

Abstract
This report investigates the homotetrameric membrane protein structure of the S31N M2 protein from Influenza A virus in the presence of a high molar ratio of lipid. The structured regions of this protein include a single transmembrane helix and an amphipathic helix. Two structures of the S31N M2 conductance domain from Influenza A virus have been deposited in the Protein Data Bank (PDB). These structures present different symmetries about the channel main axis. We present new magic angle spinning and oriented sample solid-state NMR spectroscopic data for S31N M2 in liquid crystalline lipid bilayers using protein tetramer:lipid molar ratios ranging from 1:120 to 1:240. The data is consistent with an essentially 4-fold-symmetric structure very similar to the M2 WT structure that also has a single conformation for the four monomers, except at the His37 and Trp41 functional sites when characterized in samples with a high molar ratio of lipid. While detergent solubilization is well recognized today as a nonideal environment for small membrane proteins, here we discuss the influence of a high lipid to protein ratio for samples of the S31N M2 protein to stabilize an essentially 4-fold-symmetric conformation of the M2 membrane protein. While it is generally accepted that the chemical and physical properties of the native environment of membrane proteins needs to be reproduced judiciously to achieve the native protein structure, here we show that not only the character of the emulated membrane environment is important but also the abundance of the environment is important for achieving the native structure. This is a critical finding as a membrane protein spectroscopist’s goal is always to generate a sample with the highest possible protein sensitivity while obtaining spectra of the native-like structure.
Graphical Abstract
INTRODUCTION
Multiple distinctly different structures of the M2 protein from Influenza A, an important drug target, have been reported in the literature.1–6 Here, we provide an explanation for why some of this structural heterogeneity has been observed. The tetrameric transmembrane (TM) domain of M2 forms a proton channel, while residues 47–62 form an amphipathic helix that interacts with the lipid interface and with the neighboring monomer splaying the TM helices apart at the C terminus.7–9 The result, as shown in the PDB file 2L0J, has a frustrum shape critical for viral budding and forming adequate space in the pore of the C-terminal region for the His37 and Trp41 tetrads to function in transporting and gating proton conductance.2,10,11 As a result, these two segments are together referred to as the conductance domain (M2CD) that reproduces the conductance properties of the full-length protein.12–15 During the swine flu pandemic of 2009, the variant containing an M2 S31N mutation became widespread, such that the antiflu drugs of the day were no longer effective. This mutation of a Ser to the larger Asn residue reduced the aqueous pore dimensions, preventing the drugs amantadine and rimantadine from binding in the pore and blocking proton conductance,16–19 and hence, a search has continued for new anti-influenza drugs.20–23 Consequently, it is important to have an accurate description of this mutated drug target and to understand the origin of the structural differences displayed between the wild-type and the mutant proteins.
Small helical membrane proteins are exquisitely sensitive to their environment due to the weak interactions that are largely responsible for holding the helical bundles together in the membrane environment.24–26 This is in contrast to β-barrel membrane structures that include hydrogen bonding between transmembrane strands of the protein.27 In addition, the bilayer environment forms dramatic gradients in water concentration, which influences the orientation of TM helices within the membrane, in dielectric constant, which influences the strength of hydrogen bonding in the TM helices resulting in enhanced helical uniformity, and in reduced backbone dynamics, while the lateral pressure profile of the membrane is likely to influence helical packing.28–30 Even for the more stable β-barrel proteins, solvation in an adequate lipid bilayer has been shown to be important for obtaining narrow NMR resonances.27,31
The M2 proton channel functions as an oligomer of four monomers held together primarily by numerous weak van der Waals and hydrophobic interactions in its TM domain (residues 26–46) composed of 15 hydrophobic residues (6 Leu, 5 Ile, 2 Val, and 2 Ala) and 6 hydrophilic residues (1 Gly, 1 Ser, 1His, 1 Trp, 1 Asp, and 1Arg) that line the aqueous pore.1,32 The mix of weak interactions at the helix–helix interface is very different from the mix of stronger electrostatic interactions that typically dominate water-soluble proteins along with hydrophobic interactions.33 The external surface of the TM domain faces the fatty acyl chain environment of the membrane interstices, and consequently, this interface is dominated by hydrophobic residues that interact with fatty acyl chains through weak van der Waals interactions.
It is well recognized that the use of organic solvents and detergent micelles for solubilizing these small TM helical proteins can lead to structural models that misrepresent the native structure.24,28,29 Solid-state NMR (ssNMR), in contrast to solution NMR, has been promulgated as a technology that can characterize the native structure of membrane proteins utilizing a liquid crystalline lipid bilayer environment to solvate the protein.33–37
The WT M2CD (residues 22–62) structure (PDB 2LOJ)2 was characterized primarily by oriented sample ssNMR (OS ssNMR)38,39 using a protein tetramer to lipid ratio of 1:360 where the lipid was a 4:1 ratio of DOPC:DOPE and the sample was approximately 40% by weight water, leading to a 4-fold-symmetric structure.2 OS ssNMR is also a technology that determines the orientation of membrane proteins in a lipid bilayer.2 The first S31N M2CD (residues 18–60) structure deposited in the Protein Data Bank was also approximately a 4-fold-symmetric structure (PDB 2KIH) characterized by solution NMR in short-chain diheptanoylphosphatidyl choline (DHPC) micelles.6 A second S31N M2CD (residues 18–60) structure (PDB 2N70) was derived from high-resolution magic angle spinning (MAS) ssNMR spectra of the M2 protein in a diphytanoyl-sn-glycero-3-phosphocholine (DPhPC) lipid environment having a protein tetramer to lipid ratio of 1:24.4 The 2N70 structure presents a dimer of dimers conformation (Figure 1). We seek to understand what led this homotetrameric protein in the 2N70 structure to acquire two different conformations, representing two different energy minima for the exact same amino acid sequence. In addition, why does the solution NMR structure display so much structural heterogeneity? Furthermore, why has the symmetry changed based on the S31N mutation (2N70) compared to the wild-type structure?
Figure 1.
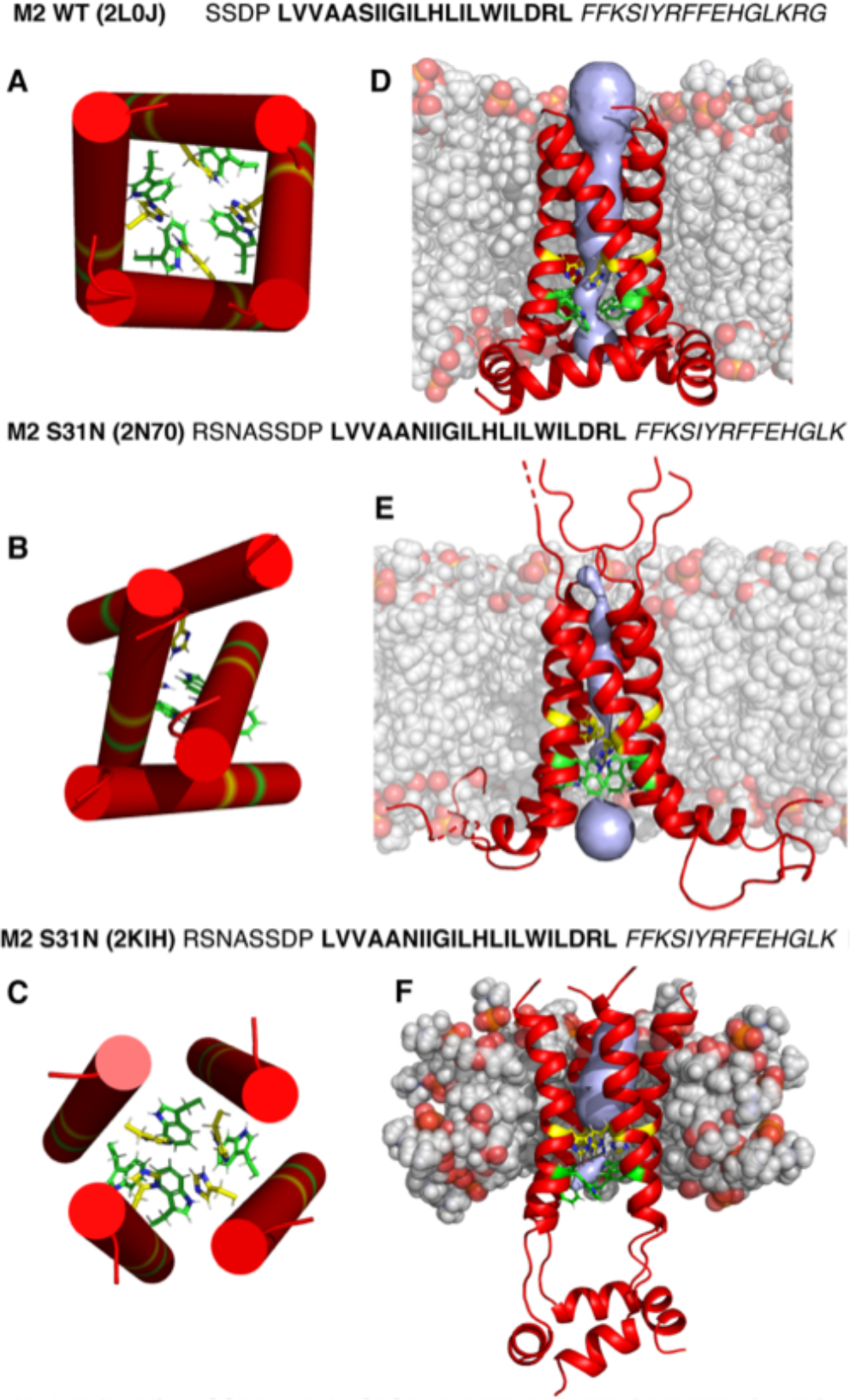
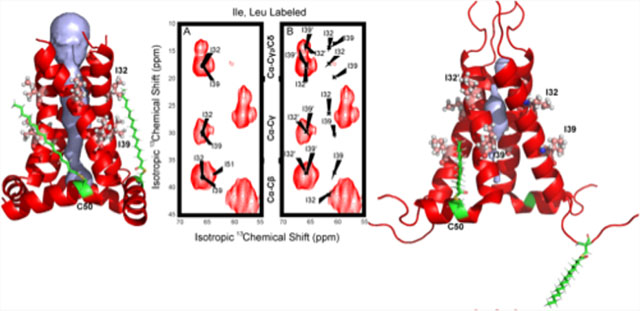
Amino acid sequence of the M2 conductance domain and structures of 2L0J (A and D), 2N70 (B and E), and 2KIH (C and F). (A–C) Cartoons of the TM domains of M2 viewed from the N-terminus and extracellular side of the membrane as cylinders illustrating approximate differences in TM helical tilt angles that influence the critical packing of His37 (yellow) and Trp41 (green)9 residues in the pore. While the lipid-solubilized structures (D and E) have relatively linear TM helices, those in a detergent environment (F) are more bent, forming an hourglass-shaped four-helix bundle. Moreover, these latter figures show different packings of the amphipathic helices in each M2 structure as well as differences in the channel pore (gray volumes) calculated with Mole2.556 (using a probe radius of 5 Å and a threshold radius of 1.4 Å). Lipids were added to the protein structures by charmm-gui without equilibration for the sake of illustration.57 Lipids are in gray with oxygens highlighted in red. In D, the lipids are DOPC and DOPE, in E they are DPhPC, and in F they are DHPC. Amino acid sequences corresponding to each structure are shown above the panels with TM segments in bold and amphipathic helices in italic.
We have recorded multiple OS ssNMR spectra, reported here, for lipid bilayer preparations of S31N M2CD that are not consistent with either of these S31N M2CD published structures. We have also obtained MAS ssNMR spectra of the S31N M2CD at higher lipid to protein ratios than those used by Andreas et al. that generate a different structural result.4 While it should now be well recognized that lipid bilayer environments are critical for the characterization of native membrane protein structures, here we show that the extent of the lipid environment is also critical for characterization of the native-like protein structure.
For the WT structure (2L0J), the binding of rimantadine and amantadine in the TM pore implies that the pore is wide enough for the amantadyl carbon cage to enter (~7 Å) and bind, resulting in blockage of the proton-conducting pore. Indeed, excellent data from DNP40 and REDOR41 defining the precise location of the amantadyl cage in the pore have been published. For the set of 2N70 S31N mutant structures, at least two of the N31 residues face into the pore. The side-chain atomic distances from the pore-facing N31 residues of opposing helices are only 3–5 Å apart, thereby preventing the amantadyl-type inhibitors from binding in the pore cavity, and hence, the effective antiflu drugs of the time became ineffective.4 Surprisingly, not only is the pore reduced in the 2N70 structure by the asparagine residues but also it is occluded for almost the entire length of the TM domain, a result of the dimer of dimers structure. Similar measurements along the 2KIH S31N M2 pore structure show typical separation values of 6–9 Å, which does not ensure that the pore can be closed to proton conductance by the amantadyl cage.5
The residues participating in the amphipathic helix that follows the TM helix vary among the different structures compared to the WT protein structure (2L0J). In 2L0J, the amphipathic helix is composed of 19 residues (47–59) making an angle of 105° for the helix axis with respect to the bilayer normal, consistent with a substantial interaction with the lipid interface. Furthermore, the hydrophobic surface of the amphipathic helix is properly oriented to interact with the hydrophobic interstices of the lipid bilayer.2 However, for the 2KIH S31N structure, the amphipathic helices are reduced in length (residues 51–58), forming a second four-helix bundle tethered to the TM domain by multiple disordered residues. This amphipathic tetrameric feature appears to be a water-soluble structure having the hydrophobic surface facing toward the other monomers, and hence, there appears to be no interaction with the modeled membrane environment.5 In the 2N70 S31N structure, just 2 of the 4 segments (residues 46–55) are shown to form amphipathic helices that interact with the modeled membrane, while the residues for the other two monomers have disordered domains.42 This asymmetry in 2N70 is consistent with the dimer of dimers structure, while the 2L0J and 2KIH structures are both 4-fold-symmetric tetramers.
Here, the new data for the S31N M2 protein from OS and MAS ssNMR samples highlight the differences that arise from a sample environment and a sample preparation protocol that includes much more lipid to fully solubilize the protein structure in its environment. The low sensitivity of ssNMR spectroscopy requires extensive signal-averaging times that can restrict whether an experiment is feasible or not. For Fast MAS ssNMR, proton detection generates the potential for high sensitivity but the small sample size lowers the sensitivity.36,37 Consequently, there is a strong motivation to maximize the amount of protein in the sample and hence minimize its other components. In this case, it means minimizing the amount of lipid for solubilizing the membrane protein, so that the protein signals per unit time can be maximized. For ssNMR spectroscopy that can characterize membrane proteins in a native-like liquid crystalline environment, it is critically important to evaluate just how much lipid is needed for the protein to reflect the native structure. Here, we identify the consequences of limiting the lipid content not only for describing the structure and understanding how these proteins function but also for structural subtleties important for drug discovery against the Influenza virus. This manuscript focuses on these important issues for the M2 protein, and in understanding these issues for M2 we recognize that they are not unique to this system but are likely to affect the characterization of many other membrane proteins.
METHODS AND MATERIALS
Protein Sequence and Constructs.
M2CD WT and S31N mutant sequences are presented in Table 1 with residue 31 highlighted in bold.
Table 1.
M2 Proton Channel Construct Sequence
| Protein | N-terminal domain | TM domain | amphipathic helix |
|---|---|---|---|
| M2CD WT | SN20ASSDP | LVVAA30S31IIGILHLIL40WILDRL | FFKS50IYRFFEHGLK60RG |
| M2CD S31N | SN20ASSDP | LVVAA30N31IIGILHLIL40WILDRL | FFKS50IYRFFEHGLK60RG |
M2CD was expressed as a fusion protein with an N-terminal His6-tagged maltose binding protein (MBP) and a tobacco etch virus42 protease cleavage site between MBP and M2CD (residues 22–62) in plasmids that conveyed ampicillin resistance.
Protein Expression and Purification.
Overexpression of WT and S31N M2CD in Escherichia coli followed previously published protocols.2,43 In summary, MBP fusion M2S31N protein was transformed into E. coli strain BL21 (DE3)-RP Codon Plus; a single colony was used to inoculate 50 mL of ampicillin (100 μg/mL) supplemented LB medium. Following overnight growth of LB at 37 °C, 25 mL was transferred into 1 L of LB medium (ampicillin 100 μg/mL). The cells were grown until OD600 = 0.7 and then pelleted, washed in M9 salts, resuspended in M9 media at a ratio of 2 L of LB to 1 L of M9, and grown at 37 °C for 2.0 h. Protein expression was induced with IPTG (400 μM). Harvested cells were resuspended in 120 mL of 20 mM Tris HCl and 500 mM NaCl at pH 8.0 and stored at −80 °C.
Protein was purified by nickel affinity chromatography. After thawing, the cell suspension was supplemented with 4 μL of benzonase nuclease and 0.25 mg/mL lysozyme and stirred for 30 min at room temperature. Cell lysis was carried out with six French Press cycles at 10 000 psi. The supernatant, containing the fusion protein, was clarified by ultracentrifugation and loaded onto a 20 mL nickel affinity column in the presence of 20 mM imidazole. The column was washed with 20 mM imidazole and 60 mM imidazole. Final elution was performed in 20 mM Tris, 500 mM NaCl, and 500 mM imidazole at pH 8.0. A typical yield of the MBP/S31N M2CD fusion protein was 250–300 mg/L of M9 media.
Immediately following purification, S31N M2CD was cleaved from the MBP expression tag using TEV at a 1:2 molar ratio and at a total protein concentration of 3 mg/mL. A high cleavage efficiency of >90% was attained after 24–36 h when checked by SDS-PAGE (Figure S1). The protein was precipitated with trichloroacetic acid (TCA) and pelleted by centrifugation at 16 000g. The protein pellet was washed three times with ultrapure water to remove excess TCA The pellet was vacuum dried overnight. S31N M2CD was extracted by washing the dry pellet with HPLC-grade methanol for 30 min. S31N M2CD extraction with methanol was terminated by another round of centrifugation at 16 000g.
Sample Preparation.
An organic solvent-mediated protocol was used for protein reconstitution into DOPC/DOPE (4:1 molar ratio) liposomes at pH 7.5 as previously described.44 A tetramer:lipid ratio of 1:240 was used for the OS ssNMR samples and of 1:120 for the MAS ssNMR samples. Chloroform lipid stocks were mixed, and lipid film formation was facilitated by a gentle stream of nitrogen gas, after which the film was left under vacuum overnight to aid in removal of residual chloroform. M2CD in methanol was added to the lipid film and gently agitated to solubilize the protein. Again, the solvent was removed under nitrogen gas, and the protein/lipid film was left under vacuum overnight. The film was then hydrated with 20 mM Tris HCI pH 7.5 and left to “age” for 3 h at room temperature in an incubator shaker at 150 rpm. Once homogenized, the proteoliposomes were dialyzed against 5 mM Tris HCI pH 7.5 for 24 h to remove traces of organic solvent and to equilibrate the pH. The final pellet was ultracentrifuged at 200 000g for 2 h. For MAS samples, the pellet was left in the ultracentrifuge tube for several days at 0 °C, after which it was packed into the rotor. For oriented sample ssNMR spectroscopy, after ultracentrifugation, the pellet was resuspended in 5 mM Tris HCl pH 7.5 buffer to a final volume of 1200 μL and deposited onto glass slides with 30 μL of the proteoliposome suspension applied to one 5.7 mm × 12 mm surface of each 60 μm thick slide (total 40 slides). The slides were dehydrated in a sealed 98% relative humidity chamber at room temperature for ~16 h. Once a film was formed on the slides, it were rehydrated with 1.5 μL of ultrapure water and stacked. The stack was incubated in the hydration chamber for an average of 7 days. Once stacked slides were no longer opaque in appearance, they were transferred to a glass sample cell and sealed with beeswax. The weight of the cell was monitored to assess potential hydration loss. Mechanical alignment of the membrane proteins for static ssNMR on glass slides has been successfully applied to many other systems in structure determination efforts.45–48
Solid-State NMR Spectroscopy.
The MAS ssNMR experiments (REDOR, NCA, and DARR) were performed at 600 MHz utilizing a low-E triple-resonance probe in a 1H–13C–15N or 1H–13C–2H configuration. The OS ssNMR spectra (PISEMA49,50 and SAMPI451) that correlate anisotropic 15N chemical shifts and 1H–15N dipolar couplings were acquired at 720 MHz with a low E 1H/15N double-resonance probe. Acquisition took place at 303 K, above the gel to liquid crystalline phase transition temperature of DOPC/DOPE lipids. Experimental parameters included a 90° pulse of 5 ms and cross-polarization contact time of 0.9 ms, a 4 s recycle delay, and SPINAL decoupling sequence.52 Twenty-two to twenty-eight t1 points were acquired with 2048 transients. Additional details are presented in the SI. Spectral processing was performed with NMRPIPE53 and plotting with SPARKY. 15N chemical shifts were referenced to a concentrated solution of (NH4)2SO4, defined as 26.8 ppm relative to liquid ammonia. Further description of spectral acquisition and data analysis can be found in the SI.
RESULTS
The amino acid sequences for WT and S31N M2CD are presented in Figure 1 with the TM helical region in bold and the C-terminal amphipathic helix in italic. The 2L0J WT M2CD structure (Figure 1A and 1D) was characterized by OS ssNMR spectroscopy using a tetramer:lipid ratio of 1:360.2 The liquid crystalline dioleoylphosphatidylcholine (DOPC) and dioleoylphosphatidylethanolamine (DOPE) lipid bilayer has a well-defined hydrophobic thickness and a substantial hydrophobic/hydrophilic interface typical of liquid crystalline lipid bilayers (modeled in Figure 1D). This bilayer has approximately the thickness of native inner membranes, and consequently, the helical tilt angles should closely reflect those of the native structure in its native membrane environment. The OS ssNMR restraints were uniquely obtained as high-resolution orientations of the 15N–1H dipolar vectors and the 15N anisotropic chemical shift tensors relative to the bilayer normal that was aligned parallel to the magnetic field. The 2KIH S31N M2CD structure (Figure 1C and 1F) was characterized using a suite of distance and torsional restraints from 1H solution NMR spectroscopy of a sample in DHPC micelles.5 The DHPC micelles have neither a well-defined hydrophobic thickness nor a sharp hydrophobic/hydrophilic interface due to the variable size of the protein–micelle complex (modeled in Figure 1F). Consequently, the tilt and uniformity of the helices are not constrained as in native lipid bilayers by this environment.28 The 2N70 S31N M2CD structure (Figure 1B and 1E) was obtained using distance and chemical shift restraints from MAS ssNMR data obtained from a DPhPC liquid crystalline lipid bilayer preparation (modeled in Figure 1E).4 These methylated lipid chains display no gel to liquid crystalline lipid bilayer phase transition over a temperature range from −120 to +120 °C, a significant advantage for the spectroscopy. However, these bilayers may be somewhat thicker than native liquid crystalline lipid bilayers for the M2 protein. For this sample, the authors used a protein tetramer to lipid molar ratio of 1:24 (equal dry weights of protein and lipid).
All three structures are tetrameric. The 2L0J structure obtained from residues 22–62 of the full-length WT M2 protein represents the structured portion of the entire protein. The helical backbone structure is a 4-fold-symmetric structure in lipid bilayers that gives rise to a single set of resonances in OS ssNMR for all of the observed sites.2 MAS ssNMR spectra published in 2012 are consistent with this view of a 4-fold-symmetric backbone but with the His37 and Trp41 side chains showing pairs of resonances for each site, documenting that these functionally critical side chains form a dimer of dimers structure.44 Indeed, recent results confirm a hydrogen bond between pairs of His37 residues.54,55 While this side-chain behavior does extend to the backbone of these His and Trp sites, it is limited to a small Ca shift in the backbone for the His37 and Trp41 sites in 2L0J2 and to our S31N M2MAS ssNMR44 (see Table S2). The N-terminal portion of the TM helix has a slight kink near Gly34, a residue that is known to facilitate small kinks in the TM helices. The majority of the helix has a tilt of ~32°. The amphipathic helix formed by residues 47–62 in 2L0J has a tilt angle of 105° and is embedded in the lipid interface.
The 2N70 structure of S31N M2 (residues 18–60) in a DPhPC lipid environment forms a dimer of dimers structure from four identical monomers. One pair of opposing helices in the dimer of dimers structure shares a large interaction surface, while the other pair has no interaction surface, as seen in Figure 1B. However, it is similar to the 2L0J structure in that the first portion of the helix appears to have a slightly larger tilt angle with respect to the axis of the helical bundle than the latter portion of the helix. In addition, one of the helical pairs has amphipathic helices in the presumed membrane interface, but the other pair of monomers has disordered C-terminal sequences.
The 2KIH structure of S31N M2 (residues 18–60) is also a tetrameric structure, but there is considerable heterogeneity in the set of TM helical structures, and the amphipathic helices form a water-soluble four-helix bundle. While this amphipathic bundle of helices does not appear to be associated with the modeled membrane environment, there may be a different micellar environment for the amphipathic helices separated from the TM helical environment.6 The TM four-helix bundle forms an hourglass shape such that the pore narrows at the center of the membrane. The shape is due to a bend in the helical axis that is highly variable both within each tetramer (~0–28°) and within the set of PDB structures, often with one helix nearly straight and the other three with bend angles of ~20°. This shape of the four-helix bundle results in a pore that narrows near the His37 residues that are packed tightly together with very little space for the dynamics that are essential for the proton transport function of this protein (Figure 1F).
TM Domain Comparison with the S31N M2CD 2N70 Structure.
In Figure 2 the MAS ssNMR 13C–13C correlation spectrum for 13C Ile- and Leu-labeled S31N M2CD in lipid bilayers is presented. Resonances were assigned with confidence based on our previous published work.44 There are 5 Ile residues in the TM domain of M2 (residues 32, 33, 35, 39, and 42) and another Ile residue in the amphipathic helix (I51) of this construct that has a somewhat upfield-shifted Ca resonance. The experimental Ca resonances observed here for all of these Ile helical sites fall into a narrow range of 65.5 ± 0.5 ppm as seen in both panels of Figure 2, which displays two copies of the same data set. These resonance frequencies are all downfield of the mean helical Ca frequency for Ile (~63.8 ppm) consistent with typical TM helical Ca chemical shift values reflecting the hydrophobic TM environment and the typical uniformity of the TM helical structure.58–60 A set of Ile Ca resonance frequencies similar to what we observe here for the S31N protein was published in 2012 for WT M2CD44 and presented in Table S1. The upfield shoulder on the observed group of Ile Ca resonances is most likely associated with I51 of the amphipathic helix having a frequency more typical of water-soluble helices than the TM Ile resonances including I32 and I39 that are further downfield near 66 ppm. The Ca Leu resonances are centered at a 13C chemical shift of 58.5 ±1.5 ppm as assigned previously by Can et al.44 The resonance cluster includes residue 26 at the start of the TM helix as well as L36, L38, L40, L43, and L46 in the TM helix. In addition, there is one Leu in the amphipathic helix (L59), which we tentatively assign to the upfield resonance near 57 ppm. These Ca frequencies have a slightly broader range than the Ile resonances, but again these Leu Ca chemical shifts reflect either helical or hyperhelical sites, as is typical of TM helices.60 Table S2 compares selected N and Ca chemical shifts obtained from a sample of S31N M2TM, the transmembrane domain of S31N M2, with the chemical shifts from 2N70.
Figure 2.
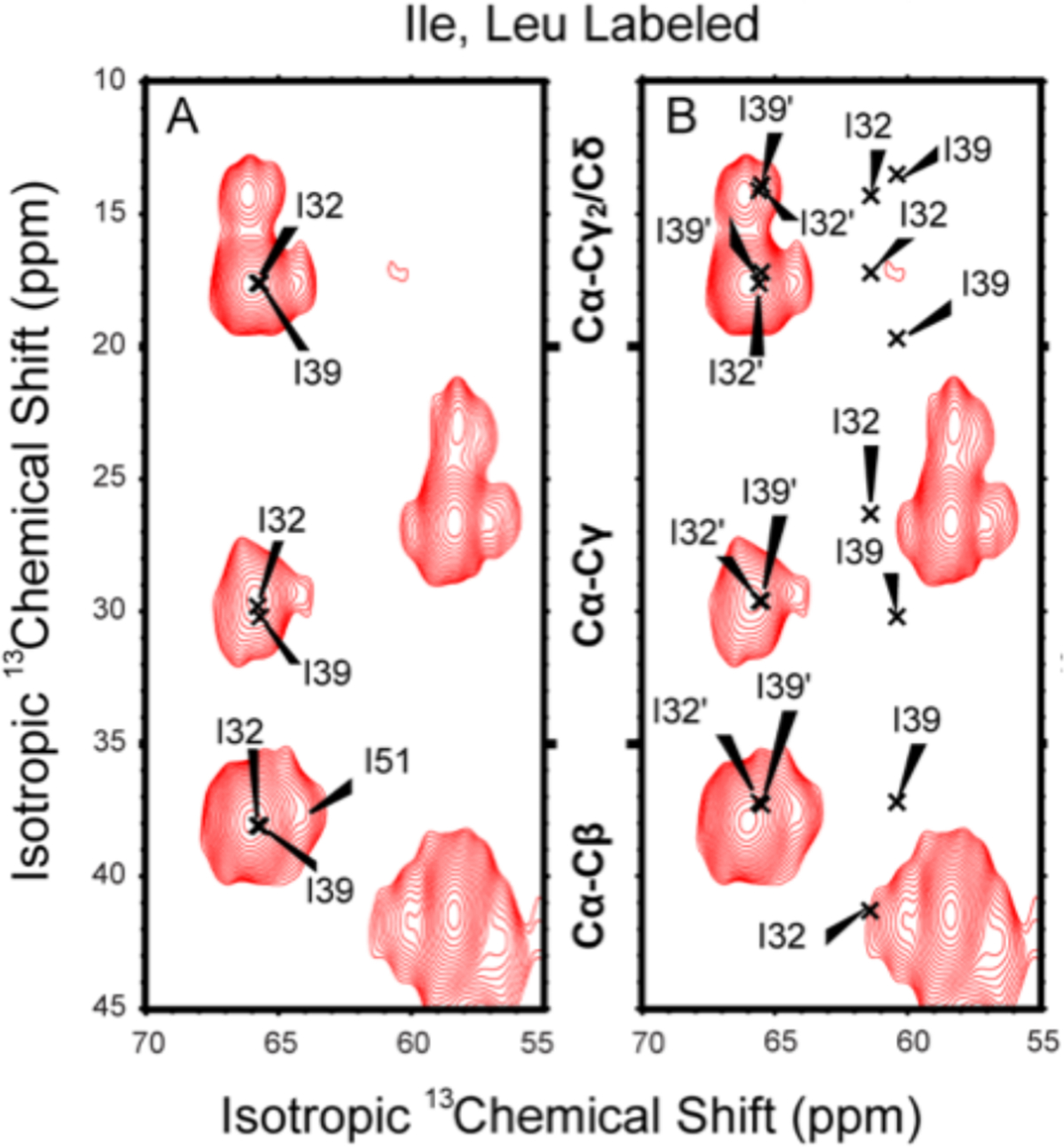
Two-dimensional 13C–13C PARIS MAS spectra of 13C,15N Ile- and Leu-labeled S31N M2CD in DOPC/DOPE lipid bilayers at pH 7.5 using a 10 ms mixing time. Both panels show the same experimental spectrum. (A) Five M2 Ile residues in the TM helix of M2CD (I32, I33, I35, I39, and I42) have Ca chemical shifts between 65.7 and 65.8 ppm as determined by Can et al. using the same lipid preparation44 I51 of the amphipathic helix has a slightly upfield-shifted resonance (~64.5 ppm). (B) Same spectrum as A, but the x’s denote resonance frequencies and assignments as determined by Andreas et al.4 Resonance frequencies and assignments for the published data from 2L0J and 2N70 are presented in Table S1.
MAS ssNMR Spectral Comparison for S31N M2CD.
The MAS ssNMR resonances observed here for S31N M2CD are compared with data from 2N70. Because the 2N70 dimer of dimers structure has two different monomer structures, one pair of monomers is denoted with a prime and the other pair without a prime. In Figure 2, while the prime frequencies are in excellent agreement with the data presented here, the nonprime frequencies from 2N70 for I32 and I39 are shifted upfield by 4.7 and 5.8 ppm (Table S1), as indicated by the x’s in the figure. This is despite the amino acid constructs for 2N70 (residues 18–60) and for our study (residues 22–62) being nearly identical, and both samples provide lipid environments for the S31N M2CD. These two residues are in I to I+7 positions in the TM helices, both facing the external environment of the tetramer, which for I32 and I39 in the 2L0J structure is the extensive lipid environment that induces downfield Ca resonance shifts having backbone φ,ψ torsion angles near −60° and −45°.58–60 However, the resonance frequencies for these residues of the nonprime helices observed by Andreas and co-workers4 are shifted upfield into a nonhelical Ca chemical shift range for Ile. From TALOS+,61 these Ca frequencies equate to φ,ψ torsion angles of −104°,−9° for I32 and −90°,−13° for I39, clearly nonhelical values that should significantly distort the helix, and yet the 2N70 structure has helical φ,ψ torsion angles of −51°,−39° and −50°,−39° for these two sites, consistent with the helical structure presented but inconsistent with the Ca frequencies (Table S1).
Interestingly, the Ca frequency rmsd for the primed helix (residues 26–43) of 2N70 versus the WT M2 values published by Can and co-workers excluding N31 is just 0.55 ppm (Table S1), and if V28 is also excluded, the rmsd over 16 residues is just 0.27 ppm. The structures of the primed helix and that of Can et al. are therefore very similar. The pair of TM nonprimed helices in 2N70 hosting the dramatically shifted I32 and I39 resonances has a Ca frequency rmsd of 0.83 ppm with the WT M2 values if the shifts for N31 (relative to S31), I32, and I39 are excluded. In other words, the dramatic nonhelical Ca shifts in this nonprimed helix are localized to just these two Ile residues. Consequently, the spectral differences between the WT helical structure44 and the S31N mutant structure4 are primarily associated with the nonprimed helical sites for I32 and I39 that face what should be the lipid environment. Since the fatty acyl lipid environment is a very uniform environment, a different environment than that of the DPhPC lipids is required to explain these results. Furthermore, since the primed helix of 2N70 generates very similar Ca frequencies compared to the WT M2 protein, this implies that the cause for the structural distortions observed in the nonprime helix has nothing to do with the amino acid sequence but with a different external influence that affects two of the four helices breaking the 4-fold helical symmetry resulting in the 2N70 dimer of dimers structure.
The 2N70 authors acknowledged a pair of cross peaks arising from interactions between a pair of tetramers (V27′g1-H37′e1, V27/28′g1-W41d1) in their manuscript.4 Such contacts that give rise to significant cross peaks document very stable interactions associated with a non-native antiparallel pairing of tetramers. Indeed, the 2-fold symmetry documents that pairs of antiparallel “tetramers” that are themselves a pair of dimers dominate the sample. These observed cross peaks allude to the realization that there exists a large and stable interaction surface between a pair of four-helix bundles, not just within the four-helix bundle. Such interactions are likely responsible for corrupting the 4-fold symmetry of each tetramer into the observed dimer of dimers structures that is presented in the 2N70 PDB file. In other words, the environment for two of the monomers is different from the other two monomers in each tetramer. Indeed, this provides the required explanation for a break in the 4-fold symmetry. Recall that it is not just these two cross peaks that break the symmetry, as only two of the amphipathic helices are structured. We note here that while Andreas et al. used a protein:lipid molar ratio of 1:24, we used a molar ratio of 1:120 for our MAS ssNMR samples, a factor of 5 more lipid. Note that a ratio 1:24 means only 12 lipids per lipid leaflet per M2 tetramer of this protein. What we document here is that when enough lipid is present, as in our samples and those for the WT M2 structure, 2L0J, there are no stable interactions between the tetramers and instead of a dimer of dimers structure, a stable 4-fold-symmetric tetramer is observed. We conclude that the protein in the samples used by Andreas et al.4 formed a 2-fold-asymmetric interaction with other M2 tetramers. In other words, strings of antiparallel tetramers formed leading to 2-fold-symmetric structures, as opposed to forming a 4-fold-symmetric array of interactions, such that all monomers would be influenced equally. These antiparallel 2-fold-symmetric interactions are non-native, as there is no evidence that M2 is inserted into the plasma membrane with an orientation other than the one having the C terminus on the cytoplasmic side of the membrane.
OS ssNMR Spectral Data for S31N M2CD.
The 2N70 authors provided no explanation for why four identical monomers should form anything other than a 4-fold-symmetric structure. It is rare in biochemistry that the same sequence can lead to two different conformations, but an example appears to be the pair of EmrE structures.62 Indeed, the His37 and Trp41 residues do break this symmetry slightly due to His-His+ hydrogen bonding between structural monomers, but in the 2N70 structure, it is clear that interactions between tetramers dramatically break the symmetry, generating an asymmetric helical bundle. Unfortunately, there is no biological situation in which the antiparallel packing of these structures could occur, and therefore, this is a non-native structure.
An additional point can be made here: in each dimer of dimers structure there are two monomers that have dramatically shifted Ile Ca (residues 32 and 39) resonances. These two helices are on opposite sides of the tetramer. Consequently, the observed structure is not a pair of antiparallel dimer of dimers as described in the text but extensive strings of antiparallel dimer of dimers.
OS ssNMR spectra of S31N M2CD are presented in Figure 3B–D from three 15N isotopically labeled samples having a protein:lipid molar ratio of 1:240 that in combination represents more than one-half of the backbone amide sites in the TM helical domain and a pair of backbone amide sites in the amphipathic helix. The OS ssNMR spectra separate signals based on the orientations of each backbone 15N–1H dipolar and 15N anisotropic chemical shift tensors relative to the magnetic field axis. These samples have the bilayer normal aligned parallel with the magnetic field axis. For a TM helix with ~3.6 residues per turn, adjacent residues should be well separated in the spectra, while the resonances for the I and I+4 residues as well as those of I and I+7 should be close together in the spectra. The line widths associated with these resonances reflect less than a 2° dispersion in orientation of the proteins with respect to the magnetic field and the bilayer normal. This demonstrates excellent alignment of the samples as well as a uniform 4-fold symmetry for these samples. Consequently, there is no uniform doubling of the resonances. Note that in these spectra there is often some intensity in the vicinity of the isotropic chemical shift (~120 ppm) with a small dipolar coupling due to imperfections in the uniform alignment of the sample (typically from some sample that is not between the glass slides responsible for aligning the sample).
Figure 3.
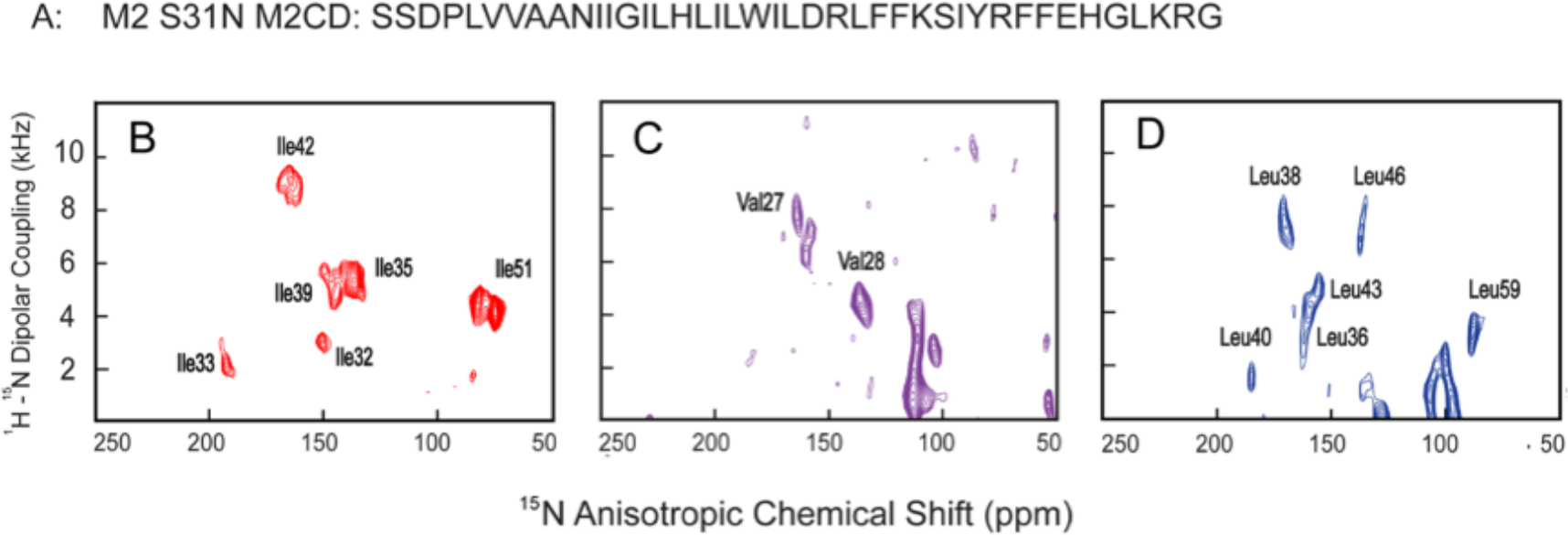
PISEMA OS ssNMR spectra of amino acid-specific-labeled S31N M2CD in aligned DOPC/DOPE lipid bilayers. (A) M2CD S31N amino acid sequence. (B) 15N Ile-labeled, (C) 15N Val-labeled, and (D) 15N Leu-labeled M2CD samples.
In Figure 4A,B independent wave analyses of the S31N M2CD data from Figure 3 are presented that reflect an oscillatory pattern of the anisotropic OS ssNMR observables with a period of 3.6 residues against the residue number in the TM helix. These plots characterize a unique helical tilt (τ = 36° ± 2°) and rotational angle (ρ = 105 ± 5°) relative to the V27 Ca site for each of the four helices.45,46 In both sets of waves there is scatter that is greater than the experimental error bars, suggesting small variations in the torsion angles along the helical axis. Considering the large size difference of the side chains and varying environment around the helical axis this is not surprising. The tilt angle is a little larger than what was observed for the WT M2CD (τ = 32° ± 2° for the N-terminal domain and τ = 22° ± 2° for the C-terminal region of the TM helix), but it has the same value of rotational angle (ρ = 105 ± 5°). The back calculation of the dipolar and anisotropic chemical shifts from the 2N70 dimer of dimers structure leads to a pair of helices consistent with the authors dimer of dimers structure (Figure 4C,D and 4E,F). The nonprimed helix (Figure 4C,D) is a kinked helix with residues 26–34 having a helical tilt and rotation angles of 29° and 150°, respectively, while residues 35–46 have a tilt of 23° and a rotation angle of 125°. However, the primed data set (Figure 4E,F) leads to a more uniform helix characterized by a single tilt of 30° and a rotation angle of 105°. This latter helical characterization is very similar to the helical structure characterized for the N-terminal domain of the 2L0J structure having a tilt of 32° and a rotation angle of 105°. Furthermore, it is similar to the values determined here of 36° and 105° for the S31N M2CD structure. Overall, it is clear that the 2N70 S31N M2CD structure is a dimer of dimers structure, while our experimental data with a much larger lipid environment for S31N M2CD clearly represents a single-helical structure generating a 4-fold-symmetric S31N M2CD structure.
Figure 4.
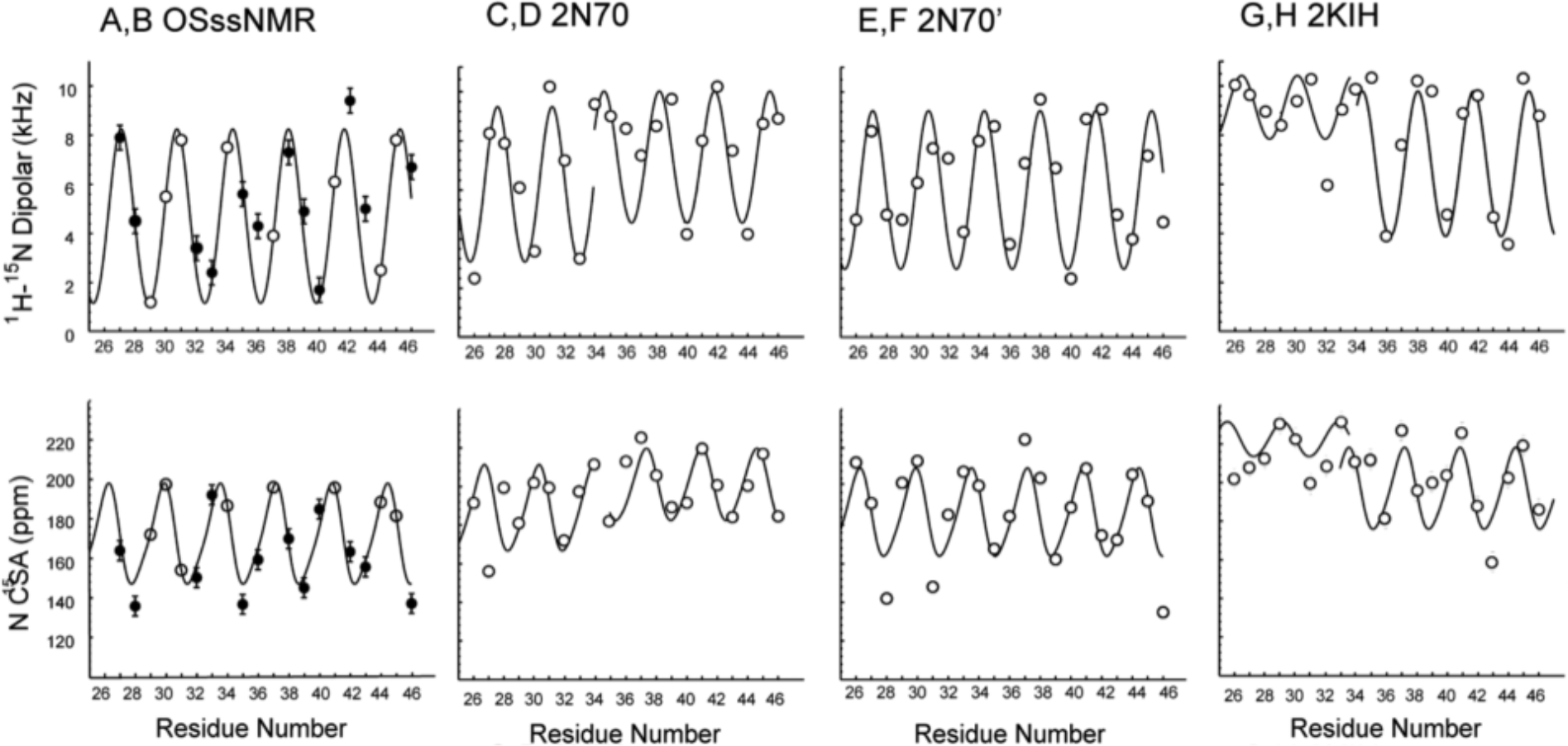
Helical tilt (τ) and rotation (ρ) analysis of the experimental OS ssNMR data for S31N M2CD from Figure 3 shown in A,B compared with predicted data from 2N70 (C,D and E,F) and 2KIH (G,H). Experimental data in A,B is shown as solid circles with an experimental error bar, and for sites without experimental data, predicted values on the curves are shown based on the modeled helix through the data set that defines a tilt and rotational orientation for the helix. For C–H, predicted values from the structural coordinates are shown as open circles and sinusoidal waves having a period of 3.6 residues are drawn as best fits to the data. All values are listed in Table S3, τ is defined relative to the bilayer normal or the symmetry axis for the structure, and ρ is set arbitrarily relative to the rotational position of V27 Ca. Top row is based on 1H–15N dipolar predictions and data, while bottom row is based on 15N anisotropic chemical shift predictions and data. From the (A,B) experimental S31N M2CD data in Figure 3 with error bars of ±0.5 kHz and ±5 ppm, a uniform value of τ = 36° and ρ = 105° is obtained. (C,D) 2N70 nonprimed helices: residues 26–34 are consistent with τ = 29° and ρ = 150°, and residues 35–46 are consistent with τ = 23° and ρ = 125°. (E,F) 2N70 primed helices, reflecting values of τ = 30° and ρ = 105°. (G,H) 2KIH data from helix B: residues 26–33 consistent with τ = 10° and ρ = 40°, and residues 34–46 are consistent with τ = 26° and ρ = 40°.
TM Domain Comparison with the S31N M2CD 2KIH Structure.
The 2KIH S31N M2 structure achieved by Chou and colleagues in 2009 using solution NMR in short-chain detergent micelles formed by DHPC was the first structure of the S31N M2 mutant to be characterized.6 These DHPC micelles have a prolate ellipsoid shape,63 and even 12 years ago there were serious concerns that this structural approach was fraught with potential troubles due to the properties of this environment.28 A weak hydrophobic environment is formed by these micelles that results in weakened hydrogen bonding, leading to less uniform and more dynamic helical structures. Extensive water penetration into the hydrophobic region also weakens the intraprotein hydrogen-bonding and van der Waals interactions in the hydrocarbon environment.28,29 Variable hydrophobic dimensions formed by detergent environments further weaken this important structural restraint on the tilt of α-helices that is induced by the well-defined hydrophobic thickness of lipid bilayers. However, the detergent environment results in short correlation times for the protein/micelle complexes, leading to an ability to observe high-resolution solution NMR spectra, and for this purpose, the authors used detergent micelles from 300 mM DHPC. The set of 10 structures deposited in the 2KIH PDB file has much greater structural variability than the structures characterized by ssNMR in lipid bilayers (2L0J or 2N70). Indeed, the PDB 2KIH set of structures illustrates a distinct hourglass shape resulting from a set of curved helices with a bend of up to 25° in the TM region of the structure. A set of dipolar and chemical shift waves for a helix from structure 1 of the PDB file with relatively well-defined tilt and rotation angles is shown in Figure 4 G,H, illustrating a small tilt angle in the N-terminal half of the TM and a much greater tilt angle in the latter half of the TM helix.
Due to the limited interactions between the four TM helices and the hydrophilicity of the micelle environment, multiple fenestrations from the pore to the detergent environment having a diameter approaching that of an alkyl chain persist through the set of 10 structures in the PDB 2KIH file. This strongly suggests that there is the potential for acyl chain penetration into the M2 pore or for water entrapment in the wall of the pore. As noted above, the pore is narrow, as would be expected for such modest tilt angles, and while it appears to be adequate for water and hydronium ions to penetrate to the His-37 residues, the C-terminal pore does not appear to be large enough to support the functional dynamics for the His37 and Trp41 side chains.15,64 Such dynamics are necessary to shuttle protons via the His37 residues to the C-terminal aqueous cavity where the indole side chains of Trp41 have been proposed to act as a gate through side-chain dynamics for proton (hydronium ion) release into the viral interior. Clearly, both of the ssNMR structures in the lipid bilayers (2L0J and 2N70) have a more tightly packed structure of the TM helices in their lipid environments.
S31N Effects on Drug Binding.
The anti-Influenza drugs amantadine and rimantadine bind in the pore of the WT M2 on the 4-fold axis in the vicinity of Ser-31. In 2013, Griffin and co-workers using DNP located the position of rimantadine in the pore with specific distances from the drug to A30 and G34,40 and in 2016, the differential binding of R- and S-rimantadine was characterized by REDOR measurements to G34,41 resulting in a characterization of the drug binding site in the same vicinity as previously proposed. The bulky native substitution of Asn for Ser lining the pore reduced the pore dimensions, such that these drugs do not have access to a pore binding site. In the DHPC environment, the WT M2 protein (2RLF) showed rimantadine bound with 4-fold symmetry at the detergent-protein interface far removed from the pore.5 In the follow-up manuscript for the S31N M2CD (2KIH) protein structure it was stated that the mutation dramatically reduced drug binding at the protein-detergent interface and that “drug-resistant mutants impair drug binding by destabilizing the helix-helix assembly”.6 This is not consistent with the set of 2KIH structures that display a broad range of helical bends and tilts; furthermore, N31 in the 2KIH structure is located at the helix–helix interface, while the Ser31 site in 2L0J or N31 in 2N70 is in a pore-lining position. The result is that the2KIH structure does not show a pore blocked by N31; instead, it remains open to the H37 residues.
S31N Effect of the Environment on Amphipathic Helices.
The amphipathic helix in the WT M2 structure 2L0J is located at the lipid interface having a dramatic hydrophobic surface including F47, F48, I51, F54, F55, and L59 with the phenylalanines forming two I to I+7 pairs. S50 in the constructs for 2L0J (Figure 5A), 2N70 (Figure 5B), and 2KIH (Figure 5C) is actually a mutation of the native C50 to avoid the formation of disulfide bonds between M2 monomers. Moreover, in the native structure these sites are palmitoylated, as shown in Figure 5, which further facilitates the tethering of the amphipathic helix to the membrane interface.
Figure 5.
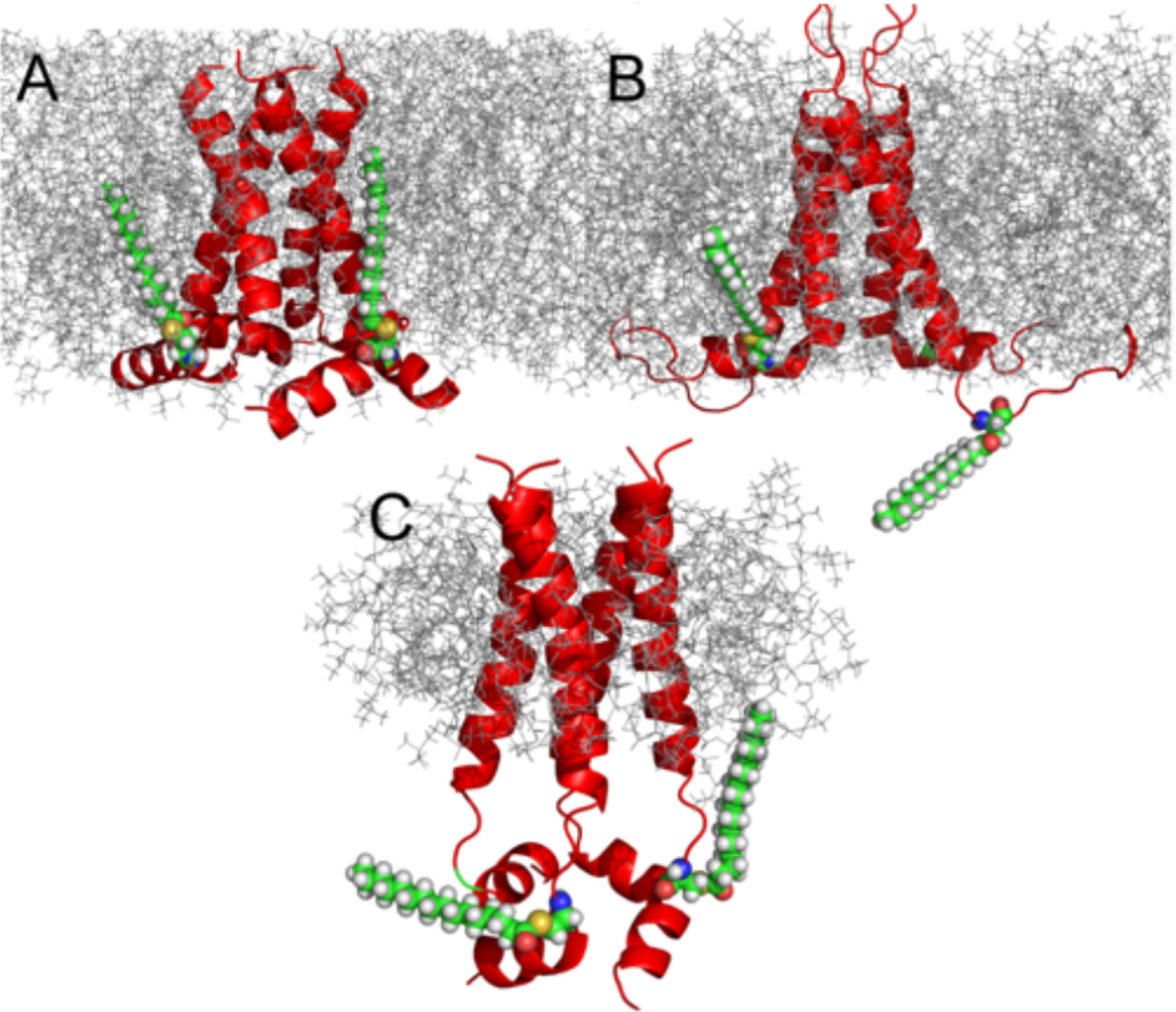
Models comparing the palmitoylation at two of the four palmitoylation sites with all atom structures bound to Cys50 in the amphipathic helices for each of the M2 structures: 2L0J (A), 2N70 (B), and 2KIH (C). Lipids and detergent micelle environments were introduced for illustration purposes using chamm-gui.57 Palmitoylation of Cys50 was also introduced using charmm-gui57 Assembly was not equilibrated; therefore, the orientation of the palmitic acyl chain is constrained by the rotameric state of Cys50. In the protein sequence for the NMR experiments, Cys50 was mutated to Ser. Here, the oxygen atom of Ser50 was exchanged for sulfur and the rotamer state was kept as obtained from the structures.
Such palmitoylation of amphipathic helices has been observed in multiple GPCRs that result in facilitating the binding of cholesterol, further tethering amphipathic helices to the lipid surface.65 This binding is often observed at the interface between two transmembrane helices. Such binding of cholesterol is also known for the M2 protein requiring the amphipathic helix to be in a position such that Cys-50 palmitoylation can facilitate cholesterol binding at the TM helix–helix interface.66 While none of the structural endeavors described in this manuscript included this palmitoylation, we model in Figure 5 such palmitoylation for just two of the four monomers. These amphipathic helices are critical for the viral budding process, since this virus does not co-opt the cell ESCRT complex to bud-off nascent virions.67,68 Mutations that lead to disruption of the amphipathic nature of this C-terminal helix have been shown to abolish viral budding and influence infectivity.66 This is because the amphipathic helices induce a frustrum-shaped structure for this tetramer, generating a broader C-terminal base in the cellular interior compared to the smaller surface on the cellular exterior. In addition, the amphipathic helices result in splaying the C-terminal ends of the TM helices, as is clear in the 2L0J structure, facilitating the functional dynamics of the bulky indole and imidazole side chains in the pore where they function in transporting and gating protons through the aqueous pore of M2.
In Figure 5B the AC monomers of the 2N70 structure have amphipathic helices that are approximately parallel to the bilayer surface, similar to those of the WT 2L0J structure, where for this later structure the tilt and rotational orientation were defined by a PISA wheel analysis characterizing a tilt of 105° with respect to the bilayer normal.2 However, the BD monomers of 2N70 have disordered residues for the amphipathic helices. This is most likely the result of interactions between not only two antiparallel tetramers, which would interfere with just one of the four amphipathic helices, but rather, the symmetry of this dimer of dimers structure appears to support an extensive string of dimer of dimers that would preserve the 2-fold symmetry for all of the dimer of dimers. This again illustrates the lack of lipids in the sample and the source of the structural distortion. Clearly, only two of the amphipathic helices are in a position that would be appropriate for palmitoylation.
In the detergent-solubilized S31N M2CD (2KIH) structure, the amphipathic helices form of a water-soluble helical tetrad (Figure 5C).5 This was also true for the WT M2CD characterized in the same DHPC micelles.5 The native interaction of the amphipathic helices with the lipid interfacial region is not supported in this detergent micelle environment.
DISCUSSION
Membrane proteins occur in cellular and viral environments where the membrane protein content is known to be high, so it may seem to be reasonable to characterize membrane proteins under conditions where the fraction of an individual protein is high. However, in cell membranes, the proteins have a unique orientation and there is a great diversity of membrane proteins that function independently and typically do not bind to each other. Indeed, the M2 tetramers function independently as proton channels, but for viral budding, the uniquely oriented tetramers are known to interact with each other in a parallel orientation, facilitating membrane curvature that is essential for the budding process.4,11 However, in membranes where this protein functions as a proton channel, these channels function independently.
Detergent-based environments for membrane proteins do not represent a good model for either the hydrophobic interstices of the lipid bilayer or the lipid interfacial region. This is well recognized in the NMR community today, and the use of lipid nanodiscs permits relatively short correlation times and the solvation of membrane proteins in a lipid environment representing a vast improvement over detergent micelle environments.25,26 This development has been a great advance for solution NMR. However, the solution NMR structures of the S31N M2CD structure were performed in detergent environments of DHPC micelles having a hydrophobic dimension that is variable unlike lipid bilayers that have a well-defined hydrophobic thickness. Furthermore, water penetration into detergent micelles is much more significant than that into a lipid bilayer where the lateral pressure at the bilayer hydrophilic/hydrophobic interface makes water penetration much more difficult. Water access to the TM helix from the detergent environment coupled with water access from the pore destabilizes the hydrogen bonds between turns of the helices, resulting in much more flexibility in the helices, a feature that is observed in the 2KIH set of structures. Consequently, more significant bends in the TM helices and a much weaker packing of the helices is observed than in a lipid bilayer environment, which leads to a more uniform helical structure having water exposure to the helix only via the pore.
The key to forming a structure, such as 2L0J, that can account for both proton conductance and facilitation of viral budding is the development of an environment that also permits a 4-fold-symmetric structure with the amphipathic helices in the lipid interface. While 2L0J is a WT structure and 2N70 and 2KIH are both mutants, this mutation does not distort the 4-fold symmetry of the backbone structure, as is clear from the new experimental data presented in this manuscript. To their credit, the authors of the 2N70 dimer of dimers structure used a lipid environment; however, their samples had a tetramer:lipid ratio of 1:24. Calculating the number of lipids necessary to solvate each tetramer with a liquid crystalline lipid environment is complex, as can be seen in the molecular dynamics simulations where the fatty acyl chains flail about. However, in our studies of the smaller M2TM domain, a 1:28 protein tetramer to lipid ratio resulted in a 24-fold increase in the rotational correlation time of the tetramer in a liquid crystalline lipid environment compared to preparations using a 1:80 ratio.11 This dramatic reduction in mobility at low molar ratios strongly suggests that the tetrameric TM domains are interacting with each other as was shown by molecular dynamics in the same manuscript. For the M2CD structure, even more lipid would need to be present to avoid interactions between the tetramers, as illustrated by MD simulations.11 This suggests that the protein:lipid ratio of 1:24 used by Andreas et al.4 was not adequate to ensure lipid solvation of individual M2CD tetramers.
In our studies presented here using a tetramer to lipid molar ratio of 1:120 for the MAS samples and 1:240 for the OS samples there was no evidence for a dimer of dimers structure. The observed individual resonances in both the MAS and the OS ssNMR spectra for single sites reflect single resonances for a 4-fold-symmetric structure. Consequently, we conclude that the 1:24 molar ratio of protein:lipid used by Griffin and co-workers leads to tetramer–tetramer interactions between antiparallel channels, which is not a state experienced by the protein in the viral membrane. Indeed, the authors acknowledged the observation of two molecular interactions between antiparallel tetramers: V27′g1-H37′e1 and V27/28′g1-W41d1. These cross peaks suggest a stable and substantial set of interactions between antiparallel tetramers throughout their sample that explains the formation of the dimer of dimers conformation. If the tetramers formed a string of tetramers as the molecular dynamics simulations showed for the M2TM studies noted above, then the result would be a dimer of dimers structure.11 Here, we document that such a dimer of dimers structure is not formed when an adequate lipid environment is provided and that the tetrameric structure is stable for S31N M2CD. Such structural sensitivity even with an equal dry weight of lipid to protein (1:24 molar ratio) might be surprising. However, unlike water-soluble protein structures, tertiary interactions (i.e., between secondary structural components) in single TM helical protein structures are almost exclusively in the form of weak van der Waals interactions strengthened somewhat by hydrophobic effects associated with the aqueous pore running through the tetrameric structure and the very low dielectric of the lipid bilayer interstices.
CONCLUSIONS
Here, we present new structural data for S31N M2CD utilizing a mix of MAS and OS ssNMR structural restraints for the polypeptide backbone. This data clearly shows that the protein is not a dimer of dimers structure but a 4-fold-symmetric structure, much like the WT M2CD structure, 2L0J, when an adequate membrane environment is present. The dimer of dimers structure of S31N M2CD is shown to be a consequence of crowding, leading to non-native antiparallel tetramer–tetramer interactions that further lead each tetramer to take on a dimer of dimers conformation. The crowding in the samples for the 2N70 structure is due to the lack of an adequate lipid environment for this tetrameric membrane protein. Consequently, the dimer of dimers conformation resulting from antiparallel tetramer interactions is induced by the sample preparation and is not a native-like conformation experienced by the M2 protein in the viral membrane. Importantly, M2 is unlikely to be the only small membrane protein sensitive to the protein:lipid ratio. Testing samples for such dimerization is something that could be done before launching a major structural project.
Detergent micelle environments are now well recognized as non-native-like environments for membrane proteins and for modeling a lipid bilayer environment. Today, the nanodisc environments can be used to solubilize a single protein or protein oligomers.25,26 Furthermore, we know much more about membrane protein biophysics today than we did a decade ago. However, these structures continue to be the only representatives of the entire structured regions of the S31N M2 protein, and consequently, they remain as important structures in the Protein Data Bank. Here, we show that the lipid content for M2 protein samples must be higher than what was used for the 2N70 structure even though high-quality spectra were obtained with less lipid. Indeed, this criteria for high-quality spectra is often used for defining a good sample. This may be particularly relevant for the large community working with fast MAS ssNMR samples prepared for the sample-limited 1.3 mm and smaller rotors, similar to the effort discussed here by Andreas et al.4 The sample volume is so small that despite 1H detection there is a need to maximize the amount of protein. In forming a protein tetramer, if the environment had uniform influences on each monomer, a tetrameric structure would presumably result, such as the 2L0J M2 WT structure. If, however, substantial interactions take place between oligomers, the symmetric balance of the intraoligomer interactions may be broken, such as this case for the non-native antiparallel interactions documented in the spectroscopy for the 2N70 structure. The 4-fold symmetry was broken, and a dimer of dimers structure was generated stabilized by intertetramer interactions, two of which were identified by the authors. These intertetramer interactions led to a 2-fold structural symmetry within each tetramer, leading to the characterization of a dimer of dimers structure.4
Furthermore, for both M2TM and M2CD domains, recently published rotational correlation data document that rotational inhibition of tetramers occurs even when the lipid content is higher11 than that used by Andreas et al.4 These results were modeled by molecular dynamics simulations, suggesting that strings of M2CD “tetramers” form that could account for the dimer of dimers structural perturbation observed in the 2N70 structure.11 The 2N70 M2CD structure illustrates just how sensitive an oligomeric membrane protein structure is to its environment. However, this result should not be surprising as the tetramer is held together almost exclusively by van der Waals and hydrophobic interactions, weak interactions that are easily distorted, and hence, the need for an adequate lipid environment to isolate such tetramers. Having an adequate lipid environment is also necessary for the amphipathic helices that pack into this lipid interface only when there is an enough lipid environment available.
Supplementary Material
ACKNOWLEDGMENTS
This work was supported in part by NIH Grants AI119178 and GM122698. NMR experiments were performed at the National High Magnetic Field Laboratory, funded by the NSF Division of Materials Research (DMR-1644779) and the State of Florida.
Footnotes
The authors declare no competing financial interest.
ASSOCIATED CONTENT
Supporting Information
The Supporting Information is available free of charge at https://pubs.acs.org/doi/10.1021/jacs.1c10174.
Solid-state NMR spectroscopy methods, details, and assignment tables (PDF)
Complete contact information is available at: https://pubs.acs.org/10.1021/jacs.1c10174
Contributor Information
Anna K. Wright, Institute of Molecular Biophysics, Florida State University, Tallahassee, Florida 32306, United States; National High Magnetic Field Laboratory, Florida State University, Tallahassee, Florida 32310, United States
Joana Paulino, Institute of Molecular Biophysics, Florida State University, Tallahassee, Florida 32306, United States; National High Magnetic Field Laboratory, Florida State University, Tallahassee, Florida 32310, United States.
Timothy A. Cross, Institute of Molecular Biophysics, Florida State University, Tallahassee, Florida 32306, United States; Department of Chemistry and Biochemistry, Florida State University, Tallahassee, Florida 32306, United States; National High Magnetic Field Laboratory, Florida State University, Tallahassee, Florida 32310, United States.
REFERENCES
- (1).Sugrue RJ; Hay AJ Structural Characteristics of the M2 Protein of Influenza A Viruses: Evidence that it forms a Tetrameric Channel. Virology 1991, 180, 617–624. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (2).Sharma M; Yi M; Dong H; Qin H; Peterson E; Busath DD; Zhou H-X; Cross T A Insight into the mechanism of the influenza A proton channel from a structure in a lipid bilayer. Science 2010, 330 (6003), 509–512. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (3).Liao SY; Fritzsching KJ; Hong M Conformational analysis of the full length M2 protein of the influenza A virus using solid-state NMR. Protein Sci. 2013, 22 (11), 1623–38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (4).Andreas LB; Reese M; Eddy MT; Gelev V; Ni QZ; Miller EA; Emsley L; Pintacuda G; Chou JJ; Griffin RG. Structure and mechanism of the influenza A M218–60 dimer of dimers. J. Am. Chem. Soc. 2015, 137 (47), 14877–14886. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (5).Schnell JR; Chou JJ Structure and Mechanism of the M2 Proton Channel of Influenza A Virus. Nature 2008, 451, 591–595. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (6).Pielak RM; Schnell JR; Chou JJ Mechanism of drug inhibition and drug resistance of influenza A M2 channel. Proc. Natl. Acad. Sci. U.SA. 2009, 106 (18), 7379–7384. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (7).Takeda M; Pekosz A; Shuck K; Pinto LH; Lamb R A Influenza a virus M2 ion channel activity is essential for efficient replication in tissue culture. J. Virol. 2002, 76 (3), 1391–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (8).Martyna A; Bahsoun B; Badham MD; Srinivasan S; Howard MJ; Rossman JS Membrane remodeling by the M2 amphipathic helix drives influenza virus membrane scission. Sci. Rep. 2017, 7, 44695. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (9).Huang S; Green B; Thompson M; Chen R; Thomaston J; DeGrado WF; Howard KP C-terminal juxtamembrane region of full-length M2 protein forms a membrane surface associated amphipathic helix. Protein Sci. 2015, 24 (3), 426–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (10).Torabifard H; Panahi A; Brooks CL M2 amphipathic helices facilitate pH dependent conformational transition in influenza A virus. Proc. Natl Acad. Sci. U.S.A. 2020, 117 (7), 3583–3591. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (11).Paulino J; Pang X; Hung I; Zhou H-X; Cross T A Influenza A M2 channel clustering at high protein/lipid ratios: Viral budding implications. Biophys. J. 2019, 116 (6), 1075–1084. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (12).Ma C; Polishchuk AL; Ohigashi Y; Stouffer AL; Schön A; Magavem E; Jing X; Lear JD; Freire E; Lamb RA; et al. Identification of the functional core of the influenza A virus A/M2 proton-selective ion channel. Proc. Natl Acad. Sci. U.S.A. 2009, 106 (30), 12283–12288. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (13).Tang Y; Zaitseva F; Lamb RA; Pinto LH The M2 Ion channel protein of influenza virus is gated by a single tryptophan residue. J. Biol Chem. 2002, 277, 39880–39886. [DOI] [PubMed] [Google Scholar]
- (14).Venkataraman P; Lamb RA; Pinto LH Chemical Rescue of Histidine Selectivity Filter Mutants of the M2 Ion Channel of Influenza A Virus. J. Biol Chem. 2005, 280, 21463–21472. [DOI] [PubMed] [Google Scholar]
- (15).Hu J; Fu R; Nishimura K; Zhang L; Zhou HX; Busath DD; Vijayvergiya V; Cross TA Histidines, heart of the hydrogen ion channel from influenza A virus: toward an understanding of conductance and proton selectivity. Proc. Natl. Acad. Sci. U.S.A. 2006, 103 (18), 6865–70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (16).Bright RA; Shay DK; Shu B; Cox NJ; Klimov AI Adamantane resistance among influenza A viruses isolated early during the 2005–2006 influenza season in the United States. JAMA 2006, 295 (8), 891–4. [DOI] [PubMed] [Google Scholar]
- (17).Krumbholz A; Schmidtke M; Bergmann S; Motzke S; Bauer K; Stech J; Durrwald R; Wutzler P; Zell R High prevalence of amantadine resistance among circulating European porcine influenza A viruses. J. Gen. Virol 2009, 90, 900–908. [DOI] [PubMed] [Google Scholar]
- (18).Simonsen L; Viboud C; Grenfell BT; Dushoff J; Jennings L; Smit M; Macken C; Hata M; Gog J; Miller MA; Holmes EC The genesis and spread of reassortment human influenza A/H3N2 viruses conferring adamantane resistance. Mol. Biol. Evol 2007, 24 (8), 1811–20. [DOI] [PubMed] [Google Scholar]
- (19).Hu J; Asbury T; Achuthan S; Li C; Bertram R; Quine JR; Fu R; Cross TA Backbone structure of the amantadine-blocked trans-membrane domain M2 proton channel from Influenza A virus. Biophys. J. 2007, 92 (12), 4335–43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (20).Kolocouris A; Tzitzoglaki C; Johnson FB; Zell R; Wright AK; Cross TA; Tietjen I; Fedida D; Busath DD Aminoadamantanes with persistent in vitro efficacy against H1N1 (2009) influenza A. J. Med. Chem. 2014, 57 (ll), 4629–4639. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (21).Kolocouris A; Spearpoint P; Martin SR; Hay AJ; López-Querol M; Sureda FX; Padalko E; Neyts J; De Clercq E Comparisons of the influenza virus A M2 channel binding affinities, anti-influenza virus potencies and NMDA antagonistic activities of 2-alkyl-2-aminoadamantanes and analogues. Bioorg. Med. Chem. Lett. 2008, 18 (23), 6156–6160. [DOI] [PubMed] [Google Scholar]
- (22).McGuire KL; Smit P; Ess DH; Hill JT; Harrison RG; Busath DD Mechanism and Kinetics of Copper Complexes Binding to the Influenza A M2 S31N and S31N/G34E Channels. Biophys. J. 2021, 120 (1), 168–177. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (23).Watkins LC; DeGrado WF; Voth GA Influenza A M2 Inhibitor Binding Understood through Mechanisms of Excess Proton Stabilization and Channel Dynamics. J. Am. Chem. Soc. 2020, 142 (41), 17425–17433. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (24).Chipot C; Dehez F; Schnell JR; Zitzmann N; Pebay-Peyroula E; Catoire LJ; Miroux B; Kunji ERS; Veglia G; Cross TA; Schanda P Perturbations of native membrane protein structure in alkyl phosphocholine detergents: a critical assessment of NMR and biophysical studies. Chem. Rev. 2018, 118 (7), 3559–3607. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (25).Lee A Lipid–protein interactions in biological membranes: a structural perspective. Biochim. Biophys. Acta Biomemhr. 2003, 1612 (1), 1–40. [DOI] [PubMed] [Google Scholar]
- (26).Lee AG Biological membranes: the importance of molecular detail. Trends Biochem. Sci. 2011, 36 (9), 493–500. [DOI] [PubMed] [Google Scholar]
- (27).Yao Y; Dutta SK; Park SH; Rai R; Fujimoto LM; Bobkov AA; Opella SJ; Marassi FM High resolution solid-state NMR spectroscopy of the Yersinia pestis outer membrane protein Ail in lipid membranes. J. Biomol. NMR 2017, 67 (3), 179–190. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (28).Zhou H-X; Cross TA Influences of membrane mimetic environments on membrane protein structures. Anna. Rev. Biophys. 2013, 42, 361–392. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (29).Cross TA; Murray DT; Watts A Helical membrane protein conformations and their environment. Eur. Biophys. J. 2013, 42 (10), 731–755. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (30).Murray DT; Li C; Gao FP; Qin H; Cross TA Membrane protein structural validation by oriented sample solid-state NMR: diacylglycerol kinase. Biophys. J. 2014, 106 (8), 1559–1569. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (31).Hagn F; Etzkorn M; Raschle T; Wagner G Optimized phospholipid bilayer nanodiscs facilitate high-resolution structure determination of membrane proteins. J. Am. Chem. Soc. 2013, 135 (5), 1919–1925. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (32).Holsinger LJ; Alams R Influenza virus M2 integral membrane protein is a homotetramer stabilized by formation of disulfide bonds. Virology 1991, 183 (l), 32–43. [DOI] [PubMed] [Google Scholar]
- (33).Cross TA; Sharma M; Yi M; Zhou HX Influence of solubilizing environments on membrane protein structures. Trends Biochem. Sci. 2011, 36, 117–125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (34).McDermott A Structure and dynamics of membrane proteins by magic angle spinning solid-state NMR. Annu. Rev. Biophys. 2009, 38, 385–403. [DOI] [PubMed] [Google Scholar]
- (35).Linser R; Dasari M; Hiller M; Higman V; Fink U; Lopez del Amo JM; Markovic S; Handel L; Kessler B; Schmieder P; et al. Proton-detected solid-state NMR spectroscopy of fibrillar and membrane proteins. Angew. Chem., Int. Ed. 2011, 50 (19), 4508–4512. [DOI] [PubMed] [Google Scholar]
- (36).Barbet Massin E; Pell AJ; Retel JS; Andreas LB; Jaudzems K; Franks WT; Nieuwkoop AJ; Hiller M; Higman V; Guerry P; et al. Rapid proton-detected NMR assignment for proteins with fast magic angle spinning. J. Am. Chem. Soc. 2014, 236 (35), 12489–12497. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (37).Xue K; Movellan KT; Zhang XC; Najbauer EE; Forster MC; Becker S; Andreas LB Towards a native environment: structure and function of native membranes in lipid bilayers by NMR. Chem. Sci. 2021, 12, 14332–14342. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (38).Das N; Murray DT; Cross TA Lipid bilayer preparations of membrane proteins for oriented and magic-angle spinning solid-state NMR samples. Nat. Protoc. 2013, 8 (11), 2256–2270. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (39).Murray DT; Das N; Cross TA Solid-state NMR strategy for characterizing native membrane protein structures. Acc. Chem. Res. 2013, 46 (9), 2172–2181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (40).Andreas LB; Barnes AB; Corzilius B; Chou JJ; Miller EA; Caporini M; Rosay M; Griffin RG Dynamic nuclear polarization study of inhibitor binding to the M2(18–60) proton transporter from influenza A. Biochem. 2013, 52 (16), 2774–82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (41).Wright AK; Batsomboon P; Dai J; Hung I; Zhou H-X; Dudley GB; Cross TA Differential binding of rimantadine enantiomers to influenza A M2 proton channel. J. Am. Chem. Soc. 2016, 138 (5), 1506–1509. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (42).Andreas LB; Eddy MT; Pielak RM; Chou J; Griffin RG Magic angle spinning NMR investigation of influenza A M2(18–60): support for an allosteric mechanism of inhibition. J. Am. Chem. Soc. 2010, 132 (32), 10958–60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (43).Stouffer AL; Acharya R; Salom D; Levine AS; Di Costanzo L; Soto CS; Tereshko V; Nanda V; Stayrook S; DeGrado WF Structural basis for the function and inhibition of an influenza virus proton channel. Nature 2008, 451 (7178), 596–599. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (44).Can TV; Sharma M; Hung I; Gor’kov PL; Brey WW; Cross TA Magic Angle Spinning and Oriented Sample Solid-State NMR Structural Restraints Combine for Influenza A M2 Protein Functional Insights. J. Am. Chem. Soc. 2012, 134 (22), 9022–9025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (45).Wang J; Denny J; Tian C; Kim S; Mo Y; Kovacs F; Song Z; Nishimura K; Gan Z; Fu R; Quine JR; Cross TA Imaging membrane protein helical wheels. J. Magn. Reson. 2000, 144 (l), 162–7. [DOI] [PubMed] [Google Scholar]
- (46).Marassi FM; Opella SJ A solid-state NMR index of helical membrane protein structure and topology. J. Magn. Reson. 2000, 144 (1), 150–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (47).Porcelli F; Buck B; Lee DK; Hallock KJ; Ramamoorthy A; Veglia G Structure and orientation of pardaxin determined by NMR experiments in model membranes. J. Biol Chem. 2004, 279 (44), 4S815–23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (48).Ramamoorthy A; Wei Y; Lee DK PISEMA Solid-State NMR Spectroscopy. Annu. Rep. NMR Spectrosc. 2004, 52, 1–52. [Google Scholar]
- (49).Wu CH; Ramamoorthy A; Opella SJ High Resolution Heteronuclear Dipolar Solid-State NMR Specroscopy. J. Magn. Reson. A 1994, 109, 270–272. [Google Scholar]
- (50).Gopinath T; Veglia G Sensitivity Enhancement in Static Solid-State NMR Experiments via Single- and Multiple-Quantum Dipolar Coherences. J. Am. Chem. Soc. 2009, 131, 5754–5756. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (51).Nevzorov AA; Opella SJ Selective averaging for high-resolution solid-state NMR spectroscopy of aligned samples. J. Magn. Reson. 2007, 185 (l), 59–70. [DOI] [PubMed] [Google Scholar]
- (52).Fung BM; Khitrin AK; Ermolaev K An Improved Broadband Decoupling Sequence for Liquid Crystals and Solids. J. Magn. Reson. 2000, 142 (l), 97–101. [DOI] [PubMed] [Google Scholar]
- (53).Delaglio F; Grzesiek S; Vuister GW; Zhu G; Pfeifer J; Bax A NMRPipe: a multidimensional spectral processing system based on UNIX pipes. J. Biomol. NMR 1995, 6 (3), 277–293. [DOI] [PubMed] [Google Scholar]
- (54).Fu R; Miao Y; Qin H; Cross TA Observation of the Imidazole-Imidazolium Hydrogen Bonds Responsible for Selective Proton Conductance in the Influenza A M2 Channel. J. Am. Chem. Soc. 2020, 142 (5), 2115–2119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (55).Movellan KT; Wegstroth M; Overkamp K; Leonov A; Becker S; Andreas LB Imidazole-Imidazole Hydrogen Bonding in the pH-Sensing Histidine Side Chains of Influenza A M2. J. Am. Chem. Soc. 2020, 142 (6), 2704–2708. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (56).Sehnal D; Svobodova Varekova R; Berka K; Pravda L; Navratilova V; Banas P; Ionescu C-M; Otyepka M; Koca J MOLE 2.0: advanced approach for analysis of biomacromolecular channels. J. Cheminform. 2013, 5 (1), 39. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (57).Jo S; Kim T; Iyer VG; Im W CHARMM-GUI: a web-based graphical user interface for CHARMM. J. Comput. Chem. 2008, 29 (11), 1859–1865. [DOI] [PubMed] [Google Scholar]
- (58).Kim S; Cross TA Uniformity, ideality, and hydrogen bonds in transmembrane α-helices. Biophys. J. 2002, 83 (4), 2084–2095. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (59).Page RC; Kim S; Cross TA Transmembrane helix uniformity examined by spectral mapping of torsion angles. Structure 2008, 16 (5), 787–797. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (60).Shen Y; Bax A Protein backbone and sidechain torsion angles predicted from NMR chemical shifts using artificial neural networks. J. Biomol. NMR. 2013, 56 (3), 227–41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (61).Shen Y; Delagho F; Cornilescu G; Bax A TALOS+: a hybrid method for predicting protein backbone torsion angles from NMR chemical shifts. J. Biomol. NMR. 2009, 44 (4), 213–23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (62).Morrison EA; DeKoster GT; Dutta S; Vafabakhsh R; Clarkson MW; Bahl A; Kem D; Ha T; Henzler-Wildman KA Antiparallel EmrE exports drugs by exchanging between asymmetric structures. Nature 2012, 481 (7379), 45–50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (63).Lipfert J; Columbus L; Chu VB; Lesley SA; Doniach S Size and shape of detergent micelles determined by small-angle X-ray scattering. J. Phys. Chem. B 2007, 111 (43), 12427–12438. [DOI] [PubMed] [Google Scholar]
- (64).Miao Y; Fu R; Zhou HX; Cross TA Dynamic Short Hydrogen Bonds in Histidine Tetrad of Full-Length M2 Proton Channel Reveal Tetrameric Structural Heterogeneity and Functional Mechanism. Structure 2015, 23 (12), 2300–2308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (65).Zheng H; Pearsall EA; Hurst DP; Zhang Y; Chu J; Zhou Y; Reggio PH; Loh HH; Law P-Y Palmitoylation and membrane cholesterol stabilize μ-opioid receptor homodimerization and G protein coupling. BMC Mol. Cell Biol. 2012, 13 (l), 6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (66).Rossman JS; Jing X; Leser GP; Balannik V; Pinto LH; Lamb RA Influenza Virus M2 Ion Channel Protein Is Necessary for Filamentous Virion Formation. J. Virol 2010, 84 (10), 5078–S088. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (67).Rossman JS; Jing X; Leser GP; Lamb RA Influenza virus M2 protein mediates ESCRT-independent membrane scission. Cell 2010, 142 (6), 902–913. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (68).Rossman JS; Lamb RA Influenza virus assembly and budding. Virology 2011, 411 (2), 229–236. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.