in silico towards CO2 insertion and epoxide ring opening at a lower computational cost.
Combined computational/experimental investigation of reaction mechanisms.
We decided to begin with calculating energies for two dissociation pathways (Scheme 2a), homolytic and heterolytic, for truncated model complexes 6 and 7. However, dissociation energies for both 6 and 7 proved to be too high, even when including effects of the toluene solvent. It is worth noting that Gibbs free energies and enthalpies are significantly (>20 kcal/mol) lower for homolytic rather than heterolytic dissociation. Steric bulkiness of substituents (i.e. Me vs Ph) appeared irrelevant: for homolytic dissociation both ΔGd and ΔHd were just ~6 kcal/mol lower for significantly bulkier 7, and for heterolytic dissociation Gibbs free energies and enthalpies for 7 were even higher than for 6. Therefore, we decided not to proceed with dissociation calculations for complex 2, a decision that later proved imprudent.
Scheme 2.
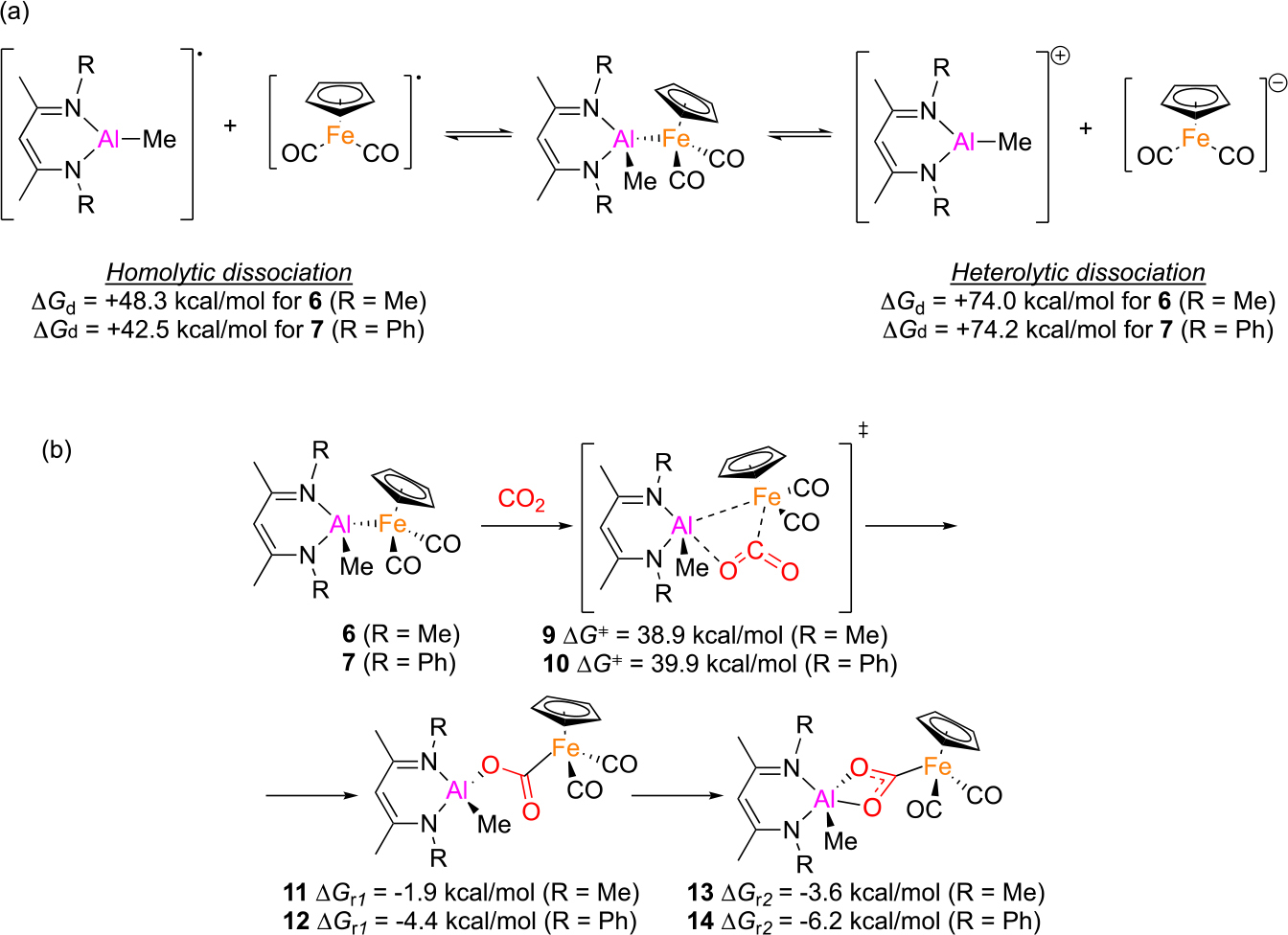
Unscaled Gibbs free energies (PBE1PBE) for (a) Al-Fe bond dissociation and (b) concerted CO2 insertion pathways.
Since calculated dissociation energies were inconsistent with reactions that occur readily at ambient conditions, we decided to explore possible mechanistic pathways beginning with the concerted CO2 insertion (Scheme 2b) via the corresponding transition state (TS). Obtained Gibbs free energies indicated that while the CO2 insertion per se is thermodynamically favorable, the concerted pathway’s high activation barrier is also inconsistent with a reaction that occurs with reasonable rate at room temperature (as observed experimentally). To make sure the obtained TS energy was not an artifact of poorly chosen DFT functional, we recalculated the transition state energy using several DFT functionals (Figure S17). Obtained Gibbs free energies are generally consistent among all employed functionals, which indicates that such a high obtained value is not an error of the chosen functional.
To gain additional insights on the mechanism, we next conducted an Eyring analysis by obtaining experimental, pseudo-first order (excess CO2) rate constants for CO2 activation by 2 across the temperature range 263–303 K (Figure 4). Fitting the data to the Eyring equation provided the experimentally determined activation parameters for CO2 insertion by complex 2: ΔH⧧ = 26 ± 2 kcal/mol, ΔS⧧ = 24 ± 7 cal/mol*K, ΔG⧧298K = 19 ± 3 kcal/mol. The activation entropy for this reaction is large and positive, which indicates a dissociative rate-determining step and is therefore inconsistent with the concerted mechanism shown in Scheme 2b (which should give ΔS⧧ < 0). Moreover, the trans-stereochemistry of the ring-opened product 4 evident by X-ray crystallography indicates anti-addition of the two metals to the epoxide and thus also testifies against any similar concerted mechanisms.
Figure 4.

Eyring analysis to extract activation parameters for the CO2 activation reaction by 2.
With these considerations in mind, we decided to revisit the dissociation pathways, this time with the model for 2 with full-sized dipp-substituted ligands (Scheme 3). To our surprise, the dissociation Gibbs free energies and enthalpies were ~15 kcal/mol lower than for closest counterpart 7. This brought the activation barrier down, closer to the experimental value and within the expected range for a room-temperature reaction. Therefore, relying on models with truncated substituents, a common approach in computational organometallic chemistry, is not always justified. It is curious that addition of just two isopropyl groups to each phenyl ring had such drastic effect on dissociation energies, which we attribute solely to significantly higher steric repulsion in complex 2 relative to 6 and 7, given their similar electronic structures. Although the homolytic dissociation energy for 2 is similar to the calculated barrier for concerted CO2 activation by 6, a dissociative mechanism would better align with the experimental Eyring analysis for CO2 reactivity of 2. Also noteworthy is the trans geometry of epoxide activation product 4, rather than the cis configuration expected from a concerted, four-membered transition state for C-O cleavage at the Al-Fe bond. Collectively, these observations also reinforce the critical importance of calibrating computed mechanisms with experimental (preferably kinetics) data.
Scheme 3.

Gibbs free energies (PBE1PBE) for Al-Fe bond dissociation of 2.
With further evidence towards homolytic dissociation of complex 2 as the origin of its reactivity and having ruled out several alternatives (Figure S17), we calculated the Gibbs free energies of interactions of both Al and Fe metalloradicals with both CO2 and cyclohexene oxide. As follows from the obtained Gibbs energies, both CO2 and cyclohexene oxide prefer to react with the Al radical [LdippAlMe]· (15) rather than with Fe radical Fp· (16), as indicated in Scheme 4. In the first case, for CO2 insertion, we could not localize a minimum corresponding to the radical [FpCO2]· complex 19. (However, we were able to localize a minimum for a similar anionic complex, which has been observed experimentally38; see SI for more details.) Instead, the localized minimum was a supramolecular complex 19’ of Fp radical 16 and CO2, held together by weak non-covalent bonds (as shown by QTAIM diagrams and calculated WBIs; Figure 5a). However, we localized a minimum for the complex 17 where a linear CO2 molecule is coordinated on the Al center of complex 15, giving a quite strong covalent bond (also Figure 5a). Such coordination is endothermic, but the subsequent interception of Fp radical 16 makes CO2 insertion a thermodynamically favorable process.
Scheme 4.

Gibbs free energy diagram for calculated pathways of CO2 activation mediated by 2.
Figure 5.

(a) Selected QTAIM paths connecting (3,−3) and (3,−1) critical points for 19′ and 17 with key all-electron Wiberg bond indices shown; (b) calculated Mulliken spin densities for selected atoms and corresponding spin density plots (isosurface value = 0.0004) for 15 and 17.
Epoxide ring opening is also an exothermic process even for initiation with the Al radical 15, while initiation by Fe radical 16 has a very high barrier (Scheme 5). Subsequent recombination of Al-epoxide complex 21 with 16 to give the observed product 4 is energetically very favorable.
Scheme 5.

Gibbs free energy diagrams for calculated pathways of cyclohexene oxide ring opening mediated by 2.
The identification of the O-bound CO2 adduct 17 along the reaction pathway is notable, as this coordination mode has rarely been observed experimentally39 and is typically proposed during (1-electron) reductive coupling of CO2 to form oxalate40 (rather than for 2-electron CO2 reduction). Moreover, it is a rare example of a formally AlII species playing an important role in reactivity.41 Thus, we chose to analyze the electronic structure of 17 and its precursor complex 15 further. Calculated Mulliken spin densities for selected atoms (Figure 5b) of these complexes indicate that the unpaired electron is mostly delocalized through the conjugated β-diketiminate ligand but not on Al itself for either complex, nor on the CO2 moiety in the case of 17. In other words, although 15 is formally a [(Ldipp)−]AlIIMe species, it is perhaps better formulated as [(Ldipp)·2−]AlIIIMe and thus possess a vacant Al-centered 3pz orbital for substrate coordination. This dual radical/acid character of 15 is critical to successful activation of CO2, which otherwise resists participating in radical chemistry due to its unfavorable one-electron reduction potential (E° = −1.9 V at pH 7).
Experimental validation of the radical pair mechanism.
Given these computational results indicating an unusual radical pair mechanism for the cooperative bond activation chemistry of 2, we sought experimental validation of the computational predictions. One notable prediction from computational modeling is that the homolytic Al-Fe bond dissociation energy in LPhAl(Me)Fp (7) should be higher than that in LdippAl(Me)Fp (2) by 18 kcal/mol. Thus, radical pair generation from 7 should not proceed under the mild conditions at which reactivity of 2 was observed. Indeed, exposure of 7 to CO2 resulted in no reaction (Scheme 6a) under conditions at which 2 activated CO2 rapidly (Scheme 1). This observation provides experimental confirmation of a key prediction of our computational modeling.
Scheme 6.

(a) Experimental validation of a computational prediction, and (b) direct observation of the purple Al-containing radical, LdippAl(Me)(OCPh2), 22.
Next, we sought further evidence for formation of metalloradicals 15 and/or 16 under relevant conditions by screening reactions with other substrates with C-O or C=O bonds. Fortuitous results were obtained from the reaction of 2 with benzophenone (Scheme 6b). Addition of benzophenone to 2 at room temperature results in an intense, purple-colored solution. Analysis of this solution by 1H NMR spectroscopy indicates stoichiometric formation of Fp2, which presumably forms from dimerization of 16. The aluminum-containing product was NMR silent, indicating a paramagnetic species. Indeed, analysis of the solution by EPR spectroscopy provided evidence for a S = 1/2 species assigned as LdippAl(Me)(OCPh2) (22), which can be viewed as the benzophenone analogue of CO2 adduct 17. Concentrating solutions of 22 or attempting to crystallize it resulted in white-colored solids that have the same empirical formula as 22 according to elemental analysis. At this time, the identity of these solids is unclear, but they likely represent a dialuminum structure resulting from homocoupling of radical 22, as has been observed previously by Thomas for a metalloradical-benzophenone system.42 Reconstitution of the white solid in toluene restores the purple color of 22, indicating that the presumed homocoupling process is reversible and is driven towards the monomeric radical in solution and the dimeric, diamagnetic species in the solid state.
Next, we analyzed the electronic structure of 22, in a manner similar to 17 and 19’. QTAIM analysis of the localized minimum for 22 revealed a highly covalent Al-O bond (Figure 6a). Calculated WBIs (also Figure 6a) showed that benzophenone’s C=O bond loses its double bond character (WBI = 1.707 < 2), which is consistent with the IR spectrum obtained for 22 (see SI for more details). The bond between Al and O (WBI = 1.294) is almost three times as covalent as a similar bond (WBI = 0.439) in the complex with CO2 (17).
Figure 6.

(a) Selected QTAIM paths connecting (3,−3) and (3,−1) critical points for 22 with key all-electron Wiberg bond indices shown; (b) calculated Mulliken spin densities for selected atoms and corresponding spin density plot (isosurface value = 0.0004) for 22.
Calculated Mulliken spin densities (Figure 6b) for selected atoms of 22 reveal that the unpaired spin resides predominantly on the benzophenone unit and not on the conjugated β-diketiminate ligand as was observed in the previous case with 17, nor on Al. Therefore, 22 can be described as LdippAlIII[(OCPh2)·−]Me, with the formerly vacant Al-centered 3pz orbital occupied by benzophenone’s oxygen lone pair electrons. The relation of these results with EPR experiments is discussed in the following section.
The purple toluene solution of 22 exhibits a room temperature (i.e., fluid solution) X-band EPR signal as shown in Figure 7a. The signal is at giso = 2.006 and comprises a Gaussian lineshape with width (hwhm) of 16 MHz. However, this does not fully describe the signal as there appears to be superimposed hyperfine splitting. This splitting can be modeled by hyperfine coupling (hfc) to one 27Al (I = 5/2, 100% abundance) with aiso = 4 MHz and three types of 1H: aiso = 22 MHz (4 equivalent nuclei), aiso = 7.5 MHz (4 equivalent nuclei), and aiso = 26 MHz (2 equivalent nuclei). The rationale for this parameter set, which is neither ideal nor unique will be given below. That the hfc is not fully (i.e., “baseline”) resolved is explained by appearance of the EPR spectrum recorded at 80 K (i.e. in frozen solution, Figure 7b), which is overall broader due to g anisotropy (see Q-band results below) and possible hfc anisotropy as well. Here we did not even attempt to simulate any possible hfc. At 300 K, there is some rotational averaging so that hfc is partially resolved, while there is none at 200 K (not shown).
Figure 7.

EPR spectra (black traces) of 22 in toluene solution with simulations (red traces): a) X-band spectrum recorded at ambient temperature; microwave frequency, 9.45999 GHz; field modulation amplitude, 0.1 mT; time constant, 20 ms; b) X-band spectrum recorded at 80 K, conditions as in (a) except microwave frequency, 9.456414 GHz; c) digital derivative of Q-band spectrum; d) (passage conditions) Q-band spectrum recorded at 2 K; microwave frequency, 35.0530 GHz; microwave power, 2 μW; field modulation amplitude, 0.2 mT; time constant, 32 ms; scan time, 120 s. Simulation parameters: a) g = 2.006, sum of W = 16 MHz (Gaussian hwhm) no hfc and W = 0.5 MHz with aiso(27Al) = 4 MHz; aiso(o-1H)×4 = 22 MHz; aiso(m-1H)×4 = 7.5 MHz; aiso(p-1H)×2 = 26 MHz; The contribution of the simulation with hfc to that without was visually scaled to match experiment and has no quantitative relevance; b) g = [2.0075, 2.0066, 2.0032], W = 25 MHz; c) and d) g = [2.0070, 2.0061, 2.0027], W = 14, 10, 10 MHz. This g tensor reflects actual g anisotropy resolved in the rigid matrix and was used in (b), with a slight shift in g values to account for magnetic field imprecision (< 0.1 mT). The features corresponding to g|| = 2.0027 and g⊥ ≈ 2.0065 are indicated.
A close analog to 22 is the complex studied by Murphy et al., LdippMg(DMAP)(OCPh2), where DMAP = 4-dimethylaminopyridine.43 This compound can be formally described as Mg(I) with coordinated benzophenone or, more realistically, as Mg(II) with coordinated benzophenone radical anion. The X-band EPR spectrum of LdippMg(DMAP)(OCPh2) in toluene gave poorly resolved hfc at 200 K (with giso = 2.004), but resolution improved at higher temperatures and already at 250 K (as well as at 298 K) gave a beautifully resolved spectrum that was perfectly simulated when accounting for accompanying ENDOR and TRIPLE (i.e., double ENDOR) spectroscopy data. In contrast, for 22, we believe that a temperature much higher than ambient would be needed to obtain complete resolution, which could allow definitive determination of hfc in this relatively bulky molecule. Murphy et al. extracted aiso values for 25Mg (I = 5/2, 10.0%) of 5.54 MHz and 14N of DMAP of 0.90 (zero from 14N of Ldipp). Additionally, aiso values for the four sets of benzophenone phenyl protons were determined as ortho-H1, 8.80 MHz; ortho-H2, 7.85 MHz; meta-H, 3.00 MHz; para-H, 10.00. This approximate ratio of benzophenone aiso(o-,m-,p-1H) values was used as a starting point to simulate the room-temperature X-band EPR spectrum of 22, after scaling up to match the breadth of hfc seen for 22. The aiso(27Al) value for 22 was estimated from ENDOR data as described in the following section. The very small value for 14N hfc in the Mg complex, combined with the computational results for 22 described above, led us to ignore any possible hfc from 14N.
An EPR spectrum of 22 in frozen toluene solution was also recorded at 35 GHz (Q-band) at 2 K. The EPR signal is under “passage” conditions and thus has an absorption lineshape as shown in Figure 7d; Figure 7c presents a digital derivative so that a standard EPR presentation is seen.44 No hfc was resolved at the Q-band, which is typical considering the increased microwave frequency and corresponding resonant magnetic field.45 More importantly, the form of 22 in frozen solution appears to be slightly different in that g = [2.0027, 2.0061, 2.007] so that gavg = 2.0053, as opposed to giso = 2.006 at high temperature. There is spin delocalization onto the 27Al center as definitively shown by ENDOR spectroscopy. EPR spectra exhibiting beautifully resolved 27Al hfc have been reported by Roesky and co-workers. These systems, however, can be more properly considered as Al(II) species than 22 as the reported compounds lack any moiety equivalent to a benzophenone ketyl radical. Accordingly, those complexes give much larger 27Al hfc in the range of 15 – 35 MHz.46 Very recently, Britt, Arnold, and co-workers reported X-band EPR, 35-GHz pulsed EPR, and (Davies) ENDOR spectra for (η5-Cp)2Ti(μ−1,2H)2Al1,2H(CTMS3), where CTMS3 = −C(SiMe3)3.47 This compound is formally Ti(III)-Al(III), but could be considered to have some Ti(IV)-Al(II) character. In contrast to 22, but as expected for Ti(III), this complex exhibits significant g anisotropy: g = [2.003, 1.992, 1.971], which allowed determination of not only aiso(27Al) = 9.4 MHz, but also A(27Al) = [6.0, 14.6, 7.6] MHz.
ENDOR spectroscopy is broad banded in that signals from all magnetically active nuclei with hfc to the paramagnetic center can in principle be observed. A wide scan ENDOR spectrum of 22 recorded at g = 2.006 is shown in Figure 8 (main figure). Signals from 1H are readily observed at the 1H Larmor frequency (νH ≈ 53 MHz). In addition to a strong signal directly at νH, which corresponds to protons with negligible hfc, there are features symmetrically disposed about it that give hfc for three types of magnetically equivalent protons, as expected for the phenyl rings of benzophenone.43 These give A(1H) ≈ 4, 10, and 15 MHz, as seen in Figure 8 (right inset). These values are smaller than the three aiso(1H) values used in the 300 K fluid solution X-band EPR simulation. This may be due to the difference between fluid and frozen solution in that the features seen by ENDOR may each be only one component (~A⊥) of an anisotropic, indeed highly dipolar, hfc tensor: A(1H) = [aiso ± (T − ρ),aiso ± (T + ρ),aiso ∓ 2T].
Figure 8.

CW 35 GHz ENDOR spectra of 22 in toluene solution. Experimental conditions: 35.0530 GHz; microwave power, 2 μW; field modulation amplitude, 0.1 mT; time constant, 32 ms; magnetic field, 1.2484 T (g = 2.0061); radiofrequency(rf) scan rate, 1 MHz/s; 3 scans, The spectra shown in the insets used the same conditions except in the 27Al spectra: field modulation, 0.2 mT; 20 scans; and the spectrum shown in blue had magnetic field, 1.2497 T (g = 2.0040).
An additional feature is seen at ~14 MHz that corresponds exactly to the 27Al Larmor frequency. This signal recorded at two field positions, corresponding roughly to g|| and g⊥, is shown in Figure 8 (left inset). This feature provides the rough estimate used for EPR simulation of aiso(27Al) = 4 MHz. This estimate as to hfc corresponds to ~0.1% spin on 27Al.50 The metal hfc found by Murphy et al., |aiso(25Mg)| = 5.54 MHz, corresponds to 1.14% 3s1 spin density.
CONCLUSIONS
Heterobimetallic Al-Fe complex, 2, reacts cleanly with CO2 and cyclohexene oxide, giving CO2 inserted product 3 and epoxide ring opening product 4, respectively. Detailed kinetic and theoretical studies were performed on these two reactions and indicated an unusual radical-pair mechanism in which Al-Fe homolytic dissociation precedes pairwise metalloradical addition to substrate. The Al-containing metalloradical coordinated by benzophenone (22) was observed directly and thoroughly characterized by EPR and ENDOR spectroscopies. Particularly novel aspects of this study include:
Aluminum-containing radicals with dual radical/acid character implicated as a reactive intermediates
An unprecedented mechanism for CO2 activation
An instructive interplay between theory and experiment that emphasizes the importance in computational organometallic chemistry of (i) validating calculated reaction pathways with kinetics measurements, and (ii) modeling complete (as opposed to truncated) ligand substituents
METHODS
Experimental methods.
All experimental manipulations were carried out under inert dinitrogen atmosphere using standard Schlenk line and glovebox techniques. All new compounds were characterized by 1H and 13C{1H} NMR spectroscopy, solid-state IR spectroscopy, and single-crystal X-ray diffraction. Detailed experimental procedures and spectral data are available as Supplementary Information, and supporting crystallographic data in the form of CIF files are available upon request from the Cambridge Crystallographic Data Centre using deposition numbers 2100519, 2100520, 2100523, and 2100525.
Computational methods.
Density Functional Theory calculations (at the PBE049a-G3BJ49b/def2-TZVP49c level of theory; SMD51d solvation model with toluene parameters), as implemented in the Gaussian 16 (Revision B.01)50 code, were employed to optimize molecular geometries and determine bonding energies, Mulliken spin densities and NBO charges (using NBO version 3.1,51 as implemented in Gaussian 16). The open-source MultiWFN52 (version 3.8) program was employed to calculate and visualize Wiberg bond indices, QTAIM charges, critical points and paths (using data derived from Gaussian DFT calculations). For more details on comprehensive computational methods please see Supplementary Information.
Supplementary Material
ACKNOWLEDGMENT
This material is based upon work supported by the U.S. Department of Energy (DOE), Office of Science, Office of Basic Energy Sciences (BES), under Award Number DE-SC0021055. We thank Prof. Brian M. Hoffman (Northwestern University) for use of a 35-GHz CW EPR/ENDOR spectrometer, which is supported by the US DOE, Office of Science, BES, under Award Number DE-SC0019342. Computational resources and services were provided by the Advanced Cyberinfrastructure for Education and Research (ACER) group at UIC. Instrumentation for X-ray crystallography at UIC was funded, in part, by the National Institutes of Health under grant R01 GM116820. The structure of complex 5 was obtained using NSF’s ChemMatCARS Sector 15 supported by the Divisions of Chemistry (CHE) and Materials Research (DMR), National Science Foundation, under grant number NSF/CHE-1834750. Use of the Advanced Photon Source, an Office of Science User Facility operated for the DOE Office of Science by Argonne National Laboratory, was supported by the U.S. DOE under Contract No. DE-AC02-06CH11357. We thank Dr. Guodong Rao, UC-Davis, and Dr. Vlasta Brezová, Slovenská technická univerzita v Bratislave (STU), Slovakia, for helpful discussions. We also thank Dr. Daniel McElheny (UIC) for assistance with X-band EPR spectroscopy.
Footnotes
ASSOCIATED CONTENT
Supporting Information
The Supporting Information is available free of charge on the ACS Publications website.
Experimental procedures, spectral data, computational data (PDF)
REFERENCES
- (1).Stephan DW The Broadening Reach of Frustrated Lewis Pair Chemistry. Science 2016, 354, 7229. [DOI] [PubMed] [Google Scholar]
- (2).(a) Gunanathan C; Milstein D Metal–Ligand Cooperation by Aromatization–Dearomatization: A New Paradigm in Bond Activation and “Green” Catalysis. Acc. Chem. Res. 2011, 44, 588–602. [DOI] [PubMed] [Google Scholar]; (b) Morris RH Exploiting Metal-Ligand Bifunctional Reactions in the Design of Iron Asymmetric Hydrogenation Catalysts. Acc. Chem. Res. 2015, 48, 1494–1502. [DOI] [PubMed] [Google Scholar]
- (3).(a) Campos J Bimetallic cooperation across the periodic table. Nat. Rev. Chem. 2020, 4, 696–702. [DOI] [PubMed] [Google Scholar]; (b) Powers IG; Uyeda C Metal-metal bonds in catalysis. ACS Catal. 2017, 7, 936–958. [Google Scholar]
- (4).Buchwalter P; Rose J; Braunstein P Multimetallic Catalysis Based on Heterometallic Complexes and Clusters. Chem. Rev. 2015, 115, 28–126. [DOI] [PubMed] [Google Scholar]
- (5).Paparo A; Okuda J Carbon Dioxide Complexes: Bonding Modes and Synthetic Methods. Coord. Chem. Rev. 2017, 334, 136–149. [Google Scholar]
- (6).(a) Burkart MD; Hazari N; Tway CL; Zeitler EL Opportunities and Challenges for Catalysis in Carbon Dioxide Utilization. ACS Catal. 2019, 9, 7937–7956. [Google Scholar]; (b) Appel AM; Bercaw JE; Bocarsly AB; Dobbek H; Dubois DL; Dupuis M; Ferry JG; Fujita E; Hille R; Kenis PJA Frontiers, Opportunities, and Challenges in Biochemical and Chemical Catalysis of CO2 Fixation. Chem. Rev. 2013, 113, 6621–6658. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (7).Gibson DH The Organometallic Chemistry of Carbon Dioxide. Chem. Rev. 1996, 9, 2063–2095. [DOI] [PubMed] [Google Scholar]
- (8).(a) Gambarotta S; Arena F; Floriani C; Zanazzi PF Carbon dioxide fixation: bifunctional complexes containing acidic and basic sites working as reversible carriers. J. Am. Chem. Soc. 1982, 104, 5082–5092. [Google Scholar]; (b) Fachinetti G; Floriani C; Zanazzi PF Bifunctional activation of carbon dioxide. Synthesis and structure of a reversible carbon dioxide carrier. J. Am. Chem. Soc. 1978, 100, 7405–7407. [Google Scholar]
- (9).(a) Hanna TA; Baranger AM; Bergman RG Reaction of Carbon Dioxide and Heterocumulenes with an Unsymmetrical Metal-Metal Bond - Direct Addition of Carbon-Dioxide across a Zirconium-Iridium Bond and Stoichiometric Reduction of Carbon Dioxide to Formate. J. Am. Chem. Soc. 1995, 117, 11363–11364. [Google Scholar]; (b) Krogman JP; Foxman BM; Thomas CM Activation of CO2 by a Heterobimetallic Zr/Co Complex. J. Am. Chem. Soc. 2011, 133, 14582–14585. [DOI] [PubMed] [Google Scholar]; (c) Mömming CM; Otten E; Kehr G; Fröhlich R; Grimme S; Stephan DW; Erker G Reversible Metal-Free Carbon Dioxide Binding by Frustrated Lewis Pairs. Angew. Chem., Int. Ed. 2009, 48, 6643–6646. [DOI] [PubMed] [Google Scholar]; (d) Schlenker K; Christensen EG; Zhanserkeev AA; McDonald GR; Yang EL; Lutz KT; Steele RP; Vanderlinden RT; Saouma CT Role of Ligand-Bound CO2 in the Hydrogenation of CO2 to Formate at a (PNP)Mn Catalyst. ACS Catal. 2021, 11, 8358–8369. [Google Scholar]; (e) Escomel L; Del Rosal I; Maron L; Jeanneau E; Veyre L; Thieuleux C; Camp C Strongly Polarized Iridiumδ−–Aluminumδ+ Pairs: Unconventional Reactivity Patterns Including CO2 Cooperative Reductive Cleavage. J. Am. Chem. Soc. 2021, 143, 4844–4856. [DOI] [PubMed] [Google Scholar]
- (10).(a) Dobbek H; Svetlitchnyi V; Gremer L; Huber R; Meyer O Crystal Structure of a Carbon Monoxide Dehydrogenase Reveals a [Ni-4Fe-5S] Cluster. Science 2001, 293, 1281–1285. [DOI] [PubMed] [Google Scholar]; (b) Mankad NP; Ghosh D Biomimetic Studies of the Mo/Cu Active Site of CO Dehydrogenase. Comprehensive Coordination Chemistry III, 2021, pp 772–789. [Google Scholar]
- (11).Karunananda MK; Mankad NP Cooperative Strategies for Catalytic Hydrogenation of Unsaturated Hydrocarbons. ACS Catal. 2017, 7, 6110–6119. [Google Scholar]
- (12).(a) Agarwal J; Fujita E; Schaefer HF III; Muckerman JT Mechanisms for CO Production from CO2 Using Reduced Rhenium Tricarbonyl Catalysts. J. Am. Chem. Soc. 2012, 134, 5180–5186. [DOI] [PubMed] [Google Scholar]; (b) Liu L; Cao LL; Shao Y; Ménard G; Stephan DW A Radical Mechanism for Frustrated Lewis Pair Reactivity. Chem. 2017, 3, 259–267. [Google Scholar]; (c) Dasgupta A; Richards E; Melen RL Frustrated Radical Pairs: Insights from EPR Spectroscopy. Angew. Chem., Int. Ed. 2021, 60, 53–65. [DOI] [PMC free article] [PubMed] [Google Scholar]; (d) Holtrop F; Jupp A; Slootweg JC Radicals in frustrated Lewis pair chemistry. Springer, Cham, 2021. pp. 361–385. [Google Scholar]
- (13).(a) Charles III RM; Brewster TP H2 and carbon-heteroatom bond activation mediated by polarized heterobimetallic complexes. Coord. Chem. Rev. 2021, 433, 213765. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Hicken A; White AJP; Crimmin MR Selective Reduction of CO2 to a Formate Equivalent with Heterobimetallic Gold-Copper Hydride Complexes. Angew. Chem., Int. Ed. 2017, 56, 15127–15130. [DOI] [PubMed] [Google Scholar]; (c) Mankad NP Selectivity Effects in Bimetallic Catalysis. Chem. - Eur. J. 2016, 22, 5822–5829. [DOI] [PubMed] [Google Scholar]; (d) Bagherzadeh S; Mankad NP Catalyst Control of Selectivity in CO2 Reduction Using a Tunable Heterobimetallic Effect. J. Am. Chem. Soc. 2015, 137, 10898–10901. [DOI] [PubMed] [Google Scholar]; (e) Chipman JA; Berry JF Paramagnetic Metal-Metal Bonded Heterobimetallic Complexes. Chem. Rev. 2020, 120, 2409–2447. [DOI] [PubMed] [Google Scholar]; (f) Power PP Main-group elements as transition metals. Nature 2010, 463, 171–177. [DOI] [PubMed] [Google Scholar]
- (14).(a) Lai Q; Bhuvanesh N; Ozerov OV Unexpected B/Al Transelementation within a Rh Pincer Complex. J. Am. Chem. Soc. 2020, 142, 20920–20923. [DOI] [PubMed] [Google Scholar]; (b) Hara N; Saito T; Semba K; Kuriakose N; Zheng H; Sakaki S; Nakao Y Rhodium Complexes Bearing PAlP Pincer Ligands. J. Am. Chem. Soc. 2018, 140, 7070–7073. [DOI] [PubMed] [Google Scholar]; (c) Ekkert O; White AJP; Toms H; Crimmin MR Addition of Aluminium, Zinc and Magnesium Hydrides to Rhodium(III). Chem. Sci. 2015, 6, 5617–5622. [DOI] [PMC free article] [PubMed] [Google Scholar]; (d) Lai Q; Cosio MN; Ozerov OV Ni Complexes of an Alane/Tris(phosphine) Ligand Built Around a Strongly Lewis Acidic Tris(N-pyrrolyl)aluminum. Chem. Commun. 2020, 56, 14845–14848. [DOI] [PubMed] [Google Scholar]
- (15).Anand BN; Krossing I; Nöth H Synthesis and X-Ray Crystal Structure of (Tmp)2Al-Fe(Cp)(CO)2: An Alanyl-Containing Iron Complex with a Tricoordinated Aluminum Atom. Inorg. Chem. 1997, 36, 1979–1981. [DOI] [PubMed] [Google Scholar]
- (16).Braunschweig H; Müller J; Ganter B Molecular Structure of [CpFe(CO)2]2AlAr (Ar = 2-[(Dimethylamino)methyl]phenyl): An Alanediyl Complex with Two Fe-Al Bonds. Inorg. Chem. 1996, 35, 7443–7444. [DOI] [PubMed] [Google Scholar]
- (17).Mears KL; Stennett CR; Taskinen EK; Knapp CE; Carmalt CJ; Tuononen HM; Power PP Molecular Complexes Featuring Unsupported Dispersion-Enhanced Aluminum-Copper and Gallium-Copper Bonds. J. Am. Chem. Soc. 2020, 142, 19874–19878. [DOI] [PubMed] [Google Scholar]
- (18).Liu H-Y; Schwamm RJ; Hill MS; Mahon MF; McMullin CL; Rajabi NA Ambiphilic Al-Cu Bonding. Angew. Chem., Int. Ed. 2021, 60, 14390–14393. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (19).Rudd PA; Liu S; Gagliardi L; Young VG; Lu CC Metal-Alane Adducts with Zero-Valent Nickel, Cobalt, and Iron. J. Am. Chem. Soc. 2011, 133, 20724–20727. [DOI] [PubMed] [Google Scholar]
- (20).Kong RY; Crimmin MR 1st row transition metal aluminylene complexes: preparation, properties and bonding analysis. Dalton Trans. 2021, 50, 7810–7817. [DOI] [PubMed] [Google Scholar]
- (21).Hicks J; Mansikkamäki A; Vasko P; Goicoechea JM; Aldridge S A Nucleophilic Gold Complex. Nat. Chem. 2019, 11, 237–241 [DOI] [PubMed] [Google Scholar]
- (22).Sorbelli D; Belpassi L; Belanzoni P Reactivity of a Gold-Aluminyl Complex with Carbon Dioxide: A Nucleophilic Gold? J. Am. Chem. Soc. 2021, 143, 14433–14437. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (23).McManus C; Hicks J; Cui X; Zhao L; Frenking G; Goicoechea JM; Aldridge S Coinage metal aluminyl complexes: probing regiochemistry and mechanism in the insertion and reduction of carbon dioxide. Chem. Sci. 2021, 12, 13458–13468. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (24).Roy MMD; Hicks J; Vasko P; Helimann A; Baston A-M; Goicoechea JM; Aldridge S Probing the Extremes of Covalency in M−Al bonds: Lithium and Zinc Aluminyl Compounds. Angew. Chem., Int. Ed. 2021, 60, 22301–22306. [DOI] [PubMed] [Google Scholar]
- (25).(a) Fischer RA; Priermeier T Transition-Metal-Substituted Alanes: Synthesis and Spectroscopic Studies and the Structure of (η5-C5H5)(CO)2Fe-Al[(CH2)3NMe2](iBu). Organometallics 1994, 13, 4306–4314. [Google Scholar]; (b) Jones C; Aldridge S; Gans-Eichler T; Stasch A Synthesis and characterisation of complexes of Group 13 metal amidinate heterocycles with the CpFe(CO)2 fragment. Dalton Trans. 2006, 5357–5361. [DOI] [PubMed] [Google Scholar]; (c) Riddlestone IM; Urbano J; Phillips N; Kelly MJ; Vidovic D; Bates JI; Taylor R; Aldridge S Salt Metathesis for the Synthesis of M-Al and M-H-Al Bonds. Dalton Trans. 2013, 42, 249–258. [DOI] [PubMed] [Google Scholar]; (d) Yanagisawa T; Mizuhata Y; Tokitoh N Dibromometallyl-iron complexes generated by the recombination of an alumanyl-iron complex with EBr3 (E = Al, Ga). Heteroat. Chem. 2018, 29, e21465. [Google Scholar]; (e) Agou T; Yanagisawa T; Sasamori T; Tokitoh N Synthesis and Structure of an Iron-Bromoalumanyl Complex with a Tri- Coordinated Aluminum Center. Bull. Chem. Soc. Jpn. 2016, 89, 1184–1186. [Google Scholar]; (f) Weiss J; Stetzkamp D; Nuber B; Fischer RA; Boehme C; Frenking G [(η5-C5Me5)Al-Fe(CO)4]-Synthesis, Structure, and Bonding. Angew. Chem., Int. Ed. Engl. 1997, 36, 70–72. [Google Scholar]
- (26).Whiteoak CJ; Kielland N; Laserna V; Escudero-Adán EC; Martin E; Kleij AW A Powerful Aluminum Catalyst for the Synthesis of Highly Functional Organic Carbonates. J. Am. Chem. Soc. 2013, 135, 1228–1231. [DOI] [PubMed] [Google Scholar]
- (27).Pinkes JR; Steffey BD; Vites JC; Cutler AR Carbon Dioxide Insertion into the Fe-Zr and Ru-Zr Bonds of the Heterobimetallic Complexes Cp(CO)2M-Zr(Cl)Cp2: Direct Production of the μ-η1(C): η2(O,O′)-CO2 Compounds Cp(CO)2M-CO2-Zr(Cl)Cp2. Organometallics 1994, 13, 21–23. [Google Scholar]
- (28).Lamb JR; Hubbell AK; MacMillan SN; Coates GW Carbonylative, Catalytic Deoxygenation of 2,3-Disubstituted Epoxides with Inversion of Stereochemistry: An Alternative AlkeneIsomerization Method. J. Am. Chem. Soc. 2020, 142, 8029–8035. [DOI] [PubMed] [Google Scholar]
- (29).Memmler H; Kauper U; Gade LH; Scowen IJ; McPartlin M Insertion of X = C=Y Heteroallenes into Unsupported Zr-M Bonds (M = Fe, Ru). Chem. Commun. 1996, 15, 1751–1752. [Google Scholar]
- (30).Friedrich HB; Onani MO; Rademeyer M Bromopropyldicarbonyl(η5-pentamethyl-cyclopentadienyl)iron(II). Acta Cryst. E 2004, 60, m551–m553. [Google Scholar]
- (31).Giannini L; SoIari E; Angelis SD; Ward TR; Floriani C; Chiesi-Villa A; Rizzoli C Migratory Aptitude of the Zr-C Functionalities Bonded to a Macrocyclic Structure: Thermally- and Solvent-assisted Intra- and Intermolecular Migrations in Dialkyl(dibenzotetramethyltetraazaannulene)zirconium(IV). J. Am. Chem. Soc. 1995, 117, 5801–5811. [Google Scholar]
- (32).Camp C; Arnold J On the non-innocence of ″Nacnacs″: ligand-based reactivity in β-diketiminate supported coordination compounds. Dalton Trans. 2016, 45, 14462–14498. [DOI] [PubMed] [Google Scholar]
- (33).(a) Foster JP; Weinhold F Natural hybrid orbitals. J. Am. Chem. Soc. 1980, 102, 7211–7218. [Google Scholar]; (b) Reed AE; Weinhold F Natural bond orbital analysis of near-Hartree-Fock water dimer. J. Chem. Phys. 1983, 78, 4066–4073. [Google Scholar]
- (34).(a) Bader RFW A quantum theory of molecular structure and its applications. Chem. Rev. 1991, 91, 893–928. [Google Scholar]; (b) Bader RFW Atoms in Molecules. A Quantum Theory (Oxford Univ. Press, Oxford, 1994), pp. 1–458. [Google Scholar]
- (35).Laidig KE; Bader RFW Properties of Atoms in Molecules: Atomic Polarizabilities. J. Chem. Phys. 1990, 93, 7213–7224. [Google Scholar]
- (36).Jayarathne U; Mazzacano TJ; Bagherzadeh S; Mankad NP Heterobimetallic Complexes with Polar, Unsupported Cu-Fe and Zn-Fe Bonds Stabilized by N-Heterocyclic Carbenes. Organometallics 2013, 32, 3986–3992. [Google Scholar]
- (37).(a) Wiberg KB Application of the pople-santry-segal CNDO method to the cyclopropylcarbinyl and cyclobutyl cation and to bicyclobutane. Tetrahedron 1968, 24, 1083–1096. [Google Scholar]; (b) Sizova OV; Skripnikov LV; Sokolov AY Symmetry decomposition of quantum chemical bond orders, J. Mol. Struct. (Theochem) 2008, 870, 1–3, 1–9. [Google Scholar]
- (38).Pinkes John R.; Masi Christopher J.; Chiulli Robert; Steffey Bryan D.; Cutler AR Carbon Dioxide Complexation: Infrared Spectroscopy of Iron and Ruthenium η5-Cyclopentadienyl Carbonyl Metallocarboxylates. Inorg. Chem. 1997, 36, 70–79. [Google Scholar]
- (39).Castro-Rodriguez I; Nakai H; Zakharov LN; Rheingold AL; Meyer K A linear, O-Coordinated η1-CO2 Bound to Uranium. Science 2004, 305, 1757–1759. [DOI] [PubMed] [Google Scholar]
- (40).Saouma CT; Lu CC; Day MW; Peters JC CO2 reduction by Fe(I): solvent control of C–O cleavage versus C–C coupling Chem. Sci. 2013, 4, 4042–4051. [Google Scholar]
- (41).Falconer RL; Nichol GS; Smolyar IV; Cockroft SL; Cowley MJ. Reversible Reductive Elimination in Aluminum(II) Dihydrides. Angew. Chem. Int. Ed, 2021, 60, 2047–2052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (42).Marquard SL; Bezpalko MW; Foxman BM; Thomas CM Stoichiometric C=O Bond Oxidative Addition of Benzophenone by a Discrete Radical Intermediate To Form a Cobalt(I) Carbene. J. Am. Chem. Soc. 2013, 135, 6018–6021. [DOI] [PubMed] [Google Scholar]
- (43).Murphy DM; McDyre LE; Carter E; Stasch A; Jones C A CW-EPR, ENDOR and special TRIPLE resonance study of a novel magnesium ketyl radical. Magn. Reson. Chem. 2011, 49, 159–163. [DOI] [PubMed] [Google Scholar]
- (44).(a) Mailer C; Hoffman BM Tumbling of an adsorbed nitroxide using rapid adiabatic passage. J. Phys. Chem. 1976, 80, 842–846. [Google Scholar]; (b) Mailer C; Taylor CPS Rapid Adiabatic Passage EPR of Ferricytochrome c: Signal Enhancement and Determination of Spin-Lattice Relaxation Time. Biochim. Biophys. Acta, Protein Struct. 1973, 322, 195–203. [DOI] [PubMed] [Google Scholar]
- (45).Hyde JS; Froncisz W The Role of Microwave Frequency in EPR Spectroscopy of Copper Complexes. Ann. Rev. of Biophys. and Bioeng. 1982. 11:1, 391–417. [DOI] [PubMed] [Google Scholar]
- (46).(a) Kundu S; Sinhababu S; Dutta S; Mondal T; Koley D; Dittrich B; Schwederski B; Kaim W; Stuckl AC; Roesky HW Synthesis and characterization of Lewis base stabilized mono- and di-organo aluminum radicals. Chem. Commun. 2017, 53, 10516–10519. [DOI] [PubMed] [Google Scholar]; (b) Li B; Kundu S; Stuckl AC; Zhu H; Keil H; Herbst-Irmer R; Stalke D; Schwederski B; Kaim W; Andrada DM; Frenking G; Roesky HW A stable neutral radical in the coordination sphere of aluminum. Angew. Chem., Int. Ed. 2017, 56, 397–400. [DOI] [PubMed] [Google Scholar]; (c) Siddiqui MM; Banerjee S; Bose S; Sarkar SK; Gupta SK; Kretsch J; Graw N; Herbst-Irmer R; Stalke D; Dutta S; Koley D; Roesky HW Cyclic (Alkyl)(Amino)Carbene-Stabilized Aluminum and Gallium Radicals Based on Amidinate Scaffolds. Inorg. Chem. 2020, 59, 11253–11258. [DOI] [PubMed] [Google Scholar]
- (47).Rao G; Altman AB; Brown AC; Tao L; Stich TA; Arnold J; Britt RD Metal Bonding with 3d and 6d Orbitals: An EPR and ENDOR Spectroscopic Investigation of Ti3++–Al and Th3++–Al Heterobimetallic Complexes. Inorg. Chem. 2019, 58, 7978–7988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (48).Morton JR; Preston KF Atomic parameters for paramagnetic resonance data. J. Magn. Reson. 1978, 30, 577–582. [Google Scholar]
- (49).(a) Adamo C; Barone V Toward reliable density functional methods without adjustable parameters: The PBE0 model. J. Chem. Phys. 1999, 110, 6158–6169. [Google Scholar]; (b) Grimme S; Ehrlich S; Goerigk L Effect of the damping function in dispersion corrected density functional theory. J. Comp. Chem. 2011, 32, 1456–1465. [DOI] [PubMed] [Google Scholar]; (c) Weigend F; Ahlrichs R Balanced basis sets of split valence, triple zeta valence and quadruple zeta valence quality for H to Rn: Design and assessment of accuracy. Phys. Chem. Chem. Phys. 2005, 7, 3297–3305. [DOI] [PubMed] [Google Scholar]; (d) Marenich AV; Cramer CJ; Truhlar DG; Universal solvation model based on solute electron density and a continuum model of the solvent defined by the bulk dielectric constant and atomic surface tensions. J. Phys. Chem. B 2009, 113, 6378–6396. [DOI] [PubMed] [Google Scholar]
- (50).Gaussian 16, Revision B.01, Frisch MJ; Trucks GW; Schlegel HB; Scuseria GE; Robb MA; Cheeseman JR; Scalmani G; Barone V; Petersson GA; Nakatsuji H; Li X; Caricato M; Marenich AV; Bloino J; Janesko BG; Gomperts R; Mennucci B; Hratchian HP; Ortiz JV; Izmaylov AF; Sonnenberg JL; Williams-Young D; Ding F; Lipparini F; Egidi F; Goings J; Peng B; Petrone A; Henderson T; Ranasinghe D; Zakrzewski VG; Gao J; Rega N; Zheng G; Liang W; Hada M; Ehara M; Toyota K; Fukuda R; Hasegawa J; Ishida M; Nakajima T; Honda Y; Kitao O; Nakai H; Vreven T; Throssell K; Montgomery JA Jr.; Peralta JE; Ogliaro F; Bearpark MJ; Heyd JJ; Brothers EN; Kudin KN; Staroverov VN; Keith TA; Kobayashi R; Normand J; Raghavachari K; Rendell AP; Burant JC; Iyengar SS; Tomasi J; Cossi M; Millam JM; Klene M; Adamo C; Cammi R; Ochterski JW; Martin RL; Morokuma K; Farkas O; Foresman JB; Fox DJ Gaussian, Inc., Wallingford CT, 2016. [Google Scholar]
- (51).NBO Version 3.1, Glendening ED; Reed AE; Carpenter JE; Weinhold F Theoretical Chemistry Institute, University of Wisconsin, Madison, 2003. [Google Scholar]
- (52).Lu T; Chen F Multiwfn: A Multifunctional Wavefunction Analyzer. J. Comput. Chem. 2012, 33, 580–592. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.