

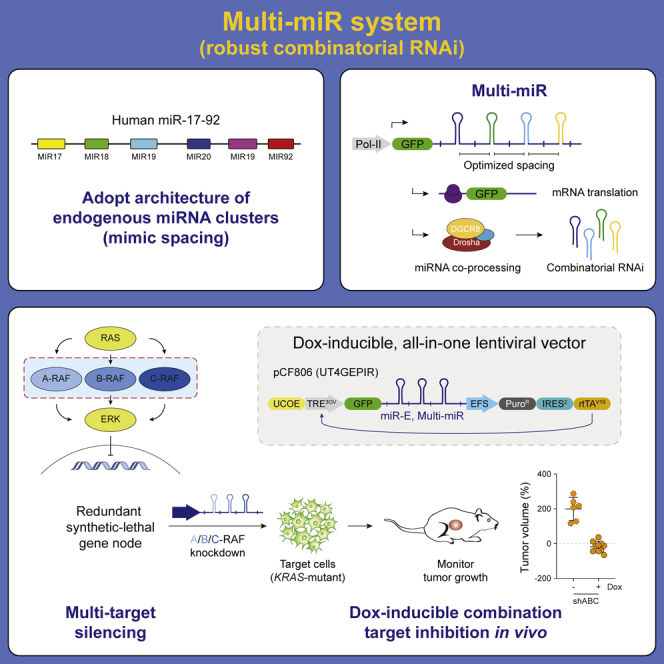
Summary
We present Multi-miR, a microRNA-embedded shRNA system modeled after endogenous microRNA clusters that enables simultaneous expression of up to three or four short hairpin RNAs (shRNAs) from a single promoter without loss of activity, enabling robust combinatorial RNA interference (RNAi). We further developed complementary all-in-one vectors that are over one log-scale more sensitive to doxycycline-mediated activation in vitro than previous methods and resistant to shRNA inactivation in vivo. We demonstrate the utility of this system for intracranial expression of shRNAs in a glioblastoma model. Additionally, we leverage this platform to target the redundant RAF signaling node in a mouse model of KRAS-mutant cancer and show that robust combinatorial synthetic lethality efficiently abolishes tumor growth.
Keywords: gene regulation, combinatorial target inhibition, RNA interference, RNAi, microRNA, cancer modeling, target discovery, target validation, viral vectors
Graphical abstract
Highlights
-
•
Multi-miR enables robust combinatorial RNAi of up to three or four targets
-
•
Tighter spacing of synthetic shRNA clusters increases knockdown efficiency
-
•
Optimized vectors facilitate inducible in vivo RNAi of redundant gene nodes
Motivation
Gene silencing is a powerful strategy to probe signaling pathways and discover drug targets. RNA interference (RNAi) using short hairpin RNAs (shRNAs) offers a number of advantages over other approaches, including its inducible and reversible nature and ability to target distinct transcript variants. However, applications of RNAi have been hampered by inefficient combinatorial target knockdown and restricted shRNA induction in vivo. To overcome these limitations, we developed Multi-miR, a robust microRNA-embedded shRNA expression platform for potent inducible combination target inhibition in vitro and in vivo.
The use of RNAi to silence genes is hampered by poor in vivo performance and limited combinatorial potential. Amen et al. present an optimized RNAi platform, Multi-miR, that overcomes these limitations and achieves effective combination knockdown of redundant synthetic lethal genes and robust tumor inhibition in a mouse model of cancer.
Introduction
Loss-of-function screens enable the decoding of mammalian signaling networks and elucidation of novel therapeutic targets. RNA-guided immune systems, including RNA interference (RNAi) and CRISPR, enable programmed regulation of gene expression (Fellmann and Lowe, 2014; Fellmann et al., 2017). While CRISPR is ideal for generating irreversible genetic edits, the reversibility and simple one-component nature of RNAi tools—and their similarity to small-molecule-based target inhibition—make them a valuable alternative for target suppression. Moreover, RNAi enables knockdown of specific isoforms such as the long variant of GABPB1 (Amen et al., 2021), which cannot be accomplished using CRISPR interference (CRISPRi) systems that work by targeting transcription start sites (TSSs) (Gilbert et al., 2013). Conversely, since many genes have multiple TSSs, especially across different cell types, a single CRISPRi guide RNA often cannot target all transcript variants (Forrest et al., 2014). RNAi triggers can be designed to inhibit specific messenger RNAs (mRNAs) or suppress all variants through targeting of common regions. Particularly for use in primary cell systems, organoids, and in vivo, RNAi provides a powerful platform for controlled gene regulation (Premsrirut et al., 2011; Zuber et al., 2011a). However, the use of RNAi has been hampered by inefficient multi-target knockdown, especially beyond two dimensions, as well as inactivation of short hairpin RNA (shRNA) expression in vivo, often requiring the establishment of monoclonal sublines for clear readouts (Amen et al., 2021; Bassik et al., 2013; Chicas et al., 2010, 2012; McJunkin et al., 2011; Zuber et al., 2011b, 2011c).
Experimental RNAi enables loss-of-function genetics by programming endogenous mammalian machinery with exogenous sources of double-stranded RNA that mimic natural triggers of the pathway (Fellmann and Lowe, 2014). While shRNAs can be expressed as simple stem-loop structures from RNA polymerase III (Pol-III) promoters, such strategies can lead to off-target effects through high precursor shRNA levels and saturation of the endogenous microRNA machinery (Grimm et al., 2006; Shalem et al., 2014; Yi et al., 2005). Use of microRNA-embedded shRNAs driven by RNA polymerase II (Pol-II) promoters overcomes these limitations (Baek et al., 2014; Boudreau et al., 2008; McBride et al., 2008), especially when used at single-copy genomic integration (Fellmann et al., 2011; Yuan et al., 2014). Since target knockdown efficiency was another constraint, we previously developed a biological high-throughput sensor assay to evaluate shRNA potency, an optimized microRNA scaffold (miR-E) for efficient shRNA biogenesis, and a sequential classification algorithm (SplashRNA) that reliably predicts highly potent sequences for virtually any gene (Fellmann et al., 2011, 2013; Pelossof et al., 2017).
Here, to enable higher-order combinatorial gene regulation, we focused on better understanding the architecture of endogenous microRNA clusters and leveraged this insight to establish a programmable platform for sustained multi-target inhibition in vivo. Using this Multi-miR system, we show efficient knockdown of up to three to four target genes from a single vector without loss of potency compared with one-shRNA constructs. Additionally, we demonstrate sensitive doxycycline (dox)-mediated shRNA induction in an intracerebral glioblastoma xenograft model as well as effective in vivo suppression of a redundant synthetic lethal gene node by concomitant inhibition of all three RAF effectors in a mouse model of KRAS-mutant non-small cell lung cancer (NSCLC). Together, the Multi-miR architecture and our optimized vector system enable robust dox-controlled expression of miR-E shRNAs for higher-order combinatorial RNAi in vivo.
Results
A backbone architecture for robust multidimensional target knockdown
Tandem miR-30 vectors, where each shRNA is simply cloned one after the other, have been constructed before (Chicas et al., 2010, 2012). However, unless functionally linked to cell survival, this setup results in decreased knockdown compared with the respective single-shRNA versions. Vectors using multiple different shRNA backbones have also been built (Choi et al., 2015; Liu et al., 2008), but they complicate the approach because each subsystem needs to be individually optimized for knockdown potency. Additionally, combination screens have been used to build mammalian genetic interaction maps but were restricted to two dimensions due to library size (Bassik et al., 2013). Hence, to establish an efficient multi-shRNA system based on the well-characterized miR-E backbone (Fellmann et al., 2013), we examined the evolutionarily conserved human and mouse miR-17-92 clusters (Figure 1A). Analysis of small RNA sequencing data (Fellmann et al., 2013; Yuan et al., 2014) revealed that microRNAs from both clusters are generally well expressed (Figure S1A). Importantly, we noticed that, compared with previous miR-30 tandem setups (Chicas et al., 2010, 2012), the natural miR-17-92 clusters arrange each microRNA in much closer proximity to its neighbors. Specifically, precursor microRNAs across both clusters are separated by a relatively narrow range of 35–96 nt (average 60–63 nt), when considering canonical Drosha cleavage.
Figure 1.

Spacing is critical for efficient co-processing of clustered shRNAs
(A) Human and mouse miR-17-92 clusters. The precursor microRNAs (miRNAs) (stem-loop structures defined by Drosha processing) across both clusters are separated by 35–96 nt.
(B) Multi-miR system for combinatorial RNAi. For co-expression of several miR-E shRNAs, we modeled the Multi-miR architecture after the human and mouse miR-17-92 clusters, with 74-nt spacing between stem-loop elements. Combinatorial shRNA expression is directly linked to a fluorescence marker. X, XhoI restriction site; E, EcoRI restriction site; E, destroyed EcoRI restriction site.
(C) Shorter spacing of concatenated shRNAs improves target knockdown efficiency. Comparison between a tandem design with wider spacing and the Multi-miR system for simultaneous expression of multiple miR-E shRNAs. Shown is immunoblotting of Pten and Bcl2 in NIH/3T3 cells transduced at single copy with the indicated single, double, or quadruple miR-E shRNA constructs. Identical shRNA sequences were either concatenated using the conventional tandem design or cloned into a tighter cluster following the Multi-miR strategy. Actb was used as loading control.
(D) Sensor assay for fluorescence-based quantification of RNAi knockdown efficiency. A reporter (sensor) construct containing shRNA target sites in the 3′ UTR of a fluorescence reporter (dTomato) was stably transduced into NIH/3T3 cells. After sorting for dTomato-high, cells were transduced with GFP-containing shRNA expression vectors. Reduction of dTomato fluorescence was quantified at day 8 post transduction by flow cytometry as a readout for RNAi efficiency.
(E) Multi-miR clusters enable dual-shRNA expression without loss of knockdown potency. Select shRNAs targeting Tnks and Tnks2 were expressed alone or in combination using the tandem or Multi-miR system. RNAi efficiency was quantified according to (D). dTomato fluorescence was normalized (norm) to a non-targeting control shRNA (shCtnnb1.S2). Error bars show the standard error of the mean (SEM) (n = 3). Significance was calculated using the two-tailed Student’s t test with alpha = 0.05. ns, not significant.
To test whether this proximity affects target knockdown efficiency, we designed the Multi-miR system by mimicking the natural spacing of the miR-17-92 clusters (Figure 1B). Previous research showed that the miR-E backbone, like most microRNAs, contains and requires a 5′-DCNNC-3′ motif in its 3′ flank for efficient processing by Drosha and its cofactors (Auyeung et al., 2013; Fellmann et al., 2013). To retain the functionality of this critical motif as well as the XhoI and EcoRI restriction sites used for cloning (Fellmann et al., 2013), while approaching the natural average, we converged on an optimized 74-nt spacing (Figure S1B). As a first step to evaluate the Multi-miR design, we assessed Pten and Bcl2 knockdown by immunoblotting in a mouse fibroblast cell line (NIH/3T3) after single-copy transduction of LEPG viral vectors (Fellmann et al., 2013) expressing one, two, or four shRNAs (Table S1). Using the conventional tandem design, we observed a strong decrease in potency of shRNAs in the two- and four-shRNA vectors compared with their single-shRNA counterparts (Figure 1C). In contrast, using the Multi-miR system, only a slight decrease in potency was observed with two shRNAs, and even four-shRNA vectors showed considerable target knockdown.
To further assess differences between the tandem and Multi-miR design, we next evaluated shRNAs targeting two redundant Tankyrase genes: Tnks (GeneID: 21951) and Tnks2 (GeneID: 74493). For accurate quantitative comparison, we adapted the RNAi sensor assay (Fellmann et al., 2011) to measure target knockdown efficiency by flow cytometry via a fluorescence reporter containing shRNA target sites in its 3′ UTR (Figure 1D). After transducing NIH/3T3 cells with the dTomato reporter constructs and sorting for high dTomato expression, we transduced the cells with the respective shRNA vectors and assessed target knockdown (Figure 1E). For both Tnks and Tnks2, dual-shRNA tandem constructs showed significantly less target knockdown than their single-shRNA counterparts. Conversely, the Multi-miR system retained full knockdown potency for double-shRNA constructs compared with single shRNAs, highlighting the importance of appropriate spacing.
In order to expand this analysis, we chose another set of four genes (Alas1, Chil1, Trp63, Cdk9) and quantified their knockdown levels by qRT-PCR in NIH/3T3s upon single-copy transduction of LEPG shRNA vectors (Fellmann et al., 2013) expressing one, two, or four miR-E shRNAs from a Multi-miR cluster (Figure 2A). As observed above, target knockdown potencies using the Multi-miR system remained nearly unchanged in the presence of additional shRNAs (Figure 2B). Specifically, seven out of seven dual-shRNA Multi-miR constructs showed target knockdown efficiencies as good as or better than their single-shRNA counterparts, and four out of seven four-shRNA Multi-miR clusters showed no significant decrease in RNAi potency compared with their respective single-shRNA constructs. Additionally, target knockdown potencies were independent of the shRNA’s position within the cluster. For example, shTrp63.1 and shTrp63.2 at position one alone showed comparable potencies with when they were placed at position three in a four-shRNA Multi-miR, and shCdk9.2 alone showed similar potency to when at the end of a four-shRNA construct. Overall, this demonstrates that the Multi-miR architecture enables robust combinatorial knockdown for up to three to four dimensions.
Figure 2.

Multi-miR systems retain knockdown efficiency of up to four shRNAs
(A) Assessment of RNAi potency when expressing miR-E shRNAs either alone or in Multi-miR clusters of two to four shRNAs. All constructs were expressed using LEPG vectors. LTR, long terminal repeat promoter. Psi (ψ+), packaging signal.
(B) Target knockdown quantification by qRT-PCR of Trp63, Cdk9, Alas1, and Chil1 mRNA levels in NIH/3T3 cells transduced at single copy with the indicated single, double, or quadruple Multi-miR constructs. mRNA expression levels were normalized to shRen.713. Error bars show the SD (n = 3). Significance was calculated using the two-tailed Student’s t test with alpha = 0.05. ns, not significant. #, shAlas1.2 targets its transcripts after a proximal alternative cleavage and polyadenylation (ApA) site, explaining why it shows no knockdown. ##, the 2-shRNA vector was slightly but significantly more efficient.
Combinatorial in vivo knockdown
To assess the ability of the Multi-miR system to concomitantly suppress several genes in vivo, we delivered vectors encoding the Myc oncogene together with vectors coding for GFP-linked shRNAs targeting the tumor suppressors Trp53, Pten, or Apc individually, dual Multi-miR clusters targeting Trp53-Pten or Trp53-Apc, and triple Multi-miR clusters targeting Trp53-Pten-Apc, through hydrodynamic tail vein injection (Figure 3A). Animals were monitored for the appearance of tumors by palpation. No liver tumors were observed in C57BL/6 control mice injected with the Myc transposon only, while shTrp53;Myc (median = 65 days), shPten;Myc (median = 109 days), shApc;Myc (median = 52 days), shTrp53-shPten;Myc (median = 59 days), shTrp53-shApc;Myc (median = 36 days), and shTrp53-shPten-shApc;Myc (median = 31 days) mice succumbed to liver tumors with progressively shorter median tumor-free survival (Figures 3B and 3C). Simultaneous Multi-miR-mediated suppression of Trp53, Pten, and Apc led to a remarkable abundance of liver tumors (Figure 3C). Importantly, Multi-miR shRNAs triggered potent knockdown of all targets in tumor samples independent of the amount of shRNAs per vector (Figure 3D) and further affected downstream signaling such as abnormally high levels of β-catenin (Ctnnb1) in nuclei upon Apc knockdown (Figure S2A). Together, this demonstrates that the Multi-miR system enables highly efficient multi-target inhibition in vivo.
Figure 3.

Multi-miR mediated inhibition of tumor suppressor genes in the mouse liver
(A) Diagram depicting the generation of autochthonous liver tumor models using hydrodynamic tail vein injection. The injection delivers transposons expressing the Myc oncogene to the liver, together with GFP-linked Multi-miR shRNAs targeting multiple tumor suppressor genes (Trp53, Pten, Apc). BL6, C57BL/6 mice.
(B) Kaplan-Meier survival curves of mice treated with hydrodynamic tail vein injections of transposon vectors expressing Myc and GFP-linked miR-E shRNAs targeting Trp53, Pten or Apc, or Multi-miR shRNAs targeting Trp53-Pten, Trp53-Apc, or Trp53-Pten-Apc (n = 5). C57BL/6 mice injected with the Myc transposon and Multi-miR shTrp53-shPten-shApc succumbed to liver tumors with a median tumor-free survival of 31 days, while others produced tumors with longer median survival (Myc: no onset of disease, shTrp53;Myc = 65 days, shPten;Myc = 109 days, shApc;Myc = 52 days, shTrp53-shPten;Myc = 59 days, shTrp53-shApc;Myc = 36 days). Statistical significance was calculated using the log rank (Mantel-Cox) test (∗p < 0.05; ∗∗p < 0.01).
(C) Representative images showing typical liver tissue and tumors (top), GFP signals (middle), and hematoxylin and eosin (H&E) staining (bottom). Scale bar, 50 μm.
(D) Quantification of target knockdown in tumor samples. Total RNA was extracted from liver tumors in two independent mice (n = 2) injected with the Myc transposon and the indicated shRNA constructs. Expression levels of Trp53, Pten, and Apc were assessed by qRT-PCR. Each qRT-PCR was done in triplicate. Error bars represent the SD.
Robust dox-inducible RNAi of essential genes in the brain
Many in vivo experiments focus on identifying and validating therapeutic targets that are essential within the testing context (e.g., synthetic lethal genes). Additionally, for cancer drug discovery, it is often necessary to induce target inhibition only after tumor initiation, requiring an inducible system. Unfortunately, such setups—where escape from the testing strategy enables cell survival—pose a particular technological challenge because escaping subpopulations will expand over time and decrease the overall effect (Amen et al., 2021; McJunkin et al., 2011). These problems are compounded for multi-target inhibition. Past mitigation strategies often involved establishing monoclonal sublines with strong individual shRNA expression (Zuber et al., 2011b, 2011c), but they come with caveats due to the monoclonal nature of the approach. To overcome these limitations, we set out to generate an optimized dox-inducible, all-in-one Tet-On lentiviral vector for single or multiplexed miR-E expression (Figure 4A).
Figure 4.

A platform for efficient dox-inducible RNAi in the brain
(A) Schematic of all-in-one Tet-On lentiviral vectors for doxycycline (dox) inducible shRNA expression. Both vectors can be used with either a single miR-E shRNA or a Multi-miR cassette. TRE, tetracycline responsive element; rtTA, reverse tetracycline-controlled transactivator; PGK, phosphoglycerate kinase promoter; EFS, EF1-alpha short promoter; UCOE, ubiquitous chromatin opening element; Puro, puromycin resistance marker; IRES, internal ribosome entry site.
(B) Flow cytometry quantification of shRNA induction (GFP+) in U251 glioblastoma (GBM) cells stably transduced with the indicated vectors and treated with various concentrations of dox for 48 h. The cells had initially been transduced at single copy (<20% GFP+), followed by selection on puromycin before the assay.
(C) Dox titration curves showing sensitivity and efficiency of shRNA induction (GFP+) for the indicated vectors. Shown are data from cells partially represented in (B). Error bars indicate the SD (n = 3).
(D) Quantification (qRT-PCR) of RPA1 knockdown in U251 GBM cells expressing the indicated shRNAs from LT3GEPIR or pCF806 vectors. Cells were transduced at single copy, followed by selection on puromycin, and treatment with various concentrations of dox for 72 h. Error bars represent the SD (n = 3). Significance was calculated using the two-tailed Student’s t test with alpha = 0.05. ns, not significant. ∗∗∗p < 0.001.
(E) Schematic of intracranial GBM xenografts. U251 GBM cells stably expressing luciferase and dox-inducible, all-in-one shRNA constructs were injected into the right frontal lobe of the brain. After tumor establishment, animals were treated with dox chow (625 mg/kg), starting at day 21 post injection, and were monitored by bioluminescence imaging.
(F) Bioluminescence brain tumor imaging of mice expressing shRPA1.844 (essential gene) or shRen.713 (negative control) from LT3GEPIR or pCF806 (UT4GEPIR) vectors. Radiance is given as photons/s/cm2/sr. Significance was calculated using the two-tailed Student’s t test.
(G) Kaplan-Meier survival curves of mice expressing an essential gene-targeting shRNA (shRPA1.844) from LT3GEPIR or UT4GEPIR (pCF806) vectors. Significance was calculated using the log rank test.
Compared with previous LT3GEPIR vectors (Fellmann et al., 2013), the pCF806 (UT4GEPIR) vectors utilize a V10 variant of the reverse Tet-transactivator that is more sensitive to dox (Zhou et al., 2006) and a ubiquitous chromatin opening element (UCOE) to protect from epigenetic inactivation (Müller-Kuller et al., 2015; Zhang et al., 2010). The pCF806 vectors also feature the improved TRE3GV promoter (Clontech), a GFP marker linked to shRNA expression, a miR-E or Multi-miR cassette, an EFS promoter-driven puromycin resistance marker, and IRES2-rtTAV10. To assess the dox response of pCF806, we transduced U251 glioblastoma (GBM) cells at single copy with either LT3GEPIR or pCF806 vectors expressing a non-targeting control shRNA (shRen.713) and treated them with increasing concentrations of dox (0 ng/mL, 1 ng/mL, 2 ng/mL, 4 ng/mL, 8 ng/mL, 16 ng/mL, 31 ng/mL, 63 ng/mL, 125 ng/mL, 250 ng/mL, 500 ng/mL, 1,000 ng/mL). Overall, pCF806 showed stronger shRNA induction (monitored by GFP expression) and over 1 log-scale higher sensitivity to dox (Figures 4B and 4C), while maintaining the same minimal leakiness in absence of dox.
Given the strong inducibility of pCF806, we next decided to test the targeting of an essential gene. Replication protein A, a heterotrimeric single-stranded DNA binding protein complex composed of RPA1/2/3, is a highly conserved key essential gene that is often used as a control for functional genomics assays, especially when developing in vivo models (Amen et al., 2021; McJunkin et al., 2011; Zuber et al., 2011b). RPA1/2/3 cannot compensate for each other, making every one of them essential. Hence, we chose RPA1, designed shRNAs against it using the SplashRNA algorithm (Pelossof et al., 2017), and cloned them into LT3GEPIR and pCF806. After transducing U251 GBM cells at single copy and selecting them on puromycin, we treated the cells with the indicated concentrations of dox for 72 h and quantified target knockdown by qRT-PCR (Figure 4D). Although both vectors led to similar RPA1 knockdown at the highest dox concentration (1,000 ng/mL), LT3GEPIR-shRPA1.393, -844, and -1578 vectors did not induce any knockdown at 16 ng/mL dox, while pCF806-shRPA1.393, -844, and -1578 vectors led to significant target inhibition. This further highlights the improved dox sensitivity of pCF806. Of note, pCF806 vectors also consistently led to higher viral titers (Figure S3A).
To assess possible in vivo benefits of pCF806, we transduced luciferase-expressing U251 cells at single copy with LT3GEPIR or pCF806 vectors expressing dox-inducible shRen.713 and shRPA1.844. After puromycin selection, we injected the cells into the right frontal lobe to establish intracerebral GBM xenografts (Figure 4E). Bioluminescence monitoring showed that dox administration, after tumor establishment, had no effects on tumors encoding a negative control shRNA (shRen.713) and only slightly suppressed tumor growth when an shRNA targeting RPA1 was expressed from LT3GEPIR (Figure 4F). In contrast, expression of the same shRPA1.844 from pCF806 led to a strong reduction in tumor signal and associated moderate increase in overall survival (Figures 4F, 4G, and S3B).
Efficient combinatorial in vivo RNAi of redundant synthetic lethal gene nodes
Encouraged by the results from the intracranial GBM xenografts, we proceeded to evaluate benefits of the Multi-miR and pCF806 system for combinatorial in vivo RNAi. RAF, a set of downstream RAS effectors, is a redundant gene node that is synthetic lethal in KRAS-mutant tumors (Yuan et al., 2014). Because the three RAF effectors are redundant, and hence can compensate for each other, only complete simultaneous inhibition of A-, B-, and C-RAF (also called RAF1) can phenocopy KRAS inhibition (Figure 5A). This need for inducible, concomitant, and robust triple inhibition thus poses a critical technological challenge for the assessment of combination strategies, making it a good benchmark for testing improved systems.
Figure 5.

Combinatorial in vivo RNAi of redundant synthetic lethal gene nodes
(A) Inducible triple-RAF inhibition in a non-small cell lung cancer (NSCLC) model. KRAS-mutant H2009 NSCLC cells stably transduced with dox-inducible Multi-miR constructs targeting all three RAF effectors (shABC2) or a negative control (shRen) were injected into the flanks of mice. After tumor establishment to ∼100 mm3 (47 days), animals were treated with dox to induce shRNA expression. Tumor volumes were longitudinally monitored.
(B) Validation of ARAF, BRAF, and CRAF (also called RAF1) knockdown by immunoblotting in KRAS-mutant NSCLC H2009 cells expressing shRNAs targeting all three RAFs (shABC2 = shARAF.169-shBRAF.1296-shCRAF.387) or a negative control (shRen.713). Indicated shRNAs were expressed from either LT3GEPIR or UT4GEPIR (pCF806) vectors at single-copy genomic integration. Cells were treated with dox (1,000 ng/mL) for 72 h to induce shRNA expression prior to harvest.
(C) Tumor progression in mice treated with LT3GEPIR or UT4GEPIR (pCF806) vectors. Shown are the means and standard deviations of tumor volumes from H2009 flank tumors expressing the indicated single or multiplexed shRNAs. After tumor establishment, all mice were treated with dox to induce shRNA expression. Significance was calculated using the Mann-Whitney U test with Holm-Sidak p value adjustment. ns, not significant (alpha = 0.05).
(D) Analysis of terminal tumor size. Shown is the change in H2009 flank tumor volume at day 15 of treatment (+/− dox) compared with day 0. shRen, shRen.713 negative control; shABC2, shARAF.169-shBRAF.1296-shCRAF.387. Significance was calculated using a non-parametric one-way ANOVA (Kruskal-Wallis test) with alpha = 0.05. ns, not significant.
To evaluate the efficiency of triple-RAF inhibition, we first cloned KRAS (shKRAS.234, 355) and RAF (shARAF.381, 169; shBRAF.2015, 1296; shCRAF.2446, 387) shRNAs, which we previously designed (Fellmann et al., 2011; Yuan et al., 2014), into pCF806 as single shRNAs or triple-shRNA Multi-miR clusters (shABC1 = shARAF.381-shBRAF.2015-shCRAF.2446; shABC2 = shARAF.169-shBRAF.1296-shCRAF.387). We then validated each vector at single copy by immunoblotting in the KRAS wild-type NSCLC cell lines PC9 and H1975 (Figures S4A and S4B). While shARAF.381 did not work well alone and failed as part of the triple construct, the pCF806-shABC2 Multi-miR vector led to very strong knockdown of all three RAFs. To further measure functional consequences of target knockdown, we used a competitive proliferation assay comparing KRAS with ABC-RAF inhibition. Surprisingly, triple ABC-RAF knockdown with the stronger set of shRNAs (shABC2) fully phenocopied KRAS knockdown in KRAS-mutant H2009 NSCLC cells (Figures S4C and S4D).
We next cloned shABC2 into LT3GEPIR to compare the two vector systems. In cell culture, triple single-copy ABC-RAF knockdown in H2009 was slightly stronger when using the pCF806 vector rather than LT3GEPIR (Figure 5B). However, this relatively modest in vitro effect translated into a critical difference when looking at the consequences on tumor progression in vivo using an NSCLC flank tumor model (Figure 5C). While some LT3GEPIR-shABC2 tumors responded well to dox treatment, others escaped and showed little overall response. In contrast, all pCF806-shABC2 tumors treated with dox showed a strong response, resulting in a significant decrease in tumor volume at endpoint (Figure 5D). At the same time, tumors expressing negative controls (LT3GEPIR-shRen.713, pCF806-shRen.713) and non-treated tumors (no dox) showed no effects and continued tumor growth. Overall, the optimized pCF806 vector and Multi-miR system enabled robust dox-inducible triple-knockdown of a redundant synthetic lethal gene node in vivo, providing a blueprint for efficient higher-order combinatorial RNAi.
Discussion
RNAi provides a valuable platform for reversible loss-of-function genetics in vitro and in vivo. However, the lack of robust strategies for multi-target inhibition, as well as subpopulations of cells escaping dox-inducible target knockdown in vivo, have hindered the use of RNAi for combinatorial approaches. Here, we demonstrate that the Multi-miR architecture and pCF806 (UT4GEPIR) vector system enable robust (efficient, durable) inducible combinatorial in vivo RNAi with minimal loss of potency up to three to four dimensions, even in the context of strong selective pressure that favors escapees. Additionally, the Multi-miR system and pCF806 vector are readily compatible with the miR-E shRNA scaffold and SplashRNA design algorithm for easy integration with existing resources (Fellmann et al., 2013; Pelossof et al., 2017).
The high sensitivity to low dox concentrations of the pCF806 system not only enables an efficient response in intracranial models encumbered by the blood-brain barrier but will also be a boon for the use of lower dox concentrations in other settings to reduce possible dox-based side effects (Das et al., 2016; Moullan et al., 2015; Zhou et al., 2006). We further showed that this system is more resistant to shRNA inactivation in vivo, minimizing the emergence of escaper populations and alleviating the need for clonal populations to achieve clean readouts. Consequently, this strategy saves time by avoiding the need for monoclonal lines and grants the option to maintain a polyclonal population that better reflects clinical scenarios. Together, the robust combinatorial RNAi platform developed here will advance functional genomics to better understand gene interaction networks and accelerate the development of combination therapies.
Limitations of the study
While the Multi-miR architecture and pCF806 (UT4GEPIR) vector system showed critical advantages over prior methods in the cell culture and xenograft settings tested here, we have not yet assessed this platform in transgenic setups. Additionally, in immunocompetent animal models, it might be necessary to replace immunogenic vector components (e.g., puromycin resistance marker, rtTA) and/or tolerize the animals to our system to achieve full activity.
STAR★Methods
Key resources table
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| Bcl2 | BioLegend | Clone BCL/10C4 |
| Pten | Cell Signaling | Cat#9188 |
| β-actin | Sigma-Aldrich | Clone AC-15 |
| A-RAF | Cell Signaling Technology | Cat#75804 |
| B-RAF | Cell Signaling Technology | Cat#14814 |
| C-RAF | Cell Signaling Technology | Cat#53745 |
| KRAS | Abnova | Cat#H00003845-M01, clone 3B10-2F2 |
| ɑ-tubulin | Sigma Aldrich | Cat#T6199 |
| Rabbit anti-GFP | Cell Signaling | Cat#2956 |
| Rabbit anti-Pten | Cell Signaling | Cat#9188 |
| Mouse anti-β-catenin | BD Biosciences | Cat#610153 |
| Anti-rabbit | Vector Laboratories | Cat#MP7401 |
| Anti-mouse | Vector Laboratories | Cat#MP2400 |
| Ex perimental models: Cell lines | ||
| 293FT | Thermo Fisher Scientific | Cat#R70007; RRID:CVCL_6911 |
| U-251 | Sigma-Aldrich | Cat#09063001; RRID:CVCL_0021 |
| NIH/3T3 | ATCC | RRID:CVCL_0594 |
| H2009 | ATCC | Cat#CRL-5911; RRID: CVCL_1514 |
| H1975 | ATCC | Cat#CRL-5908; RRID:CVCL_1511 |
| PC9 | BIDMC | RRID:CVCL_B260 |
| Experimental models: Organisms/strains | ||
| Female athymic nu/nu mice | The Jackson Laboratory | RRID:IMSR_JAX:002019 |
| Female C57BL/6 mice | Harlan Laboratories | N/A |
| Oligonucleotides | ||
| Primers, shRNAs | See Table S1 | N/A |
| Recombinant DNA | ||
| Plasmid: pCF806-reci (UT4GEPIR) | This paper (Table S1) | Addgene, pCF806-reci, Cat#186711 |
| Plasmid: pCF806-shRen.713 (UT4GEPIR) | This paper (Table S1) | Addgene, pCF806-shRen.713, Cat#186712 |
| Plasmid: pCF806-shABC1 (UT4GEPIR) | This paper (Table S1) | Addgene, pCF806-shABC1, Cat#186713 |
| Plasmid: pCF806-shABC2 (UT4GEPIR) | This paper (Table S1) | Addgene, pCF806-shABC2, Cat#186714 |
| Plasmid: LT3GEPIR | Fellmann et al. (2013) | N/A |
| Software and algorithms | ||
| SplashRNA algorithm to predict shRNA sequences | Pelossof et al. (2017) | http://splashrna.mskcc.org/ |
Resource availability
Lead contact
Further information and requests for resources and reagents should be directed to and will be fulfilled by the lead contact, Christof Fellmann (christof.fellmann@gladstone.ucsf.edu).
Materials availability
Plasmids generated in this study are deposited at Addgene (pCF806-reci, #186711; pCF806-shRen.713, #186712; pCF806-shABC1, #186713; pCF806-shABC2, #186714).
Experimental model and subject details
Mammalian cell culture
All mammalian cell cultures were maintained in a 37°C incubator at 5% CO2. Phoenix HEK293T packaging cells and HEK293T human kidney cells (293FT; Thermo Fisher Scientific, #R70007; RRID:CVCL_6911) were grown in Dulbecco’s Modified Eagle Medium (DMEM; Corning Cellgro, #10-013-CV) supplemented with 10% fetal bovine serum (FBS; Seradigm #1500-500), and 100 units/mL penicillin and 100 μg/mL streptomycin (100-Pen-Strep; Gibco, #15140-122). U-251 human glioblastoma cells (Sigma-Aldrich, #09063001; RRID:CVCL_0021) were cultured in Dulbecco’s Modified Eagle Medium/Nutrient Mixture F-12 (DMEM/F-12; Gibco, #11320-033 or Corning Cellgro, #10-090-CV) supplemented with 10% FBS and 100-Pen-Strep. NIH/3T3 (ATCC; RRID:CVCL_0594) were maintained in DMEM with 10% bovine calf serum or 10% fetal bovine serum (FBS) containing 100-Pen-Strep. H2009 (ATCC, #CRL-5911; RRID: CVCL_1514) and H1975 (ATCC, #CRL-5908; RRID:CVCL_1511) NSCLC cell lines were purchased from ATCC. The PC9 (RRID:CVCL_B260) lung cancer cell line was a gift from Dr. Stephen Soltoff of Beth Israel Deaconess Medical Center. All lung cancer cell lines were cultured in Dulbecco’s Modified Eagle Medium (DMEM; Corning Cellgro, #10-013-CV) supplemented with 10% fetal bovine serum (FBS; Cellgro, #35-010-CV) and 100 units/mL penicillin and 100 μg/mL streptomycin (100-Pen-Strep; Gibco, #15140-122).
U-251 cells were authenticated using short tandem repeat DNA profiling (STR profiling; UC Berkeley Cell Culture/DNA Sequencing facility), as previously described (Amen et al., 2021). U-251 and HEK293T were tested, and confirmed negative, for mycoplasma contamination (UC Berkeley Cell Culture facility) by fluorescence microscopy of methanol fixed and Hoechst 33,258 (Polysciences, #09460) stained samples.
Animal studies
All animal procedures were carried out in compliance with respective Institutional Animal Care and Use Committee (IACUC) guidelines approved by the Memorial Sloan Kettering Cancer Center (MSKCC) IACUC, the University of California, San Francisco (UCSF) IACUC, and the Sanford Burnham Prebys Medical Discovery Institute IACUC. Experimental mice were maintained under specific pathogen free conditions.
Hydrodynamic tail vein injection
Female C57BL/6 mice (6-8 weeks of age, Harlan Laboratories) were injected with a 0.9% NaCl solution/plasmid mix into the lateral tail vein with a total volume corresponding to 10% of body weight in 5-7 s. The plasmid/sterile 0.9% NaCl solution mix was prepared containing pT3-EF1α-GFP-miR-E (20 μg) and pT3-EF1α-Myc (5 μg) transposon vectors together with CMV-SB13 Transposase (1:5 ratio) for each injection. The pT3 transposon vector was a kind gift of Dr. Xin Chen, UCSF. The miR-E shRNAs used were sh.Trp53.1224, sh.Pten.1523, sh.Apc.8745 (Table S1).
Intracerebral glioblastoma xenograft model
U-251 cells were stably transduced with Firefly Luciferase Lentifect Purified Lentiviral Particles (Genecopoiea, #LPP-FLUC-Lv105) at an MOI of 3. 4x105 cells were injected in a 4 μL total volume into the right frontal cortex (ML:1.5 mm, AP:1 mm, DV:3.5 mm) of 6-7 week old female athymic nu/nu mice (The Jackson Laboratory, RRID:IMSR_JAX:002,019). Animals were excluded from analysis if surgical complications arose immediately (within 48 h) post-surgery, or if location of injection was deemed inaccurate via luciferase imaging. Approximate tumor size was monitored via bioluminescence imaging (IVIS Lumina S5 Imaging System, PerkinElmer) 1-2 times per week following intraperitoneal injection of luciferin reagent (GOLDBIO, #LUCK-1G) at 150 mg/kg. Animals were weighed three times per week, and once daily after initial weight loss was observed. General behavior and symptomology for all mice were recorded daily. Humane endpoints for sacrifice were hunched posture, >15% weight loss from maximum recorded weight, or neurologic symptoms, and were verified by approved UCSF veterinary technicians.
Non-small cell lung cancer (NSCLC) flank tumor model
H2009 cells were transduced at single-copy using either LT3GEPIR or UT4GEPIR (pCF806) containing shRNAs targeting all three RAFs (shABC2 = shARAF.169-shBRAF.1296-shCRAF.387) or a negative control (shRen.713). This resulted in the generation of four unique H2009 cell lines (LT3GEPIR shRen.713, LT3GEPIR shABC2, pCF806 shRen.713, and pCF806 shABC2). 1 × 106 H2009 cells per condition were mixed with matrigel in a 1:1 (v/v) ratio and kept on ice prior to injection. H2009 cell/matrigel mix were injected into both abdominal flanks of 7-8 week old female athymic nu/nu mice (The Jackson Laboratory, RRID:IMSR_JAX:002,019). Tumor establishment was determined by monitoring injection sites for formation of palpable tumors and subsequent measuring using digital calipers. At 45 days post injection, tumors reached ∼70-125mm3 and mice were randomized to doxycycline treated and untreated groups. Mice in the doxycycline treatment control group were placed on doxycycline chow (Envigo Teklad #TD.01306, 625 mg/kg). To ensure robust activation, doxycycline-treated mice were supplemented with oral gavage of doxycycline (10 mg/mL) twice weekly to achieve an effective dose of 50 μg of doxycycline per gram of mouse bodyweight. Tumor measurements were performed every other day up to 15 days post treatment initiation, using digital calipers. General behavior and changes in weight were recorded every other day to ensure animal health was maintained.
Method details
Cloning of Multi-miR vectors
New miR-E shRNA sequences were predicted using the SplashRNA algorithm (Pelossof et al., 2017). The shRNA numbers refer to the position of the shRNA, specifically the 3′ nucleotide of the guide strand, on the target transcript. Single miR-E shRNAs were cloned as previously described (Dow et al., 2012; Fellmann et al., 2013; Pelossof et al., 2017). Multi-miR expression cassettes were cloned by concatenating multiple shRNAs cloned into miR-E, using LEPG (Fellmann et al., 2013), LT3GEPIR (Fellmann et al., 2013), pT3-EF1α-GFP-miR-E (Huang et al., 2014), or similar vectors. Each shRNA was cloned behind the previous one by generating a 74 nt spacing between consecutive pre-miRNAs (Figure S1B), when considering canonical Drosha cleavage sites (Fellmann et al., 2013). Specifically, existing miR-E shRNAs were PCR amplified using the primers Multi-mini-sh-fw and Multi-sh1-rev for LEPG/LT3GEPIR/UT4GEPIR (pCF806) vectors, or Multi-mini-sh-fw and Multi-sh2-rev for pT3-EF1α-GFP-miR-E vectors. Primers were ordered from IDT (Integrated DNA Technologies, Table S1). Following cleanup (QIAquick PCR Purification Kit, Qiagen), the PCR products were digested with BbsI (NEB) and MluI (NEB) in NEBuffer 2.1 (37°C) and column purified again. Destination vectors (LEPG, LT3GEPIR, UT4GEPIR (pCF806), pT3-EF1α-GFP-miR-E, or similar), already containing one or several miR-E shRNAs, were digested with EcoRI-HF (NEB) and MluI-HF (NEB) in CutSmart buffer (37°C), CIP (NEB) treated for 45 min and column purified (QIAquick PCR Purification Kit, Qiagen). The miR-E inserts were then ligated into the destination vectors (T4 DNA ligase, NEB) at a 3:1 molecular ratio, using 150 ng destination vector per ligation. After transformation, bacterial amplification and plasmid preparation, vectors were control sequenced with the primers MSCV5′, miRseq5, PGK-rev, and EFS-rev, as appropriate.
The pCF806 (UCOE-TRE-GFP-miR-E-EFS-Puro-IRES-rtTA, UT4GEPIR) vectors (Table S1) were loosely based on the lentiviral backbone of pCF525 (Watters et al., 2018). For sustained transgene expression, a modified CBX3 ubiquitous chromatin opening element (UCOE) (Zhang et al., 2010) from pMH0001 (Adamson et al., 2016) was introduced at the beginning of the expression cassette. The TRE-3GV promoter (pLVX-TRE3G, Clontech) was derived from TRE-3G in LT3GEPIR (Fellmann et al., 2013) by PCR-based modification. GFP-miR-E cassettes were subcloned from LEPG and LT3GEPIR vectors (Fellmann et al., 2013). The EFS promoter and the puromycin resistance marker were amplified from pCF226 (Oakes et al., 2019). An improved IRES2 element, derived from the encephalomyocarditis virus (EMCV, NC_001,479.1), was cloned using gBlocks (IDT). The V10 variant of rtTA (Zhou et al., 2006) was generated using gBlocks (IDT).
RNAi sensor assay
The concept of the RNAi sensor assay was described previously (Fellmann et al., 2011). Here, the sensor assay was adapted using NIH/3T3 cells. For Tnks- and Tnks2-specific reporters, shRNA target sequences for Tnks or Tnks2, respectively, were arrayed and synthesized as gBlocks (IDT), followed by cloning into the 3′UTR of a dTomato sensor vector. Following transduction of target cells, the top 20% brightest dTomato-positive cells were isolated by FACS. Sorted cells were then transduced with shRNA-expressing viral constructs at 1:10 dilution to achieve approximately single copy integration. Reduction of dTomato fluorescence was quantified at day 8 post-transduction by flow cytometry as a readout of RNAi efficiency.
Retroviral and lentiviral transduction
Retroviral particles were produced as previously described (Fellmann et al., 2011). Lentiviral particles were produced in HEK293T cells using polyethylenimine (PEI; Polysciences #23966) based transfection of plasmids, as previously described (Amen et al., 2021; Oakes et al., 2019). In brief, lentiviral vectors were co-transfected with the lentiviral packaging plasmid psPAX2 (Addgene #12260) and the VSV-G envelope plasmid pMD2.G (Addgene, #12259). Transfection reactions were assembled in reduced serum media (Opti-MEM; Gibco, #31985-070). For lentiviral particle production on 6-well plates, 1 μg lentiviral vector, 0.5 μg psPAX2 and 0.25 μg pMD2.G were mixed in 0.4 mL Opti-MEM, followed by addition of 5.25 μg PEI. After 20-30 min incubation at room temperature, the transfection reactions were dispersed over the HEK293T cells. Media was changed 12-14 h post-transfection, and virus harvested at 42-48 h post-transfection. Viral supernatants were filtered using 0.45 μm polyethersulfone (PES) membrane filters, diluted in cell culture media as appropriate, and added to target cells. Polybrene (5 μg/mL; Sigma-Aldrich) was supplemented to enhance transduction efficiency, if necessary. Transduction efficiency was usually assessed 48 h after transduction by quantification of fluorescent reporters using flow cytometry (Guava EasyCyte, Millipore or Attune NxT, Thermo Fisher Scientific). Where a specific transduction rate was desired, transductions were carried out at different dilutions and ideal dilution ratios deduced. All shRNAs were assessed at single copy genomic integration (“single copy”) by infecting target cell population at <20% of their maximal infection rate, guaranteeing <2% cells with multiple integrations (Fellmann et al., 2011). Transduced cell populations were usually selected 24-48 h after transduction, using 1.0-2.0 μg/mL puromycin (Sigma-Aldrich or InvivoGen) or 500-2000 μg/mL G418 (Geneticin, Gibco-Invitrogen).
RNA isolation and quantitative RT-PCR
For validation of Multi-miR shRNAs in NIH/3T3 cells, total RNA from cells was extracted using The RNeasy Kit and RNase-Free DNase Set (Qiagen) according to the manufacturer’s instructions. For Multi-miR mediated inhibition of tumor suppressor genes in the mouse liver, total RNA from liver tumors was extracted using the AllPrep DNA/RNA Micro Kit and RNase-Free DNase Set (Qiagen) according to the manufacturer’s instructions. cDNA synthesis was prepared from 1 μg total RNA using Taqman reverse transcription kit (Applied Biosystems, #N808-0234) with random hexamers. For comparison of LT3GEPIR and pCF806 efficacy, RNA isolation and cDNA generation was performed using the Cells-to-CT kit (Invitrogen, #4402954), per the manufacturer’s instructions. qRT-PCR analyses were carried out in technical triplicate using SYBR green (Applied Biosystems) and specific primers (Table S1). Measurements were carried out using the ViiA seven system (Life Technologies) or a QuantStudio five Real-Time PCR machine (Thermo Fisher Scientific). The mRNA expression levels were normalized to the levels of mouse Actb mRNA, or human GUSB mRNA, and quantified using the comparative CT method.
Immunoblotting
Unless otherwise stated, cells were transduced at single copy with the constitutive retroviral vector LEPG (Fellmann et al., 2013) expressing the indicated single, double or quadruple miR-E shRNA constructs. NIH/3T3 or murine tumor cell pellets were lysed in Laemmli buffer (100 mM Tris-HCl pH 6.8, 5% glycerol, 2% SDS, 5% 2-mercaptoethanol). Equal amounts of protein were separated on SDS-polyacrylamide gels and transferred to PVDF membranes. The abundance of β-actin (ACTB, Actb) was monitored to ensure equal loading. Images were analyzed using the AlphaView software (ProteinSimple) and quantified by ImageJ. Immunoblotting was performed using antibodies for Bcl2 (1:1000, BioLegend, clone BCL/10C4), Pten (1:1000, Cell Signaling, #9188), β-actin (1:10,000, Sigma-Aldrich, clone AC-15).
NSCLC cell lines (H2009, H1975, PC9) were transduced at single-copy with lentiviral vectors LT3GEPIR and UT4GEPIR (pCF806). NSCLC total cell lysates were lysed in RIPA buffer containing 1% Triton X-100, 0.1% SDS, 150 mM NaCl, 50 mM Tris-HCl pH 7.5, and Halt protease and phosphatase inhibitor cocktail (Thermo Fisher Scientific). Protein concentration was measured using BCA assay (Thermo Fisher Scientific) and 25 μg of total cell lysate were run and separated on SDS-polyacrylamide gels. Proteins were then transferred to 0.45 μM nitrocellulose membranes, which were then probed overnight with primary antibody at 4°C. Equal protein loading was verified using ɑ-tubulin as a loading control. Immunoblotting was performed using the following antibodies: A-RAF (1:1000, Cell Signaling Technology, #75804), B-RAF (1:1000, Cell Signaling Technology, #14814), C-RAF (1:1000, Cell Signaling Technology, #53745), KRAS (1:1000, Abnova, #H00003845-M01, clone 3B10-2F2), ɑ-tubulin (1:10,000, Sigma Aldrich, #T6199).
Immunohistochemistry
Mouse liver tissues fixed in 10% neutral buffered formalin (NBF) for 24 h, were embedded in paraffin and sectioned by IDEXX RADIL (Columbia, MO). Sections were rehydrated and unmasked as described previously (Dow et al., 2015). Sections were treated with 3% H2O2 for 10 min followed by a wash in deionized water (for IHC only), washed in PBS, and blocked in TBS/0.1% Triton X-100 containing 1% BSA. Primary rabbit anti-GFP (1:200, Cell Signaling, #2956), rabbit anti-Pten (1:200, Cell Signaling, #9188) and mouse anti-β-catenin (1:200, BD Biosciences, #610153) antibodies were incubated at 4°C overnight in blocking buffer solution. Sections were then incubated with anti-rabbit (Vector Laboratories, #MP7401) or anti-mouse (Vector Laboratories, #MP2400) ImmPRESS HRP-conjugated secondary antibodies and chromogen development performed using ImmPact DAB (Vector Laboratories, #SK4105). Stained slides were counterstained with Harris' hematoxylin. Immunohistochemistry images were taken on a Zeiss Axioscope Imager Z.1 using a 10X (Zeiss NA 0.3) or 20X (Zeiss NA 0.17) objective.
Quantification and statistical analysis
For all in vivo studies, Prism 9 and Microsoft Excel software were used to calculate p values. Statistical significance was calculated using the two-tailed Student’s t test (∗, p < 0.05; ∗∗, p < 0.01; ∗∗∗, p < 0.001), unless otherwise indicated. Statistical significance for Kaplan-Meier survival curves was calculated using the log rank (Mantel-Cox) test. Significance of changes in tumor volume were calculated using the Mann-Whitney U tests with Holm-Sidak p value adjustment as control for family-wise error rate (FWER) or a non-parametric one-way ANOVA (Kruskal-Wallis test), as indicated. Other specific statistical tests were used as indicated.
Acknowledgments
We thank Jennifer A. Doudna and lab members, and Scott W. Lowe and lab members, for unwavering support and insightful discussions. We are grateful to Prem K. Premsrirut, Vaishali Sridhar, Karen Y. Zhu, Xiang Li, and Darjus F. Tschaharganeh for outstanding assistance and support. This work was supported by NIH Pathway to Independence Award NIGMS K99/R00 GM118909 (C.F.), NIH Maximizing Investigators’ Research Award (MIRA) for ESI R35 GM143124 (C.F.), NCI R01 CA237536 (B.M.E.), NCI T32 CA211036 (R.M.L.), and American Cancer Society RSG-20-064-01-TBE (B.M.E.).
Author contributions
C.F. conceived and designed the study, carried out experiments, and evaluated data. A.M.A., R.M.L., and C.-H.H. designed and carried out experiments and analyzed data. R.J.L., A.R., and Y.G. carried out experiments. B.M.E. planned experiments and evaluated data. E.M.S. and L.E.D. designed, carried out, and analyzed Tankyrase sensor experiments. C.F. wrote the manuscript with help from all authors.
Declaration of interests
C.F. is a founder, former chief scientific officer, and advisor of Mirimus Inc., a company that develops RNAi-based reagents and transgenic mice. L.E.D. is an advisor and holds equity in Mirimus Inc.
Published: June 21, 2022
Footnotes
Supplemental information can be found online at https://doi.org/10.1016/j.crmeth.2022.100239.
Supplemental information
The table contains multiple sheets (tabs). shRNAs tab: name (gene name, Entrez ID, shRNA name) and sequence (97-mer) of shRNAs used. Primers tab: name and sequence of primers used for cloning, sequencing, and qRT-PCR validation. Vectors tab: name and sequence of vectors.
Data and code availability
-
•
All data reported in this paper will be shared by the lead contact upon request.
-
•
This paper does not report original code.
-
•
Any additional information required to reanalyze the data reported in this paper is available from the lead contact upon request.
References
- Adamson B., Norman T.M., Jost M., Cho M.Y., Nuñez J.K., Chen Y., Villalta J.E., Gilbert L.A., Horlbeck M.A., Hein M.Y., et al. A multiplexed single-cell CRISPR screening platform enables systematic dissection of the unfolded protein response. Cell. 2016;167:1867–1882.e21. doi: 10.1016/j.cell.2016.11.048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Amen A.M., Fellmann C., Soczek K.M., Ren S.M., Lew R.J., Knott G.J., Park J.E., McKinney A.M., Mancini A., Doudna J.A., Costello J.F. Cancer-specific loss of TERT activation sensitizes glioblastoma to DNA damage. Proc. Natl. Acad. Sci. U S A. 2021;118 doi: 10.1073/pnas.2008772118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Auyeung V.C.C., Ulitsky I., McGeary S.E.E., Bartel D.P.P. Beyond secondary structure: primary-sequence determinants license pri-miRNA hairpins for processing. Cell. 2013;152:844–858. doi: 10.1016/j.cell.2013.01.031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baek S.T., Kerjan G., Bielas S.L., Lee J.E., Fenstermaker A.G., Novarino G., Gleeson J.G. Off-target effect of doublecortin family shRNA on neuronal migration associated with endogenous microRNA dysregulation. Neuron. 2014;82:1255–1262. doi: 10.1016/j.neuron.2014.04.036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bassik M.C., Kampmann M., Lebbink R.J., Wang S., Hein M.Y., Poser I., Weibezahn J., Horlbeck M.A., Chen S., Mann M., et al. A systematic mammalian genetic interaction map reveals pathways underlying ricin susceptibility. Cell. 2013;152:909–922. doi: 10.1016/j.cell.2013.01.030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boudreau R.L., Martins I.I., Davidson B.L. Artificial MicroRNAs as siRNA shuttles: improved safety as compared to shRNAs in vitro and in vivo. Mol. Ther. 2008;17:169–175. doi: 10.1038/mt.2008.231. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chicas A., Wang X., Zhang C., McCurrach M., Zhao Z., Mert O., Dickins R.A., Narita M., Zhang M., Lowe S.W. Dissecting the unique role of the retinoblastoma tumor suppressor during cellular senescence. Cancer. Cell. 2010;17:376–387. doi: 10.1016/j.ccr.2010.01.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chicas A., Kapoor A., Wang X., Aksoy O., Evertts A.G., Zhang M.Q., Garcia B.A., Bernstein E., Lowe S.W. H3K4 demethylation by Jarid1a and Jarid1b contributes to retinoblastoma-mediated gene silencing during cellular senescence. Proc. Natl. Acad. Sci. U S A. 2012;109:8971–8976. doi: 10.1073/pnas.1119836109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Choi J.G., Bharaj P., Abraham S., Ma H., Yi G., Ye C., Dang Y., Manjunath N., Wu H., Shankar P. Multiplexing seven miRNA-Based shRNAs to suppress HIV replication. Mol. Ther. 2015;23:310–320. doi: 10.1038/mt.2014.205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- T Das A., Tenenbaum L., Berkhout B. Tet-on systems for doxycycline-inducible gene expression. Curr. Gene. Ther. 2016;16:156–167. doi: 10.2174/1566523216666160524144041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dow L.E., Premsrirut P.K., Zuber J., Fellmann C., McJunkin K., Miething C., Park Y., Dickins R.A., Hannon G.J., Lowe S.W. A pipeline for the generation of shRNA transgenic mice. Nat. Protoc. 2012;7:374–393. doi: 10.1038/nprot.2011.446. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dow L.E., Fisher J., O’Rourke K.P., Muley A., Kastenhuber E.R., Livshits G., Tschaharganeh D.F., Socci N.D., Lowe S.W. Inducible in vivo genome editing with CRISPR-Cas9. Nat. Biotechnol. 2015;33:390–394. doi: 10.1038/nbt.3155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fellmann C., Lowe S.W. Stable RNA interference rules for silencing. Nat. Cell. Biol. 2014;16:10–18. doi: 10.1038/ncb2895. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fellmann C., Zuber J., McJunkin K., Chang K., Malone C.D., Dickins R.A., Xu Q., Hengartner M.O., Elledge S.J., Hannon G.J., Lowe S. Functional identification of optimized RNAi triggers using a massively parallel sensor assay. Mol. Cell. 2011;41:733–746. doi: 10.1016/j.molcel.2011.02.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fellmann C., Hoffmann T., Sridhar V., Hopfgartner B., Muhar M., Roth M., Lai D.Y., Barbosa I.A.M., Kwon J.S., Guan Y., et al. An optimized microRNA backbone for effective single-copy RNAi. Cell Rep. 2013;5:1704–1713. doi: 10.1016/j.celrep.2013.11.020. [DOI] [PubMed] [Google Scholar]
- Fellmann C., Gowen B.G., Lin P.-C., Doudna J.A., Corn J.E. Cornerstones of CRISPR-Cas in drug discovery and therapy. Nat. Rev. Drug Discov. 2017;16:89–100. doi: 10.1038/nrd.2016.238. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Forrest A.R.R., Kawaji H., Rehli M., Baillie J.K., de Hoon M.J.L., Haberle V., Lassmann T., Kulakovskiy I.V., Lizio M., Itoh M., et al. A promoter-level mammalian expression atlas. Nature. 2014;507:462–470. doi: 10.1038/nature13182. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gilbert L.A., Larson M.H., Morsut L., Liu Z., Brar G.A., Torres S.E., Stern-Ginossar N., Brandman O., Whitehead E.H., Doudna J.A., et al. CRISPR-mediated modular RNA-guided regulation of transcription in eukaryotes. Cell. 2013;154:442–451. doi: 10.1016/j.cell.2013.06.044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grimm D., Streetz K.L., Jopling C.L., Storm T.A., Pandey K., Davis C.R., Marion P., Salazar F., Kay M.A. Fatality in mice due to oversaturation of cellular microRNA/short hairpin RNA pathways. Nature. 2006;441:537–541. doi: 10.1038/nature04791. [DOI] [PubMed] [Google Scholar]
- Huang C.H., Lujambio A., Zuber J., Tschaharganeh D.F., Doran M.G., Evans M.J., Kitzing T., Zhu N., de Stanchina E., Sawyers C.L., et al. CDK9-mediated transcription elongation is required for MYC addiction in hepatocellular carcinoma. Genes Dev. 2014;28:1800–1814. doi: 10.1101/gad.244368.114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu Y.P., Haasnoot J., ter Brake O., Berkhout B., Konstantinova P. Inhibition of HIV-1 by multiple siRNAs expressed from a single microRNA polycistron. Nucleic. Acids. Res. 2008;36:2811–2824. doi: 10.1093/nar/gkn109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McBride J.L., Boudreau R.L., Harper S.Q., Staber P.D., Monteys A.M., Martins I., Gilmore B.L., Burstein H., Peluso R.W., Polisky B., et al. Artificial miRNAs mitigate shRNA-mediated toxicity in the brain: implications for the therapeutic development of RNAi. Proc. Natl. Acad. Sci. U S A. 2008;105:5868–5873. doi: 10.1073/pnas.0801775105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McJunkin K., Mazurek A., Premsrirut P.K., Zuber J., Dow L.E., Simon J., Stillman B., Lowe S.W. Reversible suppression of an essential gene in adult mice using transgenic RNA interference. Proc. Natl. Acad. Sci. U S A. 2011;108:7113–7118. doi: 10.1073/pnas.1104097108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moullan N., Mouchiroud L., Wang X., Ryu D., Williams E.G., Mottis A., Jovaisaite V., Frochaux M.V., Quiros P.M., Deplancke B., et al. Tetracyclines disturb mitochondrial function across eukaryotic models: a call for caution in biomedical research. Cell Rep. 2015;10:1681–1691. doi: 10.1016/j.celrep.2015.02.034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Müller-Kuller U., Ackermann M., Kolodziej S., Brendel C., Fritsch J., Lachmann N., Kunkel H., Lausen J., Schambach A., Moritz T., Grez M. A minimal ubiquitous chromatin opening element (UCOE) effectively prevents silencing of juxtaposed heterologous promoters by epigenetic remodeling in multipotent and pluripotent stem cells. Nucleic. Acids. Res. 2015;43:1577–1592. doi: 10.1093/nar/gkv019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oakes B.L., Fellmann C., Rishi H., Taylor K.L., Ren S.M., Nadler D.C., Yokoo R., Arkin A.P., Doudna J.A., Savage D.F. CRISPR-Cas9 circular permutants as programmable scaffolds for genome modification. Cell. 2019;176:254–267.e16. doi: 10.1016/j.cell.2018.11.052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pelossof R., Fairchild L., Huang C.-H., Widmer C., Sreedharan V.T., Sinha N., Lai D.-Y., Guan Y., Premsrirut P.K., Tschaharganeh D.F., et al. Prediction of potent shRNAs with a sequential classification algorithm. Nat. Biotechnol. 2017;35:350–353. doi: 10.1038/nbt.3807. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Premsrirut P.K.K., Dow L.E.E., Kim S.Y.Y., Camiolo M., Malone C.D.D., Miething C., Scuoppo C., Zuber J., Dickins R.A.A., Kogan S.C.C., et al. A rapid and scalable system for studying gene function in mice using conditional RNA interference. Cell. 2011;145:145–158. doi: 10.1016/j.cell.2011.03.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shalem O., Sanjana N.E., Hartenian E., Shi X., Scott D.A., Mikkelsen T.S., Heckl D., Ebert B.L., Root D.E., Doench J.G., Zhang F. Genome-scale CRISPR-Cas9 knockout screening in human cells. Science. 2014;343:84–87. doi: 10.1126/science.1247005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Watters K.E., Fellmann C., Bai H.B., Ren S.M., Doudna J.A. Systematic discovery of natural CRISPR-Cas12a inhibitors. Science. 2018;362:236–239. doi: 10.1126/science.aau5138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yi R., Doehle B.P., Qin Y., Macara I.G., Cullen B.R. Overexpression of exportin 5 enhances RNA interference mediated by short hairpin RNAs and microRNAs. RNA. 2005;11:220–226. doi: 10.1261/rna.7233305. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yuan T.L., Fellmann C., Lee C.-S., Ritchie C.D., Thapar V., Lee L.C., Hsu D.J., Grace D., Carver J.O., Zuber J., et al. Development of siRNA payloads to target KRAS-mutant cancer. Cancer Discov. 2014;4:1182–1197. doi: 10.1158/2159-8290.cd-13-0900. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang F., Frost A.R., Blundell M.P., Bales O., Antoniou M.N., Thrasher A.J. A ubiquitous chromatin opening element (UCOE) confers resistance to DNA methylation-mediated silencing of lentiviral vectors. Mol. Ther. 2010;18:1640–1649. doi: 10.1038/mt.2010.132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou X., Vink M., Klaver B., Berkhout B., Das A.T. Optimization of the Tet-On system for regulated gene expression through viral evolution. Gene Ther. 2006;13:1382–1390. doi: 10.1038/sj.gt.3302780. [DOI] [PubMed] [Google Scholar]
- Zuber J., Shi J., Wang E., Rappaport A.R., Herrmann H., Sison E.A., Magoon D., Qi J., Blatt K., Wunderlich M., et al. RNAi screen identifies Brd4 as a therapeutic target in acute myeloid leukaemia. Nature. 2011;478:524–528. doi: 10.1038/nature10334. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zuber J., McJunkin K., Fellmann C., Dow L.E., Taylor M.J., Hannon G.J., Lowe S.W. Toolkit for evaluating genes required for proliferation and survival using tetracycline-regulated RNAi. Nat. Biotechnol. 2011;29:79–83. doi: 10.1038/nbt.1720. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zuber J., Rappaport A.R., Luo W., Wang E., Chen C., Vaseva A.V., Shi J., Weissmueller S., Fellmann C., Taylor M.J., et al. An integrated approach to dissecting oncogene addiction implicates a Myb-coordinated self-renewal program as essential for leukemia maintenance. Genes Dev. 2011;25:1628–1640. doi: 10.1101/gad.17269211. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
The table contains multiple sheets (tabs). shRNAs tab: name (gene name, Entrez ID, shRNA name) and sequence (97-mer) of shRNAs used. Primers tab: name and sequence of primers used for cloning, sequencing, and qRT-PCR validation. Vectors tab: name and sequence of vectors.
Data Availability Statement
-
•
All data reported in this paper will be shared by the lead contact upon request.
-
•
This paper does not report original code.
-
•
Any additional information required to reanalyze the data reported in this paper is available from the lead contact upon request.