

Abstract
While aminoadamantanes are well-established inhibitors of the influenza A M2 proton channel, the mechanisms by which they are rendered ineffective against M2S31N are unclear. Solid state NMR, isothermal titration calorimetry, electrophysiology, antiviral assays, and molecular dynamics simulations suggest stronger binding interactions for amino adamantanes to M2WT compared to negligible or weak binding to M2S31N. This is due to reshaping of the M2 pore when N31 is present, which, in contrast to wild-type (WT), leads (A) to the loss of the V27 pocket for the adamantyl cage and to a predominant orientation of the ligand’s ammonium group toward the N-terminus and (B) to the lack of a helical kink upon ligand binding. The kink, which reduces the tilt of the C-terminal helical domain relative to the bilayer normal, includes the W41 primary gate for proton conductance and may prevent the gate from opening, representing an alternative view for how these drugs prevent proton conductance.
Graphical Abstract

1. INTRODUCTION
A proven vulnerability of influenza A infections is the blockage of the viral M2 proton channel. M2 is required for acidification of the virion interior during infection and neutralization of the trans-Golgi network during viral egress.1 Amantadine (Amt, 1; Scheme 1) is an established inhibitor of the influenza A/M2-mediated proton currents2 and a licensed influenza A infection therapy.3 The primary binding site of 1 is located within the pore of the tetrameric M2 transmembrane (M2TM) domain that forms the transmembrane proton transit path.4,5 However, since 2005,6 the 1-insensitive Ser-to-Asn mutation at position 31 in M2 (S31N) has become globally prevalent,7 abrogating the clinical usefulness of 18 and possibly other previously reported M2 inhibitors.9 Thus, new agents are needed to combat drug-resistant forms of influenza.
Scheme 1. Structures of Aminoadamantane Derivatives 1–10.
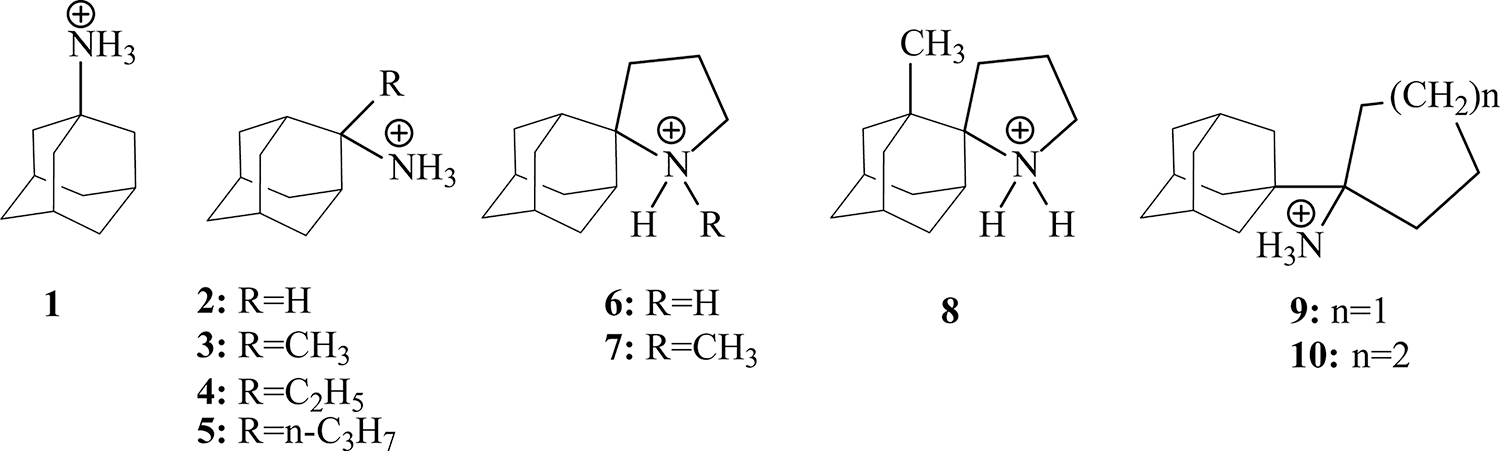
Based on the experimental solid state NMR (ssNMR) structure of the complex M2TM–1,4a,5a,b the replacement of S31 residues at the drug binding site with the more polar and bulky Asn side chains may induce the adamantyl ring to move deeper into the M2 pore, toward the C-terminus close to G34, where the pore of the helical bundle has its largest diameter, and drugs larger than 1 might be effective. We and others have searched for such larger drugs and found, for example, compounds 5–7, which are active against A/California/07/2009 (H1N1) virus encoding S31N but not against WSN/33 also bearing the same mutation.10 However, while a preliminary ssNMR experiment of the S31N M2TM domain complexed with compound 5 showed evidence of drug binding, electrophysiology (EP) studies showed no blockage for the full-length S31N M2 protein.10a These studies therefore indicate that more detailed experimental investigation and modeling are needed to explain the mechanisms by which aminoadamantanes are rendered ineffective against M2S31N in order to design effective drugs.
Recent molecular dynamics (MD) simulations showed that the ammonium group of 1 has a prevalent orientation toward the C-terminus of M2TMWT (Udorn), with the adamantane hydrocarbon cage shifted slightly toward the N-terminus of the A30/S31 Cα sites,11–18 producing a region of the pore devoid of water17–19 consistent with blockage of proton transport and in agreement with ssNMR findings.5c,20,21 In contrast, MD simulations of 1 in M2TMS31N from Gleed et al.17b and rimantadine in M2TMS31N from Alhadeff et al.16 suggested that these compounds have a variable orientation, but with a propensity to have the ammonium group toward the N-terminus17b,22 of the M2TMS31N pore (Figure 1). In this orientation water occupancy is not widely occluded in any configuration.17 These studies16,17b presented a description of the potential of mean force (PMF) curves of amantadine and rimantadine in interaction with the M2TMS31N pore, but the main structural/interaction changes due to S31N and leading to aminoadamantane resistance were not defined. In addition, no experimental evidence is presented to date for the binding of aminoadamantane ligands in M2TMS31N pore. Nevertheless, the propensity of 1’s amino group to orient toward the N-terminus was used for the successful design of 1–polar head conjugates that were shown to be potent inhibitors of WSN/33 virus and proton conductance by M2S31N.23 A ssNMR analysis of a system including one of these compounds, a phenylisoxazole derivative linked with amantadine through a methylene bridge, with M2TM in membrane bilayers was realized. The results showed that the compound’s heterocyclic ring system may be trapped by the V27 side chains at the N-terminus of the M2TM pore24 with the isoxazole group forming hydrogen bonds with the N31 amide side chains.
Figure 1.

Amt (1) orients toward the protein C-terminus in M2TMWT and toward the protein N-terminus in M2TMS31N. Superposition of final snapshots and constant temperature and pressure molecular dynamics simulations at 310 K of 1 in complex with S31 or N31 M2 [PDB ID, 2KQT] in 150 mM NaCl, water, and DMPC lipid. Compound 1 and waters are shown in yellow-green and purple in WT and S31N, respectively. Two of four M2TM backbones are shown as green ribbons. In the S31 case, the ammonium group of 1 is projecting toward the C-terminus and hydrogen bonding with four water molecules. In the N31 case, the adamantane cage (purple) is lower, and the amantadine amine projects toward the N-terminus and hydrogen bonds with the one N31 side chain and a water molecule.
We are interested to investigate how subtle changes in amantadine structure are related with the binding affinity against M2TM variants. We previously used the molecular mechanics Poisson–Boltzmann surface area (MM/PBSA) to interpret thermodynamic profiles measured using isothermal titration calorimetry (ITC) for aminoadamantanes binding to the avian M2TMWeybridge (M2TMWeybridge has two different amino acids that do not line into the pore, i.e., V28 → I28 and L38 → F38 compared to M2TMWT) in order to successfully prioritize aminoadamantane derivatives.25 We also applied rigorous free energy binding calculations by the Bennett acceptance ratio (BAR) approach to accurately predict relative binding affinities of aminoadamantane derivatives toward M2TMWT or M2TMWeybridge measured by ITC or other methods, respectively.26,27
Here we investigate the interaction of aminoadamantane derivatives (Scheme 1) with M2TMWT and M2TMS31N both through experiment and by MD simulations, with the aim of determining how the S31N mutation leads to the inability of 1 and other aminoadamantanes to block S31N M2 proton transport. First, we measured binding constants of selected aminoadamantane derivatives with ITC against M2TMWT and M2TMS31N in dodecylphosphocholine (DPC) micelles at alkaline pH. Second, we applied a series of MD simulations (80 ns trajectories) of the M2TMWT and M2TMS31N complexes with 1–10 (Scheme 1) to analyze in some detail how the polar N31 amide side chains affect binding of an aminoadamantane ligand inside the M2TM pore. To this end, we suggest that repulsive forces between N31 and the adamantane ring may influence the orientation and binding interactions of the ligand, the overall stability of the complexes, and the shape of M2TM. Third, we performed oriented sample (OS) and magic angle spinning (MAS) ssNMR experiments28–30 on representative M2TM–ligand complexes at alkaline pH using both M2TMWT and M2TMS31N sequences to characterize the structural influence of compounds on the channel. Fourth, we selected and tested new synthetic derivatives with slightly larger or similarly sized adducts, such as 8 and 10, than previously synthesized compounds 5–7 and 9 for their abilities to bind M2TMS31N. We realized EP experiments for selected compounds against both WT and S31N M2 proteins to test M2 channel blockage and additionally measured the antiviral potency of the compounds against the naturally amantadine-resistant H1N1 influenza strain A/WSN/1933 (WSN/33 with M2N31) and its reverse genetics-generated amantadine-sensitive variant with the amino acid substitution N31S in M2 (WSN/33-M2-N31S) using cytopathic effect inhibition (CPE) assays.
2. RESULTS
2.1. Binding Affinities of Aminoadamantanes to M2TM by ITC.
ITC measurements31 were determined for both M2TMWT and M2TMS31N tetramers at pH 8 corresponding to the closed state of the M2TM pore32 (Table 1). Binding data for compounds 1, 5, and 6–8 to M2TMWT measured in our previous work26 were included in Table 1. Thus, as has been published recently26 (see Table 1 and Supporting Information Table S1) compounds 1–8 bind M2TMWT. Compound 9 and the newly synthesized analogue 10, which has a cyclohexane ring instead of the cyclopentane in 9, bind M2TMWT with at least an order of magnitude lower dissociation constant (Kd) than those for 1–8, possibly because their sizes do not enable them to fit well inside M2TMWT. We note that techniques for measuring affinity against M2TM such as ITC correspond to models of molecular recognition for ligand binding to M2TM and do not necessarily reflect functional inhibition of M2 proton currents or in vitro inhibition of influenza A virus. For example, 9 inhibits M2WT-dependent currents as measured by electrophysiology with an IC50 of 10 ± 2 μM (Table 4) and inhibited influenza A virus replication in infected cells with an IC50 of 3.92 ± 2.22 μM (Table 5) while compound 10 was inactive. Nevertheless, ITC and related techniques such as surface plasmon resonance (SPR) and structural techniques such as ssNMR can provide useful insights for M2TM binding and trends in structure–activity relationships for the M2−aminoadamantane system. For example, compounds 1–4 did not bind efficiently to M2TMS31N according to ITC and previous SPR measurements for 133 while 5 and 6 with larger adducts connected to adamantane bind weakly to M2TMS31N compared to M2TMWT according to ITC (Table 1) and ssNMR. The Kd values of low-affinity binders (e.g., 5, 6, 9, and 10) against M2TMS31N, and 9 against M2TMWT, possess relatively large errors due to the limitations of the ITC method (as explained in the Supporting Information). Subsequently comparison of the relative Kd values of compounds 5, 6, 9, and 10 do not reflect relative binding affinity strength against M2TMS31N. Nevertheless, although the quantitation of the ITC results against M2TMS31N is limited for this method, the measurements suggest that the Kd values of 5 and 6 against M2TMS31N are much smaller compared to those of M2TMWT, suggesting weaker binding to M2TMS31N (Table 1). Narrower line widths and larger chemical shifts changes for 5 bound to M2TMWT compared to M2TMS31N were observed in the ssNMR spectra described below suggesting a reduction in dynamics for binding to M2TMWT and a more specific binding site, which in turn causes a significant reduction in hydration due to drug-induced desolvation of the binding pocket.31
Table 1.
Binding Constants and Other Thermodynamic Parameters Derived from ITC Measurements for Influenza A M2TMWT and M2TMS31N
| M2TMWT | ||||
| liganda | K d b | ΔGc,d | ΔHc,e | TΔSc,f |
| 1g | 2.17 ± 0.52 | −32.51 ± 0.59 | −27.87 ± 2.09 | 4.64 ± 2.18 |
| 2g | 1.60 ± 0.34 | −33.30 ± 0.54 | −29.41 ± 1.76 | 3.89 ± 1.84 |
| 3g | 0.89 ± 0.19 | −34.77 ± 0.54 | −28.41 ± 1.09 | 6.32 ± 1.21 |
| 4g | 0.62 ± 0.14 | −35.65 ± 0.54 | −29.87 ± 0.88 | 5.77 ± 1.05 |
| 5g | 0.63 ± 0.17 | −36.43 ± 0.69 | −32.62 ± 1.29 | 3.90 ± 1.45 |
| 6g | 0.36 ± 0.22 | −38.11 ± 1.84 | −21.49 ± 1.76 | 16.61 ± 2.57 |
| 7g | 0.93 ± 0.36 | −34.64 ± 0.96 | −15.98 ± 1.17 | 18.66 ± 1.51 |
| 8g | 1.30 ± 0.43 | −33.81 ± 0.84 | −21.25 ± 1.30 | 12.55 ± 1.55 |
| 9 | 14.61 ± 4.62 | −27.77 ± 0.79 | h | h |
| 10 | >10 | h | h | h |
| M2TMS31N | ||||
| liganda | K d b | ΔGc,d | ΔH | TΔS |
| 1–4 | I | |||
| 5 | >10 | h | h | h |
| 6 | 17.5 ± 8.5 | −27.95 ± 1.24 | h | h |
| 9 | 15.8 ± 5.95 | −28.22 ± 0.96 | h | h |
| 10 | 9.90 ± 4.99 | −29.41 ± 1.29 | h | h |
See Scheme 1.
Binding constant Kd in μM calculated from measured Ka in M−1 by Kd = (1/Ka) × 10−6 and error in Kd in μM determined by Kd,error = (Ka,error/Ka2) × 10−6.
In kJ mol−1.
Free energy of binding computed from Kd by ΔG = −RT ln(Kdref/Kd) with Kdref = 1 M and T = 300 K, and error in ΔG determined according to with T = 300 K.
Enthalpy of binding and error in the enthalpy of binding calculated from measured binding enthalpy and measured error by ΔH = ΔHmeasured(T/Tmeasured) with T = 300 K and the temperature at which the ITC measurements were performed Tmeasured = 293.15 K.
Entropy of binding calculated by ΔS = (−ΔG + ΔH)/T and error in ΔS computed by the equation
Measured in ref 24.
Values could not be determined reliably due to the limitations of the methods in the area of very weak binding.
No detectable binding.
Table 4.
Block of Full-Length M2-Dependent Currents by Select Compounds in Transfected HEK Cellsa
| A/California/07/2009 (H1N1) M2: N31 |
A/California/09/2009 (H1N1) M2: S31 |
|||
|---|---|---|---|---|
| compd | % block after 3 min | % block after 30 min | % block after 3 min | IC50 |
| 1b | 14 ± 2 (100 μM; 26) | (N/A) | 75 ± 9 (10 μM; 4) | 2 ± 3 μM (3) |
| 2b | 13 ± 3 (100 μM; 2) | 16 (100 μM; 1) | 95 ± 8 (10 μM; 2) | 2 ± 1 μM (2) |
| 5b | 0 ± 5 (100 μM; 2) | 9 ± 10 (100 μM, 3) | 63 ± 5 (10 μM; 2) | 7 ± 2 μM (2) |
| 6 | 4 ± 3 (100 μM; 2) | 5 ± 4 (100 μM, 2) | 66 ± 6 (10 μM; 3) | 5 ± 2 μM (3) |
| 9 | 4 ± 4 (100 μM; 2) | 4 ± 10 (100 μM; 4) | 46 ± 2 (10 μM; 2) | 10 ± 2 μM (2) |
For each compound, the percent block of pH-dependent M2 current at listed concentration (±sem) and/or IC50 (μM) is shown. Parentheses show number of replicates.
Data were also reported in ref 10a.
Table 5.
Antiviral Activity of Compounds 1–10 against Influenza Virus A/WSN/33 (H1N1) Variants in Madin–Darby Canine Kidney Cells
| IC50 (μM)a |
|||
|---|---|---|---|
| compd | A/WSN/33-M2-N31S | A/WSN/33-M2-N31 | CC50 (μM)b |
| 1 | 0.27 ± 0.16 | N.A.c | >100d |
| 2 | 0.42 ± 0.46 | N.A. | 70.50 ± 25.31 |
| 3 | 0.33 ± 0.10 | N.A. | 25.63 ± 7.65 |
| 4 | 0.34 ± 0.26 | N.A. | >100 |
| 5 | 0.34 ± 0.10 | N.A. | >100 |
| 6 | 0.34 ± 0.08 | N.A. | >100 |
| 7 | 0.90 ± 0.29 | N.A. | >100 |
| 8 | 6.12 ± 2.59 | N.A. | >100 |
| 9 | 4.34 ± 2.94 | N.A. | >100 |
| 10 | N.A. | N.A. | >100 |
| Oseltamivir | 0.02 ± 0.01 | 0.02 ± 0.01 | not determined |
Mean and standard deviations of the 50% inhibitory concentration (IC50) of at least three independent measures.
Mean and standard deviations of the 50% cytotoxic concentration (CC50) of at least three independent measures.
N.A., not active. Maximum concentration tested, 100 μM.
Exact value published in ref 46.
2.2. Solid State NMR of M2TM–Aminoadamantanes Complexes.
2.2.1. OS ssNMR Spectra.
Drug binding to the channel pore of M2TMWT and M2TMS31N was evaluated using OS ssNMR experiments. Spectral correlation observed in these data sets, between anisotropic 15N chemical shifts and 1H–15N dipolar coupling values for selectively 15N-labeled backbone amide sites, gives rise to resonance patterns known as PISA (polarity index slant angle) wheels.34,35 The shape, size, and position of the PISA wheel for α-helical membrane proteins in uniformly oriented lipid bilayer preparations is determined by the helical tilt relative to the bilayer normal and is sensitive to drug-induced structural perturbations. Previous investigations by Cross and co-workers have reported dramatic changes in anisotropic chemical shifts and dipolar coupling values for M2TMWT in the presence of 1.36 PISA wheel analysis of M2TMWT spectra in the apo state and in the presence of 136 or 5 correlates the shifts with a substantial reduction in the tilt angle for the C-terminal region of the M2TMWT helix, following ligand binding. In contrast, M2TMS31N in the presence of 5 results in a uniform 5 ± 2° decrease in the helical tilt imparting a change to the entire helix–helix interface.10a
Here, to monitor changes in the resonance frequencies and to the resulting PISA wheel in the presence of other variants of 1, a 15N–V28,A30,I42 (VAI-M2TM) labeling scheme was used. Residues 28 and 30 are in the N-terminal domain of the M2TM helix and I42 in the C-terminal region, and hence the labels effectively sample both segments. Resonance frequencies for these sites are well-resolved allowing for a PISA wheel analysis of drug binding in the channel pore that induces small structural perturbations. The signals of these three pertinent and isotopically labeled backbone amides were shifted in nearly an identical fashion when 1 or 5 bind to the M2TMWT (Figure 2). As before, changes suggest only a slight perturbation to the N-terminus and a much more significant structural perturbation to the C-terminus with a change in the tilt angle from 32° to just 22°, while the N-terminal tilt changes by only a degree or two (Table 2).4b,36 These results imply similar drug-induced structural perturbations to the TM configuration of WT M2 protein in the presence of 1 or 5.
Figure 2.

Superimposed PISEMA spectra of the M2TMWT (residues 22–46), 15N-labeled at V28, A30, and I42, in DMPC lipid bilayers uniformly aligned on glass slides with (red) and without (black) compound 1 (A) and compound 5 (B). Assignments without drug were made based on the known structure and spectra of M2TMWT.36 Assignments with drug follow the rotational orientation of the helices. Changes in the resonance frequencies of the 15N-labeled backbone amide sites are indicated with black arrows. All spectra were collected at 720 MHz, pH 7.5, and 303 K. The molar ratio of lipid/protein was 30:1. PISA wheels are drawn for helix tilts of 22° and 32° for the N-and C-terminal residues, respectively, when the drugs are bound.
Table 2.
Mean Helical Tilt Values Relative to the Bilayer Normal from MD Simulations of 1 and 5 in Complex with M2TMWT in DMPC Bilayer
For the binding studies to M2TMS31N, no observable effect was detected for binding of 1, while 5 induces smaller changes compared to those induced by the binding of 1 or 5 to M2TMWT based on the signals from the same three backbone amides (Figure 3). In addition, the linewidths are narrower when 5 is bound to M2TMWT compared to M2TMS31N, suggesting less dynamics in the WT complex implying a tighter complex.
Figure 3.
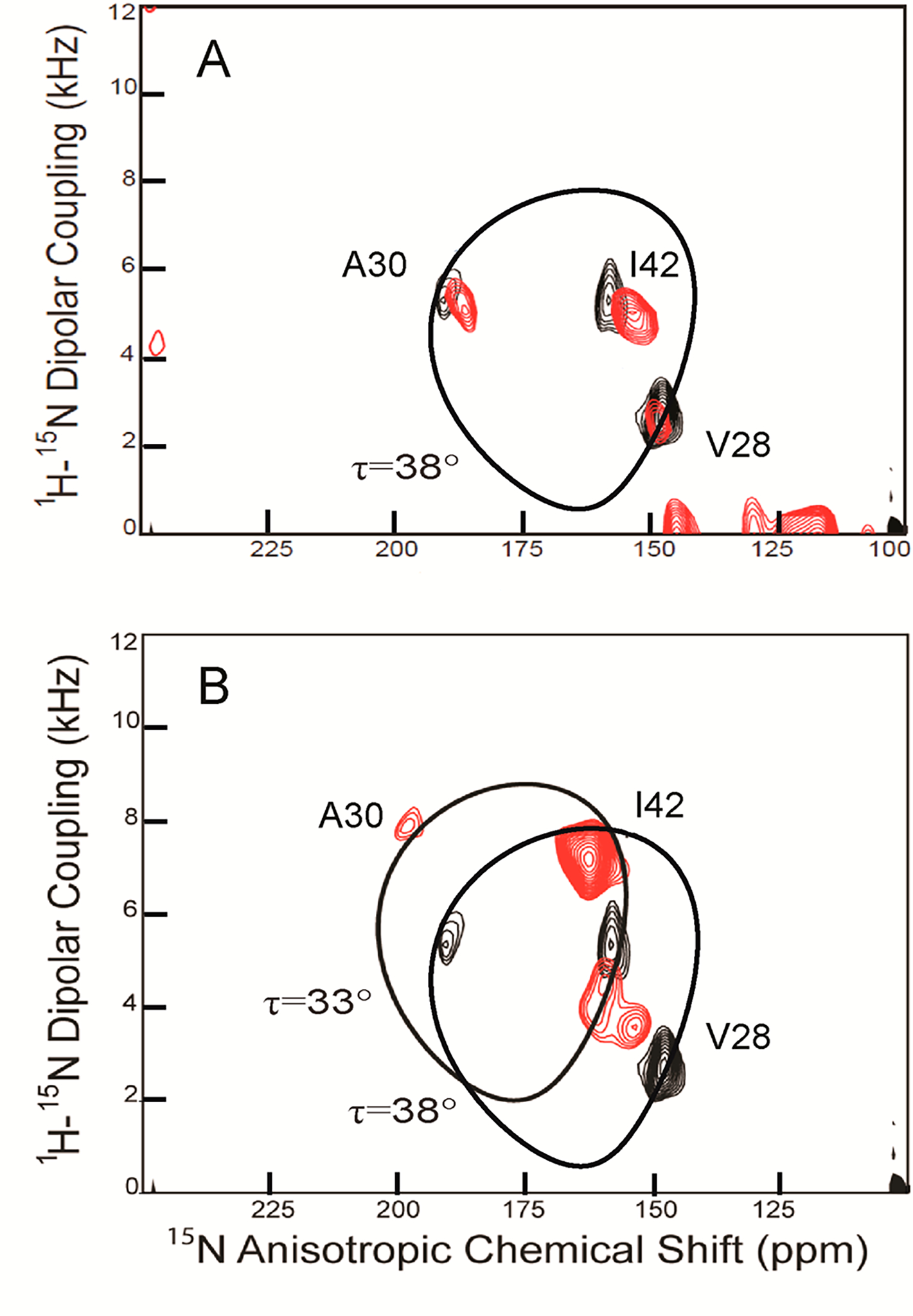
Superimposed PISEMA spectra of the M2TMS31N, 15N-labeled at residues V28, A30, and I42, in DMPC lipid bilayers uniformly aligned on glass slides with (red) and without (black) compound 1 (A) and compound 5 (B). Assignments without drug were made based on the known structure and spectra of M2TMWT. 36Assignments with drug follow the rotational orientation of the helices. Theoretical PISA wheel calculated for an ideal helix [(ϕ, φ)] = (−60°, −45°)] with a 33° tilt angle relative to the bilayer normal is superimposed on M2TMS31N resonance frequencies, which are shifted after compound 5 addition. All spectra were collected at 720 MHz, pH 7.5, and 303 K. The molar ratio of lipid/protein was 30:1.
There is a small difference in the structure of M2TMS31N versus M2TMWT without drugs in lipid bilayers (black resonances in Figure 2 versus Figure 3). The M2TMS31N data suggest a helical tilt of approximately 38°, somewhat greater than the helical tilt observed in the WT structure (32°) consistent with prior characterizations.4,36 While the binding of 1 to M2TMWT produced a 10° kink near G34 in each helix of the tetramer,4b,36 the binding of 1 produced no such structural change for the 1-resistant M2TMS31N. The shifts in the anisotropic resonance frequencies which resulted when 5 binds to M2TMS31N demonstrates a significant change in helical tilt from 38° to 33° in the tetrameric complex (Table 3). Based on these resonances that sample the N- and C-regions of the TM helix, the structural change appears to be a uniform change in tilt. Consequently, the residues facing the pore remain the same, but the interactions between helices that provide the tetrameric stability may change substantially. Upon binding of 1 or 5 to M2TMWT a kinked helix at G34 is produced but when adding these compounds to M2TMS31N helices, are not kinked at G34. Instead, when 5 binds to M2TMS31N, the entire helix–helix interface changes with the ~5° reduction in tilt for each of the four helices.
Table 3.
Mean Helical Tilt Values Relative to the Bilayer normal from MD Simulations of 5–8 in Complex with M2TMS31N in DMPC Bilayer
The structurally similar aminoadamantanes 6–8 produced similar ssNMR results when added to M2TMS31N (Figure 4) in that the change in structure always reflected a uniform change in helical tilt. Compound 8 is a new derivative, which was synthesized to test the effect of C-methylation to binding compared to N-methylation in 6. The measured helical tilt angles are 28° for 6, 31° for 7, and 33° for 8, with the latter compound yielding results that are similar to those induced by 5 (Table 3). For 5–8, there is no evidence for partial binding, since the unbound states are not observed in the spectra with drugs present. With 5, V28 appears to have multiple resonances (Figure 3) clustered on an anisotropic chemical shift of 160 ppm and a dipolar interaction of 4 kHz. The structural differences would be small, i.e., no more than 1° change in the orientation of the V27–V28 peptide plane with respect to the bilayer normal. This suggests that even with 5 on the time scale of 10–100 μs there is no significant evidence for asymmetry in this tetrameric structure; i.e., there is no evidence from these three labeled sites that M2TMS31N bound with different compounds 1 and 5–8 forms a dimer of dimer, as has been suggested for the S31N structure.37,38 If such structures are present, they must interconvert on a time scale faster than the difference in spectral frequencies in hertz. Furthermore, if this was occurring with significantly different structures, there would be a reduction in the width and height of the PISA wheel, which was not observed. The intensity of the resonances varies, again especially for 5, suggesting that the compound may have competing binding interactions with the pore leading to heterogeneity in the frequencies, the weakening of the A30 resonance, and the small dispersion of the V28 resonances as described above. These dynamics are in sharp contrast to the intense resonance for I42 for M2TMS31N bound with 5. The uniformity of the resonance intensities that result from the interaction with heterocyclic compounds 6–8 may suggest less dynamics and more potent binders than 5 to M2TMS31N.
Figure 4.

Superimposed PISEMA spectra of the M2TMS31N (residues 22–46), 15N-labeled at residues V28, A30, and I42, in DMPC bilayers uniformly aligned on glass slides with (red) and without (black) 6, 7, and 8 (panels A–C, respectively). Assignments without drug were made based on the known structure and spectra of M2TMWT. 36Assignments with drug follow the rotational orientation of the helices. Theoretical PISA wheels calculated for an ideal helix [(ϕ, φ)] = (−60°, −45°)] with varying tilt angles relative to the bilayer normal were superimposed on drug bound M2TMS31N spectra (panels A–C). All spectra were collected at 720 MHz, pH 7.5, and 303 K. The molar ratio of lipid/protein was 30:1.
2.2.2. MAS Spectra.
To evaluate further the effect of 6 binding to M2TMS31N, 2D N–Cα correlation MAS ssNMR experiments were performed using uniformly 13C,15N-labeled V7, A30, S31, and G34 M2TMS31N (13C,15N-VANG-labeled M2TMS31N). The labeling used aims to explore the binding interactions of amantadine analogues into M2TMS31N pore through measuring the effect of binding to the chemical shifts of V27, A30, G34, and N31. In M2TMWT the binding area includes the expanded area including V27, A30 and also G34, S31.4a,5b Spectra were obtained (Figure 5) for both the apo protein (blue) and for 6 bound (red) to the labeled M2TMS31N. Addition of the drug to the M2TMS31N sample resulted in chemical shift changes for N31 and G34 of 1.2 and 2.1 ppm, respectively; V27 and A30 resonances remain unchanged, suggesting that they are not involved in binding, while the isotropic chemical shift perturbation of the N31 and G34 residues suggest the drug binding site for M2TMS31N suggesting either direct hydrogen bonding with the backbone or indirect hydrogen bonding through water molecules.21 Significant 15N and/or 13Cα chemical shift changes at V27, S31, and G34 have been reported when rimantadine is bound to M2TMWT20,21 with S31 experiencing a dramatic 7 ppm shift relative to the apo state.21 Despite the observed chemical shift changes at residues N31 and G34 when 6 bound to the M2TMS31N, cross-peak intensity was not increased as was observed for the WT full-length M2 in complex with rimantadine.21 This suggests that the conformational heterogeneity of M2TMS31N was not increased for these residues in the presence of the drug. A reduction in dynamics which is in agreement with narrower line widths would be anticipated with a specific binding site or a significant reduction in hydration due to drug-induced desolvation of the binding pocket. The narrower line widths and increased anisotropic chemical shifts for the M2TMWT and M2TMS31N bound to 5 (Figures 2 and 3) illustrate reduced dynamics and weaker or less specific binding of the drug to the M2TMS31N.
Figure 5.
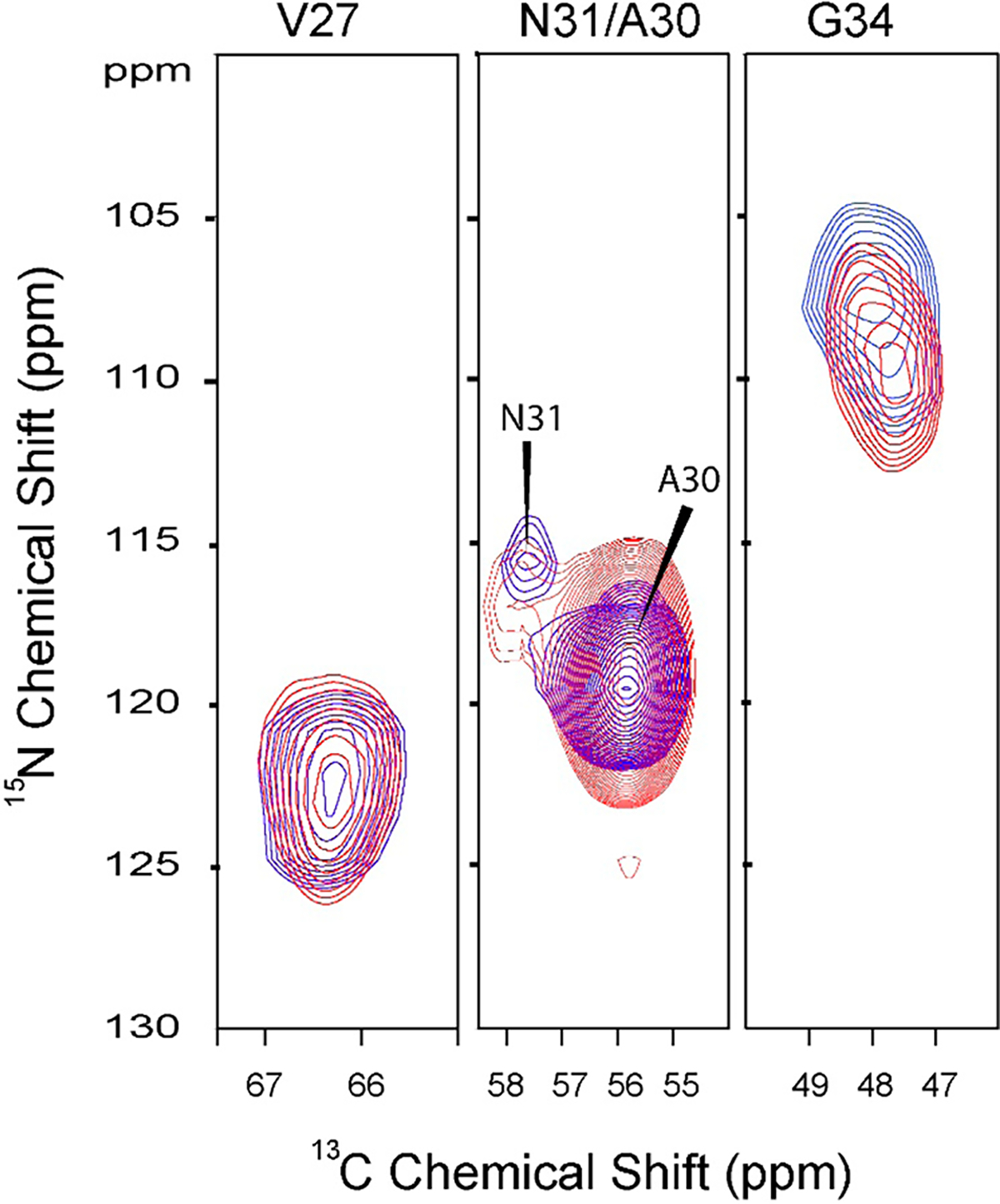
Superimposed 2D strip plots for 15N/13C (NCA) correlation spectra of 13C,15N–V27A30N31G34-labeled M2TMS31N (residues 22–46) in DMPC lipid bilayers with (red) and without (blue) compound 6. Spectra were collected at 600 MHz proton frequency, pH 7.5, 10 kHz spinning rate, and a calibrated temperature at sample of 263 K.
2.3. MD Simulations of M2TM–Aminoadamantanes Complexes.
2.3.1. Starting Structure of Protein−Ligand Complex.
MD simulations of the complexes between 1, 2-alkyl-2-aminoadamantanes 2–5, and cyclic derivatives 6–10 with both M2TMWT and M2TMS31N provide insights for the binding interactions and possible structural changes in the binding area. All of the ligands’ amino groups were considered to be protonated according to model calculations preformed previously for ligands 1 and 6.25,26 The structure of the complex M2TMWT-1 (PDB ID 2KQT4a,5b) was used as a starting structure for the simulations of M2TMWT–ligand complexes and of M2TMS31N–ligand complexes. The PDB ID 2KQT4a,5b structure was chosen since it was determined at pH 7.5 and DMPC planar bilayers and vesicles,4a,5b i.e., similar conditions to those used in our simulations. Subsequently only small structural changes are to be expected in the M2TM structure during MD simulations of its complexes with ligands, and the equilibration phase should be short. It has been shown that the stability of the binding region for 1 in the M2TM tetramer is increased considerably when using DMPC compared to other lipids.39–42 In addition, 2KQT (see ref 5b) should be considered as the best structure of the amantadine bound state of M2TM since it utilized both isotropic chemical shift restraints and all of the orientational restraints of an earlier structure characterized by Cross and co-workers (see ref 4a). The structures of the M2TMS31N–ligand (1–10) complexes were generated from M2TMWT complexes by mutating S31 to N (see also Experimental Methods).
2.3.2. Complexes of Ligands with M2TMWT.
The MD simulations of the M2TMWT–ligand complexes reached equilibration in less than 40 ns with the protein system having full flexibility. The RMSDs values (Table S2) were ≤1.6 Å for M2TM Cα-carbons with respect to the initial structure of the production period4a,5b suggesting that M2TMWT–ligand (1–8) complexes were very stable and the M2TM tetramer structure was considerably unchanged in the course of the simulation. The mean values of the N-terminal and the C-terminal helical tilt of M2TMWT for the complexes with 1 and 5 from the corresponding MD trajectories were measured to be 31°, 18° and 31°, 19°, respectively, and are in very good agreement with the values of 32°, 22° and 32°, 22° determined by OS ssNMR (Table 2). As noted above, the starting structure 2KQT4a,5b having helical domain tilt angles of 31°, 19° was determined in conditions similar to those used in our MD simulations by Cross and co-workers4a Even so these findings suggest that the ligand–M2TMWT complex was successfully equilibrated and the conditions were well-adjusted to produce consistent results. Consistent with the experimental findings5b,c and previous observations,25,26 the ammonium group of the aminoadamantane compounds was oriented toward the C-terminus with angles of 77° between the C–N bond of the ligand and the membrane’s normal (Table S2). The complexes of compounds 1–8 inside the M2TM lumen are stabilized through (a) formation of hydrogen bonds between their ammonium group and water molecules in the region between H37 residues and the ligand and (b) van der Waals interactions between the adamantyl group and a lipophilic pocket formed by V27 and A30 side chains (see Figure 6). This binding area is in accordance (a) with the 15N chemical shifts perturbations for V27, A30, and S31 in complexes between 1 or 3-azaspiro[5,5]-undecane and M2TMWT compared to the apo M2TMWT.39 and (b) with the measured distance of 4.5 Å between G34Ca and the methyl group of rimantadine from REDOR ssNMR experiments in studies of the full-length protein.21 In addition, for complexes of M2TMWT with 1–8, the distance between the adamantyl ring of the ligand and the center of mass of the four V27 (V27-Ad) varies between 4 and 4.9 Å and the distance between the adamantyl ring of the ligand and the center of mass of the four A30 (A30–Ad) varies between 0.5 and 1.4 Å (Table S2). The position of the adamantyl ring inside the lumen was similar for ligands 2–8 differing from 1 only by 0.2–0.6 Å. To account for the position of the ligands toward the C-end, we measured the distance between the center of mass of the four V27 and the ammonium nitrogen of the ligand (V27–N+) which varied between 7.1 and 7.8 Å (Table S2). According to the measures in Table S2 the hydrogen bonding ability is higher for the subset of ligands 1–5, 9, and 10 than for the subset 6, 8, or 7. Thus, 1–5, 9, and 10 can form on average three hydrogen bonds with neighboring water molecules; 6 and 8, two hydrogen bonds; and 7, one hydrogen bond, respectively (Table S2). This is what was expected since 1–5, 9, and 10 have a primary ammonium group, 6 and 8 have a secondary ammonium group, and 7 has a tertiary ammonium group. For ligands 3–8 which include a carbon substituent at the C-2 adamantane position the molecule is rotated in order to avoid repulsive van der Waals forces of the alkyl group with the symmetric M2TM pore. The average angle between the pore axis and the C–N bond vector was increased progressively according to the alkyl group size, i.e., from 13.5° for 1 to 25–35° for 2–8 (Table S2). Although all ligands 1–8 have a binding mode similar to that shown from the OS ssNMR spectra of complexes including 1 and 5 (Figure 2), a subtle compromise between hydrogen bonding and hydrophobic interactions with key pore residues, such as V27, A30, affects the ligand tilt inside the pore and its different binding strength as shown by the ITC results (Table 1, Table S1). The adamantyl ring is slightly toward the N-terminus compared to the A30/S31 Cα, producing a region without water molecules. Snapshots of the simulation complexes with 5 and 6 are shown in Figure 6.
Figure 6.

Snapshots from the simulation of ligands bound to M2TMWT. Waters within 10 Å from the ligand are shown. (A) Compound 5 bound to M2TMWT. Eight waters are shown between the ligand and H37 residues. Three hydrogen bonds between the secondary ammonium group of the ligand and three water molecules are shown (see Table S2). Hydrogen bonding together with van der Waals interactions of the adamantane core with V27 and A30 stabilize the ligand inside the pore with its ammonium group oriented toward the C-terminus. (B) Compound 6 bound to M2TMWT. Eight waters are shown between the ligand and H37 residues. Two hydrogen bonds between the secondary ammonium group of the ligand and two water molecules are shown (see also Table S2). Hydrogen bonding together with van der Waals interactions of the adamantane core with V27 and A30 stabilize the ligand inside the pore with its ammonium group oriented toward the C-terminus.
The MD simulations suggest that the M2TMWT–ligand complex stability is reduced when a sizable adduct is attached to adamantane; i.e., the M2TM pore does not efficiently accommodate the large adducts in compounds 9 and 10. This is in accordance with the weak or no binding respectively of these drugs according to the ITC results (Table 1). For example, the MD simulations showed that the stability of the complex is considerably reduced in the case of 10 where after 80 ns the ligand is shifted toward the N-terminus; the mean distances of the trajectory V27–Ad, A30–Ad, and V27–N+ are 3.5, 4.7, and 2.1 Å, respectively, suggesting a loosening of the M2TM-complex integrity at the N-terminus (a snapshot of the simulation complex with 10 is depicted in Figure S1a).
2.3.3. Complexes of Ligands with M2TMS31N.
In the starting configuration of M2TMS31N–aminoadamantane ligand complex the ammonium group points toward the C-terminus forming H-bonds with waters between the ligand and H37 residues as in the M2TMWT pore. The MD simulations of the complexes between M2TMS31N and 1–8 reach equilibration in less than 60 ns with the protein system having full flexibility. In complexes with M2TMS31N the ligand is more mobile inside the pore having a propensity to orient its ammonium group toward the N-terminus, in contrast to M2TMWT–ligand complexes where the ligand forms a strong complex with the ammonium group oriented toward the C-terminus. Thus, after a few nanoseconds of unrestrained dynamics the ligand moves toward the C-terminus by ~2 Å, probably because the adamantyl group is repelled by the polar N31 side chains resulting in the loss of the V27 lipophilic pocket, and the molecule rotates 180° through an attraction to the polar environment around N31. This finding is consistent with the narrower line widths and larger chemical shifts changes for M2TMWT when bound with compound 5 compared to M2TMS31N as observed in the ssNMR spectra suggesting a stronger binding interaction with M2TMWT. In addition waters are transferred from the region between the ligand and H37 to the area around N31 where the drug’s amine can interact with the carbonyls of the N31 amide groups and waters around the amide side chains. Snapshots of the MD simulation complexes of M2TMS31N with 1, 5, 6, and 10 are depicted in Figure 7. The MD runs showed that by progressively increasing the size of the adduct connected to the adamantyl moiety in complexes with 2–8, the ammonium group orientation turns toward the N-terminus, and the drug keeps this orientation during the entire simulation period. While this change was observed for 1 after ~20 ns of production, the time needed for the aminoadamantane ligand to rotate toward the N-terminus may be increased for some larger adducts. For example, in the case of 5 the ligand’s ammonium group keeps its orientation toward the C-terminus during the first 40 ns and then the molecule turns toward the N-terminus until the end of the 80 ns production time. While no significant conformational change was observed for M2TM31N tetramer in its complexes with 1–8 during the production period, the RMSDs for M2TMS31N Cα-carbons were ≤2.8 Å from the initial structure,4a,5b i.e., 1–1.5 Å higher than the RMSDs for M2TMWT Cα-carbons, suggesting less dynamics for M2TMWT complexes (Table S3). The mean values of the helical tilt angles for the complexes of 1 and 5–8 with M2TMS31N from the corresponding MD trajectories were measured to be ~34°, 31°, 28°, 33°, and 31° respectively, and are close to 38°, 33°, 28°, 31°, and 33° determined by OS ssNMR (Table 3). The uniformly tilted helical structure is a conversion from the initial kinked state in the 80 ns MD, where the starting structure reflects a homology model of 2KQT.4a,5b Here the major finding is the lack of kinked helices in M2TMS31N compared to the OS ssNMR structure when 1 binds M2TMWT suggesting than the MD simulations describe well the ligand–M2TMS31N structure.
Figure 7.

Snapshots from the simulation of various ligands bound to M2TMS31N. Waters within 10 Å from the ligand are shown. Several waters are shown covering the region between the mouth of the ligand and H37 residues. Few waters may be found between the ligands and the wall of the pore, suggesting a relatively free passage through the lumen. (A) Compound 1 bound to M2TMS31N. Seven waters are shown between the ligand and H37 residues and six waters between N31 and the mouth of the pore. In the depicted shapshot one hydrogen bond between the ammonium group of the ligand and one water, and one hydrogen bond between the ligand and the carbonyl group of N31 amide side chain, are shown (see also Table S3). No efficient van der Waals interactions can be formed for the adamantane core in the region close to A30, and the ligand cannot be stabilized inside the pore. (B) Compound 5 bound to M2TMS31N. Three waters are shown between the ligand and the region close to H37 residues and three waters between N31 and the mouth of the pore. One water is shown between the ligand and the wall of the pore. One hydrogen bond between the ammonium group of the ligand and one water and two hydrogen bonds between the ligand and the carbonyl groups of two N31 amide side chain are shown (see also Table S3). Hydrogen bonding together with weak van der Waals interactions between the adamantane core and the cleft between A30 and G34 can weakly stabilize the ligand inside the pore with its ammonium group oriented toward the N-terminus. (C) Compound 6 bound to M2TMS31N. Four waters are shown between the ligand and H37 residues and seven waters between N31 and the mouth of the pore. Two hydrogen bonds between the ammonium group of the ligand and the carbonyl group of N31 amide side chain and one water are shown (see also Table S3). One water is shown between the ligand and the wall of the pore. Hydrogen bonding together with weak van der Waals interactions between the adamantane core and the cleft between A30 and G34 can weakly stabilize the ligand inside the pore with its ammonium group oriented toward the N-terminus. (D) Compound 10 bound to M2TMS31N. Two waters are shown in the region between the ligand and H37 residues and six waters between N31 and the mouth of the pore. One water is shown between the ligand and the wall of the pore. Two hydrogen bonds between the ammonium group of the ligand and two waters are shown (see also Table S3). Hydrogen bonding together with weak van der Waals interactions between the adamantane core and the cleft between A30 and G34 can moderately stabilize the ligand inside the pore with its ammonium group oriented toward the N-terminus.
For most of the ligands 1–8 the mean angle between the C–N bond vector and the normal of the membrane is >140° for more than 40 ns of the simulation reflecting the propensity of the ammonium group to orient toward the N-terminus (Table S3). The distance between the center of mass of the four V27 and the adamantyl ring of the ligands 1–8 (V27–Ad) was longer than in complexes with the M2TMWT, which is consistent with an orientation of the ammonium group toward the N-terminus. On average, the adamantyl ring in M2TMS31N was found to be ~1–2 Å toward the C-terminus compared to the M2TMWT complexes. Accordingly, the distance between the center of mass of the four V27 and the ligand’s amine nitrogen in M2TMS31N complexes was shorter by 3–3.5 Å compared to M2TMWT complexes. In addition, especially for the ligands with large adducts, the adamantyl ring is positioned toward the C-terminus close to G34 (see distances A30–-Ad and G34–Ad in Table S3), whereas, in the M2TMWT complexes, the adamantyl ring is shifted toward the N-terminus embraced by V27 and A30. The sum of the H-bonds between the ammonium groups of the ligands and waters and between the ammonium groups and the N31 side chain amides was on average three hydrogen bonds for the primary ammonium groups (ligands 1–5), two hydrogen bonds for secondary (ligands 6 and 8), and one hydrogen bond for tertiary ones (ligand 7) (Table S3). Compounds 9 and 10 form three hydrogen bonds only with water molecules in the vicinity of N31 close to the wall of the pore, possibly because the ammonium group is sterically crowded between the adamantly and cycloalkane rings and cannot reach the side chains’ carbonyls of N31 (Table S3). In compound 10, due to the large hydrocarbon framework, the adamantyl ring moves more than in any other compound toward the C-terminus; i.e., the distance between the center of mass of the four V27 to the adamantyl cage was 3.7–2.3 Å longer than 1–8 to fit inside the pore close to G34, and the distance between the center of mass of the four G34 to the adamantyl cage was 2.7 Å. A snapshot of the simulation complex with ligand 10 is depicted in Figure 7.
2.3.4. Electrophysiology Results of Aminoadamantanes Blockage Using Full-Length M2.
Compound 1, its isomer 2, and three compounds with sizable adducts (5, 6, and 9) were assessed for their ability to inhibit low-pH-dependent proton currents induced by full-length M2 protein (A/California/07/2009 with M2N31) in transiently transfected, voltage clamped HEK cells (Table 4).10a,43 Data for compounds 1, 2, and 5 were reported previously.10a It was found that none of these compounds blocked inward proton currents better than 1, either following a standard 3 min exposure to compound or after prolonged exposure for 30 min. These EP-based results indicated that the compounds were unable to block the full-length M2 channel encoding N31. However, when this M2 protein was modified to encode the 1-sensitive S31 sequence (through an N31S mutation), all compounds inhibited proton currents with IC50s of 10 μM or less (Table 4); by increasing the adduct size, the blocking effect of inward proton currents is reduced.
2.3.5. In Vitro Testing of Aminoadamantanes against Influenza A Virus.
The antiviral potencies of compounds 1–10 were measured against WSN/33 (H1N1) bearing the M2 S31N mutation44 and its amantadine-sensitive variant WSN/33-M2-N31S26 in MDCK cells with CPE inhibitory assay.45,46 Compounds 1–9 showed sub-micromolar IC50 values against WSN/33-M2-N31S but were inactive against WSN/33 (Table 5). Compound 10 was inactive against both strains. The selectivity indices (CC50/IC50) of compounds 4–7 were comparable to 1. The inhibition efficiency showed stringent head-ammonium-group requirements for inhibition of amantadine-sensitive influenza A viruses—a result consistent with ITC and EP results.
3. DISCUSSION
3.1. Unraveling the Binding Differences for Amino-adamantanes to M2TMS31N and M2TMWT.
Based on the MD simulations results and experimental findings, the molecular basis for weak binding and the inability of aminoadamantanes to effectively block M2S31N is described. The results of the experimental data from ssNMR and ITC experiments directly correlate with the MD simulation results showing that aminoadamantane derivatives are weaker binders in the pore of M2TMS31N compared to M2TMWT and that 5–8 are stronger binders compared to 1–4 against M2TMS31N.
The S31N mutation of M2TM results in a shift of the hydrophobic adamantyl ring toward the C-terminus thereby losing the stabilizing hydrophobic interactions of the V27 isopropyl groups with the adamantyl ring that is present in M2TMWT. The bulky N31 side chains are oriented toward the N-terminus and the V27 side chains, and the ammonium group of the ligands is also turned toward the N-terminus to form significant hydrogen bonding interactions with the polar N31 side chains and surrounding waters. This ammonium group orientational preference of amantadine and rimantadine has been previously noted by Gleed et al.17b and Alhadeff et al.16 The hydrogen bonding interactions with N31 are consistent with the MAS experimental data performed with compound 6 showing a chemical shift perturbation for N31 and G34 compared to the apo M2TMS31N. Distance measurements from ssNMR experiments showed the preference for the ammonium group of the aminoadamantane drugs orienting toward the C-terminus in M2TMWT5c and toward the N-terminus in a complex of M2TMS31N with a conjugate of 1 having a phenylisoxazole polar head.24 In this class of conjugates23b–d additional van der Waals interactions with the N-terminus stabilizes the ligand resulting in potential anti-influenza drugs. Here, where the aminoadamantane ligands, such as 1–8, are hydrogen-bonded with the polar N31 environment, favorable van der Waals and hydrophobic interactions such as those in M2TMWT are missing. In M2TMWT the adamantyl ring is well-accommodated by the V27 and A30 side chains and sizable adducts such as ligands 5–8 additionally fill the region between A30 and G34 (Figures 1 and 6), but in M2TMS31N the adamantyl ring is close to A30 and in the vicinity of G34 (see Tables S2 and S3) lacking a favorable hydrophobic pocket. This is consistent with the absence of chemical shift perturbations for V27 in the NCA MAS spectrum of 6 bound to M2TMS31N in comparison with the apo M2TMS31N compared to the significant chemical shift changes at V27, S31, and G34 which have been reported when rimantadine is bound to M2TMWT relative to the apo state.20,21 These structural differences can be clearly observed in Figures 6 and 7. The lack of favorable van der Waals interactions results in less stable complexes and a weaker binding for aminoadamantane ligands consistent with the much smaller Kd values of 5 and 6 by ITC against M2TMS31N compared to M2TMWT (Table 1). In addition, line widths are narrower for the M2TMWT complex with 5 compared to the M2TMS31N complex suggesting less dynamics consistent with stronger interactions for the aminoadamantane derivatives in complexes with M2TMWT. The RMSDs for M2TM Cα-carbons are 1.0–1.5 Å higher for the trajectories of M2TMS31N compared to M2TMWT complexes further suggesting more dynamics and weaker interactions in the S31N complexes.
In 1 and analogues with small adducts the adamantyl ring has only a limited hydrophobic contact with A30 and is close to polar N31 side chains which exert repulsive forces on the adamantyl ring. It should be noted that trajectories sampled in the MD simulations of ligands in the pore were of 80 ns length; i.e., they are much shorter than the microsecond to millsecond time scales sampled by ssNMR. The MD runs of the complexes of 1–4 with M2TMS31N showed qualitatively that these molecules cannot bind M2TMS31N because significant favorable van der Waals interactions are missing. This can be observed from the snapshot for the complex of 1 with M2TMS31N in Figure 7. Molecules 5–8 with sizable adducts of the adamantyl ring fill effectively the region between A30 and G34, and the interactions needed for binding are slightly improved resulting in weak binding to M2TMS31N as compared to no binding for 1–4. This can be observed from snapshots for complexes of 5 and 6 with M2TMS31N in Figure 7. Indeed, 1 did not bind as documented by the OS ssNMR spectra (Figure 3), but larger adducts such as those present in compounds 5–8 appear to stabilize weak binding of the drug in the region between A30 and G34 (see Figure 7), and this is in accordance with the results from OS ssNMR spectra for weak binding of compounds 5–8 (Figures 3 and 4) to M2TMS31N. In addition, we were not able to detect any binding for 1–4 with M2TMS31N using ITC while we obtained approximate binding constants for 5, 6, 9, and 10 (Table 1). NMR data were not obtained for 9 and 10 because they produced disordered lipid bilayers. Thus, the results from the combination of simulations and experiments showed that 1 (and similarly 2–4) did not bind M2TMS31N contrary to the weak binding for 1 previously suggested from short simulations,17b but 5–8 having sizable adducts display weak binding.
While binding strength of aminoadamantanes against M2TMWT was sensitive to modification of the adduct, the M2TMWT–aminoadamantane complex stability was found to be very sensitive to the adduct size, as shown by the ITC and MD simulation results when considering that 9 and 10 produce unstable complexes. The MD simulations clearly show that 10 moves considerably toward the N-terminus of the pore losing specific binding interactions. Ligands 9 and 10 cause a dramatic reduction in affinity for M2TMWT compared to 1–8 but did not affect M2TMS31N affinity, which seems to be similar to those of 5 and 6. The forces that cause the rotation of the adamantyl ring in the M2 pore appear to be inherent in the shared amine group and are not greatly perturbed by the other ligand variations as characterized by MD and OS ssNMR for adducts 1–4 and 5–8 against M2TMS31N. Taken together, the results from the combination of MD simulations, ITC, and OS ssNMR showed no binding for 1 and similar in size analogues and only weak binding for sizable adducts. The binding is more specific as showed by the more stringent head-ammonium-group requirements in the binding pocket of M2TMWT than in the M2TMS31N according to the ITC but also as showed by the EP experiments and antiviral assay results.
Thus, EP experiments indicate that the aminoadamantanes block the S31 but not the N31 full-length M2 protein. Notably, while the secondary gate formed by the V27 residues in the M2TMWT11 has the potential to limit water access to the pore, the hydrophilic asparagine side chains make this environment less hydrophobic and diminish the effectiveness of the V27 gate in the M2TMS31N pore. The mutant waters are observed above and below the ligand and in a few snapshots between the ligand and the wall of the pore suggesting a relatively free passage through the M2TMS31N lumen despite the presence of the ligand.12,17,18 (snapshots from the simulation of the complexes of M2TMS31N with 1, 6, are 10 are depicted in Figure 7).
3.2. Changes in C-Terminus Structure–Function of M2TMS31N and M2TMWT Induced by V27 Interactions with the Adamantyl Cage of Amantadine Variants.
Furthermore, it is suggested that the 10° helical kink that reduces the tilt of the C-terminal portion of the transmembrane helix in the WT is likely induced by the formation of a strong binding pocket for the aminoadamantanes. This hydrophobic pocket formed by the V27 aliphatic side chains coupled with the aliphatic adamantyl cage of the aminoadamantanes may prevent the W41 gate from opening further, defeating proton conductance. In contrast the aminoadamantanes in M2TMS31N included a loss of V27 lipophilic pocket and have their amino group drawn toward the N-terminus by the asparagine side chains resulting in the presence of multiple water molecules in this region. In addition the weak binding of the aminoadamantanes results in no perturbation of the helical tilt and the W41 gate can function normally.
4. CONCLUSIONS
This work represents a study of the binding of amantadine variants against the proton channel formed by the tetrameric structure of the influenza A M2 protein. Significantly, we focus on aminoadamantane variants of 1 binding to M2TMS31N compared to M2TMWT aiming at investigating why these variants are ineffective in blocking proton conductance of the M2S31N channel. The results of this effort are based on a combination of experimental techniques and MD simulations both performed in liquid crystalline lipid bilayer environments. Aminoadamantane derivatives are known to be blockers of the M2 WT protein. They are known to bind in the pore and are presumed to block proton access to the H37 tetrad that is known to shuttle protons through aqueous pore into the viral interior.47 There are two gates that can inhibit conductance, the V27 tetrad, known as the secondary gate at the external entrance to the pore, and the W41 tetrad near the exit of the pore into the viral interior, known as the primary gate. These aminoadamantane derivatives were weaker binders to M2TMS31N compared to M2TMWT as observed by a reduced influence on the protein structure and by reduced amplitude of the channel dynamics as observed by both the MD and experimental data. Moreover, these aminoadamantane derivatives were ineffective against M2S31N while blocking M2WT protein. We suggest that 1 and the similar sized analogues 2–4 lack binding affinity and the larger sized analogues 5–8 showed weak binding affinity to M2TMS31N because of a lack of effective hydrophobic interactions as a result of reshaping of the cavity when N31 is present which included loss of the V27 lipophilic pocket. In contrast V27 interactions are present with 1–8 in M2TMWT pore and these ligands are effective binders to M2TMWT. All ligands 1–8 have a tight binding for M2TMWT, and the binding is more specific as showed by the more stringent head-ammonium-group requirements in the binding pocket of M2TMWT than in the M2TMS31N according to the ITC, EP, and CPE results. The weak binding of 5–8 to M2TMS31N was significant enough to induce observable changes in the helix tilt angles characterized by the experimental data and by the MD simulations.
It is interesting to note that the blockage of M2TMWT by both 1 and 5 involved a 10° kink in the TM helix and a very significant change in the helix orientation in the C-terminal half of the TM. This is likely due to the V27 side chains—adamantane hydrophobic interactions that are not possible in M2TMS31N. While aminoadamantane ligand binding causes a kink in the C-terminal half of M2TMWT and a blockage for proton conductance by the M2 channel, aminoadamantane ligand interactions with M2TMS31N did not result in a helix kink in the TM helix and proton conductance was not blocked. While the helix kink has been associated with blockage and potential disabling of the opening of the W41 gate in M2TMWT previously,48 here, the explanation for the helix kink in the M2TMWT and its absence in M2TMS31N has been suggested to be induced by the V27 interactions with the adamantyl cage in M2TMWT and the absence of such significant interactions in M2TMS31N.
5. EXPERIMENTAL METHODS
5.1. Synthesis of Aminoadamantane Ligands.
The procedures applied for the synthesis of the new derivatives 8 and 10 are depicted in Schemes 2 and 3. The description of synthetic procedures for 8 and 10, experimental details, and compound characterization can be found in the Supporting Information. All compounds’ purity was ≥95% as determined by elemental analysis (see Supporting Information).
Scheme 2. Synthesis of 1′-Methylspiro[pyrrolidine-2,2′-adamantane] (8)a.
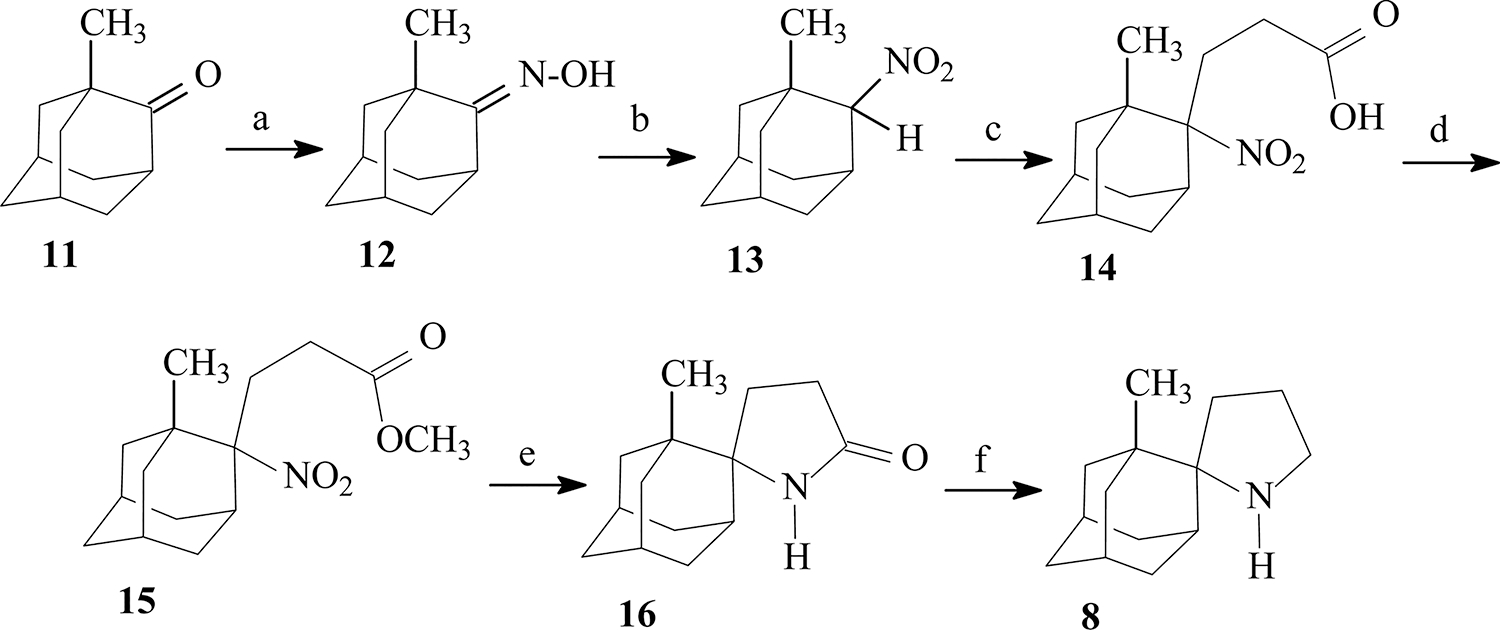
aReagents and conditions: (a) H2NOH·HCl, Na2CO3 90 °C, 40 min (93%). (b) (i) NBS, NaHCO3, dioxane/water, 10 °C, 40 min; (ii) HNO3, pentane, 0 °C, 15 min; (iii) NaBH4, MeOH/H2O. (c) (i) CH2═CHCO2Et, Triton-B, t-BuOH, 70 °C, 8 h. (ii) 1 N NaOH, 3:1 EtOH–H2O, 70 °C, 8 h (89%). (d) MeOH/HCl(g), 60 °C, 4 h, and then overnight at room temperature (r.t.) (79%). (e) H2/Ni-Raney, EtOH, 50 psi, r.t., 24 h (84%). (f) LiAlH4, THF, reflux, 48 h (60%).
Scheme 3. Synthesis of 1-(1-Adamantyl)cyclohexanamine (10)a.

aReagents and conditions: (a) Li, dry THF, cyclohexanone, sonication, 0 °C, 5 h (70%); (b) NaN3, TFA, CH2Cl2, 0 °C, 5 h, then r.t. 24 h (35%). (c) LiAlH4, dry ether, reflux, 5 h, then H2O, NaOH 10% (65%).
5.2. M2TM Peptide Synthesis.
M2TMWT peptides corresponding to residues 22–46 of the Udorn (A/Udorn/307/72) sequence of M2 (C-terminally amidated SSDPLVVAASIIGILHLILWILDRL) and of the S31N mutant peptide (C-terminally amidated SSDPLVVAANII-GILHLILWILDRL) were synthesized by standard Fmoc (9-fluorenylmethoxycarbonyl) solid phase peptide synthesis using an aminomethyl polystyrene resin loaded with the amide linker and purified by reverse phase HPLC before being used for the ITC experiments. Additional quantities of these needed peptides were purchased from Centic Biotec, Heidelberg, Germany. For ssNMR experiments M2TMWT and M2TMS31N (22–46) peptides with 15N labeled at V28, A30, and I42 and M2TMS31N (22–46) peptides with 13C, 15N labeled at structurally important residues V27, A304a,5b and N31, G34 (13C,15N-VANG) were synthesized using Fmoc chemistry. Fmoc-[15N]-Val, Fmoc-[15N]-Ala, Fmoc-[15N]-Ile, and Fmoc-[13C,15N]-Val, Fmoc-[13C,15N]-Ala, Fmoc-[13C,15N]-Ser, and Fmoc-[13C,15N]-Gly were purchased from Cambridge Isotope Laboratory (Andover, MA, USA). Solid phase syntheses of M2TM peptides (0.25 mmol) were performed on an Applied Biosystems 430A peptide synthesizer as previously described.49,50 The peptide was cleaved from the resin by the treatment with ice cold 95% TFA, 2.5% H2O, 1.25% ethanedithiol, and 1.25% thioanisole and precipitated from TFA using ice cold ether. Following centrifugation, the supernatant was discarded and the pellet was washed with cold ether again. The precipitated peptide was dried under vacuum. A purification procedure previously described51 and modified50 was used. Peptide purity and identity was confirmed using ESI mass spectrometry (positive ion mode). The final peptide purity was 98%.
5.3. ITC Measurements of Aminoadamantane Ligands Binding to M2TM.
Binding affinities of the aminoadamantane derivatives 1–5, 6, 9, and 10 for M2TMS31N were measured by ITC experiments52,53 in DPC micelles at pH 8. Compounds 7 and 8 were not measured. M2TM fragments form stable tetramers at this pH, in contrast to low-pH (<6.5) conditions.54 Furthermore, experimental data indicate that 1 binds with higher affinity at alkaline pH to M2TM, where the pore of the M2 channel is in the closed state, than at low pH, where the open state of M2TM is prevalent.54a,2a
All measurements were performed with a TAM 2277 (TA Instruments) at pH 8 and 20 °C in a buffer of 50 mM NaH2PO4 and 100 mM NaCl. The peptide and the aminoadamantane derivative were dissolved in a freshly prepared DPC solution with a concentration of 13 mmol L−1. Measurements against M2TMWT were conducted using 2 mL of 125 μM peptide (corresponding to 31.25 μM M2TMWT tetramer) as has been described previously.26 For the low-affinity ligands 9 and 10 a 250 μM peptide concentration was used. For M2TMS31N measurements the concentration of the peptide used was 167 μM, which was increased to 500 μM when 5 was tested in order to get a curve adequate for measurements. A concentration of 1.1 mM of the ligand was used for the titrant, of which 7.6 μL (equivalent to 8.4 nmol) was dispensed in the peptide/DPC solution with each injection. The time interval between two injections was set to at least 6 min. The first injection was not used due to dilution effects.
Synthetic M2TMS31N (residues 22–46) was reconstituted in DPC micelles at pH 8 at a 1:40 monomer/DPC ratio which guarantees the quantitative formation of M2TM tetramers54 by dissolving and sonicating 334 nmol of M2TM with the 40-fold amount of DPC in the aforementioned buffer system (for M2TMWT a 1:26 ratio was applied for the measurements of ligands 9 and 10 compared to 1:57 applied previously26). Solutions of the ligands in the buffer were titrated into the calorimetric cell at 20 °C. The released heat of binding was derived by subtracting the heat of dilution from the heat of reaction.55,56
Describing in more detail, to determine the heat of dilution, ligand and receptor (M2TM) were dissolved in buffer containing DPC. Two reference experiments were carried out; i.e., (a) ligand was titrated into the buffer containing DPC, and (b) receptor was titrated into buffer containing DPC. The obtained signals showed that the released heat of each injection is very small and stays constant during the experiment. No interaction is detected between ligand and DPC containing buffer or receptor and DPC containing buffer, respectively. The released heat is the heat of dilution, which occurs when the two solutions are mixed. The heat of dilution was estimated for each titration experiment individually. At the end of each measurement, the receptor is titrated with the ligand being in an excess (the ratio for ligand/receptor after 19 injections is 2:1). If all binding sites are saturated, ligand cannot bind to the receptor anymore. The released heat is then equivalent to the heat of dilution. As mentioned, the heat of dilution is calculated as an average from the last five titration steps, when the heat is constant. This heat is taken into account as a Q correction. It is subtracted from all titration steps in this measurement.
Data evaluation, including the integration of the peaks, was carried out with Digitam for Windows v4.1. The measured heat per amount of substance against the molar ratio of titrant to peptide tetramer was plotted, and the affinity constants were calculated by nonlinear regression of the measured heat per injection using Origin 8.057 and have been included in Table 1. Compounds 1–8 have been measured against M2TMWT in a previous work (Table S1).26 For the calculation, the concentration of the peptide was kept variable because the M2TM tetramer formation was not complete. The fit function involves three parameters. One of them is a factor for the correction of the peptide concentration (the difference between the concentration, which has been weighted in, and the active concentration). The cell volume is fixed at 2 mL. The concentration of the ligand in the solution is known, because pure substance was weighted in and the stoichiometry of the binding of ligand to receptor was assumed to be 1:1. If these quantities are set, the concentration of active receptor is obtained by fitting the measured data points. From the calculations, it can be seen that the concentration of active receptor is lower than the concentration of receptor that has been weighted in. The binding affinity of 1 measured against M2TMWT in a previous work26 was 2.17 ± 0.52 μM and is comparable with the value of 12 μM measured using analytical ultracentrifugation54c and the value of 9 ± 2 μM derived based on kinetic studies in electrophysiological experiments.2a For the M2TM peptide investigated in this study, the solubility in the DPC micelles limits the concentration of M2TM that can be tested. Consequently, affinity constants of low-affinity binders, e.g., amino-adamantanes against M2TMS31N and 9 and 10 against M2TMWT, possess relatively large errors.
5.4. Sample Preparation for Solid State NMR Spectroscopy.
15N–V28A30I42 M2TMWT or M2TMS31N was codissolved in trifluor-oethanol (TFE) with DMPC in a 1:30 molar ratio. (The molar ratio of one protein tetramer to 120 DMPC lipids was used. The molecular weight of the M2TMWT peptide is MW = 2729 g/mol, and the lipid is MW = 678 g/mol.) The solvent was removed under a stream of nitrogen gas to yield a lipid film and then dried to remove residual organic solvent under vacuum for 12 h. Thoroughly dried lipid film was hydrated with 10 mM HEPES buffer at pH 7.5 to form multilamellar vesicles containing M2TM in the tetrameric state. This suspension was bath sonicated, dialyzed against 2 L of 10 mM HEPES pH 7.5 buffer for 1 day, and centrifuged at 196000g to harvest unilamellar proteoliposomes. The pellet was resuspended in a 1 mL aliquot of the decanted supernatant containing the ligand, resulting in a 1:6 molar ratio of the M2TM tetramer to drug. Typically a preparation for solid state NMR would include 5 mg of protein and 37 mg of DMPC. For samples that included drug the molar ratio of drug to tetramer was 6:1. For 1 with a molecular weight of MW = 187.5 g/mol that would mean the addition 0.52 mg for a 5 mg protein sample. (There is sufficient evidence in the literature to demonstrate that M2 structure is not perturbed by drug concentration. The most obvious example in this work is the absence of structural changes in the S31N spectra in the presence of 1.) Following overnight incubation at 37 °C, the pellet was deposited on 5.7 × 10 mm glass strips (Matsunami Trading, Osaka, Japan). The bulk of the water from the sample was removed during a 2 day period in a 98% relative humidity environment at 298 K. Rehydration of the slides, before stacking and sealing into a rectangular sample cell, increased the sample weight by 40–50%. Compounds 1 and 5 were used for ssNMR experiments against M2TMWT; compounds 1 and 5–8 were used for ssNMR experiments against M2TMS31N; compounds 9 and 10 produce disordered lipid bilayers according to the 31P spectra (not shown).
5.5. Solid State NMR Experiments of M2TM–Amino-adamananes Complexes.
5.5.1. OS ssNMR Spectra.
PISEMA28 and SAMPI429,30 spectra were acquired at 720 MHz utilizing a low-E 1H/15N double resonance probe.58 Acquisition took place at 303 K, above the gel to liquid crystalline phase transition temperature of DMPC lipids. Experimental parameters included a 90° pulse of 5 μs and cross-polarization contact time of 0.8–1 ms, a 4 s recycle delay, and a SPINAL decoupling sequence.58 For the spectrum of 15N–V28A30I42 M2TMWT with compounds 1 and 5, 32 t1 increments were obtained, and nine t1, for the sample of 15N–V28A30I42 M2TMWT without drug; 16–28 t1 increments were obtained for the spectrum of 15N–V28A30I42 M2TMS31N with compounds 1 and 5–8, and nine t1 increments, for the sample of 15N–V28A30I42 M2TMS31N without drug. Spectral processing was done with NMRPIPE59 and plotting with SPARKY. 15N chemical shifts were referenced to a concentrated solution of N2H8SO4, defined as 26.8 ppm relative to liquid ammonia.
5.5.2. NCA MAS Spectra.
15N–13Cα correlation experiments were performed on a Bruker Avance 600 MHz NMR spectrometer with an NHMFL 3.2 mm low-E-field triple resonance probe.60,61 The 13C chemical shifts were referenced using the published chemical shifts of adamantane relative to DSS62, and 15N chemical shifts were calculated with IUPAC relative frequency ratios between the DSS (13C) and liquid ammonia (15N).63,64 Spectra were acquired at MAS frequency of 10–12 kHz and a calibrated sample temperature of −10 °C. Thirty points were collected in the 15N dimension for an acquisition time of 5–6.25 ms, while in the direct dimension the acquisition time was 10.2 ms. In all experiments, 92 kHz of proton decoupling was used. To get one bond 15N–13C correlation, a mixing time of 5 ms was used. Spectra were processed with Topspin.
5.6. MD Simulations of M2TM–Aminoadamantane Complexes.
5.6.1. Docking Calculations.
The ligands in their ammonium forms were built by means of Maestro 8.565 and were then minimized by means of Macromodel 9.6 and the MMFF94 force field66 implemented with Macromodel 9.6 using the CG method and a distance-dependent dielectric constant of 4.0 until a convergence value of 0.0001 kJ Å−1 mol−1 was reached. The M2TMWT–1 complex structure (PDB ID 2KQT4a,5b) served as a model structure for M2TMWT with bound ligands. N- and C-termini of the M2TM model systems were capped by acetyl and methylamino groups. After applying the protein preparation module of Maestro, all hydrogens of the protein complex were minimized with the AMBER* force field by means of Maestro/Macromodel 9.6 using a distance-dependent dielectric constant of 4.0. The molecular mechanics minimizations were performed with a conjugate gradient (CG) method and a threshold value of 0.0001 kJ Å−1 mol−1 as the convergence criterion. The structures of the protein and ligand 1 were saved separately and were used for the subsequent docking calculations. The ligands minimized in this manner were docked into the M2TMWT binding site. Docking poses of the aminoadamantane derivatives 1–10 in the M2TMWT bound state were generated with GOLD 5.267,68 considering five water molecules located between the ammonium group of 1 and H37 within the M2TMWT pore-binding site and applying the ChemPLP implemented in the software.69,70 The option “toggle” was used to let the algorithm decide whether to take into account a water molecule or neglect it based on an empirical desolvation penalty. The region of interest used by GOLD was defined to contain the atoms that were within ~15 Å of the 1 binding site in the receptor structure. The “allow early termination” command was deactivated. For all of the other parameters, GOLD default values were used. Ligands were submitted to 30 genetic algorithm runs. Ten docking poses were produced for each ligand which were visually inspected using the UCSF Chimera package.71 The docking pose with the best ChemPLP score was used for the subsequent MD simulations with M2TMWT and M2TMS31N structures created as described below.
5.6.2. MD Simulations.
Models of M2TMS31N–aminoadamantane complexes were generated from M2TMWT aminoadamantane complexes by mutating amino acids S31–N31 with Maestro65 and preparing the structure as described above; i.e., N- and C-termini of the M2TM peptides were capped by acetyl- and methylamino groups, respectively. For N31, the side chain rotamers may have χ1 angles of −160° or −80° corresponding to N31 side chains placed at the interface between helices or inside the lumen, respectively. Structures of M2TMS31N(18–60) in DPC micelles solved by solution NMR spectroscopy show residue 31 in the helix–helix interface72 while MAS ssNMR studies in 1,2-diphytanoyl-sn-glycero-3-phosphocholine (DPhPC) bilayers showed the side chain of the N31 residue oriented toward the pore in two helices and toward an adjacent helix in the other two, with neighboring N31 side chains close enough to form polar contacts.37 A just-released X-ray structure shows that N31 residues are oriented into the channel pore forming a hydrogen-bonding network,73 and it was suggested that this may prevent drug from entering the channel. Preliminary OS ssNMR results in liquid crystalline lipid bilayers confirm that all four of the M2S31N N31 residues are oriented toward the pore. Simulations of M2TMS31N–ligands were run (a) with N31 side chains placed at the interface between helices72 (χ1 angle is −80°) to avoid a biased starting conformer in which N31 repels adamantane and (b) with N31 pointing toward the pore in two helices (χ1 angle is ~−160°) and toward an adjacent helix in the other two37 (χ1 angle is ~−80°). For comparison reasons a few MD simulations with 1, 5, and 6 were also performed with the starting structure of M2TMS31N having N31 residues pointing into the center of the channel pore (χ1 angle is −160°).73 It should be mentioned that when the starting structure has all four N31 side chains placed at the interface between helices, after a few nanoseconds of simulation the side chains of at least two N31 residues change orientation pointing inside the pore lumen. This was also observed with the apo protein M2TMS31N after a few nanoseconds. Configurations with different N31 rotamers produced MD trajectories with similar behavior for aminoadamantane ligands. MD simulations were run in triplicate or more for 1, 2, 5, and 6 to test the reproducibility of the behavior of the system.
The M2TMWT complexes or M2TMS31N complexes were embedded in a DMPC lipid bilayer extending 10 Å beyond the solutes. Complex and ligand systems were solvated using the TIP3P74 water model. Na+ and Cl− ions were placed in the water phase to neutralize the systems and to reach the experimental salt concentration of 0.150 M NaCl. Membrane creation and system solvation were conducted with the “System Builder” utility of Desmond.75,76 The M2TMWT–1 complex structure in the hydrated DMPC bilayer with ions included 18617 atoms.
The OPLS 2005 force field77–79 was used to model all protein and ligand interactions, and the TIP3P model74 was used for water. The particle mesh Ewald method (PME)80,81 was employed to calculate long-range electrostatic interactions with a grid spacing of 0.8 Å. van der Waals and short-range electrostatic interactions were smoothly truncated at 9.0 Å. The Nosé–Hoover thermostat82 was utilized to maintain a constant temperature in all simulations, and the Martyna–Tobias–Klein method82 was used to control the pressure. Periodic boundary conditions were applied (50 × 50 × 80 Å3). The equations of motion were integrated using the multistep RESPA integrator83 with an inner time step of 2 fs for bonded interactions and nonbonded interactions within a cutoff of 9 Å. An outer time step of 6.0 fs was used for nonbonded interactions beyond the cutoff.
Each system was equilibrated in MD simulations with a modification of the default protocol provided in Desmond, which consists of a series of restrained minimizations and MD simulations designed to relax the system, while not deviating substantially from the initial coordinates. First, two rounds of steepest descent minimization were performed with a maximum of 2000 steps with harmonic restraints of 50 kcal mol−1 Å−2 applied on all solute atoms, followed by 10000 steps of minimization without restraints. The first simulation was run for 200 ps at a temperature of 10 K in the NVT (constant number of particles, volume, and temperature) ensemble with solute heavy atoms restrained with a force constant of 50 kcal mol−1 Å−2. The temperature was then raised during a 200 ps MD simulation to 310 K in the NVT ensemble with the force constant retained. The temperature of 310 K was used in our MD simulations in order to ensure that the membrane state was above the melting temperature state of 297 K for DMPC lipids.84
The heating was followed by equilibration runs. First, two stages of NPT equilibration (constant number of particles, pressure, and temperature) were performed, one with the heavy atoms of the system restrained for 1 ns and one for solvent and lipids for 10 ns, with a force constant of 10 kcal mol−1 Å−2 for the harmonic constraints, respectively. A NPT simulation followed with the Cα atoms restrained for 1 ns with a force constant of 2 kcal mol−1 Å−2. The above-mentioned equilibration was followed by an 80 ns NPT simulation without restraints. Within this time, the total energy and the RMSD reached a plateau, and the systems were considered equilibrated.
5.7. Electrophysiology Experiments of M2 Blockage by Aminoadamantanes.
Electrophysiology was performed as previously described.10a pcDNA3 vectors encoding the full-length A/California/07/2009 (H1N1) M2 protein containing either an N31 or an S31 mutation was cotransfected with a pcDNA3 vector encoding eGFP into TSA-201 (HEK parental) cells using standard transfection protocols (Lipofectamine 2000, Life Technologies). This construct was previously annotated as A/England/195/2009 (H1N1)10a but is identical in amino acid sequence to A/California/07/2009 (H1N1). Macroscopic ionic currents were recorded in the whole-cell configuration from GFP-positive cells 24–48 h after transfection. Cells were perfused continuously at 3–5 mL min−1 with external (bath) solution containing (mM) 150 NMG, 10 HEPES, 10 D-glucose, 2 CaCl2, and 1 MgCl2 buffered at pH 7.4 with HCl. For low-pH (pH = 5.5) solution, HEPES was replaced by MES. Patch electrodes were pulled from thin-walled borosilicate glass (World Precision Instruments, Sarasota, FL, USA) and fire-polished before filling with standard pipet solution containing (mM) 140 NMG, 10 EGTA, 10 MES, and 1 MgCl2 buffered at pH 6.0 with HCl. Voltage–clamp experiments were performed with an Axopatch 200B amplifier (Molecular Devices, Sunnyvale, CA, USA) connected to a Digidata 1322A 16-bit digitizer. Data were acquired with the pCLAMP8.0 software (Molecular Devices, CA) sampled at 10 kHz and low-pass-filtered at 5 kHz. Cells were held at −40 mV. The voltage protocol consisted of a 100 ms pulse to −80 mV followed by a 300 ms ramp to +40 mV and a 200 ms step to 0 mV before stepping back to −40 mV, which was repeated every 4 s. All drugs were prepared as DMSO stocks (50 or 100 mM) and diluted with external solution to desired concentrations. To measure a block of M2 currents by compounds, cells were recurrently treated with pH 7.4 and pH 5.5 solutions until stable and pH-dependent inward currents were reproducibly observed, followed by treatment with compound at pH 5.5 for 2–30 min. At the end of each experiment, cells were treated with a 100 μM solution of 1.
5.8. Cells and Viruses.
Madin–Darby canine kidney (MDCK) cells (Cat. No. RIE 328, Friedrich-Loeffler Institute, Riems, Germany) were propagated as monolayer in Eagle’s minimum essential medium (EMEM) supplemented with 10% fetal bovine serum, 1% nonessential amino acids (NEAA), 1 mM sodium pyruvate, and 2 mM L-glutamine. Amantadine-resistant WSN/33 (with N31 in M2) and its amantadine-sensitive variant WSN/33-M2-N31S44 were used in this study. For the generation of WSN/33-M2-N31S44 the plasmid pHW187-M2-N31 was altered by site-directed mutagenesis PCR and afterward used as part of a plasmid set for virus recovery.44 Both WSN/33-variants were propagated on MDCK cells in serum-free EMEM supplemented with 2 mM L-glutamine, 2 μg/mL trypsin, and 0.1% sodium bicarbonate (test medium). Virus containing supernatant was harvested after about 48 h of incubation at 37 °C when cytopathic effect became microscopically visible. Aliquots were stored at −80 °C until use. The M2 gene identity of all recombinant viruses was verified by sequencing.
5.9. CPE Inhibition Assay of Influenza A Viruses by Aminoadamantanes.
Cytotoxicity and CPE inhibition studies were performed on two-day-old confluent monolayers of MDCK cells grown in 96-well plates as published.46 Cytotoxicity was analyzed 72 h after compound addition (2-fold or half-log dilutions; at least two parallels per concentration; maximum concentration, 100 μM). In CPE inhibition assay, 50 μL of 2-fold compound dilutions in test medium and a constant multiplicity of infection of test virus (0.045 for WSN/33 and 0.04 for WSN/33-M2-N31S) in a volume of 50 μL of the test medium were added to cells. Then, plates were incubated at 37 °C with 5% CO2 for 48 h until the untreated, infected control showed maximum cytopathic effect. Crystal violet staining and optical density determination were performed as described before to determine the percentage of antiviral activity of the tests compounds.45,46 After log transformation of compound concentrations, linear regression was used to determine the 50% cytotoxic (CC50) and the 50% inhibitory concentration (IC50) (Table 5). At least three independent assays were conducted to calculate the mean CC50 as well as IC50s and their standard deviations.
Supplementary Material
ACKNOWLEDGMENTS
The NMR experiments were performed at the National High Magnetic Field Laboratory, which is supported by a National Science Foundation Cooperative Agreement (Grant DMR-1157490) with the State of Florida. A.K. thanks Chiesi Hellas for supporting the Ph.D. research of C.T. A.K. also thanks Harris Ioannidis and Antonios Drakopoulos (previous M.Sc. students) for running a few preliminary MD runs. This work was in part supported by NIH Grant AI023007 and a Grand Challenges Stars in Global Health grant to I.T. and D.F. D.F. thanks Daniel Kwan for technical assistance.
ABBREVIATIONS USED
- BAR
Bennett acceptance ratio
- CPE
cytopathic effect
- DMPC
1,2-dimyristoyl-sn-glycero-3-phosphocholine
- DPhPC
1,2-diphytanoyl-sn-glycero-3-phosphocholine
- EP
electrophysiology
- HEK
human embryonic kidney
- ITC
isothermal titration calorimetry
- M2TM
M2 transmembrane domain
- MAS
magic angle spinning
- MD
molecular dynamics
- MM/PBSA
molecular mechanics Poisson–Boltzmann surface area
- NCA
15N–13Cα
- OS
oriented sample
- PMF
potential of mean force
- ssNMR
solid state NMR
- WT
wild-type
Footnotes
The authors declare no competing financial interest.
ASSOCIATED CONTENT
Supporting Information
The Supporting Information is available free of charge on the ACS Publications website at DOI: 10.1021/acs.jmed-chem.6b01115.
Detailed synthesis information, information for ITC method limitations, tables of structural and dynamic measures, snapshots of additional MD runs, RMSD plots from MD runs, and relevant references (PDF)
Molecular formula strings (CSV)
REFERENCES
- (1).Helenius A Unpacking the incoming influenza virus. Cell 1992, 69, 577–578. [DOI] [PubMed] [Google Scholar]
- (2).(a) Wang C; Takeuchi K; Pinto LH; Lamb RA Ion channel activity of influenza A virus M2 protein: characterization of the amantadine block. J. Virol. 1993, 67, 5585–5594. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Chizhmakov IV; Geraghty FM; Ogden DC; Hayhurst A; Antoniou M; Hay AJ Selective proton permeability and pH regulation of the influenza virus M2 channel expressed in mouse erythroleukaemia cells. J. Physiol. 1996, 494, 329–336. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (3).Hayden FG Clinical applications of antiviral agents for chemophrophylaxis and therapy of respiratory viral infections. Antiviral Res. 1985, 5, 229–239. [DOI] [PubMed] [Google Scholar]
- (4).(a) Hu J; Asbury T; Achuthan S; Li C; Bertram R; Quine JR; Fu R; Cross TA Backbone structure of the amantadine-blocked transmembrane domain M2 proton channel from influenza A virus. Biophys. J. 2007, 92, 4335–4343. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Hu J; Fu R; Cross TA The chemical and dynamical influence of the anti-viral drug amantadine on the M2 proton channel transmembrane domain. Biophys. J. 2007, 93, 276–283. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (5).(a) Stouffer AL; Acharya R; Salom D; Levine AS; Di Costanzo L; Soto CS; Tereshko V; Nanda V; Stayrook S; DeGrado WF Structural basis for the function and inhibition of an influenza virus proton channel. Nature 2008, 451, 596–599. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Cady SD; Schmidt-Rohr K; Wang J; Soto CS; Degrado WF; Hong M Structure of the amantadine binding site of influenza M2 proton channels in lipid bilayers. Nature 2010, 463, 689–692. [DOI] [PMC free article] [PubMed] [Google Scholar]; (c) Cady SD; Wang J; Wu Y; DeGrado WF; Hong M Specific binding of adamantane drugs and direction of their polar amines in the pore of the influenza M2 transmembrane domain in lipid bilayers and dodecylphosphocholine micelles determined by NMR spectroscopy. J. Am. Chem. Soc. 2011, 133, 4274–4284. [DOI] [PMC free article] [PubMed] [Google Scholar]; (d) Jing X; Ma C; Ohigashi Y; Oliveira FA; Jardetzky TS; Pinto LH; Lamb RA Functional studies indicate amantadine binds to the pore of the influenza A virus M2 proton-selective ion channel. Proc. Natl. Acad. Sci. U. S. A. 2008, 105, 10967–10972. [DOI] [PMC free article] [PubMed] [Google Scholar]; (e) Pielak RM; Oxenoid K; Chou JJ Structural investigation of rimantadine inhibition of the AM2-BM2 chimera channel of influenza viruses. Structure 2011, 19, 1655–1663. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (6).Bright RA; Medina MJ; Xu X; Perez-Oronoz G; Wallis TR; Davis XM; Povinelli L; Cox NJ; Klimov AI Incidence of adamantane resistance among influenza A (H3N2) viruses isolated worldwide from 1994 to 2005: a cause for concern. Lancet 2005, 366, 1175–1181. [DOI] [PubMed] [Google Scholar]
- (7).(a) Bright RA; Shay DK; Shu B; Cox NJ; Klimov AI Adamantane resistance among influenza A viruses isolated early during the 2005–2006 influenza season in the United States. J. Am. Med. Assoc. 2006, 295, 891–894. [DOI] [PubMed] [Google Scholar]; (b) Lan Y; Zhang Y; Dong L; Wang D; Huang W; Xin L; Yang L; Zhao X; Li Z; Wang W; Li X; Xu C; Yang L; Guo J; Wang M; Peng Y; Gao Y; Guo Y; Wen L; Jiang T; Shu Y A comprehensive surveillance of adamantane resistance among human influenza A virus isolated from mainland China between 1956 and 2009. Antiviral Ther. 2010, 15, 853–859. [DOI] [PubMed] [Google Scholar]
- (8).High levels of adamantane resistance among influenza A (H3N2) viruses and interim guidelines for use of antiviral agents--United States, 2005–06 influenza season. High Levels of Adamantane Resistance Among Influenza A (H3N2) Viruses and Interim Guidelines for Use of Antiviral Agents—United States, 2005–06 Influenza Season MMWR Morb. Mortal. Wkly. Rep. 2006, 55, 44–46. [PubMed] [Google Scholar]
- (9).(a) See for example: Kolocouris N; Foscolos GB; Kolocouris A; Marakos P; Pouli N; Fytas G; Ikeda S; De Clercq E Synthesis and antiviral activity evaluation of some aminoadamantane derivatives. J. Med. Chem. 1994, 37, 2896–2902. [DOI] [PubMed] [Google Scholar]; (b) Kolocouris N; Kolocouris A; Foscolos GB; Fytas G; Neyts J; Padalko E; Balzarini J; Snoeck R; Andrei G; De Clercq E Synthesis and antiviral activity evaluation of some new aminoadamantane derivatives. 2. J. Med. Chem. 1996, 39, 3307–3318. [DOI] [PubMed] [Google Scholar]; (c) Kolocouris A; Spearpoint P; Martin SR; Hay AJ; Lopez-Querol M; Sureda FX; Padalko E; Neyts J; De Clercq E Comparisons of the influenza virus A M2 channel binding affinities, anti-influenza virus potencies and NMDA antagonistic activities of 2-alkyl-2-aminoadamantanes and analogues. Bioorg. Med. Chem. Lett. 2008, 18, 6156–6160. [DOI] [PubMed] [Google Scholar]
- (10).(a) Kolocouris A; Tzitzoglaki C; Johnson FB; Zell R; Wright AK; Cross TA; Tietjen I; Fedida D; Busath DD Aminoadamantanes with persistent in vitro efficacy against H1N1 (2009) influenza A. J. Med. Chem. 2014, 57, 4629–4639. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Torres E; Fernández R; Miquet SP; Font-Bardia M; Vanderlinden E; Naesens L; Vázquez S Synthesis and anti-influenza A virus activity of 2,2-dialkylamantadines and related compounds. ACS Med. Chem. Lett. 2012, 3, 1065–1069. [DOI] [PMC free article] [PubMed] [Google Scholar]; (c) Torres E; Duque MD; Vanderlinden E; Ma C; Pinto LH; Camps P; Froeyen M; Vazquez S; Naesens L Role of the viral hemagglutinin in the anti-influenza virus activity of newly synthesized polycyclic amine compounds. Antiviral Res. 2013, 99, 281–291. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (11).Yi M; Cross TA; Zhou H-X A secondary gate as a mechanism for inhibition of the M2 proton channel by amantadine. J. Phys. Chem. B 2008, 112, 7977–7979. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (12).Laohpongspaisan C; Rungrotmongkol T; Intharathep P; Malaisree M; Decha P; Aruksakunwong O; Sompornpisut P; Hannongbua S Why amantadine loses its function in influenza M2 mutants: MD Simulations. J. Chem. Inf. Model. 2009, 49, 847–852. [DOI] [PubMed] [Google Scholar]
- (13).Khurana E; Devane RH; Dal Peraro M; Klein ML Computational study of drug binding to the membrane-bound tetrameric M2 peptide bundle from influenza A virus. Biochim. Biophys. Acta, Biomembr. 2011, 1808, 530–537. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (14).Wang J; Ma C; Fiorin G; Carnevale V; Wang T; Hu F; Lamb RA; Pinto LH; Hong M; Klein ML; DeGrado WF Molecular dynamics simulation directed rational design of inhibitors targeting drug-resistant mutants of influenza A virus M2. J. Am. Chem. Soc. 2011, 133, 12834–12841. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (15).Gu R-X; Liu LA; Wang Y-H; Xu Q; Wei D-Q Structural comparison of the wild-type and drug-resistant mutants of the influenza A M2 proton channel by molecular dynamics simulations. J. Phys. Chem. B 2013, 117, 6042–6051. [DOI] [PubMed] [Google Scholar]
- (16).Alhadeff R; Assa D; Astrahan P; Krugliak M; Arkin IT Computational and experimental analysis of drug binding to the influenza M2 channel. Biochim. Biophys. Acta, Biomembr. 2014, 1838, 1068–1073. [DOI] [PubMed] [Google Scholar]
- (17).(a) Gleed ML; Busath DD Why bound amantadine fails to inhibit proton conductance according to simulations of the drug-resistant influenza A M2 (S31N). J. Phys. Chem. B 2015, 119, 1225–12231. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Gleed ML; Ioannidis H; Kolocouris A; Busath DD Resistance-mutation (N31) effects on drug orientation and channel hydration in amantadine-bound influenza A M2. J. Phys. Chem. B 2015, 119, 11548–11559. [DOI] [PubMed] [Google Scholar]
- (18).Llabres S; Juarez-Jimenez J; Masetti M; Leiva R; Vazquez S; Gazzarrini S; Moroni A; Cavalli A; Luque FJ Mechanism of the pseudoirreversible binding of amantadine to the M2 proton channel. J. Am. Chem. Soc. 2016, 138, 15345–15358. [DOI] [PubMed] [Google Scholar]
- (19).Gianti E; Carnevale V; DeGrado WF; Klein ML; Fiorin G Hydrogen-bonded water molecules in the M2 channel of the influenza A virus guide the binding preferences of ammonium-based inhibitors. J. Phys. Chem. B 2015, 119, 1173–1183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (20).(a) Andreas LB; Barnes AB; Corzilius B; Chou JJ; Miller EA; Caporini M; Rosay M; Griffin RG Dynamic nuclear polarization study of inhibitor binding to the M218–60 proton transporter from influenza A. Biochemistry 2013, 52, 2774–2782. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Andreas LB; Eddy MT; Pielak RM; Chou J; Griffin RG Magic angle spinning NMR investigation of influenza A M2(18–60): support for an allosteric mechanism of inhibition. J. Am. Chem. Soc. 2010, 132, 10958–10960. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (21).Wright AK; Batsomboon P; Dai J; Hung I; Zhou H-X; Dudley GB; Cross TA Differential binding of rimantadine enantiomers to influenza A M2 proton channel. J. Am. Chem. Soc. 2016, 138, 1506–1509. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (22).Rey-Carrizo M; Barniol-Xicota M; Ma C; Frigolé-Vivas M; Torres E; Naesens L; Llabrés S; Juárez-Jiménez J; Luque FJ; DeGrado WF; Lamb RA; Pinto LH; Vázquez S Easily Accessible Polycyclic Amines that Inhibit the Wild-Type and Amantadine-Resistant Mutants of the M2 Channel of Influenza A Virus. J. Med. Chem. 2014, 57, 5738–5747. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (23).(a) Zhao X; Jie Y; Rosenberg MR; Wan J; Zeng S; Cui W; Xiao Y; Li Z; Tu Z; Casarotto MG; Hu W Design and synthesis of pinanamine derivatives as anti-influenza A M2 ion channel inhibitors. Antiviral Res. 2012, 96, 91–99. [DOI] [PubMed] [Google Scholar]; (b) Wang J; Wu Y; Ma C; Fiorin G; Wang J; Pinto LH; Lamb RA; Klein ML; DeGrado WF Structure and inhibition of the drug-resistant S31N mutant of the M2 ion channel of influenza A virus. Proc. Natl. Acad. Sci. U. S. A. 2013, 110, 1315–1320. [DOI] [PMC free article] [PubMed] [Google Scholar]; (c) Wang J; Ma C; Wang J; Jo H; Canturk B; Fiorin G; Pinto LH; Lamb RA; Klein ML; DeGrado WF Discovery of novel dual inhibitors of wild-type and the most prevalent drug-resistant mutant, S31N, of M2 proton channel from influenza A virus. J. Med. Chem. 2013, 56, 2804–2812. [DOI] [PMC free article] [PubMed] [Google Scholar]; (d) Wu Y; Canturk B; Jo H; Ma C; Gianti E; Klein ML; Pinto LH; Lamb RA; Fiorin G; Wang J; DeGrado WF Flipping in the pore: Discovery of dual inhibitors that bind in different orientations to the wild-type versus the amantadine-resistant S31N mutant of the influenza A virus M2 proton channel. J. Am. Chem. Soc. 2014, 136, 17987–17995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (24).Williams JK; Tietze D; Wang J; Wu Y; DeGrado WF; Hong M Drug-induced conformational and dynamical changes of the S31N mutant of the influenza M2 proton channel investigated by solid-state NMR. J. Am. Chem. Soc. 2013, 135, 9885–9897. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (25).Homeyer N; Ioannidis H; Kolarov F; Gauglitz G; Zikos C; Kolocouris A; Gohlke H Interpreting thermodynamic profiles of aminoadamantane compounds inhibiting the M2 proton channel of influenza A by free energy calculations. J. Chem. Inf. Model. 2016, 56, 110–126. [DOI] [PubMed] [Google Scholar]
- (26).Ioannidis H; Drakopoulos A; Tzitzoglaki C; Homeyer N; Kolarov F; Gkeka P; Freudenberger K; Liolios C; Gauglitz G; Cournia Z; Gohlke H; Kolocouris A Alchemical free energy calculations and isothermal titration calorimetry measurements of aminoadamantanes bound to the closed state of influenza A/M2TM. J. Chem. Inf. Model. 2016, 56, 862–876. [DOI] [PubMed] [Google Scholar]
- (27).Gkeka P; Eleftheratos S; Kolocouris A; Cournia Z Free energy calculations reveal the origin of binding preference for aminoadamantane blockers of influenza A/M2TM pore. J. Chem. Theory Comput. 2013, 9, 1272–1281. [DOI] [PubMed] [Google Scholar]
- (28).(a) Wu CH; Ramamoorthy A; Opella SJ High-resolution heteronuclear dipolar solid-state NMR spectroscopy. J. Magn. Reson., Ser. A 1994, 109, 270–272. [Google Scholar]; (b) Ahuja S; Jahr N; Im S-C; Vivekanandan S; Popovych N; Le Clair SV; Huang R; Soong R; Xu J; Yamamoto K; Nanga RP; Bridges A; Waskell L; Ramamoorthy A A model of the membrane-bound cytochrome b5-cytochrome P450 complex from NMR and mutagenesis data. J. Biol. Chem. 2013, 288, 22080–22095. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (29).Nevzorov AA; Opella SJ Selective averaging for high-resolution solid-state NMR spectroscopy of aligned samples. J. Magn. Reson. 2007, 185, 59–70. [DOI] [PubMed] [Google Scholar]
- (30).De Simone A; Mote KR; Veglia G Structural dynamics and conformational equilibria of SERCA regulatory proteins in membranes by solid-state NMR restrained simulations. Biophys. J. 2014, 106, 2566–2576. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (31).Rühmann EH; Rupp M; Betz M; Heine A; Klebe G Boosting affinity by correct ligand preorganization for the S2 pocket of thrombin: a study by isothermal titration calorimetry, molecular dynamics, and high-resolution crystal structures. ChemMedChem 2016, 11, 309–319. [DOI] [PubMed] [Google Scholar]
- (32).Ma C; Polishchuk AL; Ohigashi Y; Stouffer AL; Schon A; Magavern E; Jing X; Lear JD; Freire E; Lamb RA; DeGrado WF; Pinto LH Identification of the functional core of the influenza A virus A/M2 proton-selective ion channel. Proc. Natl. Acad. Sci. U. S. A. 2009, 106, 12283–12288. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (33).Rosenberg MR; Casarotto MG Coexistence of two adamantane binding sites in the influenza A M2 ion channel. Proc. Natl. Acad. Sci. U. S. A. 2010, 107, 13866–13871. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (34).Marassi FM; Opella SJ A solid-state NMR index of helical membrane protein structure and topology. J. Magn. Reson. 2000, 144, 150–155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (35).Wang J; Denny J; Tian C; Kim S; Mo Y; Kovacs F; Song Z; Nishimura K; Gan Z; Fu R; Quine JR; Cross TA Imaging membrane protein helical wheels. J. Magn. Reson. 2000, 144, 162–167. [DOI] [PubMed] [Google Scholar]
- (36).Li C; Qin H; Gao FP; Cross TA Solid-state NMR characterization of conformational plasticity within the transmembrane domain of the influenza A M2 proton channel. Biochim. Biophys. Acta, Biomembr. 2007, 1768, 3162–3170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (37).Andreas LB; Eddy MT; Chou JJ; Griffin RG Magic-angle-spinning NMR of the drug resistant S31N M2 proton transporter from influenza A. J. Am. Chem. Soc. 2012, 134, 7215–7218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (38).Andreas LB; Reese M; Eddy MT; Gelev V; Ni Q-Z; Miller EA; Emsley L; Pintacuda G; Chou JJ; Griffin RG Structure and mechanism of the influenza A M218–60 dimer of dimers. J. Am. Chem. Soc. 2015, 137, 14877–14886. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (39).(a) Cady SD; Luo WB; Hu F; Hong M Structure and function of the influenza M2 proton channel. Biochemistry 2009, 48, 7356–7364. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Cady SD; Hong M Effects of amantadine binding on the dynamics of bilayer-bound influenza A M2 transmembrane peptide studied by NMR relaxation. J. Biomol. NMR 2009, 45, 185–196. [DOI] [PMC free article] [PubMed] [Google Scholar]; (c) Hu F; Luo W; Cady SD; Hong M Conformational plasticity of the influenza A M2 transmembrane helix in lipid bilayers under varying pH, drug binding and membrane thickness. Biochim. Biophys. Acta, Biomembr. 2011, 1808, 415–423. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (40).Cristian L; Lear JD; DeGrado WF Use of thiol-disulfide equilibria to measure the energetics of assembly of transmembrane helices in phospholipid bilayers. Proc. Natl. Acad. Sci. U. S. A. 2003, 100, 14772–14777. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (41).Cady S; Wang T; Hong M Membrane-dependent effects of a cytoplasmic helix on the structure and drug binding of the influenza virus M2 protein. J. Am. Chem. Soc. 2011, 133, 11572–11579. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (42).Saotome K; Duong-Ly K; Howard KP Influenza A M2 protein conformation depends on choice of model membrane. Biopolymers 2015, 104, 405–411. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (43).(a) Chizhmakov IV; Geraghty FM; Ogden DC; Hayhurst A; Antoniou M; Hay AJ Selective proton permeability and pH regulation of the influenza virus M2 channel expressed in mouse erythroleukaemia cells. J. Physiol. 1996, 494, 329–336. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Balannik V; Carnevale V; Fiorin G; Levine BG; Lamb RA; Klein ML; DeGrado WF; Pinto LH Functional studies and modeling of pore-lining residue mutants of the influenza A virus M2 ion channel. Biochemistry 2010, 49, 696–708. [DOI] [PMC free article] [PubMed] [Google Scholar]; (c) Duque MD; Ma C; Torres E; Wang J; Naesens L; Juarez-Jimenez J; Camps P; Luque FJ; DeGrado WF; Lamb RA; Pinto LH; Vazquez S Exploring the size limit of templates for inhibitors of the M2 ion channel of influenza A virus. J. Med. Chem. 2011, 54, 2646–2457. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (44).Schade D; Kotthaus J; Riebling L; Kotthaus J; Müller-Fielitz H; Raasch W; Hoffmann A; Schmidtke M; Clement B Zanamivir Amidoxime- and N-Hydroxyguanidine-Based Prodrug Approaches to Tackle Poor Oral Bioavailability. J. Pharm. Sci. 2015, 104, 3208–3219. [DOI] [PubMed] [Google Scholar]
- (45).Torres E; Duque MD; Vanderlinden E; Ma C; Pinto LH; Camps P; Froeyen M; Vázquez S; Naesens L Role of the viral hemagglutinin in the anti-influenza virus activity of newly synthesized polycyclic amine compounds. Antiviral Res. 2013, 99, 281. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (46).Schmidtke M; Schnittler U; Jahn B; Dahse H-M; Stelzner A A rapid assay for evaluation of antiviral activity against coxsackie virus B3, influenza virus A, and herpes simplex virus type 1. J. Virol. Methods 2001, 95, 133–143. [DOI] [PubMed] [Google Scholar]
- (47).Miao Y; Fu R; Zhou HX; Cross TA Dynamic short hydrogen bonds in histidine tetrad of full-length M2 proton channel reveal tetrameric structural heterogeneity and functional mechanism. Structure 2015, 23, 2300–2308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (48).Yi M; Cross TA; Zhou HX Conformational heterogeneity of the M2 proton channel and a structural model for channel activation. Proc. Natl. Acad. Sci. U. S. A. 2009, 106, 13311–13316. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (49).Kovacs FA; Cross TA Transmembrane four-helix bundle of influenza A M2 protein channel: structural implications from helix tilt and orientation. Biophys. J. 1997, 73, 2511–2517. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (50).Kolocouris A; Zikos C; Broadhurst RW 19F NMR detection of the complex between amantadine and the receptor portion of the influenza A M2 ion channel in DPC micelles. Bioorg. Med. Chem. Lett. 2007, 17, 3947–3952. [DOI] [PubMed] [Google Scholar]
- (51).Hansen RK; Broadhurst RW; Skelton PC; Arkin IT Hydrogen/deuterium exchange of hydrophobic peptides in model membranes by electrospray ionization mass spectrometry. J. Am. Soc. Mass Spectrom. 2002, 13, 1376–87. [DOI] [PubMed] [Google Scholar]
- (52).Ladbury JE; Klebe G; Freire E Adding calorimetric data to decision making in lead discovery: a hot tip. Nat. Rev. Drug Discovery 2010, 9, 23–27. [DOI] [PubMed] [Google Scholar]
- (53).Perozzo R; Folkers G; Scapozza L Thermodynamics of protein-ligand interactions: history, presence, and future aspects. J. Recept. Signal Transduction Res. 2004, 24, 1–52. [DOI] [PubMed] [Google Scholar]
- (54).(a) Salom D; Hill BR; Lear JD; DeGrado WF pH-dependent tetramerization and amantadine binding of the transmembrane helix of M2 from the influenza A virus. Biochemistry 2000, 39, 14160–14170. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Stouffer AL; Ma C; Cristian L; Ohigashi Y; Lamb RA; Lear JD; Pinto LH; DeGrado WF The interplay of functional tuning, drug resistance, and thermodynamic stability in the evolution of the M2 proton channel from the influenza A virus. Structure 2008, 16, 1067–1076. [DOI] [PMC free article] [PubMed] [Google Scholar]; (c) Stouffer AL; Nanda V; Lear JD; DeGrado WF Sequence determinants of a transmembrane proton channel: an inverse relationship between stability and function. J. Mol. Biol. 2005, 347, 169–179. [DOI] [PubMed] [Google Scholar]
- (55).Wiseman T; Williston S; Brandts JF; Lin L-N Rapid measurement of binding contstants and heats of binding using a new titration calorimeter. Anal. Biochem. 1989, 179, 131–137. [DOI] [PubMed] [Google Scholar]
- (56).Doyle ML Characterization of binding interactions by isothermal titration calorimetry. Curr. Opin. Biotechnol. 1997, 8, 31–35. [DOI] [PubMed] [Google Scholar]
- (57).Origin 8.1G SR1, v8.1.13.88; OriginLab, Northampton, MA, USA, 2009. [Google Scholar]
- (58).Fung BM; Khitrin AK; Ermolaev K An improved broadband decoupling sequence for liquid crystals and solids. J. Magn. Reson. 2000, 142, 97–101. [DOI] [PubMed] [Google Scholar]
- (59).Delaglio F; Grzesiek S; Vuister GW; Zhu G; Pfeifer J; Bax A NMRPipe: a multidimensional spectral processing system based on UNIX pipes. J. Biomol. NMR 1995, 6, 277–293. [DOI] [PubMed] [Google Scholar]
- (60).Gor'kov PL; Chekmenev EY; Li C; Cotten M; Buffy JJ; Traaseth NJ; Veglia G; Brey WW Using low-E resonators to reduce RF heating in biological samples for static solid-state NMR up to 900 MHz. J. Magn. Reson. 2007, 185, 77–93. [DOI] [PubMed] [Google Scholar]
- (61).McNeill SA; Gor'kov PL; Shetty K; Brey WW; Long JR A low-E magic angle spinning probe for biological solid state NMR at 750 MHz. J. Magn. Reson. 2009, 197, 135–144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (62).Morcombe CR; Zilm KW Chemical shift referencing in MAS solid state NMR. J. Magn. Reson. 2003, 162, 479–486. [DOI] [PubMed] [Google Scholar]
- (63).Markley JL; Bax A; Arata Y; Hilbers CW; Kaptein R; Sykes BD; Wright PE; Wüthrich K Recommendations for the presentation of NMR structures of proteins and nucleic acids. J. Mol. Biol. 1998, 280, 933–952. [DOI] [PubMed] [Google Scholar]
- (64).Harris RK; Becker ED; Cabral de Menezes SM; Goodfellow R; Granger P NMR nomenclature: nuclear spin properties and conventions for chemical shifts. IUPAC recommendations 2001. Solid State Nucl. Magn. Reson. 2002, 22, 458–483. [DOI] [PubMed] [Google Scholar]
- (65).Maestro, version 8.5; Schrodinger: New York, NY, USA, 2008. [Google Scholar]
- (66).(a) Halgren TA Merck molecular force field. V. Extension of MMFF94 using experimental data, additional computational data, and empirical rules. J. Comput. Chem. 1996, 17, 616–641. [Google Scholar]; (b) Halgren TA MMFF VII. Characterization of MMFF94, MMFF94s, and other widely available force fields for conformational energies and for intermolecular-interaction energies and geometries. J. Comput. Chem. 1999, 20, 730–748. [DOI] [PubMed] [Google Scholar]
- (67).Jones G; Willett P; Glen RC; Leach AR; Taylor R Development and validation of a genetic algorithm for flexible docking. J. Mol. Biol. 1997, 267, 727–748. [DOI] [PubMed] [Google Scholar]
- (68).Verdonk ML; Chessari G; Cole JC; Hartshorn MJ; Murray CW; Nissink JW; Taylor RD; Taylor R Modeling water molecules in protein-ligand docking using GOLD. J. Med. Chem. 2005, 48, 6504–6515. [DOI] [PubMed] [Google Scholar]; (c) Korb O; Stützle T; Exner TE Empirical scoring functions for advanced protein-ligand docking with PLANTS. J. Chem. Inf. Model. 2009, 49, 84–96. [DOI] [PubMed] [Google Scholar]
- (69).Mooij WT; Verdonk ML General and targeted statistical potentials for protein-ligand interactions. Proteins: Struct., Funct., Genet. 2005, 61, 272–287. [DOI] [PubMed] [Google Scholar]
- (70).Korb O; Stützle T; Exner TE Empirical scoring functions for advanced protein-ligand docking with PLANTS. J. Chem. Inf. Model. 2009, 49, 84–96. [DOI] [PubMed] [Google Scholar]
- (71).Pettersen EF; Goddard TD; Huang CC; Couch GS; Greenblatt DM; Meng EC; Ferrin TE UCSF Chimera–a visualization system for exploratory research and analysis. J. Comput. Chem. 2004, 25, 1605–1612. [DOI] [PubMed] [Google Scholar]
- (72).Pielak RM; Schnell JR; Chou JJ Mechanism of drug inhibition and drug resistance of influenza A M2 channel. Proc. Natl. Acad. Sci. U. S. A. 2009, 106, 7379–7384. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (73).Thomaston JL; DeGrado WF Crystal structure of the drug-resistant S31N influenza M2 proton channel. Protein Sci. 2016, 25, 1551–1554. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (74).Jorgensen WL; Chandrasekhar J; Madura JD; Impey RW; Klein ML Comparison of simple potential functions for simulating liquid water. J. Chem. Phys. 1983, 79, 926–935. [Google Scholar]
- (75).(a) Desmond Molecular Dynamics System, version 3.0; D. E. Shaw Research: New York, NY, USA, 2011. [Google Scholar]; (b) Desmond Molecular Dynamics System, version 3.1; D. E. Shaw Research: New York, NY, USA, 2012. [Google Scholar]; (c) Bowers KJ; Chow E; Xu H; Dror RO; Eastwood MP; Gregersen BA; Klepeis JL; Kolossvary I; Moraes MA; Sacerdoti FD; Salmon JK; Shan Y; Shaw DE Scalable algorithms for molecular mynamics simulations on commodity clusters. In Proceedings of the 2006 ACM/IEEE Conference on Supercomputing (SC’06), article 84, Tampa, FL, USA; Association for Computing Machinery: New York, NY, USA, 2006; DOI: 10.1145/1188455.1188544 [DOI] [Google Scholar]
- (76).Maestro-Desmond Interoperability Tools, version 3.1; Schrodinger: New York, NY, USA, 2012. [Google Scholar]
- (77).(a) Jorgensen WL; Maxwell DS; Tirado-Rives J Development and testing of the OPLS all-atom force field on conformational energetics and properties of organic liquids. J. Am. Chem. Soc. 1996, 118, 11225–11236. [Google Scholar]; (b) Rizzo RC; Jorgensen WL OPLS all-atom model for amines: resolution of the amine hydration problem. J. Am. Chem. Soc. 1999, 121, 4827–4836. [Google Scholar]
- (78).Kaminski G; Friesner RA; Tirado-Rives J; Jorgensen WL Evaluation and reparametrization of the OPLS-AA force field for proteins via comparison with accurate quantum chemical calculations on peptides. J. Phys. Chem. B 2001, 105, 6474–6487. [Google Scholar]
- (79).Shivakumar D; Williams J; Wu Y; Damm W; Shelley J; Sherman W Prediction of absolute solvation free energies using molecular dynamics free energy perturbation and the OPLS force field. J. Chem. Theory Comput. 2010, 6, 1509–1519. [DOI] [PubMed] [Google Scholar]
- (80).Darden T; York D; Pedersen L Particle mesh Ewald: an N log(N) method for Ewald sums in large systems. J. Chem. Phys. 1993, 98, 10089–10092. [Google Scholar]
- (81).Essmann U; Perera L; Berkowitz ML; Darden T; Lee H; Pedersen LG A Smooth particle mesh Ewald method. J. Chem. Phys. 1995, 103, 8577–8593. [Google Scholar]
- (82).Martyna GJT; Tobias DJ; Klein ML Constant pressure molecular-dynamics algorithms. J. Chem. Phys. 1994, 101, 4177–4189. [Google Scholar]
- (83).Humphreys DD; Friesner RA; Berne BJ A multiple-time-step molecular-dynamics algorithm for macromolecules. J. Phys. Chem. 1994, 98, 6885–6892. [Google Scholar]
- (84).Koynova R; Caffrey M Phases and phase transitions of the phosphatidylcholines. Biochim. Biophys. Acta, Rev. Biomembr. 1998, 1376, 91–145. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.