

Abstract
Purpose of review
Metabolic diseases, including dyslipidemia, diabetes mellitus, and chronic inflammation are risk factors for clinically significant thrombotic events. Thrombosis in these settings is multifaceted with coordinated mechanisms between platelet activation and the hemostatic pathways. This review focuses on recent advances in platelet procoagulant and apoptotic signaling with emphasis on the pathophysiologic mechanisms induced by platelet CD36 in dyslipidemia, and the key unaddressed questions relating to the field.
Recent findings
CD36 promotes platelet activation and increases the risk for thrombosis through signaling events. These include generation of reactive oxygen species, activation of redox-sensitive MAP kinase ERK5, and promotion of a pro-thrombotic phenotype. CD36 promotes phosphatidylserine externalization leading to a procoagulant function downstream from MAP kinase ERK5 that is separate from a pro-aggregatory function. Phosphatidylserine externalization requires maladaptive caspase activation, promotes assembly of the factor tenase and prothrombinase complex, and promotes fibrin formation. It is distinct from the canonical pathways mediating platelet procoagulant function by strong physiologic stimuli or by the platelet apoptotic-like Bak/Bax-mediated pathway for cellular clearance.
Summary
Understanding CD36 signaling in the context of dyslipidemia, or other metabolic diseases will identify important and novel signaling hubs that could be potential therapeutic targets for intervention without impacting hemostasis.
Keywords: apoptotic signaling, Cluster of Differentiation 36, dyslipidemia, ERK5, procoagulant, thrombosis
INTRODUCTION
Clinically relevant thrombotic events remain the leading cause of disability and mortality in the United States [1]. These include arterial thrombotic diseases, such as myocardial infarction and stroke, and venous thromboembolic diseases, such as deep vein thrombosis and pulmonary embolism. The risks for these events are context-dependent but include metabolic disorders, such as dyslipidemia [2■■,3■■,4,5], chronic inflammation [6], obesity [7], and diabetes mellitus [8]. Although current therapeutic regimens are effective at preventing platelet activation, these treatments are limited by increased risk for bleeding complications and incomplete efficacy in preventing recurrent thrombosis [9].
In this review, we address physiologic events that promote loss of membrane asymmetry (e.g. exposure of phosphatidylserine from the inner leaflet of the membrane to the outer leaflet of the membrane) in platelets, a process intricately linked to platelet procoagulant function in a growing thrombus. Furthermore, we also highlight advances in platelet membrane asymmetry as they relate to platelet survival. We then focus the review on the pathophysiologic signaling mechanisms leading to loss of membrane asymmetry in platelets during dyslipidemia and the association of this phenotype with scavenger receptor Cluster of Differentiation 36 (CD36).
PLATELET ACTIVATION PROMOTES THROMBOSIS
Hemostasis and thrombosis are multifaceted processes that have been excellently reviewed elsewhere (see reference [10]). In brief, the steps of thrombosis at the sites of vascular injury include: platelet adhesion onto the exposed subendothelial surface following vessel injury; platelet aggregation induced by activated integrin αIIbβ3 present on the cell surface; and fibrin clot formation, which is dependent on the activity of thrombin to cleave soluble fibrinogen to fibrin. In the context of a growing thrombus, physiologic agonists promote the pro-aggregatory functions of platelets. Stalker et al. [11] reviewed the detailed mechanisms of platelet activation by classic agonists, all of which contribute to this function by calcium flux, dense and alpha granule secretion and integrin activation. However, the functions of platelets are not limited to a pro-aggregatory state and are also linked to coagulation through secondary hemostasis.
PLATELETS ARE MEDIATORS OF SECONDARY HEMOSTASIS THROUGH PROCOAGULANT FUNCTIONS
The signaling events by which platelets are linked to hemostasis are an active area of research. Activation of platelets by strong physiologic agonists induces generation of a distinct subpopulation of platelets with dysregulated membrane asymmetry [12,13]. These so-called procoagulant platelets, coated platelets, or ‘ballooned’ platelets are a link between primary and secondary hemostasis [12]. The exact nature and definition of a procoagulant platelet are not fully understood as indicated by a variety of nomenclature to define its characteristic properties (reviewed in Hua and Chen [12]). The undisputed characteristic property of the procoagulant platelet subpopulation is externalization of anionic phosphatidylserine and subsequent capacity to support thrombin generation. Loss of membrane asymmetry, that is, the exposure of phosphatidylserine, is mediated by coordinated activity among scramblases, flippases, and floppases to promote translocation of anionic phosphatidylserine lipids residing predominantly in the inner leaflet of the cellular membrane to the outer leaflet [14]. In the procoagulant pathway, it was proposed that TMEM16F (also called Anoctamin 6 [15]) is the major scramblase to mediate these changes in membrane asymmetry [16]. Phosphatidylserine externalization presents a negatively charged surface conducive for the assembly of tenase and prothrombinase complexes to generate factor Xa and thrombin, respectively, leading to fibrin formation [14].
Strong physiologic stimuli, including thrombin and collagen, PAR and Glycoprotein VI (GPVI) receptor agonists, respectively, are the best studied activators of these pathways leading to procoagulant phosphatidylserine externalization. Strong stimuli are necessary to induce calcium flux, loss of mitochondrial membrane potential, and cyclophilin D-mediated sensitization of the mitochondrial permeability transition pore (mPTP) [17,18]. These events are also thought to be associated with inability of integrins to activate because of calcium-mediated, calpain-dependent cleavage of the domains for integrin inside-out signaling [19]. Furthermore, elegant single cell microscopy studies show that strong stimuli induce ‘ballooning’ of the membrane with phosphatidylserine detection highly localized to the sites of membrane disfiguration [20]. In some cases, strong physiologic stimuli promote generation of subpopulation of necrotic-like platelets that share procoagulant properties with coated platelets [21,22]. Necrotic-like platelets undergo depletion of ATP and calcium flux to levels cytotoxic to the cells. These events are followed by a rise in mitochondrial reactive oxygen species generation and a loss of plasma membrane integrity [22]. Although sharing the characteristic property of phosphatidylserine externalization, the surface of coated platelets is highly enriched with alpha-granular proteins and show high fibrinogen binding to activated integrins [23]. Phenotypic and functional differences between these subpopulations of procoagulant platelets were previously described (reviewed by Jackson and Schoenwaelder in [22]).
Dynamic studies of procoagulant platelets in a growing thrombus show a distinct spatial distribution. Nechipurenko et al. [24■] reported that the procoagulant platelet is not static during a thrombus growth. Using in silico modeling, intravital and confocal microscopy, and electron micro-imaging, they showed convincing evidence that procoagulant platelets are spatially distributed towards the exterior of a thrombus. This mechanism is mediated by nonmuscle myosin heavy chain IIa, as targeted genetic deletion prevented the movement of procoagulant platelets towards the exterior of the thrombus. Kholmukhamedov and Jobe [25] suggested this ‘squeezing’ of procoagulant platelet towards the thrombus shell is a mechanism to limit thrombus growth. This is based on the assumption that procoagulant platelets induced by strong physiologic stimuli lose sensitivity to JON/A or PAC1 binding, which are antibodies specific to the activated form of integrin αIIbβ3 [26,27]. The mechanisms of clot formation induced by phosphatidylserine exposure in procoagulant platelets could be context-dependent.
Although platelet procoagulant function requires calcium flux and sensitization of the mPTP by cyclophilin D [17], the signaling events initiating this pathway are incompletely defined and require further investigation. Disruption of inner mitochondrial membrane potential required for phosphatidylserine externalization is related to mitochondrial dysfunction [19]. With relevance to mPTP activation, it was shown that the calcium uniporter was present and functional in platelet mitochondria to regulate procoagulant function [28■]. Genetic deletion of this calcium uniporter limited calcium entry into the mitochondria and desensitized mPTP formation. This prevented phosphatidylserine externalization and the subsequent procoagulant phenotype. Agbani and co-workers showed that the Aquaporin 1 was also functionally present in the open canalicular system of platelets and regulates phosphatidylserine externalization [29,30■]. They showed that mice that do not express platelet Aquaporin 1 did not show defects in platelet adhesion, granule secretion, and spreading, but had diminished platelet procoagulant function and decreased thrombosis in the FeCl3-induced arterial thrombosis model.
Further characterization of the signaling events that contribute to procoagulant phosphatidylserine externalization are warranted in order to fully appreciate its multifaceted phenotype in relationship to the membrane asymmetry events associated with apoptotic signaling.
APOPTOTIC-LIKE CELL DEATH IMPACTS PLATELET SURVIVAL
It can be argued that platelets do not undergo classic apoptosis as they are anucleate and they do not have the capacity to go through a classic ‘programmed’ cell death pathway involving nuclear membrane disintegration. Nonetheless, the apoptotic machinery is present in platelets and functional with mechanisms distinct from procoagulant or pro-aggregatory properties.
Similar to its procoagulant counterpart, apoptotic-like platelets promote phosphatidylserine externalization (presumably through the scramblase Xkr8 [31]) as shown in the seminal studies by Mason et al. [32]. They showed that genetic haploinsufficiency of antiapoptotic BcL-XL or the use of a BH3-mimetic compound ABT-737 promoted platelet cell death as indicated by an increase in caspase activation, phosphatidylserine externalization, and platelet clearance. However, apoptotic platelets do not have the capacity to promote thrombin activity. Schoenwaelder et al. showed convincingly that platelets with genetic deletion of Bak and Bax neither supported fibrin formation capacity nor impacted agonist-mediated or BH3-mimetic ABT-737-induced phosphatidylserine externalization [33]. Thus, phosphatidylserine externalization mediated through BcL-XL mediators Bak/Bax is an indicator of platelet life span and promote cellular clearance. As such, phosphatidylserine externalization by ‘aged’ platelets by Bak/Bax decreases the half-life of circulating platelets.
Although Bak and Bax are mediators of phosphatidylserine externalization, additional signaling effectors control the fate of platelet cell death and survival. Protein kinase A (PKA) was shown by Zhao et al. [34■] to regulate the initiation of the apoptosome. They found that PKA activity determined the fate of platelet survival or apoptosis through a complex balance between cAMP availability and phosphodiesterase activity. Thrombin signaling decreased cAMP levels and the activity of PKA dependency on phosphodiesterases. The decrease in cAMP levels promoted interaction of BAD with the antiapoptotic regulator BCL-XL through dephosphorylation of BAD at S155 and the release of 14–3–3, which promoted cell death leading to a decrease in platelet lifespan and survival.
This study further suggested that apoptotic-like signaling by BAK/BAX-mediating platelet clearance is physiologically distinct from procoagulant signaling by the cyclophilin D/mPTP pathway. Detailed understanding of platelet apoptosis and its relevance to the procoagulant phenotype in the setting of hemostasis are yet to be determined. Furthermore, determining the differences between apoptotic clearance of platelets and that of the clearance mechanisms by desialylation and the Ashwell–Morrell receptors [35] are important to determine therapeutic targets that promote or inhibit the platelet storage defect, half-life, thrombocytosis and cytopenic conditions, all of which are context-dependent.
PLATELET CD36 PROMOTES ARTERIAL THROMBOSIS IN DYSLIPIDEMIA
Our recent work describing the prothrombotic function of platelet scavenger receptor CD36 during dyslipidemia surprisingly revealed that pathways generating procoagulant and apoptotic platelets were linked. Platelet CD36 is a pattern recognition receptor of 88 kDa that is variably expressed on the membrane surface with an average copy number of 17000–20000 [36]. It recognizes a diverse array of danger-associated molecular patterns (DAMP) [37■], which are endogenous or exogenously derived structures that are present in conditions of tissue injury, infection, inflammation, oxidant stress and metabolic dysfunction. Of its ligands, the most studied in the platelet system are thrombospondin 1, oxidized lipids in oxidized low-density lipoprotein particles [4,38], myeloid-related protein 14, a member of the S100 family of calcium binding proteins, [39], advanced glycated end products [8], and cell-derived microparticles [6]. The signaling mechanisms of platelet CD36 downstream of these ligands share characteristic effectors.
We and others have characterized the platelet CD36 signaling pathway ever since the seminal publication by Podrez et al. [4] showing that CD36-linked hyperlipidemia and oxidative stress to a prothrombotic phenotype. The first reported evidence of platelet CD36 signal transduction was from studies showing Src family kinases Fyn, Lyn, and Yes were co-precipitated by an antibody to the receptor [40]. Since these publications, we and others found that there are multiple coordinated signaling events that induce platelet activation or blunt platelet inhibitory pathways when CD36 recognizes its ligands. In particular, CD36 promotes activation of Src family kinases, predominantly Fyn and Lyn [41], nonreceptor tyrosine kinase Syk [42,43], Vav-guanine nucleotide exchange factors [44], phospholipase c gamma 2 leading to activation of protein kinase C and NADPH oxidase [5,43], and activation of MAP kinase JNK2 [41] and ERK5 [3■■]. We also showed that ERK5 mediated caspase activation [2■■], and increased expression of the GTPase Rac [37■]. Platelet CD36 was also shown to put the ‘brakes’ on platelet inhibitory signaling including that of the nitric oxide/cGMP/PKG pathway, thus promoting platelet activation [43,45].
Recently, we found that the activation of ERK5 by specific reactive oxygen species could be a signaling hub for platelet activation by CD36 (modeled in Fig. 1) [3■■]. Specific reactive oxygen species, particularly superoxide radical anion and hydrogen peroxide, accumulated when platelets were incubated with the CD36 model ligand oxidized lipids. These species were detected using high performance liquid chromatography with fluorescence detection. The fluorescent probe hydroethidium was used to detect the superoxide-radical anion-specific fluorescent oxidation product, 2-hydroxyethidine. An analytical approach was used as hydroethidium undergoes various nonspecific oxidation products [46]. We also used a boronate-based fluorescent probe, coumarin boronic acid, to detect the peroxide-specific oxidation product 7-hydroxycoumarin by oxidized lipid signaling. We and others confirmed that the pharamacologic inhibition of NADPH oxidase with the small molecule inhibitor VAS2870, or with the inhibitory peptide gp91ds-tat prevented the generation of these species and activation of platelets by oxidized lipids [3■■,43]. These studies show selectivity towards the redox-sensitive MAP kinase ERK5 to promote platelet activation, adhesion on immobilized collagen under arterial shear flow, and platelet aggregation.
FIGURE 1.
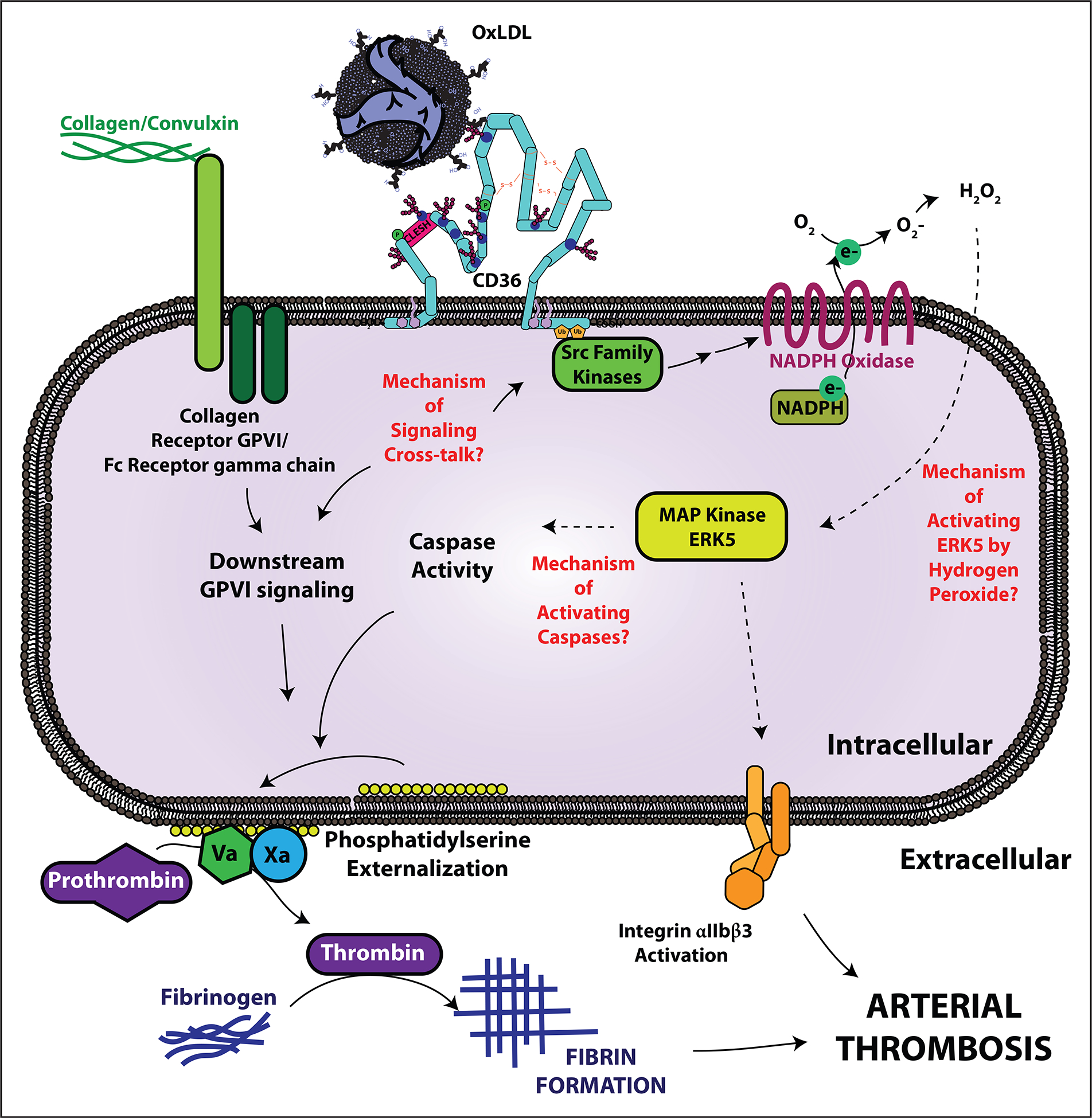
Knowledge gaps in CD36 and ERK5-mediated platelet prothrombotic signaling. Circulating oxidized lipids are recognized by platelet scavenger receptor CD36. Src family kinases, particularly Fyn and Lyn, associate with CD36 upon ligand recognition and promote downstream signaling events, including generation of reactive oxygen species superoxide anion and hydrogen peroxide from NADPH oxidase. Hydrogen peroxide in turn activates the redox-sensitive MAP kinase ERK5 through a yet to be defined mechanism. ERK5 links CD36 to a prothrombotic platelet phenotype by activating integrin αIIbβ3 for platelet aggregation and promoting caspase-dependent procoagulant phosphatidylserine externalization. The mechanisms of ERK5-mediated caspase activation are unclear. Furthermore, CD36 participates in signaling crosstalk with the collagen receptor GPVI to amplify phosphatidylserine externalization. The signaling crosstalk mechanisms are not clear and may involve specific family members of Src kinases. CD36, CD36.
The in-vivo relevance of MAP kinase ERK5 activation by the CD36 pathway was studied by generating chimeric apoe null mice that either expressed or did not have platelet ERK5 in bone marrow platelet precursors [3■■]. In the ferric chloride-induced carotid artery thrombosis model, the chimeric mice expressing platelet ERK5 showed reduced time to occlusion of the vessel after high-fat dieting, indicating a pro-thrombotic state, compared with control animals. However, high-fat diet-fed chimeric mice that did not express platelet ERK5 normalized the decreased occlusion time to that observed in control diet conditions, suggesting that ERK5 was necessary to accelerate arterial thrombosis in high-fat diet conditions (Fig. 2a). Importantly, these studies were replicated using an arterial thrombosis model that does not rely on oxidants generated, for example, by transition metal-mediated Fenton chemistry in the ferric chloride model [47–49] or singlet oxygen by the photo-oxidizable Rose Bengal model [50]. In this model, a segment of the epigastric artery is removed and then inserted into the lumen of the carotid artery [51], presenting the collagen-rich adventitia to flowing blood. This model was shown to be sensitive to diet-induced arterial thrombosis as the chimeric ERK5-expressing mice showed significantly enhanced thrombosis with embolism that was not observed with control diet. Like the ferric chloride model, specific platelet ERK5 deletion prevented enhanced arterial thrombosis in the high-fat diet condition. These findings show the significance of oxidative stress in platelet redox-sensitive signaling as related to dyslipidemic conditions.
FIGURE 2.
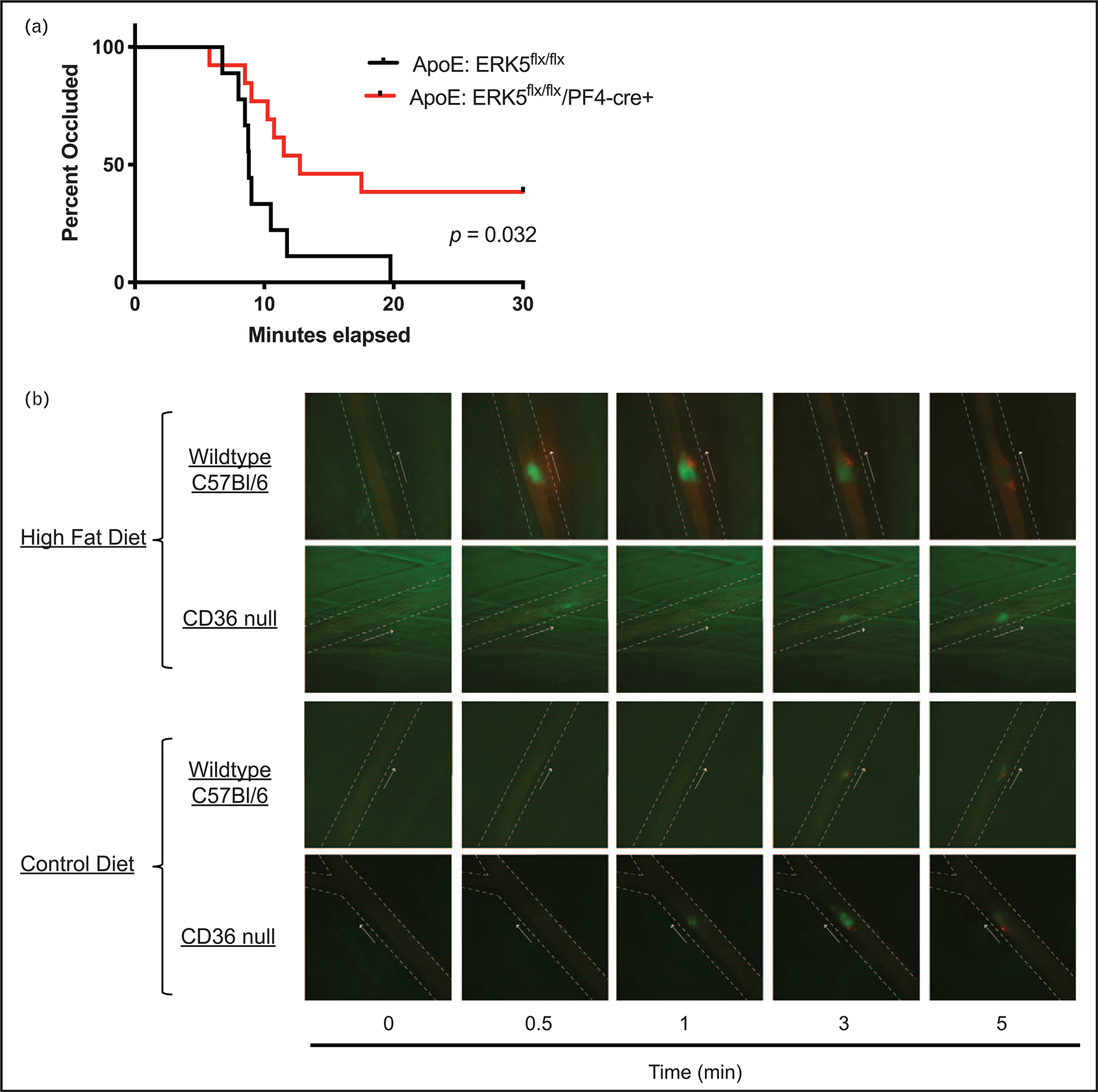
CD36 and ERK5 promote arterial thrombosis in dyslipidemic conditions. (a) apoE null mice were lethally irradiated followed by transplantation of bone marrows from ERK5 expressing or platelet ERK5 deleted mice. The chimeric mice were allowed to recover from bone marrow transplantation before feeding control or high-fat diet for at least 6 weeks. Kaplan–Meier analyses of the ferric chloride-induced in-vivo thrombosis on the carotid artery with time to occlusion as the censored analysis. (b) Wild type C57Bl/6 or CD36 null mice at 8 weeks of age were fed control or high-fat diet for at least 6 weeks before performing laser-induced in-vivo arterial thrombosis on the cremasteric artery. Representative video microscopy images of platelet accumulation (in green) and fibrin accumulation (in red) in real time up to 5 min are shown. The arrows indicate direction of blood flow and the arrow on the side indicates the side of vessel injury. CD36, CD36.
PLATELET CD36 SIGNALING THROUGH ERK5 MEDIATES MALADAPTIVE CASPASE ACTIVATION AND A PROCOAGUALNT PHENOTYPE
In a surprising finding, we found that CD36 and ERK5 promote maladaptive caspase activity and procoagulant phosphatidylserine externalization through crosstalk with the collagen receptor GPVI pathway [2■■,37■]. This signaling mechanism is dependent on Src family kinases, generation of hydrogen peroxide, and intracellular calcium; pharmacologic interruption of any of these molecules, which includes inhibiting CD36, ERK5, and caspases, prevented oxLDL-sensitization of phosphatidylserine externalization by the GPVI receptor agonist convulxin. Additionally, these studies were confirmed using platelets from murine models with genetic deficiency in CD36, platelet ERK5, or Bak/Bax. This crosstalk substantially magnifies procoagulant phosphatidylserine externalization by activation of GPVI or CD36 alone.
We also showed using a sensitive spectrophotometric method developed by Campbell et al. [52] that CD36-ERK5-caspase signaling cooperates with the GPVI pathway to promote fibrin formation ex vivo. In these studies, phosphatidylserine externalization mediated by oxLDL alone was not enough to support fibrin formation. However, when platelets were pretreated with oxLDL before stimulating with GPVI agonist convulxin, the onset time for fibrin formation was significantly reduced compared with convulxin alone. These studies suggest that there could be a threshold for phosphatidylserine externalization required to support fibrin formation. Preventing CD36, ERK5, and caspase activation inhibits oxLDL-sensitized fibrin formation.
Using the transplantation model of thrombosis described above as well as the laser-injury model, we found that high-fat diet significantly augmented fibrin formation in vivo. Animals with absence of CD36 or platelet ERK5 did not show enhanced fibrin accumulation in the hyperlipidemic environment. In fact, in the laser injury model, fibrin formation by the CD36 pathway supported sustained thrombi growth whereas in control diet conditions or in the absence of CD36 thrombi resolved over time (Fig. 2b). These studies are the first to link the apoptotic phosphatidylserine externalization mechanism to platelet procoagulant function.
CONCLUSION
Although the signaling events leading to phosphatidylserine externalization to promote classic procoagulant activity and apoptotic platelet clearance are an active area of research, the relevance of these two pathways under metabolic disease conditions are not well understood. Unlike the apoptotic-like pathway important for platelet survival or the procoagulant pathway mediated by cyclophilin D-dependent sensitization of mPTP, our studies support the hypothesis that in dyslipidemia, CD36 signaling through ERK5 links apoptotic-like phosphatidylserine externalization to a procoagulant phenotype to support pathophysiologic arterial thrombosis. These studies suggest that conditions associated with chronic inflammation and oxidative stress integrate with known classic agonist pathways to induce aberrant signaling to increase the risk for clinically significant thrombosis.
Key knowledge gaps still remain to be addressed. In particular, it is not clear how ERK5 promotes maladaptive caspase activation as its functions are typically thought to be related to pro-survival and pro-proliferative mechanisms. Furthermore, it is plausible that CD36 and ERK5 coordinate activation of specific caspases to promote the procoagulant phenotype. We also believe that the crosstalk between CD36 and GPVI should be investigated especially as GPVI is critical for platelet adhesion to collagen. Cross talk could involve selective activation of Src family members in both pathways, which could not be directly investigated in our studies because of a lack of specific inhibitors. In the context of dyslipidemia, it could be important to determine the inter-relationship between platelet phosphatidylserine externalization by CD36 with platelet lifespan, particularly, because of the intricate link between dyslipidemia and increased platelet production, which is a risk factor for thrombosis [53].
A major technical challenge in the field is imaging phosphatidylserine externalization events without inhibiting its procoagulant function. Advances in chemical tools to detect phosphatidylserine externalization without impacting the function of the prothrombinase complex could reveal the relevance of membrane asymmetry in a spatial-temporal manner in vivo. These tools could involve synthesis of small molecule fluorescent probes that could selectively ‘tag’ phosphatidylserine. In fact, some studies attempted to address this knowledge gap by studying the function of cyclic peptides related to lactadherin [54] or by using synthetic scramblases to promote membrane asymmetry [55]. These studies, however, require robust measures to show that factor tenase and prothrombinase could assemble and generate thrombin both in vitro and in vivo.
It is expected that understanding CD36 signaling in the context of dyslipidemia, or other metabolic diseases will identify important and novel signaling hubs that could be potential therapeutic targets for intervention without impacting hemostasis.
KEY POINTS.
Platelets promote secondary hemostasis through cyclophilin D-sensitized mPTP formation, which increases procoagulant activity through phosphatidylserine externalization.
Phosphatidylserine externalization mediated through BcL-XL mediators Bak/Bax is an indicator of platelet life span, promote cellular clearance, and has not been associated with platelet procoagulant function.
Platelet CD36 promotes arterial thrombosis in dyslipidemic conditions by activating redox-sensitive MAP kinase ERK5.
ERK5 is a signaling hub for CD36 to initiate caspase-dependent phosphatidylserine externalization and subsequent fibrin formation, which is the first evidence linking apoptotic signaling to procoagulant function.
Understanding the mechanisms of CD36 and ERK5-mediated prothrombotic events could identify potential targets for therapeutic intervention in metabolic disorders associated with risks for arterial thrombosis.
Acknowledgements
This work was supported by NIH NHLBI grants R01 HL111614 (R.L.S.), T32 HL134643 (M.Y.), R01 HL111614-S (M.Y.), and the Medical College of Wisconsin Cardiovascular Center’s A. O. Smith Fellowship Scholars Program (M.Y.).
Footnotes
Financial support and sponsorship
None.
Conflicts of interest
There are no conflicts of interest.
REFERENCES AND RECOMMENDED READING
Papers of particular interest, published within the annual period of review, have been highlighted as:
■ of special interest
■■ of outstanding interest
- 1.Wendelboe AM, Raskob GE. Global burden of thrombosis: epidemiologic aspects. Circ Res 2016; 118:1340–1347. [DOI] [PubMed] [Google Scholar]
- 2. Yang M, Kholmukhamedov A, Schulte ML, et al. Platelet CD36 signaling through ERK5 promotes caspase-dependent procoagulant activity and fibrin deposition in vivo. Blood Adv 2018; 2:2848–2861. ■■ In this article, CD36 recognition of oxidized lipids promote a signaling pathway leading to MAP kinase ERK5 activation. ERK5 is a signaling hub for CD36 to link the physiologic procoagulant and apoptotic-like mechanisms of phosphatidylserine externalization to fibrin formation in dyslipidemia. This is the first report of a procoagulant function through activated caspases and is mediated by crosstalk between the CD36 signaling axis and that of the collagen receptor GPVI pathway.
- 3. Yang M, Cooley BC, Li W, et al. Platelet CD36 promotes thrombosis by activating redox sensor ERK5 in hyperlipidemic conditions. Blood 2017; 129:2917–2927. ■■ The authors in this article used an analytical approach to measure specific reactive oxygen species generated in the platelet system by CD36. These reactive oxygen species then activate redox-sensitive MAP kinase ERK5. ERK5 activation promotes platelet activation, adhesion, and aggregation in the setting of arterial shear flow. ERK5 also promotes thrombosis in dyslipidemic conditions.
- 4.Podrez EA, Byzova TV, Febbraio M, et al. Platelet CD36 links hyperlipidemia, oxidant stress and a prothrombotic phenotype. Nat Med 2007; 13:1086–1095. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Berger M, Wraith K, Woodward C, et al. Dyslipidemia-associated atherogenic oxidized lipids induce platelet hyperactivity through phospholipase Cgam-ma2-dependent reactive oxygen species generation. Platelets 2019; 30:467–472. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Ghosh A, Li W, Febbraio M, et al. Platelet CD36 mediates interactions with endothelial cell-derived microparticles and contributes to thrombosis in mice. J Clin Invest 2008; 118:1934–1943. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Coutinho T, Goel K, Correa de Sa D, et al. Central obesity and survival in subjects with coronary artery disease: a systematic review of the literature and collaborative analysis with individual subject data. J Am Coll Cardiol 2011; 57:1877–1886. [DOI] [PubMed] [Google Scholar]
- 8.Zhu W, Li W, Silverstein RL. Advanced glycation end products induce a prothrombotic phenotype in mice via interaction with platelet CD36. Blood 2012; 119:6136–6144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Majithia A, Bhatt DL. Novel antiplatelet therapies for atherothrombotic diseases. Arterioscler Thromb Vasc Biol 2019; 39:546–557. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Kapoor S, Jain MK, Nayak L. Thrombosis. Concise guide to hematology. Springer, Cham, 2019, pp. 149–161. [Google Scholar]
- 11.Stalker TJ, Newman DK, Ma P, et al. Platelet signaling. Handb Exp Pharmacol 2012; (210):59–85. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Hua VM, Chen VM. Procoagulant platelets and the pathways leading to cell death. Semin Thromb Hemost 2015; 41:405–412. [DOI] [PubMed] [Google Scholar]
- 13.Jobe SM. Not dead yet. Blood 2015; 126:2774–2775. [DOI] [PubMed] [Google Scholar]
- 14.Bevers EM, Williamson PL. Getting to the outer leaflet: physiology of phosphatidylserine exposure at the plasma membrane. Physiol Rev 2016; 96:605–645. [DOI] [PubMed] [Google Scholar]
- 15.Pedemonte N, Galietta LJ. Structure and function of TMEM16 proteins (anoctamins). Physiol Rev 2014; 94:419–459. [DOI] [PubMed] [Google Scholar]
- 16.van Kruchten R, Mattheij NJ, Saunders C, et al. Both TMEM16F-dependent and TMEM16F-independent pathways contribute to phosphatidylserine exposure in platelet apoptosis and platelet activation. Blood 2013; 121:1850–1857. [DOI] [PubMed] [Google Scholar]
- 17.Jobe SM, Wilson KM, Leo L, et al. Critical role for the mitochondrial permeability transition pore and cyclophilin D in platelet activation and thrombosis. Blood 2008; 111:1257–1265. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Choo HJ, Saafir TB, Mkumba L, et al. Mitochondrial calcium and reactive oxygen species regulate agonist-initiated platelet phosphatidylserine exposure. Arterioscler Thromb Vasc Biol 2012; 32:2946–2955. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Choo HJ, Kholmukhamedov A, Zhou C, Jobe S. Inner mitochondrial membrane disruption links apoptotic and agonist-initiated phosphatidylserine externalization in platelets. Arterioscler Thromb Vasc Biol 2017; 37:1503–1512. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Agbani EO, Williams CM, Hers I, Poole AW. Membrane ballooning in aggregated platelets is synchronised and mediates a surge in microvesiculation. Sci Rep 2017; 7:2770. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Hua VM, Abeynaike L, Glaros E, et al. Necrotic platelets provide a procoagulant surface during thrombosis. Blood 2015; 126:2852–2862. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Jackson SP, Schoenwaelder SM. Procoagulant platelets: are they necrotic? Blood 2010; 116:2011–2018. [DOI] [PubMed] [Google Scholar]
- 23.Mattheij NJ, Swieringa F, Mastenbroek TG, et al. Coated platelets function in platelet-dependent fibrin formation via integrin alphaIIbbeta3 and transglutaminase factor XIII. Haematologica 2016; 101:427–436. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Nechipurenko DY, Receveur N, Yakimenko AO, et al. Clot contraction drives the translocation of procoagulant platelets to thrombus surface. Arterioscler Thromb Vasc Biol 2019; 39:37–47. ■ In this article, procoagulant platelets are not static in a growing thrombus. Procoagulant platelets translocate towards the exterior of the thrombus through a mechanism in part by nonmuscle myosin, as genetic deficiency of the nonmuscle myosin gene MYH9 abrogated this phenotype. This mechanism could be because of the proaggregatory platelets ‘squeezing’ the procoagulant platelets towards the exterior of the thrombus, as suggested by Kholmukhamedov and Jobe.
- 25.Kholmukhamedov A, Jobe S. Procoagulant platelets get squeezed to define the boundaries of the hemostatic plug. Arterioscler Thromb Vasc Biol 2019; 39:5–6. [DOI] [PubMed] [Google Scholar]
- 26.Liu F, Gamez G, Myers DR, et al. Mitochondrially mediated integrin alphaIIbbeta3 protein inactivation limits thrombus growth. J Biol Chem 2013; 288:30672–30681. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Mattheij NJ, Gilio K, van Kruchten R, et al. Dual mechanism of integrin alphaIIbbeta3 closure in procoagulant platelets. J Biol Chem 2013; 288:13325–13336. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Kholmukhamedov A, Janecke R, Choo HJ, Jobe SM. The mitochondrial calcium uniporter regulates procoagulant platelet formation. J Thromb Hae-most 2018; 16:2315–2321. ■ The authors in this article identified the calcium uniporter being present and functional on mitochondria as a way to sensitize activation of the peptidylprolyl isomerase cyclophilin D. Cyclophilin D activation then promotes the formation of the mitochondrial permeability transition pore required for procoagulant phosphatidylserine externalization. This article bridges a connection between calcium flux and activation of cyclophilin D.
- 29.Agbani EO, Poole AW. Procoagulant platelets: generation, function, and therapeutic targeting in thrombosis. Blood 2017; 130:2171–2179. [DOI] [PubMed] [Google Scholar]
- 30. Agbani EO, Williams CM, Li Y, et al. Aquaporin-1 regulates platelet procoa gulant membrane dynamics and in vivo thrombosis. JCI Insight 2018; 3:; pii: 99062. ■ Aquaporin-1 was shown to be present in internal membrane structures (likely open canalicular system) to support procoagulant platelet formation. Deletion of Aquaporin-1 in platelets does not affect normal platelet pro-aggregatory functions. Using a number of imaging techniques, the authors show that Aquaporin-1 deletion in mice did not impact membrane ballooning, a signature event in procoagulant formation. However, full procoagulant responses were blunted with reduced thrombus formation in in-vivo models of arterial thrombosis.
- 31.Suzuki J, Denning DP, Imanishi E, et al. Xk-related protein 8 and CED-8 promote phosphatidylserine exposure in apoptotic cells. Science 2013; 341:403–406. [DOI] [PubMed] [Google Scholar]
- 32.Mason KD, Carpinelli MR, Fletcher JI, et al. Programmed anuclear cell death delimits platelet life span. Cell 2007; 128:1173–1186. [DOI] [PubMed] [Google Scholar]
- 33.Schoenwaelder SM, Yuan Y, Josefsson EC, et al. Two distinct pathways regulate platelet phosphatidylserine exposure and procoagulant function. Blood 2009; 114:663–666. [DOI] [PubMed] [Google Scholar]
- 34. Zhao L, Liu J, He C, et al. Protein kinase A determines platelet life span and survival by regulating apoptosis. J Clin Invest 2017; 127:4338–4351. ■ In this article, Zhao et al. showed through elegant signaling studies that aged platelets show reduced protein kinase A (PKA) activity, leading to enhanced apoptosis and platelet clearance. Protease-activated receptor agonist thrombin decreased PKA activity that promotes apoptotic-like activity. Inducing PKA activity through small chemical activators decreased platelet clearance in vivo and protected platelets from storage lesions.
- 35.Grozovsky R, Begonja AJ, Liu K, et al. The Ashwell-Morell receptor regulates hepatic thrombopoietin production via JAK2-STAT3 signaling. Nat Med 2015; 21:47–54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Ghosh A, Murugesan G, Chen K, et al. Platelet CD36 surface expression levels affect functional responses to oxidized LDL and are associated with inheritance of specific genetic polymorphisms. Blood 2011; 117:6355–6366. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Yang M, Silverstein RL. CD36 signaling in vascular redox stress. Free Radic Biol Med 2019; 136:159–171. ■ In this review, the authors discuss CD36 function as it relates to redox signaling in vascular smooth muscle cells, macrophages, and platelets. The mechanisms of redox-regulated signaling pathways are context-dependent. The authors also discuss CD36 structure and genetics that are related to the receptor’s function as antiangiogenic, pro-atherosclerotic, and pro-thrombotic.
- 38.Podrez EA, Poliakov E, Shen Z, et al. Identification of a novel family of oxidized phospholipids that serve as ligands for the macrophage scavenger receptor CD36. J Biol Chem 2002; 277:38503–38516. [DOI] [PubMed] [Google Scholar]
- 39.Wang Y, Fang C, Gao H, et al. Platelet-derived S100 family member myeloid-related protein-14 regulates thrombosis. J Clin Invest 2014; 124:2160–2171. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Huang MM, Bolen JB, Barnwell JW, et al. Membrane glycoprotein IV (CD36) is physically associated with the Fyn, Lyn, and Yes protein-tyrosine kinases in human platelets. Proc Natl Acad Sci U S A 1991; 88:7844–7848. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Chen K, Febbraio M, Li W, Silverstein RL. A specific CD36-dependent signaling pathway is required for platelet activation by oxidized low-density lipoprotein. Circ Res 2008; 102:1512–1519. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Wraith KS, Magwenzi S, Aburima A, et al. Oxidized low-density lipoproteins induce rapid platelet activation and shape change through tyrosine kinase and Rho kinase-signaling pathways. Blood 2013; 122:580–589. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Magwenzi S, Woodward C, Wraith KS, et al. Oxidised LDL activates blood platelets through CD36-NADPH oxidase-mediated inhibition of the cGMP/Protein kinase G signalling cascade. Blood 2015; 125:2693–2703. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Chen K, Li W, Major J, et al. Vav guanine nucleotide exchange factors link hyperlipidemia and a prothrombotic state. Blood 2011; 117:5744–5750. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Silverstein RL. Disabling the platelet’s brakes to promote thrombosis. Blood 2015; 125:2591–2593. [DOI] [PubMed] [Google Scholar]
- 46.Zielonka J, Vasquez-Vivar J, Kalyanaraman B. Detection of 2-hydroxyethidium in cellular systems: a unique marker product of superoxide and hydroethidine. Nat Protoc 2008; 3:8–21. [DOI] [PubMed] [Google Scholar]
- 47.Ciciliano JC, Sakurai Y, Myers DR, et al. Resolving the multifaceted mechanisms of the ferric chloride thrombosis model using an interdisciplinary microfluidic approach. Blood 2015; 126:817–824. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Li W, Nieman M, Sen Gupta A. Ferric chloride-induced murine thrombosis models. J Vis Exp 2016. doi: 10.3791/54479. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Li W, McIntyre TM, Silverstein RL. Ferric chloride-induced murine carotid arterial injury: a model of redox pathology. Redox Biol 2013; 1:50–55. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Cooley BC. Murine arterial thrombus induction mechanism influences subsequent thrombodynamics. Thromb Res 2015; 135:939–943. [DOI] [PubMed] [Google Scholar]
- 51.Cooley BC. Collagen-induced thrombosis in murine arteries and veins. Thromb Res 2013; 131:49–54. [DOI] [PubMed] [Google Scholar]
- 52.Campbell RA, Overmyer KA, Selzman CH, et al. Contributions of extravascular and intravascular cells to fibrin network formation, structure, and stability. Blood 2009; 114:4886–4896. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Wang W, Tang Y, Wang Y, et al. LNK/SH2B3 Loss of Function Promotes Atherosclerosis and Thrombosis. Circ Res 2016; 119:e91–e103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Zheng H, Wang F, Wang Q, Gao J. Cofactor-free detection of phosphatidylserine with cyclic peptides mimicking lactadherin. J Am Chem Soc 2011; 133:15280–15283. [DOI] [PubMed] [Google Scholar]
- 55.Smith BA, O’Neil EJ, Lampkins AJ, et al. Evaluation of fluorescent phosphatidylserine substrates for the aminophospholipid flippase in mammalian cells. J Fluoresc 2012; 22:93–101. [DOI] [PMC free article] [PubMed] [Google Scholar]