

Summary
Here, we present a methodology based on multiplexed fluorescence screening of two- or three-dimensional cell cultures in a newly designed multichambered microwell chip, allowing direct assessment of drug or immune cell cytotoxic efficacy. We establish a framework for cell culture, formation of tumor spheroids, fluorescence labeling, and imaging of fixed or live cells at various magnifications directly in the chip together with data analysis and interpretation. The methodology is demonstrated by drug cytotoxicity screening using ovarian and non-small cell lung cancer cells and by cellular cytotoxicity screening targeting tumor spheroids of renal carcinoma and ovarian carcinoma with natural killer cells from healthy donors. The miniaturized format allowing long-term cell culture, efficient screening, and high-quality imaging of small sample volumes makes this methodology promising for individualized cytotoxicity tests for precision medicine.
Keywords: precision medicine, drug screening, cell therapy, immunotherapy, natural killer cells, tumor spheroids, tumor on a chip, combinatorial drug therapy, microwell
Graphical abstract
Highlights
-
•
A multichambered microwell chip is presented for miniaturized live cell assays
-
•
Assays combine long-term, multiplexed screening with high-resolution imaging
-
•
Drug and immune cell cytotoxicity are evaluated in 2D and 3D cancer cell cultures
-
•
The method is aimed at precision medicine, cell therapy, or drug development
Motivation
Personalized testing of drugs or immunotherapy is vital for the implementation of precision medicine. Although the field has developed tremendously in recent years, there is still a lack of methods that take advantage of benefits, such as low consumption of patient material or reagents, offered by miniaturized in vitro analysis systems.
In this work, Sandström et al. have developed a miniaturized method for high-content screening of drug and immune cell sensitivity of cancer cells. It is advantageous for accurate, time-resolved, and high-resolution analysis of dynamic cellular events and is well suited for applications within personalized medicine.
Introduction
The goal of precision medicine is to provide healthcare with individualized treatments based on analysis of patient-specific biomarkers (Tsimberidou et al., 2020) that predict susceptibility to anticancer drugs or immunotherapy. Data-driven searches for new biomarkers based on sequencing or proteomics data will not replace functional test but instead generate new hypotheses that will require in vitro validation, increasing the need for high-content screening with readouts based on functional responses. One area where it has proven challenging to predict patient responses using biomarkers is immunotherapy (Ameratunga et al., 2018; Chyuan et al., 2021; Fujii et al., 2018; Maleki Vareki, 2018; Maleki Vareki et al., 2017), where most treatments show significant percentages of non-responders across multiple cancer types (Borcoman et al., 2019; Sharma et al., 2017). Therefore, development of new methods for validating individualized treatments is urgently needed to personalize immunotherapy and to advance precision medicine.
A key to reach this goal is the development of improved cell culture tools that recreate in vivo environment and allow direct assays using primary tumor and immune cells (Navin et al., 2020). Three-dimensional (3D) cell cultures like tumor spheroids have potential to mimic the tumor microenvironment and thus better recapitulate and predict the response of anticancer therapy (Hu et al., 2021; Kitaeva et al., 2020). There are multiple approaches to culture tumor spheroids, but the majority of techniques are based on gravity, low adhesion, and curved surfaces, such as ultra-low attachment plates and hanging drop cultures, or more active approaches, such as magnetic aggregation or stirring culture vessels (Nath and Devi, 2016). More advanced models that also include components of the tumor stroma have been developed for evaluating drug and immune cell responses, although with limited screening capabilities (Ayuso et al., 2021; Cui et al., 2020; Nashimoto et al., 2020). Despite recent developments of sophisticated cell culture tools combined with live cell imaging readout (Ayuso et al., 2019, 2021; Berg et al., 2019; Gopal et al., 2021; Lemon and McDole, 2020; Lin et al., 2020; Olofsson et al., 2018; Park et al., 2019; Shelton et al., 2021; Wen et al., 2020; Zhou et al., 2020), it is still rare with assays that manage to combine high-content screening over long periods of time with sufficient imaging quality to examine cellular behavior with single-cell resolution.
Cytotoxicity screening is often done in different variants of multiwell plates, which benefit from a standardized format and compatibility with imaging instruments and liquid-handling robots. They combine ease of use with possibilities for multiplexing and screening. However, plastic multiwell plates and U-shaped wells that are designed for 3D spheroid cultures often suffer from low optical quality. Furthermore, the large bottom area of the wells limits live cell imaging analysis of dynamic cell events since cells of interest can move out of the field of view (FoV) between time points. Moreover, most assays consume relatively large volumes of expensive reagents and precious samples, such as primary cells from tumor biopsies or blood that can be difficult to retrieve and may only exist in low quantities.
The development of miniaturized systems, i.e., microwell and microfluidic lab-on-chip devices, has enabled advanced live-cell analyses in controlled microenvironments (Sinha et al., 2018). Microwells in particular feature stationary and geometrically confined environments where single cells (Guldevall et al., 2010, 2016; Kim et al., 2019; Varadarajan et al., 2011; Yuan and Sims, 2016; Zhou et al., 2020) or small populations of cells (Chao et al., 2020; Christakou et al., 2013; Fang et al., 2019; Prager et al., 2019; Yang et al., 2017) are contained and imaged. In contrast to conventional multiwell plates, the confined space available in microwells facilitates localization, monitoring, and tracking of the cells. This is particularly useful for efficient high-throughput screening and time-lapse imaging at the single-cell level (Desalvo et al., 2020; Zhou et al., 2020). In addition, manual handling of the chip or rapid microscopy stage movements during screening have little or no influence on the position of cells inside the microscale wells, where the liquid remains stationary. However, many microwell devices have limitations in terms of long-term (>24 h), live cell assays (Desalvo et al., 2020; Kim et al., 2019; Zhou et al., 2020); simultaneous testing of multiple or multiplexed liquid conditions (Desalvo et al., 2020; Fang et al., 2019; Guldevall et al., 2016; Kim et al., 2019; Olofsson et al., 2018; Varadarajan et al., 2011; Yang et al., 2017; Zhou et al., 2020); optical quality of the chip (Chao et al., 2020; Desalvo et al., 2020; Fang et al., 2019; Varadarajan et al., 2011; Yang et al., 2017; Zhou et al., 2020); and possibility for both 2D and 3D cell cultures (Chao et al., 2020; Desalvo et al., 2020; Fang et al., 2019; Guldevall et al., 2016; Kim et al., 2019; Varadarajan et al., 2011; Zhou et al., 2020).
Here, we present a set of miniaturized cytotoxicity screening assays based on a new multichambered microwell chip that offers the unique and flexible combination of rapid and multiplexed cytotoxicity screening at low magnification of 2D and 3D cell cultures with detailed analysis of single cells or intracellular proteins of interest at high resolution, all in a single experiment. Four different screening assays for drug and immune cell cytotoxicity are reported. First, we demonstrate concentration response of an ovarian cancer cell line treated with the commonly used microtubule-stabilizing drug paclitaxel. Secondly, a long-term multiplexed drug cytotoxicity screen is performed using tumor spheroids from ovarian or non-small cell lung cancer cell lines, with time-resolved analysis of viability and apoptosis. Thereafter, we show long-term cytotoxicity screening of primary immune cells against renal carcinoma spheroids. Lastly, we perform combinatorial treatments of ovarian cancer spheroids to evaluate the synergetic anticancer effects between different drugs and primary immune cells. With this work, we demonstrate a methodology with potential applications in drug development and precision medicine.
Results
Chip design for multiplexed assays in 2D and 3D live cell cultures
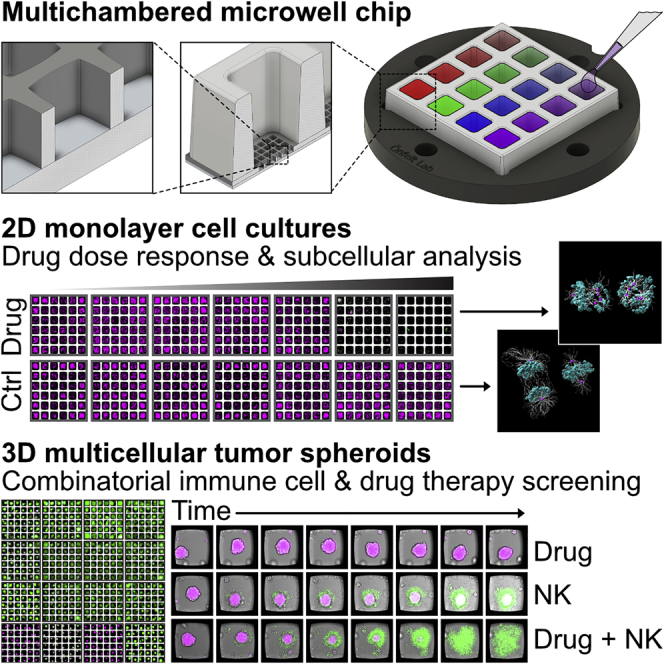
The multichambered microwell chip presented herein consists of two parts: a silicon (Si)-glass microwell array chip and a matching multichambered polymer frame. The microwell chip with frame has a footprint of 22 × 22 mm2 and includes 16 separate liquid media chambers (Figure 1A). Each chamber has a bottom width of 3.2 mm, a height of 5 mm, and a total volume of 58 μL (Figure 1B). The liquid volume used in each chamber in this work was typically 40 μL. The bottom of each chamber contains an array with 6 × 6 microwells, giving a total number of 576 wells per chip. The microwells have a width of 350 μm, a height of 300 μm, and a volume of 34 nL. Every microwell can accommodate a cell culture that is physically confined but shares the same liquid as the neighboring microwells in the chamber. Thus, the chip can provide 36 physically separated cell culture replicates in 16 different liquid conditions. In contrast, a conventional 384-well plate accommodates a single cell culture per well, in a liquid volume that is similar or larger compared with what is used for the 36 cultures in a chamber on the chip.
Figure 1.

Design of the microwell chip platform
(A) Illustration of the multichambered microwell chip, including numbers and letters to indicate the columns and rows of the 4 × 4 chambers.
(B) Cross-section illustrations of the chip with notations of dimensions and materials. Left: a single chamber (position D1) with a microwell array at the bottom is shown. Middle: a microwell array is shown. Right: a single microwell is shown.
(C) Illustration showing the chip placed in a Ø 35-mm-round chip holder (the colored chambers illustrate the use of different experimental liquid conditions in each chamber). The chambers are accessible for pipetting from above, and imaging is done from below by an inverted microscope.
(D) Illustration of the chip holder alone, with its 22-mm-wide slot for the chip visible in the middle of the holder.
(E and F) Photographs of the multichambered microwell chip (E) and of the Si-glass microwell chip without a bonded polymer frame (F).
(G and H) Scanning electron micrographs of a 6 × 6 microwell array in the bottom of a chamber on the chip (G) and of a single 350 μm microwell (H).
(I) Illustrations with a top-perspective view of the formation of 2D and 3D cell cultures in a microwell. From left to right: seeding of cells into a microwell, a 2D monolayer cell culture, formation of a cell aggregate using USW in the microwell, and the resulting 3D spheroid cell culture are shown.
The flat bottom of the microwell chip is made of 175 μm glass with comparable optical quality to #1.5H microscopy coverslips, which enables high-resolution imaging. The well array layout on the chip was designed for rapid and efficient screening, but if a specific experimental condition needs to be more closely examined, the chip allows for imaging at high magnification of selected wells.
The chip is placed in a chip holder of a standard format that fits in common microscopy stage inserts and mounting frames (Figure 1C). The holder has a central slot to accommodate the microwell chip, which rests on a small step around a centrally positioned open window in the holder (Figure 1D). The window enables direct access to the underside of the chip for low- or high-magnification objectives of an inverted microscope.
An assembled multichambered microwell chip is shown in Figure 1E, and a Si-glass microwell chip without a bonded polymer frame is shown in Figure 1F. Scanning electron micrographs of the microwell arrays (Figure 1G) and individual microwells (Figure 1H) confirmed that the arrays of wells were uniform. The glass floor and Si walls of the microwells had no visible defects, apart from the typical surface structures on the Si sidewalls from the etching process. The chips were cleaned and re-used multiple times for the experiments in this work, with no observed chemical and biological residuals from previous experiments or deterioration of the chip.
The microwell chips can be used for either 2D or 3D cell cultures (Figure 1I) with typically 5–20 × 103 cells seeded in each chamber, corresponding to a few hundred cells per microwell, depending on the application. The well shape and chip materials were designed for ultrasound-assisted formation of tumor spheroids (Christakou et al., 2015; Olofsson et al., 2018; Vanherberghen et al., 2010). Time-averaged acoustic radiation forces in an ultrasonic standing wave (USW) rapidly move the cells into a centrally located aggregate, which matures into a tumor spheroid over time. Addition of 2-methcryloxypropyl phosphorylcholine (MPC), 3-methacryloxypropyl trimethoxysilane (MPTMSi), and 3-(methacryloyloxy)propyl-tris(trimethylsilyloxy) silane (MPTSSi), a non-adherent coating of the chip, helps the formation of symmetrical spheroids that do not stick to the bottom. This coating method has shown to be very efficient and non-toxic for forming spheroids of various cell types (Olofsson et al., 2018), and it also produces bioinert and drug-repellant surface properties of polydimethylsiloxane (PDMS) (Nagahashi et al., 2015; Uen et al., 2020).
Cell seeding and optimization of multiplex staining of 2D cell cultures
Renal carcinoma A498-red fluorescent protein (RFP) cells were seeded in each chamber of the microwell chip, fixed, and stained for F-actin and nuclei. In the same experiment, immunostaining of microtubules was performed by combining the titration of primary and secondary antibodies. The chip was screened by confocal microscopy, and the number of nuclei was assessed in each well in each chamber (Figures S1A and S1B). No significant differences in the number of nuclei and in the fluorescence intensity of RFP, F-actin, and DAPI were observed across wells and chambers, demonstrating that the cells distributed evenly in the multichambered microwell chip (Figures S1B and S1C). The fluorescence signal from α-tubulin was measured, the optimal antibody concentration was assessed (Figures S1D and S1E), and high-resolution multi-color imaging provided a detail visualization of the microtubule and actin filaments (Figure S1F). To assess the benefit of having multiple wells in each chamber, we performed statistical analysis, where increasing numbers of wells were included (Figures S2A–S2D). These experiments showed that the chip could be used for robust cell seeding and culture, multiplexed staining, screening, and detailed analysis at cellular and subcellular levels. It also provided an optimized protocol for immunostaining of microtubules that was used in the assay described in the next section.
Concentration response assay for 2D monolayers of ovarian cancer cells treated with paclitaxel
Next, we performed a drug-response assay targeting ovarian cancer cells with different concentrations of paclitaxel, a microtubule-targeting agent that is commonly used to treat several types of cancers, including ovarian or breast carcinoma and non-small cell lung carcinoma (Ma et al., 2021; Weaver, 2014). Its primary mechanism of action is to prevent microtubule depolymerization with consequent cell-cycle arrest and death. A secondary mechanism of action is apoptosis due to metabolic stress and mitochondrial damage (Gallego-Jara et al., 2020; Nawara et al., 2021).
We exposed OVCAR8 cells, cultured as 2D monolayers in the microwells, to paclitaxel (Figure 2A). The response to treatment was evaluated by live-cell imaging for 72 h, assessing cell viability by measuring the intensity of the dyes tetramethylrhodamine, methyl ester (TMRM) (viability) (Lemasters et al., 1999) and caspase-3/7 (apoptosis) (Cen et al., 2008) over time. At the 72 h endpoint of the assay, drastic decrease of TMRM intensity was observed for paclitaxel concentrations higher than 1 × 10−7 M (Figure 2A). We also noticed decreased TMRM signals in the control with highest DMSO concentration (1 v/v%; Figure 2A), so this condition was excluded from further analysis. The background corrected and normalized TMRM signal was used to define the specific viability. A concentration response curve of the specific viability at 72 h fitted to the Hill equation gave an inhibitory concentration (IC50) of 1 × 10−7.8 M (Figure 2B).
Figure 2.

Concentration response assay for 2D monolayers of ovarian cancer cells treated with paclitaxel
(A) Representative images of OVCAR8 2D monolayer culture in the multichambered microwell chip 72 h after the addition of paclitaxel (upper panel) or DMSO control (lower panel) at the concentrations indicated. Cells were stained with the viability dye TMRM (magenta) and the apoptosis indicator caspase-3/7 (green).
(B) Specific viability after 72 h for OVCAR8 cells treated with paclitaxel at increasing concentration fitted with the Hill equation to evaluate IC50. Dotted line represents the predicted curve with R2 = 0.99, LogIC50 = −7.77, and H value = 1.34.
(C) Time course of specific viability of OVCAR8 cells exposed to different paclitaxel concentrations for 72 h.
(D) Time course of the specific apoptotic index (A.U., arbitrary unit).
(E) Confocal images of OVCAR8 cells cultured for 8 h with 1 × 10−6 M paclitaxel (upper panel) or without drug (lower panels). 3D rendering was performed in Imaris using conventional settings (left panel) showing microtubules (α-tubulin, gray) and nuclei (DAPI, cyan) or surface rendering with filament analysis (right panel) to visualize the poles of the mitotic spindles (dots in magenta).
(F) Fraction of multipolar OVCAR8 cells after 8 h cell culture in increasing concentrations of paclitaxel (n = 36 per concentration). Statistical method was one-way ANOVA followed by Dunnett’s post hoc test (∗∗∗∗p ≤ 0.0001).
(G) Example images of tubulin and nuclei staining of OVCAR8 cells cultured for 8 h in different concentrations of paclitaxel.
Plotting the specific viability over time showed that paclitaxel doses ≥1 × 10−8 M reduced OVCAR8 viability to different extents and with different timing (Figure 2C). Untreated cells, as well as cells exposed to paclitaxel concentrations lower than 1 × 10−8 M, kept proliferating, indicated by increased signals of TMRM over time. In fact, small doses of paclitaxel (≤1 × 10−9 M) seemed to increase proliferation and thus the specific viability relative to controls (Figure 2C). The apoptotic index, calculated from the background corrected and normalized caspase-3/7 signal, showed transient signals of various amplitudes for paclitaxel concentrations ≥1 × 10−7 M, while little apoptosis was observed for lower concentrations (Figure 2D).
Chromosome mis-segregation is one of the main mechanisms of paclitaxel-induced cytotoxicity (Nawara et al., 2021; Zasadil et al., 2014). To quantify this, OVCAR8 cells treated with paclitaxel for 8 h, i.e., before the onset of substantial cell death, were stained for α-tubulin and DNA following the optimized protocol from the previous section (Figure S1) and imaged by confocal microscopy. Cells treated with 1 × 10−6 M of paclitaxel showed up to five spindle poles, distributed in multiple directions causing aberrant chromosomal segregation, which was not observed on control cells (Figure 2E; Videos S1 and S2). Consistent with the viability data, the highest fractions of multipolar cells were observed for paclitaxel concentrations ≥1 × 10−7 M (Figures 2F and 2G).
3D rendered confocal images of OVCAR8 cells cultured for 8 h with 1 × 10−6 M paclitaxel. Rendering was performed in Imaris using conventional settings showing microtubules (α-tubulin, gray) and nuclei (DAPI, cyan), followed by surface rendering with filament analysis to visualize the poles of the mitotic spindles (dots in magenta).
3D rendered confocal images of OVCAR8 cells cultured for 8 h without paclitaxel. Rendering was performed in Imaris using conventional settings showing microtubules (α-tubulin, gray) and nuclei (DAPI, cyan), followed by surface rendering with filament analysis to visualize the poles of the mitotic spindles (dots in magenta).
Here, we performed a concentration-response drug assay, quantifying the viability and the apoptotic index of tumor cells exposed to paclitaxel. We demonstrated that our multichambered microwell chip is compatible with live imaging over 72 h, allowing the possibility to study the dynamics of drug-induced cytotoxicity as well as high-resolution imaging to characterize drug-induced phenotypic abnormalities in treated cells.
Ultrasound-assisted tumor spheroid formation followed by multiplexed drug screening
To perform drug screening assays with 3D cell cultures, we used USW-assisted formation of tumor spheroids previously developed in our labs (Christakou et al., 2015; Olofsson et al., 2018, 2021; Vanherberghen et al., 2010; Wiklund et al., 2014). A498 renal carcinoma cells were seeded and exposed to ultrasound for 24 h, followed by 24 h of standard cell culture. Viable spheroids were obtained over the whole multichambered microwell chip, and analysis of three independent experiments revealed insignificant differences in A498 spheroid size and viability between different chambers and wells (Figures S3A and S3B). Very low numbers of dead tumor cells were detected when dissociated spheroids were analyzed by flow cytometry, indicating that the spheroids contained no necrotic cores (Figure S3C). This observation is consistent with the small size of the spheroids used here (∼155 μm in diameter), since for most cancer cell lines, necrotic cores become more prevalent in spheroids with diameter >500 μm (Nath and Devi, 2016).
Tumor spheroids from two non-small cell lung cancer cell lines (NCIH-1975 and HCC827) and two ovarian adenocarcinoma cell lines (Kuramochi and OVCAR8) were formed in the chip and treated with the following drugs: A-1155463, a highly potent and selective BCL-xl inhibitor; gefitinib, an epidermal growth factor receptor (EGFR)-specific tyrosine kinase inhibitor (TKI); and paclitaxel, the microtubule-stabilizing agent used in the previous section. This combination of cell lines and drugs, together with the concentrations to use, was based on drug sensitivity data previously collected from 2D cell cultures, drug sensitivity databases, and literature research (Figure S3D; https://www.cancerrxgene.org/; Lee et al., 2012). Our 2D data predicted sensitivity to gefitinib for HCC827, to A-1155463 for Kuramochi, and to paclitaxel for OVCAR8 (Figure S3D). NCIH-1975 was predicted to be less sensitive to gefitinib compared with HCC827 (Lee et al., 2012). An experimental scheme is shown in Figure S3E. While tumor spheroids from all cell lines showed high degree of viability at the beginning of the experiment, various extent of cell death was evident after 48 h (Figures 3A and 3B). Partial cell death was observed in NCIH-1975 treated with A-1155463 and gefitinib after 48 h, while paclitaxel had no effect. As predicted, high sensitivity to gefitinib was found for HCC827, with almost no live cells detected in the wells after 48 h. Kuramochi showed partial response to A-1155463 and gefitinib. OVCAR8 responded to A-1155463 and paclitaxel (Figures 3A and 3B).
Figure 3.

Multiplexed drug screening targeting tumor spheroids
(A) Mosaic image of tumor spheroids labeled with TMRM (magenta) and caspase-3/7 (green) distributed in the multichambered microwell chip 48 h after the addition of drugs. Spheroids of the cell lines NCIH-1975, HCC827, Kuramochi, and OVCAR8 were cultured for 48 h before exposed to A-1155463 (10 μM), gefitinib (10 μM), paclitaxel (1 μM), or DMSO for another 48 h.
(B) Images of representative wells showing spheroid viability before the addition of the drugs and after 48 h.
(C) Time course of specific viability for tumor spheroids treated with A-1155463 (pink), gefitinib (teal), and paclitaxel (violet), with each cell line plotted in a separate graph.
(D) Time course of apoptotic index for tumor spheroids treated with A-1155463 (pink), gefitinib (teal), and paclitaxel (violet), with each cell line plotted in a separate graph.
(E) Total apoptosis for each drug organized in bar plots for each cell line. Values of the specific viability, apoptotic index, and total apoptosis represent means from 36 tumor spheroids, and the error bars represent SEM. Statistical analysis was done using one-way ANOVA followed by Dunnett’s post hoc test (n = 36, ∗∗p ≤ 0.01; ∗∗∗∗p ≤ 0.0001; ns, not significant).
Following the specific viability and apoptotic index over time revealed that A-1155463 in general induced fast responses (within a few hours) in the sensitive cell lines (NCIH-1975, Kuramochi, and OVCAR8), while the responses to gefitinib (NCIH-1975, HCC827, and Kuramochi) and paclitaxel (OVCAR8) were delayed (Figures 3C and 3D). The apoptotic index reflects the number of apoptotic cells present at a given time point, making the signal transient and therefore sensitive to the timing of assessment. As an alternative quantification of apoptotic cell death, we estimated the total apoptosis as the area under the curve of the caspase-3/7 signal during the 48 h (Figure 3E). This confirmed that NCIH-1975, Kuramochi, and OVCAR8 were sensitive to A-1155463, while HCC827 was sensitive to gefitinib. However, it also highlighted differences between the two reporter dyes, as the specific viability at 48 h suggests that NCIH-1975 is equally sensitive to A-1155463 as gefitinib (Figure 3C), while the total apoptosis only shows the sensitivity towards A-1155463 (Figure 3E). We used the HCC827 total apoptosis data to study how the number of analyzed wells affected the statistical significance of the data (Figures S2E–S2H). Whereas only a few wells were needed to reach significant differences between gefitinib and DMSO, higher numbers of wells were needed before combinations of wells indicating significant differences appeared for the other conditions.
The high-quality imaging data from the microwells allowed us to determine that the caspase-3/7 signal was homogeneously distributed in OVCAR8 spheroids treated with A-1155463 for 48 h (Figure S3F), confirmed by 3D confocal imaging of optically cleared spheroids (Video S3). Control spheroids showed low caspase-3/7 signal throughout (Video S4). This shows that the control spheroids were viable without a necrotic core, and it indicates that A-1155463 can diffuse into the spheroid and kill cells in the center. In contrast, paclitaxel-induced apoptosis was confined to the periphery of OVCAR8 spheroids (Figure S3G). Using image-analysis-based DNA content quantification, we have previously shown that cells in an active cell cycle stage are more common in the cell layers close to the spheroid surface (Olofsson et al., 2021). Thus, it is possible that paclitaxel-induced apoptosis is preferentially observed in dividing cells in the periphery of the spheroids.
3D rendered OVCAR8 spheroids after incubation with A-1155463. Spheroids were treated for 48 h, followed by fixation, DAPI counterstaining and spheroid clearing. 3D imaging was performed by confocal microscopy and rendering was done in Imaris using conventional settings to visualize the distribution of apoptotic cells (Caspase-3/7, green) among the total cells (DAPI, gray) within the spheroids. Apoptotic cells were uniformly distributed in the tumor spheroid treated with A-1155463.
3D rendered OVCAR8 spheroids after incubation with DMSO. Spheroids were treated for 48 h, followed by fixation, DAPI counterstaining and spheroid clearing. 3D imaging was performed by confocal microscopy and rendering was done in Imaris using conventional settings to visualize the distribution of apoptotic cells (Caspase-3/7, green) among the total cells (DAPI, gray) within the spheroids. Low and sparse caspase signal was detected in the sample, indicating the absence of necrotic core formation in the spheroids.
Here, we demonstrated the possibility of combining the production of tumor spheroids from various cell lines with direct testing of multiple drugs. By following the dynamics of target cell viability and apoptosis complemented with optical clearing and 3D confocal imaging, we could get insight into the mechanisms of drug-induced cell death.
Immune cell killing of renal carcinoma multicellular tumor spheroids over 64 h
Immunotherapy is one of the major breakthroughs in cancer therapy in the last decade. One of the main challenges is to improve the efficacy of immunotherapy in solid tumors, and to do so, relevant in vitro model systems and assays are needed. For this purpose, we developed a long-term live-imaging 3D immune cell cytotoxicity assay in our multichambered microwell chip.
Natural killer (NK) cells were chosen as effector cells, due to their ability to rapidly recognize and kill tumor cells without histocompatibility leukocyte antigen (HLA) class I restriction, and A498 renal carcinoma spheroids were used as target cells. Figure S4A gives a schematic overview of the experiment. In presence of NK cells (Figure S4B), A498 spheroids gradually lost the TMRM signal, while the caspase-3/7 signal increased (Figure 4A; Video S5). In the absence of NK cells, no apparent variations of TMRM and caspase-3/7 signal were observed (Figure 4A; Video S6). Due to the high image quality reached in the chip, it was possible to get detailed views of individual NK cells inducing target cell apoptosis in the outer layer of the A498 spheroids (Figure 4B). NK-cell-mediated A498 spheroid cytotoxicity was visually detected to various degrees in the 36 spheroids, while no death was observed in the control chamber (Figure 4C).
Figure 4.

NK-cell-mediated cytotoxicity of renal carcinoma spheroids
(A) Selected images from time-lapse sequences from two representative wells showing representative A498 spheroids cultured either in the presence of IL-15-activated NK cells (upper panel) or alone (lower panel). Images were recorded every hour for 64 h. Time point “0 h” corresponds to the pre-screen image taken before the addition of fresh new medium with or without NK cells. At that point, the tumor spheroids had grown in the chip for 48 h. TMRM (magenta) and caspase-3/7 (green) were used as viability and apoptotic markers, respectively.
(B) Two examples of NK cell interaction and killing at the periphery of A498 spheroid. Left: an NK cell (arrowhead) in contact with an apoptotic A498 cell at the external layer of the tumor spheroid is shown. Right: three NK cells (arrowheads) in contact with a single apoptotic A498 tumor cell are shown. The unlabeled NK cells are readily detected in the transmitted light channel, and the apoptotic A498 cell has clear caspase-3/7 signal from the cell nucleus.
(C) Overview of the 36 wells in two chambers at the end of the assay. Cell death, seen as decreased TMRM signal (magenta) and increased caspase-3/7 signal (green), was observed in multiple wells from the chamber containing a mix of tumor and NK cells (upper panel), while cell death was not detectable in the chamber containing tumor cells alone.
(D) Apoptotic index plotted over time for A498 spheroids alone (black) or A498 spheroids co-cultured with NK cells (pink). Error bars indicate SD.
(E) Viability plotted over time for A498 spheroids alone (black) or A498 spheroids co-cultured with NK cells (pink) (n = 36). Error bars indicate SD.
(F) Total apoptosis between A498 spheroids cultured alone (black bar) and A498 spheroids co-cultured with NK cells (pink bar). Error bars indicate SEM. Statistical analysis was by unpaired t test (n = 36, ∗∗∗∗p ≤ 0.0001).
(G) Viability of individual A498 spheroids alone (black) or A498 spheroids co-cultured with NK cells (pink) for 64 h. Error bars indicate SD. Statistical analysis was by unpaired t test (n = 36, ∗∗∗∗p ≤ 0.0001).
(H) Time-lapse sequences from two representative wells showing low NK-cell-mediated killing (well no. 23, upper panel) and high NK-cell-mediated killing (well no. 15, lower panel).
Time-lapse sequences from a representative well showing A498 spheroids cultured in the presence of IL-15 activated NK cells (Video S5). Images were recorded every hour for 64 h. Movies include pre-screen images taken before the addition of NK cells when the spheroids had grown in the chip for 48 h. TMRM (magenta) and Caspase- 3/7 (green) were used as viability and apoptotic markers. NK cells are visible in the bright-field channel.
Time-lapse sequences from a representative well showing A498 spheroids cultured without NK cells. Images were recorded every hour for 64 h. Movies include pre-screen images taken before the addition of fresh cell culture medium when the spheroids had grown in the chip for 48 h. TMRM (magenta) and Caspase- 3/7 (green) were used as viability and apoptotic markers. NK cells are visible in the bright-field channel.
The apoptotic index values significantly increased within a few hours from NK cell addition, while only a small gradual increase was detected in the control chamber (Figure 4D). The apoptotic index reached a maximum around 26 h, after which the signal declined (Figure 4D). The viability decreased with similar timing, while A498 spheroids cultured alone tended to accumulate TMRM dye over time, resulting in a small rise of the viability signal (Figure 4E). About 50% viability was reached within 24 h, and at 54 h, a plateau, corresponding to approximately 30% of target cells alive, was reached (Figure 4E). This could reflect that NK cells became exhausted (Bi and Tian, 2017), leading to decreased or a complete stop of killing.
The total apoptosis in NK-spheroid co-culture was significantly larger than for A498 spheroids alone (Figure 4F). A remarkably high variation of A498 spheroid viability was observed after 64 h of NK exposure (Figures 4G and S4C). Whereas most of the spheroids showed a percentage of viability between 20% and 40%, a few spheroids had high viability (around 70%), and others were almost completely dead (around 6%–10% viability). Examples of this heterogeneity can be found in Figure 4H and Videos S7 and S8, showing spheroids with low and high NK-cell-mediated cytotoxicity. No significant correlation was found between initial spheroid size and viability at 64 h (Figure S4D), suggesting that this heterogeneity was driven by more complex mechanisms.
Time-lapse sequences from a representative well showing low NK cell-mediated killing, using the same experimental settings as in Videos S5–S6.
Time-lapse sequences from a representative well showing high NK cell-mediated killing, using the same experimental settings as in Videos S5–S6.
In this section, we demonstrated an immune-cell-mediated spheroid cytotoxicity assay in the microwell chip. Together with screening of NK cytotoxicity, this also allowed the detection of single NK cells interacting with tumor cells and visualization of the details of tumor spheroid killing. The possibility of screening 36 wells for each condition led us to detect heterogeneity of the NK cell response, showing the strength of acquiring many datapoints per condition in a single experiment.
NK cell killing of ovarian multicellular tumor spheroids in combination with drug therapy
We have demonstrated the possibility to test multiple monotherapies in our microchip, adding a single type of treatment per chamber. Combinatorial therapy, defined as a treatment modality that combines the use of multiple therapeutic approaches, is emerging as an effective strategy against cancer. In the last years, particular attention has been directed toward the use of cellular immunotherapy in combination with antibody-based treatments or more standard approaches, such as chemo- and radiotherapy.
To enable in vitro analysis of combinatorial treatments, we developed a miniaturized assay that combined NK-cell-based immunotherapy with antibody treatment and standard chemotherapy against OVCAR8 in the microwell chip. OVCAR8 spheroids were exposed to human peripheral blood NK cells obtained from three different donors with or without A-1155463, cetuximab, or paclitaxel. A-1155463 and paclitaxel were partially effective against OVCAR8 spheroids in the previous 3D screening assay (Figure 3). Since gefitinib did not show activity against OVCAR8 (Figure 3), it was replaced with cetuximab. Gefitinib and cetuximab are both EGF inhibitors but with different mechanisms of action. Gefitinib acts intracellularly, inhibiting the phosphorylation of tyrosine kinases associated to the EGFR signaling pathway. Cetuximab is a chimeric monoclonal antibody directed towards the extracellular domains of EGFR. Its mechanisms of action include blocking of EGF binding and EGFR dimerization, leading to inhibition of proliferation and tumor growth, as well as promotion of NK and T cell anti-tumor activity mediated by antibody-dependent cellular cytotoxicity (ADCC). Therefore, if not effective by directly inhibiting the EGFR pathway, cetuximab can still increase NK-mediated cytotoxicity.
Flow cytometry showed that OVCAR8 cells were positive for EGFR (Figure S5A). No significant differences in NK cells viability or expression of activation markers were found on NK cells treated with either A-1155463 or paclitaxel (Figure S5B). Interleukin-15 (IL-15)-activated NK cells and drugs were added to chambers containing pre-formed OVCAR8 spheroids, and viability was monitored over 48 h by live-cell imaging (Figure 5A; see experimental scheme in Figure S5C). Consistent with previous experiments (Figure 3), A-1155463 and paclitaxel treatment on their own induced OVCAR8 death, while cetuximab did not (Figure S5D). In the absence of drugs, NK cells from all three donors were effective in killing OVCAR8 spheroids, showing reduction of viability by 50%–60% (Figures 5B and S5E). Consistent with previous results (Figure 4), heterogeneity in NK cell responses was observed between individual wells. NK cells isolated from donor 2 were slightly less effective compared with donor 1 and 3 in killing OVCAR8 in the absence of drugs (Figures 5B and S5E). The NK cell populations that were used in the screening assays were analyzed by flow cytometry to check for phenotypic signatures that could indicate differences in NK cell activation and maturation (Figure S5F). Among the three donors analyzed, NK cells obtained from donor 2 showed a more immature phenotype, with the highest percentage of CD56Bright CD16− NK cells and lowest levels of the early activation marker CD69. NK cells obtained from donor 1 showed a more mature phenotype, expressing high amounts of CD69.
Figure 5.

NK cell killing of OVCAR8 spheroids in combination with drug therapy
(A) Images of OVCAR8 spheroids labeled with TMRM (magenta) and caspase-3/7 (green) after 48 h of treatment with a combination of drugs and NK cells. Tumor spheroids were grown in the chip for 48 h before drugs (one drug per column) and NK cells (one donor per row) were added. From top to bottom: NK donor 1 (NK D1), NK donor 2 (NK D2), NK donor 3 (NK D3), or no NK cells (no NK) are shown. From left to right: DMSO, A-1155463 (10 μM), cetuximab (10 μM), and paclitaxel (1 μM) are shown.
(B) Dot plot showing the specific viability of each OVCAR8 spheroid (small symbol) and the mean values for each donor (big symbols) after 48 h from the addition of DMSO (circles), A1155463 (squares), cetuximab (inverted triangles), and paclitaxel (triangles) in the presence of NK cells from donor 1 (orange), donor 2 (yellow), and donor 3 (blue). Mean values from all donors are shown as black bars. Representative images for each treatment and donor are shown below the plot. Statistical test was by one-way ANOVA with Dunnett’s post hoc test (n = 3, ∗p ≤ 0.05).
(C) Time course of specific spheroid viability during combinatorial toxicity assay with DMSO (black), A-1155463 (pink), cetuximab (teal), and paclitaxel (violet). Each NK cell donor has been plotted in a separate graph in the order donor 1 (left), donor 2 (middle), and donor 3 (right).
(D) Time-lapse images from representative wells showing OVCAR8 spheroids treated with cetuximab (upper panel), NK cells from donor 1 (middle panel), or both cetuximab and NK cells from donor 1 (lower panel).
(E) Time course of the specific viability of OVCAR8 spheroids treated with cetuximab in the presence of NK cells from donor 1 (orange), donor 2 (yellow), and donor 3 (blue) individually fitted to an exponential decay function. Best fits are shown as dotted lines, and the corresponding values for time constant (), plateau (), and R2 were and for donor 1; , , for donor 2; and , , for donor 3.
Next, we analyzed OVCAR8 viability when combining NK cells and drugs. The effects of A-1155463 and paclitaxel in combination with NK cells were small, reducing OVCAR8 viability around 10 percentage points compared with NK cells alone, while the combination of NK cells with cetuximab led to almost complete OVCAR8 death (Figure 5B). Differences between donors could be observed also in the response to drugs (Figures 5B and S5G). For instance, the combination with paclitaxel was very effective for donor 3, while it had smaller effects for donors 1 and 2. The donor variation was minimal for cetuximab (Figures 5B and S5G).
The effect of combinatorial therapy compared with NK cells alone was detected after 4–8 h from the beginning of the assay (Figures 5C and 5D). An exception was paclitaxel, where we observed delayed cytotoxicity in all three donors compared with the other treatments. Measurements of NK degranulation against OVCAR8 showed that 48 h exposure to paclitaxel reduced the levels of the degranulation marker CD107a on the cell surface (Figure S5H). Thus, this indicates that there could be an effect of paclitaxel on the NK cell killing machinery causing the delay of cytotoxicity (Chuang et al., 1994; Mentlik et al., 2010), even if previous experiments showed minor effects on viability and phenotype (Figure S5B).
Although there were only small variations between donors in the outcome of the combinatorial treatment with cetuximab at 48 h (Figures 5B and S5G), the different donors showed different killing dynamics (Figure 5C). For instance, the effect was faster for donors 2 and 3 compared with donor 1. To better quantify such differences, we performed curve fitting using a one-phase exponential decay model and estimated a time constant τ and a predicted plateau (interpreted as the maximum effect) for each donor (Figure 5E). We obtained the highest τ value and lowest predicted plateau for donor 1. In other words, the long-term efficacy was predicted to be highest for donor 1, while short-term efficacy was higher for the other donors. This shows that time-resolved data could be important to understand cytotoxic mechanisms and synergism between treatments.
Assessing the susceptibility to different treatments for different individuals is a key concept in precision medicine. Here, we applied the same concept to cell-based immunotherapy and demonstrated the possibility to use the microwell chip to test drug responses of NK cells from different donors.
Discussion
In this work, we have presented a methodology for evaluation of anticancer chemo- and immunotherapies based on four miniaturized cytotoxicity screening assays in a new multichambered microwell chip. Compared with conventional cell culture substrates and miniaturized devices, the chip is unique by its design and properties for long-term 2D or 3D cell culture, efficient multiplexed screening, and high-resolution microscopy, making it possible to collect longitudinal, high-content data and to make detailed observations at the subcellular level within a single experiment. The chip enables significantly reduced consumption of cells and reagents compared with standard multiwell plates, yet it allows multiplexing and screening of several experimental conditions. The high number of replicates that the microwells provide in each chamber of the chip increases the possibility to resolve statistically significant differences between conditions. It also allowed us to capture heterogeneity in NK cell response between individual wells. The number of simultaneous conditions could be further increased by using multiple chips in the same holder; a multiwell plate format would fit 22 chips, i.e., 352 chambers and 12,672 microwells. In fact, the small footprint of the chip enables it to be adapted using bespoke holders to most standard formats available on the market today, including cell culture dishes, slides, and multiwell plates, making it flexible for use in various imaging instruments.
First, we demonstrated a drug concentration response of the microtubule-targeting agent paclitaxel in 2D cell cultures of ovarian cancer cells. The concentration where we saw effects were in the same range as paclitaxel concentrations found intratumorally (1 × 10−6 M to 0.9 × 10−5 M) and in the plasma (2.8 × 10−7 M to 0.8 × 10−7) of treated patients (Nawara et al., 2021). High-resolution confocal imaging directly in the chip showed that higher concentrations of paclitaxel induced multipolar mitotic spindles and aberrant chromosomal aggregation. Thus, this assay demonstrated both a drug concentration screen over multiple days and high-resolution phenotypic cell characterization for better mechanistic understanding of the drug effect. Since 3D cell cultures can reproduce factors of the tumor microenvironment important for drug efficacy, such as drug diffusion through multiple cell layers, cell metabolisms, and selective pressure exerted by the microenvironment, they are rapidly becoming the standard for drug screening. Therefore, we designed the second assay for multiplexed drug screening with tumor spheroids generated from different target cell lines. This showed high susceptibility of HCC827 and partial resistance of NCIH-1975 to gefitinib and efficient killing of Kuramochi and OVCAR8 by A-1155463, which was expected from previous experiments, corroborating robustness of the method.
By following drug-induced cell death over time, we could capture dynamic differences. For instance, Kuramochi spheroids showed similar levels of specific viability after 48 h of A-1155463 and gefitinib treatments, while their levels of specific apoptosis differed. The amplitude of the apoptosis signal from caspase-3/7 was transient and very dependent on the time of observation, showing the importance of time-resolved measurements as opposed to screening at a single time point when using this dye. The total apoptosis, calculated by integrating the caspase-3/7 signal over time, could be used to get an overview of the apoptotic death seen in different conditions. The signal from TMRM, on the other hand, could be used in endpoint assay to immediately assess cytotoxicity. However, valuable mechanistical insight of the target cells death can be gained by looking at both viability and apoptosis in parallel over time.
The high optical quality of the chip allowed us to show that paclitaxel-induced apoptosis was localized mostly at the periphery of OVCAR8 spheroids, whereas A-1155463 rapidly caused cell death throughout the spheroids. This could be coupled to limited diffusion of paclitaxel into the central parts of the spheroids, which is further supported by our observation and previous reports that killing with the drug was less efficient in 3D compared with 2D cultures (Hirst et al., 2018). Another contributing factor could be that cells in mitosis, and therefore more sensitive to paclitaxel, are more often observed in the outer layers of the spheroids. Overall, this assay showed the complexity of drug cytotoxicity in the tumor microenvironment and highlighted the importance of robust, time-resolved, high-quality imaging of live cells to get dynamic and mechanistic insight into drug cytotoxicity.
NK cell immunotherapies have emerged as a viable option to T cell therapies for treating cancer with a reduced risk of graft-versus-host disease and cytokine release syndrome (Shimasaki et al., 2020). Different approaches are explored to enhance the number, potency, and functionality of NK cells (Fang et al., 2017), including for example engineered cells like chimeric antigen receptor (CAR)-NK cells (Navin et al., 2020; Xie et al., 2020). Nevertheless, problems such as development of resistance frequently occur for patients receiving NK cell therapy, and the efficacy is typically specific to each patient and disease (Sordo-Bahamonde et al., 2020), which highlights the need for personalized immunotherapy. To fully harness the potential of cell therapies for cancer, it is critical to elucidate the immunosubversive mechanisms of cancer cells and heterogeneity among donors and individual immune cells within populations (Vanherberghen et al., 2013).
To address this, we developed a miniaturized NK cell cytotoxicity screening assay. Importantly, only 2 × 104 NK cells and the same amount of target cells were used in a single chamber for cytotoxicity testing on 36 separate tumor spheroids. The low number of primary cells needed is beneficial in clinical contexts, where the availability of both tumor and blood samples is often scarce.
Despite high initial activity of NK cells against A498 spheroids, killing slowed down or stopped completely, although a population of tumor cells remained alive, indicating exhaustion of the NK cells or a resistant subpopulation of target cells (Bi and Tian, 2017). We also observed high variation in NK cell killing efficiency between individual wells. At this point, we can only speculate, but we think that this could be due to small variations in NK cell number leading to a disproportionately heterogeneous response across wells or heterogeneity among the NK cells, combined with intricate interplay between feedback loops triggering or dampening responses. NK cells consist of different subsets, which might show different properties in terms of proliferation, activation, cytokine production, cytotoxic activity, and migration during tumor recognition (Castriconi et al., 2018; Cooper et al., 2001; Stabile et al., 2017). It is possible that the presence of a few particularly active NK cells can drive early onset of killing that reshape the tumor microenvironment, which in turn triggers activation of more NK cells, leading to effective killing of the spheroid. On the other hand, the lack of early NK cell killing and downstream activation may lead to low overall killing. These complicated interplays highlight the need for efficient in vitro model systems and assays for detailed evaluation of the factors shaping the tumor microenvironment and how immune cells respond to it.
The fourth and final assay in this work was designed for evaluation of combinatorial treatments with NK cells and different drugs. This provided the possibility to observe variations between NK cell donors and discriminate between short-term and long-term efficacy. Such information was also combined with flow cytometry analysis of the NK cells used in the drug toxicity assay, providing the possibility to correlate the cell phenotype with the response to treatment. Thus, these experiments provided information that could be valuable for optimizing a treatment strategy involving selection of specific cell donors and combinatorial drug treatment.
The miniaturized cell assay platform presented in this work was designed for imaging-based cytotoxicity assays targeting 2D or 3D tumor cell cultures combining multiplexing, screening, and high-quality imaging. The interplay between drugs, immune cells, and cancer cells is complex, even for in vitro cell cultures, and heterogeneity among both cancer cells and immune cells can contribute to results that are challenging to interpret. However, with the development of new technologies that can resolve and reveal the finer details of these interactions, we have great opportunities to improve anticancer therapies. We hope that the multichambered microwell chip platform presented here can contribute to establishing precision medicine for drug- and immune-cell-based anticancer therapy.
Limitations of the study
The fabrication of the Si-glass microwell chips required semiconductor processing tools and clean room facilities, not available to everyone. In contrast to conventional multiwell plates and slides, the chip was of a non-standardized format, requiring the use of chip holders to fit it in commercial imaging instruments, but that also made it highly adaptable. However, one limitation with the current design was that it did not directly allow usage of multi-pipettes. The focus of this study was on high-content screening, and therefore, the spheroid tumor cultures had limitations in comparison to some in vitro models that prioritize in vivo-like culture conditions over screening capabilities. For example, the spheroids contained no stroma components or vascularization, and there was no continuous perfusion of the spheroids. Instead, the addition of drugs and other reagents primarily relied on convective mixing by pipetting and diffusion, as in multiwell plates. The spheroids used in this study were composed of individual tumor cell lines, but the methodology allows usage of primary cells, addition of other cell types like stromal or immune cells, or components of the extracellular matrix, which would further shape the tumor microenvironment. No necrotic cores were found by confocal microscopy, and low fractions of dead tumor cells were detected in flow cytometry of the spheroids used in this study. A necrotic core can provide a hypoxic environment that can affect drug potency as well as immune cell activity and function, which may be important factors to consider and include in some solid-tumor models. In future work with different cell lines or primary tumor cells, it will be important to investigate under what circumstances necrotic cores are formed.
STAR★Methods
Key resources table
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| Mouse anti-CD45 FITC | BD Biosciences | Cat#561865; RRID:AB_10896120 |
| Mouse anti-CD56 PE | Biolegend | Cat#318306; RRID:AB_604101 |
| Mouse anti-CD3 BV605 | BD Biosciences | Cat#563219; RRID:AB_2738076 |
| Mouse anti-CD69 V450 | BD Biosciences | Cat#560740; RRID:AB_1727512 |
| Mouse anti-CD16 APC-H7 | BD Biosciences | Cat#560195; RRID:AB_1645466 |
| Mouse anti-CD25 Pe-Cy7 | BD Biosciences | Cat#557741; RRID:AB_396847 |
| Mouse anti-granzyme B AF647 | BD Biosciences | Cat#561999; RRID:AB_10897997 |
| Mouse anti-EGF-R BV421 | BD Biosciences | Cat#566254; RRID:AB_2739632 |
| Mouse anti-ɑ-tubulin | Sigma-Aldrich | Cat#T6074; RRID:AB_477582 |
| Donkey anti-mouse IgG (H + L) AF647 | Thermo Fisher Scientific | Cat#A-31571; RRID:AB_162542 |
| Mouse anti-CD107a AF647 | BD Biosciences | Cat#562622; RRID:AB_2737684 |
| Biological samples | ||
| Buffy-coats obtained from anonymous healthy donors | Karolinska University Hospital, Universitets Laboratoriet | https://www.karolinska.se/for-vardgivare/karolinska-universitetslaboratoriet/ |
| Chemicals, peptides, and recombinant proteins | ||
| RPMI 1640 supplemented with L-glutamine | Thermo Fisher Scientific | Cat#11875093 |
| 100x MEM non-essential amino acid solution | Sigma-Aldrich | Cat#M7145 |
| HEPES | Sigma-Aldrich | Cat#H0887; CAS:7365-45-9 |
| Fetal Bovine Serum | Sigma-Aldrich | Cat#F7524 |
| 0.05% Trypsin, 0.02% EDTA solution | Sigma-Aldrich | Cat#59417C |
| Ficoll-Hypaque | GE-Healthcare | Cat#17-1440-03 |
| Recombinant Human IL-15 Protein | R&D System | Cat#247-ILB |
| A-1155463 Bcl-xL selective inhibitor | Selleck Chemicals | Cat# S7800 |
| Paclitaxel | Sigma-Aldrich | Cat#T7191 |
| Gefitinib | Selleck Chemicals | Cat#S1025 |
| Cetuximab | InvivoGen | Cat#hegfr-mab1 |
| DMSO | Sigma-Aldrich | Cat#41639; CAS:67-68-5 |
| Bovine Serum Albumin | Sigma-Aldrich | Cat#A7906; CAS:9048-46-8 |
| Fixable Viability Stain 510 | BD Biosciences | Cat#564406 |
| 16% Formaldehyde (w/v) methanol-free | Thermo Fisher Scientific | Cat#28906 |
| Triton X-100 Surfact-Amps detergent solution | Thermo Fisher Scientific | Cat#28314 |
| Oregon Green 448 Phalloidin | Thermo Fisher Scientific | Cat#O7466 |
| DAPI | Thermo Fisher Scientific | Cat#D1306; CAS:28718-90-3 |
| TMRM | Thermo Fisher Scientific | Cat#T668 |
| BioTracker NucView 488 Green Caspase-3 Dye | EMD Millipore | Cat#SCT101 |
| Critical commercial assays | ||
| NK Cell Isolation Kit, human | Miltenyi Biotec | Cat#130-092-657 |
| BD CompBeads | BD Biosciences | Cat# 552843 |
| Protein-repellent polymer | Nagahashi, et al., 2015 | |
| Iohexol | GE-Healthcare | Cat#019112 |
| Deposited data | ||
| MATLAB code for statistical analysis | This paper | http://10.5281/zenodo.6645210 |
| Experimental models: Cell lines | ||
| Human: A498 cells | ATCC | HTB-44 |
| Human: HCC827 cells | ATCC | CRL-2868 |
| Human: NCIH-1975 cells | ATCC | CRL-5908 |
| Human: Kuramochi cells | JCRB | JCRB0098 |
| Human: OVCAR8 cells | NCI | NCI60 |
| Recombinant DNA | ||
| pLenti-CMV-MCS-RFP-SV-puro vector | Addgene | Cat#19319 |
| Software and algorithms | ||
| MATLAB (2021), Version 9.10.0 (R2021a) | The MathWorks Inc | https://se.mathworks.com/products/matlab.html |
| ImageJ2 (2021), Version 2.3.0/1.53f | Open-source | https://imagej.nih.gov/ij/ |
| FlowJo for Mac (2021), Version 10.8.1 | Becton, Dickinson and Company | https://www.flowjo.com/ |
| GraphPad Prism, Version 9.3.1 | GraphPad Software | https://www.graphpad.com/ |
| Imaris, Version 9.7 | Oxford Instruments | https://imaris.oxinst.com/ |
| Watershed algorithm | Soille and Vincent, 1990 | https://www.spiedigitallibrary.org/conference-proceedings-of-spie/1360/1/Determining-watersheds-in-digital-pictures-via-flooding-simulations/10.1117/12.24211.short?SSO=1 |
| “Analyze particle” algorithm | https://imagej.nih.gov/ij/docs/menus/analyze.html | |
Resource availability
Lead contact
Further information and requests for resources and reagents should be directed to and will be fulfilled by the lead contact, Björn Önfelt (onfelt@kth.se).
Material availability
This study describes the use of custom-made, multichambered microwell chips. Information regarding the fabrication of these chips is available in the method details and Figure S6.
Experimental models and subject details
Cell lines and culture
The following cell lines were used: A498 renal carcinoma (ATCC, HTB-44), HCC827 lung adenocarcinoma (ATCC, CRL-2868), NCIH-1975 lung adenocarcinoma (ATCC, CRL-5908), Kuramochi ovarian adenocarcinoma and OVCAR8 ovarian carcinoma (both provided by the FIMM Research group, Helsinki University, Finland). A498 cell line has been validated using PCR-based single-locus technology. HCC827, NCIH-1975, Kuramochi and OVCAR8 cell lines have been authenticated using the Cell Line Authentication Service provided by ATCC.
Cells were cultured in complete RPMI 1640 with L-glutamine medium (ThermoFisher Scientific) supplemented with 10% heat-inactivated fetal bovine serum (Sigma Aldrich), 1x MEM Non-Essential Amino Acid Solution (Sigma-Aldrich) and 10 mM HEPES (Sigma-Aldrich). All cell lines were maintained at 37°C in 5% CO2 and passaged before they reached confluency. The stable A498 cell line expressing RFP (A498-RFP) was obtained using a pLenti-CMV-MCS-RFP-SV-puro vector as previously described (Edwards et al., 2020). A498-RFP cells were cultured in complete medium with 2 μg/mL puromycin.
Peripheral blood mononuclear cells (PBMCs) were isolated from buffy-coats from anonymous healthy donors (Karolinska University Hospital) by Ficoll-Hypaque gradient separation (GE-Healthcare). NK cells were separated from PBMCs by negative selection using NK cell Isolation Kit (Miltenyi Biotec), according to the manufacturer’s instructions. Purified NK cells were immediately cultured in RPMI 1640 with L-glutamine (ThermoFisher Scientific) supplemented with 10% heat-inactivated fetal bovine serum (Sigma-Aldrich), 1x MEM Non-Essential Amino Acid Solution (Sigma-Aldrich) and 10 mM HEPES (Sigma-Aldrich) and maintained at 37°C in 5% CO2 overnight in the presence of 10 ng/mL IL-15 (R&D System). Cell isolation from anonymous donors requires no ethical permit according to local regulations. The purity (percentage of CD56+CD3− cells) and phenotype of enriched polyclonal NK cells was assessed by flow cytometry. To analyze the effect of A-1155463 and paclitaxel on NK cells, overnight IL-15 activated NK cells were harvested and re-suspended in complete medium (106 cells/mL) in presence of 10 μM of A-1155463 (Selleckchem), 1 μM of paclitaxel (Sigma-Aldrich) or DMSO (Sigma-Aldrich). Flow cytometry analysis was performed after 24 and 48 h.
Method details
Fabrication of multichambered microwell chips
The Si/glass microwell array chip was fabricated by the SciLifeLab Customized Microfluidics facility using standard cleanroom processes for semiconductor and MEMS devices, which have been described in previous work (Frisk et al., 2011). In short, a 4-inch 300 μm Si wafer was patterned by lithography and through-holes were formed by deep reactive ion etching (DRIE). Thereafter, the Si wafer was oxidized and anodically bonded to a 175 μm BF33 glass wafer, transforming the holes into microwells. Lastly, the wafer stack was diced into nine 22 × 22 mm2 Si-glass microwell array chips (Figure S6A).
The multichambered polymer frame was fabricated by manual reaction injection molding (Figure S6B). A two-piece mold was made of a polyoxymethylene (POM) substrate and a glass plate. The POM substrate was milled to form injection and outlet ports, channels (sprue + runner) and a structured mold cavity. The mold cavity was closed by clamping together the POM substrate with the glass plate. A thermosetting polymer, polydimethylsiloxane (PDMS, Sylgard 184, DOW), was mixed in a 1:10 ratio and degassed in a desiccator. The pre-polymer solution was then manually injected into the injection port of the mold using a syringe until the mold cavity and the outlet channel was filled. The pre-polymer solution in the mold was thermally polymerized in an oven at 65°C for at least 1 h. Thereafter, the polymerized part in the mold cavity was released by separating the glass plate from the POM mold. The PDMS part was then manually lifted off from the glass and the polymer residues from the mold channels were trimmed off manually using a scalpel.
Before assembly, the bottom side of the molded PDMS frames and the top side of the Si-glass chips were exposed to oxygen plasma (50 W, 5 bar and 9 sccm of oxygen for 30 s) to activate the surfaces prior to bonding (Figure S6C). Lastly, the stacks were aligned and gently pressed together to ensure a conformal contact at the interfaces and then thermally treated in an oven at 110°C for 5 min to aid the bonding. The bonded chips were then stored in room temperature until use. During experiments, the chips were placed in custom designed, machined and black anodized Ø 35 mm aluminum holders (Figure 1). After experiments, the chips were cleaned with FacsClean (BD Bioscience) followed by rinsing in MilliQ water. A sonication step (10 min in MilliQ) was used if debris or particles were still found in the microwells. The cleaning protocol allowed reusing the chip around 5 times before recoating. If any biological debris remained in the chip after normal cleaning, it could be removed by first delaminating the PDMS frame and then clean the chip with piranha solution, however, that was never needed in this work.
2D drug screening assay and assessment of paclitaxel-induced tubulin deformation
OVCAR8 cells were treated with 0.05% Trypsin-EDTA (Sigma-Aldrich), re-suspended in complete medium (2 × 105 cells/mL) and 40 μL of the cell suspension was transferred into each chamber of the microwell chip. The microwell chip was maintained in sterile conditions at 37°C in 5% CO2. After overnight incubation, OVCAR8 cells were incubated 1 h with 200 nM TMRM (ThermoFisher Scientific) and 5 μM BioTracker NucView Capsase-3 Dye (Caspase-3/7 for short; Sigma-Aldrich). Serial dilutions of paclitaxel (Sigma-Aldrich) ranging from 1 × 10−11 M to 1 × 10−5 M were prepared in 40 μL complete culture medium containing TMRM (200 nM) and Capsase-3/7 (5 μM), to maintain constant level of the dyes and capture death events during live cell imaging. Serial dilutions of DMSO (Sigma-Aldrich) were prepared following the same procedure, starting with the lowest dilution of 1:100 vol/vol (corresponding to the highest paclitaxel concentration used). Live-cell imaging was performed for 72 h in a widefield microscope equipped with environmental control (37°C, 5% CO2 and humidity), recording one frame every 4 h. To analyze the changes of tubulin architecture after paclitaxel exposure, OVCAR8 cells were cultured overnight in the multichambered microwell chip and exposed to serial dilutions of paclitaxel and DMSO for 8 h before proceeding to tubulin immunostaining, following the protocol described in section “Immunostaining for confocal microscopy”.
Ultrasound-assisted spheroid formation
To form tumor spheroids, a single cell suspension was added to each chamber (20 μL, 106 cells/mL) where cells were allowed to settle at the bottom of the microwells. Two washes with fresh cell medium removed all cells settled on the surface above the microwells. Two plastic coverslips (22 × 11 mm2, Nunc Thermanox, Thermofisher) were placed on top of the polymer frame to protect the culture from contamination and evaporation. The microwell chip with cells was placed on a ring transducer consisting of a round piezoceramic plate mounted in an aluminum frame and connected to an SMB-connector with thin copper wires. USWs were introduced into the microwells by connecting the transducer to a signal generator and amplifier to excite the transducer with a sinusoidal wave corresponding to the half wavelength criterion in the microwells. A frequency modulation scheme was applied to the signal during actuation (2.47MHz ±50 kHz at a 1 kHz sweep rate) and the actuation amplitude was 15 Vpp. The resulting acoustic radiation forces pushed all cells into aggregates in the center of the microwells. Continuous acoustic trapping for 24 h ensured stable cell aggregates that could be further cultured in a regular incubator until use. To prevent the cell adhesion to the glass, Si or PDMS, a protein-repellent polymer coating composed of 2-methcryloxypropyl phosphorylcholine (MPC), 3-methacryloxypropyl trimethoxysilane (MPTMSi) and 3-(methacryloyloxy)propyl-tris(trimethylsilyloxy) silane (MPTSSi) described in detail elsewhere was used (Nagahashi et al., 2015). In short, the cleaned and dried microwell chip was plasma treated using a corona arch discharger (BD-10ASV, ETP, IL, USA) for 60 s before 40 μL of coating solution was added to each chamber. After 2 h, the solution was aspirated from the chambers and the chip was dried in a desiccator for 30 min followed by another drying step in an oven set to 60°C. The concentration of the coating solution was 0.10 wt%, which prevented cell adhesion while being thin enough to not interfere with the USW (Olofsson et al., 2018). Gentle washing of the spheroids during labeling ensured that they were not displaced from the wells.
Tumor spheroid drug screening assay
After 24 h of USW-exposure, spheroids of NCIH-1975, HCC827, Kuramochi and OVCAR8 were incubated overnight with TMRM (200 nM; ThermoFisher Scientific) and Caspase-3/7 (5 μM; Sigma-Aldrich) to ensure complete dye penetration into the spheroids. The day after, the spheroids were imaged by widefield fluorescence microscopy to evaluate the background death and spheroid size before the addition of the drugs. Based on data from 2D screening, drug compounds were diluted in 40 μL of complete culture medium at the following final concentrations: 10 μM of A-1155463 (Selleckchem), 10 μM of gefitinib (Selleckchem) and 1 μM of paclitaxel (Sigma-Aldrich). DMSO (Sigma-Aldrich) was used as control at a final dilution of 1:1000, corresponding to the highest DMSO concentration introduced in the drug treatment. TMRM and Capsase-3/7 were added to maintain constant levels of the dyes for the duration of the assay. Live cell imaging was performed using a widefield microscope equipped with environmental control (37°C, 5% CO2 and humidity) for 48 h, with image frames acquired every 4 h.
NK-tumor spheroid cytotoxicity assays
A498 renal carcinoma cells were seeded into the microchip 48 h before the NK killing assay and cultured for 24 h under USW-exposure to form spheroids. After 24 h, formed spheroids were incubated overnight with TMRM (200 nM; ThermoFisher Scientific) and Caspase-3/7 (5 μM; Sigma-Aldrich) to ensure complete dye penetration into the spheroids. The following day, a pre-screening image of A498 spheroids was taken by widefield fluorescence microscopy to evaluate their viability and size before the addition of NK cells. Overnight IL-15 activated NK cells were harvested and re-suspended in complete medium at the concentration of 106 cells/mL. Spheroid supernatant was replaced with 20 μL of NK suspension in the chambers except for control chambers where only complete medium was added. Additional 20 μL of complete medium containing TMRM and Capsase-3/7 were added in all chambers to maintain constant level of the dyes for the duration of the assay. Where indicated, the following drugs were added: 10 μM of A-1155463 (Selleckchem), 10 μM of cetuximab (Invivogen), 1 μM of paclitaxel (Sigma-Aldrich) and DMSO (Sigma-Aldrich). After addition of drugs and dyes and seeding of the NK cells, the multichambered microwell chip was exposed to a short (1 h) round of USW to focus the NK cells onto the tumor spheroids. Then the microwell chip was transferred to a widefield microscope equipped with environmental control (37°C, 5% CO2 and humidity) for live cell time-lapse imaging acquiring image frames every hour for 64 h.
Immunostaining for flow cytometry
The following antibodies were used for flow cytometry analysis: CD45 FITC (BD Biosciences, clone H130), CD56 PE (Biolegend, clone HCD56), CD3 BV605 (BD Biosciences, clone SK7), CD69 V450 (BD Biosciences, clone FN50), CD16 APC-Cy7 (BD Biosciences, clone 3G8), CD25 PE-Cy7 (BD Biosciences, clone M-A251), Granzyme B AF647 (BD Biosciences, clone GB11) and EGFR BV421 (BD Bioscience, clone EGFR.1). Cells were incubated with antibodies diluted in staining buffer (PBS +2% BSA, both Sigma Aldrich) for 25 min at 4°C in the dark. Cells were washed twice and stained with Fixable Viability Stain 510 (BD Bioscience) for 15 min at room temperature for live/dead cell discrimination. After two washes, NK cells were analyzed by flow cytometry (BD FACSCanto IVD 10, standard 3-lasers, 10 colors, BD Biosciences). Unstained control sample was used to set up PMT voltages. Single stain controls with BD CompBeads compensation particles (BD Biosciences) were used to optimize fluorescence compensation settings. Data analysis was performed with FlowJo Software v10.7.1 (FlowJ0, Becton, Dickinson and Company).
CD107a degranulation assay
Overnight IL-15 activated NK cells were harvested and re-suspended in complete medium (106 cells/mL) in the presence of DMSO or paclitaxel. After 48 h incubation, NK cells were harvested, washed and counted. 1 × 105 NK cells were transferred into a 96 V-bottom well plate and co-cultured with 1 × 105 OVCAR8 cells (E:T ratio = 1:1) Alexa Fluor 647-conjugated anti-CD107a antibody (BD Bioscience, clone H4A3) was added to each well from the onset of co-culture. To detect spontaneous NK degranulation, control samples without OVCAR8 cells were included. All samples were analyzed in duplicates. Cells were incubated at 37°C, 5% CO2 in the dark for 4 h before cells were collected, washed twice in staining buffer and labeled for CD45, CD56, CD3, CD69, CD16, CD25 and CD107a for 25 min at 4°C in the dark. Fixable Viability Stain 510 (BD Bioscience) was used for live/dead exclusion. Flow cytometry analysis was performed immediately after immunostaining.
Immunostaining for confocal microscopy
A498-RFP cells treated with 0.05% Trypsin, 0.02% EDTA (Sigma-Aldrich) were re-suspended in complete medium (2 × 105 cells/mL) and 40 μL of the cell suspension was transferred into each chamber of the microwell chip. The microwell chip was maintained in sterile conditions at 37°C, 5% CO2 overnight before immunostaining. Cells were washed three times with PBS (Sigma-Aldrich) and incubated in 4% (w/v) methanol-free formaldehyde (ThermoFisher Scientific) for 15 min at room temperature in the dark. They were then washed three times with PBS and permeabilized with 0.5% Triton X-100 Surfact-Amps detergent (ThermoFisher Scientific) solution in PBS for 5 min. A blocking step was performed adding PBS containing 5% (w/v) bovine serum albumin (BSA, Sigma-Aldrich) for 5 min at room temperature. Serial dilutions of mouse anti-ɑ-tubulin monoclonal antibody (clone B-5-1-2, Sigma-Aldrich) were prepared in PBS to obtain the following final concentrations: 20 μg/mL, 2 μg/mL, 0.2 μg/mL. Primary antibody staining was performed by adding 30 μL/chamber of each dilution in each row of the multichambered microwell chip. PBS was used as control. After an hour incubation at room temperature, cells were washed three times with PBS. A second step of permeabilization and blocking was performed before proceeding with secondary antibody staining. The secondary polyclonal Alexa Fluor 647-conjugated donkey anti-mouse IgG (H + L) antibody (Thermo Fisher Scientific) was diluted in PBS at the following final concentrations: 100 μg/mL, 10 μg/mL, 1 μg/mL, 0.1 μg/mL. Oregon Green 448 Phalloidin (Sigma-Aldrich) was added at a final concentration of 60 nM in each solution to achieve uniform F-actin staining. Secondary antibody and F-actin staining was performed by adding 30 μL of secondary antibody and phalloidin solution per chamber, with increasing concentration of the secondary antibody for every column. After an hour incubation at room temperature, cells were washed three times with PBS and incubated with 2 μg/mL of DAPI (ThermoFisher Scientific) for 10 min at room temperature for nuclear staining. Cells were washed in PBS three times before imaging by confocal microscopy. For the optical clearing experiments, tumor spheroids were washed three times with PBS (Sigma-Aldrich) and incubated in 4% (w/v) methanol-free formaldehyde (ThermoFisher Scientific) for 15 min at room temperature in the dark. They were then washed three times with PBS and incubated with increasing concentrations of refractive index matching solution Iohexol (GE Healthcare) before proceeding with confocal imaging directly on the multichambered microwell chip.
Microscopy
Fixed cells were imaged by confocal microscopy (Zeiss LSM 880) using either a 10×/0.45 Plan-Apochromat Air objective (Zeiss) or a 40×/1.20 C-Apochromat Water objective (Zeiss). At 10x magnification, the image size was 1024× 1024 pixels with a pixel size of 1.38 × 1.38 μm2 and 20 optical sections were acquired with a 2.5 μm step size. At 40x magnification, the image size was 2048 × 2048 pixels with pixel size of 0.10 × 0.10 μm2 and 10 optical sections were acquired with a 0.45 μm step size. Time-lapse live cell microscopy was performed with a widefield Zeiss Axio Observer Z1 seven microscope equipped with an incubation chamber with environmental control (37°C, 5% CO2, and humidity). Autofocused images were acquired every 4 h for 48 h (drug compound screening) or every hour for 64 h (NK killing assay) using a Fluar 10×/0.50 Air objective and a Hamamatsu ORCA-flash 4.0 camera. Image size was 1024 × 1024 pixels with pixel size of 1.3 × 1.3 μm.
Quantification and statistical analysis
Image analysis of antibody titration and mitotic spindles
To evaluate the best signal-to-noise ratio for the combination of ⍺-tubulin primary antibody and AlexaFluor647 secondary antibody, Z-stacks were projected in 2D using the “Sum slices” command in ImageJ and the total fluorescence intensity was measured in each microwell of the chip. The staining index was calculated as the ratio between the signal from chambers stained with both primary and the signal from the corresponding chambers stained with secondary antibody alone. To calculate the number of nuclei in each well, nuclear segmentation was performed by applying manual thresholding on the DAPI channel followed by a Watershed algorithm to separate adjacent nuclei (Soille and Vincent, 1990). The “Analyze particle” plugin in ImageJ was used to automatically count the number of nuclei in individual wells (Schindelin et al., 2012). The number of cells that appeared to have abnormal chromosome and mitotic spindle arrangement was assessed manually. For the visualization of chromosome and mitotic spindle poles during cell division we used the volume rendering and filament tracer modules of the Imaris 9.7 software (Oxford Instruments).
Image analysis of drug toxicity and NK killing
Live cell time-lapse sequences were analyzed as follows. The mean of the TMRM background was subtracted from each pixel and the viability was defined as the TMRM total intensity normalized to the TMRM total intensity at timepoint 0 h (Figures S7A and S7B). In some experiments the TMRM signal increased shortly after the addition of fresh media and treatment, or in the long-term due to cell proliferation causing the viability index to reach values exceeding 100%. Where indicated, the specific viability, defined as the viability normalized to the mean value of the individual measurements from the chamber containing the corresponding control, was calculated (Figure S7C). In NK-A498 killing assays, the variations of the TMRM background signal over time were used to correct for photobleaching. Similarly, the mean of the Caspase-3/7 background was subtracted from each pixel before calculating the Caspase-3/7 total intensity in all the experiments. As the effects of photobleaching were negligible, no further photobleaching correction was performed in the Caspase-3/7 analysis. The total apoptosis was calculated as the area under the curve of the time-course of the mean Caspase-3/7 total intensity from whole chambers using the GraphPad Prism software. Apoptotic index, defined as the ratio between the Caspase-3/7 total intensity at each timepoint and the TMRM signal at timepoint 0, was calculated to be able to compare drug effects between cell lines having different spheroid dimensions (Figures S7D and S7E). Where indicated, the specific apoptotic index was calculated by subtracting the mean apoptotic index of the control chamber from the apoptotic index of the condition of interest. Spheroid segmentation was performed by applying an intensity threshold on the TMRM channel followed by the “Analyze particle” algorithm in ImageJ to identify a single object in each well by using a size threshold to exclude single cells and small particles. Spheroid area and perimeter were calculated using ImageJ.
Statistical analysis and curve fitting
Statistical analysis and curve fitting were performed using GraphPad Prism. Comparison between two or multiple conditions was performed using unpaired t-test, One-way ANOVA followed by Dunnett’s Post Hoc Test, or two-way ANOVA followed by Tukey’s Post Hoc Test as indicated. The total apoptosis data were log transformed before statistical analysis. For some graphs statistical differences were calculated between readings in the 36 microwells in individual chambers of the microchip, and sometimes between median values from experimental repeats. Data groups were considered not significantly (ns) different when p > 0.05 or significantly different with the following annotation: p ≤ 0.05 (∗), p ≤ 0.01 (∗∗), p ≤ 0.001 (∗∗∗); p ≤ 0.0001 (∗∗∗∗).
To investigate how the number of analyzed wells influenced the statistics, a MATLAB script was designed to perform 500 statistical tests between samples containing n = 3–36 randomly picked wells from the two different conditions to be compared. The same well could be picked multiple times within in one test to stimulate a larger dataset. For the statistical tests on the HCC827 caspase data, the AUC was calculated for each individual spheroid before using the inbuilt ANOVA function in MATLAB. This data treatment differs from the analysis in Figure 3E where an ANOVA was performed on the AUC calculated from mean and standard deviation for each time point using GraphPad Prism. The differences in data treatment between the MATLAB and the GraphPad Prism approach resulted in slightly different significances.
Curve fitting analysis was performed with GraphPad Prism using a sigmoidal dose-response curve (the Hill equation):
| (Equation 1) |
Where spV is the specific viability, spV0 is the specific viability without drug, is the specific viability at infinitely high drug concentrations, C is the drug concentration, IC50 is the inhibitory concentration (50%) and H is the Hill slope.
Or an exponential decay:
| (Equation 2) |
Where V is the specific viability, V0 is the specific viability at the starting point, is a plateau value representing specific viability at longer times, t is time and is the time constant.
Acknowledgments
We thank The Knut and Alice Wallenberg Foundation (grant no. 2018.0106), The Swedish Research Council (grant no. 2019-04925), The Swedish Foundation for Strategic Research (grant no. SBE13-0092), The Swedish Childhood Cancer Foundation (grant no. MT2019-0022), and The Swedish Cancer Foundation (grant no. 19 0540 Pj) for financial support; and the VirusTech Core Facility at Karolinska Institutet for generating the pLenti-CMV-MCS-RFP-SV-puro viral particles; the FIMM Research group at Helsinki University for providing Kuramochi and OVCAR8 cell lines; and Aman Russom, KTH Royal Institute of Technology, for access to equipment for producing polymer frames.
Author contributions
Conceptualization, N.S., V.C., K.O., M.W., and B.Ö.; methodology, N.S., V.C., K.O., H.V.O., Q.V., T.F., M.W., and B.Ö.; software, V.C., K.O., H.V.O., and Q.V.; validation, N.S., V.C., and K.O.; formal analysis, N.S., V.C., and K.O.; investigation, N.S., V.C., K.O., P.A.S., E.M.-L., B.S.-L., and T.F.; resources, E.M.-L., B.S.-L., M.T., M.W., P.Ö., and B.Ö.; data curation, N.S., V.C., and K.O.; writing – original draft and review and editing, N.S., V.C., K.O., and B.Ö.; visualization, N.S., V.C., and B.Ö.; supervision, M.W., P.Ö., and B.Ö.; project administration, B.Ö.; funding acquisition, B.Ö.
Declaration of interests
B.Ö. is a consultant, has ownership in Vycellix, and receives research funding from Affimed.
Published: July 18, 2022
Footnotes
Supplemental information can be found online at https://doi.org/10.1016/j.crmeth.2022.100256.
Supplemental information
Data and code availability
-
•
All data reported in this paper will be shared by the lead contact upon request.
-
•
All original code has been deposited at Zenodo and is publicly available as of the date of publication. DOI is listed in the key resources table.
-
•
Any additional information required to reanalyze the data reported in this paper is available from the lead contact upon request.
References
- Ameratunga M., Xu W., Lopez J. Personalized cancer immunotherapy: today’s challenge and tomorrow’s promise. J. Immunother. Precis. Oncol. 2018;1:56–67. doi: 10.4103/jipo.jipo_13_18. [DOI] [Google Scholar]
- Ayuso J.M., Truttschel R., Gong M.M., Humayun M., Virumbrales-Munoz M., Vitek R., Felder M., Gillies S.D., Sondel P., Wisinski K.B., et al. Evaluating natural killer cell cytotoxicity against solid tumors using a microfluidic model. Oncoimmunology. 2019;8:1553477. doi: 10.1080/2162402X.2018.1553477. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ayuso J.M., Rehman S., Virumbrales-Munoz M., McMinn P.H., Geiger P., Fitzgerald C., Heaster T., Skala M.C., Beebe D.J. Microfluidic tumor-on-a-chip model to evaluate the role of tumor environmental stress on NK cell exhaustion. Sci. Adv. 2021;7:eabc2331. doi: 10.1126/sciadv.abc2331. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Berg S., Kutra D., Kroeger T., Straehle C.N., Kausler B.X., Haubold C., Schiegg M., Ales J., Beier T., Rudy M., et al. ilastik: interactive machine learning for (bio)image analysis. Nat. Methods. 2019;16:1226–1232. doi: 10.1038/s41592-019-0582-9. [DOI] [PubMed] [Google Scholar]
- Bi J., Tian Z. NK cell exhaustion. Front. Immunol. 2017;8:760. doi: 10.3389/fimmu.2017.00760. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Borcoman E., Kanjanapan Y., Champiat S., Kato S., Servois V., Kurzrock R., Goel S., Bedard P., Le Tourneau C. Novel patterns of response under immunotherapy. Ann. Oncol. 2019;30:385–396. doi: 10.1093/annonc/mdz003. [DOI] [PubMed] [Google Scholar]
- Castriconi R., Carrega P., Dondero A., Bellora F., Casu B., Regis S., Ferlazzo G., Bottino C. Molecular mechanisms directing migration and retention of natural killer cells in human tissues. Front. Immunol. 2018;9:2324. doi: 10.3389/fimmu.2018.02324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cen H., Mao F., Aronchik I., Fuentes R.J., Firestone G.L. DEVD-NucView488: a novel class of enzyme substrates for real-time detection of caspase-3 activity in live cells. FASEB J. 2008;22:2243–2252. doi: 10.1096/fj.07-099234. [DOI] [PubMed] [Google Scholar]
- Chao C., Ngo L.P., Engelward B.P. SpheroidChip: patterned agarose microwell compartments harboring HepG2 spheroids are compatible with genotoxicity testing. ACS Biomater. Sci. Eng. 2020;6:2427–2439. doi: 10.1021/ACSBIOMATERIALS.9B01951. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Christakou A.E., Ohlin M., Vanherberghen B., Khorshidi M.A., Kadri N., Frisk T., Wiklund M., Önfelt B. Live cell imaging in a micro-array of acoustic traps facilitates quantification of natural killer cell heterogeneity. Integr. Biol. 2013;5:712–719. doi: 10.1039/c3ib20253d. [DOI] [PubMed] [Google Scholar]
- Christakou A.E., Ohlin M., Önfelt B., Wiklund M. Ultrasonic three-dimensional on-chip cell culture for dynamic studies of tumor immune surveillance by natural killer cells. Lab Chip. 2015;15:3222–3231. doi: 10.1039/C5LC00436E. [DOI] [PubMed] [Google Scholar]
- Chuang L.T., Lotzová E., Heath J., Cook K.R., Munkarah A., Morris M., Wharton J.T., MunkArAh A. Alteration of lymphocyte microtubule assembly, cytotoxicity, and activation by the anticancer drug taxol. Cancer Res. 1994;54:1286–1291. [PubMed] [Google Scholar]
- Chyuan I.T., Chu C.L., Hsu P.N. Targeting the tumor microenvironment for improving therapeutic effectiveness in cancer immunotherapy: focusing on immune checkpoint inhibitors and combination therapies. Cancers. 2021;13:1188. doi: 10.3390/cancers13061188. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cooper M.A., Fehniger T.A., Caligiuri M.A. The biology of human natural killer-cell subsets. Trends Immunol. 2001;22:633–640. doi: 10.1016/S1471-4906(01)02060-9. [DOI] [PubMed] [Google Scholar]
- Cui X., Ma C., Vasudevaraja V., Serrano J., Tong J., Peng Y., Delorenzo M., Shen G., Frenster J., Morales R.-T.T., et al. Dissecting the immunosuppressive tumor microenvironments in Glioblastoma-on-a-Chip for optimized PD-1 immunotherapy. Elife. 2020;9:e52253. doi: 10.7554/eLife.52253. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Desalvo A., Bateman F., James E., Morgan H., Elliott T. Time-resolved microwell cell-pairing array reveals multiple T cell activation profiles. Lab Chip. 2020;20:3772–3783. doi: 10.1039/D0LC00628A. [DOI] [PubMed] [Google Scholar]
- Edwards S.J., Carannante V., Kuhnigk K., Ring H., Tararuk T., Hallböök F., Blom H., Önfelt B., Brismar H. High-resolution imaging of tumor spheroids and organoids enabled by expansion microscopy. Front. Mol. Biosci. 2020;7:208. doi: 10.3389/fmolb.2020.00208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fang F., Xiao W., Tian Z. NK cell-based immunotherapy for cancer. Semin. Immunol. 2017;31:37–54. doi: 10.1016/j.smim.2017.07.009. [DOI] [PubMed] [Google Scholar]
- Fang G., Lu H., Law A., Gallego-Ortega D., Jin D., Lin G. Gradient-sized control of tumor spheroids on a single chip. Lab Chip. 2019;19:4093–4103. doi: 10.1039/C9LC00872A. [DOI] [PubMed] [Google Scholar]
- Frisk T.W., Khorshidi M.A., Guldevall K., Vanherberghen B., Önfelt B. A silicon-glass microwell platform for high-resolution imaging and high-content screening with single cell resolution. Biomed. Microdevices. 2011;13:683–693. doi: 10.1007/s10544-011-9538-2. [DOI] [PubMed] [Google Scholar]
- Fujii T., Naing A., Rolfo C., Hajjar J. Biomarkers of response to immune checkpoint blockade in cancer treatment. Crit. Rev. Oncol. Hematol. 2018;130:108–120. doi: 10.1016/j.critrevonc.2018.07.010. [DOI] [PubMed] [Google Scholar]
- Gallego-Jara J., Lozano-Terol G., Sola-Martínez R.A., Cánovas-Díaz M., de Diego Puente T. A compressive review about Taxol®: history and future challenges. Molecules. 2020;25:5986. doi: 10.3390/molecules25245986. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gopal S., Kwon S.J., Ku B., Lee D.W., Kim J., Dordick J.S. 3D tumor spheroid microarray for high-throughput, high-content natural killer cell-mediated cytotoxicity. Commun. Biol. 2021;4:893. doi: 10.1038/s42003-021-02417-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guldevall K., Vanherberghen B., Frisk T., Hurtig J., Christakou A.E., Manneberg O., Lindström S., Andersson-Svahn H., Wiklund M., Önfelt B. Imaging immune surveillance of individual natural killer cells confined in microwell arrays. PLoS One. 2010;5:15453. doi: 10.1371/journal.pone.0015453. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guldevall K., Brandt L., Forslund E., Olofsson K., Frisk T.W., Olofsson P.E., Gustafsson K., Manneberg O., Vanherberghen B., Brismar H., et al. Microchip screening platform for single cell assessment of NK cell cytotoxicity. Front. Immunol. 2016;7:119. doi: 10.3389/fimmu.2016.00119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hirst J., Pathak H.B., Hyter S., Pessetto Z.Y., Ly T., Graw S., Koestler D.C., Krieg A.J., Roby K.F., Godwin A.K. Licofelone enhances the efficacy of paclitaxel in ovarian cancer by reversing drug resistance and tumor stem-like properties. Cancer Res. 2018;78:4370–4385. doi: 10.1158/0008-5472.CAN-17-3993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hu Y., Sui X., Song F., Li Y., Li K., Chen Z., Yang F., Chen X., Zhang Y., Wang X., et al. Lung cancer organoids analyzed on microwell arrays predict drug responses of patients within a week. Nat. Commun. 2021;12:2581. doi: 10.1038/s41467-021-22676-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim S.-E., Kim H., Doh J. Single cell arrays of hematological cancer cells for assessment of lymphocyte cytotoxicity dynamics, serial killing, and extracellular molecules. Lab Chip. 2019;19:2009–2018. doi: 10.1039/C9LC00133F. [DOI] [PubMed] [Google Scholar]
- Kitaeva K.V., Rutland C.S., Rizvanov A.A., Solovyeva V.V. Cell culture based in vitro test systems for anticancer drug screening. Front. Bioeng. Biotechnol. 2020;8:322. doi: 10.3389/fbioe.2020.00322. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee M.S., Kim H.P., Kim T.Y., Lee J.W. Gefitinib resistance of cancer cells correlated with TM4SF5-mediated epithelial-mesenchymal transition. Biochim. Biophys. Acta Mol. Cell Res. 2012;1823:514–523. doi: 10.1016/j.bbamcr.2011.11.017. [DOI] [PubMed] [Google Scholar]
- Lemasters J.J., Qian T., Bradham C.A., Brenner D.A., Cascio W.E., Trost L.C., Nishimura Y., Nieminen A.L., Herman B. Mitochondrial dysfunction in the pathogenesis of necrotic and apoptotic cell death. J. Bioenerg. Biomembr. 1999;31:305–319. doi: 10.1023/A:1005419617371. [DOI] [PubMed] [Google Scholar]
- Lemon W.C., McDole K. Live-cell imaging in the era of too many microscopes. Curr. Opin. Cell Biol. 2020;66:34–42. doi: 10.1016/j.ceb.2020.04.008. [DOI] [PubMed] [Google Scholar]
- Lin S., Schorpp K., Rothenaigner I., Hadian K. Image-based high-content screening in drug discovery. Drug Discov. Today. 2020;25:1348–1361. doi: 10.1016/j.drudis.2020.06.001. [DOI] [PubMed] [Google Scholar]
- Ma Y., Yu S., Ni S., Zhang B., Kung A.C.F., Gao J., Lu A., Zhang G. Targeting strategies for enhancing paclitaxel specificity in chemotherapy. Front. Cell Dev. Biol. 2021;9:626910. doi: 10.3389/fcell.2021.626910. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maleki Vareki S. High and low mutational burden tumors versus immunologically hot and cold tumors and response to immune checkpoint inhibitors. J. Immunother. Cancer. 2018;6:157. doi: 10.1186/s40425-018-0479-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maleki Vareki S., Garrigós C., Duran I. Biomarkers of response to PD-1/PD-L1 inhibition. Crit. Rev. Oncol. Hematol. 2017;116:116–124. doi: 10.1016/j.critrevonc.2017.06.001. [DOI] [PubMed] [Google Scholar]
- Mentlik A.N., Sanborn K.B., Holzbaur E.L., Orange J.S. Rapid lytic granule convergence to the MTOC in natural killer cells is dependent on dynein but not cytolytic commitment. Mol. Biol. Cell. 2010;21:2241–2256. doi: 10.1091/mbc.e09-11-0930. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nagahashi K., Teramura Y., Takai M. Stable surface coating of silicone elastomer with phosphorylcholine and organosilane copolymer with cross-linking for repelling proteins. Colloids Surf. B Biointerfaces. 2015;134:384–391. doi: 10.1016/j.colsurfb.2015.07.040. [DOI] [PubMed] [Google Scholar]
- Nashimoto Y., Okada R., Hanada S., Arima Y., Nishiyama K., Miura T., Yokokawa R. Vascularized cancer on a chip: the effect of perfusion on growth and drug delivery of tumor spheroid. Biomaterials. 2020;229:119547. doi: 10.1016/J.BIOMATERIALS.2019.119547. [DOI] [PubMed] [Google Scholar]
- Nath S., Devi G.R. Three-dimensional culture systems in cancer research: focus on tumor spheroid model. Pharmacol. Ther. 2016;163:94–108. doi: 10.1016/j.pharmthera.2016.03.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Navin I., Lam M.T., Parihar R. Design and implementation of NK cell-based immunotherapy to overcome the solid tumor microenvironment. Cancers. 2020;12:3871. doi: 10.3390/cancers12123871. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nawara H.M., Afify S.M., Hassan G., Zahra M.H., Seno A., Seno M. Paclitaxel-based chemotherapy targeting cancer stem cells from mono- to combination therapy. Biomedicines. 2021;9:500. doi: 10.3390/biomedicines9050500. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Olofsson K., Carannante V., Ohlin M., Frisk T., Kushiro K., Takai M., Lundqvist A., Önfelt B., Wiklund M. Acoustic formation of multicellular tumor spheroids enabling on-chip functional and structural imaging. Lab Chip. 2018;18:2466–2476. doi: 10.1039/c8lc00537k. [DOI] [PubMed] [Google Scholar]
- Olofsson K., Carannante V., Takai M., Önfelt B., Wiklund M. Single cell organization and cell cycle characterization of DNA stained multicellular tumor spheroids. Sci. Rep. 2021;11:17076. doi: 10.1038/s41598-021-96288-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Park D., Son K., Hwang Y., Ko J., Lee Y., Doh J., Jeon N.L. High-throughput microfluidic 3D cytotoxicity assay for cancer immunotherapy (CACI-ImpacT platform) Front. Immunol. 2019;10:1133. doi: 10.3389/fimmu.2019.01133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Prager I., Liesche C., van Ooijen H., Urlaub D., Verron Q., Sandström N., Fasbender F., Claus M., Eils R., Beaudouin J., et al. NK cells switch from granzyme B to death receptor–mediated cytotoxicity during serial killing. J. Exp. Med. 2019;216:2113–2127. doi: 10.1084/jem.20181454. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schindelin J., Arganda-Carreras I., Frise E., Kaynig V., Longair M., Pietzsch T., Preibisch S., Rueden C., Saalfeld S., Schmid B., et al. Fiji: an open-source platform for biological-image analysis. Nat. Methods. 2012;9:676–682. doi: 10.1038/nmeth.2019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sharma P., Hu-Lieskovan S., Wargo J.A., Ribas A. Primary, adaptive, and acquired resistance to cancer immunotherapy. Cell. 2017;168:707–723. doi: 10.1016/j.cell.2017.01.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shelton S.E., Nguyen H.T., Barbie D.A., Kamm R.D. Engineering approaches for studying immune-tumor cell interactions and immunotherapy. iScience. 2021;24:101985. doi: 10.1016/j.isci.2020.101985. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shimasaki N., Jain A., Campana D. NK cells for cancer immunotherapy. Nat. Rev. Drug Discov. 2020;19:200–218. doi: 10.1038/s41573-019-0052-1. [DOI] [PubMed] [Google Scholar]
- Sinha N., Subedi N., Tel J. Integrating immunology and microfluidics for single immune cell analysis. Front. Immunol. 2018;9:2373. doi: 10.3389/fimmu.2018.02373. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Soille P., Vincent L.M. In: Visual Communications and Image Processing ’90: Fifth in a Series. Kunt M., editor. 1990. Determining watersheds in digital pictures via flooding simulations; pp. 240–250. [Google Scholar]
- Sordo-Bahamonde C., Vitale M., Lorenzo-Herrero S., López-Soto A., Gonzalez S. Mechanisms of resistance to NK cell immunotherapy. Cancers. 2020;12:893. doi: 10.3390/cancers12040893. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stabile H., Fionda C., Gismondi A., Santoni A. Role of distinct natural killer cell subsets in anticancer response. Front. Immunol. 2017;8:293. doi: 10.3389/fimmu.2017.00293. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tsimberidou A.M., Fountzilas E., Nikanjam M., Kurzrock R. Review of precision cancer medicine: evolution of the treatment paradigm. Cancer Treat. Rev. 2020;86:102019. doi: 10.1016/j.ctrv.2020.102019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Uen T., Kushiro K., Hibino H., Takai M. Surface functionalization of carbon-based sensors with biocompatible polymer to enable electrochemical measurement in protein-rich environment. Sensors Actuators B Chem. 2020;309:127758. doi: 10.1016/j.snb.2020.127758. [DOI] [Google Scholar]
- Vanherberghen B., Manneberg O., Christakou A., Frisk T., Ohlin M., Hertz H.M., Önfelt B., Wiklund M. Ultrasound-controlled cell aggregation in a multi-well chip. Lab Chip. 2010;10:2727. doi: 10.1039/c004707d. [DOI] [PubMed] [Google Scholar]
- Vanherberghen B., Olofsson P.E., Forslund E., Sternberg-Simon M., Khorshidi M.A., Pacouret S., Guldevall K., Enqvist M., Malmberg K.J., Mehr R., Önfelt B. Classification of human natural killer cells based on migration behavior and cytotoxic response. Blood. 2013;121:1326–1334. doi: 10.1182/blood-2012-06-439851. [DOI] [PubMed] [Google Scholar]
- Varadarajan N., Julg B., Yamanaka Y.J., Chen H., Ogunniyi A.O., McAndrew E., Porter L.C., Piechocka-Trocha A., Hill B.J., Douek D.C., et al. A high-throughput single-cell analysis of human CD8+ T cell functions reveals discordance for cytokine secretion and cytolysis. J. Clin. Invest. 2011;121:4322–4331. doi: 10.1172/JCI58653. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weaver B.A. How Taxol/paclitaxel kills cancer cells. Mol. Biol. Cell. 2014;25:2677–2681. doi: 10.1091/mbc.E14-04-0916. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wen L., Fan Z., Mikulski Z., Ley K. Imaging of the immune system - towards a subcellular and molecular understanding. J. Cell Sci. 2020;133:jcs234922. doi: 10.1242/jcs.234922. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wiklund M., Christakou A.E., Ohlin M., Iranmanesh I., Frisk T., Vanherberghen B., Önfelt B. Ultrasound-induced cell-cell interaction studies in a multi-well microplate. Micromachines. 2014;5:27–49. doi: 10.3390/mi5010027. [DOI] [Google Scholar]
- Xie G., Dong H., Liang Y., Ham J.D., Rizwan R., Chen J. CAR-NK cells: a promising cellular immunotherapy for cancer. EBioMedicine. 2020;59:102975. doi: 10.1016/j.ebiom.2020.102975. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang W., Yu H., Li G., Wei F., Wang Y., Liu L. Mask-free fabrication of a versatile microwell chip for multidimensional cellular analysis and drug screening. Lab Chip. 2017;17:4243–4252. doi: 10.1039/C7LC01101F. [DOI] [PubMed] [Google Scholar]
- Yuan J., Sims P.A. An automated microwell platform for large-scale single cell RNA-seq. Sci. Rep. 2016;6:33883. doi: 10.1038/srep33883. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zasadil L.M., Andersen K.A., Yeum D., Rocque G.B., Wilke L.G., Tevaarwerk A.J., Raines R.T., Burkard M.E., Weaver B.A. Cytotoxicity of paclitaxel in breast cancer is due to chromosome missegregation on multipolar spindles. Sci. Transl. Med. 2014;6:229ra43. doi: 10.1126/scitranslmed.3007965. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou Y., Shao N., Bessa de Castro R., Zhang P., Ma Y., Liu X., Huang F., Wang R.-F.F., Qin L. Evaluation of single-cell cytokine secretion and cell-cell interactions with a hierarchical loading microwell chip. Cell Rep. 2020;31:107574. doi: 10.1016/j.celrep.2020.107574. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
3D rendered confocal images of OVCAR8 cells cultured for 8 h with 1 × 10−6 M paclitaxel. Rendering was performed in Imaris using conventional settings showing microtubules (α-tubulin, gray) and nuclei (DAPI, cyan), followed by surface rendering with filament analysis to visualize the poles of the mitotic spindles (dots in magenta).
3D rendered confocal images of OVCAR8 cells cultured for 8 h without paclitaxel. Rendering was performed in Imaris using conventional settings showing microtubules (α-tubulin, gray) and nuclei (DAPI, cyan), followed by surface rendering with filament analysis to visualize the poles of the mitotic spindles (dots in magenta).
3D rendered OVCAR8 spheroids after incubation with A-1155463. Spheroids were treated for 48 h, followed by fixation, DAPI counterstaining and spheroid clearing. 3D imaging was performed by confocal microscopy and rendering was done in Imaris using conventional settings to visualize the distribution of apoptotic cells (Caspase-3/7, green) among the total cells (DAPI, gray) within the spheroids. Apoptotic cells were uniformly distributed in the tumor spheroid treated with A-1155463.
3D rendered OVCAR8 spheroids after incubation with DMSO. Spheroids were treated for 48 h, followed by fixation, DAPI counterstaining and spheroid clearing. 3D imaging was performed by confocal microscopy and rendering was done in Imaris using conventional settings to visualize the distribution of apoptotic cells (Caspase-3/7, green) among the total cells (DAPI, gray) within the spheroids. Low and sparse caspase signal was detected in the sample, indicating the absence of necrotic core formation in the spheroids.
Time-lapse sequences from a representative well showing A498 spheroids cultured in the presence of IL-15 activated NK cells (Video S5). Images were recorded every hour for 64 h. Movies include pre-screen images taken before the addition of NK cells when the spheroids had grown in the chip for 48 h. TMRM (magenta) and Caspase- 3/7 (green) were used as viability and apoptotic markers. NK cells are visible in the bright-field channel.
Time-lapse sequences from a representative well showing A498 spheroids cultured without NK cells. Images were recorded every hour for 64 h. Movies include pre-screen images taken before the addition of fresh cell culture medium when the spheroids had grown in the chip for 48 h. TMRM (magenta) and Caspase- 3/7 (green) were used as viability and apoptotic markers. NK cells are visible in the bright-field channel.
Time-lapse sequences from a representative well showing low NK cell-mediated killing, using the same experimental settings as in Videos S5–S6.
Time-lapse sequences from a representative well showing high NK cell-mediated killing, using the same experimental settings as in Videos S5–S6.
Data Availability Statement
-
•
All data reported in this paper will be shared by the lead contact upon request.
-
•
All original code has been deposited at Zenodo and is publicly available as of the date of publication. DOI is listed in the key resources table.
-
•
Any additional information required to reanalyze the data reported in this paper is available from the lead contact upon request.