

Abstract
Leukemic blasts are immune cells gone awry. We hypothesized that dysregulation of inflammatory pathways contributes to the maintenance of their leukemic state and can be exploited as a cell-intrinsic, self-directed immunotherapy. To this end, we applied genome-wide screens to discover genetic vulnerabilities in acute myeloid leukemia (AML) cells implicated in inflammatory pathways. We identified the immune modulator interferon regulatory factor 2 binding protein 2 (IRF2BP2) as a selective AML dependency. We validated AML cell dependency on IRF2BP2 with genetic and protein degradation approaches in vitro and genetically in vivo. Chromatin and global gene expression studies demonstrated that IRF2BP2 represses IL-1ß/TNFα signaling via NF-κB, and IRF2BP2 perturbation results in an acute inflammatory state leading to AML cell death. These findings elucidate a hitherto unexplored AML dependency, reveal cell-intrinsic inflammatory signaling as a mechanism priming leukemic blasts for regulated cell death, and establish IRF2BP2-mediated transcriptional repression as a mechanism for blast survival.
Introduction
Immunotherapy has revolutionized the treatment of some malignancies. CD19-directed Chimeric Antigen Receptor (CAR) T-cell therapies in B-cell Acute Lymphoblastic Leukemia (B-ALL) (1) and immune checkpoint inhibitors in adult malignancies, such as melanoma (2) and non-small cell lung cancer (3), highlight transformative advances in cancer treatment. Nevertheless, some forms of cancer, such as acute myeloid leukemia (AML), remain poorly treated despite improvements in targeted therapy. AML has yet to be successfully addressed by newer immunotherapy approaches beyond hematopoietic stem cell transplantation. In AML, however, an alternative therapeutic approach might be to exploit innate inflammatory pathways intrinsic to the leukemia cells.
Genetic events perturbing hematopoietic stem or myeloid progenitor cells can give rise to AML blasts and prevent maturation to terminally differentiated immune cells, which normally possess a limited life span (4). We hypothesized that dysregulation of inflammatory pathways could contribute to the maintenance of the leukemic state in these immune cells gone awry. Moreover, we sought to decipher whether the rewiring of cell inherent inflammation can be exploited as cell-intrinsic, self-directed immunotherapy. Myeloid leukocytes represent the dominant cellular components of the innate immune system (5). Their mission is to protect the organism, sometimes at the expense of their own survival. Leukemic blasts, however, exhibit rapid clonal expansion at an immature cell differentiation state (6).
The innate immune response constitutes the first line of defense against foreign pathogens. Activation of innate immune cells relies on the discrimination between self and foreign antigens, and inflammation is the immune system’s response to the detection of harmful stimuli. While significant discoveries have broadened our understanding of inflammatory signaling in mature myeloid immune cells, the effects of enhanced or repressed intrinsic inflammation within an AML cell have yet to be fully elucidated. Several studies in B cell malignancies, however, hint at potential therapeutic opportunities for perturbing inflammatory signaling. For example, the activating L265P mutation in the common Toll-like receptor (TLR) adaptor Myeloid Differentiation primary response 88 (MyD88), prevalent in multiple lymphoid malignancies, leads to the activation of nuclear factor κB (NF-κB) via Bruton’s tyrosine kinase (BTK) and the interleukin-1 receptor associated kinases (IRAK1 and IRAK4) (7, 8). Targeting BTK with Ibrutinib has shown high response rates in patients with Waldenström’s macroglobulinemia with MyD88 mutations (9). Intriguingly, direct activation of members of the TLR family has also been reported to induce apoptosis in subsets of multiple myeloma and ALL (10, 11).
A strong interconnection between inflammatory signaling and leukemia also comes in the discoveries of a role for Tet methylcytosine dioxygenase 2 (TET2) mutations in association with both AML and increased risk for atherosclerosis. Loss-of-function mutations in TET2, an epigenetic regulator, are present in approximately 15% of myeloid malignancies, causing DNA hypermethylation and enhanced self-renewal in hematopoietic stem cells. In addition, TET2 is one of the most commonly mutated genes in clonal hematopoiesis of indeterminate potential (CHIP) (12, 13). In the context of CHIP, TET2 mutations increase the risk of subsequent development of a hematologic malignancy but are also strongly associated with atherosclerotic cardiovascular disease and inflammation (14). For instance, Tet2-deficient macrophages exhibit an increase in NOD-, LRR-, and pyrin domain-containing protein 3 (NLRP3) inflammasome-mediated interleukin-1β secretion (15). Similarly, plasma interleukin-8, a CXC chemokine not present in mice, was increased in humans with TET2 mutations (14).
Interferon Regulatory Factor 2 Binding Protein 2 (IRF2BP2) provides another example of a gene both involved in cancer as well as inflammatory conditions. IRF2BP2 was initially identified as a co-repressor molecule for Interferon Regulatory Factor-2 (IRF2) in a yeast two-hybrid library screen (16). Subsequently, it was found to be involved in fusion oncoproteins in a number of malignancies, including AML, and implicated in inflammatory diseases. Studies on IRF2BP2 and leukemia are restricted to a handful of case reports (17–21) identifying IRF2BP2 as a fusion partner to RARA, leading to the phenotype of acute promyelocytic leukemia (APML). In addition, IRF2BP2 was reported to be fused to CDX1 in mesenchymal chondrosarcoma (22). Interestingly, a small number of studies also link IRF2BP2 to inflammation in cardiovascular disease and sepsis (23, 24).
To test the hypothesis that AML cells depend on proper regulation of inflammatory networks, we analyzed large data collections from primary AML samples and functional genomic screens. Applying CRISPR-Cas9 genome editing technology, the Broad Institute Cancer Dependency Map has screened close to 800 cancer cell lines with a genome-scale sgRNA (small guide RNA) library (25, 26). We integrated data from the Cancer Dependency Map with two independent genome-wide screens, one using RNAi (27, 28), and a second CRISPR-Cas9 screen with a unique library (29). An advantage to performing large-scale screens across hundreds of cancer cell line models is the ability to identify disease selective versus pan-essential targets, which should increase the therapeutic index of identified targets. We thus focused on novel dependencies that are stronger in AML than in other cancer models screened. Herein, we identified and validated IRF2BP2 as a strong immune-related dependency in AML and elucidated its mechanism of action as a gatekeeper to prevent suicidal inflammatory signaling in AML cells.
Results
Identification of IRF2BP2 as a strong immune dependency enriched in AML
To test the hypothesis that inflammatory pathways are inherent to AML blasts we performed single sample gene set enrichment analysis (ssGSEA) across large collections of primary AML samples versus the hallmark gene set collection of the Molecular Signature Database (MSigDB). ssGSEA is an extension of GSEA that calculates separate enrichment scores for each pairing of a sample and gene set. Each ssGSEA enrichment score represents the degree to which the genes in a particular gene set are coordinately up- or down-regulated within a sample. Unsupervised clustering applied to the ssGSEA projection of the 179 primary AML samples from The Cancer Genome Atlas (TCGA) in the space of Hallmark gene sets, identified an AML subgroup of approximately 40% of all samples enriched for immune and inflammatory pathways (Figure 1A–C). Quantitatively, the ssGSEA Z-score enrichment for the canonical pathway HALLMARK_INFLAMMATORY_RESPONSE was significantly higher in these immune/inflammatory samples when comparing them to all other AML samples within the TCGA collection (Figure 1D). Moreover, we were able to fully recapitulate these findings in three additional primary AML collections, namely from the European Hovon study group (GSE14468), the Beat AML trial, and the Target AML data collection from children with AML (Figure S1A, S1B, S1C). We estimated the lineage enrichment within AML blasts in cohorts of primary AML samples using a gene expression deconvolution method implemented in CIBERSORT (30). We identified a positive correlation between the enrichment of inflammatory response and enrichment of a monocytic lineage signature in these primary AML samples (Figure S1D). Consistently, the AML group with high immune/inflammatory response displayed a significant enrichment for FAB M4/M5 samples as compared to the non-inflammatory counterpart (Figure S1E).
Figure 1. Enrichment analysis for inflammatory pathways identifies an AML subgroup and uncovers IRF2BP2 as a selective immune dependency.
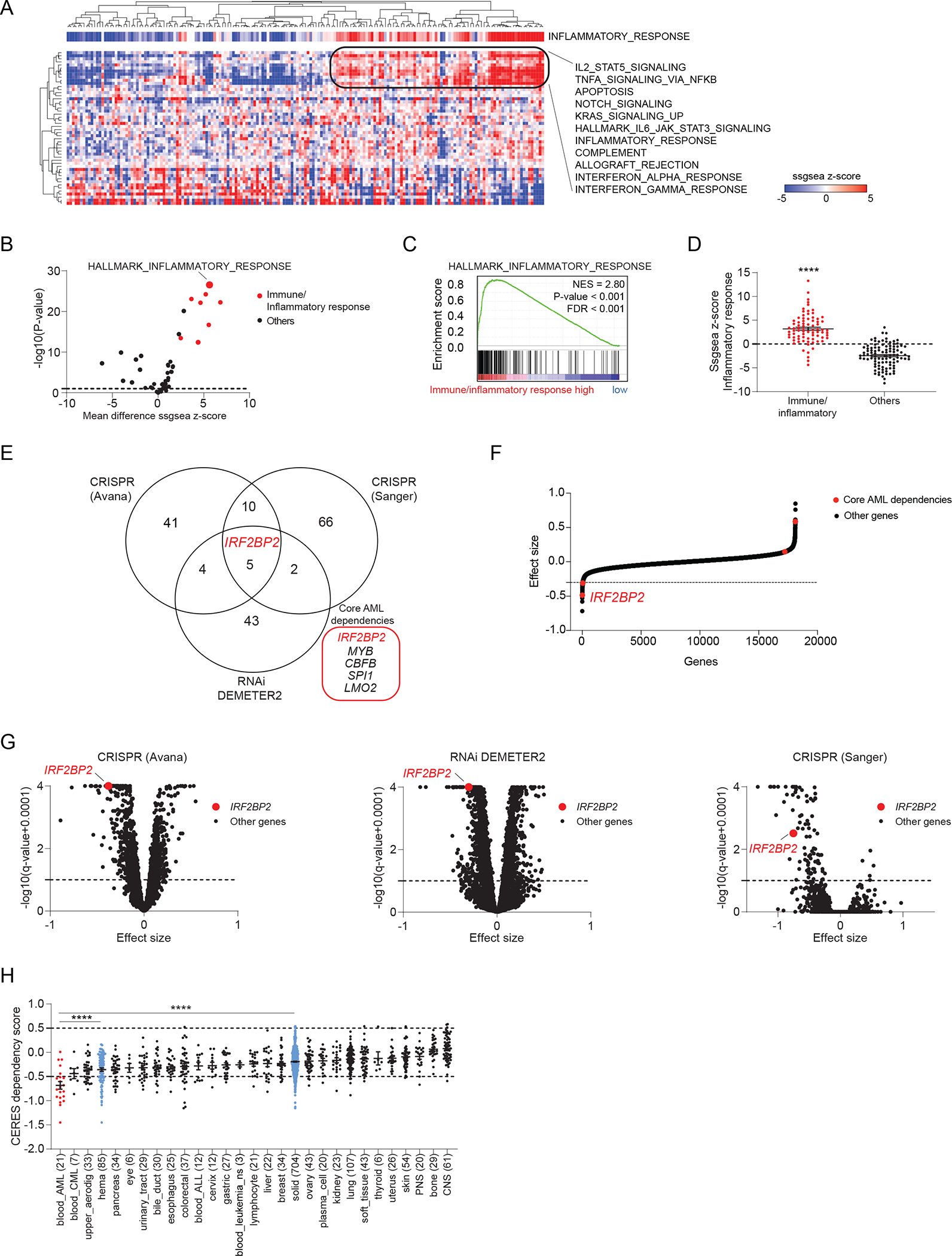
A, Heatmap depicting the single sample GSEA (ssGSEA) projection of the Cancer Genome Atlas (TCGA) Acute Myeloid Leukemia (LAML) expression data for 179 AML samples on the collection of 50 hallmark gene sets (MSigDB v7.1), defining a cluster of AML samples enriched for immune/inflammatory pathways. AML samples are annotated with the ssGSEA scores for HALLMARK_INFLAMMATORY_RESPONSE. Data are clustered according to the hierarchical clustering for Spearman rank correlation. Top scoring hallmark gene sets within the cluster with strong enrichment for immune/inflammatory response are listed next to the heatmap.
B, Volcano plot depicting the differential enrichment of the ssGSEA projection on hallmark gene sets for AML samples enriched for immune/inflammatory pathways (defined as shown in panel A) compared to all other AML samples within TCGA LAML collection. Highlighted in red are the immune/inflammatory hallmarks gene sets. Limma eBayes, |effect size| ≥ 0.5, p-value ≤ 0.10.
C, GSEA plot for HALLMARK_INFLAMMATORY_RESPONSE enrichment in the genome-wide list of genes ranked by differential expression in AML samples enriched for immune/inflammatory pathways (defined as shown in panel A) compared to all other AML samples within the TCGA LAML collection. GSEA significance cut-offs: |Normalized Enrichment Score (NES)| ≥ 1.3, p-value ≤ 0.05, FDR ≤ 0.25.
D, Scatter dot plots depicting the HALLMARK_INFLAMMATORY_RESPONSE ssGSEA scores for AML samples enriched for immune/inflammatory pathways (defined as shown in panel A) compared to all other AML samples within TCGA LAML. T-test with Welch correction, **** p < 0.0001.
E, Venn-diagram depicting the overlap between gene dependencies enriched in AML identified in three independent data sets: 60 AML dependencies identified in the CRISPR (Avana) 20Q3 public data on 789 cell lines, 54 AML dependencies identified in the Combined RNAi (Broad, Novartis, Marcotte) 20Q3 DEMETER2 public data on 712 cell lines, and 83 AML dependencies identified in the CRISPR (Sanger) 20Q3 public data on 318 cell lines. The number of AML cell lines among all screened cancer cell lines is indicated for each data set. AML enriched dependencies were identified by limma eBayes, |effect size| ≥ 0.3, q-value ≤ 0.1.
F, Hockey stick plot depicting the differential CERES dependency scores in AML cell lines with a strong monocytic signature compared to all other AML cell lines in the CRISPR (Avana) data. Core dependencies are highlighted in red. IRF2BP2 scores as the strongest core dependency (most negative score) among monocytic AML cell lines. Significance |effect size| ≥ 0.3, q-value ≤ 0.1.
G, Volcano plots depicting the gene dependency status for AML versus other lineages across three independent screens. Left: CRISPR (Avana) 20Q3 CERES data set; middle: Combined RNAi 20Q3 DEMETER2 data set; right: CRISPR (Sanger) 20Q3 data set. Effect sizes of all other genes within AML are shown as black; IRF2BP2 is highlighted in red (limma eBayes, |effect size| ≥ 0.3, q-value ≤ 0.10).
H, Boxplots depicting the IRF2BP2 CERES dependency scores across the cell line lineages in the CRISPR (Avana) 20Q3 data. Lineages are ranked by increasing mean IRF2BP2 CERES dependency scores. The lower the CERES score, the greater the dependency; a score below −0.5 indicates gene dependency. Differential dependencies were assessed by one-way ANOVA, Tukey’s multi-comparison test, **** p < 0.0001.
As part of the Broad Institute Cancer Dependency Map v20q3, 23 AML cell lines were screened with a genome-wide CRISPR-Cas9 library and compared to 766 non-AML cancer cell lines. Twenty-one days post transduction, we sequenced the sgRNAs and quantified their relative abundance as CERES dependency scores (31). This drop-out screen allowed us to identify sgRNAs depleted relative to the input, suggesting that the genes targeted by those sgRNAs are genetic dependencies. We selected as top hits those sgRNAs that were more highly depleted in AML cell lines relative to other cancer cell lines and identified 60 genes on which AML cells were significantly more dependent than other cancer cell lines (based on the empirical Bayes statistics for differential dependency with significance cut-offs: effect size ≤ −0.3, adjusted p-value ≤ 0.10; Figure 1E and Table S1A). GSEA performed for the genome-wide differential dependency scores in AML versus non-AML cancer cell lines identified several immune and inflammatory Gene Ontology gene sets associated with depletion in AML (Figure S1F). To enhance the robustness of our AML dependency findings, we interrogated two additional genome-wide screens for AML enriched dependencies and intersected these results: only five genes were significantly enriched in all three screens using these strict criteria (Figure 1E and Supplementary Tables S1A to C). Within this core group of five genes, IRF2BP2 stood out as the strongest when comparing the differential CERES scores in the CRISPR (Avana) AML cell lines with high enrichment for monocytic lineage compared to all other AML cell lines (Figure 1F). A dependency on IRF2BP2 correlated strongly with enrichment for the monocytic lineage in both CRISPR (Avana) and RNAi (DEMETER2) screens (Figure S1G). Of note, IRF2BP2 was also the single unexplored dependency amongst the other well-characterized candidates, namely MYB, CBFB, SPI1, and LMO2 scoring in all three data sets (Figure 1E and 1G).
Comparing the dependency scores of all AML cell lines with either other hematopoietic or solid tumor cell lines within the CRISPR (Avana) screen, AML cells were significantly more dependent on IRF2BP2 than any other group, representing the only cancer type with a mean dependency score of less than −0.5 (Figure 1H). Gene expression is one of the strongest predictive biomarkers of dependencies (25). Indeed, when profiling IRF2BP2 expression across cancer cell types, we found that IRF2BP2 expression was highest in AML cell lines (Figure S1H) and in AML patient samples (Figure S1I). However, we did not observe a significant correlation between IRF2BP2 expression and dependency on IRF2BP2 in AML cell lines across independent dependency screens (Figure S1J), likely due to high IRF2BP2 expression with a low variation in AML. Moreover, we discovered that IRF2BP2 was marked by a super-enhancer in all 71 AML samples in a collection published by McKeown et al. consistent with its high level of expression in AML (32) (Figure S1K and S1L).
Validation of IRF2BP2 as a dependency in AML in vitro and in vivo
We next sought to validate IRF2BP2 as a dependency in vitro and in vivo. We transduced the AML cell lines MV4-11, U937, MOLM13, THP1 and NB4 with IRF2BP2-directed CRISPR guides used in the genome-wide screen and confirmed guide efficacy by western blot (Figure 2A and S2A). We observed a marked decrease in the growth of cells infected with IRF2BP2 targeting guides in comparison to non-targeting CRISPR guides (Figure 2A and S2A). In addition, using an orthogonal shRNA approach, we validated the dependency on IRF2BP2 using the shRNAs from the screen (Figure 2B), and by targeting IRF2BP2 with unique doxycycline-inducible hairpins (Figure S2B and S2C). To account for potential off-target effects of these inducible hairpins, we performed site-directed mutagenesis of an open reading frame (ORF) for IRF2BP2, introducing a silent point mutation at the binding site of the shRNAs. We then expressed this ORF in MV4-11 cells, followed by knock-down of IRF2BP2 (Figure 2C). When these cells were exposed to doxycycline, we no longer observed any effect on viability (Figure 2C), confirming that the observed effect of the shRNA is on-target for IRF2BP2. We next transduced luciferase-expressing MV4-11 cells (MV4-11-luc) with these inducible hairpins and the resistant, mutated IRF2BP2 ORF. These cells were then injected into NOD/SCID/IL2rγnull (NSG) mice. After documenting leukemia engraftment by bioluminescence, the mice were fed a doxycycline-containing diet for two weeks. We then assessed for leukemia burden by bioluminescence imaging (Figure 2D and 2E), and by determining the chimerism of the human isoform of the pan-leukocyte marker CD45 in peripheral blood (PB) and bone marrow (BM) by flow cytometry (Figure S2D and 2F). Both approaches resulted in a significant reduction in leukemia burden in mice with leukemia cells containing IRF2BP2 targeting hairpins compared to non-targeting shRNAs or mice injected with leukemia cells containing the rescue ORF and the IRF2BP2 targeting shRNAs (Figure 2E and 2F).
Figure 2. IRF2BP2 validates as a dependency in vitro and in vivo.
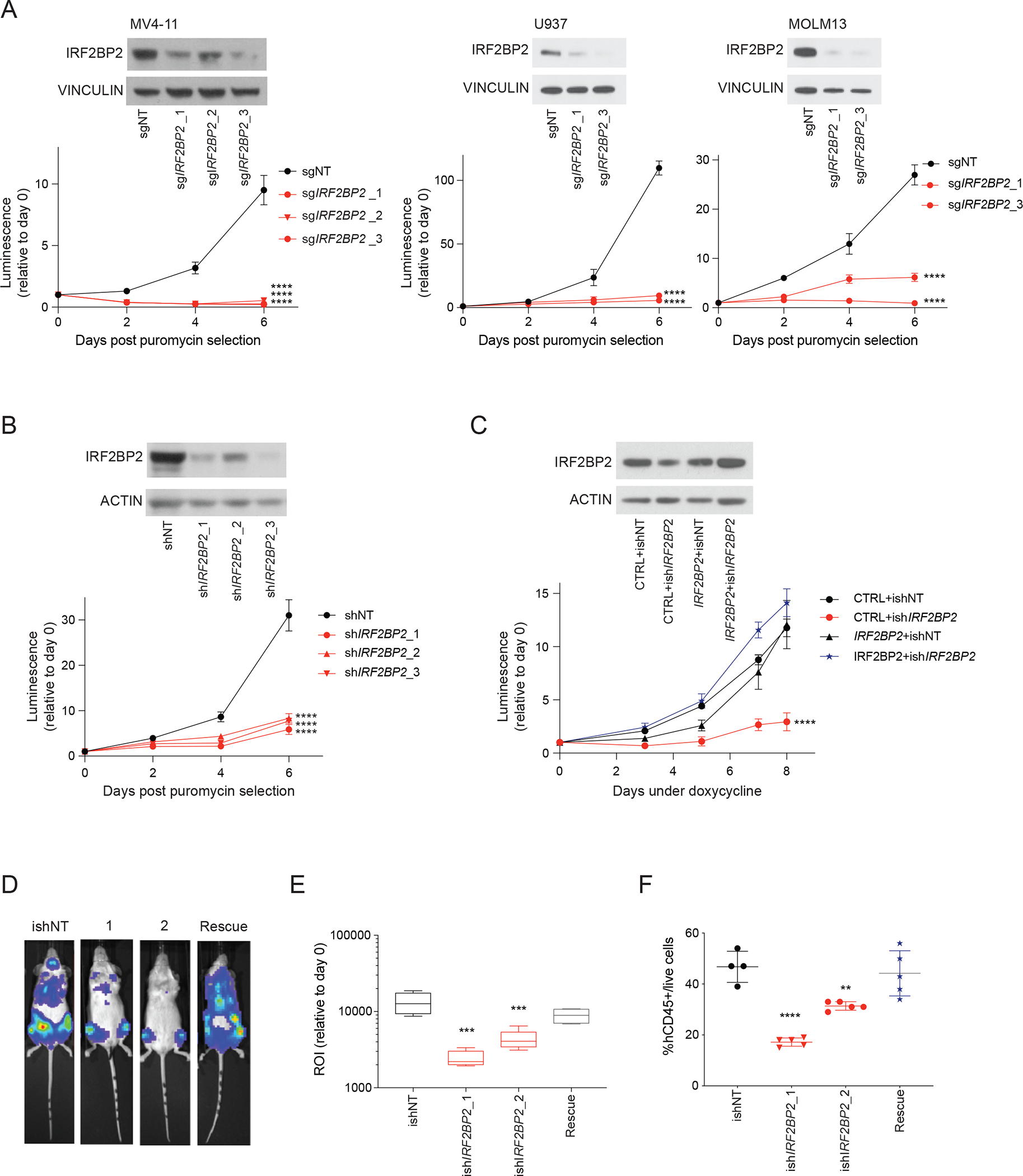
A, Upper left panel: western blot analysis showing IRF2BP2 protein levels after CRISPR knock-out in MV4-11 cells post puromycin selection; vinculin was used as a loading control. Lower left panel: viability measured in MV4-11 cells following lentiviral infection with three CRISPR guides against IRF2BP2 (sgIRF2BP2_1 to 3) and one non-targeting control guide (sgNT) using an ATP-based CellTiter-Glo assay. Middle and right upper panel: western blot analysis showing IRF2BP2 protein levels after CRISPR knock-out with two single guides in U937 and MOLM13 cells post puromycin selection; vinculin was used as a loading control. Middle and right lower panel: CellTiter-Glo viability assay in U937 and MOLM13 cells following lentiviral infection with two CRISPR guides against IRF2BP2 (sgIRF2BP2_1 and 3) and one non-targeting control guide (sgNT). Two-way ANOVA, **** p < 0.0001.
B, Upper panel: western blot analysis showing levels of IRF2BP2 protein using the IRF2BP2-targeting shRNAs from the genome-wide screen in MV4-11 cells, infected with control hairpin (shNT) or hairpins against IRF2BP2 (shIRF2BP2_1/2/3). Lower panel: CellTiter-Glo viability assay in MV4-11 cells with knock-down of IRF2BP2 (shIRF2BP2_1/2/3) or control hairpin (shNT), using the hairpins from the shRNA screen. Two-way ANOVA, **** p < 0.0001.
C, Upper panel: Western blot analysis for IRF2BP2 in MV4-11 cells double infected with a CTRL (plx3.17 GFP) or IRF2BP2 mutant ORF resistant to the shRNA and either an inducible shRNA control (ishCTRL) or an inducible shRNA against IRF2BP2 (ishIRF2BP). Actin was used as a loading control. Lower panel: CellTiter-Glo viability assay in MV4-11 cells double infected with a CTRL ORF or IRF2BP2 mutant ORF and either ishNT or ishIRF2BP2 as described above. Two-way ANOVA, **** p < 0.0001.
D, Bioluminescence signal for one representative mouse per group: NSG mice (n = 4 to 5 animals per study group) injected with luciferase-expressing MV4-11 cells infected with control, or IRF2BP2-directed inducible hairpins, or a rescue IRF2BP2 silent mutated ORF plus the hairpin ishIRF2BP2_2, respectively, were imaged to assess bioluminescence intensity as a surrogate marker for leukemia burden after a doxycycline diet for two weeks.
E, Quantification of the bioluminescence signal intensity on day 14. One-way ANOVA, Dunnett’s multiple comparison test, **** p < 0.0001, *** p < 0.001.
F, Flow cytometry analysis for hCD45+ cells/live cells on bone marrow samples of all mice included in the study (n = 4 to 5 animals per study group; every symbol represents one animal). One-way ANOVA, Dunnett’s multiple comparison test, **** p < 0.0001, ** p < 0.01.
Degradation of IRF2BP2 leads to annexin V positive cell death
To study the acute loss of IRF2BP2, we leveraged an IRF2BP2 degrader system applying a previously reported technology known as the degradation tag (dTAG) system to selectively degrade FKBP12F36V-tagged fusion proteins (33). This degradation approach relies on the small molecule dTAG-13 that binds to the FKBP12F36V-tag and recruits the E3 ubiquitin ligase cereblon, thereby inducing proteasome-mediated degradation of the FKBP12F36V-tagged target. We generated AML cells with degradable IRF2BP2 by co-infection with FKBP12F36V-tagged IRF2BP2 and an endogenous IRF2BP2-targeting sgRNA. Exogenous tagged IRF2BP2 was expressed at comparable levels to the endogenous IRF2BP2 expression level with effective depletion of endogenous IRF2BP2 by sgIRF2BP2 (Figure 3A and S3A). We found IRF2BP2 degradation to be maximal at 100 to 200 nM dTAG-13 with maximal loss after 12 hours of treatment with dTAG-13 (Figure 3A and 3B). Additionally, fractionation of cellular compartments revealed degradation of IRF2BP2 in the cytoplasmic and nuclear extracts, including the chromatin-bound and soluble fractions (Figure S3B). Degradation of IRF2BP2 translated into a reduction of AML cell viability and decreased colony formation capacity (Figure 3C and 3D). AML cells tolerated expression of exogenous IRF2BP2 without depletion of endogenous IRF2BP2 and showed a comparable growth rate to cells expressing an ORF for GFP (Figure 3C). Mechanistically, cells treated with dTAG-13 underwent cell death with hallmarks of apoptosis, as evidenced by an increase in annexin V/PI-positive cells (Figure 3E) and an induction of cleaved caspase 3 (Figure 3F) and cleaved caspase 8 (Figure 3G). Similarly, we also saw evidence of apoptosis using CRISPR-based knock-out of IRF2BP2 (Figure S3C and S3D). We did not observe markers of pyroptosis (cleaved GSDMD) nor necroptosis (phospho-MLKL) over 24 hours post degradation of IRF2BP2 (Figure S3E and S3F).
Figure 3. Degradation of IRF2BP2 impairs viability and colony formation in AML and induces apoptosis.
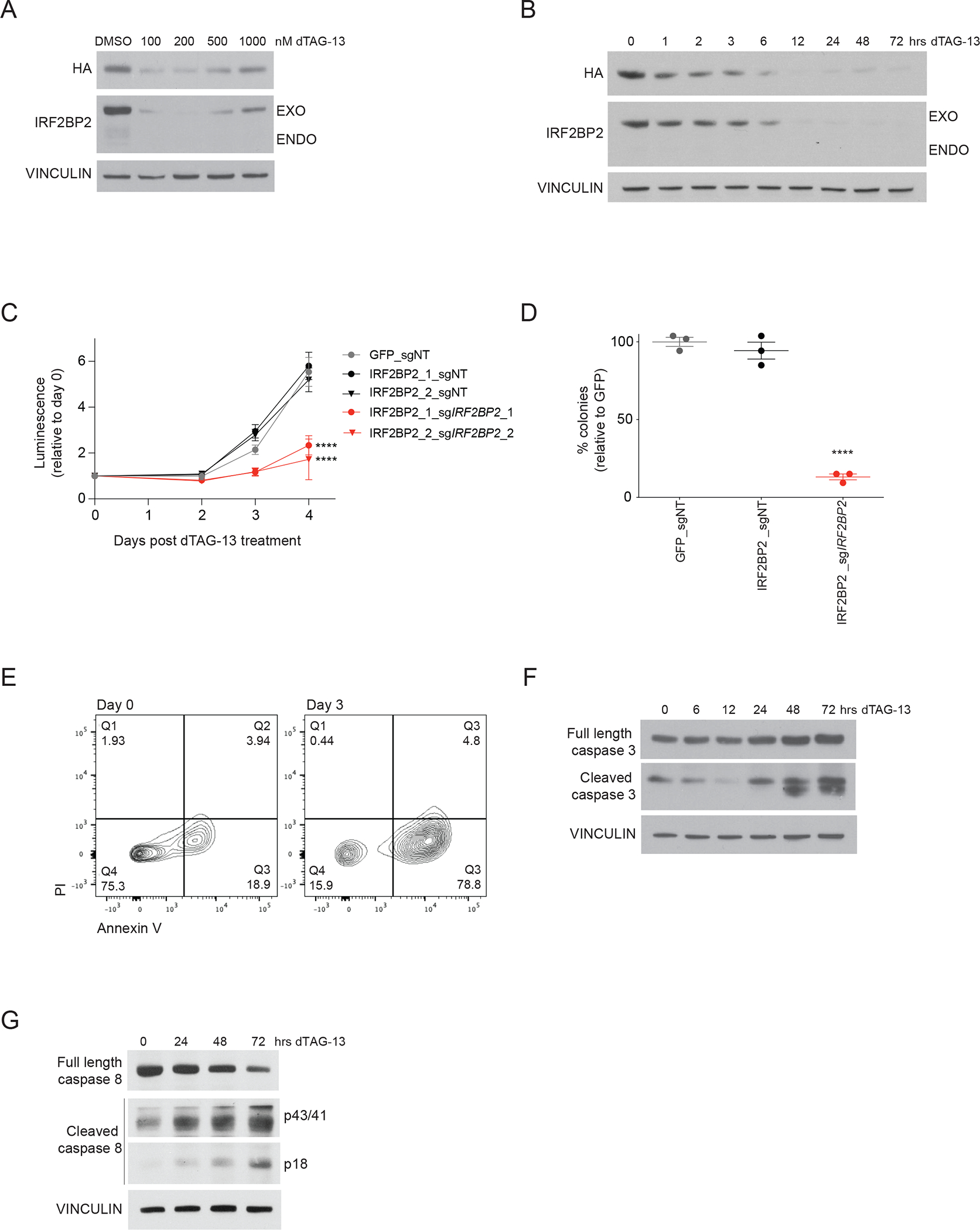
A, Western blot analysis of a dose-response experiment after 24 hours of treatment with dTAG-13 for HA, IRF2BP2 and vinculin in MV4-11 cells with a degradable N-terminally tagged FKBP12F36V-HA-IRF2BP2-fusion and knock-out of endogenous IRF2BP2. A hook effect is observed at the highest concentrations. An HA antibody was used to detect the IRF2BP2-fusion.
B, Western blot analysis of a time-course experiment showing IRF2BP2 protein levels following treatment with dTAG-13 in MV4-11 cells. Vinculin was used as a loading control.
C, CellTiter-Glo viability assay in MV4-11 cells overexpressing a GFP CTRL-ORF or an IRF2BP2 N-dTAG-construct, double-infected with non-targeting (sgNT) or a CRISPR guide targeting endogenous IRF2BP2 (sgIRF2BP2_1, sgIRF2BP2_2) following treatment with 100 nM dTAG-13. Two-way ANOVA, **** p < 0.0001.
D, Colony formation capacity is assessed in MV4-11 cells overexpressing a GFP CTRL-ORF or IRF2BP2 N-dTAG-construct, double-infected with non-targeting (sgNT) or CRISPR guide targeting endogenous IRF2BP2 (sgIRF2BP2) treated with 100 nM dTAG-13 for six days. One-way ANOVA, Dunnett’s multiple comparisons test, **** p < 0.0001.
E, Flow cytometry analysis for Annexin V/PI in MV4-11 cells with degradable IRF2BP2 treated with 100 nM dTAG-13 for 72 hours (right panel) and untreated control cells (left panel).
F, Western blot analysis for full length and cleaved caspase 3 in MV4-11 cells with degradable IRF2BP2 following treatment with dTAG-13 for 0 to 72 hours. Vinculin was used as a loading control.
G, Western blot analysis for full length and cleaved caspase 8 in MV4-11 cells with degradable IRF2BP2 following treatment with dTAG-13 for 0 to 72 hours. Vinculin was used as a loading control.
Knock-down of IRF2BP2 in PDX cells reduces leukemia burden and prolongs survival
We next determined if there is a therapeutic window between targeting IRF2BP2 in AML cells versus normal hematopoietic progenitor cells. Because of the low efficiency of lentiviral transduction of Cas9 into CD34+ hematopoietic stem and progenitor cells we nucleofected human CD34+ healthy bone marrow cells with Cas9-sgRNA ribonucleoprotein complexes (RNPs) to enable genome editing as previously reported (34) and subsequently performed colony forming assays and serial cell counting to evaluate their progenitor activity. We achieved complete loss of IRF2BP2 protein (Figure 4A). Colony numbers on day 10 were not significantly different in human CD34+ bone marrow cells with IRF2BP2-targeting or a control guide, while knock-out of the essential gene RPA3 led to a significant reduction in colony number (Figure 4B). Consistently, serial cell counting in human CD34+ bone marrow cells showed a stable number of cells with IRF2BP2-targeting guides (Figure S4A).
Figure 4. Knock-down of IRF2BP2 in PDX cells reduces leukemia burden and prolongs survival.
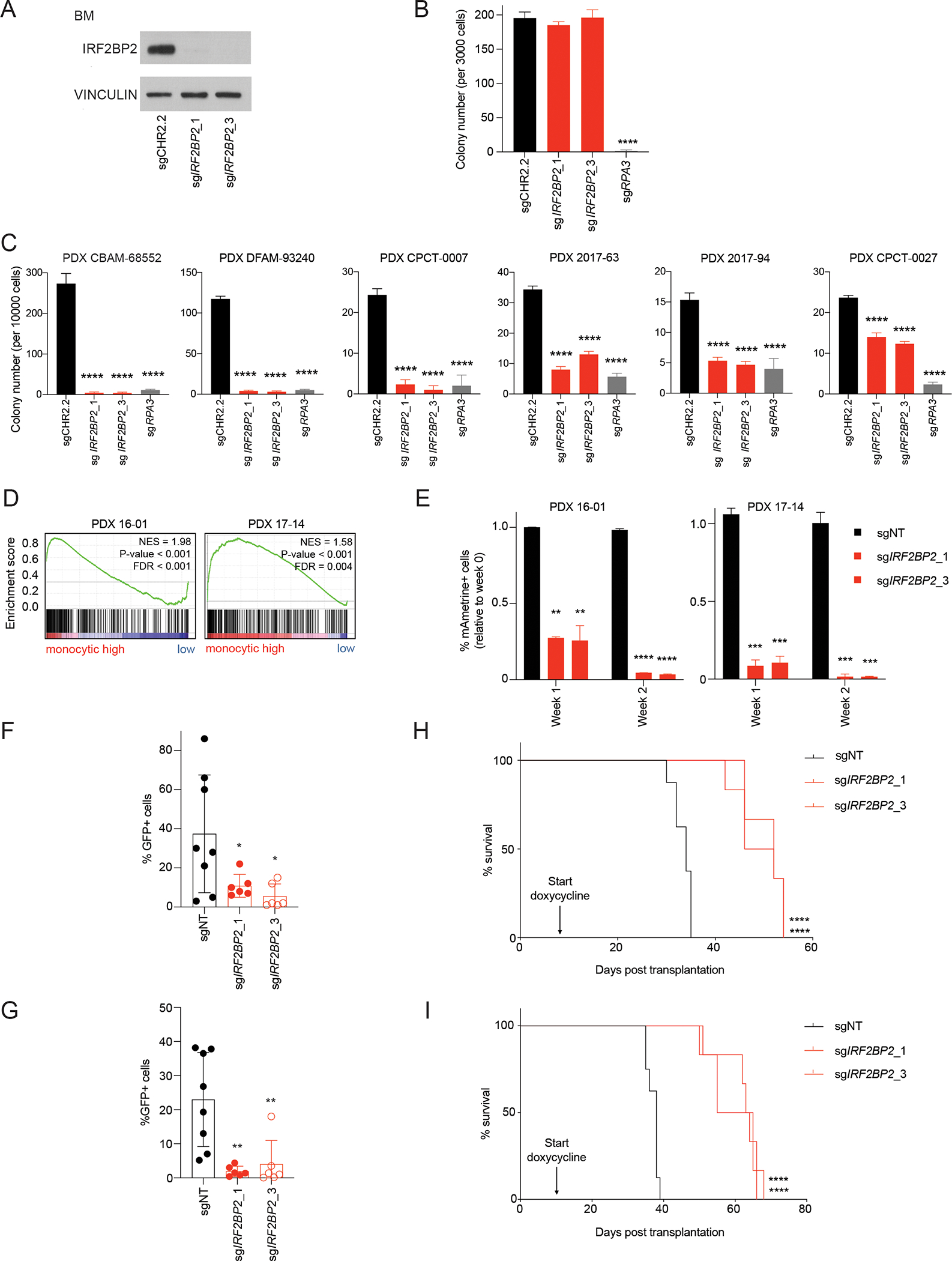
A, Western blot analysis for IRF2BP2 in hCD34+ bone marrow cells nucleofected with synthetic IRF2BP2-targeting guides or a non-targeting control guide. Vinculin was used as a loading control.
B, Quantification of colony numbers of hCD34+ cells nucleofected with synthetic IRF2BP2-targeting guides, a non-targeting control guide or a positive control guide targeting an essential gene (RPA3) on day 10 post seeding. One-way ANOVA, Dunnett’s multiple comparison test (per week), **** p < 0.0001.
C, Quantification of colony numbers of six PDX models (for molecular characteristics see Table 1) nucleofected with synthetic IRF2BP2-targeting guides, a non-targeting control guide or a positive control guide targeting an essential gene (RPA3) on day 10 post seeding. One-way ANOVA, Dunnett’s multiple comparison test (per week), **** p < 0.0001.
D, GSEA for human monocytic lineage gene markers (35) run per individual PDX sample on the genome-wide genes ranked by log2(TPM+1) expression. NES >/= 1.3, p-value </= 0.05, FDR </= 0.25.
E, PDX cells from PDX model 16-01 (left) and 17-14 (right) were infected with mAmetrine-positive CRISPR guides targeting IRF2BP2 or a non-targeting control guide (sgNT). Flow-cytometry for mAmetrine-positive cells was performed 72 hours post infection, and then weekly. The fraction of mAmetrine-positive cells relative to week 0 was assessed at one and two weeks. One-way ANOVA, Dunnett’s multiple comparison test (per week), ** p < 0.01, *** p < 0.001, **** p < 0.0001.
F, Flow cytometry analysis for GFP-positive cells on bone marrow aspirates from sub-lethally irradiated NSG mice transplanted with PDX 16-01 cells infected with doxycycline-inducible CRISPR guides against IRF2BP2 or a non-targeting control guide after a two-week doxycycline diet (n = 6 to 8 mice per group, each dot represents one animal). One-way ANOVA, Dunnett’s multiple comparison test, * p < 0.05.
G, Flow cytometry analysis for GFP-positive cells on bone marrow aspirates from sub-lethally irradiated NSG mice transplanted with PDX 17-14 cells infected with doxycycline-inducible CRISPR guides against IRF2BP2 or a non-targeting control guide (sgNT) after a two-week doxycycline diet (n = 6 to 8 mice per group, each dot represents one animal). One-way ANOVA, Dunnett’s multiple comparison test, ** p < 0.01.
H, Kaplan-Meier curves depicting survival of NSG mice transplanted with PDX 16-01 cells infected with doxycycline-inducible CRISPR guides against IRF2BP2 or a non-targeting control guide under continuous doxycycline diet. The median survival of the mice transplanted with PDX 17-14 sgIRF2BP2_1 and sgIRF2BP2_3 cells was 49 and 52 days, respectively, as compared to 34 days for mice who received sgNT PDX cells. Log-rank Mantel-Cox test, **** p < 0.0001.
I, Kaplan-Meier curves depicting survival of NSG mice transplanted with PDX 17-14 cells infected with doxycycline-inducible CRISPR guides against IRF2BP2 or a non-targeting control guide under continuous doxycycline diet. The median survival of the mice transplanted with PDX 17-14 sgIRF2BP2_1 and sgIRF2BP2_3 was 63.5 and 60 days respectively, as compared to 38 days for mice who received sgNT PDX cells. Log-rank Mantel-Cox test, **** p < 0.0001.
To assess response to IRF2BP2 loss in primary patient models of AML, we selected six patient-derived xenograft (PDX) samples (Table 1; (35, 36)), confirmed their colony forming capacity and performed nucleofection as previously reported (34). We then assessed colony numbers 10 days post seeding nucleofected PDX cells. We found a significant reduction in colony forming capacity in PDX cells that had received IRF2BP2-targeting guides (Figure 4C). Interestingly, the effect was least pronounced in PDX cells with a CBFA2T3-GLIS2 fusion (PDX CPCT-0027) (Figure 4C). ssGSEA for monocytic lineage markers (37) in these PDX samples found significant enrichment for monocytic lineage markers in all PDX models but PDX CPCT-0027 (Figure S4B). In addition, to study the acute effects of IRF2BP2 loss in PDX cells, we generated a PDX model with degradable IRF2BP2 and validated IRF2BP2 degradation (Figure S4C), and impaired cell growth with IRF2BP2 degradation (Figure S4D).
Table 1:
Characteristics of PDX models
| ID | Disease type | Karyotype | Mutations |
|---|---|---|---|
| 16–01 (34) | relapsed AML | CALM-AF10, complex karyotype | NF1, TP53, PHF6 |
| 17–14 (34) | tAML, relapsed | MLL-AF10, complex karyotype | KRAS |
| CBAM-68552 (35) | primary AML | MLL-AF6 | none detected |
| CPCT-0007 (36) | refractory AML | MLL-CALM | NRAS |
| CPCT-0027 (36) | primary AML | CBFA2T3-GLIS2 | NRAS, WT1 |
| DFAM-93240 (35) | primary AML | AML with inv(16) CBFB-MYH11 | TP53 |
| 2017–63 | relapsed, refractory AML | MNX1-ETV6 | none detected |
| 2017–94 | relapsed, refractory AML | complex karyotype | CDKN1A/B |
To validate the relevance of the IRF2BP2 dependency in primary patient AML cells in vivo we next assessed the effects of knock-out of IRF2BP2 in PDX blasts on leukemia burden and survival. To first test our experimental system, we selected two monocytic PDX models that tolerated stable Cas9 expression and can be passaged for a limited time in vitro (PDX 16-01 with a CALM-AF10 fusion and PDX 17-14 with a MLL-AF10 fusion; see also Figure 4D and extended information Table 1). We infected these Cas9-expressing PDX cells with sgIRF2BP2_1 and sgIRF2BP2_3 cloned into a vector with mAmetrine as a selection marker. When we assessed the percentage of mAmetrine-positive cells over time, we found a rapid depletion of cells infected with guides targeting IRF2BP2, while the percentage of cells infected with non-targeting guides was stable over the course of two weeks (Figure 4E). We then infected PDX 16-01 and PDX 17-14 cells with a control guide or CRISPR guides against IRF2BP2 under a doxycycline-inducible promoter. Flow cytometry for GFP-positive cells expressing the CRISPR guides revealed a rapid decline in GFP-positive cells in PDX cells infected with sgIRF2BP2 over time. In contrast, the relative number of GFP-positive cells remained stable in cells infected with the non-targeting control (Figure S4E). PDX 16-01 and PDX 17-14 cells infected with IRF2BP2-directed CRISPR guides or a control guide were flow-sorted for GFP to increase purity and then injected by tail vein into irradiated NSG mice. Western blot analysis on day four post doxycycline-induction showed decreased IRF2BP2 protein in these sorted cells in vitro (Figure S4F and S4G). Upon detection of at least 1% GFP-positive cells in the peripheral blood on day 9 (PDX 16-01) and day 11 (PDX 17-14) post transplantation, all mice received a doxycycline-containing diet to induce knock-out of IRF2BP2. When we performed bone marrow aspirations after two weeks on doxycycline, we detected a significant reduction of leukemia burden in mice transplanted with PDX cells with IRF2BP2 targeting guides compared to those with a control guide (Figure 4F and 4G). When we then followed these mice for survival under continuous doxycycline diet and knock-out of IRF2BP2, we observed a significantly prolonged survival in mice transplanted with PDX cells with the targeting guides as compared to control guides: the median survival was 49 (PDX 16-01; sgIRF2BP2_1) and 63.5 (PDX 17-14; sgIRF2BP2_1) and 52 (PDX 16-01; sgIRF2BP2_2) and 60 (PDX 17-14; sgIRF2BP2_2) days respectively in mice transplanted with PDX cells with IRF2BP2 targeting guides as compared to 34 (PDX 16-01; sgNT) and 38 days (PDX 17-14; sgNT) in mice receiving PDX cells with a control guide (Figure 4H and 4I). All mice showed a high leukemia burden with %hCD45+/live cells >/= 95% at the time when they met endpoint criteria. When we assessed IRF2BP2 protein levels in bone marrow cells of mice transplanted with PDX cells transduced with IRF2BP2-targeting guides, these cells had regained IRF2BP2 expression (Figure S4H).
IRF2BP2 binds to promoter and enhancer regions
Having validated IRF2BP2 as a strong dependency in AML, we next evaluated the underlying mechanisms explaining this dependency. We were particularly eager to understand how IRF2BP2 was implicated in inflammatory pathways. IRF2BP2 has been reported to be involved in transcriptional control (38), prompting us to study the localization of IRF2BP2 on chromatin. We performed chromatin immunoprecipitation (ChIP) for IRF2BP2 in MV4-11 cells using three different antibodies against IRF2BP2 followed by sequencing. We identified 6,269 binding sites that were identified in at least two out of three experiments, defining a set of IRF2BP2 binding targets (Figure 5A and S5A). Approximately 2300 of these peaks were found in promoter regions and 2900 marked enhancers defined by H3K27ac binding (Figure 5A). H3K4me1 marks coincided with binding sites defined as gene enhancers, and the histone mark H3K4me3 was found in H3K27ac-marked promoter regions. Additionally, we identified H3K27ac-marked promotor regions to coincide with RNA polymerase II binding. We assigned genes to genome-wide IRF2BP2 chromatin binding peaks using the model-based analysis of the ChIP-seq (macs2) algorithm for the merged IRF2BP2 ChIP-peaks. GSEA for all genes bound by IRF2BP2 identified immune response pathways to be the most significantly enriched (Figure 5B); the top gene set was TNFα signaling via NF-κB (Figure 5B). To validate these findings, we performed ChIP for HA-tagged IRF2BP2 and H3K27ac in two PDX models and one additional AML cell line. We consistently found genome-wide IRF2BP2 binding in H3K27ac-marked promoter and enhancer regions (Figure 5A, heatmaps 6 and 7 and S5B). We identified core promoter and enhancer IRF2BP2 binding regions across all four models (Figure S5C) with 2097 genes bound in all four experiments (Figure S5D). When we performed GSEA for all genes bound by IRF2BP2 in PDX 16-01, THP1, and PDX17-14 we identified TNFα signaling via NF-κB as the top enriched gene set (Figure 5B). TNFα signaling via NF-κB is also a top enriched pathway in the IRF2BP2 “core” bound gene set (Figure 5B and S5D).
Figure 5. IRF2BP2 represses immune response genes in AML.
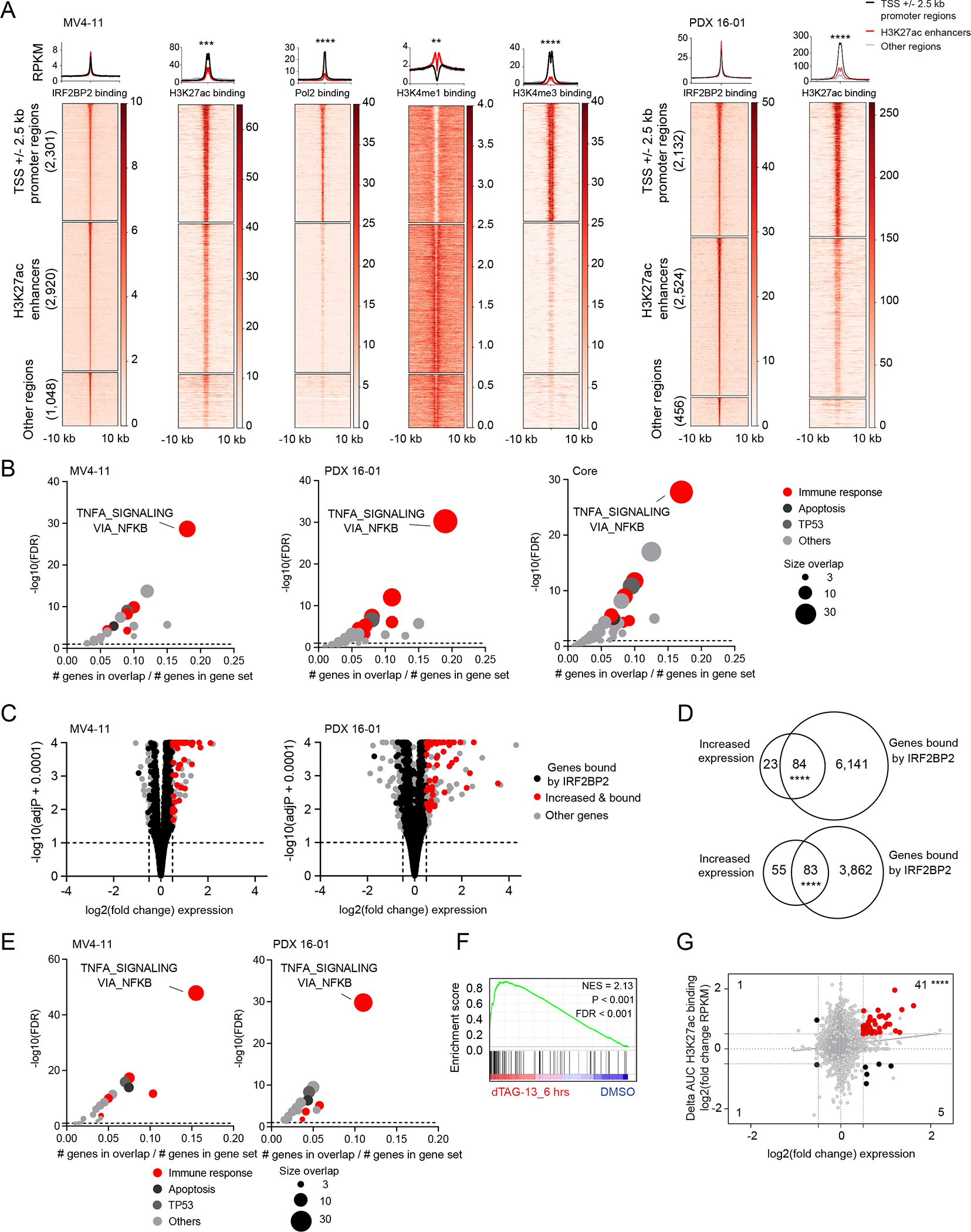
A, Clustered heatmaps and metaplots showing genome-wide IRF2BP2 chromatin binding +/− 10 kb regions in MV4-11 cells (left) and PDX 16-01 (right). Heatmaps showing area under the curve RPKM normalized signal for IRF2BP2, H3K27ac, Pol2, H3K4me1 and H3K4me3 binding for MV4-11 cells, and area under the curve RPKM normalized signal for IRF2BP2 and H3K27ac binding in PDX 16-01. The IRF2BP2 binding sites were grouped into three clusters based on the promotor/enhancer status: promoter regions (TSS +/− 2.5kb), enhancers, and other regions, depicting peaks not classified as promotors or enhancers. Clustered regions were ranked by IRF2BP2 signal. Read density metaplots are showing average RPKM normalized signal for IRF2BP2, H3K27ac, Pol2, H3K4me1 and H3K4me3 in promotor regions (black), H3K27ac enhancers (red) and other regions (gray). Differential read density in promotor versus H3K27ac enhancer regions was evaluated by unpaired t-test with Welch’s correction, **** p < 0.0001, *** p < 0.001, ** p < 0.01.
B, Bubble plots summarizing the significant enrichments of the MSigDB v7.1 collection of 50 hallmark pathways within the top 500 nearest genes bound by IRF2BP2 (macs2, FDR ≤ 0.01), defined by normalized area under the curve signal ranking. Left panel: IRF2BP2 binding in MV4-11 cells, middle panel: IRF2BP2 binding in PDX 16-01 cells, right panel: core intersection of IRF2BP2 binding regions in MV4-11, THP1, PDX 16-01 and PDX 17-14. Enriched gene sets are clustered in functional categories indicated by the color code; red indicates immune response signatures. The bubble size indicates the number of overlapping genes. Hypergeometric test, p ≤ 0.05, FDR ≤ 0.05.
C, Volcano plots depicting the gene-level differential transcriptional status after degradation of IRF2BP2 for 6 hours in MV4-11 cells (left) and PDX 16-01 cells (right). Red dots represent genes that are bound by IRF2BP2 and increased in expression. Black dots represent all other genes that are bound by IRF2BP2. Grey dots depict all other genes. Shown per gene are log2(fold change expression) versus -log10(adjusted p-value + 0.0001).
D, Venn diagram depicting the overlap of genes with increased expression following degradation of IRF2BP2 for 6 hours (fold change expression ≥ 1.5, adjusted p-value ≤ 0.10) and genes bound by IRF2BP2 (ChIP-seq binding sites, defined by macs2, FDR ≤ 0.10) in MV4-11 cells (upper panel) and PDX 16-01 (lower panel). Two-tailed Fisher exact test, **** p < 0.0001. E, Bubble plots summarizing the significant enrichments in MSigDB v7.1 collection of 50 hallmark pathways within the IRF2BP2 bound genes with increased expression following degradation of IRF2BP2 for 6 hours in MV4-11 cells (left) and PDX 16-01 cells (right). Enriched gene sets are clustered in functional categories indicated by the color code; red indicates immune response signatures. The bubble size indicates the number of overlapping genes. Hypergeometric test, p ≤ 0.05, FDR ≤ 0.05.
F, GSEA plot for HALLMARK_INFLAMMATORY_RESPONSE enrichment within IRF2BP2 bound and differentially expressed genes following degradation of IRF2BP2 after treatment with dTAG-13 for 6 hours in MV4-11 cells. NES > 1.3, p-value < 0.001, FDR < 0.001.
G, Scatter plot depicting the overlap between the 1,678 genes with significant changes in H3K27ac binding (Delta(area under curve signal) ≥ 1.5) following treatment with dTAG-13 for 24 hours and genes with increased or decreased expression in MV4-11 cells following treatment with dTAG-13 for 6. Red dots in the right upper quadrant represent 107 genes with increased expression and gain in H3K27ac marking. Statistical significance was tested by two-tailed Fisher exact test, **** p < 0.0001.
Gene expression changes following degradation of IRF2BP2
We next explored whether genes bound by IRF2BP2 were also differentially expressed following degradation of IRF2BP2. We performed RNA-seq in MV4-11 and PDX 16-01 cells with degradable IRF2BP2 after 6 hours, and in MV4-11 cells after 24 hours of dTAG-13 treatment. Significantly more genes were increased than decreased in expression at both time points (Figure 5C, S5E and S5F), in line with IRF2BP2’s proposed primary function as a transcriptional repressor. Strikingly, IRF2BP2 bound most of the upregulated genes at 6 hours (Figure 5C and 5D) and at 24 hours (Figure S5F and S5G). Virtually none of the genes with decreased expression at 6 hours (Figure 5C), and only a minor percentage at 24 hours (Figure S5F) were bound by IRF2BP2. In contrast, there was a significant overlap in genes upregulated and bound by IRF2BP2 at 6 and 24 hours (Figure S5H). In addition, differentially expressed genes following degradation of IRF2BP2 at 6 hours were enriched within bound and upregulated genes at 24 hours post degradation of IRF2BP2 and vice versa (Figure S5I, upper panel). When we analyzed enriched gene sets within the bound and differentially expressed genes at 6 hours in MV4-11 and PDX 16-01 cells, we again found immune response signatures to be the top enriched gene sets at both time points (Figure 5E), with genes regulated by NF-κB in response to TNFα being most significantly enriched (Figure 5E, S5I lower panel). Of note, we also found a strong enrichment for the immune/inflammatory high AML cluster defined by HALLMAK_INFLAMMATORY RESPONSE gene set within bound and upregulated genes following treatment with dTAG-13 for 6 hours (Figure 5F). Moreover, when we performed ChIP-seq for H3K27ac after degradation of IRF2BP2 for 24 hours in MV4-11 cells, we observed a significant increase in genome-wide peaks assigned to genes that had increased expression upon IRF2BP2 degradation for 6 hours (Figure 5G). These results again support a role for IRF2BP2 as a transcriptional repressor in AML cells. When we performed GSEA for the 41 genes with increased expression and increased H3K27ac binding after dTAG-13 treatment (Figure 5F right upper quadrant), we again identified TNFα signaling via NF-κB as the top gene set (Figure S5J).
IRF2BP2 controls NF-κB signaling
Results from the gene expression and ChIP-seq profiling led us to hypothesize that IRF2BP2 is a repressor of NF-κB mediated TNFα signaling that, when acutely perturbed, leads to leukemia cell death. To test this hypothesis, we first established an NF-κB-luciferase reporter assay in MV4-11 cells with degradable IRF2BP2. When we degraded IRF2BP2 with dTAG-13 treatment, we observed an acute upregulation of NF-κB signaling at 6 and 24 hours followed by a decrease in signaling at 72 hours (Figure 6A and Figure S6A). Cells treated with human TNFα as a positive control showed a similar pattern over time. Consistent with the reporter assay results, nuclear NF-κB (RELA) protein levels increased at 24 hours with dTAG-13-induced IRF2BP2 degradation in MV4-11 cells (Figure 6B) and PDX 16-01 cells (Figure S6B). Moreover, we observed an increased enrichment for NF-κB (RELA) chromatin binding upon degradation of IRF2BP2 as compared to the DMSO control when performing ChIP PCR for the canonical RELA binding site at the NFKBA promoter (Figure 6C). If cell death induced by perturbation of IRF2BP2 was mediated by de-repressing NF-κB signaling, then super-repression of NF-κB should rescue this phenotype. To test this hypothesis, we overexpressed the wildtype form of the inhibitor of NF-κB (IκBα), a mutant “super-repressor” allele of IκBα (IκBα MUT) harboring two amino acid substitutions (S32A/S36A) that render the mutated IκBα resistant to phosphorylation and degradation by IκB kinase (IKK) (39), or a GFP control ORF, in AML cells (Figure S6C). When we treated these MV4-11 cells with TNFα to activate NF-κB signaling, we detected an increase in IκB phosphorylation in cells expressing the GFP control or wildtype IκBα ORF, while there was no phosphorylation in cells expressing IκBα MUT (Figure S6D). We next used CRISPR-Cas9 to knock-out IRF2BP2 in MV4-11 cells with the control plasmid, IκBα MUT, and IκBα wildtype (WT) (Figure 6D). Mutated IκBα expression led to a complete rescue of the impaired cell growth with IRF2BP2 deletion in MV4-11 cells, and a greater tolerance of IRF2BP2 knock-out was also evidenced by western blot (Figure 6D and E). The overexpression of IκBα WT led to partial rescue of cell death (Figure 6E), consistent with its repressive effect and high levels of expression. Consistently, doublings over time were significantly increased in cells expressing mutated IκBα compared to cells with a GFP control vector upon IRF2BP2 knock-out, the latter which underwent cell death (Figure S6E). Similarly, the mutant “super-repressor” allele of IκBα rescued the effects of IRF2BP2 knock-out in PDX 17-14 cells (Figure S6F and S6G). Of note, when we treated MV4-11 cells overexpressing IκBα or a GFP control with JQ1 or THZ1, two compounds that have been shown to perturb oncogenic transcription, we did not observe any significant difference in the IC50 for these drugs when comparing IκBα WT, mutant or GFP expressing cells (Figure S6H). All told, these data are consistent with the cell death associated with IRF2BP2 loss being mediated through activation of NF-κB signaling.
Figure 6. IRF2BP2 controls NF-κB signaling in AML.
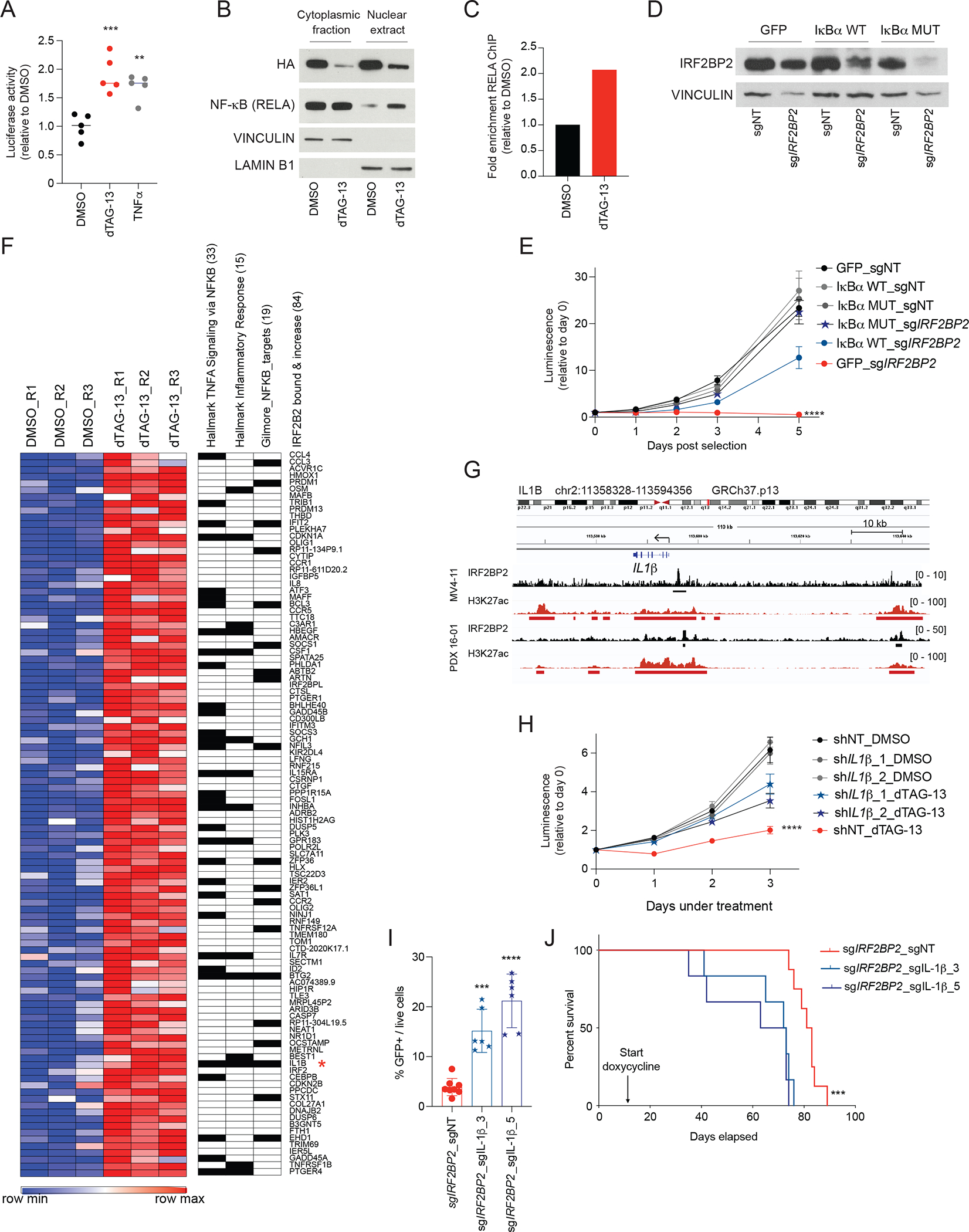
A, NF-κB reporter assay showing luciferase activity following degradation of IRF2BP2 at 6 hours after treatment with dTAG-13 or TNFα, which is used as a positive control. One-way ANOVA, Dunnett’s multiple comparison test, *** p < 0.001, ** p < 0.01.
B, Western blot analysis showing NF-κB protein (RELA) in the cytoplasmic fraction and nuclear extracts following degradation of IRF2BP2 after treating MV4-11 cells with degradable IRF2BP2 with dTAG-13 for 24 hours. Vinculin indicates the cytoplasmic fraction; lamin B1 is found in nuclear extracts.
C, ChIP PCR assessing enrichment for NF-κB (RELA) chromatin binding 24 hours post DMSO and dTAG-13 treatment in MV4-11 cells with degradable IRF2BP2 (amplification of NFKBA promotor region).
D, Western blot analysis for IRFBP2 protein in MV4-11 cells overexpressing plx317 GFP control ORF (GFP), plx317 IκBα (IκBαWT), or plx317 IκBα(S32A/S36A) (IκBαMUT), double-infected with either non-targeting CRISPR guide (sgNT), or sgIRF2BP2.
E, Viability assay in MV4-11 cells overexpressing plx317 GFP control ORF, plx317 IκBα, or plx317 IκBα(S32A/S36A), double-infected with either non-targeting CRISPR guide (sgNT), or an IRF2BP2 targeting guide (sgIRF2BP2). Two-way ANOVA, **** p < 0.0001.
F, Heatmap of gene expression profiling by RNA-seq showing 84 IRF2BP2 bound genes with increased expression after 6 hours of treatment with dTAG-13 compared to DMSO in triplicates (fold change ≥ 1.5, adjusted p ≤ 0.1; left panel). Genes are ranked by fold change expression. IL-1ß is marked with a red star. Leading edge genes for the top two enriched immune response hallmark gene sets and for the Gilmore NF-κB target gene set are annotated. The number of overlapping genes is shown in parentheses.
G, Integrated genomics viewer plots (GRCh37/hg19) showing IRF2BP2 and H3K27ac ChIP-seq RPKM binding signal in the IL-1ß neighborhood region in MV4-11 cells and PDX 16-01.
H, Viability assay in MV4-11 cells with degradable IRF2BP2 and knock-down of IL-1ß with two different hairpins (shIL-1ß_1/2) under DMSO or dTAG-13 treatment. Two-way ANOVA, **** p < 0.0001.
I, Flow cytometry analysis for GFP-positive cells on bone marrow aspirates from sub-lethally irradiated NSG mice transplanted with PDX 16-01 cells infected with doxycycline-inducible CRISPR guides against IRF2BP2 plus sgIL-1β or a non-targeting control guide after a two-week doxycycline diet (n = 6 to 8 mice per group). One-way ANOVA, Dunnett’s multiple comparison test, *** p = 0.0002, **** p < 0.0001.
J, Kaplan-Meier curves depicting survival of NSG mice transplanted with PDX 16-01 cells transduced with doxycycline-inducible CRISPR guides against IRF2BP2 plus sgIL-1β or a non-targeting control guide under continuous doxycycline diet. The median survival of the mice transplanted with PDX 16-01 sgIRF2BP2 plus sgIL-1β_3/5 was 72.5 and 68 days as compared to 82 days for mice who received sgIRF2BP2 plus sgNT PDX 16-01 cells. Log-rank Mantel-Cox test, *** p = 0.0004.
IL-1ß is a mediator of NF-κB signaling following IRF2BP2 degradation
To identify mediators of NF-κB activation upon perturbation of IRF2BP2, we further explored genes bound by IRF2BP2 that were differentially expressed at 6 hours post degradation of IRF2BP2 in the RNA-seq data. Evaluation of the top differentially expressed IRF2BP2 targets revealed IL-1ß bound by IRF2BP2 (Figure 6F, 6G and S6I) and increased in expression with IRF2BP2 degradation at 6 hours, also scoring as one out of two genes in three different gene sets for immune response and TNFα signaling (Figure 6F). These results were validated by quantitative PCR; IL-1ß was upregulated at 6 hours, returning to near baseline by 24 hours after IRF2BP2 degradation (Figure S6J).
If IL-1ß is a key positive regulator of NF-κB signaling upon perturbation of IRF2BP2, then loss of IL-1ß function should attenuate NF-κB signaling and the negative effects of IRF2BP2 degradation on cell growth. When we knocked down IL-1ß in MV4-11 cells (Figure S6K) expressing degradable IRF2BP2, we indeed observed attenuation of NF-κB nuclear translocation (Figure S6L) and rescue of the viability reduction following degradation of IRF2BP2 (Figure 6H). Consistently, we observed a diminished and delayed induction of cleaved caspase 3 (Figure S6M), supporting the hypothesis that IL-1ß enhances an inflammatory response upon perturbation of IRF2BP2 that results in death of AML blasts. Moreover, to test whether constitutive abrogation of IL-1ß activity would influence cell growth in the context of IRF2BP2 degradation, we pre-treated MV4-11 cells with degradable IRF2BP2 for 12 hours with a neutralizing anti-h-IL-1ß-antibody followed by dTAG-13. Pre-treatment with an anti-h-IL-1ß-antibody abrogated the activation of NF-κB signaling in our reporter cells (Figure S6N). When we assessed cell viability, anti-h-IL-1ß-antibody treated cells showed superior viability compared to cells pre-treated with the antibody solvent water when degrading IRF2BP2, resulting in a rescue of the IRF2BP2 dTAG phenotype (Figure S6O).
We next sought to assess if we could rescue the enhancing effect of IL-1ß upon IRF2BP2 perturbation in PDX cells in vivo. We transduced Cas9 expressing PDX 16-01 cells with an inducible IRF2BP2-targeting CRISPR guide, and, in a second step, knocked-out IL-1ß with two different sgRNAs in these cells (Figure S6P). We observed a partial rescue of the viability effect in vitro, while knockout of IL-1β alone did not increase proliferation (Figure S6Q). We next transplanted sub-lethally irradiated NSG mice with the above engineered PDX 16-01 cells with IRF2BP2-targeting guides. Upon confirmation of engraftment, the mice received a doxycycline containing diet to induce knock-out of IRF2BP2. We performed bone marrow aspirations on all mice after two weeks on doxycycline and observed significantly less reduction of leukemia burden in mice transplanted with PDX 16-01 cells with knock-out of IL-1ß (Figure 6I). This difference in disease burden translated into prolonged median survival in mice transplanted with IRF2BP2 knock-out PDX 16-01 cells as compared with mice with concurrent IRF2BP2 and IL-1ß knock-out (Figure 6J). All told, our data support a role for IRF2BP2 in controlling IL-1ß-mediated NF-κB signaling. Its de-repression leads to an acute inflammatory signaling prompting AML cell death.
Discussion
This study was designed to identify key players at the crossroads of immune and inflammatory signaling and selective dependencies in AML. To this end we applied genome-wide CRISPR-Cas9-dropout screens for the unbiased identification of novel AML dependencies. By intersecting hits from three independent screens using complementary RNAi and CRISPR-Cas9 approaches, we increased the confidence in prioritized hits and reduced the likelihood of off-target effects confounding hit selection. We identified IRF2BP2 as a strong dependency in AML among a small group of well-established genes, including MYB and CBFB, providing additional confidence in this unexplored target. AML cell lines were more dependent on IRF2BP2 than any other cancer type, nominating IRF2BP2 as a selective enriched dependency in AML. Pathway enrichment analysis identified a subset of AML with a strong enrichment for immune/inflammatory pathways. We found the strongest enrichment in samples from patients with AML that expressed a monocytic signature. IRF2BP2 scored as the top dependency within monocytic AML cell lines when we assessed AML selective, core dependencies.
Our study explored the mechanistic underpinnings of IRF2BP2 in its dual role as a strong dependency and transcriptional regulator. While most other studies focus on the role of transcriptional activators as cancer dependencies (e.g., BRD4, MYB, WT1, MYC) (40–43), our data provide evidence that IRF2BP2 acts primarily as a transcriptional repressor in AML. Genome-wide chromatin studies revealed that IRF2BP2 binds both in enhancer and promoter regions of AML cells. Assessing global gene expression changes following degradation of IRF2BP2, however, the majority of differentially expressed genes upregulated at 6 and 24 hours were bound by IRF2BP2 at baseline. Moreover, we observed a significant gain in H3K27ac in upregulated IRF2BP2 targets following degradation of IRF2BP2, consistent with its role as a transcriptional repressor.
Our study focused on the molecular function of IRF2BP2 suggests the repression of inflammation as a key strategy for AML cell survival. While tumor evasion from immune surveillance is an established hallmark of cancer, to our knowledge, cell-intrinsic transcriptional repression of inflammatory signaling within cancer cells has not been extensively studied. Specifically, by acting as a transcriptional repressor, IRF2BP2 puts a break on NF-κB mediated signaling. Previous studies suggest that cells require a precise set point of NF-κB signaling (44), and our results indicate a similar requirement in AML cells. Our data suggest that de-repression of NF-κB signaling by perturbation of IRF2BP2 in leukemia cells induces an overwhelming inflammatory response. In this model, IRF2BP2 suppresses NF-κB signaling as a cell-intrinsic innate immune response that would otherwise be detrimental for the leukemia cell. Thus, perturbation of IRF2BP2 serves to release the safety break, resulting in an inflammatory burst and driving the AML cell toward cell death.
Over-expression of an IκBα-super-repressor mutant (Figure S6C), which suppresses NF-κB signaling, rescued AML cells with IRF2BP2 knock-out from death. Importantly, overexpression of an IκBα-super-repressor mutant in MV4-11 cells did not influence the IC50 of transcription perturbing compounds such as JQ1 and THZ1 (Figure S6H), suggesting that the hyperactivation of NF-κB signaling is not a non-specific mechanism leading to cell death with transcriptional perturbation.
TNFα acts as a pleiotropic cytokine inducing various cellular responses, ranging from inflammatory cytokine production to cell death (45). By activating the strongly pro-inflammatory transcription factor NF-κB, TNFα plays an essential role in the acute phase reaction, modulating fever and inflammation (46). Our understanding of its functions in leukemic blasts, however, remains limited. It is well established that in resting cells, NF-κB resides in the cytoplasm, where it is controlled by inhibitory kappa B (IκB) proteins. Upon stimulation (e.g., by IL-1ß), IκB is phosphorylated by the IκB kinase complex (IKK), which leads to the rapid degradation of IκB and the release of NF-κB. NF-κB subsequently translocates into the nucleus, where it controls gene expression (47). Accordingly, with IRF2BP2 degradation, we observed an upregulation of NF-κB signaling based on an NF-κB reporter, global gene expression changes, and an increase in nuclear NF-κB protein. While at first glance a paradoxical pairing, activation of TNFα-mediated NF-κB signaling can also lead to apoptotic cell death. Specifically, it has been reported that TNFα-induced caspase 8 activation promotes apoptosis in cells treated either with cycloheximide or a SMAC mimetic (48). With IRF2BP2 repression in AML, we observed an increase in annexin V/PI-positive cells, induction of cleaved caspase 3 (Figure 3E and F and S3D), and an increase in cleaved caspase 8 (Figure 3G), all consistent with apoptotic cell death.
In exploring key mediators of TNFα signaling, we found IL-1ß to be a molecular protagonist in the early phase following the degradation of IRF2BP2. When we reduced IL-1ß expression in cells and subsequently degraded IRF2BP2, we observed less profound and more delayed cell death (Figure 6H and S6M). Moreover, we observed a partial rescue of the effect on leukemia burden and survival in PDX cells when we compared knock-out of IRF2BP2 with or without additional loss of IL-1β in vivo (Figure 6I and J).
Several studies have examined the effects of an inflamed micro-environment on hematopoietic stem cells (HSCs) and leukemogenesis (49–52). For example, some groups have focused on transcription factors that activate inflammatory pathways; in this context GATA2 has been reported to positively regulate both IL-1β and CXCL-2 expression in AML (53). Others have explored the concept of an inflamed milieu providing advantage for selective clones. For example, a comprehensive study by Hormaechea-Agulla et al. showed the clonal advantage of Dnmt3a loss-of-function clonal hematopoiesis under chronic mycobacterial infection and INFγ treatment (54). Avagyan and colleagues showed in a zebrafish model that clonal fitness of a mutant CHIP clone can be driven by enhanced resistance to extrinsic inflammatory signals (55). While our study focused on a role for IRF2BP2 as a repressor of cell-intrinsic inflammation, cell-extrinsic, environmental factors might also contribute to the inflammatory phenotype of leukemic cells and are the subject of ongoing investigations. One hypothesis in light of these recent findings is that in a steady-state situation, IRF2BP2 controls cell-intrinsic inflammatory signaling, allowing leukemic blasts to tolerate more environmentally-mediated inflammatory signals, conferring a selective advantage.
The role of IL-1β, as one major mediator of inflammatory signaling in hematopoietic cells, is complex. Collectively, published studies suggest a dose- and time-dependent (acute versus chronic exposure) response to IL-1ß. Importantly, some studies assess cell-intrinsic effects, while others are focused on cell-extrinsic mechanisms; some focus on impact on normal hematopoietic progenitors and others on leukemic cells. For example, Yang and collegues observed lower IL-1ß expression in CD34+/CD38− cells from patients with AML compared to CD34+ cells from healthy volunteers. Moreover, they were able to induce apoptosis by higher concentrations of IL-1ß treatment, and forced expression of IL-1ß, which also significantly diminished the self-renewal capacity of IL-1ß transduced CD34+/CD38− cells. Furthermore, when they transplanted IL-1ß over-expressing blasts into immunocompromised mice, the engraftment and AML reconstitution were impaired (56). Carey et al. (57), however, described an expansion of myeloid progenitors in longer-term assays in a subgroup of samples from patients with AML when culturing them with one-tenth the IL-1ß concentration (10 ng/ml) used in the study led by Yang et al. (56). Work by Pietras et al. on the effects of IL-1β in normal hCD34+ cells further support a time-dependent function of IL-1β; while IL-1β supports inflammation as an emergency signal in an acute setting, chronic IL-1β exposure restricts lineage output of hematopoietic stem cells (58).
While our data suggest that cell-intrinsic IL-1ß acts as a key enhancer of an inflammatory burst following the depletion of IRF2BP2, we do not think that IL-1ß is the sole mediator of the observed hyper-inflammation. First, knock-down of IL-1ß does not fully rescue the phenotype of IRF2BP2 perturbation. Second, our gene expression and chromatin studies suggest that IRF2BP2 is a direct transcriptional repressor of other members of TNFα-mediated NF-κB signaling, such as TNFR2, suggesting NF-κB signaling activation on multiple levels. Third, it is well established that canonical NF-κB signaling can be activated by numerous, and even multiple parallel stimuli, feeding into and maintaining a highly dynamic circuit, with NF-κB at the crossroads as the master regulator of innate immune response (59). Our mechanistic model suggests IL-1ß as a direct target of IRF2BP2, which is de-repressed upon IRF2BP2 perturbation to activate NF-κB signaling. IL-1ß is also an established target gene of NF-κB, which will then limit its expression in a negative feedback loop, consistent with our observations of an early increase in IL-1ß expression that trends down by 24 hours.
Tumor necrosis factor (TNF), first described by O’Malley et al. (60), was initially reported to induce programmed cell death or apoptosis. Following depletion of IRF2BP2, we consistently observed activation of caspase-8 and caspase-3, which were previously thought to trigger apoptosis exclusively. With an increasing understanding of distinct forms of regulated cell death (61), caspase 8 and caspase 3 were more recently found to mediate both apoptotic and pyroptotic cell death. Pyroptosis is a rapid, lytic form of cell death, induced by caspase 8 or caspase 3 cleavage of gasdermin D (GSDMD) leading to pore-forming fragments of GSDMD (62, 63), resulting in the release of inflammatory cytokines. Necroptosis, yet another form of regulated cell death, is associated with an increase in cytokine levels, promoting inflammation by the release of cytokines from dying cells, including IL-1ß, executed by oligomerization of mixed lineage kinase domain-like (MLKL) and subsequent plasma membrane rupture (64–66). Within 24 hours following degradation of IRF2BP2 we did not observe induction of cleaved GSDMD nor phospho-MLKL as hallmarks of pyroptosis or necroptosis (Figure S3E and S3F). Whether pyroptotic or necroptotic cell death might be contributing to the phenotypic consequences of IRF2BP2 loss as secondary effects at later time points remains to be determined but will be the focus of future studies.
The kinetics of CRISPR- or RNAi-based approaches to target repression are slow, limiting the assessment of target perturbation on acute gene expression changes. In contrast, our study leveraged a degradation technology that allowed us to study immediate, direct changes in global gene expression (33). Moreover, chemical degradation more closely approximates treatment with a small molecule and can better enable preclinical target validation in the absence of a small molecule inhibitor.
Using patient-derived xenograft models, we demonstrated that depletion of IRF2BP2 is a viable and highly efficacious therapeutic strategy for AML. In an in vivo study approximating a clinical scenario where we induced knock-out of IRF2BP2 in patient-derived leukemia cells after the mice had developed leukemia, we observed a significant reduction in leukemia burden and an increase in median survival in mice that had received AML cells with IRF2BP2-targeting guides. When mice transplanted with AML cells with IRF2BP2-targeting guides did eventually die, they died of leukemia with re-gained IRF2BP2 expression (Figure S4H). The therapeutic efficacy of single target perturbation of IRF2BP2 positions this transcriptional repressor as a promising target for future clinical translation. While there are no small molecule inhibitors of IRF2BP2, one might envision a natural glue-like degrader or proteolysis targeting chimera (PROTAC) strategy for the repression of IRF2BP2 therapeutically. Importantly we confirmed in hCD34+ bone marrow cells that targeting IRF2BP2 will not indiscriminately kill normal hematopoietic progenitor cells (Figure 4B) suggesting potential for a high therapeutic window.
Myeloid blasts have been described as abnormal white blood cells over 200 years ago, inspiring Rudolf Virchow to name this condition of an excessive number of white blood cells as leukemia (“white blood”) in 1845 (67), yet, there are many unexplored aspects of the blast intrinsic immune programs. Our study revealed a subgroup of AML strongly enriched for immune inflammatory pathways. Within this “immune/inflammatory response high” subgroup, we observed more samples with a monocytic signature than in the control group. We exploited this observation to uncover immune dependencies that led to the identification of IRF2BP2. Intriguingly, recent studies highlight a monocytic AML state associated with resistance to venetoclax-based treatment (68) – not only supporting the need for alternative or combination treatment strategies for this AML subgroup, but also suggesting the immune differentiation status of AML as a biomarker for clinical decision making.
In summary, we identified IRF2BP2 as a dependency in AML and determined that IRF2BP2 acts as a transcriptional repressor, controlling NF-κB mediated TNFα signaling to keep AML cells alive. Perturbation of IRF2BP2 induces an acute inflammatory response, enhanced by IL-1ß, which promotes leukemic blast cell death. More broadly, the functional characterization of IRF2BP2 reveals transcriptional repression as a strategy for leukemia cell survival. This discovery provides a basis for leveraging acute inflammation as self-directed immuno-therapy and a therapeutic approach to control leukemic blasts.
METHODS
EXPERIMENTAL MODEL AND SUBJECT DETAILS
Cell lines and patient-derived xenograft samples
The AML cell lines MV4-11, U937, MOLM13, NB4, and THP1 cells were cultured in RPMI 1640 (Cellgro) supplemented with 10% fetal bovine serum (FBS, Sigma-Aldrich) and 1% penicillin-streptomycin (PS, Cellgro). HEK293T cells were cultured in DMEM with 10% FBS and 1% PS. All cell lines were STR-profiled. Primary patient samples were acquired following written informed consent in accordance with the Declaration of Helsinki, and patient-derived xenografts (PDX) models were established under protocols approved by Dana-Farber Cancer Institute and Cincinnati Children’s Hospital Medical Center institutional review boards. Characteristics of the PDX models are provided in the table below. For short-term in vitro culture, PDX cells were maintained in IMDM containing 20% FBS and 1% PS, and supplemented with 10 ng/mL human SCF, TPO, FLT3L, IL3 and IL6 (PeproTech 300–07, 300–18, 300–19, 200–03 and 200–06).
QUANTIFICATION AND STATISTICAL ANALYSIS
GSEA v4.2.0 (details below), GraphPad PRISM 9, R 4.0.1 and Python 3.7.4 software packages were used to perform the statistical analyses. Statistical tests used are specified in the figure legends. Errors bars represent standard deviation, unless otherwise stated. The threshold for statistical significance is p-value ≤ 0.05, unless otherwise specified.
METHOD DETAILS
Genomic screens
The CRISPR screen was performed on 789 cancer cell lines including 21 AML at the Broad Institute using the Avana sgRNA library, containing 73,372 guides with an average of 4 guides per gene (69, 70). The combined RNAi (Broad, Novartis, Marcotte) screen included 712 cancer cell lines with 23 AML cell lines as previously described (27, 28). The CRISPR Sanger screen was performed at the Sanger Institute (29, 71) on 317 cancer cell lines including 2 AML cell lines, using a 90,709 sgRNA library targeting 18,009 genes (~5 sgRNAs/gene) as previously described (72).
Plasmids and reagents
To knock-out/knock-down IRF2BP2 the sgRNAs/shRNAs from the CRISPR(Avana)/Broad RNAi library were used. Inducible hairpins targeting IRF2BP2 were purchased from Dharmacon (TRIPZ lentiviral inducible shRNA collection). To rescue these hairpins, we inserted silent point mutations (A to G at bp position 1056, A to G at 1059 bp, and A to C at 1062 bp) at the hairpin binding sites in the IRF2BP2 ORF (Precision LentiORF, Dharmacon; OHS5897-202624069). The pLX317-GFP ORF was obtained from the Broad Institute. MV4-11 cells were transduced with pMMP-luc-neo retrovirus (the pMMP-luc-neo vector was a kind gift from Andrew Kung) and selected with neomycin at 1 mg/mL. MV4-11-luc cells were then stably infected with either a non-targeting or an IRF2BP2-directed doxycycline-inducible hairpin (RHS4740-EG359938, Dharmacon).
The lentiviral Cas9 vectors co-expressing a blasticidin resistance gene or GFP (pXPR_BRD101 and pXPR_BRD104) and the lentiviral sgRNA expression vectors co-expressing a puromycin resistance gene, mCherry or mAmetrine (pXPR_BRD003, pXPR_BRD043 and pXPR_BRD052) were provided by the Genetic Perturbation Platform (GPP) at the Broad Institute. The Cas9-T2A-mCherry and DOX-inducible sgRNA vectors were obtained from Addgene (70182 and 70183). sgRNAs were cloned into BsmBI-digested vectors.
IRF2BP2 was cloned into pLEX_305-N-dTAG using gateway recombination cloning strategies (Invitrogen) as previously described (Nabet et al., 2018) and dTAG-13 was synthesized as previously described (73). To knock-out endogenous IRF2BP2 sgIRF2BP2_1 (sgRNA IRF2BP2_1: CCTCGTAGTTGACGCAGCCG) and sgIRF2BP2_2 (sgRNA IRF2BP2_2: TCTCGATGACGAACTCGACG) from the AVANA library were used. To rescue the knock-out of endogenous IRF2BP2 by sgIRF2BP2_1 and 2 we introduced the following point mutation to the respective PAM sites: PAM_1: C to T at bp position 111 and PAM_2: C to T at bp position 141.
PGreenFire1-NF-κB (EF1a-puro) Lentivector was purchased from SBI System Biosciences (TR012PA-P). Luminescence was detected by the Dual-Luciferase Reporter Assay System and Dual-Glo Luciferase Assay System (E1919 and E2920, Promega) following the manufacturer’s protocol.
IκBα-WT (pDONR223_NFKBIA_WT) was purchased from Addgene (81833) and cloned into pLEX_307 (Addgene, 41392) using gateway cloning following the manufacturer’s protocol. To generate the super-repressor mutant (IκBα-MUT) two amino acids were substituted (S32A/S36A).
Lentiviral human shRNA targeting IL-1ß (RHS4531-EG355) and a non-targeting control (empty vector, RHS4349) were purchased from Dharmacon. To knock-out IL-1β the sgRNAs from the CRISPR(Avana) library were used.
An anti-hIL-1β-IgG monoclonal antibody was purchased from InvivoGen (mabg-hil1b-3).
For subcellular fractionations a subcellular protein fractionation kit for cultured cells (Thermo Fischer Scientific, Cat. No. PI78840) was used following the manufacturer’s instructions.
Nucleofection
Nucleofection was prepared using the P3 Primary Cell 4D-Nucleofector X Kit S (Lonza V4XP-3032) for CD34+ HSPCs and PDX cells. Per transfection, 6 μg purified Cas9 nuclease (Integrated DNA Technologies #1081058) was mixed with 100 pmol of chemically modified synthetic sgRNA (Synthego) in P3 buffer and incubated at room temperature for 10 minutes to form RNP. Thawed CD34+ HSPCs were cultured in StemSpan SFEM II medium (StemCell Technologies #09605) supplemented with 100 ng/mL human SCF, TPO and FLT3L, and 10 ng/mL IL-3 and IL-6, for two days before nucleofection. For each nucleofection, 2–3 ×105 CD34+ cells or PDX cells were harvested, washed once with PBS, then mixed with the RNP and nucleofected using the Lonza 4D-nucleofector X Unit (program DZ-100). Two days after nucleofection cells were seeded to assess colony forming capacity.
Colony forming assay
Nucleofected cells were resuspended in MethoCult Express methylcellulose medium (StemCell Technologies #04437) and plated at the indicated numbers; colonies were scored on day 10 post seeding.
Flow cytometry
Apoptosis was assessed with an APC Annexin V Apoptosis Detection Kit (BioLegend) following the manufacturer’s protocol. Mouse samples from bone marrow and peripheral blood were stained with human CD45 (Invitrogen Cat. No. MHCD4528) and murine CD45 (Biolegend Cat. No. 103113) antibodies, unless the cells had been previously infected with a fluorescence marker (GFP, mcherry or mAmetrine). Flow cytometry was performed on a LSR Fortessa or FACS Celesta flow cytometer. Cells were sorted on a FACSAria II flow cytometer (BD Biosciences). The data were analyzed with FloJo software (TreeStar).
Western blotting
Proteins were extracted using Lysis Buffer (Cell Signaling Technology) supplemented with Complete, Ethylenediaminetetraacetic Acid (EDTA)-free Protease Inhibitor Cocktail (Roche Diagnostics) and Phosphatase Inhibitor Cocktail (Roche Diagnostics). Protein samples were separated by SDS-PAGE and subsequently transferred to polyvinylidene difluoride (PVDF) membranes, which were blocked in 5% BSA and incubated with primary antibodies against IRF2BP2 (Proteintech, Cat. No. 18847-1-AP), vinculin (Cell Signaling Technology, Cat. No. 4650S), HA (Cell Signaling Technology, Cat. No. 3724S), caspase 3 (Cell Signaling Technology, Cat. No. 9662S), cleaved caspase 3 (Cell Signaling Technology, Cat. No. 9664S), caspase 8 (Cell Signaling Technology, Cat. No. 9746S), cleaved caspase 8 (Cell Signaling Technology, Cat. No. 9496S), actin (Cell Signaling Technology, Cat. No. 3700S), lamin B1 (Abcam, Cat. No. ab16048), H3K4me3 (Cell Signaling Technology, Cat. No. 9733S), NF-κB p65 (Cell Signaling Technology, Cat. No. 8242S), IL-1ß (Novus Biologicals, Cat. No. NB600-633), phospho-IκBα (Cell Signaling Technology, Cat. No. 9246L), IκBα (Cell Signaling Technology, Cat. No. 9242S), Gasdermin D (Cell Signaling Technology, Cat. No. 96458), Cleaved Gasdermin D (Cell Signaling Technology, Cat. No. 36425), MLKL (Abcam, Cat. No. ab184718), phospho-MLKL (FabGennix, Cat. No. PMLKL-140AP); Phospho-MLKL Positive Control from FabGennix (Cat. No. PC-MLKL) was used as a positive control. For immunoblots showing phosphorylation and total protein content, the same protein lysates were run on separate blots to present one experiment and were normalized to its own loading control. Membranes were washed in TBS-T and incubated with the appropriate horseradish peroxidase-conjugated secondary antibodies. Signal was detected by enhanced chemiluminescence (ThermoFisher Scientific).
Cell viability assays
Cells were suspended at a concentration of 15,000 cells/mL and seeded at 40 μl/well into 384-well plates. Cells were assessed for cell viability on day 0 and subsequent time points using the Cell-TiterGlo luminescent assay kit (Promega) according to the manufacturer’s protocol. Luminescence was read on a Fluostar Omega Reader (BMG Labtech), and the viability was normalized to day 0.
Lentivirus production and transduction
Virus was produced using HEK293T cells transfected with lentiviral expression vectors, together with envelope VSVG and the gag-pol psPAX2 constructs. For transduction, AML cells were mixed with viral supernatant and 4–8 μg/mL polybrene. In some experiments, cells were centrifuged in viral supernatant at 1000 g for 1hr at 33 °C to enhance the transduction efficiency.
Xenograft transplantation
All in vivo studies were conducted under the auspices of protocols approved by the Dana-Farber Cancer Institute Animal Care and Use Committee. Six- to eight-week-old NOD/SCID/IL2rγnull (NSG) mice (The Jackson Laboratory) were injected with 0.5 to 1×106 cells. Prior to the transplantation of patient-derived cells, mice were conditioned with sub-lethal radiation. For bioluminescence imaging mice were injected with 75 mg/kg i.p. d-Luciferin (Promega), anesthetized with 2–3% isoflurane, and imaged on an IVIS Spectrum (Caliper Life Sciences). A standardized region of interest (ROI) encompassing the entire mouse was used to determine total bioluminescence flux. To assess leukemia burden, bone marrow was harvested from femur, tibia and spine, and red blood cells were lysed (BD PharmLyse) prior to analysis by flow cytometry.
ChIP-sequencing
25 million (for histone marks) to 100 million AML cells (for IRF2BP2) per condition were crosslinked with 1% formaldehyde in cell culture media for 10 minutes and then quenched with 0.125 mM glycine. Cells were lysed in Nuclei EZ Isolation buffer (Sigma EZ Prep Nuclear Isolation Kit, Cat No. NUC-101) supplemented with protease inhibitor. Chromatin was sheared using a Covaris instrument. Subsequently, 0.5% of the lysate was removed as the input control. The remaining lysate was incubated with 30 μl (for histone marks) or 100 μl (for IRF2BP2) of protein G dynabeads (Thermofisher) and 5 μg of H3K27Ac (ab4729 abcam; H3K4me1: ab8895, H3K4me3: ab8580) or 10 μg of IRF2BP2 antibody (r1: abcam_ab114997, r2: Bethyl_A303-189A-T, r3: Bethyl_A303-190A), HA (Cell Signaling Technology_3724S), or NF-κB (RELA) antibody (Sigma-Aldrich_17-10060) at 4 degrees C overnight (antibodies and beads were incubated together for 30 minutes at 4 degrees C prior to the addition of cell lysate). The precipitated lysate was then washed sequentially in two washes of each of the following buffers: ice cold low salt buffer (20 mM Tris-HCl pH 8.1, 150 mM NaCl, 2 nM EDTA, 1% Triton-X100, 0.1% SDS), high salt buffer (20 mM Tris-HCl pH 8.1, 2 mM EDTA, 500 mM NaCl, 1% Triton-X100, 0.1% SDS), LiCl buffer (10 mM Tris-HCl pH 8.1, 0.25 M LiCl, 1 mM EDTA, 1% deoxycholic acid, 1% IGEPAL CA-630) and TE buffer (10 mM Tris-HCl ph8.1, 1 mM EDTA). Samples were eluted in elution buffer (1x TE pH 7.4, 1% SDS, 150 mM NaCl, 5 mM DTT). RNase A, proteinase K, and 0.2 M NaCl were added to the elution buffer and samples were incubated at 65 degrees C for at least four hours to reverse cross-linking. DNA was purified using AMPure XP beads (Agencourt). ChIP-seq libraries were prepared using Swift S2 Acel reagents on a Beckman Coulter Biomek i7 liquid handling platform from approximately 1 ng of DNA according to manufacturer’s protocol and 14 cycles of PCR amplification. Finished sequencing libraries were quantified by Qubit fluorometer and Agilent TapeStation 2200. Library pooling and indexing were evaluated with shallow sequencing on an Illumina MiSeq. Subsequently, libraries were sequenced on a NovaSeq targeting 40 million 100 bp read pairs for H3K27ac, and 20 to 40 million 50 bp reads for H3K4me1, H3K4me3 and IRF2BP2 by the Molecular Biology Core facilities at Dana-Farber Cancer Institute.
RNA-sequencing
All experiments were performed in technical replicates for each time point and condition. ERCC RNA spike-in mix was added to all samples following the manufacturer’s instructions (ThermoFischer, 4456740). RNA was extracted from cells with the RNeasy Kit and on-column DNA digestion (Qiagen). For RNA-sequencing of MV4-11 and PDX 16-01 cells, polyA mRNA was isolated and libraries were prepared using the TruSeq Stranded mRNA Kit (Illumina) according to the manufacturer’s protocol. All samples were sequenced on an Illumina NovaSeq 6000 instrument with paired-end 150 bp reads to a depth of 40–60 million reads per sample.
RNA-sequencing for all parental PDX samples was performed in one replicate following the above-described protocol except for PDX 2017-63 and 2017-94. For these two models RNA was extracted with the mirVana miRNA Isolation Kit (Thermo Fisher, Grand Island, NY) following the manufacturer’s protocol. The library for RNA-sequencing was prepared by using NEBNext Ultra II Directional RNA Library Prep kit (New England BioLabs, Ipswich, MA). Sequencing was performed with single-end 51 bp reads at the University of Cincinnati.
Dependency data analysis
The CRISPR Avana 20Q3 public data were downloaded from the depmap.org portal. https://depmap.org/portal/download/. The gene effect scores summarizing the guide depletion were determined based on the CERES algorithm (26). The combined RNAi 20Q3 public data were downloaded from the depmap.org portal https://depmap.org/portal/download/. The effect of gene knock-down on cell line viability was inferred based on the DEMETER2 model (28). The Sanger CRISPR public data were downloaded from the Sanger Score Project site https://score.depmap.sanger.ac.uk/. The genetic differential dependencies enriched in the AML cell lines were identified separately for each of the three screens based on the eBayes empirical Bayes linear-model implemented in the limma v3.42.2 R package available in Bioconductor v3.9 (74) by performing a two-tailed t-test for the difference in distribution of gene dependency scores in AML compared to all other screened cell lines. Statistical significance for differential gene dependency was calculated as a q-value derived from the p-value corrected for multiple hypothesis testing using the Benjamini and Hochberg method (75) (limma eBayes |effect size| ≥ 0.3, q-value ≤ 0.1). Genes that scored as AML enriched dependencies in all three screens were defined as Core AML dependencies. Statistical significance of overlapping AML enriched dependency hits identified in pairs of screens was calculated by two-tailed Fisher exact test (p-value ≤ 0.05).
Gene set enrichment analysis (GSEA)
GSEA v4.2.0 software was used to identify functional associations of genome-wide molecular profiles (76, 77) within the MSigDB v7.1 database collection of gene sets (http://www.gsea-msigdb.org/gsea/downloads.jsp) and http://www.bu.edu/nf-kb/gene-resources/target-genes/. Significance cut-offs were assessed based on the GSEA standard recommendations: absolute Normalized Enrichment Score (NES) ≥ 1.3, p-value ≤ 0.05, Benjamini-Hochberg false discovery rate ≤ 0.25.
Single-sample GSEA (ssGSEA) is an extension of GSEA that calculates separate enrichment scores for each pairing of a sample and gene set. Each ssGSEA enrichment score represents the degree to which the genes in a particular gene set are coordinately up- or down-regulated within a sample.
ChIP-sequencing data analysis
Quality control tests for unmapped sequences were performed based on the FastQC v.0.11.5 software (http://www.bioinformatics.babraham.ac.uk/projects/fastqc/). The ChIP-sequences were aligned to the GRCh37/hg19 genome using bowtie2-2.3.5(78). PCR duplicates were removed with the Picard v2.18.2 Mark Duplicates tool (https://broadinstitute.github.io/picard/). The mapped reads were normalized in units of reads per kilobase per million (RPKM or rpm/bp) and coverage tracks for the RPKM signal were created as bigwig files for bins of size 20 base pairs by using the bam coverage tool available in deepTools v2.5.3 (79). Peak calling was performed against the input control using the model-based MACS2 v2.1.1.20160309 software (80), FDR ≤ 0.01. Area under the curve (AUC) RPKM normalized signal across genomic regions was computed with the bwtool software(81). Peaks with low area under curve coverage (< 300 RPKM) were disregarded and the ENCODE black-listed regions for hg19 (available at https://www.encodeproject.org/annotations/ENCSR636HFF/) were removed from each set of peak regions. Quality control tests for the mapped reads were performed by using the ChIPQC library available from Bioconductor v3.9 (82). The pairwise correlations between IRF2BP2 replicates were estimated based on the multiBamSummary function available from the deepTools v2.5.3 (79) and visualized on correlation heatmaps and PCA plots. The IRF2BP2 binding sites were identified for the merged data (macs2, FDR ≤ 0.01). The peaks were annotated with the closest hg19 genes by using the annotatePeaks function implemented in the Homer v4.11 platform (83) and the GREAT annotation platform(84). Binding peaks and normalized binding signal were visualized on the Integrative Genomic Viewer (IGV) v2.4.0 platform (85). Gene promoter regions were defined as the +/− 2.5 kb intervals around the hg19 gene transcription start site (TSS). Enhancer regions were defined as the H3K27ac binding regions outside TSS +/− 2.5 kb gene promoter regions. The BEDTools v2.27 suite (86) was used to perform various genomic region analyses (sorting, intersection, merging). Heatmaps of AUC ChIP-seq normalized signal occupancy on genomic regions were created using the computeMatrix and the plotHeatmap tools available in deepTools v2.5.3. The plotProfile tool from deepTools v2.5.3 was used to create metaplots based on the average normalized scores across genomic regions. Differential genome-wide mark binding in between treatment conditions was quantified based on the unpaired t-test with Welch correction (cutoff P-value ≤ 0.05) for the area under curve (AUC) RPKM normalized signal across the genome-wide regions in the two conditions. For a specific genomic region, the changes in signal were classified as increase, decrease (or unchanged) based on the absolute cut-off 1.5 for the delta area under curve scores.
RNA-sequencing data analysis
The spiked-in human reads were mapped to the GRCh37/hg19 human genome using STAR v2.7.3 (87). The ERCC92 mix1 spike-in sequences were mapped using STAR v2.7.3 according to the manufacturer’s protocol. Gene level reads were summarized by using the featureCounts v1.6.3 method implemented in the Subread v2.0.0 package (http://subread.sourceforge.net) (88). The technical quality metrics of the spiked-in mapped reads were assessed using the standard methods implemented in the erccdashboard package (89). Additional quality control tests for the mapped reads and for replicate reproducibility were performed using SARTools v1.7.3 (90). Gene counts were normalized and used to quantify differentially expressed genes between the experimental and control conditions using the DESeq2 v1.24.0 method implemented in Bioconductor v3.9 (91). Genes with ≥10 reads across at least 3 samples were annotated as expressed. Differentiability for expressed genes was assessed with DESeq2 based on the robust shrunken log2 fold change scores and the approximate posterior estimation for GLM coefficients (apeglm v1.6, (92) method for effect size). The cutoffs for differentially expressed genes were |fold change expression|≥1.5 and adjusted p-value ≤ 0.10. Heat maps for transcriptional data visualization was created by using the Morpheus software platform (https://software.broadinstitute.org/morpheus/) based on the log2(TPM+1) DESeq2 normalized counts data.
RESOURCE AVAILIBILITY
Materials availability
Plasmids generated in this study are being deposited to Addgene.
Data and code availability
ChIP-seq and RNA-seq data generated during this study are available at GEO (GSE168649). The ChIP-seq Pol2 data was downloaded from the GEO repository (GSE80779). We analyzed the following publicly AML expression datasets: Tumor Cancer Genome Atlas (TCGA) LAML (179 AML samples, (93)), GSE14468 (526 AML samples, (94)), Beat AML (451 samples (95)), Cancer Cell Line Encyclopedia (CCLE) v20Q3 (96), TARGET AML (232 samples (97)).
Supplementary Material
Statement of significance.
This study exploits inflammatory programs inherent to AML blasts to identify genetic vulnerabilities in this disease. In doing so, we determined that AML cells are dependent on the transcriptional repressive activity of IRF2BP2 for their survival, revealing cell-intrinsic inflammation as a mechanism priming leukemic blasts for regulated cell death.
Acknowledgments
This work was supported by the Swiss National Science Foundation (JME), the Lady Tata Memorial Trust (JME), the Pediatric Cancer Research Foundation (JME), The Helen Gurley Brown Presidential Initiative (The Pussycat Foundation) (JME), the National Cancer Institute (NCI) K99 CA263161 (SL), the NCI R50 CA211404 (MW), the NCI K08 CA222684 (YP), the NCI K22 CA258805 (BN), the NCI R35 CA210030 (KS) and P50 CA206963 (KS), a St. Baldrick’s Foundation Robert J Arceci Innovation Award (KS), St. Baldrick’s Foundation Consortium Grant (KS), the Four C’s Fund (KS) and Team Crank (KS). SL is a Fellow of the Leukemia & Lymphoma Society. SHO is an Investigator of the Howard Hughes Medical Institute. We thank Zach Herbert for contributing to the methods of this manuscript. We thank Peter Libby and Amélie Vromman for sharing their expertise on IL-1β targeting therapies. We thank all members of the Stegmaier Lab, Charles Hatton, and Haihua Chu for important discussions. We express our deep gratitude to all patients and families who contributed to the research in this study.
Footnotes
Competing interest statement
BN is an inventor on patent applications related to the dTAG system (WO/2017/024318, WO/2017/024319, WO/2018/148440, WO/2018/148443 and WO/2020/146250). Nathanael S. Gray is a founder, science advisory board member (SAB) and equity holder in Gatekeeper, Syros, Petra, C4, Allorion, Jengu, B2S, Inception and Soltego (board member). The Gray lab receives or has received research funding from Novartis, Takeda, Astellas, Taiho, Jansen, Kinogen, Arbella, Deerfield and Sanofi. KS has funding from Novartis Institute of Biomedical Research and Kronos Bio, consults for and has stock options in Auron Therapeutics and previously consulted for Kronos Bio and AstraZeneca. All other authors declare no potential conflict of interest.
References
- 1.Park JH, Riviere I, Gonen M, Wang X, Senechal B, Curran KJ, et al. Long-Term Follow-up of CD19 CAR Therapy in Acute Lymphoblastic Leukemia. N Engl J Med. 2018;378(5):449–59. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Hodi FS, O’Day SJ, McDermott DF, Weber RW, Sosman JA, Haanen JB, et al. Improved survival with ipilimumab in patients with metastatic melanoma. N Engl J Med. 2010;363(8):711–23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Herbst RS, Baas P, Kim DW, Felip E, Perez-Gracia JL, Han JY, et al. Pembrolizumab versus docetaxel for previously treated, PD-L1-positive, advanced non-small-cell lung cancer (KEYNOTE-010): a randomised controlled trial. Lancet. 2016;387(10027):1540–50. [DOI] [PubMed] [Google Scholar]
- 4.Reya T, Morrison SJ, Clarke MF, Weissman IL. Stem cells, cancer, and cancer stem cells. Nature. 2001;414(6859):105–11. [DOI] [PubMed] [Google Scholar]
- 5.Palmer L, Briggs C, McFadden S, Zini G, Burthem J, Rozenberg G, et al. ICSH recommendations for the standardization of nomenclature and grading of peripheral blood cell morphological features. Int J Lab Hematol. 2015;37(3):287–303. [DOI] [PubMed] [Google Scholar]
- 6.Hanahan D, Weinberg RA. The hallmarks of cancer. Cell. 2000;100(1):57–70. [DOI] [PubMed] [Google Scholar]
- 7.Ngo VN, Young RM, Schmitz R, Jhavar S, Xiao W, Lim KH, et al. Oncogenically active MYD88 mutations in human lymphoma. Nature. 2011;470(7332):115–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Yang G, Zhou Y, Liu X, Xu L, Cao Y, Manning RJ, et al. A mutation in MYD88 (L265P) supports the survival of lymphoplasmacytic cells by activation of Bruton tyrosine kinase in Waldenstrom macroglobulinemia. Blood. 2013;122(7):1222–32. [DOI] [PubMed] [Google Scholar]
- 9.Treon SP, Tripsas CK, Meid K, Warren D, Varma G, Green R, et al. Ibrutinib in previously treated Waldenstrom’s macroglobulinemia. N Engl J Med. 2015;372(15):1430–40. [DOI] [PubMed] [Google Scholar]
- 10.Cheng YS, Xu F. Anticancer function of polyinosinic-polycytidylic acid. Cancer Biol Ther. 2010;10(12):1219–23. [DOI] [PubMed] [Google Scholar]
- 11.Rolf N, Kariminia A, Ivison S, Reid GS, Schultz KR. Heterodimer-specific TLR2 stimulation results in divergent functional outcomes in B-cell precursor acute lymphoblastic leukemia. Eur J Immunol. 2015;45(7):1980–90. [DOI] [PubMed] [Google Scholar]
- 12.Delhommeau F, Dupont S, Della Valle V, James C, Trannoy S, Masse A, et al. Mutation in TET2 in myeloid cancers. N Engl J Med. 2009;360(22):2289–301. [DOI] [PubMed] [Google Scholar]
- 13.Jaiswal S, Fontanillas P, Flannick J, Manning A, Grauman PV, Mar BG, et al. Age-related clonal hematopoiesis associated with adverse outcomes. N Engl J Med. 2014;371(26):2488–98. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Jaiswal S, Natarajan P, Silver AJ, Gibson CJ, Bick AG, Shvartz E, et al. Clonal Hematopoiesis and Risk of Atherosclerotic Cardiovascular Disease. N Engl J Med. 2017;377(2):111–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Fuster JJ, MacLauchlan S, Zuriaga MA, Polackal MN, Ostriker AC, Chakraborty R, et al. Clonal hematopoiesis associated with TET2 deficiency accelerates atherosclerosis development in mice. Science. 2017;355(6327):842–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Childs KS, Goodbourn S. Identification of novel co-repressor molecules for Interferon Regulatory Factor-2. Nucleic Acids Res. 2003;31(12):3016–26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Shimomura Y, Mitsui H, Yamashita Y, Kamae T, Kanai A, Matsui H, et al. New variant of acute promyelocytic leukemia with IRF2BP2-RARA fusion. Cancer Sci. 2016;107(8):1165–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Yin CC, Jain N, Mehrotra M, Zhagn J, Protopopov A, Zuo Z, et al. Identification of a novel fusion gene, IRF2BP2-RARA, in acute promyelocytic leukemia. J Natl Compr Canc Netw. 2015;13(1):19–22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Jovanovic JV, Chillon MC, Vincent-Fabert C, Dillon R, Voisset E, Gutierrez NC, et al. The cryptic IRF2BP2-RARA fusion transforms hematopoietic stem/progenitor cells and induces retinoid-sensitive acute promyelocytic leukemia. Leukemia. 2017;31(3):747–51. [DOI] [PubMed] [Google Scholar]
- 20.Liu Y, Xu F, Hu H, Wen J, Su J, Zhou Q, et al. A rare case of acute promyelocytic leukemia with IRF2BP2-RARA fusion; and literature review. Onco Targets Ther. 2019;12:6157–63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Mazharuddin S, Chattopadhyay A, Levy MY, Redner RL. IRF2BP2-RARA t(1;17)(q42.3;q21.2) APL blasts differentiate in response to all-trans retinoic acid. Leuk Lymphoma. 2018;59(9):2246–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Nyquist KB, Panagopoulos I, Thorsen J, Haugom L, Gorunova L, Bjerkehagen B, et al. Whole-transcriptome sequencing identifies novel IRF2BP2-CDX1 fusion gene brought about by translocation t(1;5)(q42;q32) in mesenchymal chondrosarcoma. PLoS One. 2012;7(11):e49705. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Chen HH, Keyhanian K, Zhou X, Vilmundarson RO, Almontashiri NA, Cruz SA, et al. IRF2BP2 Reduces Macrophage Inflammation and Susceptibility to Atherosclerosis. Circ Res. 2015;117(8):671–83. [DOI] [PubMed] [Google Scholar]
- 24.Li T, Luo Q, He L, Li D, Li Q, Wang C, et al. Interferon Regulatory Factor-2 Binding Protein 2 Ameliorates Sepsis-Induced Cardiomyopathy via AMPK-Mediated Anti-Inflammation and Anti-Apoptosis. Inflammation. 2020. [DOI] [PubMed] [Google Scholar]
- 25.Tsherniak A, Vazquez F, Montgomery PG, Weir BA, Kryukov G, Cowley GS, et al. Defining a Cancer Dependency Map. Cell. 2017;170(3):564–76 e16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Meyers RM, Bryan JG, McFarland JM, Weir BA, Sizemore AE, Xu H, et al. Computational correction of copy number effect improves specificity of CRISPR-Cas9 essentiality screens in cancer cells. Nat Genet. 2017;49(12):1779–84. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Wang T, Yu H, Hughes NW, Liu B, Kendirli A, Klein K, et al. Gene Essentiality Profiling Reveals Gene Networks and Synthetic Lethal Interactions with Oncogenic Ras. Cell. 2017;168(5):890–903 e15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.McFarland JM, Ho ZV, Kugener G, Dempster JM, Montgomery PG, Bryan JG, et al. Improved estimation of cancer dependencies from large-scale RNAi screens using model-based normalization and data integration. Nat Commun. 2018;9(1):4610. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Behan FM, Iorio F, Picco G, Goncalves E, Beaver CM, Migliardi G, et al. Prioritization of cancer therapeutic targets using CRISPR-Cas9 screens. Nature. 2019;568(7753):511–6. [DOI] [PubMed] [Google Scholar]
- 30.Newman AM, Steen CB, Liu CL, Gentles AJ, Chaudhuri AA, Scherer F, et al. Determining cell type abundance and expression from bulk tissues with digital cytometry. Nat Biotechnol. 2019;37(7):773–82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Aguirre AJ, Meyers RM, Weir BA, Vazquez F, Zhang CZ, Ben-David U, et al. Genomic Copy Number Dictates a Gene-Independent Cell Response to CRISPR/Cas9 Targeting. Cancer Discov. 2016;6(8):914–29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.McKeown MR, Corces MR, Eaton ML, Fiore C, Lee E, Lopez JT, et al. Superenhancer Analysis Defines Novel Epigenomic Subtypes of Non-APL AML, Including an RARalpha Dependency Targetable by SY-1425, a Potent and Selective RARalpha Agonist. Cancer Discov. 2017;7(10):1136–53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Nabet B, Roberts JM, Buckley DL, Paulk J, Dastjerdi S, Yang A, et al. The dTAG system for immediate and target-specific protein degradation. Nat Chem Biol. 2018;14(5):431–41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Lin S, Larrue C, Scheidegger NK, Seong BKA, Dharia NV, Kuljanin M, et al. An In Vivo CRISPR Screening Platform for Prioritizing Therapeutic Targets in AML. Cancer Discov. 2021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Townsend EC, Murakami MA, Christodoulou A, Christie AL, Koster J, DeSouza TA, et al. The Public Repository of Xenografts Enables Discovery and Randomized Phase II-like Trials in Mice. Cancer Cell. 2016;29(4):574–86. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Pikman Y, Tasian SK, Sulis ML, Stevenson K, Blonquist TM, Apsel Winger B, et al. Matched Targeted Therapy for Pediatric Patients with Relapsed, Refractory, or High-Risk Leukemias: A Report from the LEAP Consortium. Cancer Discov. 2021;11(6):1424–39. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Uhlen M, Karlsson MJ, Zhong W, Tebani A, Pou C, Mikes J, et al. A genome-wide transcriptomic analysis of protein-coding genes in human blood cells. Science. 2019;366(6472). [DOI] [PubMed] [Google Scholar]
- 38.Stadhouders R, Cico A, Stephen T, Thongjuea S, Kolovos P, Baymaz HI, et al. Control of developmentally primed erythroid genes by combinatorial co-repressor actions. Nat Commun. 2015;6:8893. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Brown K, Gerstberger S, Carlson L, Franzoso G, Siebenlist U. Control of I kappa B-alpha proteolysis by site-specific, signal-induced phosphorylation. Science. 1995;267(5203):1485–8. [DOI] [PubMed] [Google Scholar]
- 40.Felsher DW, Bishop JM. Reversible tumorigenesis by MYC in hematopoietic lineages. Mol Cell. 1999;4(2):199–207. [DOI] [PubMed] [Google Scholar]
- 41.Ellisen LW, Bird J, West DC, Soreng AL, Reynolds TC, Smith SD, et al. TAN-1, the human homolog of the Drosophila notch gene, is broken by chromosomal translocations in T lymphoblastic neoplasms. Cell. 1991;66(4):649–61. [DOI] [PubMed] [Google Scholar]
- 42.Yamagami T, Sugiyama H, Inoue K, Ogawa H, Tatekawa T, Hirata M, et al. Growth inhibition of human leukemic cells by WT1 (Wilms tumor gene) antisense oligodeoxynucleotides: implications for the involvement of WT1 in leukemogenesis. Blood. 1996;87(7):2878–84. [PubMed] [Google Scholar]
- 43.Mushinski JF, Potter M, Bauer SR, Reddy EP. DNA rearrangement and altered RNA expression of the c-myb oncogene in mouse plasmacytoid lymphosarcomas. Science. 1983;220(4599):795–8. [DOI] [PubMed] [Google Scholar]
- 44.Baratin M, Foray C, Demaria O, Habbeddine M, Pollet E, Maurizio J, et al. Homeostatic NF-kappaB Signaling in Steady-State Migratory Dendritic Cells Regulates Immune Homeostasis and Tolerance. Immunity. 2015;42(4):627–39. [DOI] [PubMed] [Google Scholar]
- 45.Varfolomeev EE, Ashkenazi A. Tumor necrosis factor: an apoptosis JuNKie? Cell. 2004;116(4):491–7. [DOI] [PubMed] [Google Scholar]
- 46.Van Antwerp DJ, Martin SJ, Verma IM, Green DR. Inhibition of TNF-induced apoptosis by NF-kappa B. Trends Cell Biol. 1998;8(3):107–11. [DOI] [PubMed] [Google Scholar]
- 47.Bloom MJ, Saksena SD, Swain GP, Behar MS, Yankeelov TE, Sorace AG. The effects of IKK-beta inhibition on early NF-kappa-B activation and transcription of downstream genes. Cell Signal. 2019;55:17–25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Wang L, Du F, Wang X. TNF-alpha induces two distinct caspase-8 activation pathways. Cell. 2008;133(4):693–703. [DOI] [PubMed] [Google Scholar]
- 49.Baldridge MT, King KY, Boles NC, Weksberg DC, Goodell MA. Quiescent haematopoietic stem cells are activated by IFN-gamma in response to chronic infection. Nature. 2010;465(7299):793–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Baldridge MT, King KY, Goodell MA. Inflammatory signals regulate hematopoietic stem cells. Trends Immunol. 2011;32(2):57–65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Dong L, Yu WM, Zheng H, Loh ML, Bunting ST, Pauly M, et al. Leukaemogenic effects of Ptpn11 activating mutations in the stem cell microenvironment. Nature. 2016;539(7628):304–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Essers MA, Offner S, Blanco-Bose WE, Waibler Z, Kalinke U, Duchosal MA, et al. IFNalpha activates dormant haematopoietic stem cells in vivo. Nature. 2009;458(7240):904–8. [DOI] [PubMed] [Google Scholar]
- 53.Katsumura KR, Ong IM, DeVilbiss AW, Sanalkumar R, Bresnick EH. GATA Factor-Dependent Positive-Feedback Circuit in Acute Myeloid Leukemia Cells. Cell Rep. 2016;16(9):2428–41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Hormaechea-Agulla D, Matatall KA, Le DT, Kain B, Long X, Kus P, et al. Chronic infection drives Dnmt3a-loss-of-function clonal hematopoiesis via IFNgamma signaling. Cell Stem Cell. 2021;28(8):1428–42 e6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Avagyan S, Henninger JE, Mannherz WP, Mistry M, Yoon J, Yang S, et al. Resistance to inflammation underlies enhanced fitness in clonal hematopoiesis. Science. 2021;374(6568):768–72. [DOI] [PubMed] [Google Scholar]
- 56.Yang J, Ikezoe T, Nishioka C, Nobumoto A, Yokoyama A. IL-1beta inhibits self-renewal capacity of dormant CD34(+)/CD38(−) acute myelogenous leukemia cells in vitro and in vivo. Int J Cancer. 2013;133(8):1967–81. [DOI] [PubMed] [Google Scholar]
- 57.Carey A, Edwards DKt, Eide CA, Newell L, Traer E, Medeiros BC, et al. Identification of Interleukin-1 by Functional Screening as a Key Mediator of Cellular Expansion and Disease Progression in Acute Myeloid Leukemia. Cell Rep. 2017;18(13):3204–18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Pietras EM, Mirantes-Barbeito C, Fong S, Loeffler D, Kovtonyuk LV, Zhang S, et al. Chronic interleukin-1 exposure drives haematopoietic stem cells towards precocious myeloid differentiation at the expense of self-renewal. Nat Cell Biol. 2016;18(6):607–18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Dorrington MG, Fraser IDC. NF-kappaB Signaling in Macrophages: Dynamics, Crosstalk, and Signal Integration. Front Immunol. 2019;10:705. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.O’Malley WE, Achinstein B, Shear MJ. Action of Bacterial Polysaccharide on Tumors. Iii. Repeated Response of Sarcoma 37, in Tolerant Mice, to Serratia Marcescens Endotoxin. Cancer Res. 1963;23:890–5. [PubMed] [Google Scholar]
- 61.Galluzzi L, Vitale I, Aaronson SA, Abrams JM, Adam D, Agostinis P, et al. Molecular mechanisms of cell death: recommendations of the Nomenclature Committee on Cell Death 2018. Cell Death Differ. 2018;25(3):486–541. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Orning P, Weng D, Starheim K, Ratner D, Best Z, Lee B, et al. Pathogen blockade of TAK1 triggers caspase-8-dependent cleavage of gasdermin D and cell death. Science. 2018;362(6418):1064–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Sarhan J, Liu BC, Muendlein HI, Li P, Nilson R, Tang AY, et al. Caspase-8 induces cleavage of gasdermin D to elicit pyroptosis during Yersinia infection. Proc Natl Acad Sci U S A. 2018;115(46):E10888–E97. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Chen W, Zhou Z, Li L, Zhong CQ, Zheng X, Wu X, et al. Diverse sequence determinants control human and mouse receptor interacting protein 3 (RIP3) and mixed lineage kinase domain-like (MLKL) interaction in necroptotic signaling. J Biol Chem. 2013;288(23):16247–61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Lawlor KE, Khan N, Mildenhall A, Gerlic M, Croker BA, D’Cruz AA, et al. RIPK3 promotes cell death and NLRP3 inflammasome activation in the absence of MLKL. Nat Commun. 2015;6:6282. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Yabal M, Muller N, Adler H, Knies N, Gross CJ, Damgaard RB, et al. XIAP restricts TNF- and RIP3-dependent cell death and inflammasome activation. Cell Rep. 2014;7(6):1796–808. [DOI] [PubMed] [Google Scholar]
- 67.Virchow R Weisses Blut. Frorieps Notizen. 1845;35:151–6. [Google Scholar]
- 68.Pei S, Pollyea DA, Gustafson A, Stevens BM, Minhajuddin M, Fu R, et al. Monocytic Subclones Confer Resistance to Venetoclax-Based Therapy in Patients with Acute Myeloid Leukemia. Cancer Discov. 2020;10(4):536–51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Doench JG, Fusi N, Sullender M, Hegde M, Vaimberg EW, Donovan KF, et al. Optimized sgRNA design to maximize activity and minimize off-target effects of CRISPR-Cas9. Nat Biotechnol. 2016;34(2):184–91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Sanjana NE, Shalem O, Zhang F. Improved vectors and genome-wide libraries for CRISPR screening. Nat Methods. 2014;11(8):783–4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Dwane L, Behan FM, Goncalves E, Lightfoot H, Yang W, van der Meer D, et al. Project Score database: a resource for investigating cancer cell dependencies and prioritizing therapeutic targets. Nucleic Acids Res. 2021;49(D1):D1365–D72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Tzelepis K, Koike-Yusa H, De Braekeleer E, Li Y, Metzakopian E, Dovey OM, et al. A CRISPR Dropout Screen Identifies Genetic Vulnerabilities and Therapeutic Targets in Acute Myeloid Leukemia. Cell Rep. 2016;17(4):1193–205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Erb MA, Scott TG, Li BE, Xie H, Paulk J, Seo HS, et al. Transcription control by the ENL YEATS domain in acute leukaemia. Nature. 2017;543(7644):270–4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Ritchie ME, Phipson B, Wu D, Hu Y, Law CW, Shi W, et al. limma powers differential expression analyses for RNA-sequencing and microarray studies. Nucleic Acids Res. 2015;43(7):e47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Hochberg Y, Benjamini Y. More powerful procedures for multiple significance testing. Stat Med. 1990;9(7):811–8. [DOI] [PubMed] [Google Scholar]
- 76.Subramanian A, Tamayo P, Mootha VK, Mukherjee S, Ebert BL, Gillette MA, et al. Gene set enrichment analysis: a knowledge-based approach for interpreting genome-wide expression profiles. Proc Natl Acad Sci U S A. 2005;102(43):15545–50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Mootha VK, Lindgren CM, Eriksson KF, Subramanian A, Sihag S, Lehar J, et al. PGC-1alpha-responsive genes involved in oxidative phosphorylation are coordinately downregulated in human diabetes. Nat Genet. 2003;34(3):267–73. [DOI] [PubMed] [Google Scholar]
- 78.Langmead B, Salzberg SL. Fast gapped-read alignment with Bowtie 2. Nat Methods. 2012;9(4):357–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Ramirez F, Ryan DP, Gruning B, Bhardwaj V, Kilpert F, Richter AS, et al. deepTools2: a next generation web server for deep-sequencing data analysis. Nucleic Acids Res. 2016;44(W1):W160–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Zhang Y, Liu T, Meyer CA, Eeckhoute J, Johnson DS, Bernstein BE, et al. Model-based analysis of ChIP-Seq (MACS). Genome Biol. 2008;9(9):R137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Pohl A, Beato M. bwtool: a tool for bigWig files. Bioinformatics. 2014;30(11):1618–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Carroll TS, Liang Z, Salama R, Stark R, de Santiago I. Impact of artifact removal on ChIP quality metrics in ChIP-seq and ChIP-exo data. Front Genet. 2014;5:75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Heinz S, Benner C, Spann N, Bertolino E, Lin YC, Laslo P, et al. Simple combinations of lineage-determining transcription factors prime cis-regulatory elements required for macrophage and B cell identities. Mol Cell. 2010;38(4):576–89. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.McLean CY, Bristor D, Hiller M, Clarke SL, Schaar BT, Lowe CB, et al. GREAT improves functional interpretation of cis-regulatory regions. Nat Biotechnol. 2010;28(5):495–501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Thorvaldsdottir H, Robinson JT, Mesirov JP. Integrative Genomics Viewer (IGV): high-performance genomics data visualization and exploration. Brief Bioinform. 2013;14(2):178–92. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Quinlan AR, Hall IM. BEDTools: a flexible suite of utilities for comparing genomic features. Bioinformatics. 2010;26(6):841–2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Dobin A, Davis CA, Schlesinger F, Drenkow J, Zaleski C, Jha S, et al. STAR: ultrafast universal RNA-seq aligner. Bioinformatics. 2013;29(1):15–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Liao Y, Smyth GK, Shi W. featureCounts: an efficient general purpose program for assigning sequence reads to genomic features. Bioinformatics. 2014;30(7):923–30. [DOI] [PubMed] [Google Scholar]
- 89.Munro SA, Lund SP, Pine PS, Binder H, Clevert DA, Conesa A, et al. Assessing technical performance in differential gene expression experiments with external spike-in RNA control ratio mixtures. Nat Commun. 2014;5:5125. [DOI] [PubMed] [Google Scholar]
- 90.Varet H, Brillet-Gueguen L, Coppee JY, Dillies MA. SARTools: A DESeq2- and EdgeR-Based R Pipeline for Comprehensive Differential Analysis of RNA-Seq Data. PLoS One. 2016;11(6):e0157022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Love MI, Huber W, Anders S. Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol. 2014;15(12):550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Zhu A, Ibrahim JG, Love MI. Heavy-tailed prior distributions for sequence count data: removing the noise and preserving large differences. Bioinformatics. 2019;35(12):2084–92. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Cancer Genome Atlas Research N, Ley TJ, Miller C, Ding L, Raphael BJ, Mungall AJ, et al. Genomic and epigenomic landscapes of adult de novo acute myeloid leukemia. N Engl J Med. 2013;368(22):2059–74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Wouters BJ, Lowenberg B, Erpelinck-Verschueren CA, van Putten WL, Valk PJ, Delwel R. Double CEBPA mutations, but not single CEBPA mutations, define a subgroup of acute myeloid leukemia with a distinctive gene expression profile that is uniquely associated with a favorable outcome. Blood. 2009;113(13):3088–91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Tyner JW, Tognon CE, Bottomly D, Wilmot B, Kurtz SE, Savage SL, et al. Functional genomic landscape of acute myeloid leukaemia. Nature. 2018;562(7728):526–31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Ghandi M, Huang FW, Jane-Valbuena J, Kryukov GV, Lo CC, McDonald ER 3rd, et al. Next-generation characterization of the Cancer Cell Line Encyclopedia. Nature. 2019;569(7757):503–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Bolouri H, Farrar JE, Triche T Jr., Ries RE, Lim EL, Alonzo TA, et al. The molecular landscape of pediatric acute myeloid leukemia reveals recurrent structural alterations and age-specific mutational interactions. Nat Med. 2018;24(1):103–12. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
ChIP-seq and RNA-seq data generated during this study are available at GEO (GSE168649). The ChIP-seq Pol2 data was downloaded from the GEO repository (GSE80779). We analyzed the following publicly AML expression datasets: Tumor Cancer Genome Atlas (TCGA) LAML (179 AML samples, (93)), GSE14468 (526 AML samples, (94)), Beat AML (451 samples (95)), Cancer Cell Line Encyclopedia (CCLE) v20Q3 (96), TARGET AML (232 samples (97)).