

Abstract
The need for safer pain-management therapies with decreased abuse liability inspired a novel drug design that retains μ-opioid receptor (MOR)-mediated analgesia, while minimizing addictive liability. We recently demonstrated that targeting dopamine D3 receptors (D3R) with highly selective antagonists/partial agonists can reduce opioid self-administration and reinstatement to drug seeking in rodent models without diminishing antinociceptive effects. The identification of D3R as a target for the treatment of opioid use disorders, prompted the idea of generating a class of ligands presenting bitopic or bivalent structures, allowing the dual-target binding of MOR and D3R. Structure-activity relationship studies using computationally aided drug-design, and in vitro binding assays led to the identification of potent dual-target leads (23, 28, and 40), based on different structural templates and scaffolds, with moderate (sub-micromolar) to high (low nanomolar/sub-nanomolar) binding affinities. BRET-based functional studies revealed MOR agonist-D3R antagonist/partial agonist efficacies that suggest potential for maintaining analgesia with reduced opioid-abuse liability.
Keywords: Dopamine D3R Receptors, μ-Opioid Receptors, Bitopic Ligands, Bivalent Ligands, Opioid Use Disorders
Graphical Abstract
INTRODUCTION
Over the last decade, the United States has faced a devastating opioid epidemic with an estimate of over 130 people dying from opioid overdose every day.1 The misuse and often consequent addiction to opioids (e.g., prescription pain relievers, and synthetic opioids in general) is a serious national health, social, and economic emergency. Coupled with the novel coronavirus pandemic, the mortality rate involving opioid overdose is rising, and the need for new therapeutic strategies is more urgent than ever2, 3. According to recent reports4, >20% of patients being treated for chronic pain will misuse their opioid prescriptions5, and 8–12% develop opioid use disorders (OUD)5. Ultimately, an estimated 5% of patients is reported to transition from misuse of prescription opioids to heroin6–8, or other synthetic opioids.
The National Institutes of Health (NIH), launched the Helping to End Addiction Long-TermSM (HEAL) initiative to promote and support innovative research addressing this national health emergency. Moreover, the National Institute on Drug Abuse (NIDA) recently proposed a list of the “ten most wanted” medication development strategies to tackle the opioid epidemic/crisis9. Among them, dopamine D3 receptor (D3R) antagonists and partial agonists are a “new” proposed class of ligands as therapeutics to attenuate opioid self-administration.
In the past, D3R selective antagonists, such as GSK598809, have been investigated as potential treatments for psychostimulant use disorder (e.g., cocaine)10, however, the potentiation of hypertensive effects observed in dogs produced by cocaine in the presence of this selective D3R antagonist prevented further development11 and suggested this may be a class effect. We recently demonstrated that our novel D3R antagonists and partial agonists look promising for the treatment of OUD.12–14 Highly selective antagonists, such as VK4–116 (1) and VK4–40 (2) (Figure 1), attenuate oxycodone self-administration and reinstatement to drug seeking, without compromising oxycodone’s antinociceptive effects, in rodents. Importantly, these D3R antagonists/partial agonists do not potentiate the cardiovascular effects induced by cocaine or oxycodone in rats.15 In combination, these studies support the development of D3R antagonists/partial agonists to reduce the risk of opioid misuse and the consequent development of opioid use disorders.
Figure 1.
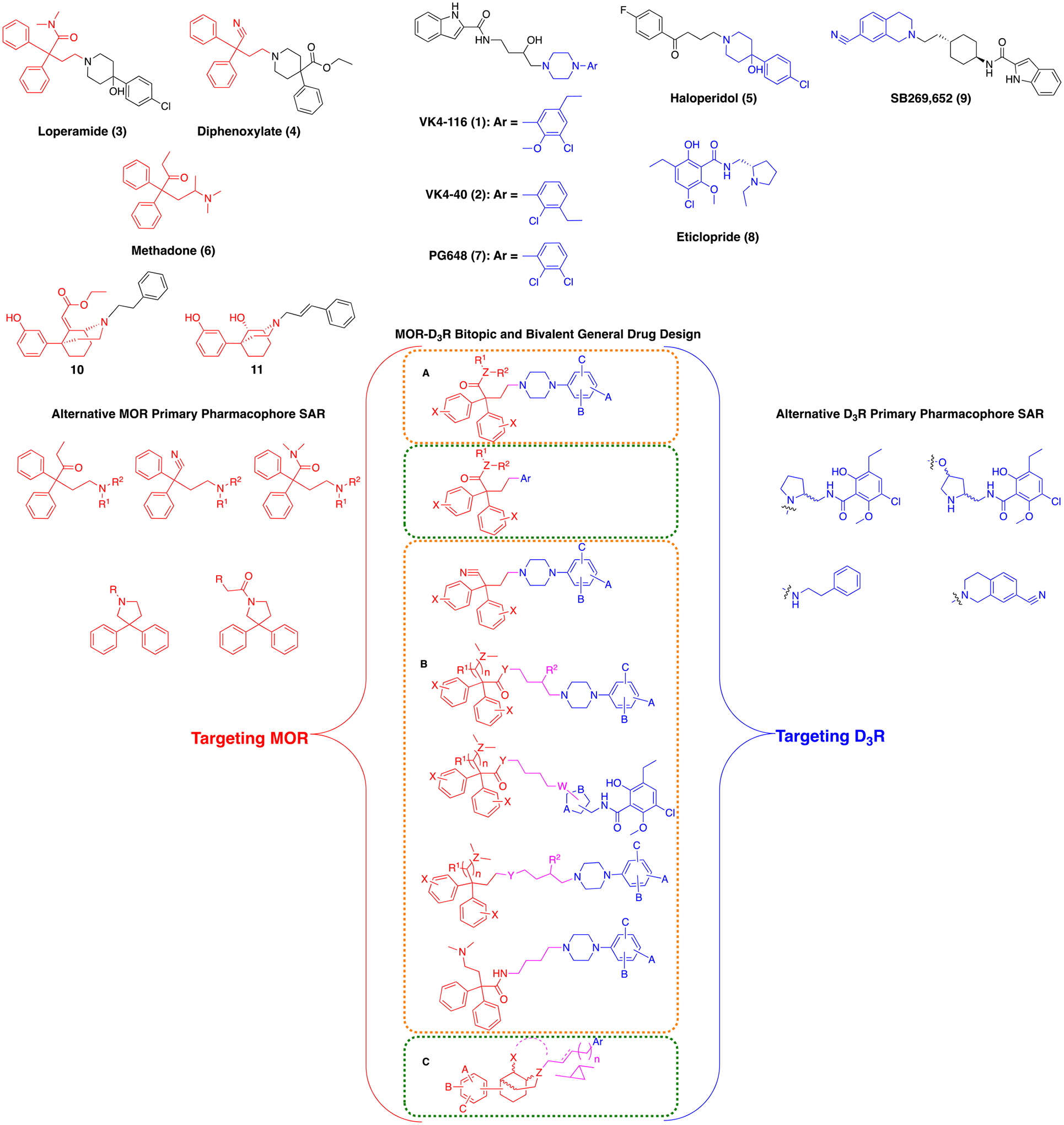
Drug design based on structural modification of canonical synthons inspired by agonists, antagonists, and partial agonists selectively targeting MOR and D3R. Orange boxes denote bivalent templates, green boxes denote bitopic templates. Variables A, B, and C correspond to the aromatic substitutions seen in compounds 1, 2, 7, 10 or 11. General Ar group corresponds to the aromatic substituents in compounds 7, 9, 10, and 11. General R1 and R2 groups represent H, Me or O. Variables Y and Z represent C or N, variable X is H, OMe, or O.
Until now, the main target for pain-management therapies and drug development has been the opioid system, and in particular the μ-opioid receptors (MOR), belonging to the G-protein coupled receptors (GPCR) family. However, due to the abuse liability and development of tolerance associated with the most common MOR agonists used in pain therapy, including chronic pain, for which opioid agonists are largely ineffective in the long-term16–18, research efforts have been directed toward identifying specific physiological responses associated with MOR agonists’ cellular activation pathways. Functionally biased agonists have been posited to reduce the side effect profile of classic opioid analgesics and augment their utility not only as analgesics, but also in the treatment of OUD19–22.
Design, synthesis, and pharmacological characterization of signaling-pathway biased agonists targeting the μ-opioid receptors (MOR) allowed the identification of highly selective G-protein biased agonists, with limited activation of β-arrestin pathways, highlighting different physiological effects mediated by each independent pathway19, 23. It has been suggested that the MOR G-protein pathway seems to be the predominant mediator of the analgesic effects of MOR agonists, meanwhile it has been posited that the simultaneous hindering of β-arrestin recruitment might reduce the respiratory depression and other side effects, such as constipation, associated with opioid-like drugs24–28. However, this has been a topic of intensive investigation, and recently it has been reported that classical MOR agonists, such as morphine and fentanyl, induce dose-dependent respiratory depression and constipation in β-arrestin2 knock-out mice, similarly to what is observed in wild-type mice29–31. Subsequent pharmacological evaluation has also shown that MOR G-protein biased agonists still have abuse potential29, 32–34. It is still unclear whether the optimal outcome and pharmacotherapeutic potential of MOR agonists can be gained through selective activation of a particular downstream signaling pathway (functional selectivity) or whether an optimal level partial agonism at multiple pathways may instead provide a route for the development of safer opioids31, 35. Independent of the signaling pathways and cellular mechanisms associated with respiratory depression and constipation, abuse liability remains as a serious concern that must be addressed with different drug design approaches.
The recognition of D3R antagonism/partial agonism as an alternative and non-opioid approach for treatment of OUD, modulating the abuse potential of common prescription opioids (e.g., oxycodone)12, 13, 15, combined with the well-established antinociceptive properties of MOR agonists, prompted the idea of generating a novel class of dual-target ligands directed to both MOR and D3R (Figure 1). In theory, compounds that are both MOR agonists/partial agonists and D3R antagonist/partial agonists would have analgesic activity without concomitant abuse liability. Our approach aimed at maintaining the analgesic effects of the classic MOR agonists, while reducing the rewarding properties and subsequent abuse liability as a result of D3R antagonism. This drug design may lead to the development of safer dual-target drugs, bridging the most promising pharmacological effects of two classes of molecules/targets previously developed independently. Of note, our drug design was to develop new small molecules endowed with differing ranges of affinities for both targets, independently modulating their physiology/pharmacology rather than towards simultaneous binding of MOR and D3R when in close proximity or in a heteromeric conformation. Nevertheless, if MOR-D3R heteromers are demonstrated to exist and are physiologically relevant, these molecules may be interesting tools to probe their pharmacology.
Furthermore, several well-known MOR agonists, such as loperamide (3) (peripherally limited potent MOR agonist, FDA approved for anti-diarrhea treatment), and diphenoxylate (4), share similar structural motifs with the highly potent non-selective D2R/D3R antagonist haloperidol (5) (Figure 1)36. Substituted phenyl-piperazine and/or phenyl-piperidine synthons, common to both classes of ligands, can exploit the structural similarities between MOR and D3R proteins, thus achieving dual-target binding. Of note, a similar approach was taken previously toward more effective peripherally limited analgesics, based on loperamide, although implementing a bivalent or bitopic drug design or binding to D3R was not described or presumably intended37.
Moreover, methadone (6), (Figure 1), also showed low micromolar D2R and D3R affinity in our binding assays (Table 1), supporting the hypothesis that some of its structural fragments, could be used to target not only the MOR orthosteric binding site (OBS), but the D2R and D3R as well. Unlike 6, the synthetic opioid fentanyl, with its completely different structural features, showed an interesting moderate affinity for the dopamine D4R subtype, but total lack of recognition for either D2R or D3R (Table 1). This template was not considered in our drug design for this reason and also because, recently, bivalent ligands using fentanyl have been reported to lack antinociceptive activity38, 39.
Table 1.
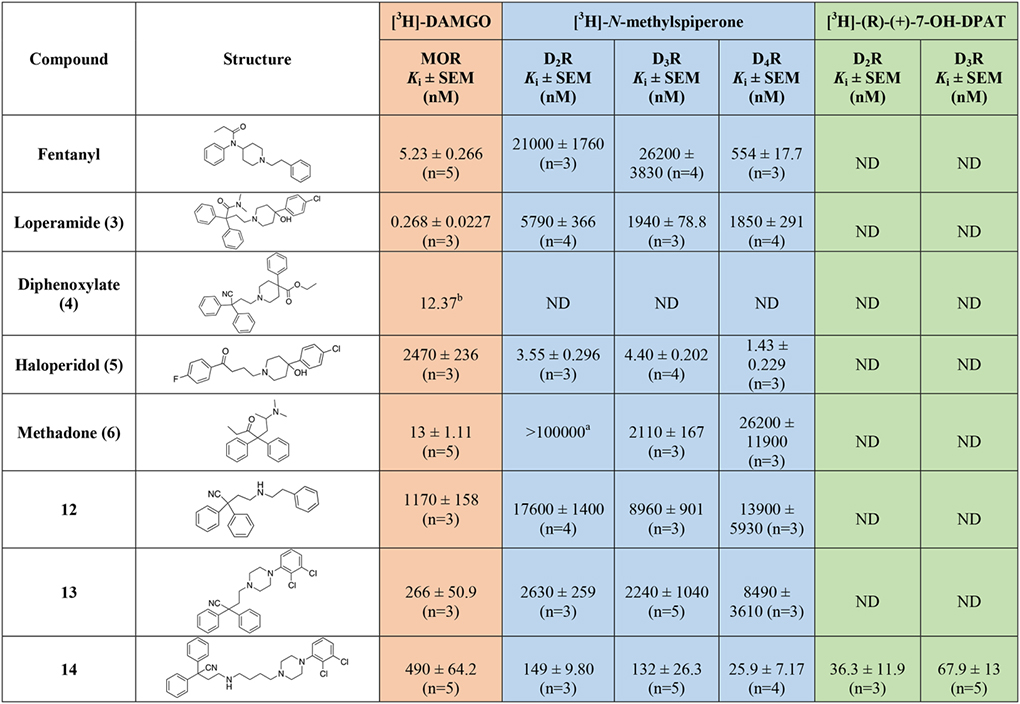
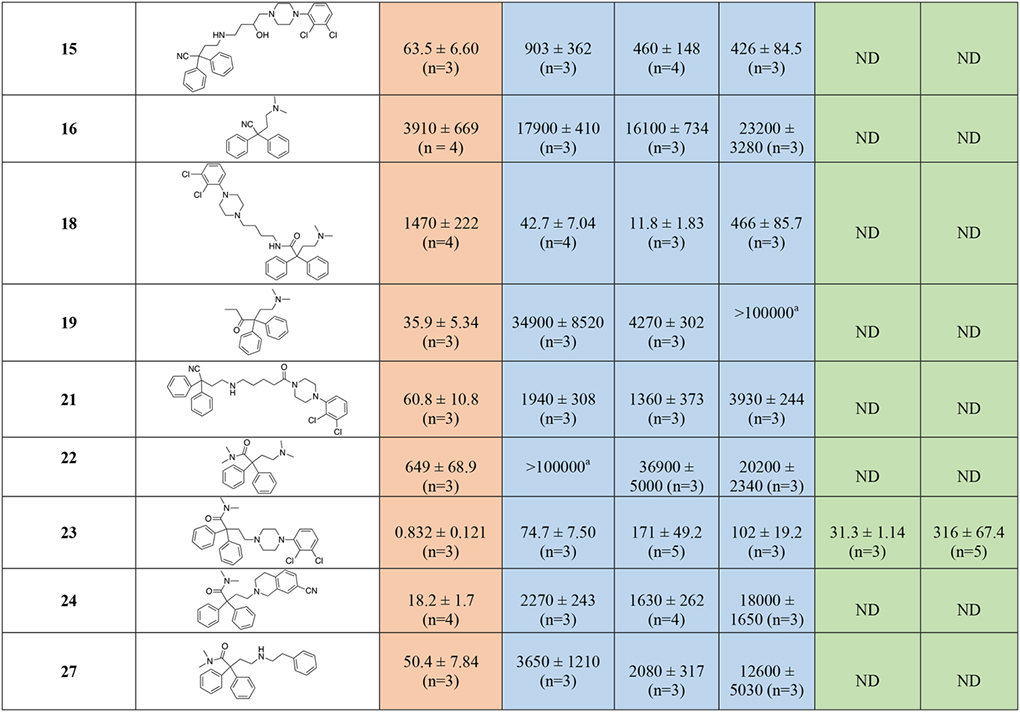
Radioligand competition binding affinity data, for all the MOR diphenyl PP analogs based on 3, 4, and 6 at hMOR and hD2-like receptor subtypes. All the affinity values are expressed as Ki ± SEM, derived from IC50 values using the Cheng-Prusoff equation63, and calculated as the mean of at least three independent experiments (n = number of independent experiments), each performed in triplicate.
|
|
|
|
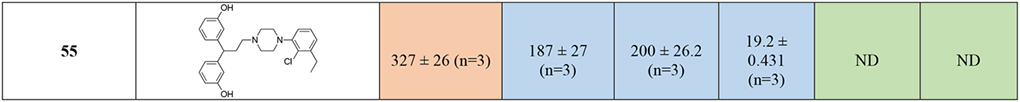
|
ND = Not Determined.
No inhibition of specific radioligand binding was observed at the highest tested concentration in one to three independent experiments, each performed in triplicate.
Ki value obtained from reference61.
Due to the limited availability of reported structure-activity relationships (SAR) for dual-target MOR-D3R ligands38, 40–42, we used a fragment-based drug design approach, supported by molecular docking, computer-aided drug design (CADD), and extensive in-vitro pharmacology to guide SAR, hit optimization and lead identification.
Herein, we refer to bivalent dual-target analogs when a MOR agonist primary pharmacophore (PP) is tethered with a D3R antagonist PP; both can bind their respective OBS, eliciting their corresponding opioid agonist and dopaminergic antagonist effects. Consistent with our definition of bitopic ligands43, we classify these new analogs as bitopic when they incorporate a MOR agonist PP, targeting its corresponding OBS that also has structural features suitable for D3R OBS recognition, tethered to a D3R secondary pharmacophore (SP), identifying the D3R secondary binding pocket (SBP)44. Of note, this D3R SP may also elicit binding interactions within the MOR SBP. All new compounds represent carefully designed linkers, as tethering fragments, with specific SAR focusing on the linkers’ regiochemistry, stereochemistry and substituents43.
All newly synthesized analogs were tested for their on-target and off-target affinities at MOR, D2R, D3R, and D4R, in a combination of agonist and antagonist radioligand competition binding assays. Compounds selected as hits, for their promising dual-target and sub-micromolar affinities, were further evaluated in functional bioluminescence resonance energy transfer (BRET) studies, to assess their agonist and/or antagonist potencies for the target of interest, and possible functional selectivity (biased agonism) for specific signaling pathways.
RESULTS AND DISCUSSION
Chemistry
This novel class of compounds can be subdivided, as depicted in Figure 1 (A–C), in three general templates: A) the N,N-dimethyl-2,2-diphenylacetamide, 2,2-diphenylacetonitrile, and 1,1-diphenylbutan-2-one MOR PPs, derived from 3, 4, and 6 respectively (Figure 1), were tethered with suitably substituted PPs, inspired by selective and non-selective D3R antagonists (e.g., 1, 2, PG648 (7), eticlopride (8) and SB269,652 (9); Figure 1), via short ethyl linker chain; B) the same MOR OBS-binding agonist PPs were linked with D3R OBS antagonist PPs via a longer and more complex butyl linker, substituted with a 3-hydroxyl group, or the piperazine or pyrrolidine basic function in several regiochemical combinations; C) the MOR agonist PPs were replaced with the more rigid, stereochemically complex and hindered ethyl-2-(-5-(3-hydroxyphenyl)-2-azabicyclo[3.3.1]nonan-9-ylidene)acetate and 5-(3-hydroxyphenyl)-2-azabicyclo[3.3.1]nonan-9-ol PP (10 and 11; Figure 1), with structural features reminiscent of previously published D3R ligands45, 46 that we have found to have low micromolar affinity for the MOR as well. This new MOR PP presented key functional groups for extending linkers of multiple lengths, substitutions, rigidity and chirality, and tethering D3R PPs (for bivalent dual-target compounds) or D3R SPs (e.g., 2-indoleamide for bitopic dual-target compounds). When possible and appropriate, a complete resolution of the chiral centers at PP, SP or linkers was performed, and the stereochemical properties of the new analogs have been taken into consideration when generating detailed SAR.
The first series of compounds with the 2,2-diphenylbutanenitrile as the MOR PP, inspired by 4, were prepared as depicted in Scheme 1. Starting from the commercially available 4-bromo-2,2-diphenylbutanenitrile, simple N-alkylation under basic conditions yielded the first group of bivalent MOR-D3R hybrids, where the D3R PP was represented by 2-phenylethan-1-amine (12), 1-(2,3-dichlorophenyl)piperazine (13), 4-(4-(2,3-dichlorophenyl)piperazin-1-yl)butan-1-amine (14), and 4-amino-1-(4-(2,3-dichlorophenyl)piperazin-1-yl)butan-2-ol (15). This provided initial SAR deduction, by increasing the structural complexity of the D3R PP, and simultaneously modifying the linker length and substitution. To investigate the optimal regiochemistry for introducing the linker and D3R PP, compound 18 was prepared, where the butyl-4-(2,3-dichlorophenyl)piperazine synthon was introduced to replace the nitrile, while maintaining the MOR 4-(dimethylamino)-2,2-diphenylbutanamide moiety, as seen in both 3 and 6 (Figure 1). In detail, 4-bromo-2,2-diphenylbutanenitrile was first N-alkylated with dimethylamine hydrochloride (Me2NH·HCl) under basic conditions, followed by hydrolysis of the nitrile in the presence of 48% HBr (aq solution) to yield the HBr salt of amino acid 17. Subsequent amidation mediated by EDC, HOBt, and 4-(4-(2,3-dichlorophenyl)piperazin-1-yl)butan-1-amine47 yielded the desired product 18. Despite the importance of the α-methyl group of 6 being well reported in the literature48, favoring the optimal binding pose within the MOR OBS, we decided to study SAR of structurally simplified analogs. Thus, via simple Grignard addition to the nitrile 16, we prepared the desmethyl-methadone 19.
Scheme 1.
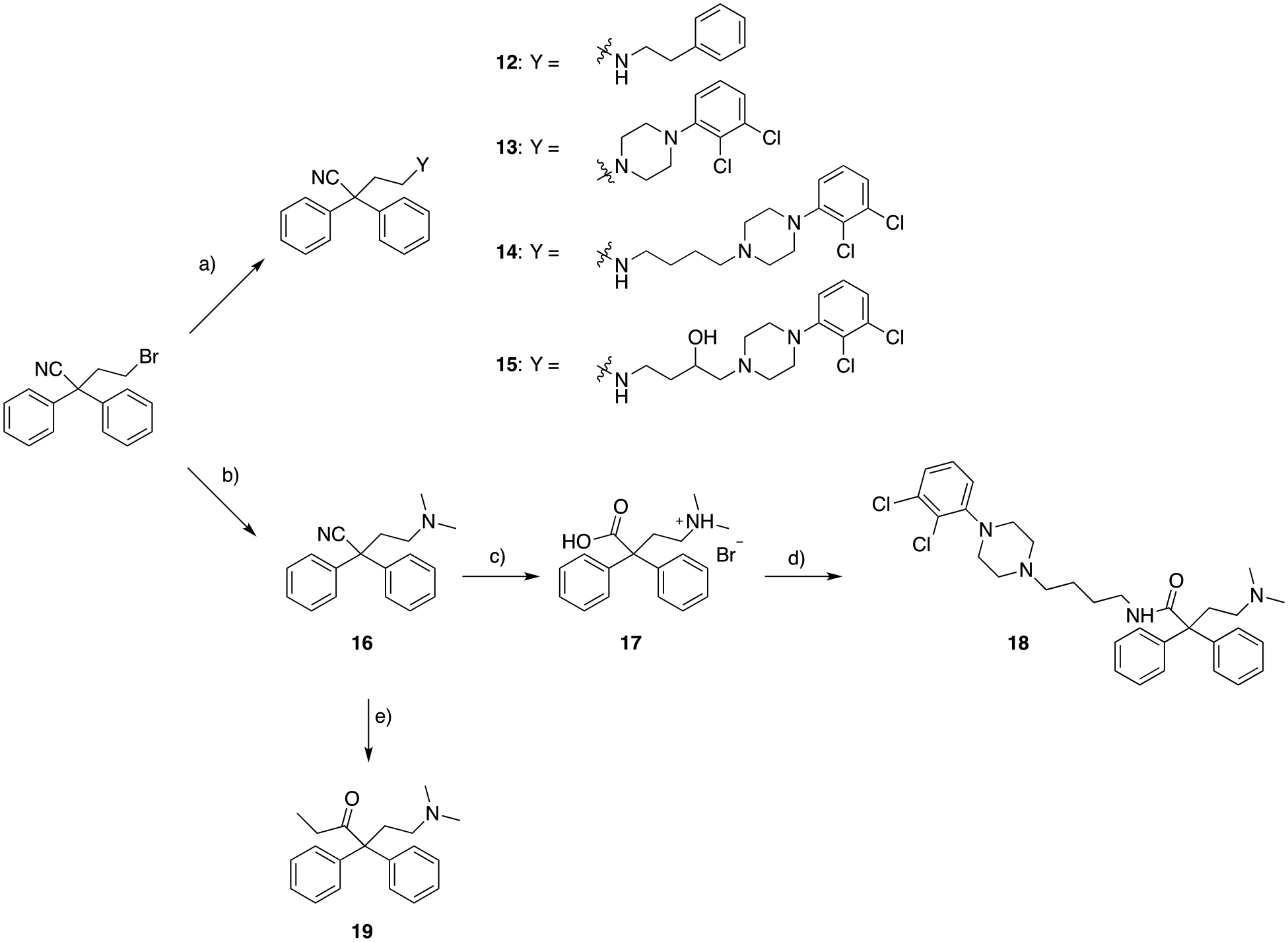
a) appropriate primary or secondary amine, K2CO3, acetonitrile (ACN), 130 ° C, overnight, 8 – 53%; b) Me2NH·HCl, K2CO3, ACN, 130 ° C, overnight, 79%; c) HBr 48% in H2O, 100 ° C, overnight; d) N-(3-dimethylaminopropyl)-N′-ethylcarbodiimide hydrochloride (EDC), 1-hydroxybenzotriazole hydrate (HOBt), 4-(4-(2,3-dichlorophenyl)piperazin-1-yl)butan-1-amine47, N,N-diisopropylethylamine (Hünig’s base, DIPEA), dichloromethane (DCM), 25 ° C, overnight, 6%; e) ethyl magnesium bromide (EtMgBr) 3M in diethyl ether (Et2O), toluene, 0 -> 110 ° C, 3 hrs, 59%.
In Scheme 2, in order to investigate the structural requirement of the D3R pharmacophore, and in particular the effect of replacing the basic butyl-4-(2,3-dichlorophenyl)piperazine, with the corresponding amide analog, HCTU mediated amide coupling of 5-((tert-butoxycarbonyl)amino)pentanoic acid, followed by removal of the Boc-protecting group, and consequent mono N-alkylation with 4-bromo-2,2-diphenylbutanenitrile afforded the desired product, 21. The longer 5 carbon atom linker, instead of the canonical butyl chain, was chosen because of the increased rigidity of the cyclic amide function and need for extending the tethered PP to an optimal distance.
Scheme 2.
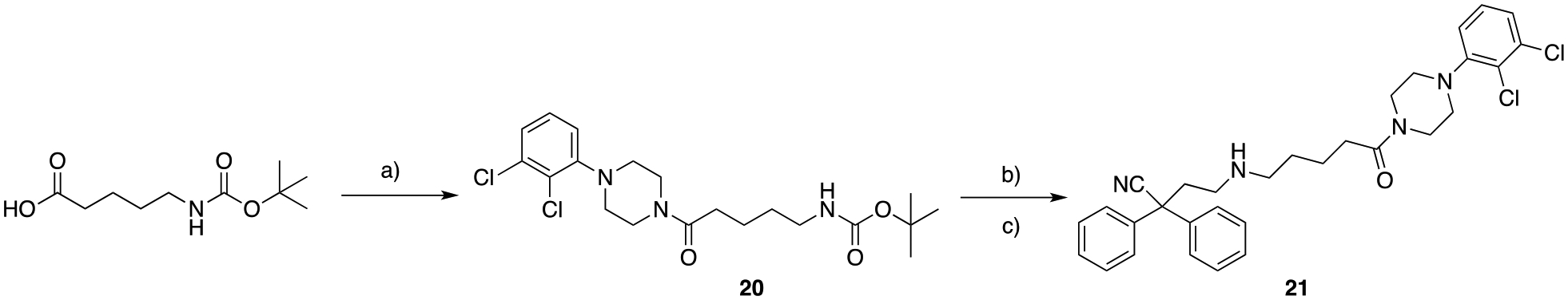
a) 1-(2,3-dichlorophenyl)piperazine, 2-(6-chloro-1-H-benzotriazole-1-yl)-1,1,3,3-tetramethylaminium hexafluorophosphate (HCTU), DCM, 25 ° C, 3 hrs, 59%; b) trifluoroacetic acid (TFA), DCM, 25 ° C, 24 hrs; c) 4-bromo-2,2-diphenylbutanenitrile, K2CO3, ACN, 82 ° C, overnight, 3% (over two steps).
The switch from 2,2-diphenylbutanenitrile (4-like analogs) to N,N-dimethyl-2,2-diphenylbutanamide (3-like analogs) as the MOR PP was achieved as described in Scheme 3. Starting from the commercially available N-(3,3-diphenyldihydrofuran-2(3H)-ylidene)-N-methylmethanaminium bromide, simple ring opening with the appropriate primary or secondary amines afforded compounds 22, presenting the 6-like dimethylamino function, 23, where the 2,3-dichlorophenyl piperazine D3R scaffold was introduced, as well as 24, whose 1,2,3,4-tetrahydroisoquinoline-7-carbonitrile is another common D2R-D3R antagonist PP fragment, present in well-characterized ligands, such as 949–52.
Scheme 3.
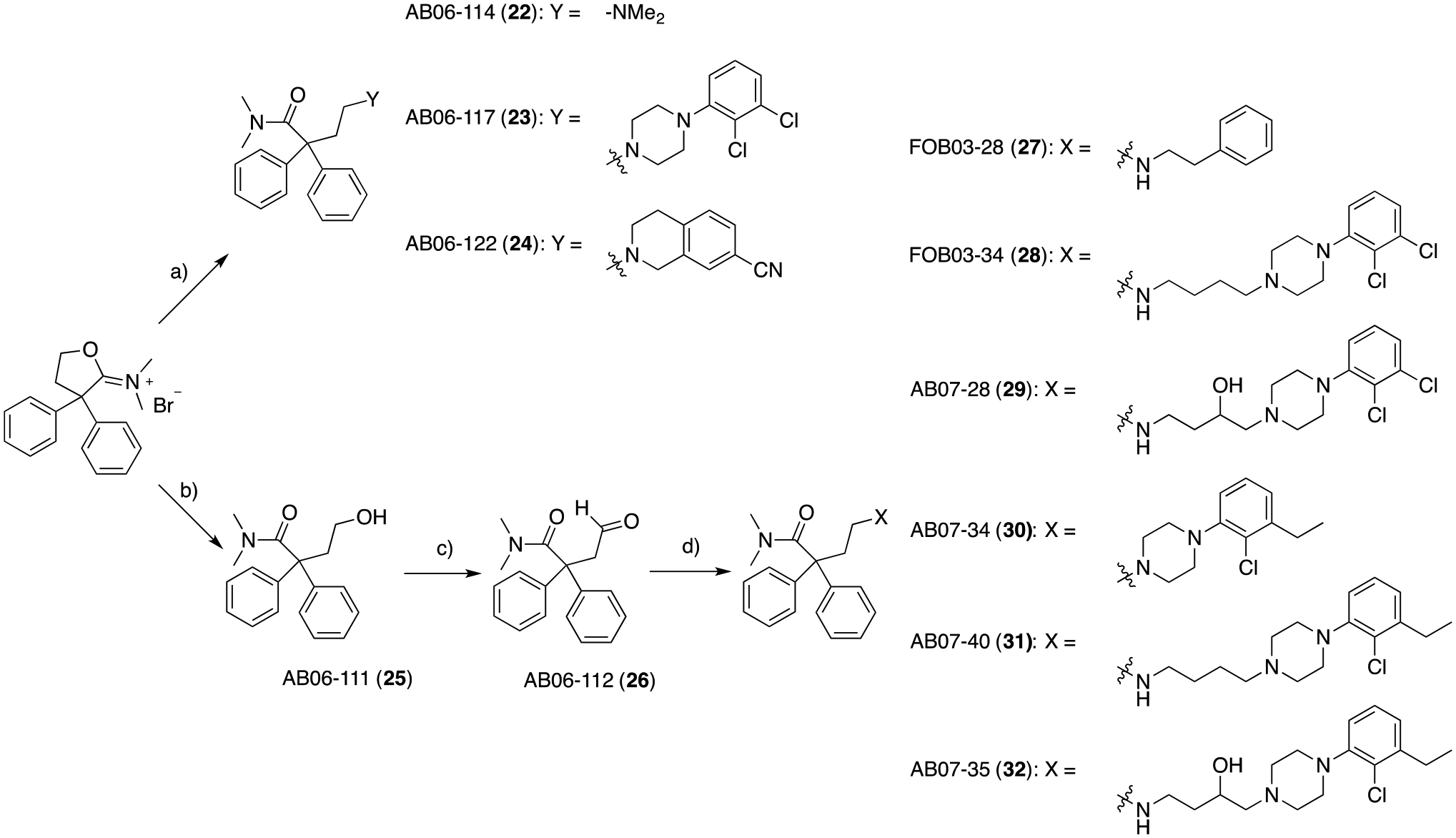
a) appropriate secondary amine, K2CO3, DIPEA (for 23 and 24), ACN, tert-butyl methyl ether (TBME), reflux, 24 hrs, 16 – 91%; b) 2N NaOH in H2O, 25 ° C, 5 min, 100%; c) Dess-Martin periodinane (DMP), DCM, 0 to 25 °C, 1 hr, 60%; d) appropriate primary or secondary amine, catalytic acetic acid (cat. AcOH), sodium triacetoxyborohydride (STAB), 1,2-dichloroethane (DCE), 25 ° C, 2.5 hrs, 12 – 100%.
To investigate the effect of the linker length and substitution on the bivalent hybrid analogs, N-(3,3-diphenyldihydrofuran-2(3H)-ylidene)-N-methylmethanaminium bromide was simply washed with 2N NaOH in H2O, and extracted with DCM, to obtain the alcohol intermediate 25, which was then oxidized to the aldehyde using Dess-Martin periodinane and then mono-N-alkylated via reductive amination conditions with the appropriate primary and secondary amines. This small library of compounds (27, 28, 29, 30, 31, and 32) covered a large structural variety of canonical D3R antagonist PPs, as well as butyl and hydroxyl substituted linkers, known for increasing D3R subtype selectivity53.
Compound 34, containing the 1,2,3,4-tetrahydroisoquinoline-7-carbonitrile pharmacophore was synthesized according to Scheme 4, via preparation of intermediate 33, and reductive amination with 26.
Scheme 4.
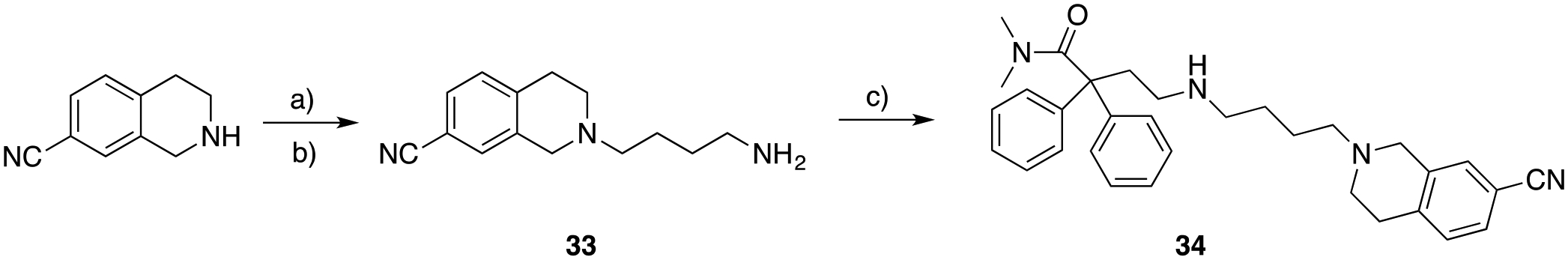
a) N-(4-bromobutyl)phthalimide, K2CO3, cat. KI, ACN, 82 ° C, overnight; b) hydrazine (NH2NH2), ethanol (EtOH), 80 ° C, 3 hrs, 94% (over 2 steps); c) N,N-dimethyl-4-oxo-2,2-diphenylbutanamide (26), cat. AcOH, STAB, DCE, 25 ° C, 2.5 hrs, 46%.
In order to expand the library towards different D3R PP, the canonical piperazine and 1,2,3,4-tetrahydroisoquinoline, inspired by selective D3R antagonists and partial agonists (1, 2, 7, and 9; Figure 1), were replaced in Scheme 5 with the highly decorated phenyl-N-(pyrrolidin-2-ylmethyl)benzamide derived from the eticlopride pharmacophore (8, Figure 1). Alkylation of (S)-nor-eticlopride (obtained from the NIDA Drug Supply Program) with 4-bromo-2,2-diphenylbutanenitrile yielded compound 35, introducing the 2,2-diphenyl-nitrile MOR PP, tethered by an ethyl linker. Meanwhile, formation of intermediate 36, allowed the synthesis of analog 37, tethered via the longer butyl-amino linker.
Scheme 5.
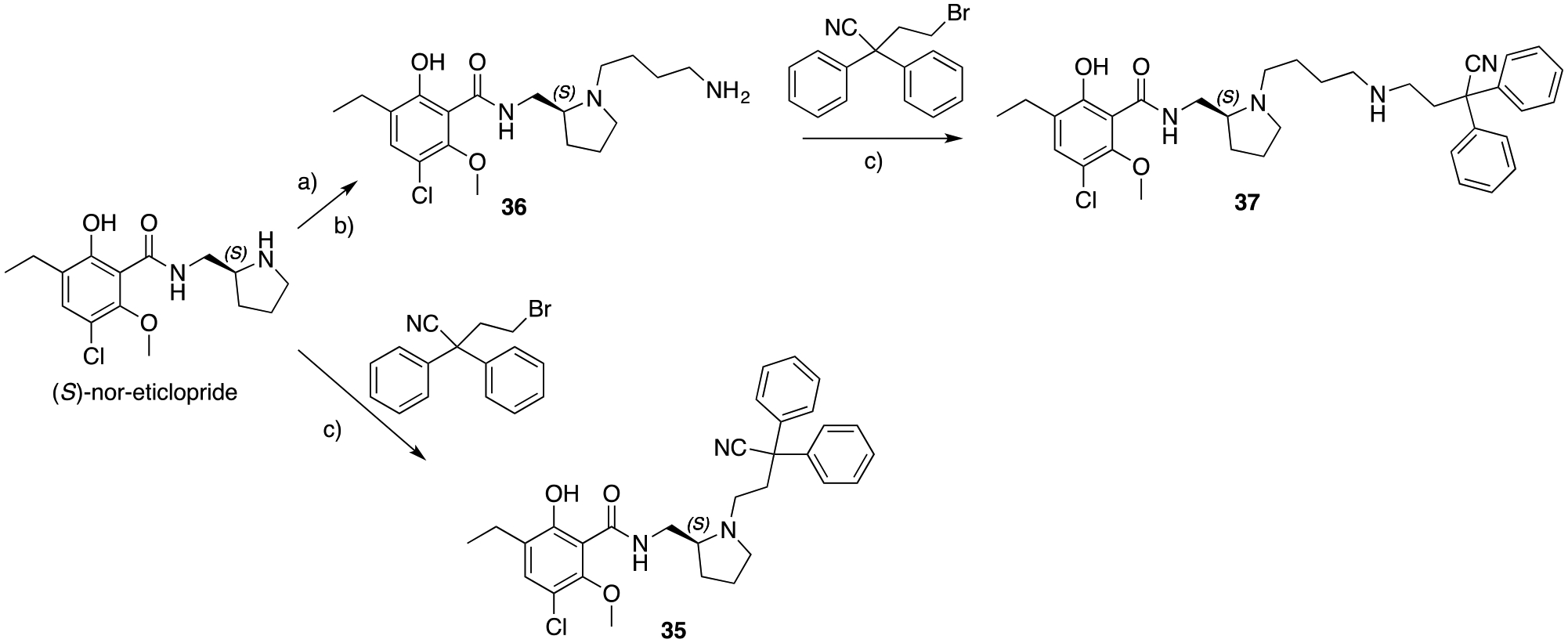
a) N-(4-bromobutyl)phthalimide, K2CO3, ACN, 82 ° C, overnight; b) NH2NH2, EtOH, 80 ° C, 3 hrs, 36% (over 2 steps); c) K2CO3, ACN, 130 ° C, overnight, 10 – 23%.
Analogous to the approach described above for the phenylpiperazine series, we investigated the replacement of the diphenyl nitrile MOR PP, with the 3-like N,N-dimethylamide functional group for the 8-based bivalent analogs. In Scheme 6, the substituted benzoic acid intermediate was prepared following previous literature procedures14, 54. The benzoic acid was coupled with 2-(4-(2-(aminomethyl)pyrrolidin-1-yl)butyl)isoindoline-1,3-dione, which was freshly prepared in situ via selective Boc- deprotection of 38, prior to the HCTU mediated amide coupling. The phthalimide group in intermediate 39 was removed and the resulting primary amine was mono-N-alkylated via reductive amination to yield 40 as the racemic mixture.
Scheme 6.
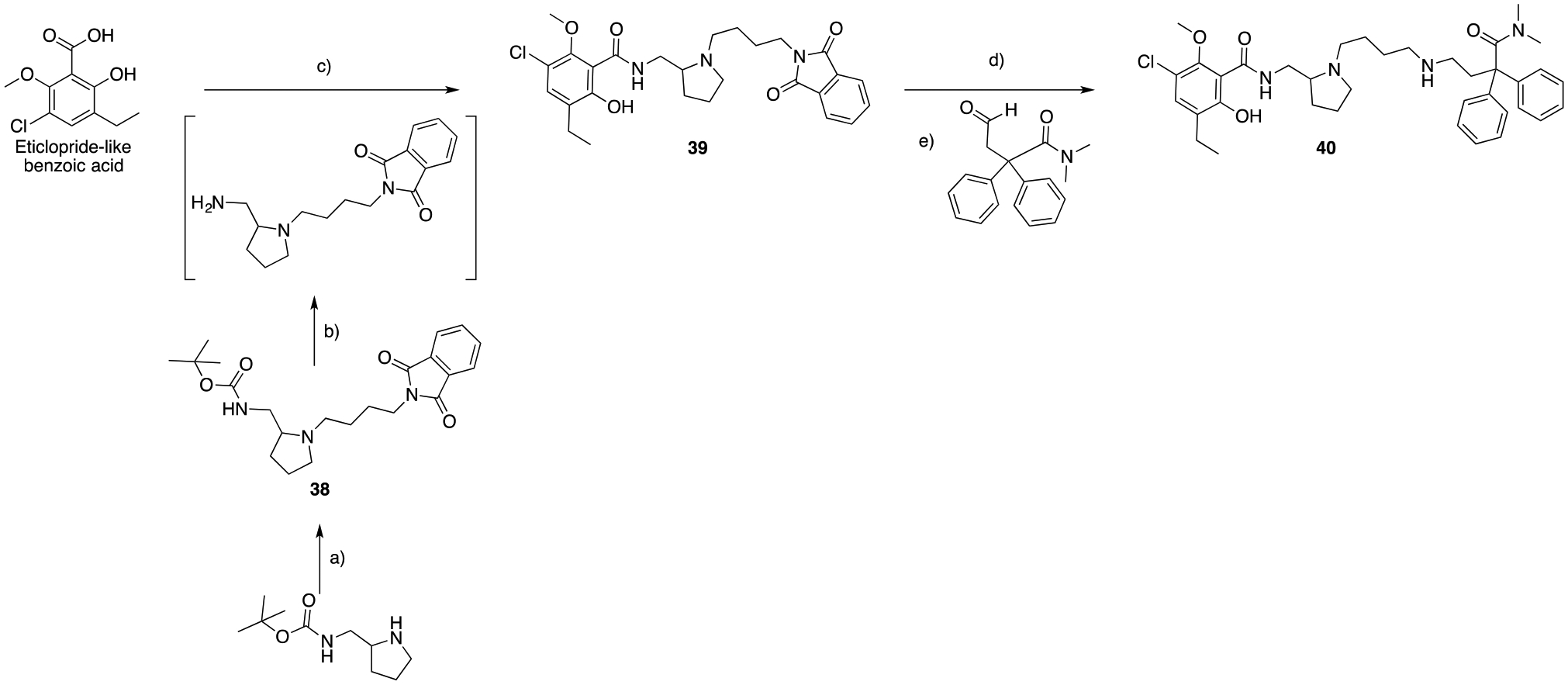
a) N-(4-bromobutyl)phthalimide, K2CO3, ACN, 82 ° C, overnight, 85%; b) TFA, DCM, 25 ° C, 3 hrs; c) HCTU, DCM, 25 ° C, 48 hrs, 26%; d) NH2NH2, EtOH, 80 ° C, 3 hrs; e) cat. AcOH, STAB, DCE, 25 ° C, 12 hrs, 21% (over two steps).
In this case, we decided to synthesize the racemic mixture, starting from the commercially available tert-butyl (pyrrolidin-2-ylmethyl)carbamate, because although 8 favors the (S) absolute configuration at the pyrrolidine ring, we didn’t want to exclude the possibility of different stereochemical requirements for this bivalent analog, as we have seen in another series of bivalent/bitopic D3R ligands46.
SAR for an alternative MOR PP, with a 3,3-diphenyl substituted pyrrolidine, were investigated through the synthesis of analogs in Scheme 7. The common starting material 4-bromo-2,2-diphenylbutanenitrile was reacted with lithium aluminum hydride (LAH), to give the primary amine, and consequent ring-closure in a one-pot step. Intermediate 41 was either methylated via treatment with methyl chloroformate followed in situ LAH reduction to afford 42, or reacted with propionyl chloride to yield the cyclic amide synthon 43. This rather simple PP allowed us to evaluate the effect of structural rigidity in 6- and 3-like MOR PP, and the effect of replacing the protonatable cyclic amine to a cyclic amide. To prepare the bivalent analog 46, 41 was initially acylated with 2-chloroacetyl chloride. Subsequent alkylation of racemic tert-butyl (pyrrolidin-2-ylmethyl)carbamate yielded 45. Finally, Boc- deprotection and amide coupling afforded the desired product.
Scheme 7.
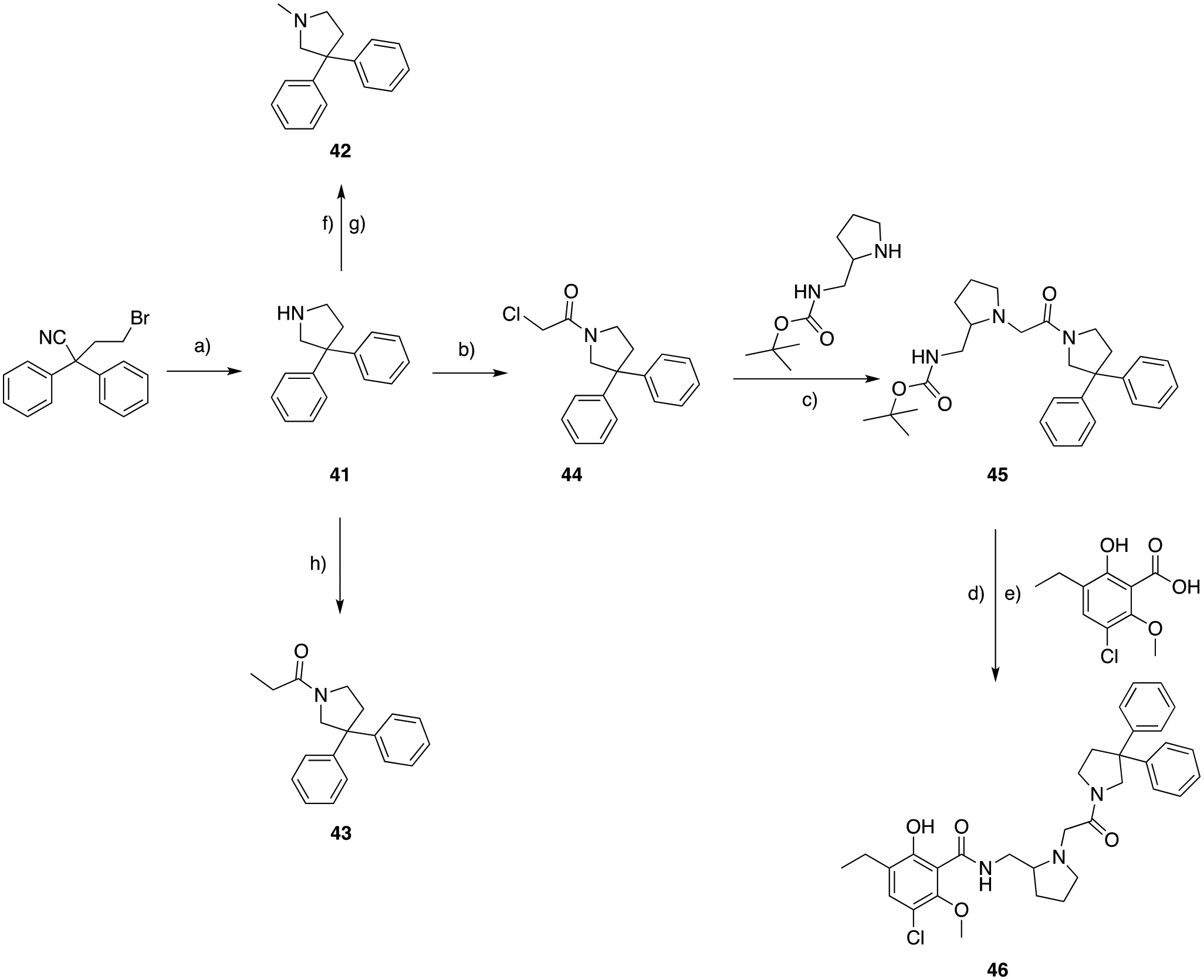
a) LAH, THF, 0 to 25 °C, 15 hrs, 40%; b) 2-chloroacetyl chloride, DIPEA, THF, 0 to 25 °C, 1 hr; c) cat. KI, K2CO3, ACN, 82 °C, 3 hrs, 74% (over two steps); d) TFA, DCM, 25 °C, 2 hrs; e) HCTU, DIPEA, DCM, 25 °C, 3 hrs, 15% (over two steps); f) methyl chloroformate, DIPEA, DCM, 25 °C, 1 hr; g) LAH, THF, 0 to 25 °C, 56% (over two steps); h) propionyl chloride, DIPEA, DCM, 40 °C, overnight, 29%.
As consistently observed in previous work43, when generating bitopic or bivalent ligand SAR, it is essential to study structural requirements not only for the PP and/or SP, but regiochemistry and stereochemistry of the linkers, which can play a crucial role in their biological activity.
In Scheme 8, we approached a modification of the regiochemistry for the pyrrolidine D3R PP scaffold. The final compound 48 presents the MOR diphenyl-N,N-dimethyl amide PP tethered to the D3R PP, via a butyl ether linker fused in postion-4 of the pyrrolidine nucleus, in a rel-trans stereochemistry configuration with respect to the 8 amide PP appended in position-2. This was easily introduced as shown in Scheme 8, starting from 47 via reductive amination and Boc-deprotection to ultimately yield 48.
Scheme 8.
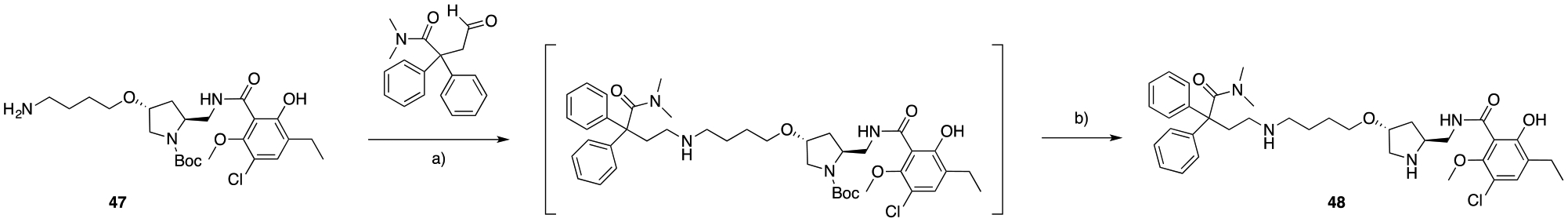
A) cat. AcOH, STAB, DCE, 25 °C, 4 hrs; b) TFA, DCM, 25 °C, overnight, 8.5% (over two steps).
Extensive in silico docking studies and optimization (see the section below) were directed towards improving the dual-target affinity of these new MOR-D3R analogs and guided us to the synthesis of 51 (Scheme 9). In particular, the hydroxy substitution of the MOR diphenyl PP was based on the in silico observation that the meta-hydroxy groups will engage a water-mediated hydrogen bond network observed in several known opioid ligands while being well-tolerated by the D3R SP binding site.
Scheme 9.
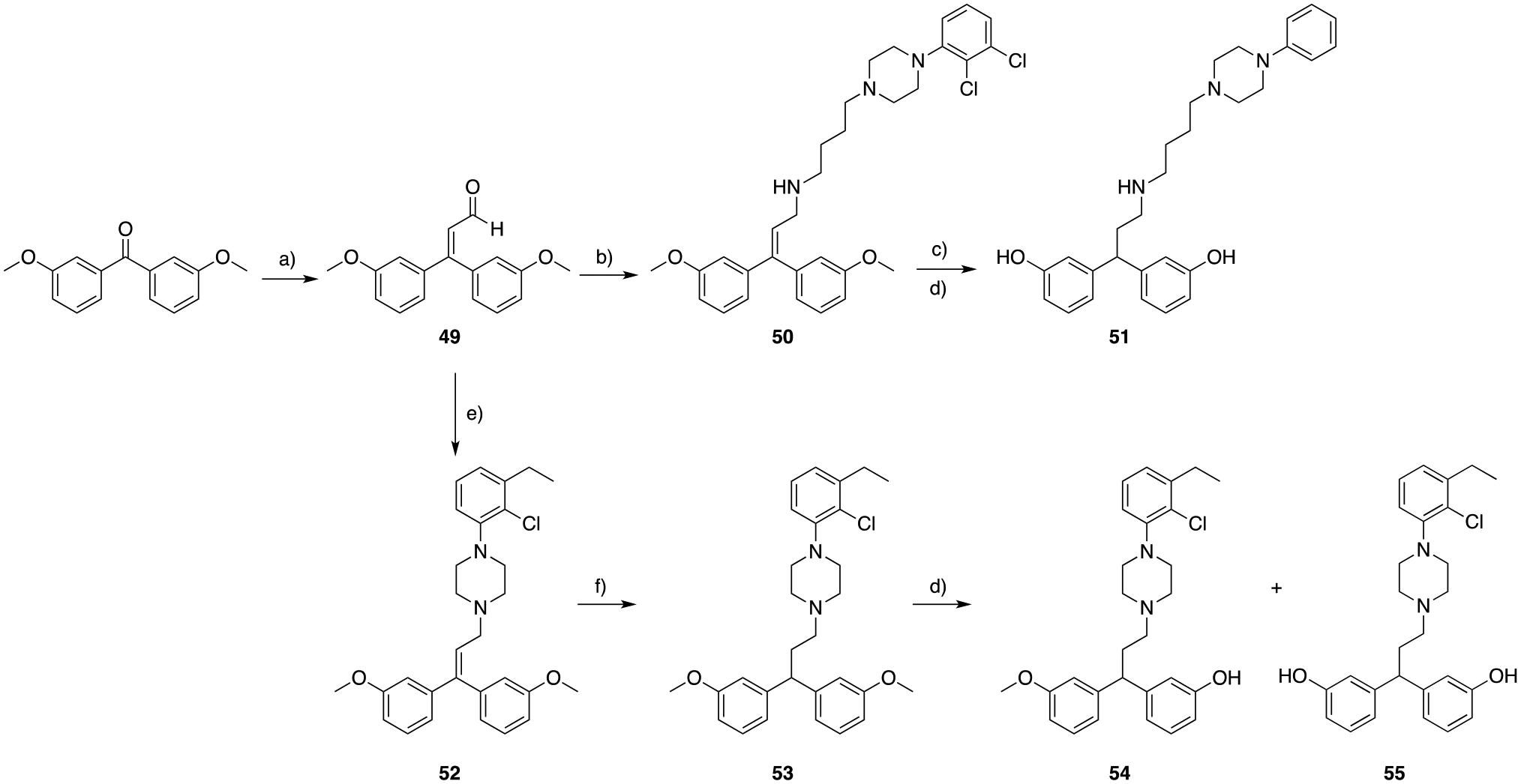
a) titanium tetrachloride (TiCl4), triethylamine (TEA), DCM, 0 to 25 ° C, overnight, 58%; b) 4-(4-(2,3-dichlorophenyl)piperazin-1-yl)butan-1-amine, cat. AcOH, STAB, DCE, 25 °C, 4 hrs, 88%; c) H2 (50 psi), Pd/C (20% wet support), EtOH, 25 ° C, 12 hrs; d) HBr 33% in AcOH; 118 ° C, 48 hrs, 13 – 31%; e) 1-(2-chloro-3-ethylphenyl)piperazine14, cat. AcOH, STAB, DCE, 25 ° C, 4 hrs, 88%; f) H2 (30 psi), Pd/C (20% wt.), EtOAc:EtOH (2:1), 25 ° C, 3 hrs, 75%.
Homologation of the commercially available bis(3-methoxyphenyl)methanone in presence of TiCl4 and triethylamine55, allowed the formation of the α,β-unsaturated aldehyde 49. Reductive amination in presence of 2,3-dichlorophenyl piperazine yielded 50, which was hydrogenated in the presence of Pd/C. The crude mixture was then subsequently O-demethylated with 33% HBr in AcOH (Scheme 9).
High resolution mass spectroscopy (HRMS) and NMR analyses revealed the loss of both chlorine atoms in final product 51. Retrospective analyses of the previous synthetic steps and intermediates showed that the loss of both chlorine atoms occurred during hydrogenation while reducing the styrenyl olefin. Unfortunately, dehalogenation and concomitant hydrogenation appears to occur faster than the reduction of the desired olefin. Despite replacing Pd/C with PtO2, solvent (from EtOH to EtOAc), and H2 pressure (from 50 psi to 15–20 psi), the same reaction outcome was observed. Nevertheless, we proceeded in testing 51, as a proof of concept to validate the biological activity of the newly proposed bis-phenol PP to target MOR.
The 1-(2-chloro-3-ethylphenyl)piperazine scaffold proved to be resistant to hydrogenation conditions14, and was introduced as the D3R PP tethered to the MOR bis-phenol scaffold (Scheme 9). Intermediate 52 was readily prepared by reductive amination, followed by milder hydrogenation conditions (30 psi H2, using a mixture of EtOAc:EtOH 3:1 as solvent, for 3 h of total reaction time) to obtain the saturated analog 53. Partial and total O-demethylation was again achieved in the presence of 33% HBr in AcOH, and respective products 54 (obtained and tested as the racemic mixture) and 55 were isolated and tested in vitro.
To further expand SAR on the MOR PP, in Scheme 10 we synthesized a new library of bitopic analogs presenting different phenylmorphan nuclei as PPs to target MOR, and the well-known 2-indoleamide SP to target D3R SBP. We were motivated to use this combination of pharmacophores because phenylmorphans are a well-characterized scaffold for obtaining highly potent and selective MOR ligands56, and they also structurally resemble phenyl/pyridine morpholino moieties that we extensively studied as a D3R OBS ligands45. Moreover, we recently demonstrated how the pyridine morpholine scaffold designed as a D3R PP, can also be structurally tweaked via the bitopic approach and linker tethering to target other families of GPCRs, including MOR46. This suggested that simple structural modification of the phenylmorphan scaffold might also exploit affinity for both targets, and ultimately be suitable for the generation of bitopic and bivalent hybrids for both MOR and D3R.
Scheme 10.
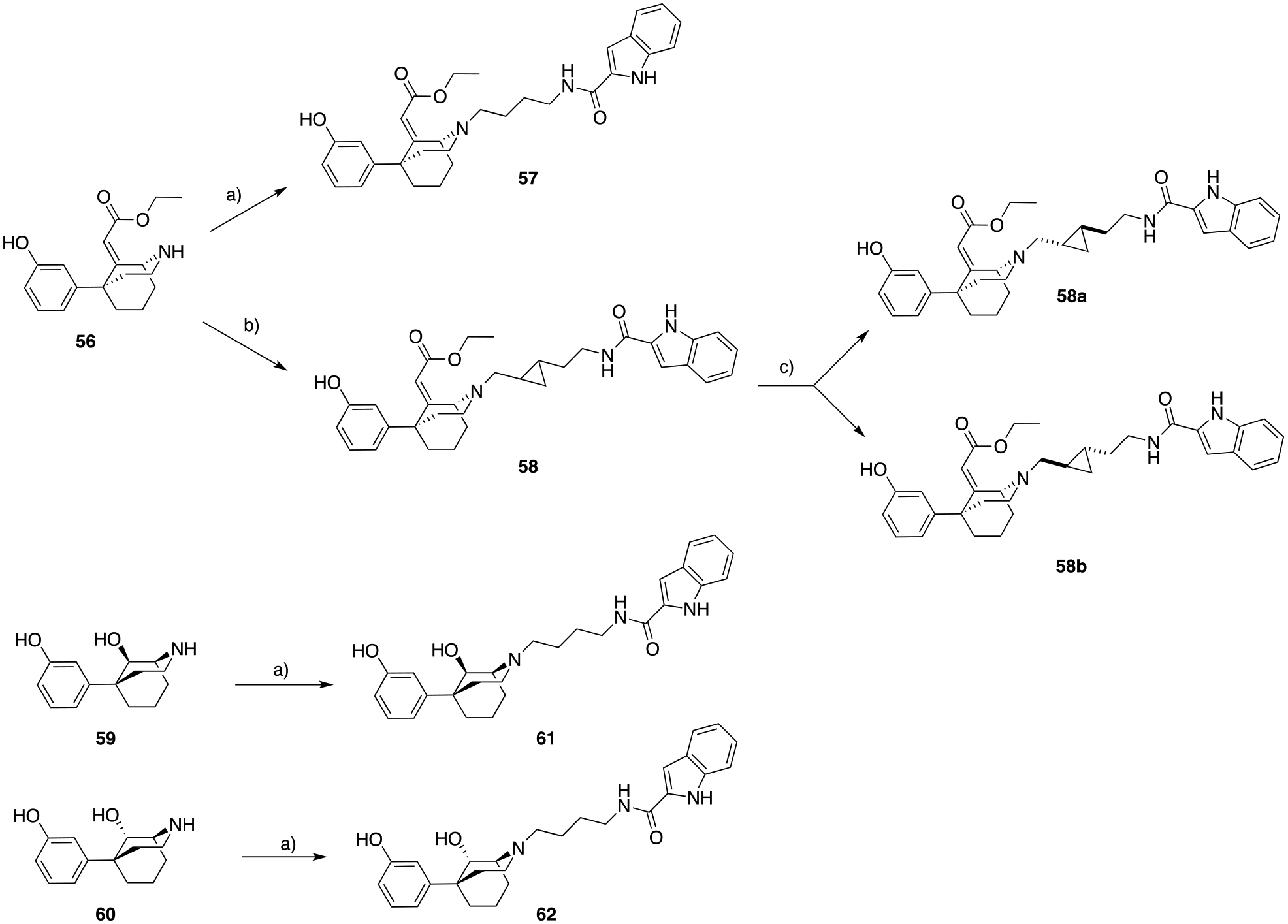
a) N-(4-oxobutyl)-1H-indole-2-carboxamide57, cat. AcOH, STAB, DCE, 25 ° C, 1.5 hrs, 26 – 88%; b) trans-N-(2-(2-formylcyclopropyl)ethyl)-1H-indole-2-carboxamide45, cat. AcOH, STAB, DCE, 25 ° C, 1.5 hrs, 65%; c) preparative enantiontioselective high-performance liquid chromatography (PREP-HPLC), ChiralPak AD-H column.
In Scheme 10, nor-56 was N-alkylated via reductive amination with N-(4-oxobutyl)-1H-indole-2-carboxamide57, and trans-N-(2-(2-formylcyclopropyl)ethyl)-1H-indole-2-carboxamide45, to obtain 57 and 58, respectively. We have previously published45 the importance of the rigid cyclopropyl linker to achieve unique pharmacological profiles, and its stereochemistry is essential in modulating favorable poses for D3R target recognition, binding affinity, selectivity and functional efficacy. Thus, both trans enantiomers of 58 were resolved via preparative chiral HPLC (58a and 58b), and pharmacologically evaluated. Due to the lack of significant differences between their biological profiles we didn’t proceed any further in assigning the absolute configuration of each trans-cyclopropyl enantiomers for these analogs.
In silico docking predicted that replacing the ethyl acrylate on the phenylmorphan ring with a less sterically hindered group (e.g., ketone or hydroxyl group), would be tolerated within the MOR OBS and could increase the affinity of the new hybrids for D3R. Moreover, since the bitopic analogs 57 and 58 showed high affinity for MOR, but moderate to low binding at D2-like receptors (Table 2), we aimed to study and invert the stereochemistry of the phenylmorphan ring, while simultaneously evaluating all the possible stereochemical combinations with the hydroxyl group. We synthesized both diastereoisomers 61 and 62, starting from the fully resolved 59 and 60, to study not only the effect of the hydroxyl substitution, but of its stereochemistry too, in the dual-target binding profile.
Table 2.
Radioligand competition binding affinity data, for all the MOR substituted phenylmorphan PP analogs, at hMOR and hD2-like receptor subtypes. All the affinity values are expressed as Ki ± SEM, derived from IC50 values using the Cheng-Prusoff equation63, and calculated as the mean of at least three independent experiments (n = number of independent experiments), each performed in triplicate.
|
|
ND = Not Determined.
No inhibition of specific radioligand binding was observed at the highest tested concentration in one to three independent experiments, each performed in triplicate.
Lastly, as shown in Scheme 11, we prepared the bivalent analog of this phenylmorphan series, 65, presenting both the MOR PP and the canonical D3R PP 2,3-dichlorophenyl piperazine for D3R OBS, instead of the 2-indoleamide for SBP binding. The alcohol intermediate 63 was prepared starting from 1-(2,3-dichlorophenyl)piperazine hydrochloride and 2-bromoethan-1-ol, under basic conditions. Tosylation of the hydroxy group and subsequent nucleophilic substitution with 56 yielded the desired product 65.
Scheme 11.
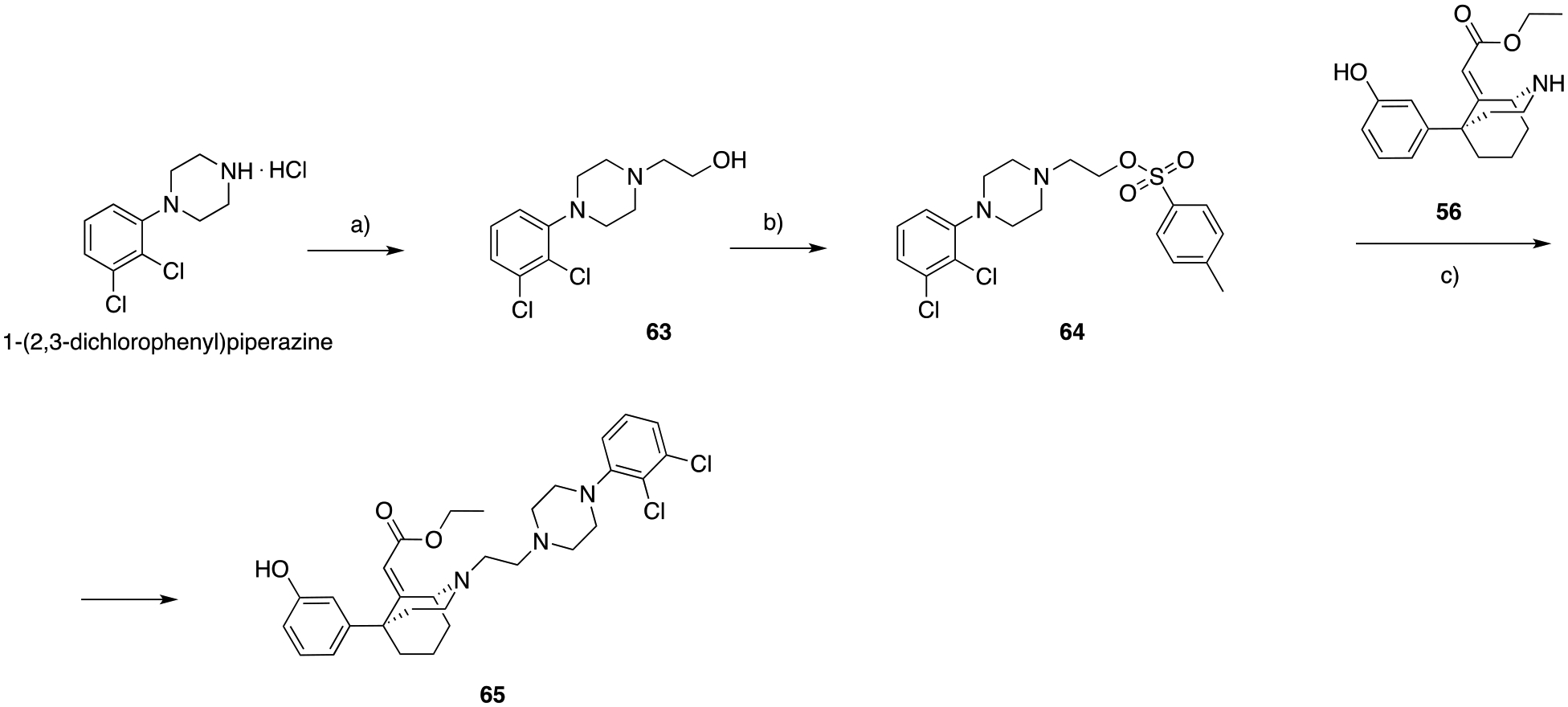
A) 2-bromoethan-1-ol, K2CO3, cat. KI, ACN, 82 ° C, overnight, 60%; b) p-toluenesulfonyl chloride (p-TsCl), DIPEA, DCM, 25 °C, overnight, 36%; c) NaHCO3, ACN, 82 ° C, 6 hrs, 43%.
Molecular Docking and Computer Aided Drug Design (CADD)
We employed structure-based molecular modeling methods to guide the rational design of our dual-target compounds. All atom docking studies on inactive state D3R (PDB: 3PBL)44, and active state MOR (PDB: 5C1M)58 were performed, followed by local optimization of receptor-ligand interactions via energy-based Monte Carlo minimization protocols59 using ICM-Pro (Molsoft LLC). Figure 2 shows optimized binding modes of 5 and 3 inside D3R and MOR, respectively, indicating salt bridge interactions between basic nitrogen and conserved D3.32 of the receptors. The fluorophenyl butanone moiety of 5 occupies the OBS between TM3, TM5, and TM6 in the D3R, lined by hydrophobic residues such as V1113.33, V1895.39, F3456.51, F3466.52, and H3496.55. In MOR, diphenyl butanamide moiety of 3 also occupies the OBS pocket similarly to canonical MOR ligands such as morphine and methadone, interacting with residues Y1503.33, M1533.35, V2385.42, W2956.48, I2986.51, H2996.52, V3026.55, W3207.35, I3247.39, and Y3287.43. Though the chlorophenyl and hydroxy substituents on the piperidine rings are common for both 5 and 3, these moieties occupy distinct SBPs and interact with different residues in their cognate receptors. Attempts at crossdocking these ligands, i.e. dock 5 to MOR and 3 to D3R were unsuccessful, corroborating selectivity of the PPs to their corresponding OBS pockets.
Figure 2.
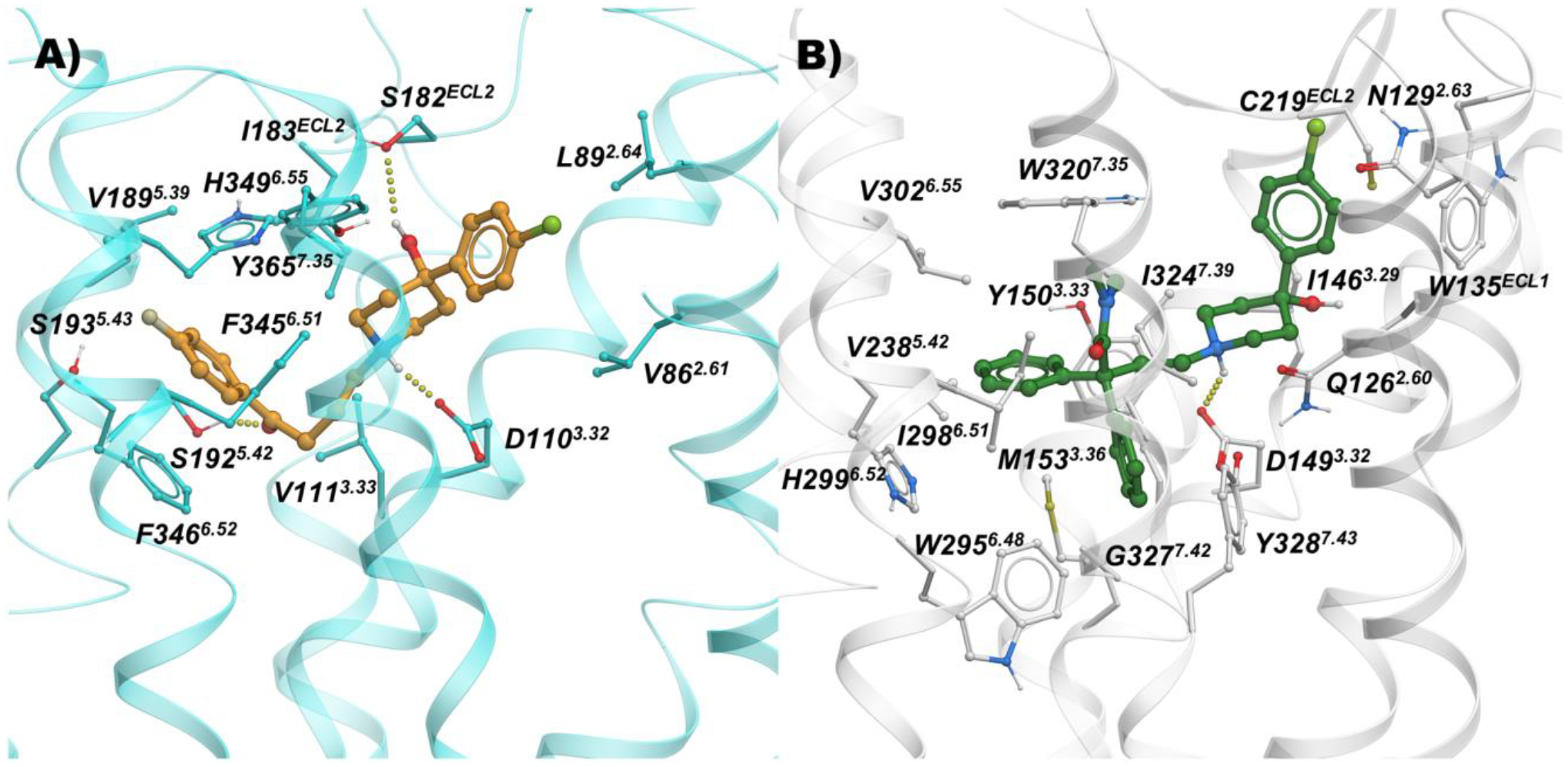
Binding mode of 5 (orange sticks) inside A) D3R (cyan; PDB: 3PBL), and B) binding mode of 3 (green sticks) MOR (white; PDB: 5C1M).
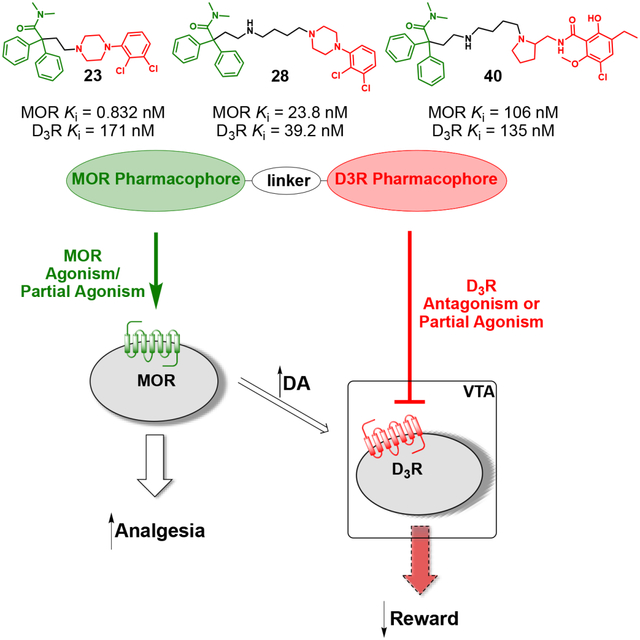
Figure 3 shows an overlay of MOR and D3R docking modes of 23, a compound with the N,N-dimethyl-diphenylbutanamide PP of MOR tethered to a 2,3-dichlorophenyl-piperazine PP of canonical D3R antagonists/partial agonists. In docking to MOR (Figure 3b), we observed a perfect fit of the MOR PP in the corresponding OBS, while the D3R PP of 23 was accommodated in the secondary site of MOR. In D3R, the lack of flexibility of the N-linked 2,3-dichlorophenyl of 23 prevented this D3R PP from reaching its OBS site (Figure 3a). However, the 2,3-dichlorophenyl moiety still comfortably fit the hydrophobic pocket lined by V1113.33, W3426.48, F3456.51, F3466.52, and H3496.55. The MOR PP moiety of 23, N,N-dimethyl-diphenylbutanamide, is placed in a hydrophobic region of D3R surrounded by V862.61, L892.64, C1033.25, F1063.28, C181ECL2, Y3657.35, and T3697.39, and makes additional interactions resulting in comparable docking scores for MOR and D3R, supporting the synthesis of this compound.
Figure 3.
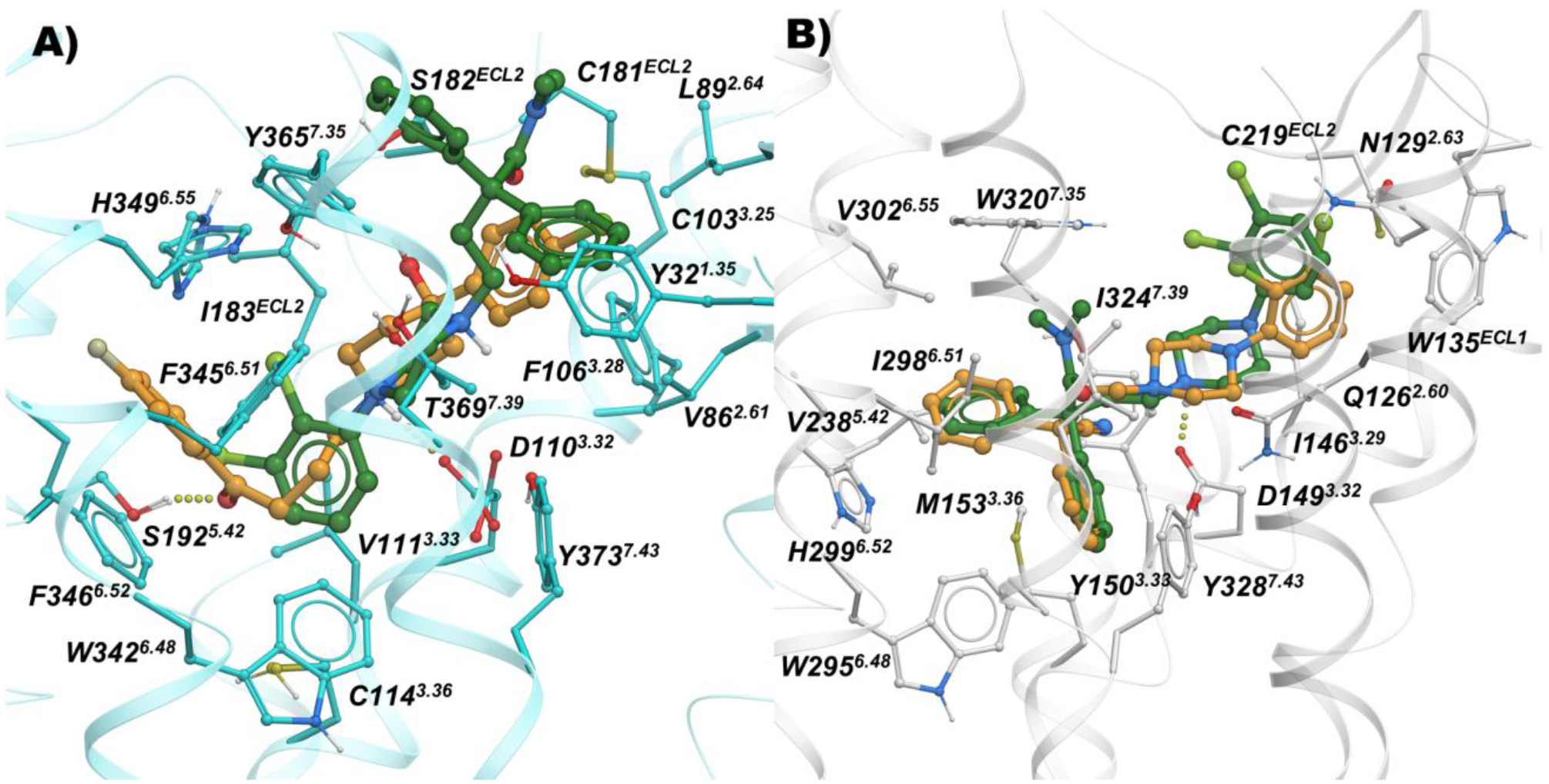
Binding modes of 23 (green) and 5 (orange) inside A) D3R (cyan; PDB: 3PBL), and B) Docking mode of 23 (green) and 13 (orange) inside MOR (white; PDB: 5C1M).
In contrast, despite D3R SAR that supported introduction of the butyl linker between the tethered N,N-dimethyl-diphenylbutanamide or 2,2-diphenylbutanenitrile MOR PP and the 2,3-dichlorophenyl-piperazine, as seen in compounds such as 14 and 28, this design had no effect on the placement of the compounds in the OBS of D3R, while the corresponding SP motif moves further towards the extracellular region, away from the hydrophobic residues of TM2, TM3 and ECL2 (Figure 4).
Figure 4.
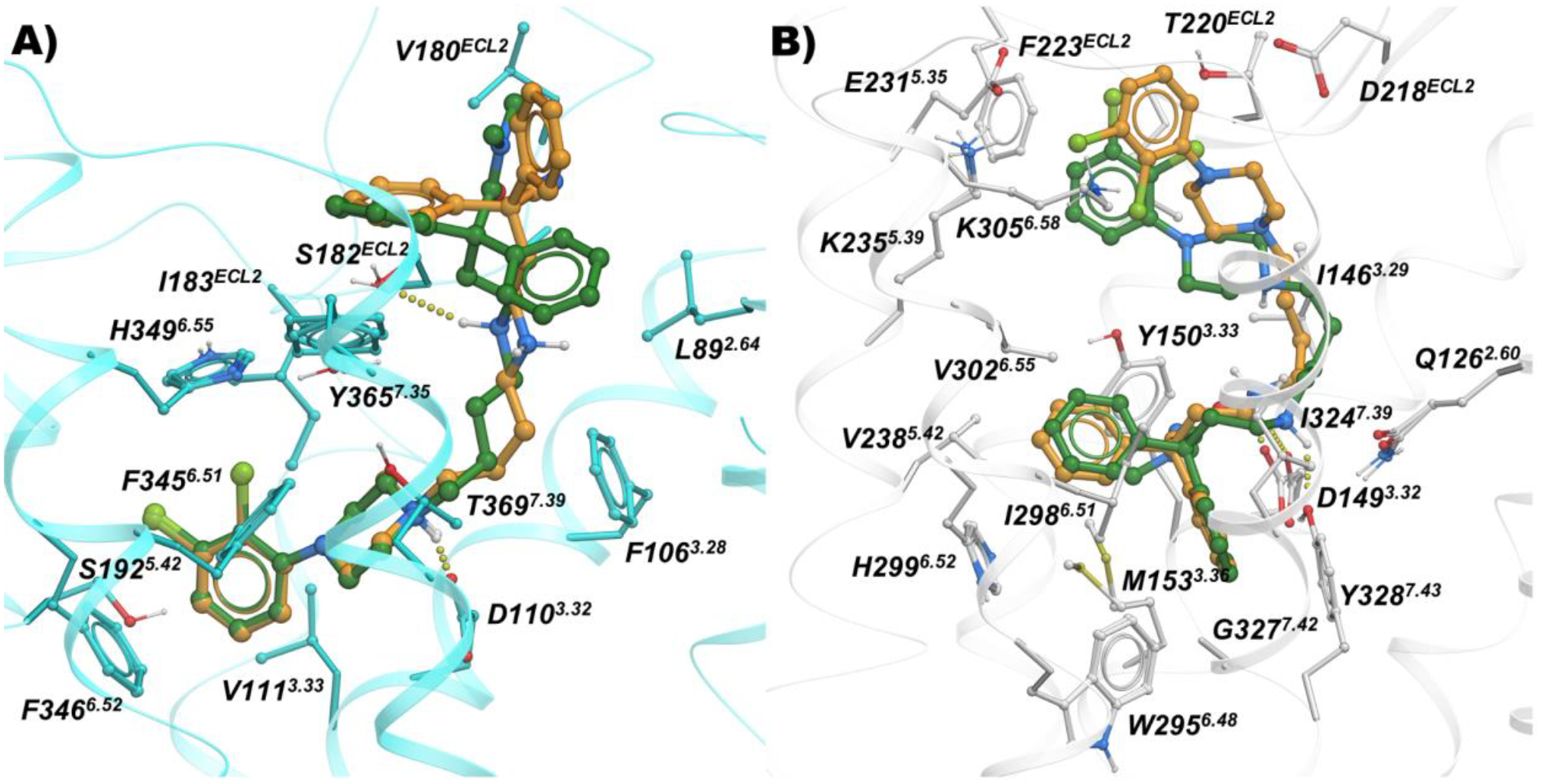
Binding mode of 14 (orange sticks) and 28 (green sticks) inside A) D3R (cyan; PDB: 3PBL), and B) MOR (white; PDB: 5C1M).
The meta-hydroxy compounds were designed to avail the water mediated hydrogen bonding network close to H2996.52 and Y1503.33 in MOR, such as 51, 54 and 55, indeed indicate the presence of those predicted hydrogen bonds, while the 2-chloro-3-ethyl-substituted phenyl-piperazine D3R PP motif is tolerated in the voluminous and solvent accessible MOR pocket (Figure S1). Further, the highly decorated 8-based D3R PP of compounds 40 and 48 are placed expectedly in the OBS of D3R and like the substituted phenyl analogs, the N,N-dimethyl-diphenylbutanamide motif occupies hydrophobic sub-pockets between TM2, TM3, ECL2 and TM7 (Figure 5).
Figure 5.
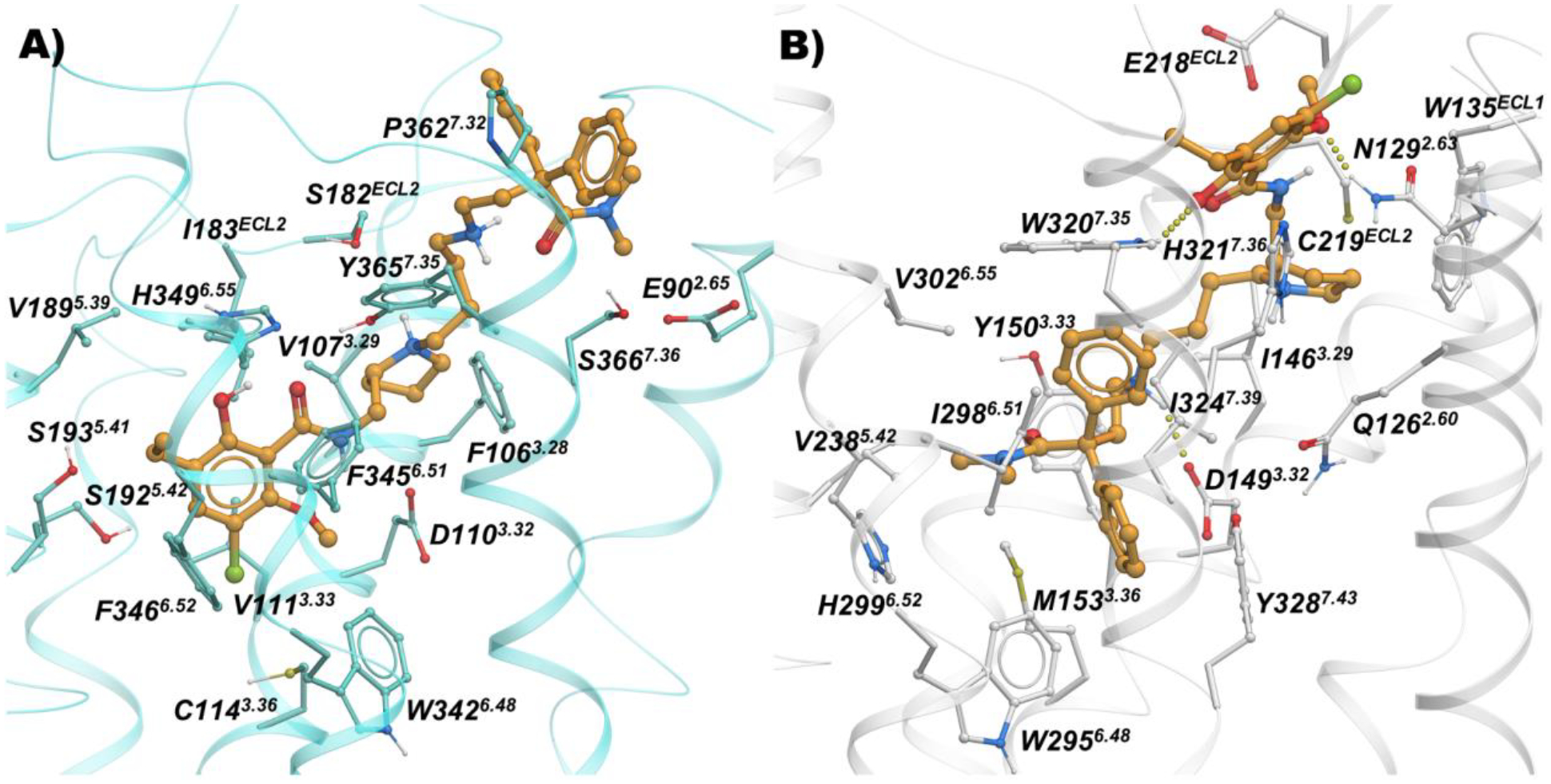
Binding mode of 40 (orange sticks) inside A) D3R (cyan; PDB: 3PBL), and B) MOR (white; PDB: 5C1M).
The conformationally restricted ethyl-2-(-5-(3-hydroxyphenyl)-2-azabicyclo[3.3.1]nonan-9-ylidene)acetate MOR PP motif of 10 expectedly docks in the MOR hydrophobic pocket formed between TM3-TM5-TM6-TM7 and 3-hydroxyphenyl forms water mediated hydrogen bonding interactions with H2996.52 and Y1503.33. The ethyl phenyl tail of 10 shares the same hydrophobic sub-pocket between TM2-TM3 engaged by fentanyl-like compounds. The docking mode of 65 overlaps completely with the binding mode of 10, except the extended 2,3-dichlorophenyl piperazine of 65 moves slightly towards the extracellular region and is placed in the sub-pocket between TM2-TM3-ECL2. The 2,3-dichlorophenyl piperazine of 65 is docked in the OBS of D3R, while the ethyl-2-(-5-(3-hydroxyphenyl)-2-azabicyclo[3.3.1]nonan-9-ylidene)acetate moiety is placed in the hydrophobic SBP region (Figure 6).
Figure 6.
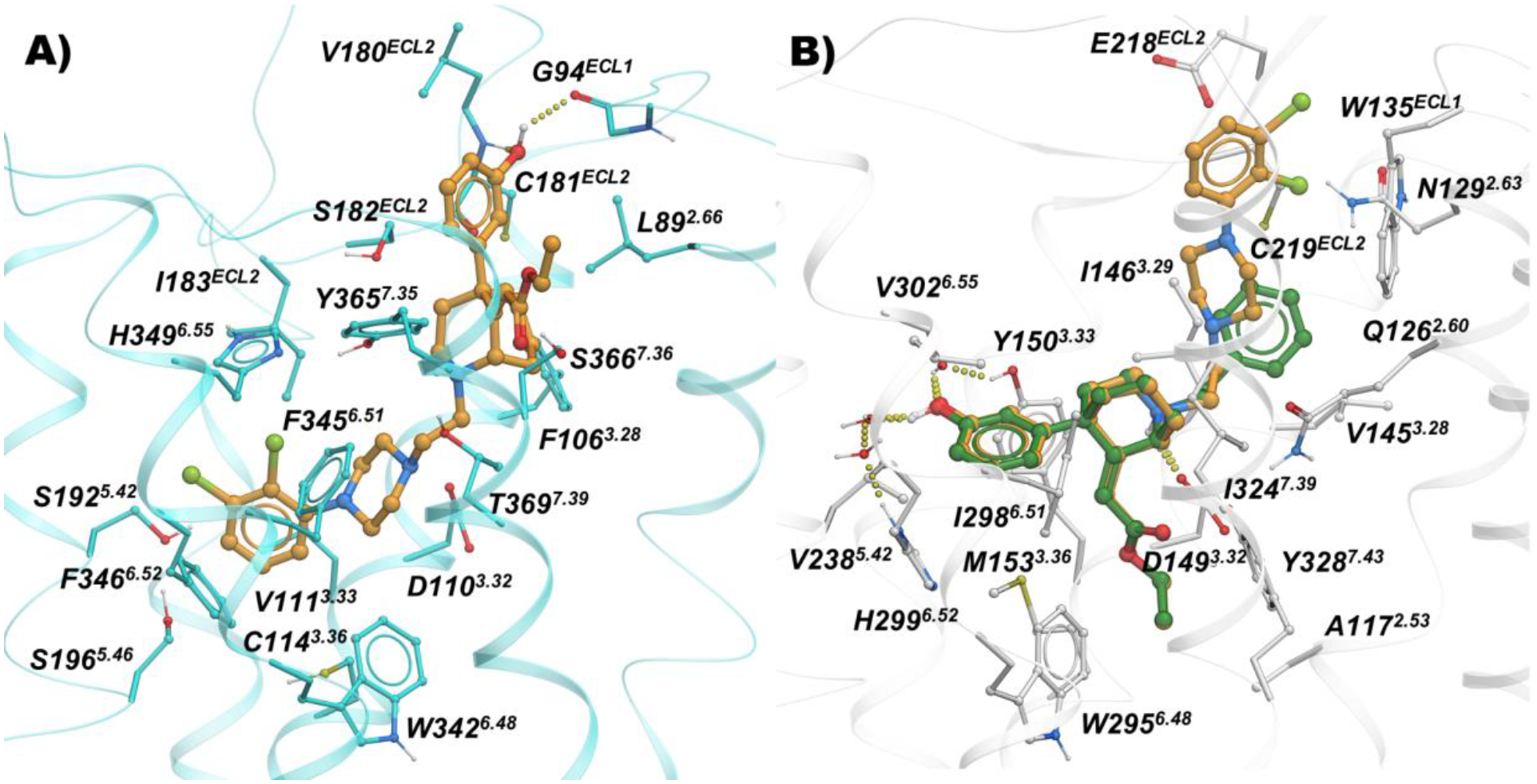
Binding mode of 65 (orange sticks) and 10 (green sticks) inside A) D3R (cyan; PDB: 3PBL), and B) MOR (white; PDB: 5C1M)
Binding Studies and Structure-Activity Relationships (SAR)
All the newly synthesized compounds were tested for their binding affinities at hMOR (in competition with [3H]-DAMGO), hD2R, hD3R, and hD4R (in competition with [3H]-N-methylspiperone ([3H]-NMSP) for all the hD2-like subtypes). Moreover, a subset of selected hits were further studied at hD2R and hD3R using the agonist [3H]-(R)-(+)-7-OH-DPAT as the competing radioisotope. We have previously observed and reported that differences in affinity due to the radioligand being an agonist or antagonist can predict functional efficacy profiles for the tested compounds45, 60.
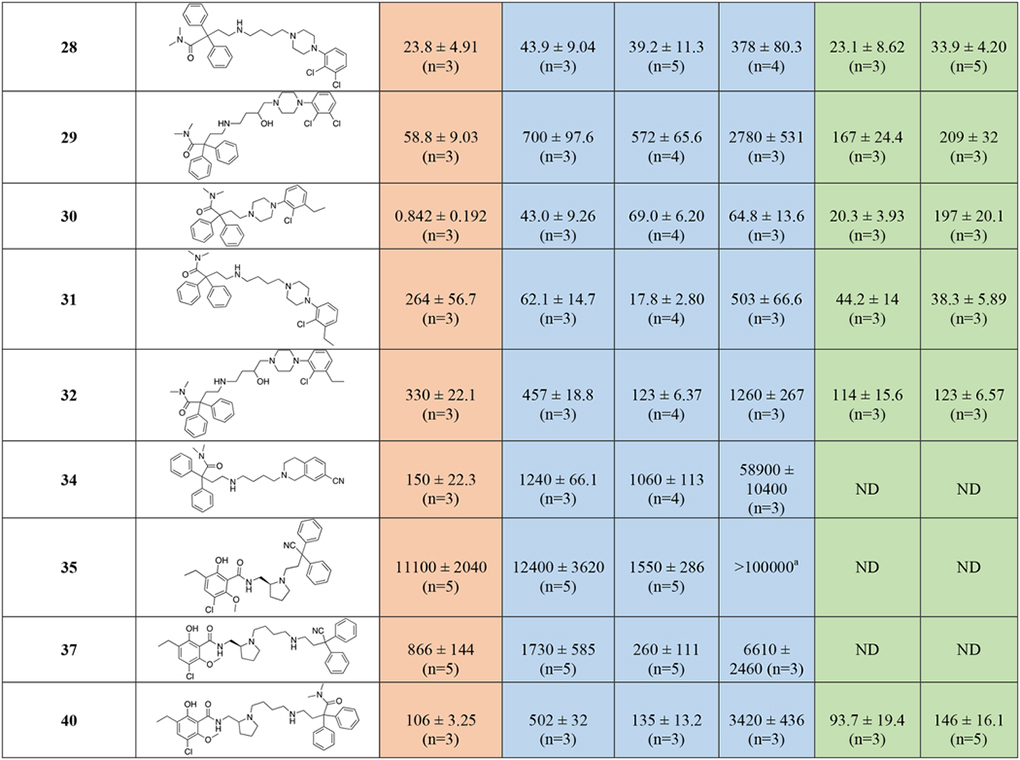
In Table 1 the binding data are reported for the first series of MOR-D3R hybrid analogs, based on, 3-, 4-, and 6-like PPs. Reference compounds, including fentanyl, 3, 4, 5, and 6 are reported in the table for useful comparisons. Among the reference compounds, it was interesting to observe how 3, a well-known potent MOR agonist, despite presenting an SP identical to the D2-like antagonist 5 (Figure 1), binds with low micromolar affinity to all the D2-like subtypes, as predicted in the CADD studies. Fentanyl, 4, and 6, all have low nanomolar affinities for MOR, as expected and consistent with the literature61, 62. Fentanyl, however, presents moderate affinity for D4R (Ki = 554 nM), while being inactive at both D2R and D3R (Kis >10,000 nM), meanwhile 6 is endowed with a preferential low micromolar affinity for D3R, being completely inactive at D2R and >10-fold selective over D4R. These data highlight how subtle structural changes in well-characterized MOR agonists, can induce different binding profiles and subtype selectivity for the D2-like dopamine receptors, and that binding affinities can be directed toward dual-target profiles with well-designed structural modifications.
The docking studies showed that the MOR PP motifs (N,N-dimethyl-diphenylbutanamide and 2,2-diphenylbutanenitrile) occupy primarily hydrophobic pockets in both D3R and MOR. In case of MOR, this pocket formed between TM3, TM5, TM6, and TM7, is an accumulation of three sub-pockets lined by: a) V2385.42, M1533.33, H2996.52, I2986.51, and V3026.55 b) M1533.33, W2956.48, and Y3287.43 c) W3207.35, I3247.39, I2986.51, and V3026.55. In D3R, these MOR PP motifs occupy a hydrophobic SBP between TM2, TM3, and ECL2 (Figure 3 and 4). In general, replacing the nitrile group with the 3-like N,N-dimethylamide synthon significantly increased the affinity profiles of all the analogs. In particular, 23 presents one of the highest MOR affinities among all the new analogs (MOR Ki = 0.832 nM), and despite the shorter linker, which is generally less favorable for D2-like receptors affinity, we still obtained a potent D2-like ligand (D2R Ki = 74.7 nM, D3R Ki = 171 nM, and D4R Ki = 102 nM). A similar nanomolar binding profile across D2R and D3R was also confirmed when 23 was tested in the presence of [3H]-(R)-(+)-7-OH-DPAT. The analogous nitrile compound, 13, showed reduced MOR binding (~320-fold; Ki = 266 nM) and reduced D3R binding (~13-fold; Ki = 2,240 nM).
The docking studies of butyl linked compounds show that, in D3R, the N,N-dimethyl-diphenylbutanamide and 2,2-diphenylbutanenitrile MOR PP motifs move further towards the extracellular region, away from the hydrophobic residues of TM2, TM3, and ECL2 (Figure 4). Perhaps, because of this conformational change, N,N-dimethylamide to cyano substitutions on the extended linker molecules such as 14 (D2R Ki = 149 nM, D3R Ki = 132 nM) are better tolerated at D3R than the shorter linker compounds such as 13 (D2R Ki = 2630 nM, D3R Ki = 2240 nM). As in D3R, the extended linker compounds are reasonably well tolerated inside MOR. However, in contrast to its binding profile at D3R, even the extended linker molecule with nitrile substitutions 14 shows reduced MOR binding (Ki = 490 nM). This was also observed with the nitrile analogues, 35 and 37. Compound 28, an analogous compound with substituted N,N-dimethylamide in the MOR PP motif, shows significant improvement in both D3R affinity (Ki = 39.2 nM) and a MOR binding (Ki = 23.8 nM). This was the first hit analog in this series to show a low nanomolar dual-target affinity for both the MOR and the D2-like receptors. In contrast, compound 18 showed similarly high affinity for the D2-like receptors, but MOR affinity was diminished (Ki = 1470 nM). Whereas compounds 15, 19, 21, 24 and 27 showed the opposite profile, having higher affinities for MOR than D2R or D3R. Compounds 16 and 22 were poorly active at all receptors tested, reflecting an inability to bind the OBS of either MOR or the D2-like receptors.
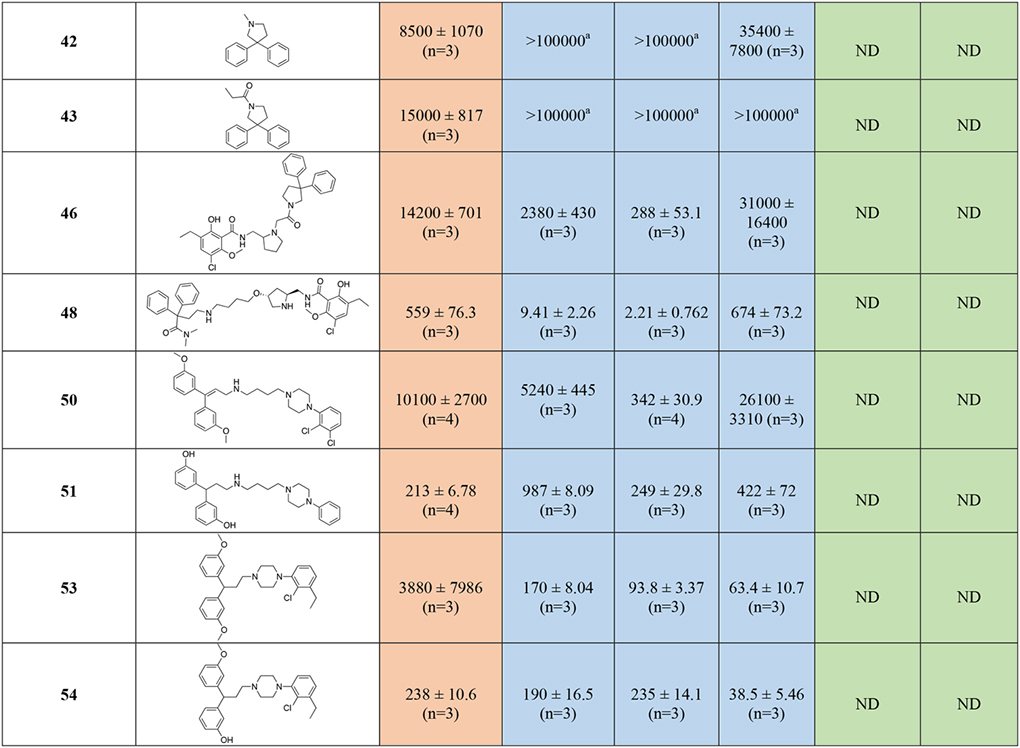
Introduction of the hydroxy substituent in the butylamine linker (compounds 29 and 32), as well as replacement of the 2,3-dichlorophenyl piperazine, with the 2-chloro-3-ethyl-phenylpiperazine (compounds 31 and 32) either maintained or slightly decreased the overall affinity for all the D2-like receptor subtypes, when compared to 28 and shown in Figure S2. The introduction of 1,2,3,4-tetrahydroisoquinoline-7-carbonitrile as D3R PP (34), decreased affinity for the D2-like receptors into the micromolar range. None of the diphenyl-pyrrolidine analogues (compounds 42, 43 and 46) were active. However, the only bivalent compound 46 did have moderate affinity for D3R (Ki = 288 nM)
Removal of either nitrile or amide functions from the diphenyl MOR PP, and introduction of the meta-hydroxy substituents to the bis-phenyl system, 51, was tolerated as suggested from the docking predictions (Figure S1). Despite the presence of a simple unsubstituted phenylpiperazine pharmacophore, 51 still maintained moderate affinities at both MOR (Ki = 213 nM) and D3R (Ki = 249 nM).
When the bis-meta-phenol MOR PP was used in combination with the 2-chloro,3-ethyl-phenylpiperazine D3R PP, tethered via the shorter (two methylene units) linker (55), moderate affinity for all the targets of interest was maintained, but this analogue was not significantly different from the longer linker analog 51. The presence of the meta-hydroxy substituents also retained D2-like affinity for 55, similar to that observed for 23 and 30 (containing the 3-like MOR PP). In a continuation of a trend noted previously, we observed a significant loss of affinity at MOR, with 55 presenting a binding Ki >300-fold less than 30 and 23. These results suggest that while the bis-phenol MOR PP is well-tolerated as an alternative to the more canonical 3-like di-phenyl-N,N-dimethylamide, the binding profile is still dependent on the linker length, rigidity and overall substitutions on the D3R PP as well.
In agreement with the CADD, the meta-substitutions in bis-phenyl containing compounds are important for MOR recognition. The methoxy analogs result in partial loss of MOR affinity with a general binding profile of 53 (di-methoxy) < 54 (mono-methoxy) ~ 55 (di-hydroxy). This, however, doesn’t apply for D2-like binding, where all three analogs show moderate affinities for all the subtypes independent of meta-methoxy or meta-hydroxy substitutions on the bis-phenyl rings, interestingly with higher affinities at D4R.
Shifting from a phenylpiperazine-based D3R PP, to a highly decorated 8-based D3R PP, to develop SAR around the pyrrolidine scaffold, 40, containing a racemic pyrrolidin-2-ylmethyl-amide linker, and 48, presenting a butyl ether linker chain in position 4 of the trans-pyrrolidine nucleus were synthesized. Compound 48 showed the highest D2R/D3R affinity among all the new analogs (D2R Ki = 9.41 nM; D3R Ki = 2.21 nM), however the regio- and stereochemistry of the substituted pyrrolidine ring was detrimental for MOR binding, with a Ki of 559 nM. On the other hand, 40 emerged as our third lead, alongside 23 and 28, with its almost identical affinities for both MOR (Ki = 106 nM) and D3R (Ki = 135 nM), ~4- and ~25-fold selectivity over D2R and D4R respectively. This profile distinguished 40 as one of the most promising dual-target MOR-D3R compounds in the series.
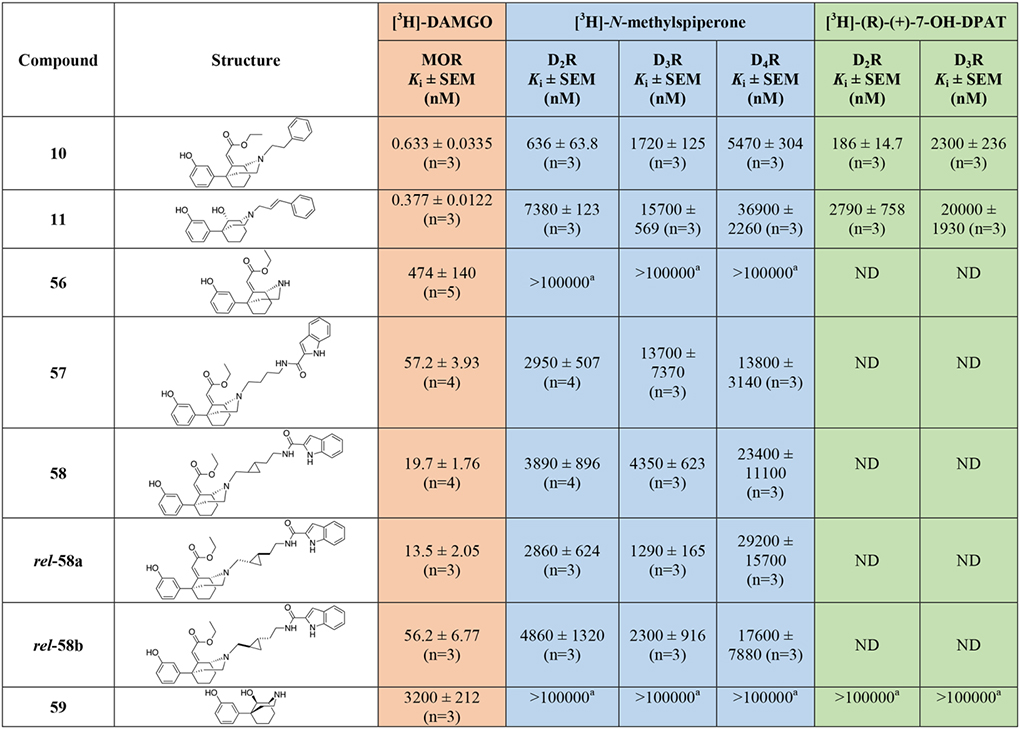
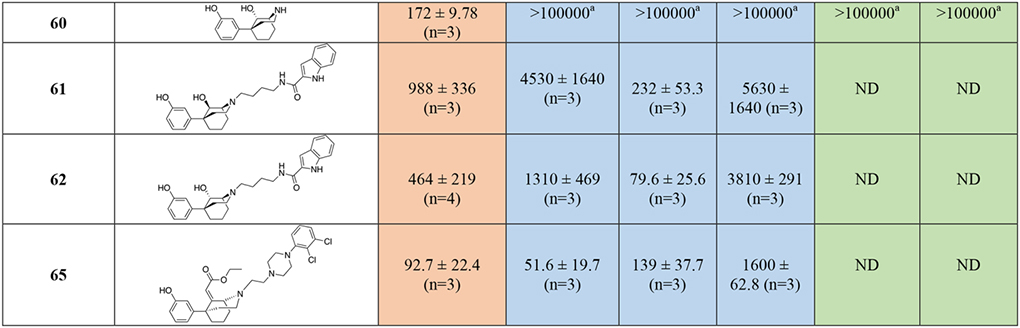
The binding data for the phenylmorphan analogs are reported in Table 2. Compounds 10, 11, and the nor-analogs 56, 59 and 60 were tested as reference compounds, since most of them were key MOR PP building blocks for our bivalent and bitopic drug design. Compound 10 showed the highest MOR affinity as an OBS PP, with Ki = 0.633 nM, similar to the affinities of 3 and the hybrid bivalent analogue, 23. Interestingly, 10 also showed moderately low micromolar and sub-micromolar affinities for D2R and D3R, supporting the hypothesis that the phenylmorphan ring structurally resembles more flexible phenyl-morpholino moieties, canonical scaffolds for D3R ligands45.
Docking studies suggested that the steric clash between the ethyl acrylate group and the backbone of the D3R receptor would limit and potentially challenge the development of bitopic ligands 57 and 58 targeting D3R, despite being tolerated in the MOR OBS. Indeed, bitopic analogs 57, 58, and the resolved enantiomers 58a and 58b, all showed high affinities at MOR (Kis ranging from 13.5 nM to 57 nM), but micromolar Kis for all the D2-like subtypes, independent of linker rigidity or stereochemistry. The shorter bivalent analog 65, with a simple ethyl linker chain (n=2 was predicted by docking studies to be the optimal spacer to resolve steric clashes of the MOR PP motif in D3R; Figure 6). Indeed, tethering the MOR phenylmorphan PP and the D3R 2,3-dichlorophenyl piperazine PP presented one of the most interesting dual-target candidates with equivalent affinities at MOR (Ki = 92.7 nM) and D3R (Ki = 139 nM).
Replacement of the sterically hindered ethyl acrylate group with a smaller hydroxy substituent allowed validation of the docking observation that a small substituent would significantly improve D3R binding. Overall, 61 and 62 adopt the same docking as 65 inside MOR but lose hydrophobic interactions of the ethyl acrylate moiety with M1533.36 and Y3287.43 (Figure S3). The hydrogen-bonding interactions between the 9-OH and Y3287.43 provide limited compensation for 62. However, for 61, this hydrogen bond is accompanied by negative interactions due to proximity of the carboxylate oxygen of D1493.32 (3.2 Å compared to 4.5 Å of S-chiral 62). Further, 61 and 62 adopt an interesting conformation inside D3R with the indole carboxamide ring situated in the OBS. In the absence of positively charged nitrogen, the nitrogen of the amide forms a hydrogen bond with the D1103.32 residue. Interestingly, the 9-OH is placed very close to the backbone of TM7 for 62 and TM2 for 61. Therefore, the substitution of this position with ethyl acrylate would cause steric clashes with D3R, as indicated by the loss of affinity of 57. Bitopic analog 62, with all the resolved stereochemical configurations around the phenylmorphan moiety inverted with respect to 57, showed a D3R Ki of 79.6 nM, ~172-fold improved affinity with respect to the ethyl acrylate analog 57, maintaining a moderate affinity (Ki = 464 nM) for MOR.
Across all the tested compounds, no significant differences were observed in the D2-like affinities determined using [3H]-NMSP and [3H]-(R)-(+)-7-OH-DPAT binding assays were observed, unlike our previous observations for efficacious agonist ligands43, 45, consistent with our hypothesis that all the new analogs are likely antagonists or low efficacy partial agonists at D3R.
Based on their binding profiles, a select group of hits were tested in functional assays, to determine their agonist and antagonist potencies for the multiple GPCR-related signaling pathways, as well as to validate and confirm the MOR agonism and D3R antagonism/partial agonism profile we were seeking with these new hybrid molecules.
BRET Functional Studies at MOR and D3R
With the binding studies and SAR established, we then characterized the action of selected ligands to signal through both MOR and D3R; these results are shown in Figure 7 and Table 3. The action of these ligands was assessed through arrestin-3 (or β-arrestin-2) recruitment at MOR and G protein activation (GPA) at MOR (Gαi2) and D3R (GαoA) assays. In addition, the ability of the ligands to induce the active state of the MOR was determined by measuring recruitment of a conformationally selective nanobody that recognizes and binds to the active conformation of MOR, nanobody 33 (Nb33)64. Seven of the newly synthesized MOR-D3R hybrids (14, 23, 28, 40, 57, 58a and 58b) were tested together with 10. The efficacious agonists DAMGO (D-Ala2, N-MePhe4, Gly5-ol-enkephalin), quinpirole and dopamine were used as reference agonists to normalize data at the MOR and D3R, respectively. We included the MOR partial agonist morphine to illustrate the relative coupling efficiency and amplification of the different assays (Figure 7A,B and D). Both known antagonists, naloxone (MOR) and 5 (D3R), inhibited agonist-stimulated GPA in a concentration-dependent manner (Figure 7C and F).
Figure 7. Functional profiles of selected MOR-D3R hybrids.
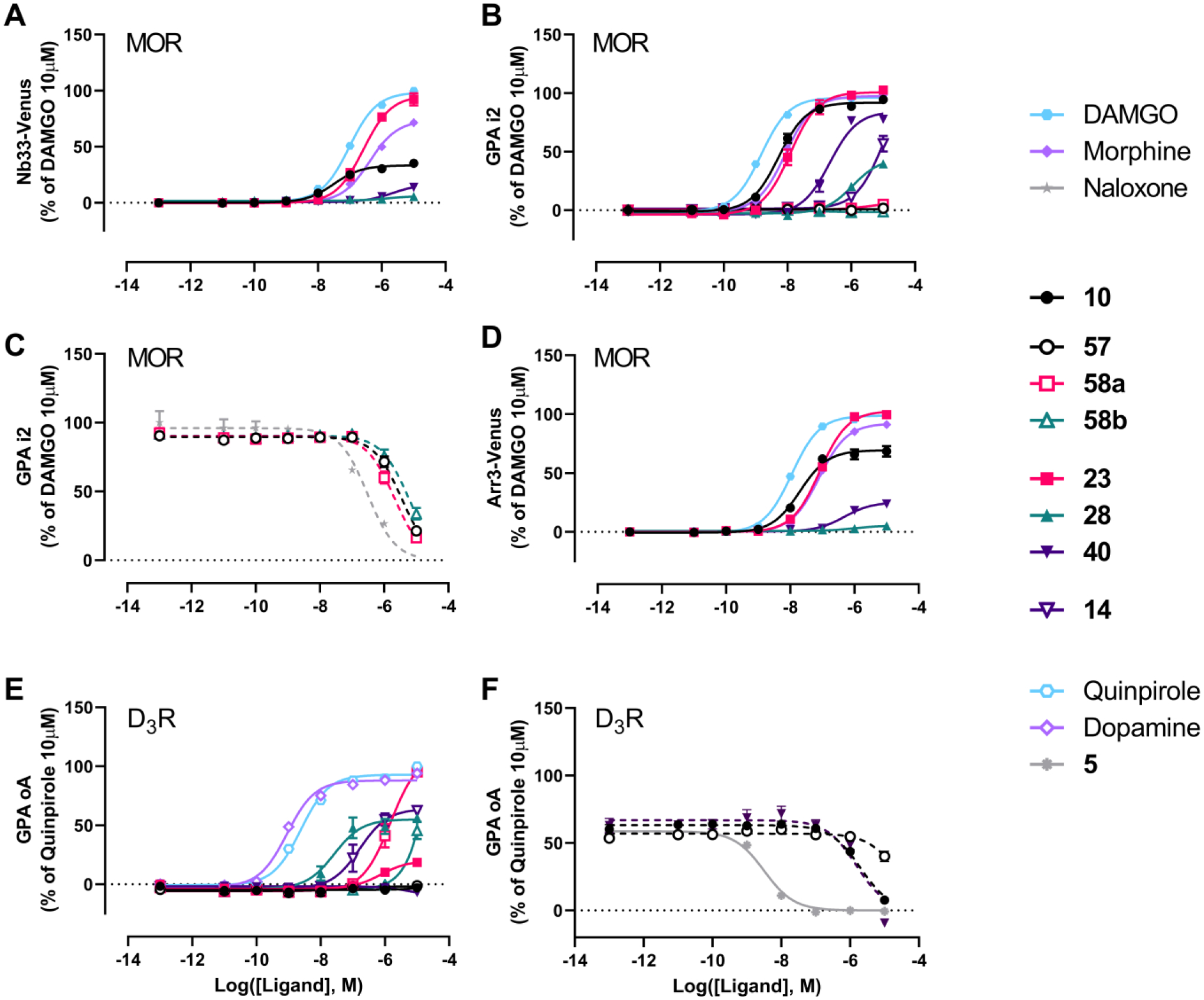
Each panel shows a different signaling readout: A. Nb33 recruitment at MOR, B. MOR-mediated Gαi2 protein activation, C. Antagonism at MOR using GPA in the presence of 100 nM of DAMGO, D. Arrestin-3 recruitment at MOR in the presence of overexpressed GRK2, E. D3R-mediated GαoA protein activation and F. Antagonism at D3R using GPA in the presence of 3 nM of quinpirole. In order to ensure that test ligands achieved maximal receptor occupancy possible before agonist addition, all ligands were added to the cells 3h prior to activating D3R with quinpirole and measuring BRET signals with the exception of 5. Dotted lines represent fits where the bottom asymptote was constrained to be 0%.
Table 3.
Potencies and efficacies at MOR and D3R. All data represent the mean of at least three independent experiments (n = number of independent experiments), each performed in duplicate. Potency values are expressed as pEC50 ± SEM with the corresponding EC50 in nM in brackets. Efficacy values are calculated as a percentage of a reference ligand (DAMGO or quinpirole for MOR and D3R respectively) and expressed as Emax ± SEM (%).
| Compound | MOR Nb33 recruitment | MOR Gi2 activation | MOR arr3 recruitment (+ GRK2) | D3R GoA activation | ||||||
|---|---|---|---|---|---|---|---|---|---|---|
| pEC50 ± SEM [nM] | Emax ± SEM (%) | pEC50 ± SEM [nM] | Emax ± SEM (%) | pIC50 ± SEM [nM] | pEC50 ± SEM [nM] | Emax ± SEM (%) | pEC50 ± SEM [nM] | Emax ± SEM (%) | pIC50 ± SEM [nM] | |
| DAMGO | 7.02 ± 0.04 [95.9] (n=3) | 100 | 8.83 ± 0.03 [1.50] (n=9) | 100 | - | 7.97 ± 0.03 [11.1] (n=3) | 100 | - | - | - |
| Morphine | 6.38 ± 0.05 [416] (n=3) | 73.4 ± 1.7 | 8.06 ± 0.03 [8.66] (n=6) | 97.4 ± 1.1 | - | 7.10 ± 0.04 [79.8] (n=3) | 92.1 ± 1.4 | - | - | - |
| Naloxone | - | - | - | - | 6.52 ± 0.1 [305] (n=3) | - | - | - | - | - |
| Quinpirole | - | - | - | - | - | - | - | 8.62 ± 0.05 [2.39] (n=7) | 100 | - |
| Dopamine | - | - | - | - | - | - | - | 9.05 ± 0.05 [0.896] (n=3) | 88.0 ± 1.3 | - |
| Haloperidol (5) | - | - | - | - | - | - | - | - | - | 8.46 ± 0.07 [3.49] (n=3) |
| 10 | 7.51 ± 0.10 [31.3] (n=3) | 33.3 ± 1.2 | 8.28 ± 0.05 [5.29] (n=9) | 91.9 ± 1.3 | - | 7.70 ± 0.07 [20.0] (n=3) | 69.3 ± 1.7 | NA | NA | 5.70 ± 0.06 [1997] (n=3) |
| 14 | NA | NA | 4.98 ± 0.29 [10530] (n=6) | 114.9 ± 39.2 | - | NA | NA | 6.78 ± 0.15 [165] (n=4) | 63.8 ± 4.2 | - |
| 23 | 6.58 ± 0.06 [266] (n=3) | 95.5 ± 2.7 | 7.91 ± 0.06 [12.3] (n=3) | 100.8 ± 2.1 | - | 7.08 ± 0.03 [83.3] (n=3) | 102.7 ± 1.3 | 6.13 ± 0.16 [747] (n=3) | 20.2 ± 2.1 | - |
| 28 | NA | NA | 5.96 ± 0.09 [1107] (n=3) | 44.5 ± 2.4 | - | NA | NA | 7.58 ± 0.16 [26.4] (n=3) | 55.0 ± 3.2 | - |
| 40 | 5.67 ± 0.20 [2136] (n=3) | 16.6 ± 2.1 | 6.68 ± 0.08 [211] (n=3) | 84.4 ± 2.9 | - | 6.32 ± 0.11 [484] (n=3) | 25.3 ± 1.3 | NA | NA | 5.82 ± 0.15 [1515] (n=3) |
| 57 | NA | NA | NA | NA | 5.45 ± 0.05 [3522] (n=5) | NA | NA | NA | NA | >5 (n=3) |
| 58a | NA | NA | NA | NA | 5.68 ± 0.05 [2102] (n=6) | NA | NA | 5.83 ± 0.08 [1498] (n=4) | 110.6 ± 6.0 | - |
| 58b | NA | NA | NA | NA | 5.24 ± 0.06 [5770] (n=6) | NA | NA | 45.6 % of DA at 10μM (n=4) | ND | - |
NA = Not Active, the compound presents no agonist activity at the highest tested concentration.
The three substituted phenylmorphan MOR PP analogs (57, 58a and 58b), despite showing improved affinities for MOR compared to 56, did not activate MOR in any of the three pathways tested (Figure 7A,B and D). Thus, we tested the ability of these bitopic compounds to inhibit the action of 100 nM of DAMGO in the GPA i2 assay. As shown in Figure 7C, all three ligands were able to inhibit DAMGO-mediated GPA to the same extent as naloxone albeit with lower potencies. When tested at the D3R, 57 failed to demonstrate any D3R activity (up to 10 μM, Figure 7E–F), which is likely due to the low affinity of this compound for this receptor (Table 2). Compounds 58a and 58b showed agonist activity at D3R but with low (micromolar) potencies that reflect their affinity for the D3R (Figure 7E, Table 3). Thus, these two phenylmorphan hybrids display MOR antagonism and D3R agonism, rather than the MOR agonist/D3R antagonist or partial agonist profile we targeted.
In contrast all four MOR diphenyl PP analogs tested (14, 23, 28 and 40), showed agonist activity at MOR, with 23 being the most potent and efficacious compound. Compound 14 showed low potency MOR agonism that could only be detected at the highest concentration used of 10 μM in the most amplified and sensitive GPA i2 assay. 40 displayed higher potency and efficacy in assays of MOR activation than 28, with 28 displaying no detectable agonism in the less amplified Nb33 and arrestin-3 recruitment assays, but an Emax of 50% that of DAMGO in the GPA i2 assay. Compound 40 gave a robust response (Emax = 84.4% of DAMGO) in the GPA i2 assay but much weaker responses in the arrestin and Nb33 assays, indicating it is a less efficacious partial agonist than morphine. All four bivalent compounds share a similar MOR diphenyl PP based on 3 and 4, the N,N-dimethyl-diphenylbutanamide PP being more favorable than the diphenylbutanenitrile PP for MOR agonism. The major structural differences are present in the D3R PP as well as the type and length of the linker between the two pharmacophores: a shorter linker being more favorable for MOR agonism. In general, across the series of compounds and morphine we observe higher maximal effects (Emax) and potencies in the GPA i2 assay as compared to that in the arrestin-3 recruitment and Nb33 assays. Such behavior is consistent with the action of partial agonists at signaling endpoints with different levels of amplification and coupling efficiency of the pathway. In agreement with this, when these data were analyzed using the Black and Leff operational model of agonism and assessed for biased agonism using DAMGO as the reference ligand, none of the compounds displayed significant bias between these two pathways relative to the action of DAMGO (Supplementary Table S2).
When these four MOR diphenyl PP ligands were tested for their ability to activate the D3R (Figure 7E–F), 14 and 28 showed similar efficacies (64% and 55% of dopamine, respectively), with 28 being the most potent compound in agreement with their relative affinities (Table 1). Although 14 and 40 display similar affinities for D3R, 40 acted as an antagonist with micromolar potency (IC50 = 1.5 μM), whereas 14 acted as a robust partial agonist (Emax = 63.8% of quinpirole). Lastly, 23, which was the most potent MOR agonist and shares the same D3R PP structure as 28 but with a shorter linker, displayed weak partial agonism (Emax = 20% of quinpirole) at the D3R and sub-micromolar potency consistent with its binding affinity.
CONCLUSIONS
The data obtained highlight a series of hit to lead candidates as MOR-D3R dual-target ligands. We have synthesized multiple combinations of bivalent or bitopic ligands based on carefully designed structural modifications and in silico guided SAR around the MOR PP, D3R PP and SP, as well as linkers, with a particular focus on regio- and stereochemistry. Importantly, we have identified compounds with a range of sub-nanomolar to sub-micromolar binding affinities for each receptor of interest and thus provide a new approach to modulate the pharmacological profiles of highly selective MOR agonists through concomitant dual-target D3R antagonism.
The functional studies revealed three lead analogs, 23, 28 and 40, which are partial agonists at MOR and partial agonists or antagonists at D3R. We and others have suggested that low intrinsic efficacy could explain the improved therapeutic window observed on the most recent MOR agonists, such as PZM21 and TRV-13031 and our three lead compounds fit this desired functional profile with the added feature of D3R low efficacy partial agonism or antagonism, which may prove beneficial in avoiding the addictive liability of opioid receptor targeted drugs. Further, evaluation and thus a better understanding of the desired kinetic profiles at both targets for optimal pharmacological effect will be crucial in the development of future generations of dual-target MOR-D3R ligands. Indeed, current drug design is focused on improving drug-like characteristics as well as blood brain barrier penetrability as the current lead compounds have central nervous system multiparameter optimization (MPO)65, 66 scores of ~2 and are predicted to be peripherally limited. This indeed was what Komoto and colleagues37 found with their loperamide analogues, which is perhaps unsurprising. Nevertheless, with proof-of-concept in hand, we have laid the groundwork for an alternative pharmacological approach, using bivalent drug design to engage both MOR and D3R in the pursuit of a novel class of opioid analgesics with lower abuse potential.
EXPERIMENTAL METHODS
Chemistry
All chemicals and solvents were purchased from chemical suppliers unless otherwise stated and used without further purification. All melting points were determined (when obtainable) on an OptiMelt automated melting point system and are uncorrected. The 1H and 13C NMR spectra were recorded on a Varian Mercury Plus 400 instrument. Proton chemical shifts are reported as parts per million (δ ppm) relative to tetramethylsilane (0.00 ppm) as an internal standard, or to deuterated solvents. Coupling constants are measured in Hz. Chemical shifts for 13C NMR spectra are reported as parts per million (δ ppm) relative to deuterated CHCl3 or deuterated MeOH (CDCl3 77.5 ppm, CD3OD 49.3 ppm). Chemical shifts, multiplicities and coupling constants (J) have been reported and calculated using Vnmrj Agilent-NMR 400Mr or MNova 9.0 software. Gas chromatography-mass spectrometry (GC/MS) data were acquired (where obtainable) using an Agilent Technologies (Santa Clara, CA) 7890B GC equipped with an HP-5MS column (cross-linked 5% PH ME siloxane, 30 m × 0.25 mm i.d. × 0.25 μm film thickness) and a 5977B mass-selective ion detector in electron-impact mode. Ultrapure grade helium was used as the carrier gas at a flow rate of 1.2 mL/min. The injection port and transfer line temperatures were 250 and 280 °C, respectively, and the oven temperature gradient used was as follows: the initial temperature (70 °C) was held for 1 min and then increased to 300 °C at 20 °C/min and maintained at 300 °C for 4 min, total run time 16.5 min. Column chromatography was performed using a Teledyne Isco CombiFlash RF flash chromatography system, or a Teledyne Isco EZ-Prep chromatography system. Preparative thin layer chromatography was performed on Analtech silica gel plates (1000 μm). When %DMA is reported as eluting system, it stands for % of methanol in DCM, in presence of 0.5%−1% NH4OH. Preparative chiral HPLC was performed using a Teledyne Isco EZ-Prep chromatography system with DAD (Diode Array Detector) and ELS detectors. HPLC analysis was performed using an Agilent Technologies 1260 Infinity system coupled with DAD (Diode Array Detector). For each analytical HPLC run multiple DAD λ absorbance signals were measured in the range of 210–280 nm. Separation of the analyte, purity and enantiomeric/diastereomeric excess determinations were achieved at 40 °C using the methods reported in each detailed reaction description. Preparative and analytical HPLC columns were purchased from Daicel corporation or Phenomenex. HPLC methods and conditions are reported in the descriptions of the chemical reactions where they were applied. Microanalyses were performed by Atlantic Microlab, Inc. (Norcross, GA) and agree with ± 0.4% of calculated values. HRMS (mass error within 5 ppm) and MS/MS fragmentation analysis were performed on a LTQ-Orbitrap Velos (Thermo-Scientific, San Jose, CA) coupled with an ESI source in positive ion mode to confirm the assigned structures and regiochemistry. Unless otherwise stated, all the test compounds were evaluated to be >95% pure on the basis of combustion analysis, NMR, GC-MS, and HPLC-DAD. The detailed analytical results are reported in the characterization of each final compound.
4-(Phenethylamino)-2,2-diphenylbutanenitrile (12).
A suspension of 4-bromo-2,2-diphenylbutanenitrile (500 mg, 1.67 mmol), 2-phenylethan-1-amine (605 mg, 5 mmol), and K2CO3 (230 mg, from 1.67 mmol up to 10 equivalents) in ACN (50 mL), was heated at 130 ° C in a sealed vessel overnight. The mixture was filtered, the solvent removed under vacuum, and the residue purified by flash chromatography eluting with 15% DMA. The desired product was obtained as a colorless oil (300 mg, 53% yield). 1H NMR (400 MHz, CDCl3) δ 7.36 – 7.18 (m, 15H), 3.74 (t, J = 7.1 Hz, 2H), 3.07 (t, J = 6.2 Hz, 2H), 2.95 (t, J = 7.2 Hz, 2H), 2.60 (t, J = 6.2 Hz, 2H); 13C NMR (101 MHz, CDCl3) δ 171.34, 142.74, 139.55, 128.86, 128.57, 128.46, 128.38, 128.36, 128.12, 126.98, 126.18, 61.47, 53.40, 46.66, 46.18, 37.35, 33.47. The free base was converted into the corresponding oxalate salt. HRMS (C24H24N2 + H+) calculated 341.20123, found 341.20048 (error −0.7 ppm). CHN (C24H24N2 + 1.5 H2C2O4 + H2O) calculated C 65.71, H 5.92, N 5.68; found C 65.76, H 5.77, N 5.57. mp: Salt too hygroscopic to determine melting point.
4-(4-(2,3-Dichlorophenyl)piperazin-1-yl)-2,2-diphenylbutanenitrile (13).
The reaction was performed following the same procedure described for 12, starting from 1-(2,3-dichlorophenyl)piperazine (250 mg, 0.9 mmol) and 4-bromo-2,2-diphenylbutanenitrile (350 mg, 1.17 mmol). The desired product was purified by flash chromatography eluting with hexanes/ethyl acetate (hex/EtOAc 5:5), and obtained as a colorless oil (30 mg, 7.5% yield). 1H NMR (400 MHz, CDCl3) δ 7.45 – 7.23 (m, 10H), 7.19 – 7.08 (m, 2H), 6.94 (dd, J = 6.8, 2.8 Hz, 1H), 3.04 (br s, 4H), 2.65 (dd, J = 10.5, 4.3 Hz, 6H), 2.57 – 2.49 (m, 2H). The free base was converted into the corresponding oxalate salt. CHN (C26H25N3Cl2 + H2C2O4 + 0.25 H2O) calculated C 61.71, H 5.09, N 7.71; found C 61.67, H 5.09, N 7.85. mp: 202–207 ° C.
4-((4-(4-(2,3-Dichlorophenyl)piperazin-1-yl)butyl)amino)-2,2-diphenylbutanenitrile (14).
The reaction was performed following the same procedure described for 12, starting from 4-(4-(2,3-dichlorophenyl)piperazin-1-yl)butan-1-amine (200 mg, 0.7 mmol) and 4-bromo-2,2-diphenylbutanenitrile (105 mg, 0.35 mmol). The desired product was purified by flash chromatography eluting with 15% DMA, and obtained as a colorless oil (58 mg, 31% yield). 1H NMR (400 MHz, CDCl3) δ 7.37 – 7.29 (m, 10H), 7.29 – 7.18 (m, 2H), 6.95 (dd, J = 6.4, 3.2 Hz, 1H), 4.84 (br s, 1H), 3.52 (t, J = 7.2 Hz, 2H), 3.26 (t, J = 6.2 Hz, 2H), 3.05 (t, J = 4.8 Hz, 4H), 2.71 (t, J = 6.2 Hz, 2H), 2.59 (br s, 4H), 2.44 (dd, J = 8.2, 6.7 Hz, 2H), 1.70 – 1.53 (m, 4H); 13C NMR (101 MHz, CDCl3) δ 171.46, 151.32, 142.37, 133.99, 128.51, 128.45, 127.48, 127.39, 127.15, 124.48, 118.56, 61.65, 58.24, 53.24, 51.31, 46.34, 44.72, 37.36, 25.14, 24.26. The free base was converted into the corresponding oxalate salt. HRMS (C30H34N4Cl2 + H+) calculated 521.22333, found 521.22363 (error 0.3 ppm). CHN (C30H34N4Cl2 + 3 H2C2O4 + 0.5 H2O) calculated C 54.01, H 5.16, N 7.00; found C 53.97, H 5.33, N 7.15. mp: Salt decomposes above 80 ° C.
4-((4-(4-(2,3-Dichlorophenyl)piperazin-1-yl)-3-hydroxybutyl)amino)-2,2-diphenylbutanenitrile (15).
The reaction was performed following the same procedure described for 12, starting from 4-amino-1-(4-(2,3-dichlorophenyl)piperazin-1-yl)butan-2-ol47 (223 mg, 0.7 mmol) and 4-bromo-2,2-diphenylbutanenitrile (105 mg, 0.35 mmol). The desired product was purified by flash chromatography eluting with 15% DMA, and obtained as a colorless oil (53 mg, 28% yield). 1H NMR (400 MHz, CDCl3) δ 7.48 – 7.32 (m, 6H), 7.24 – 7.10 (m, 6H), 6.95 (dd, J = 7.2, 2.3 Hz, 1H), 4.22 – 4.06 (m, 3H), 3.75 – 3.67 (m, 2H), 3.12 (br s, 4H), 2.96 (tt, J = 8.1, 4.2 Hz, 4H), 2.76 (d, J = 10.7 Hz, 2H), 2.67 (dd, J = 12.5, 3.2 Hz, 1H), 2.61 – 2.50 (m, 1H), 2.01 (s, 1H), 1.82 (ddd, J = 16.4, 13.9, 7.1 Hz, 1H); 13C NMR (101 MHz, CDCl3) δ 169.96, 150.78, 138.44, 138.40, 134.10, 129.50, 129.48, 128.91, 128.89, 127.88, 127.78, 127.58, 127.43, 124.83, 118.64, 64.45, 63.72, 62.31, 53.36, 50.83, 50.62, 45.63, 37.82, 31.51. HRMS (C30H34ON4Cl2 + H+) calculated 537.21824, found 537.21935 (error 1 ppm). HPLC analysis method A: Chiralpak AD-H analytical column (4.5mm × 250mm – 5 μm particle size); mobile phase: isocratic 30% 2-PrOH in hexanes; flow rate: 1 mL/min; injection volume: 20 μL; sample concentration: ~1 mg/mL; multiple DAD λ absorbance signals measured in the range of 210–280 nm, Rt 4.944 min, purity >94.3% (absorbance at 254 nm). HPLC analysis method B: Chiralpak OZ-H analytical column (4.5mm × 250mm – 5 μm particle size); mobile phase: isocratic 30% 2-PrOH in hexanes; flow rate: 1 mL/min; injection volume: 20 μL; sample concentration: ~1 mg/mL; multiple DAD λ absorbance signals measured in the range of 210–280 nm, Rt 9.072 and 10.862 min, purity >95%, er 43:57 (absorbance at 254 nm).
4-(Dimethylamino)-2,2-diphenylbutanenitrile (16).
The reaction was performed following the same procedure described for 12, starting from dimethylamine hydrochloride (2 g, 25 mmol) and 4-bromo-2,2-diphenylbutanenitrile (5 g, 16.7 mmol). The desired product was purified by flash chromatography eluting with 5% DMA, and obtained as a colorless oil (3.5 g, 79% yield). 1H NMR (400 MHz, CDCl3) δ 7.66 – 6.75 (m, 10H), 2.68 – 2.50 (m, 2H), 2.50 – 2.33 (m, 2H), 2.25 (s, 6H). GC/MS (EI), Rt 10.499 min; 264.1 (M+), purity >95%. The free base was converted into the corresponding oxalate salt. CHN (C18H20N2 + H2C2O4) calculated C 67.78, H 6.26, N 7.90; found C 67.66, H 6.43, N 7.96. mp: 163–169 ° C.
3-Carboxy-N,N-dimethyl-3,3-diphenylpropan-1-aminium bromide (17).
Compound 16 (2 g, 7.6 mmol) was dissolved in 48% HBr aq solution (50 mL) and stirred under reflux overnight. The solution was concentrated to half-volume, decanted and the residue washed multiple times with Et2O. The dried crude material was used in the next step without further purification.
N-(4-(4-(2,3-Dichlorophenyl)piperazin-1-yl)butyl)-4-(dimethylamino)-2,2-diphenylbutanamide (18).
A solution of 17 (460 mg, 1.26 mmol), EDC hydrochloride (240 mg, 1.26 mmol), HOBt (170 mg, 1.26 mmol) and DIPEA (2.2 mL; 12.6 mmol) in DCM (20 mL) was stirred at RT for 1 h, followed by dropwise addition of 4-(4-(2,3-dichlorophenyl)piperazin-1-yl)butan-1-amine (380 mg, 1.26 mmol) in DCM (20 mL). The mixture was stirred at RT overnight, the solvent evaporated under vacuum and the residue purified by flash chromatography eluting with 10% DMA. The desired product was obtained as a yellow oil (40 mg, 6% yield). 1H NMR (400 MHz, CDCl3) δ 7.36 – 7.20 (m, 10H), 7.18 – 7.08 (m, 2H), 6.94 (dd, J = 6.6, 3.0 Hz, 1H), 6.74 (br s, 1H), 3.31 – 3.14 (m, 2H), 3.03 (br s, 4H), 2.62 – 2.53 (m, 6H), 2.40 – 2.27 (m, 2H), 2.19 (m, 8H), 1.53 – 1.36 (m, 4H); 13C NMR (101 MHz, CDCl3) δ 173.99, 151.26, 143.81, 133.99, 128.74, 128.25, 127.47, 127.40, 126.81, 124.51, 118.55, 60.22, 58.03, 55.98, 53.23, 51.28, 45.53, 45.42, 44.87, 39.71, 36.80, 29.40, 27.36, 24.09. The free base was converted into the corresponding oxalate salt. HRMS (C32H40ON4Cl2 + H+) calculated 567.26519, found 567.26524 (error 1.3 ppm). CHN (C32H40ON4Cl2 + 2 H2C2O4 + 3 H2O) calculated C 53.93, H 6.29, N 6.99; found C 53.89, H 5.90, N 7.31. mp: Salt too hygroscopic to determine melting point.
6-(Dimethylamino)-4,4-diphenylhexan-3-one (19).
Ethyl magnesium bromide (3 M solution in diethyl ether, 10 mmol) was added dropwise to a solution of 16 (2 g, 7.5 mmol) in toluene (30 mL) at 0 °C. The mixture was slowly warmed to RT and then stirred at reflux for 3 h. The reaction was quenched with 10 mL of 2N HCl (aq solution) at 0 °C and stirred at reflux for for 30 min. The suspension was basified with 2N NaOH at 0 °C, the toluene removed under vacuum and the aq phase extracted with DCM/2-PrOH (3:1). The organic layers were combined, dried over Na2SO4, filtered and dried under vacuum to afford the crude material, which was purified by flash chromatography eluting with 10% DMA. The desired product was obtained as a colorless oil (1.3 g, 59%). 1H NMR (400 MHz, CDCl3) δ 7.51 – 7.06 (m, 10H), 2.52 (m, 2H), 2.30 (m, 2H), 2.15 (d, J = 0.9 Hz, 6H), 1.95 (m, 2H), 0.88 (td, J = 7.3, 0.9 Hz, 3H); 13C NMR (101 MHz, CDCl3) δ 211.02, 141.48, 129.14, 128.26, 126.94, 65.04, 55.67, 45.52, 35.41, 32.47, 9.05. The free base was converted into the corresponding oxalate salt. HRMS (C20H25NO + H+) calculated 296.20089, found 296.20150 (error 0.4 ppm). CHN (C20H25NO + H2C2O4) calculated C 68.55, H 7.06, N 3.63; found C 68.51, H 7.15, N 3.62. mp: 161–165 ° C.
tert-Butyl (5-(4-(2,3-dichlorophenyl)piperazin-1-yl)-5-oxopentyl)carbamate (20).
A solution of 5-((tert-butoxycarbonyl)amino)pentanoic acid (470 mg, 2.16 mmol), 1-(2,3-dichlorophenyl)piperazine (500 mg, 2.16 mmol) and HCTU (895 mg, 2.16 mmol) in DCM (25 mL) was stirred at RT for 3 h. The solvent was removed under vacuum and the residue purified by flash chromatography eluting with hex/EtOAc (40/60). The desired product was obtained as a yellow oil (550 mg, 59% yield). 1H NMR (400 MHz, CDCl3) δ 7.23 – 7.11 (m, 2H), 6.92 (dd, J = 7.7, 1.8 Hz, 1H), 4.63 (br s, 1H), 3.79 (t, J = 4.9 Hz, 2H), 3.68 – 3.60 (m, 2H), 3.15 (q, J = 6.6 Hz, 2H), 3.00 (dq, J = 10.7, 5.2 Hz, 4H), 2.39 (t, J = 7.4 Hz, 2H), 1.76 – 1.62 (m, 2H), 1.60 – 1.51 (m, 2H), 1.43 (s, 9H).
4-((5-(4-(2,3-Dichlorophenyl)piperazin-1-yl)-5-oxopentyl)amino)-2,2-diphenylbutanenitrile (21).
Trifluoroacetic acid (1 mL, 12.8 mmol) was added to a solution of 20 (550 mg, 1.28 mmol) in DCM (10 mL). The mixture was stirred at RT for 24 h, basified with 2N NaOH and extracted with DCM. The organic layers were combined, dried over Na2SO4, filtered and dried under vacuum to afford the crude primary amine intermediate, which was dissolved in ACN (20 mL), followed by addition of 4-bromo-2,2-diphenylbutanenitrile (384 mg, 1.28 mmol) and K2CO3 (10 equivalents). The reaction mixture was stirred at reflux overnight, filtered and the solvent removed under vacuum. The residue was purified by flash chromatography eluting with 15% DMA, and the desired product obtained as a yellow oil (20 mg, 3% yield). 1H NMR (400 MHz, CDCl3) δ 7.37 – 7.10 (m, 12H), 6.90 (dd, J = 7.8, 1.8 Hz, 1H), 3.78 (d, J = 5.8 Hz, 2H), 3.63 (t, J = 4.9 Hz, 2H), 3.59 – 3.51 (m, 2H), 3.28 (t, J = 6.2 Hz, 2H), 2.97 (dt, J = 9.3, 5.0 Hz, 4H), 2.72 (t, J = 6.2 Hz, 2H), 2.50 – 2.41 (m, 2H), 1.71 (m, 4H); 13C NMR (101 MHz, CDCl3) δ 171.48, 171.42, 150.64, 142.20, 134.16, 128.51, 128.47, 128.43, 127.74, 127.50, 127.25, 125.13, 118.75, 61.72, 51.69, 51.20, 46.41, 45.80, 44.41, 41.73, 37.36, 32.69, 26.68, 22.47. HRMS (C31H34ON4Cl2 + H+) calculated 549.21824, found 549.21825 (error 0.0 ppm). HPLC analysis method: Chiralpak AD-H analytical column (4.5mm × 250mm – 5 μm particle size); mobile phase: isocratic 30% 2-PrOH in hexanes; flow rate: 1 mL/min; injection volume: 20 μL; sample concentration: ~1 mg/mL; multiple DAD λ absorbance signals measured in the range of 210–280 nm, Rt 27.693 min, purity >95% (absorbance at 254 nm).
4-(Dimethylamino)-N,N-dimethyl-2,2-diphenylbutanamide (22).
Dimethylamine hydrochloride (1.18 g, 14.5 mmol) was added to a suspension of N-(3,3-diphenyldihydrofuran-2(3H)-ylidene)-N-methylmethanaminium bromide (500 mg, 1.45 mmol) and K2CO3 (2.0 g, 14.5 mmol) in TBME/ACN (25 mL/10 mL). The reaction mixture was heated in a sealed vessel for 24 h, the solvent removed under vacuum, and the residue purified by flash chromatography eluting with EtOAc/MeOH (95/5). The desired product was obtained as a colorless oil (70 mg, 16% yield). 1H NMR (400 MHz, CDCl3) δ 7.43 – 7.24 (m, 10H), 2.97 (br s, 3H), 2.57 – 2.46 (m, 2H), 2.30 (m + br s, 2H + 9H). GC/MS (EI), Rt 11.256 min; 310.1 (M+), purity >95%. The free base was converted into the corresponding oxalate salt. HRMS (C20H26ON2 + H+) found 311.21098 (error −2.4 ppm). CHN (C20H26ON2 + 1.5 H2C2O4 + 0.75 H2O) calculated C 60.19, H 6.70, N 6.10; found C 60.09, H 6.36, N 6.13. mp: 174–178 ° C.
4-(4-(2,3-Dichlorophenyl)piperazin-1-yl)-N,N-dimethyl-2,2-diphenylbutanamide (23).
1-(2,3-Dichlorophenyl)piperazine hydrochloride (400 mg, 1 mmol) was added to a suspension of N-(3,3-diphenyldihydrofuran-2(3H)-ylidene)-N-methylmethanaminium bromide (500 mg, 1 mmol), K2CO3 (1 g, 7 mmol) and DIPEA (1 mL, 7 mmol) in ACN (20 mL). The reaction mixture was stirred at reflux overnight, the solvent removed under vacuum and the residue purified by flash chromatography eluting with 5% DMA. The desired product was obtained as a colorless oil (640 mg, 91% yield). 1H NMR (400 MHz, CDCl3) δ 7.46 – 7.34 (m, 10H), 7.34 – 7.20 (m, 2H), 7.15 – 7.03 (m, 1H), 2.97 (br s, 6H), 2.56 – 2.43 (m, 8H), 2.19 – 2.10 (m, 2H), 1.69 (s, 2H); 13C NMR (101 MHz, CDCl3) δ 173.41, 151.42, 140.76, 133.89, 128.35, 128.10, 127.44, 127.30, 126.70, 124.29, 118.54, 59.70, 55.71, 53.16, 51.33, 42.24. The free base was converted into the corresponding oxalate salt. HRMS (C28H31ON3Cl2 + H+) calculated 496.19169, found 496.19052 (error −2.3 ppm). CHN (C28H31ON3Cl2 + 1.5 H2C2O4) calculated C 58.96, H 5.43, N 6.65; found C 58.69, H 5.33, N 6.65. mp: 199–205 ° C.
4-(7-Cyano-3,4-dihydroisoquinolin-2(1H)-yl)-N,N-dimethyl-2,2-diphenylbutanamide (24).
The reaction was performed following the same procedure described for 23, starting from 1,2,3,4-tetrahydroisoquinoline-7-carbonitrile (100 mg, 0.63 mmol). The crude material was purified by flash chromatography eluting with 5% DMA and the desired product was obtained as a colorless oil (130 mg, 40% yield). 1H NMR (400 MHz, CDCl3) δ 7.46 – 7.18 (m, 12H), 7.10 (d, J = 7.9 Hz, 1H), 3.49 (br s, 2H), 2.98 (br s, 3H), 2.82 (t, J = 5.9 Hz, 2H), 2.63 (t, J = 5.9 Hz, 2H), 2.55 – 2.46 (m, 2H), 2.40 – 2.26 (br s, 3H), 2.26 – 2.17 (m, 2H); 13C NMR (101 MHz, CDCl3) δ 173.44, 140.62, 140.46, 136.71, 130.29, 129.39, 129.33, 128.42, 128.39, 128.07, 126.80, 119.18, 109.08, 59.74, 55.45, 55.42, 50.18, 42.84, 29.55. The free base was converted into the corresponding oxalate salt. HRMS (C28H29ON3 + H+) calculated 424.23834, found 424.23765 (error −1.6 ppm). CHN (C28H29ON3 + 1.5 H2C2O4 + 0.5 H2O) calculated C 65.60, H 5.86, N 7.40; found C 65.76, H 5.87, N 7.29. mp: 178–181 ° C.
4-Hydroxy-N,N-dimethyl-2,2-diphenylbutanamide (25).
N-(3,3-Diphenyldihydrofuran-2(3H)-ylidene)-N-methylmethanaminium bromide (700 mg, 2 mmol) was suspended in 2N NaOH (15 mL aq solution) and then stirred at RT for 5 min. The mixture was extracted with DCM, the organic layers were combined, dried over Na2SO4, filtered and evaporated under vacuum to afford the pure desired product in quantitative yield, as a colorless oil. 1H NMR (400 MHz, CDCl3) δ 7.48 – 7.25 (m, 10H), 3.17 (q, J = 5.0 Hz, 2H), 3.04 – 2.95 (m, 3H), 2.57 – 2.49 (m, 2H), 2.35 – 2.27 (m, 3H). GC/MS (EI), Rt 11.847 min; 283.1 (M+).
N,N-Dimethyl-4-oxo-2,2-diphenylbutanamide (26).
DMP was added portion-wise to a solution of 25 (200 mg, 0.71 mmol) in DCM (10 mL) at 0 °C. The reaction mixture was slowly warmed to RT and stirred for 1 h. The suspension was washed with 10% NaHCO3 (aq solution), the organic layer dried over Na2SO4, filtered and evaporated under vacuum. The residue was purified by flash chromatography eluting with Hex/EtOAc (50/50) to afford the desired product as a white solid (120 mg, 60% yield). 1H NMR (400 MHz, CDCl3) δ 9.17 (t, J = 2.2 Hz, 1H), 7.46 – 7.25 (m, 10H), 3.11 – 2.99 (m, 6H), 2.33 (s, 2H). GC/MS (EI), Rt 11.744 min; 281.1 (M+).
N,N-Dimethyl-4-(phenethylamino)-2,2-diphenylbutanamide (27).
A solution of 26 (90 mg, 0.32 mmol), 2-phenylethan-1-amine (77 mg, 0.64 mmol) and cat. AcOH in DCE (5 mL) was stirred at RT for 30 min. STAB (97 mg, 0.48 mmol) was added portion-wise and the mixture stirred for additional 2 h. The solvent was removed under vacuum and the residue purified by flash chromatography eluting with 5% DMA. The desired product was obtained as a colorless oil (quantitative yield). 1H NMR (400 MHz, CDCl3) δ 7.39 – 7.10 (m, 15H), 2.96 (m, 3H), 2.75 (br s, 7H), 2.48 – 2.28 (m + br s, 4H + 1H); 13C NMR (101 MHz, CDCl3) δ 176.89, 173.88, 140.40, 139.18, 128.82, 128.73, 128.54, 128.45, 128.04, 126.94, 126.22, 126.17, 60.51, 50.11, 46.28, 44.40, 43.46, 40.00, 35.13, 23.25. The free base was converted into the corresponding oxalate salt. HRMS (C26H30ON2 + H+) calculated 387.24309, found 387.24226 (error −2.1 ppm). CHN (C26H30ON2 + 1.5 H2C2O4 + 0.1 NH4OH) calculated C 66.33, H 6.43, N 5.60; found C 66.10, H 6.61, N 5.91. mp: 112–117 ° C.
4-((4-(4-(2,3-Dichlorophenyl)piperazin-1-yl)butyl)amino)-N,N-dimethyl-2,2-diphenylbutanamide (28).
The reaction was performed following the same procedure described for 27, starting from 4-(4-(2,3-dichlorophenyl)piperazin-1-yl)butan-1-amine (272 mg, 0.9 mmol). The desired product was purified by flash chromatography eluting with 25% DMA and obtained as a colorless oil (300 mg, 65% yield). 1H NMR (400 MHz, CDCl3) δ 7.41 – 7.33 (m, 7H), 7.28 – 7.26 (m, 3H), 7.15 – 7.13 (m, 2H), 7.00 – 6.90 (m, 1H), 3.05 (s, 4H), 2.98 (s, 3H), 2.62 (br s, J = 6.0 Hz, 3H), 2.51 (t, J = 6.8 Hz, 6H), 2.45 – 2.37 (m, 4H), 2.29 (br s, 3H), 1.62 – 1.49 (m, 4H); 13C NMR (101 MHz, CDCl3) δ 178.09, 173.97, 151.17, 139.95, 133.94, 128.69, 128.65, 127.97, 127.48, 127.38, 127.15, 124.55, 118.73, 60.48, 57.93, 53.14, 51.06, 50.67, 47.97, 45.70, 43.25, 25.94, 24.19, 23.98. The free base was converted into the corresponding oxalate salt. HRMS (C32H40ON4Cl2 + H+) calculated 567.26519, found 567.26458 (error −1.1 ppm). CHN (C32H40ON4Cl2 + 2.5 H2C2O4 + H2O) calculated C 54.82, H 5.84, N 6.91; found C 54.89, H 5.68, N 6.83. mp: initial decomposition ~119 ° C, complete melting 150–160 ° C.
4-((4-(4-(2,3-Dichlorophenyl)piperazin-1-yl)-3-hydroxybutyl)amino)-N,N-dimethyl-2,2-diphenylbutanamide (29).
The reaction was performed following the same procedure described for 27, starting from 4-amino-1-(4-(2,3-dichlorophenyl)piperazin-1-yl)butan-2-ol (130 mg, 0.41 mmol). The desired product was purified by flash chromatography eluting with 25% DMA and obtained as a colorless oil (80 mg, 34% yield). 1H NMR (400 MHz, CDCl3) δ 7.37 (m, 8H), 7.31 – 7.22 (m, 2H), 7.16 – 7.07 (m, 2H), 6.93 (dd, J = 6.5, 3.1 Hz, 1H), 3.82 (m, 1H), 3.04 (br s, 6H), 2.97 (br s, 1H), 2.83 – 2.67 (m, 4H), 2.59 (dt, J = 11.8, 6.1 Hz, 4H), 2.50 – 2.30 (m, 4H), 2.30 – 2.17 (m, 4H), 1.56 – 1.32 (m, 2H); 13C NMR (101 MHz, CDCl3) δ 173.53, 151.32, 140.75, 140.71, 133.98, 128.38, 128.33, 128.12, 128.09, 128.04, 127.48, 127.36, 126.71, 126.68, 124.45, 118.56, 71.43, 68.18, 64.64, 62.95, 59.92, 53.04, 52.17, 51.35, 47.60, 47.30, 45.64, 43.84, 34.82, 34.04. HPLC analysis method A: Chiralpak AD-H analytical column (4.5mm × 250mm – 5 μm particle size); mobile phase: isocratic 20% 2-PrOH in hexanes; flow rate: 1 mL/min; injection volume: 20 μL; sample concentration: ~1 mg/mL; multiple DAD λ absorbance signals measured in the range of 210–280 nm, Rt 11.906 and 12.953 min, purity >99%, er 38:62 (absorbance at 254 nm). HPLC analysis method B: Chiralcel OD-H analytical column (4.5mm × 250mm – 5 μm particle size); mobile phase: isocratic 20% 2-PrOH in hexanes; flow rate: 1 mL/min; injection volume: 20 μL; sample concentration: ~1 mg/mL; multiple DAD λ absorbance signals measured in the range of 210–280 nm, Rt 12.011 min, purity >99% (absorbance at 254 nm). HPLC analysis method C: Chiralcel OZ-H analytical column (4.5mm × 250mm – 5 μm particle size); mobile phase: isocratic 20% 2-PrOH in hexanes; flow rate: 1 mL/min; injection volume: 20 μL; sample concentration: ~1 mg/mL; multiple DAD λ absorbance signals measured in the range of 210–280 nm, Rt 12.172 min, purity >95% (absorbance at 254 nm). The free base was converted into the corresponding oxalate salt. HRMS (C32H40O2N4Cl2 + H+) calculated 583.26011, found 583.26204 (error 2.3 ppm). CHN (C32H40O2N4Cl2 + 2 H2C2O4 + 1.5 H2O) calculated C 54.69, H 5.99, N 7.09; found C 54.75, H 5.71, N 7.16. mp: Salt decomposes above 116 ° C.
4-(4-(2-Chloro-3-ethylphenyl)piperazin-1-yl)-N,N-dimethyl-2,2-diphenylbutanamide (30).
The reaction was performed following the same procedure described for 27, starting from 1-(2-chloro-3-ethylphenyl)piperazine14 (120 mg, 0.53 mmol). The desired product was purified by flash chromatography eluting with 15% DMA and obtained as a colorless oil (130 mg, 50% yield). 1H NMR (400 MHz, CDCl3) δ 7.44 – 7.34 (m, 8H), 7.29 (m, 2H), 7.13 (t, J = 7.8 Hz, 1H), 6.91 (ddd, J = 19.7, 7.8, 1.5 Hz, 2H), 3.07 (br s, 6H), 2.99 (br s, 2H), 2.74 (br s + q, 4H + 2H), 2.61 – 2.52 (m, 2H), 2.38 (m, 4H), 1.20 (t, J = 7.5 Hz, 3H); 13C NMR (101 MHz, CDCl3) δ 175.46, 173.36, 148.98, 143.16, 140.12, 128.63, 128.54, 128.49, 128.06, 126.98, 126.87, 124.19, 118.14, 59.68, 55.36, 52.67, 50.17, 40.52, 27.39, 22.19, 14.03. The free base was converted into the corresponding oxalate salt. HRMS (C30H36ON3Cl + H+) found 490.26239 (error 0.9 ppm). CHN (C30H36ON3Cl + 1.5 H2C2O4) calculated C 63.40, H 6.29, N 6.72; found C 63.35, H 6.46, N 6.67. mp: 183–186 ° C.
4-((4-(4-(2-Chloro-3-ethylphenyl)piperazin-1-yl)butyl)amino)-N,N-dimethyl-2,2-diphenylbutanamide (31).
The reaction was performed following the same procedure described for 27, starting from 4-(4-(2-chloro-3-ethylphenyl)piperazin-1-yl)butan-1-amine (390 mg, 1.33 mmol). The desired product was purified by flash chromatography eluting with 15% DMA and obtained as a colorless oil (90 mg, 12% yield). 1H NMR (400 MHz, CDCl3) δ 7.43 – 7.30 (m, 8H), 7.30 – 7.21 (m, 2H), 7.13 (t, J = 7.8 Hz, 1H), 6.92 (ddd, J = 8.1, 6.6, 1.6 Hz, 2H), 3.03 (br s, 6H), 2.97 (br s, 2H), 2.76 (q, J = 7.5 Hz, 2H), 2.48 – 2.38 (m, 4H), 2.38 – 2.30 (m, 6H), 2.30 – 2.22 (m, 4H), 1.51 – 1.40 (m, 2H), 1.40 – 1.31 (m, 2H), 1.21 (t, J = 7.5 Hz, 3H); 13C NMR (101 MHz, CDCl3) δ 173.52, 149.69, 143.14, 140.85, 128.66, 128.32, 128.07, 126.83, 126.64, 123.85, 117.99, 59.90, 58.56, 53.42, 51.55, 49.76, 47.50, 45.75, 28.20, 27.46, 24.72, 23.03, 14.09. The free base was converted into the corresponding oxalate salt. HRMS (C34H45ON4Cl + H+) calculated 561.33547, found 561.33350 (error −4.4 ppm). CHN (C34H45ON4Cl + 2 H2C2O4 + 1.75 H2O) calculated C 59.06, H 6.85, N 7.25, found C 59.09, H 6.65, N 7.17.
4-((4-(4-(2-Chloro-3-ethylphenyl)piperazin-1-yl)-3-hydroxybutyl)amino)-N,N-dimethyl-2,2-diphenylbutanamide (32).
The reaction was performed following the same procedure described for 27, starting from 4-amino-1-(4-(2-chloro-3-ethylphenyl)piperazin-1-yl)butan-2-ol (166 mg, 0.53 mmol). The desired product was purified by flash chromatography eluting with 25% DMA and obtained as a colorless oil (110 mg, 36% yield). 1H NMR (400 MHz, CDCl3) δ 7.43 – 7.32 (m, 7H), 7.32 – 7.22 (m, 3H), 7.13 (t, J = 7.8 Hz, 1H), 6.92 (ddd, J = 11.9, 7.8, 1.6 Hz, 2H), 3.82 (m, 1H), 3.03 (s, 6H), 2.98 (s, 2H), 2.82 – 2.69 (m, 5H), 2.61 (ddd, J = 11.7, 8.1, 5.3 Hz, 3H), 2.47 – 2.19 (m, 8H), 1.58 – 1.34 (m, 2H), 1.22 (t, J = 7.5 Hz, 3H); 13C NMR (101 MHz, CDCl3) δ 173.55, 149.65, 143.17, 140.71, 128.67, 128.39, 128.05, 126.82, 126.72, 123.88, 117.98, 67.91, 64.64, 59.97, 53.73, 51.60, 47.48, 47.22, 45.49, 34.03, 27.45, 14.07. HPLC analysis method A: Chiralpak AD-H analytical column (4.5mm × 250mm – 5 μm particle size); mobile phase: isocratic 20% 2-PrOH in hexanes; flow rate: 1 mL/min; injection volume: 20 μL; sample concentration: ~1 mg/mL; multiple DAD λ absorbance signals measured in the range of 210–280 nm, Rt 8.242 and 9.055 min, purity >99%, er 37:63 (absorbance at 254 nm). HPLC analysis method B: Chiralcel OD-H analytical column (4.5mm × 250mm – 5 μm particle size); mobile phase: isocratic 20% 2-PrOH in hexanes; flow rate: 1 mL/min; injection volume: 20 μL; sample concentration: ~1 mg/mL; multiple DAD λ absorbance signals measured in the range of 210–280 nm, Rt 8.516 and 9.788 min, purity >99%, er 57:43 (absorbance at 254 nm). HPLC analysis method C: Chiralcel OZ-H analytical column (4.5mm × 250mm – 5 μm particle size); mobile phase: isocratic 20% 2-PrOH in hexanes; flow rate: 1 mL/min; injection volume: 20 μL; sample concentration: ~1 mg/mL; multiple DAD λ absorbance signals measured in the range of 210–280 nm, Rt 8.886 and 9.524 min, purity >99%. er 50:50 (absorbance at 254 nm). The free base was converted into the corresponding oxalate salt. HRMS (C34H45O2N4Cl + H+) found 577.33059 (error 0.4 ppm). CHN (C34H45O2N4Cl + 2 H2C2O4 + H2O) calculated C 58.87, H 6.63, N 7.23, found C 59.06, H 6.46, N 7.14. mp: 126–130 ° C.
2-(4-Aminobutyl)-1,2,3,4-tetrahydroisoquinoline-7-carbonitrile (33).
A suspension of 1,2,3,4-tetrahydroisoquinoline-7-carbonitrile (300 mg, 1.9 mmol), N-(4-bromobutyl)phthalimide (535 mg, 1.9 mmol), cat. KI (3.15 mg, 19 μmol) and K2CO3 (2.6 g, 19 mmol) in ACN (20 mL) was stirred under reflux overnight. The reaction mixture was cooled down to RT, filtered, and the solvent removed under vacuum and the residue dissolved in EtOH (10 mL), followed by addition of hydrazine (0.175 mL). The solution was stirred under reflux for 3 h, EtOH was evaporated, the residue diluted with 20% K2CO3 aq solution and extracted with DCM. The organic layers were combined, dried over Na2SO4, filtered and evaporated under vacuum. The crude material was used in the next step without further purification (300 mg, 94% yield).
4-((4-(7-Cyano-3,4-dihydroisoquinolin-2(1H)-yl)butyl)amino)-N,N-dimethyl-2,2-diphenylbutanamide (34).
The reaction was performed following the same procedure described for 27, starting from 33 (300 mg, 1.31 mmol) and 26 (368 mg, 1.31 mmol). The desired product was purified by flash chromatography eluting with 10% DMA and obtained as a colorless oil (300 mg, 46% yield). 1H NMR (400 MHz, CDCl3) δ 7.42 – 7.21 (m, 12H), 7.16 (d, J = 7.9 Hz, 1H), 3.57 (s, 2H), 3.47 (d, J = 0.7 Hz, 6H), 2.99 – 2.87 (m, 4H), 2.68 (t, J = 5.9 Hz, 2H), 2.49 – 2.35 (m, 7H), 1.50 (q, J = 7.7 Hz, 2H), 1.41 (q, J = 7.3 Hz, 2H); 13C NMR (101 MHz, CDCl3) δ 173.55, 140.74, 140.36, 136.37, 130.37, 129.50, 129.46, 128.35, 128.04, 126.69, 119.13, 109.29, 59.89, 58.00, 55.45, 50.77, 50.10, 49.57, 47.45, 45.64, 29.41, 27.93, 24.82. The free base was converted into the corresponding oxalate salt. HRMS (C32H38ON4 + H+) found 495.31212 (error 0.5 ppm). CHN (C34H45O2N4Cl + 2 H2C2O4 + 1.5 H2O) calculated C 61.61, H 6.46, N 7.98, found C 61.59, H 6.36, N 7.98. mp: Salt decomposes above 134 ° C.
(S)-3-Chloro-N-((1-(3-cyano-3,3-diphenylpropyl)pyrrolidin-2-yl)methyl)-5-ethyl-6-hydroxy-2-methoxybenzamide (35).
The reaction was performed following the same procedure described for 12, starting from (S)-nor-eticlopride (250 mg, 0.8 mmol) and 4-bromo-2,2-diphenylbutanenitrile (240 mg, 0.8 mmol). The desired product was purified by flash chromatography eluting with hex/EtOAc (60/40) and obtained as a colorless oil (98 mg, 23% yield). 1H NMR (400 MHz, CDCl3) δ 13.78 (s, 1H), 8.74 (s, 1H), 7.44 – 7.19 (m, 11H), 3.86 (s, 3H), 3.62 (ddd, J = 14.0, 6.9, 2.6 Hz, 1H), 3.29 – 3.12 (m, 2H), 2.89 – 2.78 (m, 1H), 2.72 – 2.55 (m, 5H), 2.49 – 2.37 (m, 1H), 2.28 (q, J = 8.8 Hz, 1H), 1.90 (dq, J = 12.3, 8.1 Hz, 1H), 1.75 (t, J = 7.8 Hz, 2H), 1.67 – 1.51 (m, 1H), 1.32 – 1.15 (m, 3H); 13C NMR (101 MHz, CDCl3) δ 169.44, 160.15, 152.40, 140.02, 139.45, 132.90, 130.91, 128.95, 128.01, 127.97, 126.64, 122.00, 116.06, 108.18, 62.15, 61.44, 54.04, 50.33, 50.08, 40.43, 38.52, 29.68, 28.12, 22.71, 22.55, 13.43. CHN (C31H34N3O3Cl + 0.4 hexanes) calculated C 70.81, H 7.05, N 7.42; found C 70.49, H 7.15, N 7.08. HPLC analysis method A: Chiralpak AD-H analytical column (4.5mm × 250mm – 5 μm particle size); mobile phase: gradient from 10% to 40% 2-PrOH in hexanes; flow rate: 1 mL/min; injection volume: 20 μL; sample concentration: ~1 mg/mL; multiple DAD λ absorbance signals measured in the range of 210–280 nm, Rt 7.799 min, purity >99%, ee >99% (absorbance at 254 nm). HPLC analysis method B: Chiralpak AD-H analytical column (4.5mm × 250mm – 5 μm particle size); mobile phase: isocratic 10% 2-PrOH in hexanes; flow rate: 1 mL/min; injection volume: 20 μL; sample concentration: ~1 mg/mL; multiple DAD λ absorbance signals measured in the range of 210–280 nm, Rt 9.043 min, purity >99%, ee >99% (absorbance at 254 nm). HRMS (C31H34N3O3Cl + H+) calculated 532.23615, found 532.23691 (error 0.4 ppm).
(S)-N-((1-(4-Aminobutyl)pyrrolidin-2-yl)methyl)-3-chloro-5-ethyl-6-hydroxy-2-methoxybenzamide (36).
The reaction was performed following the same procedure described for 33, starting from (S)-3-chloro-5-ethyl-6-hydroxy-2-methoxy-N-(pyrrolidin-2-ylmethyl)benzamide ((S)-nor-eticlopride 250 mg, 0.8 mmol). The crude material was used in the next step without further purification (110 mg, 36% yield).
(S)-3-Chloro-N-((1-(4-((3-cyano-3,3-diphenylpropyl)amino)butyl)pyrrolidin-2-yl)methyl)-5-ethyl-6-hydroxy-2-methoxybenzamide (37).
The reaction was performed following the same procedure described for 12, starting from 36 (110 mg, 0.29 mmol) and 4-bromo-2,2-diphenylbutanenitrile (78 mg, 0.26 mmol). The desired product was purified by flash chromatography eluting with 10% DMA and obtained as a colorless oil (15 mg, 10% yield). 1H NMR (400 MHz, CDCl3 + CD3OD) δ 7.39 – 7.23 (m, 6H), 7.18 – 7.06 (m, 5H), 3.83 (s, 3H), 3.82 – 3.50 (m, 6H), 3.36 – 3.26 (m, 6H), 2.82 (p, J = 6.9 Hz, 2H), 2.53 (q, J = 7.5 Hz, 2H), 1.74 (m, 5H), 1.11 (ddd, J = 8.2, 7.1, 0.8 Hz, 3H); 13C NMR (101 MHz, CDCl3) δ 174.69, 163.12, 156.56, 143.10, 142.99, 137.03, 134.45, 132.70, 132.67, 132.08, 132.06, 131.89, 131.85, 120.19, 112.47, 67.26, 66.39, 64.97, 57.87, 54.60, 49.82, 43.72, 42.16, 31.41, 27.77, 27.47, 26.07, 25.77, 16.57. HPLC analysis method A: Chiralpak AD-H analytical column (4.5mm × 250mm – 5 μm particle size); mobile phase: gradient from 10% to 40% 2-PrOH in hexanes; flow rate: 1 mL/min; injection volume: 20 μL; sample concentration: ~1 mg/mL; multiple DAD λ absorbance signals measured in the range of 210–280 nm, Rt 22.362 min, purity >95%, ee >99% (absorbance at 254 nm). HPLC analysis method B: Chiralpak AD-H analytical column (4.5mm × 250mm – 5 μm particle size); mobile phase: isocratic 30% 2-PrOH in hexanes; flow rate: 1 mL/min; injection volume: 20 μL; sample concentration: ~1 mg/mL; multiple DAD λ absorbance signals measured in the range of 210–280 nm, Rt 14.389 min, purity >99%, ee >99% (absorbance at 254 nm). HRMS (C35H43N4O3Cl + 2H)2+ found 302.15901; (C35H43N4O3Cl + H+) calculated 603.30965, found 603.31020 (error 0.4 ppm).
tert-Butyl ((1-(4-(1,3-dioxoisoindolin-2-yl)butyl)pyrrolidin-2-yl)methyl)carbamate (38).
A suspension of tert-butyl (pyrrolidin-2-ylmethyl)carbamate (500 mg, 2.5 mmol), N-(4-bromobutyl)phthalimide (775 mg, 2.75 mmol), cat. KI (4.15 mg, 25 μmol) and K2CO3 (3.45 g, 25 mmol) in ACN (20 mL) was stirred under reflux overnight. The reaction mixture was cooled down to RT, filtered, and the solvent removed under vacuum, the desired product, presenting both N-Boc and N-phthalimide protecting groups, was purified by flash chromatography eluting with 10% DMA and obtained as a yellow oil (940 mg, 85% yield). 1H NMR (400 MHz, CDCl3) δ 7.84 (dd, J = 5.5, 3.0 Hz, 2H), 7.71 (dd, J = 5.5, 3.1 Hz, 2H), 5.00 – 4.95 (m, 1H), 3.77 – 3.64 (m, 2H), 3.27 (d, J = 10.9 Hz, 1H), 3.15 – 2.99 (m, 2H), 2.73 (dt, J = 11.9, 8.1 Hz, 1H), 2.47 (br s, 1H), 2.23 – 2.02 (m, 2H), 1.89 – 1.43 (m, 8H), 1.43 (s, 9H).
3-Chloro-N-((1-(4-(1,3-dioxoisoindolin-2-yl)butyl)pyrrolidin-2-yl)methyl)-5-ethyl-6-hydroxy-2-methoxybenzamide (39).
A solution of 38 (940 mg, 2.34 mmol) and TFA (2.5 mL) in DCM (15 mL) was stirred at RT for 3 h. The reaction was quenched with NaHCO3 (sat. aq Solution) and extracted with DCM/2-PrOH (3:1). The organic layers were combined, dried over Na2SO4, filtered and evaporated under vacuum. The crude material was dissolved in DCM (15 mL), followed by dropwise addition of a solution of 3-chloro-5-ethyl-6-hydroxy-2-methoxybenzoic acid54 (647 mg, 2.81 mmol) and HCTU (1.16 g, 2.81 mmol) in DCM (15 mL). The reaction mixture was stirred at RT for 48 h, the solvent was removed under vacuum and the crude material purified by flash chromatography eluting with 5% DMA. The desired product was obtained as a brown oil (310 mg, 26% yield). 1H NMR (400 MHz, CDCl3) δ 13.12 (s, 1H), 9.11 (s, 1H), 7.80 (dd, J = 5.4, 3.0 Hz, 2H), 7.69 (dd, J = 5.5, 3.0 Hz, 2H), 7.26 – 7.15 (m, 1H), 3.96 – 3.78 (m, 5H), 3.72 (m, J = 14.0 Hz, 4H), 3.57 (br s, 1H), 3.48 (br s, 1H), 3.26 (br s, 2H), 2.66 – 2.51 (m, 3H), 2.22 (dt, J = 15.0, 7.6 Hz, 1H), 1.99 (tt, J = 13.4, 6.9 Hz, 2H), 1.80 – 1.60 (m, 3H), 1.18 (dt, J = 10.7, 7.5 Hz, 3H).
3-Chloro-N-((1-(4-((4-(dimethylamino)-4-oxo-3,3-diphenylbutyl)amino)butyl)pyrrolidin-2-yl)methyl)-5-ethyl-6-hydroxy-2-methoxybenzamide (40).
Hydrazine (0.2 mL, 50–60% wt. in H2O) was added to a solution of 39 (310 mg, 0.6 mmol) in EtOH (20 mL) and the solution was stirred at reflux for 3 h. The solvent was removed under vacuum, the residue diluted with 20% K2CO3 aq solution and extracted with DCM. The organic layers were combined, dried over Na2SO4, filtered and evaporated under vacuum. The obtained crude material was dissolved in DCE (10 mL) and added to a solution of 26 (169 mg, 0.6 mmol) and catalytic AcOH in DCE (10 mL). The mixture was stirred for 10 min at RT and STAB (190 mg, 0.9 mmol) was added portionwise. The reaction was stirred for additional 12 h, the solvent was removed under vacuum and the residue purified by flash chromatography eluting with 10% DMA. The desired product was obtained as a colorless oil (80.5 mg, 21% yield). 1H NMR (400 MHz, CDCl3) δ 8.79 (br s, 1H), 7.40 – 7.22 (m, 11H), 3.82 (s, 3H), 3.72 (dt, J = 12.1, 4.0 Hz, 2H), 3.28 – 3.19 (m, 1H), 3.14 (dt, J = 9.5, 4.5 Hz, 1H), 2.95 (br s, 3H), 2.74 – 2.52 (m, 3H), 2.47 – 2.35 (m, 4H), 2.25 (d, J = 14.1 Hz, 4H), 2.12 (q, J = 8.6 Hz, 3H), 1.93 – 1.80 (m, 1H), 1.72 (d, J = 7.8 Hz, 2H), 1.69 – 1.52 (m, 1H), 1.40 (dt, J = 16.4, 8.3 Hz, 4H), 1.20 (dt, J = 27.3, 7.3 Hz, 3H); 13C NMR (101 MHz, CDCl3) δ 173.62, 169.49, 160.11, 152.51, 140.71, 140.65, 132.82, 130.67, 128.37, 128.17, 128.16, 128.03, 126.72, 116.01, 108.17, 62.27, 61.41, 60.04, 54.01, 53.80, 49.38, 47.15, 45.23, 40.32, 28.10, 27.73, 26.57, 22.58, 22.51, 13.41. HPLC analysis method A: Chiralpak AD-H analytical column (4.5mm × 250mm – 5 μm particle size); mobile phase: isocratic 30% 2-PrOH in hexanes; flow rate: 1 mL/min; injection volume: 20 μL; sample concentration: ~1 mg/mL; multiple DAD λ absorbance signals measured in the range of 210–280 nm, Rt 9.538 and 10.664 min, purity >99%, er 46:54 (absorbance at 254 nm). HPLC analysis method B: Chiralpak AD-H analytical column (4.5mm × 250mm – 5 μm particle size); mobile phase: isocratic 15% 2-PrOH in hexanes; flow rate: 1 mL/min; injection volume: 20 μL; sample concentration: ~1 mg/mL; multiple DAD λ absorbance signals measured in the range of 210–280 nm, Rt 22.250 and 25.814 min, purity >99%, er 50:50 (absorbance at 254 nm). HRMS (C37H49N4O4Cl + 2H)2+ found 325.18021, (C37H49N4O4Cl + H+) calculated 649.35151, found 649.35221 (error 1.0 ppm).
3,3-Diphenylpyrrolidine (41).
A suspension of LAH (0.56 g, 14.8 mmol) in THF (50 mL) was cooled to 0 °C and a solution of 4-bromo-2,2-diphenylbutanenitrile (1.5 g, 5 mmol) in THF (20 mL) was added dropwise. The mixture was stirred at RT for 15 h, quenched with MeOH (5 mL) and sat. aq NaHCO3 solution (5 mL), filtered over celite and concentrated under vacuum. The residue was suspended in DCM and washed with sat. Na2CO3 solution. The organic layer was dried over Na2SO4, filtered and evaporated under vacuum. The crude material was purified by flash chromatography eluting with 10% DMA. The desired product was obtained as a yellow oil (0.450 g, 40% yield). 1H NMR (400 MHz, CDCl3) δ 7.34 – 7.22 (m, 8H), 7.22 – 7.13 (m, 2H), 3.52 (s, 2H), 3.13 (t, J = 7.2 Hz, 2H), 2.80 – 2.70 (br s, 1H), 2.52 (t, J = 7.2 Hz, 2H). GC/MS (EI), Rt 9.987 min; 223.2 (M+).
1-Methyl-3,3-diphenylpyrrolidine (42).
Methyl chloroformate (85 mg, 69 μL, 0. 9 mmol) was added dropwise to a solution of 41 (100 mg, 0.45 mmol) in THF (10 mL), followed by excess of DIPEA (5 eq.). The mixture was stirred at RT for 1 h, the solvent evaporated under vacuum and the residue dissolved in THF (10 mL). This solution was added dropwise to a suspension of LAH (17 mg, 0.45 mmol) in THF (10 mL), at 0 °C. The mixture was warmed to RT, quenched with MeOH/2N aq NaOH (1:1 ratio, 2 mL), filtered over celite, and the solvents were evaporated under vacuum. The crude material was purified by flash chromatography eluting with 10% DMA to afford the desired product as a colorless oil (60 mg, 56% yield). 1H NMR (400 MHz, CDCl3) δ 7.26 (d, J = 4.6 Hz, 8H), 7.20 – 7.11 (m, 2H), 3.22 (s, 2H), 2.82 (td, J = 7.0, 0.6 Hz, 2H), 2.60 (dd, J = 7.7, 6.7 Hz, 2H), 2.42 (s, 3H). GC/MS (EI), Rt 9.650 min; 237.2 (M+), purity >99%.
1-(3,3-Diphenylpyrrolidin-1-yl)propan-1-one (43).
A solution of 41 (70 mg, 0.31 mmol), propionyl chloride (58 mg, 0.63 mmol) and DIPEA (5 eq) in DCM (10 mL) was stirred under reflux overnight. The solvent was removed under vacuum and the residue purified by flash chromatography eluting with hex/EtOAc (70/30). The desired product was obtained as a colorless oil (25 mg, 29% yield). 1H NMR (400 MHz, CDCl3) rotamer conformations observed (60:40; rotamer A:rotamer B) δ 7.34 – 7.14 (m, 10H), 4.16 (s, 2H, rotamer A), 4.04 (s, 2H, rotamer B), 3.53 (t, J = 6.9 Hz, 2H, rotamer B), 3.42 (t, J = 6.7 Hz, 2H, rotamer A), 2.63 (t, J = 6.7 Hz, 2H, rotamer A), 2.52 (t, J = 6.9 Hz, 2H, rotamer B), 2.41 (q, J = 7.5 Hz, 2H, rotamer B), 2.26 (q, J = 7.5 Hz, 2H, rotamer A), 1.21 (t, J = 7.5 Hz, 3H, rotamer B), 1.16 (t, J = 7.5 Hz, 3H, rotamer A). GC/MS (EI), Rt 12.154 min; 279.1 (M+), purity >99%.
2-Chloro-1-(3,3-diphenylpyrrolidin-1-yl)ethan-1-one (44).
2-Chloroacetyl chloride (126 mg, 1.12 mmol) was added dropwise to a solution of 41 (250 mg, 1.12 mmol) in THF (10 mL) at 0 °C, followed by dropwise addition of DIPEA (0.3 mL, 1.68 mmol). The mixture was allowed to warm to RT and stirred for 1 h. The solvent was removed under vacuum and the residue used in the following step without further purification. GC/MS (EI), Rt 12.740 min; 299.1 (M+).
tert-Butyl ((1-(2-(3,3-diphenylpyrrolidin-1-yl)-2-oxoethyl)pyrrolidin-2-yl)methyl)carbamate (45).
A mixture of 44 (290 mg, 0.97 mmol), tert-butyl (pyrrolidin-2-ylmethyl)carbamate (194 mg, 0.97 mmol), KI (161 mg, 0.97 mmol) and K2CO3 (1.34 g, 9.67 mmol) in ACN (25 ml) was stirred under reflux for 3 h. The mixture was filtered, the solvent evaporated under vacuum and the residue purified by flash chromatography eluting with 10% DMA. The desired product was obtained as a yellow oil (330 mg, 74% yield). 1H NMR (400 MHz, CDCl3) rotamer conformations observed δ 7.34 – 7.15 (m, 10H), 5.30 (m, 1H), 3.69 – 3.38 (m, 4H), 3.29 – 2.93 (m, 3H), 2.71 – 2.17 (m, 4H), 1.90 (tt, J = 19.2, 8.6 Hz, 2H), 1.73 (m, J = 17.3, 8.5 Hz, 4H), 1.50 – 1.38 (m, 9H).
3-Chloro-N-((1-(2-(3,3-diphenylpyrrolidin-1-yl)-2-oxoethyl)pyrrolidin-2-yl)methyl)-5-ethyl-6-hydroxy-2-methoxybenzamide (46).
TFA (0.3 mL) was added to a solution of 45 (330 mg, 0.71 mmol) in DCM (10 mL). The mixture was stirred at RT for 2 h, basified with NH4OH (28% aq solution) and extracted with DCM. The organic layers were combined, dried over Na2SO4, filtered and evaporated under vacuum to afford the crude material, which was filtered over a silica pad, eluting and washing with 25% DMA to isolate the desired primary amine intermediate. The amine was dissolved in DCM (10 mL) and added dropwise to a solution of 3-chloro-5-ethyl-6-hydroxy-2-methoxybenzoic acid (70 mg, 0.3 mmol), HCTU (0.2 g, 0.33 mmol) and DIPEA (1.5 eq) in DCM (10 mL). The mixture was stirred at RT for 3 h, the solvent was removed under vacuum and the residue purified by flash chromatography eluting with 5% DMA. The desired product was obtained as a colorless oil (26 mg, 15% yield). 1H NMR (400 MHz, CDCl3) rotamer conformations observed δ 13.68 (s, 1H), 8.82 (s, 1H), 7.31 – 7.12 (m, 11H), 4.23 (t, J = 11.0 Hz, 1H), 4.02 (dd, J = 26.1, 11.5 Hz, 1H), 3.85 (ds, J = 17.4 Hz, 3H), 3.79 – 3.68 (m, 1H), 3.62 – 3.21 (m, 6H), 2.96 (br s, 1H), 2.68 – 2.52 (m, 4H), 2.14 – 1.97 (m, 1H), 1.92 – 1.66 (m, 4H), 1.28 – 1.14 (m, 3H); 13C NMR (101 MHz, CDCl3) rotamer conformations observed δ 169.75, 160.07, 160.04, 152.47, 152.46, 144.94, 144.88, 144.79, 133.07, 130.79, 130.77, 128.57, 128.53, 128.51, 126.67, 126.62, 126.59, 126.58, 126.52, 116.16, 116.11, 107.98, 61.57, 61.49, 56.60, 56.14, 56.12, 55.10, 54.87, 54.68, 54.45, 53.40, 52.22, 44.57, 44.39, 40.88, 40.46, 37.52, 35.57, 29.68, 28.38, 28.26, 23.05, 22.86, 22.51, 13.39. HPLC analysis method A: Chiralpak AD-H analytical column (4.5mm × 250mm – 5 μm particle size); mobile phase: isocratic 10% 2-PrOH in hexanes; flow rate: 1 mL/min; injection volume: 20 μL; sample concentration: ~1 mg/mL; multiple DAD λ absorbance signals measured in the range of 210–280 nm, Rt 20.539 and 23.859 min, purity >99%, er 51:49 (absorbance at 254 nm). HRMS (C33H38N3O4Cl + H+) calculated 576.26236, found 576.26297 (error 1.0 ppm).
3-Chloro-N-(((2S,4R)-4-(4-((4-(dimethylamino)-4-oxo-3,3-diphenylbutyl)amino)butoxy)pyrrolidin-2-yl)methyl)-5-ethyl-6-hydroxy-2-methoxybenzamide (48).
A solution of 47 (400 mg, 0.8 mmol), 26 (225 mg, 0.8 mmol) and cat. AcOH (0.05 eq) in DCE (20 mL) was stirred at RT for 1 h, followed by portion-wise addition of STAB (339 mg, 1.6 mmol). The mixture was stirred at RT for 3 h, basified with 10% NH4OH in MeOH, the solvent evaporated under vacuum and the residue purified by flash chromatography eluting with 10% DMA. The obtained intermediate was dissolved in DCM (20 mL) and TFA (10 mL), and the solution was stirred at RT overnight. The excess of TFA was removed under vacuum, the residue resuspended in aq NH4OH (pH 9) and extracted with DCM/2-PrOH (3:1). The organic layers were combined, dried over Na2SO4, filtered and evaporated to afford the crude material, which was purified by flash chromatography eluting with 25% DMA. The desired product was obtained as a colorless oil (45 mg, 8.5% yield). 1H NMR (400 MHz, CDCl3) δ 8.92 (s, 1H), 7.43 – 7.21 (m, 11H), 3.97 (t, J = 4.9 Hz, 1H), 3.89 (s, 3H), 3.57 (dq, J = 9.6, 4.7 Hz, 3H), 3.36 (dt, J = 5.8, 2.8 Hz, 3H), 3.22 (tt, J = 8.9, 4.5 Hz, 2H), 3.02 – 2.90 (m, 5H), 2.69 – 2.55 (m, 4H), 2.45 (dd, J = 21.5, 5.7 Hz, 4H), 2.30 (br s, 2H), 2.00 (dd, J = 13.7, 6.9 Hz, 1H), 1.70 – 1.50 (m, 5H), 1.31 – 1.14 (m, 3H); 13C NMR (101 MHz, CDCl3) δ 174.57, 169.21, 160.06, 152.48, 139.97, 132.89, 130.72, 128.78, 127.96, 127.93, 127.26, 116.16, 108.17, 80.67, 68.17, 61.57, 56.10, 51.78, 43.34, 36.07, 27.16, 22.50, 21.93, 13.41. HPLC analysis method: Chiralpak AD-H analytical column (4.5mm × 250mm – 5 μm particle size); mobile phase: isocratic 20% 2-PrOH in hexanes; flow rate: 1 mL/min; injection volume: 20 μL; sample concentration: ~1 mg/mL; multiple DAD λ absorbance signals measured in the range of 210–280 nm, Rt 18.518 min, purity >95%, ee >95% (absorbance at 210 nm). The free base was converted into the corresponding oxalate salt. HRMS (C37H49N4O5Cl + 2H)2+ calculated 333.17685, found 333.17656 (error 0.9 ppm), (C37H49N4O5Cl + H)+ calculated 665.34642, found 665.34552 (error 1.4 ppm). CHN (C37H49N4O5Cl + 2 H2C2O4 + 1.5 H2O) calculated C 56.45, H 6.47, N 6.42; found C 56.25, H 6.24, N 6.40. mp: Salt decomposes above 90 ° C.
3,3-Bis(3-methoxyphenyl)acrylaldehyde (49).
A solution of bis(3-methoxyphenyl)methanone (1 g, 4.13 mmol) and TiCl4 (1.0 M solution in DCM, 16.5 mL, 16.5 mmol) in DCM was cooled to 0 °C. TEA (2.3 mL, 16.5 mmol) was added dropwise, the mixture was warmed to RT and stirred overnight55. The reaction was quenched with sat. NH4Cl aq solution and stirred 30 min. The mixture was extracted with DCM, the organic layers were combined, dried over Na2SO4, filtered and concentrated under vacuum. The crude material was purified by flash chromatography eluting with hex/EtOAc (95/5). The desired product was obtained as a colorless oil (645 mg, 58% yield). 1H NMR (400 MHz, CDCl3) δ 9.60 – 9.53 (m, 1H), 7.42 – 7.25 (m, 2H), 7.07 – 6.77 (m, 6H), 6.64 – 6.56 (m, 1H), 3.89 – 3.74 (m, 6H). GC/MS (EI), Rt 11.647 min; 268.1 (M+).
N-(3,3-Bis(3-methoxyphenyl)allyl)-4-(4-(2,3-dichlorophenyl)piperazin-1-yl)butan-1-amine (50).
A solution of 49 (302 mg, 1.12 mmol), 4-(4-(2,3-dichlorophenyl)piperazin-1-yl)butan-1-amine (340 mg, 1.12 mmol) and cat. AcOH (0.1 eq) in DCE (20 mL) was stirred for 1 h at RT, followed by portion-wise addition of STAB (715 mg, 3.37 mmol). The reaction was stirred for 3 h, the solvent evaporated under vacuum and the residue purified by flash chromatography eluting with 5% DMA. The desired product was obtained as a colorless oil (550 mg, 88% yield). 1H NMR (400 MHz, CDCl3) δ 7.26 (m, 1H), 7.16 (q, J = 7.6 Hz, 2H), 6.90 – 6.79 (m, 3H), 6.79 – 6.67 (m, 4H), 6.67 – 6.60 (m, 1H), 6.20 – 6.11 (m, 1H), 3.74 (d, J = 5.4 Hz, 6H), 3.38 – 3.33 (m, 4H), 3.08 (br s, 4H), 2.74 (m, 2H), 2.58 (br s, 2H), 2.42 (m, 2H), 1.47 (m, 2H), 1.28 (m, 2H); 13C NMR (101 MHz, CDCl3) δ 175.73, 159.49, 159.43, 150.75, 142.64, 140.20, 134.01, 129.39, 129.11, 127.48, 127.47, 124.83, 122.01, 119.88, 118.71, 115.22, 113.32, 112.98, 112.85, 57.48, 55.20, 55.16, 53.39, 52.57, 52.48, 51.42, 50.27, 30.90, 23.33, 23.10, 21.27. HPLC analysis method: Chiralpak AD-H analytical column (4.5mm × 250mm – 5 μm particle size); mobile phase: isocratic 20% 2-PrOH in hexanes; flow rate: 1 mL/min; injection volume: 20 μL; sample concentration: ~1 mg/mL; multiple DAD λ absorbance signals measured in the range of 210–280 nm, Rt 6.538 min, purity >95% (absorbance at 254 nm). HRMS (C31H37N3O2Cl2 + H)+ calculated 554.23356, found 554.23390.
3,3’-(3-((4-(4-Phenylpiperazin-1-yl)butyl)amino)propane-1,1-diyl)diphenol (51).
A suspension of 50 (250 mg, 0.45 mmol) and Pd/C (20% wt. wet, 0.05 eq) in EtOH (10 mL), was shaken in a Parr apparatus under 50 psi pressure of hydrogen gas for 12 h. The mixture was filtered through a pad of celite, the solvent evaporated under vacuum and the residue purified trough a pad of silica eluting with 5% DMA. This intermediate was dissolved in 33% HBr (solution in AcOH, 2 mL) and stirred under reflux for 48 h. The solvent was removed under vacuum and the residue basified with 10% v/v NH4OH solution in methanol. The crude material was purified by flash chromatography eluting with 25% DMA. The desired product was obtained as a yellow oil (6.8 mg, 29% yield). 1H NMR (400 MHz, CDCl3) δ 7.28 – 7.19 (m, 1H), 7.09 (t, J = 7.8 Hz, 2H), 6.90 – 6.80 (m, 2H), 6.71 – 6.60 (m, 4H), 6.55 (s, 2H), 5.44 (br s, 2H), 3.81 (t, J = 7.9 Hz, 1H), 3.09 (t, J = 5.0 Hz, 4H), 2.61 (s, 2H), 2.49 (dt, J = 11.8, 5.4 Hz, 6H), 2.25 (p, J = 7.5 Hz, 4H), 1.30 – 1.16 (m, 4H); 13C NMR (101 MHz, CDCl3) δ 172.49, 157.37, 150.94, 145.43, 129.75, 129.07, 119.90, 119.43, 116.11, 114.54, 114.47, 65.83, 58.11, 52.97, 34.26, 26.57, 25.34, 24.12, 22.59, 15.25. HPLC analysis method: Chiralpak AD-H analytical column (4.5mm × 250mm – 5 μm particle size); mobile phase: isocratic 20% 2-PrOH in hexanes +0.1% DEA; flow rate: 1 mL/min; injection volume: 20 μL; sample concentration: ~1 mg/mL; multiple DAD λ absorbance signals measured in the range of 210–280 nm, Rt 38.741 min, purity >95% (absorbance at 254 nm). HRMS (C29H37N3O2 + H)+ found 460.29628.
1-(3,3-Bis(3-methoxyphenyl)allyl)-4-(2-chloro-3-ethylphenyl)piperazine (52).
The reaction was performed following the same procure described for 50, starting from 1-(2-chloro-3-ethylphenyl)piperazine14 (180 mg, 0.78 mmol). The desired product was obtained as a colorless oil (330 mg, 88% yield). 1H NMR (400 MHz, CDCl3) δ 7.24 – 7.11 (m, 3H), 7.00 – 6.66 (m, 8H), 6.31 (t, J = 7.0 Hz, 1H), 3.79 (d, J = 14.0 Hz, 6H), 3.34 (d, J = 6.9 Hz, 2H), 3.14 (br s, 4H), 3.00–2.76 (br s, 4H), 2.76 (q, J = 7.5 Hz, 2H), 1.21 (t, J = 7.5 Hz, 3H).
1-(3,3-Bis(3-methoxyphenyl)propyl)-4-(2-chloro-3-ethylphenyl)piperazine (53).
A suspension of 52 (330 mg, 0.69 mmol) and Pd/C (20% wt. wet, 0.05 eq) in EtOAc:EtOH (10:5 mL), was shaken in a Parr apparatus under 30 psi pressure of H2 for 3 h. The mixture was filtered through a pad of celite, the solvent evaporated under vacuum and the desired product obtain as a colorless oil (250 mg, 75% yield). 1H NMR (400 MHz, CDCl3) δ 7.18 (dt, J = 20.9, 7.8 Hz, 3H), 6.94 (ddd, J = 11.1, 7.8, 1.5 Hz, 2H), 6.90 – 6.67 (m, 6H), 3.96 – 3.86 (m, 1H), 3.82 – 3.67 (br s, 6H), 3.10 (br s, 4H), 2.76 (br s + q, J = 7.5 Hz, 6H), 2.50 (dd, J = 9.9, 5.8 Hz, 2H), 2.06 (m, 2H), 1.29 – 1.15 (t, J = 7.5 Hz, 3H). HRMS (C29H35ClN2O2 + H)+ calculated 479.24598, found 479.24505 (error 1.9 ppm). The free base (50 mg) was converted into the corresponding oxalate salt. mp: Salt decomposes above 100 ° C.
3-(3-(4-(2-chloro-3-ethylphenyl)piperazin-1-yl)-1-(3-methoxyphenyl)propyl)phenol (54) and 3,3’-(3-(4-(2-chloro-3-ethylphenyl)piperazin-1-yl)propane-1,1-diyl)diphenol (55)
A solution of 53 (200 mg, 0.42 mmol) in 33% HBr (solution in AcOH, 5 mL) and DCM (10 mL) was stirred under reflux for 24 h. The solvent was removed under vacuum, the residue basified with 28–30% v/v aq NH4OH and extracted with DCM:2-PrOH (3:1). The crude material was purified by flash chromatography eluting with 10% DMA. 54 eluted first as a yellow oil (60 mg, 31% yield): 1H NMR (400 MHz, CDCl3) δ 7.20 – 7.07 (m, 3H), 6.94 (dd, J = 7.6, 1.5 Hz, 1H), 6.91 – 6.66 (m, 4H), 6.64 (d, J = 1.0 Hz, 1H), 6.64 – 6.47 (m, 2H), 3.83 (t, J = 7.6 Hz, 1H), 3.73 (s, 3H), 3.01 (br s, 4H), 2.76 (br s + q, J = 7.5 Hz, 6H), 2.49 – 2.36 (m, 2H), 2.35 – 2.15 (m, 2H), 1.36 – 1.17 (t, J = 7.5 Hz, 3H). HRMS (C28H33ClN2O2 + H)+ calculated 465.23033, found 465.22945 (error 1.9 ppm). 55 eluted second as a yellow oil (25 mg, 13% yield): 1H NMR (400 MHz, CDCl3) δ 7.09 (td, J = 7.8, 2.4 Hz, 3H), 6.93 (dd, J = 7.6, 1.5 Hz, 1H), 6.85 – 6.69 (m, 3H), 6.63 (ddd, J = 8.1, 2.5, 0.9 Hz, 2H), 6.55 (t, J = 1.9 Hz, 2H), 3.75 (q, J = 7.9 Hz, 1H), 2.97 (br s, 4H), 2.75 – 2.60 (br s + q, J = 7.6 Hz, 6H), 2.43 (br s, 2H), 2.26 – 2.15 (m, 2H), 1.30 – 1.16 (t, J = 7.5 Hz, 3H). HRMS (C27H31ClN2O2 + H)+ calculated 451.21468, found 451.21429 (error 0.9 ppm).
trans-Ethyl (Z)-2-((1S,5S)-2-(4-(1H-indole-2-carboxamido)butyl)-5-(3-hydroxyphenyl)-2-azabicyclo[3.3.1]nonan-9-ylidene)acetate (57).
A solution of 56 (20 mg, 66 μmol), N-(4-oxobutyl)-1H-indole-2-carboxamide57 (18 mg, 80 μmol) and catalytic AcOH (0.01 eq) in DCE (10 mL) was stirred at RT for 1 h, followed by portion-wise addition of STAB (30 mg, 66 μmol). The reaction was stirred for additional 30 min and basified with 10% NH4OH solution in MeOH (10 mL), the solvent was evaporated under vacuum and the residue purified by flash chromatography eluting with 20% DMA. The desired product was obtained as a colorless oil (30 mg, 88% yield). 1H NMR (400 MHz, CDCl3) δ 9.30 (s, 1H), 7.61 (dd, J = 8.0, 1.0 Hz, 1H), 7.46 – 7.38 (m, 1H), 7.32 – 7.08 (m, 3H), 6.97 – 6.78 (m, 4H), 6.78 – 6.70 (m, 1H), 5.30 (s, 1H), 5.16 (d, J = 17.9 Hz, 2H), 4.13 – 3.92 (m, 2H), 3.63 – 3.44 (m, 2H), 3.20 (ddd, J = 13.2, 8.9, 4.8 Hz, 1H), 2.73 (ddt, J = 17.8, 12.3, 5.8 Hz, 3H), 2.46 (ddd, J = 14.4, 8.7, 6.0 Hz, 1H), 2.27 – 2.00 (m, 5H), 1.90 – 1.50 (m, 6H), 1.14 (t, J = 7.1 Hz, 3H); 13C NMR (101 MHz, CDCl3) δ 167.91, 166.96, 161.79, 155.61, 148.65, 136.14, 131.04, 129.25, 127.68, 124.33, 121.83, 120.55, 119.46, 115.00, 114.38, 113.60, 111.89, 102.22, 59.92, 55.45, 53.31, 48.76, 45.69, 40.19, 39.34, 38.73, 30.66, 27.22, 24.50, 20.48, 14.13. HPLC analysis method A: Chiralpak AD-H analytical column (4.5mm × 250mm – 5 μm particle size); mobile phase: isocratic 20% 2-PrOH in hexanes; flow rate: 1 mL/min; injection volume: 20 μL; sample concentration: ~1 mg/mL; multiple DAD λ absorbance signals measured in the range of 210–280 nm, Rt 30.677 min, purity >99%, ee >99% (absorbance at 254 nm). HPLC analysis method B: Chiralpak AD-H analytical column (4.5mm × 250mm – 5 μm particle size); mobile phase: isocratic 30% 2-PrOH in hexanes; flow rate: 1 mL/min; injection volume: 20 μL; sample concentration: ~1 mg/mL; multiple DAD λ absorbance signals measured in the range of 210–280 nm, Rt 13.495 min, purity >99%, ee >99% (absorbance at 254 nm). HRMS (C31H37O4N3 + H+) calculated 516.28568, found 516.28475 (error −1.8 ppm).
Trans-Ethyl (Z)-2-((2S,6R)-6-(((2-(2-(1H-indole-2-carboxamido)ethyl)cyclopropyl)methyl)(ethyl)amino)-2-(3-hydroxyphenyl)-2-methylcyclohexylidene)acetate (58).
The reaction was performed following the same procure described for 57, starting from 56 (20 mg, 66 μmol) and N-(2-(2-formylcyclopropyl)ethyl)-1H-indole-2-carboxamide45 (17 mg, 66 μmol). The desired product was obtained as a colorless oil (23 mg, 65% yield). 1H NMR (400 MHz, CDCl3) mixture of diastereomers observed δ 9.33 (s, 1H, d.r. 60:40), 7.62 (dd, J = 8.1, 3.8 Hz, 1H), 7.47 – 7.39 (m, 1H), 7.34 – 7.25 (m, 1H), 7.23 – 7.08 (m, 2H), 6.93 (d, J = 10.1 Hz, 1H), 6.89 – 6.69 (m, 3H), 6.60 (s, 1H), 5.25 – 5.10 (m, 2H), 4.13 – 3.95 (m, 2H), 3.72 – 3.49 (m, 2H), 3.16 (d, J = 12.3 Hz, 1H), 2.96 – 2.74 (m, 2H), 2.46 (dd, J = 14.2, 6.9 Hz, 1H), 2.37 – 2.27 (m, 1H), 2.25 – 1.98 (m, 7H), 1.64 – 1.49 (m, 4H), 1.38 – 1.24 (m, 1H), 1.15 (dt, J = 8.2, 7.1 Hz, 3H), 0.89 (d, J = 62.8 Hz, 1H, d.r. 60:40), 0.69 (d, J = 52.6 Hz, 1H, d.r. 60:40), 0.32 (dt, J = 17.8, 5.9 Hz, 1H); 13C NMR (101 MHz, CDCl3) δ 166.85, 166.83, 161.95, 161.69, 156.18, 155.88, 136.29, 136.20, 130.95, 130.50, 129.23, 129.13, 127.69, 127.56, 124.62, 124.40, 121.90, 121.85, 120.74, 120.58, 115.31, 115.13, 113.95, 113.82, 112.01, 111.90, 102.62, 60.90, 60.56, 59.87, 59.81, 55.32, 54.77, 50.85, 48.21, 48.08, 45.64, 45.60, 40.24, 40.17, 40.09, 39.74, 33.47, 33.42, 30.86, 20.19, 18.92, 17.04, 16.55, 15.72, 14.17, 14.15, 10.91, 9.23. HPLC analysis method: Chiralpak AD-H analytical column (4.5mm × 250mm – 5 μm particle size); mobile phase: isocratic 20% 2-PrOH in hexanes; flow rate: 1 mL/min; injection volume: 20 μL; sample concentration: ~1 mg/mL; multiple DAD λ absorbance signals measured in the range of 210–280 nm, Rt 22.572 and 27.023 min, purity >99%, dr 45:55 (absorbance at 254 nm). HRMS (C33H39O4N3 + H+) calculated 542.30133, found 542.29988 (error −3.6 ppm). The two diastereoisomers 58a and 58b were resolved and separated by preparative chiral HPLC (Chiralpak AD-H 21 mm × 250 mm × 5 μm): mobile phase: gradient from 10% to 40% 2-PrOH in hexanes; temperature: 25 °C; flow rate: 15–18 mL/min; injection volume: 3 mL (~15–20 mg/mL sample concentration); detection at λ 230 nm and 254 nm with the support of ELS detector. 58a eluted first; HPLC analysis method: Chiralpak AD-H analytical column (4.5mm × 250mm – 5 μm particle size); mobile phase: isocratic 20% 2-PrOH in hexanes; flow rate: 1 mL/min; injection volume: 20 μL; sample concentration: ~1 mg/mL; multiple DAD λ absorbance signals measured in the range of 210–280 nm, Rt 28.905 min, purity >99%, de >99% (absorbance at 254 nm). 58b eluted second; HPLC analysis method: Chiralpak AD-H analytical column (4.5mm × 250mm – 5 μm particle size); mobile phase: isocratic 20% 2-PrOH in hexanes; flow rate: 1 mL/min; injection volume: 20 μL; sample concentration: ~1 mg/mL; multiple DAD λ absorbance signals measured in the range of 210–280 nm, Rt 34.205 min, purity >99%, de >99% (absorbance at 254 nm).
N-(4-((1S,5S,9R)-9-Hydroxy-5-(3-hydroxyphenyl)-2-azabicyclo[3.3.1]nonan-2-yl)butyl)-1H-indole-2-carboxamide (61).
The reaction was performed following the same procure described for 57, starting from (1S,5S,9R)-5-(3-hydroxyphenyl)-2-azabicyclo[3.3.1]nonan-9-ol (59; 40 mg, 0.17 mmol). The desired product was purified by flash chromatography eluting with 20% DMA and obtained as a white solid (20 mg, 26% yield). 1H NMR (400 MHz, CD3OD) δ 7.57 (ddt, J = 9.0, 8.0, 1.0 Hz, 2H), 7.41 (dt, J = 8.2, 0.8 Hz, 2H), 7.24 – 6.94 (m, 4H), 6.89 – 6.73 (m, 2H), 6.66 – 6.58 (m, 1H), 4.25 (m, 1H), 4.07 (s, 1H), 3.74 (d, J = 0.5 Hz, 2H), 3.46 (d, J = 6.1 Hz, 1H), 3.44 – 3.25 (m, 3H), 2.96 (d, J = 9.8 Hz, 1H), 2.70 (br s, 3H), 2.48 (td, J = 13.5, 7.8 Hz, 2H), 2.31 (d, J = 12.8 Hz, 1H), 2.23 – 2.08 (m, 1H), 1.93 – 1.61 (m, J = 4.2, 3.2 Hz, 6H), 1.28 (br s, 1H). HPLC analysis method: Chiralpak AD-H analytical column (4.5mm × 250mm – 5 μm particle size); mobile phase: isocratic 50% 2-PrOH in hexanes + 0.1% DEA; flow rate: 1 mL/min; injection volume: 20 μL; sample concentration: ~1 mg/mL; multiple DAD λ absorbance signals measured in the range of 210–280 nm, Rt 29.790, purity >99%, ee >99% (absorbance at 254 nm). HRMS (C27H34O3N3 + H+) calculated 448.25947, found 448.25979.
N-(4-((1S,5S,9S)-9-Hydroxy-5-(3-hydroxyphenyl)-2-azabicyclo[3.3.1]nonan-2-yl)butyl)-1H-indole-2-carboxamide (62).
The reaction was performed following the same procure described for 57, starting from (1S,5S,9S)-5-(3-hydroxyphenyl)-2-azabicyclo[3.3.1]nonan-9-ol (60; 40 mg, 0.17 mmol). The desired product was purified by flash chromatography eluting with 20% DMA and obtained as a white solid (20 mg, 26% yield). 1H NMR (400 MHz, CD3OD) δ 7.62 – 7.53 (m, 2H), 7.42 (t, J = 8.3 Hz, 2H), 7.25 – 7.12 (m, 2H), 7.12 – 6.80 (m, 4H), 6.40 (m, 1H), 4.30 (m, 1H), 4.07 (s, 1H), 3.75 (s, 2H), 3.49 – 3.37 (m, 4H), 2.98 (br s, 1H), 2.20 – 1.93 (m, 7H), 1.73 – 1.66 (m, 5H), 1.28 (br s, 2H). HPLC analysis method: Chiralpak AD-H analytical column (4.5mm × 250mm – 5 μm particle size); mobile phase: isocratic 50% 2-PrOH in hexanes + 0.1% DEA; flow rate: 1 mL/min; injection volume: 20 μL; sample concentration: ~1 mg/mL; multiple DAD λ absorbance signals measured in the range of 210–280 nm, Rt 32.837, purity >99%, ee >99% (absorbance at 254 nm). HRMS (C27H34O3N3 + H+) calculated 448.25947, found 448.25982.
2-(4-(2,3-Dichlorophenyl)piperazin-1-yl)ethan-1-ol (63).
A solution of 1-(2,3-dichlorophenyl)piperazine hydrochloride (500 mg, 1.87 mmol) and K2CO3 (1.29 g, 9.34 mmol) in ACN (30 mL) was stirred for 25 min under gentle heating, followed by dropwise addition of 2-bromoethan-1-ol (257 mg, 2.06 mmol) and cat. KI. The suspension was stirred under reflux overnight, filtered, and the solvent evaporated under vacuum to afford the crude material. The desired product was purified by flash chromatography eluting with 100% EtOAc and obtained as a yellow oil (310 mg, 60% yield). 1H NMR (400 MHz, CDCl3) δ 7.21 – 7.10 (m, 2H), 6.96 (dd, J = 6.7, 2.8 Hz, 1H), 3.72 – 3.62 (m, 2H), 3.08 (br s, 4H), 2.71 (br s, 4H), 2.67 – 2.60 (m, 2H).
2-(4-(2,3-Dichlorophenyl)piperazin-1-yl)ethyl 4-methylbenzenesulfonate (64)
p-toluenesulfonyl chloride (p-TsCl, 200 mg, 1.05 mmol) was added portion wise to a solution of 63 (240 mg, 0.87 mmol) and DIPEA (0.310 mL, 225 mg, 1.74 mmol) in DCM (15 mL). The mixture was stirred at RT overnight, the solvent evaporated under vacuum, and the crude residue purified by flash chromatography eluting with hex/EtOAc (1/1). The desired product was obtained as a white solid (135 mg, 36% yield). 1H NMR (400 MHz, CDCl3) δ 7.81 (d, J = 7.9 Hz, 2H), 7.34 (d, J = 7.9 Hz, 2H), 7.18 – 7.08 (m, 2H), 6.91 (dd, J = 6.8, 2.9 Hz, 1H), 4.16 (t, J = 5.7 Hz, 2H), 3.06 – 2.99 (br s, 4H), 2.72 (t, J = 5.8 Hz, 2H), 2.61 (br s, 4H), 2.44 (s, 3H).
Ethyl (Z)-2-((1S,5S)-2-(2-(4-(2,3-dichlorophenyl)piperazin-1-yl)ethyl)-5-(3-hydroxyphenyl)-2-azabicyclo[3.3.1]nonan-9-ylidene)acetate (65).
A suspension of 56 (50 mg, 0.175 mmol), 64 (71.5 mg, 0.166 mmol) and NaHCO3 (100 mg, 1.2 mmol) in ACN (7 mL) was heated and stirred in a sealed vessel for 6 h. The reaction mixture was filtered, the solvent evaporated under vacuum and the residue purified by flash chromatography eluting with 10% DMA. The desired product was obtained as a yellow oil (40 mg, 43% yield). 1H NMR (400 MHz, CD3OD) δ 7.33 – 7.14 (m, 3H), 7.14 – 7.05 (m, 1H), 6.92 (m, 2H), 6.65 (m, 1H), 5.48 (s, 1H), 4.08 (dq, J = 21.4, 7.1 Hz, 2H), 3.34 – 3.21 (m, 2H), 3.08 (br s, 4H), 2.97 – 2.70 (m, 10H), 2.56 – 2.42 (m, 1H), 2.22 – 1.98 (m, 4H), 1.74 (m, 1H), 1.65 (s, 1H), 1.19 (t, J = 7.2 Hz, 3H); 13C NMR (101 MHz, CD3OD) δ 168.38, 167.92, 158.33, 152.58, 149.48, 134.91, 130.18, 129.03, 128.40, 125.82, 120.17, 119.71, 116.02, 115.67, 114.48, 111.42, 59.50, 57.16, 56.04, 54.80, 54.16, 52.08, 46.63, 41.34, 39.27, 31.73, 21.35, 14.54. The free base was converted into the corresponding oxalate salt. HRMS (C30H37N3O3Cl2 + H+) calculated 558.22847, found 558.22925 (error 1.4 ppm). CHN (C30H37N3O3Cl2 + 2.5 H2C2O4 + 2 H2O) calculated C 51.29, H 5.66, N 5.13; found C 51.07, H 5.28, N 5.21. mp: Salt decomposes above 110 ° C.
Radioligand Binding Studies
hD2R, hD3R, and hD4R.
Radioligand binding assays were conducted similarly as previously described45, 67. HEK293 cells stably expressing human D2LR or D3R or D4.4 were grown in a 50:50 mix of DMEM and Ham’s F12 culture media, supplemented with 20 mM HEPES, 2 mM L-glutamine, 0.1 mM non-essential amino acids, 1X antibiotic/antimycotic, 10% heat-inactivated fetal bovine serum, and 200 μg/mL hygromycin (Life Technologies, Grand Island, NY) and kept in an incubator at 37 °C and 5% CO2. Upon reaching 80–90% confluence, cells were harvested using premixed Earle’s balanced salt solution with 5 mM EDTA (Life Technologies) and centrifuged at 3000 rpm for 10 min at 21 °C. The supernatant was removed, and the pellet was resuspended in 10 mL hypotonic lysis buffer (5 mM MgCl2, 5 mM Tris, pH 7.4 at 4 °C) and centrifuged at 14 500 rpm (~25000g) for 30 min at 4 °C. The pellet was then resuspended in binding buffer. Bradford protein assay (Bio-Rad, Hercules, CA) was used to determine the protein concentration. For [3H]-N-methylspiperone binding studies membranes were diluted to 500 μg/mL, in fresh EBSS binding buffer made from 8.7 g/L Earle’s Balanced Salts without phenol red (US Biological, Salem, MA), 2.2 g/L sodium bicarbonate, pH to 7.4, and stored in a −80 °C freezer for later use. For [3H]-(R)-(+)-7- OH-DPAT binding studies, membranes were harvested fresh; the binding buffer was made from 50 mM Tris, 10 mM MgCl2, 1 mM EDTA, pH 7.4. On the test day, each test compound was diluted into half-log serial dilutions using the 30% dimethyl sulfoxide (DMSO) vehicle. When it was necessary to assist solubilization of the drugs at the highest tested concentration, 0.1% AcOH (final concentration v/v) was added alongside the vehicle. Membranes were diluted in fresh binding buffer. Radioligand competition experiments were conducted in 96-well plates containing 300 μL fresh binding buffer, 50 μL of the diluted test compound, 100 μL of membranes (for [3H]-N-methylspiperone assays: 10–20, 10–20, and 20–30 μg/well total protein for hD2LR, hD3R, and hD4.4R, respectively; for [3H]-(R)-(+)-7- OH-DPAT assays: 40–80, and 20–40 μg/well total protein for hD2LR, and hD3R, respectively), and 50 μL of radioligand diluted in binding buffer ([3H]-N-methylspiperone: 0.4 nM final concentration for all the hD2-like receptor subtypes; [3H]-(R)-(+)-7-OH-DPAT: 1.5 nM final concentration for hD2L, and 0.5 nM final concentration for hD3; Perkin Elmer). Aliquots of radioligands solution were also quantified accurately in each experiment replicate, to determine how much radioactivity was added, taking in account the experimentally determined counter efficiency. Nonspecific binding was determined using 10 μM (+)-butaclamol (Sigma-Aldrich, St. Louis, MO), and total binding was determined with the 30% DMSO vehicle. All compound dilutions were tested in triplicate, and the reaction incubated for 60 min ([3H]-N-methylspiperone assays) or 90 min ([3H]-(R)-(+)-7-OH-DPAT assays) at RT. The reaction was terminated by filtration through PerkinElmer Uni-Filter-96 GF/B, presoaked for the incubation time in 0.5% polyethylenimine, using a Brandel 96-Well Plates Harvester Manifold (Brandel Instruments, Gaithersburg, MD). The filters were washed thrice with 3 mL (3 Å~ 1 mL/well) of ice-cold binding buffer. PerkinElmer MicroScint 20 Scintillation Cocktail (65 μL) was added to each well, and filters were counted using a PerkinElmer MicroBeta Microplate Counter. IC50 values for each compound were determined from dose–response curves, and Ki values were calculated using the Cheng–Prusoff equation63. Kd values were determined via separate homologous competitive binding experiments. When a complete inhibition could not be achieved at the highest tested concentrations, Ki values have been extrapolated by constraining the bottom of the dose–response curves (=0% residual specific binding) in the nonlinear regression analysis. These analyses were performed using GraphPad Prism version 8 for Macintosh (GraphPad Software, San Diego, CA). All results were rounded to the third significant figure. Ki values were determined from at least three independent experiments and are reported as the mean ± standard error of the mean (SEM).
hMOR.
Radioligand binding experiments were conducted, and the results analyzed, as described above, and similarly as previously reported62. HEK293 cells stably expressing hMOR were grown in a DMEM medium, supplemented with 10% FBS, 2 mM L-glutamine, 1% penicillin-streptomycin (or antibiotic/antimycotic) and hygromycin B (50 μg/mL). Upon reaching confluence the cells were harvested and the membranes prepared as detailed before. The binding buffer was made of 50 mM Tris and 5 mM MgCl2 at pH 7.4. The experiments were performed in presence of [3H]-DAMGO (final concentration 3 nM; Perkin Elmer) and 30 μg/well of membranes (final concentration). The reactions were incubated for 60 min at RT and terminated by rapid filtration through Perkin Elmer Uni-Filter-96 GF/B, presoaked for 60 min in 0.5% polyethylenimine. The non-specific binding was determined using 10 μM C-TOP or cold DAMGO. The radioligand Kd was measured via radioligand saturation experiments.
Bioluminescence Resonance Energy Transfer (BRET) Studies
All reagents were purchased from Sigma Aldrich-Merck unless otherwise stated. BRET experiments were performed in transiently transfected human embryonic kidney 293 T (HEK 293T) cells, as described previously.31, 68 Briefly, cells were grown and maintained at 37 °C in 5 % CO2 in Dulbecco’s modified eagle medium (DMEM) supplemented with 10 % (v/v) fetal bovine serum (FBS). Cells were seeded in 10 cm Petri dishes (2.5 × 106 cells per dish) and allowed to grow overnight in media at 37 °C, 5 % CO2. The following day, cells were transiently transfected in media supplemented with antibiotics (100 U/mL penicillin and 100 μg/mL streptomycin, Gibco) using a 1:6 total DNA to PEI (PolySciences Inc) ratio. BRET constructs were as follows: 4 μg of Nb33-Venus and 1 μg of mMOR-Rluc8 for Nb33 recruitment, 2 μg of WT-Gα (i2 or oA), 1 μg of Gβ1-Venus(156–239), 1 μg of Gγ2-Venus(1–155), 1 μg of masGRK3ct-Rluc8 and 1 μg of receptor (SNAP-mMOR or hD3R) for GPA69 and 4 μg of arrestin-3-Venus, 2 μg of WT-GRK2 and 1 μg of mMOR-Rluc8 for arrestin-3 recruitment. Cells were then allowed to grow overnight at 37 °C, 5 % CO2. The next day, cells were plated in Greiner poly-D-lysine-coated 96-well plates (SLS) in media and allowed to grow overnight. On the day of the assay (48h post-transfection), cells were washed once with D-PBS (Lonza, SLS) and incubated in D-PBS for 30 min at 37 °C. For the antagonist-mode assays that require pre-incubation, the cells were washed once with D-PBS and incubated with ligands in D-PBS (supplemented with 10 mM glucose) at 37 °C, 5 % CO2 for 3h prior to starting the assay. The Rluc substrate coelenterazine h (NanoLight) was added to each well (final concentration of 5 μM) and cells were incubated for 5 minutes at 37 °C. After 5 minutes, ligands (final concentration from 10 μM to 1 nM in D-PBS) were added to the plate and cells were incubated for a further 10 minutes at 37 °C before reading the plate in a PHERAstar FSX microplate reader (Venus and Rluc emission signals at 535 and 475 nm respectively, BMG Labtech). The ratio of Venus:Rluc counts was used to quantify the BRET signal in each well. Data were normalized to the wells containing 10 μM DAMGO/quinpirole or no drug for maximal or minimal response, respectively and as indicated in the figure legends. All experiments were performed in duplicate and at least three times independently. All data points represent the mean and error bars represent the standard error of the mean (SEM) and were fitted using the built-in log(agonist) vs. response (three parameters) model in Prism 8.0 (GraphPad software Inc., San Diego, CA).For agonist-mode assays data was fitted to a three parameter concentration-response model where EC50 is the concentration of the agonist needed to elicit half the maximal response of the particular agonist, defined as Emax. For the antagonist-mode assays, data points were fitted using a three-parameter concentration-response model where IC50 is the concentration required to inhibit half the maximum response of the agonist used at a particular concentration. Values of pEC50 or pIC50 plus/minus error are given as the error has a gaussian distribution whereas the error associated with the antilog value does not. For some ligands for which the lower asymptote of the curve was not well defined within the concentration range (represented by a dotted line) the bottom was constrained to be equal to 0%.
Molecular Docking and Computer Aided Drug Design
The receptor structures corresponding to PDB accession code 6CM4, 3PBL, and 5C1M were extracted from RCSB for inactive state D2R, inactive state D3R, and active state MOR, respectively. All the objects except the receptor protein subunit, the crystallized ligand, and in case of active state MOR three crystallographic waters were deleted, and this was followed by the addition of hydrogens and optimization of the side chain residues. Ligands were sketched, assigned formal charges, and energy-optimized prior to docking. The ligand docking box for potential grid docking was defined as the whole extracellular half of the protein, and all-atom docking was performed using the energy minimized structures for all ligands with a thoroughness value of 10. The best-scored docking solutions were further optimized by several rounds of minimization and Monte Carlo sampling of the ligand conformation, including the surrounding side-chain residues (within 5 A° of the ligand) and the three crystallographic waters in the MOR orthosteric sites. All the above molecular modeling operations were performed in ICM-Pro v3.8–5 molecular modeling package.
Supplementary Material
ACKNOWLEDGMENTS
This project was supported by the National Institute on Drug Abuse – Intramural Research Program (ZIADA000609; ZIADA000424), as well as grant funding R33DA038858 for V.K. The authors thank Drs. Ludovic Muller and Shelley Jackson from the Structural Biology Core at NIDA-IRP for high-resolution MS/MS analyses, Dr. Ning Sheng Cai from the Integrative Neurobiology Section at NIDA-IRP for assistance with cell lines and tissue culture protocols, and Dr. Arthur Jacobson from the Drug Design and Synthesis Section at NIDA-IRP for insightful suggestions to the manuscript.
ABBREVIATIONS
- DA
dopamine
- D2-likeR
dopamine D2-like receptors
- OUD
opioid use disorders
- CADD
computer aided drug design
- BRET
bioluminescence resonance energy transfer
- HEK 293 cells
human embryonic kidney 293 cells
- HPLC
high performance liquid chromatography
- HRMS
high resolution mass spectrometry
- DMA
dichloromethane, methanol, ammonium hydroxide
- EDC
1-Ethyl-3-(3-dimethylaminopropyl)carbodiimide
- HOBt
hydroxybenzotriazole
- HCTU
O-(1H-6-Chlorobenzotriazole-1-yl)-1,1,3,3-tetramethyluronium hexafluorophosphate
- DIPEA
N,N-diisopropylethylamine
- STAB
sodium triacetoxyborohydride
- DMP
dess–martin periodinane
- ACN
acetonitrile
- LAH
lithium aluminium hydride
- GPA
G-protein Activation
Footnotes
The Supporting Information is available free of charge on the ACS Publications website.
Analytical data summary of the products (combustion elemental analyses, HRMS-MS/MS, GC/MS and HPLC), supplementary table reporting bias agonism analysis at MOR signaling pathways, PDB files of all the ligand/target complexes studied (with supplementary figures) and molecular formula strings.
The authors declare no competing financial interest.
REFERENCES
- 1.CDC/NCHS National Vital Statistics System, Mortality. CDC WONDER, Atlanta, GA: US Department of Health and Human Services, CDC. https://www.cdc.gov/nchs/nvss/deaths.htm. [Google Scholar]
- 2.Roy L. https://www.forbes.com/sites/lipiroy/2020/05/23/collision-of-crises-how-covid-19-will-propel-drug-overdose-from-bad-to-worse/#5cbc00837d3a.
- 3.Silva MJ; Kelly Z, The escalation of the opioid epidemic due to COVID-19 and sesulting lessons about treatment alternatives. Am. J. of Manag. Care 2020, 26, e202–e204. [DOI] [PubMed] [Google Scholar]
- 4.NIH/NIDA. https://www.drugabuse.gov/drugs-abuse/opioids/opioid-overdose-crisis.
- 5.Vowles KE; McEntee ML; Julnes PS; Frohe T; Ney JP; van der Goes DN, Rates of opioid misuse, abuse, and addiction in chronic pain: a systematic review and data synthesis. Pain 2015, 156, 569–576. [DOI] [PubMed] [Google Scholar]
- 6.Muhuri PK; Gfroerer JC; Davies MC, Associations of nonmedical pain reliever use and initiation of heroin use in the United States. CBHSQ Data Rev August 2013. [Google Scholar]
- 7.Cicero TJ; Ellis MS; Surratt HL; Kurtz SP, The changing face of heroin use in the United States: a retrospective analysis of the past 50 years. JAMA Psychiatry 2014, 71, 821–826. [DOI] [PubMed] [Google Scholar]
- 8.Carlson RG; Nahhas RW; Martins SS; Daniulaityte R, Predictors of transition to heroin use among initially non-opioid dependent illicit pharmaceutical opioid users: A natural history study. Drug Alcohol Depend 2016, 160, 127–134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Rasmussen K; White DA; Acri JB, NIDA’s medication development priorities in response to the opioid crisis: ten most wanted. Neuropsychopharmacology 2019, 44, 657–659. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Keck TM; John WS; Czoty PW; Nader MA; Newman AH, Identifying medication targets for psychostimulant addiction: unraveling the dopamine D3 receptor hypothesis. J. Med. Chem 2015, 58, 5361–5380. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Appel NM; Li SH; Holmes TH; Acri JB, Dopamine D3 receptor antagonist (GSK598809) potentiates the hypertensive effects of cocaine in conscious, freely-moving dogs. J. Pharmacol. Exp. Ther 2015, 354, 484–492. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Jordan CJ; Humburg B; Rice M; Bi GH; You ZB; Shaik AB; Cao J; Bonifazi A; Gadiano A; Rais R; Slusher B; Newman AH; Xi ZX, The highly selective dopamine D3R antagonist, R-VK4–40 attenuates oxycodone reward and augments analgesia in rodents. Neuropharmacology 2019, 158, 107597. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.You ZB; Bi GH; Galaj E; Kumar V; Cao J; Gadiano A; Rais R; Slusher BS; Gardner EL; Xi ZX; Newman AH, Dopamine D3R antagonist VK4–116 attenuates oxycodone self-administration and reinstatement without compromising its antinociceptive effects. Neuropsychopharmacology 2019, 44, 1415–1424. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Kumar V; Bonifazi A; Ellenberger MP; Keck TM; Pommier E; Rais R; Slusher BS; Gardner E; You ZB; Xi ZX; Newman AH, Highly selective dopamine D3 receptor (D3R) antagonists and partial agonists based on eticlopride and the D3R crystal structure: new leads for opioid dependence treatment. J. Med. Chem 2016, 59, 7634–7650. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Jordan CJ; Humburg BA; Thorndike EB; Shaik AB; Xi ZX; Baumann MH; Newman AH; Schindler CW, Newly developed dopamine D3 receptor antagonists, R-VK4–40 and R-VK4–116, do not potentiate cardiovascular effects of cocaine or oxycodone in rats. J. Pharmacol. Exp. Ther 2019, 371, 602–614. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Rosenblum A; Marsch LA; Joseph H; Portenoy RK, Opioids and the treatment of chronic pain: controversies, current status, and future directions. Exp. Clin. Psychopharmacol 2008, 16, 405–416. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Kaye AD; Jones MR; Kaye AM; Ripoll JG; Galan V; Beakley BD; Calixto F; Bolden JL; Urman RD; Manchikanti L, Prescription opioid abuse in chronic pain: an updated review of opioid abuse predictors and strategies to curb opioid abuse: part 1. Pain Physician 2017, 20, S93–S109. [PubMed] [Google Scholar]
- 18.Morrone LA; Scuteri D; Rombola L; Mizoguchi H; Bagetta G, Opioids resistance in chronic pain management. Curr. Neuropharmacol 2017, 15, 444–456. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Schmid CL; Kennedy NM; Ross NC; Lovell KM; Yue Z; Morgenweck J; Cameron MD; Bannister TD; Bohn LM, Bias factor and therapeutic window correlate to predict safer opioid analgesics. Cell 2017, 171, 1165–1175 e13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Grim TW; Acevedo-Canabal A; Bohn LM, Toward directing opioid receptor signaling to refine opioid therapeutics. Biol. Psychiatry 2020, 8, 15–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Bossert JM; Hoots JK; Fredriksson I; Adhikary S; Zhang M; Venniro M; Shaham Y, Role of mu, but not delta or kappa, opioid receptors in context-induced reinstatement of oxycodone seeking. Eur. J. Neurosci 2019, 50, 2075–2085. [DOI] [PubMed] [Google Scholar]
- 22.Grim TW; Schmid CL; Stahl EL; Pantouli F; Ho JH; Acevedo-Canabal A; Kennedy NM; Cameron MD; Bannister TD; Bohn LM, A G protein signaling-biased agonist at the mu-opioid receptor reverses morphine tolerance while preventing morphine withdrawal. Neuropsychopharmacology 2020, 45, 416–425. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Kennedy NM; Schmid CL; Ross NC; Lovell KM; Yue Z; Chen YT; Cameron MD; Bohn LM; Bannister TD, Optimization of a series of mu opioid receptor (MOR) agonists with high G protein signaling bias. J. Med. Chem 2018, 61, 8895–8907. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Imam MZ; Kuo A; Ghassabian S; Cai Y; Qin Y; Li T; Smith MT, Intracerebroventricular administration of CYX-6, a potent mu-opioid receptor agonist, a delta- and kappa-opioid receptor antagonist and a biased ligand at mu, delta & kappa-opioid receptors, evokes antinociception with minimal constipation and respiratory depression in rats in contrast to morphine. Eur. J. Pharmacol 2020, 871, 172918. [DOI] [PubMed] [Google Scholar]
- 25.Kliewer A; Schmiedel F; Sianati S; Bailey A; Bateman JT; Levitt ES; Williams JT; Christie MJ; Schulz S, Phosphorylation-deficient G-protein-biased mu-opioid receptors improve analgesia and diminish tolerance but worsen opioid side effects. Nat. Commun 2019, 10, 367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Montandon G; Ren J; Victoria NC; Liu H; Wickman K; Greer JJ; Horner RL, G-protein-gated inwardly rectifying potassium channels modulate respiratory depression by opioids. Anesthesiology 2016, 124, 641–650. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Levitt ES; Abdala AP; Paton JF; Bissonnette JM; Williams JT, mu opioid receptor activation hyperpolarizes respiratory-controlling Kolliker-Fuse neurons and suppresses post-inspiratory drive. J. Physiol 2015, 593, 4453–4469. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Galligan JJ; Akbarali HI, Molecular physiology of enteric opioid receptors. Am. J. Gastroenterol. Suppl 2014, 2, 17–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Kliewer A; Gillis A; Hill R; Schmidel F; Bailey C; Kelly E; Henderson G; Christie MJ; Schulz S, Morphine-induced respiratory depression is independent of beta-arrestin2 signalling. Br. J. Pharmacol 2020, 177, 2923–2931. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Imam MZ; Kuo A; Ghassabian S; Smith MT, Progress in understanding mechanisms of opioid-induced gastrointestinal adverse effects and respiratory depression. Neuropharmacology 2018, 131, 238–255. [DOI] [PubMed] [Google Scholar]
- 31.Gillis A; Gondin AB; Kliewer A; Sanchez J; Lim HD; Alamein C; Manandhar P; Santiago M; Fritzwanker S; Schmiedel F; Katte TA; Reekie T; Grimsey NL; Kassiou M; Kellam B; Krasel C; Halls ML; Connor M; Lane JR; Schulz S; Christie MJ; Canals M, Low intrinsic efficacy for G protein activation can explain the improved side effect profiles of new opioid agonists. Sci. Signal 2020, 13, eaaz3140. [DOI] [PubMed] [Google Scholar]
- 32.Negus SS; Freeman KB, Abuse potential of biased mu opioid receptor agonists. Trends Pharmacol. Sci 2018, 39, 916–919. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Altarifi AA; David B; Muchhala KH; Blough BE; Akbarali H; Negus SS, Effects of acute and repeated treatment with the biased mu opioid receptor agonist TRV130 (oliceridine) on measures of antinociception, gastrointestinal function, and abuse liability in rodents. J. Psychopharmacol 2017, 31, 730–739. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Austin Zamarripa C; Edwards SR; Qureshi HN; Yi JN; Blough BE; Freeman KB, The G-protein biased mu-opioid agonist, TRV130, produces reinforcing and antinociceptive effects that are comparable to oxycodone in rats. Drug Alcohol Depend 2018, 192, 158–162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Julle D; Gondin AB; Von Zastrow ME; Canals M, Opioid pharmacology under the microscope. Mol. Pharmacol 2020, 98, 425–432. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Stanley TH; Egan TD; Van Aken H, A tribute to Dr. Paul A. J. Janssen: entrepreneur extraordinaire, innovative scientist, and significant contributor to anesthesiology. Anesth. Analg 2008, 106, 451–462. [DOI] [PubMed] [Google Scholar]
- 37.Komoto T; Okada T; Sato S; Niino Y; Oka T; Sakamoto T, New mu-opioid receptor agonists with piperazine moiety. Chem. Pharm. Bull. (Tokyo) 2001, 49, 1314–20. [DOI] [PubMed] [Google Scholar]
- 38.Jevtić II; Penjišević JP; Ivanović MD; Kostić-Rajačić SV, Synthetic route towards potential bivalent ligands possessing opioid and D2/D3 pharmacophores. J. Serb. Chem. Soc 2019, 84, 639–647. [Google Scholar]
- 39.Jevtić II; Penjišević JZ; Savić-Vujović KR; Srebro DP; Vučković SM; Ivanović MD; Kostić-Rajačić SV, mi-Opioid/D2 dopamine receptor pharmacophore containing ligands: Synthesis and pharmacological evaluation. J. Serb. Chem. Soc 2020, 85, 711–720. [Google Scholar]
- 40.Bao X; Liu D; Jin Y; Yang Y, A facile synthesis for novel loperamide analogs as potential μ opioid receptor agonists. Molecules 2012, 17 (12), 14288–14297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Feinberg AP; Creese I; Snyder SH, The opiate receptor: a model explaining structure-activity relationships of opiate agonists and antagonists. Proc. Natl. Acad. Sci. U S A 1976, 73, 4215–4219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Qian M; Vasudevan L; Huysentruyt J; Risseeuw MDP; Stove C; Vanderheyden PML; Van Craenenbroeck K; Van Calenbergh S, Design, synthesis, and biological evaluation of bivalent ligands targeting dopamine D2-like receptors and the mu-opioid receptor. ChemMedChem 2018, 13, 944–956. [DOI] [PubMed] [Google Scholar]
- 43.Newman AH; Battiti FO; Bonifazi A, 2016 Philip S. Portoghese medicinal chemistry lectureship: designing bivalent or bitopic molecules for G-protein coupled receptors. The whole is greater than the sum of its parts. J. Med. Chem 2020, 63, 1779–1797. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Chien EY; Liu W; Zhao Q; Katritch V; Han GW; Hanson MA; Shi L; Newman AH; Javitch JA; Cherezov V; Stevens RC, Structure of the human dopamine D3 receptor in complex with a D2/D3 selective antagonist. Science 2010, 330, 1091–1095. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Battiti FO; Cemaj SL; Guerrero AM; Shaik AB; Lam J; Rais R; Slusher BS; Deschamps JR; Imler GH; Newman AH; Bonifazi A, The significance of chirality in drug design and synthesis of bitopic ligands as D3 receptor (D3R) selective agonists. J. Med. Chem 2019, 62, 6287–6314. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Battiti FO; Newman AH; Bonifazi A, Exception that proves the rule: investigation of privileged stereochemistry in designing dopamine D3R bitopic agonists. ACS Med. Chem. Lett 2020, 11, 1956–1964. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Michino M; Boateng CA; Donthamsetti P; Yano H; Bakare OM; Bonifazi A; Ellenberger MP; Keck TM; Kumar V; Zhu C; Verma R; Deschamps JR; Javitch JA; Newman AH; Shi L, Toward understanding the structural basis of partial agonism at the dopamine D3 receptor. J. Med. Chem 2017, 60, 580–593. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Goldberg JS, Stereochemical basis for a unified structure activity theory of aromatic and heterocyclic rings in selected opioids and opioid peptides. Perspect. Medicin. Chem 2010, 4, 1–10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Kumar V; Moritz AE; Keck TM; Bonifazi A; Ellenberger MP; Sibley CD; Free RB; Shi L; Lane JR; Sibley DR; Newman AH, Synthesis and pharmacological characterization of novel trans-cyclopropylmethyl-linked bivalent ligands that exhibit selectivity and allosteric pharmacology at the dopamine D3 receptor (D3R). J. Med. Chem 2017, 60, 1478–1494. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Kopinathan A; Draper-Joyce C; Szabo M; Christopoulos A; Scammells PJ; Lane JR; Capuano B, Subtle modifications to the indole-2-carboxamide motif of the negative allosteric modulator N-((trans)-4-(2-(7-Cyano-3,4-dihydroisoquinolin-2(1 H)-yl)ethyl)cyclohexyl)-1 H-indole-2-carboxamide (SB269652) yield dramatic changes in pharmacological activity at the dopamine D2 receptor. J. Med. Chem 2019, 62, 371–377. [DOI] [PubMed] [Google Scholar]
- 51.Draper-Joyce CJ; Verma RK; Michino M; Shonberg J; Kopinathan A; Klein Herenbrink C; Scammells PJ; Capuano B; Abramyan AM; Thal DM; Javitch JA; Christopoulos A; Shi L; Lane JR, The action of a negative allosteric modulator at the dopamine D2 receptor is dependent upon sodium ions. Sci. Rep 2018, 8, 1208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Verma RK; Abramyan AM; Michino M; Free RB; Sibley DR; Javitch JA; Lane JR; Shi L, The E2.65A mutation disrupts dynamic binding poses of SB269652 at the dopamine D2 and D3 receptors. PLoS Comput. Biol 2018, 14, e1005948. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Newman AH; Grundt P; Cyriac G; Deschamps JR; Taylor M; Kumar R; Ho D; Luedtke RR, N-(4-(4-(2,3-dichloro- or 2-methoxyphenyl)piperazin-1-yl)butyl)heterobiarylcarboxamides with functionalized linking chains as high affinity and enantioselective D3 receptor antagonists. J. Med. Chem 2009, 52, 2559–2570. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.De Paulis T; Hall H; Ogren SO; Wagner A; Stensadn B; Csoregh I, Synthesis, crystal structure and anti-dopaminergic properties of eticlopride (FLB 131). Eur. J. Med. Chem 1985, 20, 273–279. [Google Scholar]
- 55.Bharathi P; Periasamy M, Metalation of iminium ions formed in the reaction of tertiary amines with TiCl4. Org. Lett 1999, 1, 857–859. [Google Scholar]
- 56.Hiebel AC; Lee YS; Bilsky E; Giuvelis D; Deschamps JR; Parrish DA; Aceto MD; May EL; Harris LS; Coop A; Dersch CM; Partilla JS; Rothman RB; Cheng K; Jacobson AE; Rice KC, Probes for narcotic receptor mediated phenomena. 34. Synthesis and structure-activity relationships of a potent mu-agonist delta-antagonist and an exceedingly potent antinociceptive in the enantiomeric C9-substituted 5-(3-hydroxyphenyl)-N-phenylethylmorphan series. J. Med. Chem 2007, 50, 3765–3776. [DOI] [PubMed] [Google Scholar]
- 57.Bonifazi A; Yano H; Guerrero AM; Kumar V; Hoffman AF; Lupica CR; Shi L; Newman AH, Novel and potent dopamine D2 receptor Go-protein biased agonists. ACS Pharmacol. Transl. Sci 2019, 2, 52–65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Huang W; Manglik A; Venkatakrishnan AJ; Laeremans T; Feinberg EN; Sanborn AL; Kato HE; Livingston KE; Thorsen TS; Kling RC; Granier S; Gmeiner P; Husbands SM; Traynor JR; Weis WI; Steyaert J; Dror RO; Kobilka BK, Structural insights into micro-opioid receptor activation. Nature 2015, 524, 315–321. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Totrov M; Abagyan R, Flexible protein-ligand docking by global energy optimization in internal coordinates. Proteins 1997, Suppl 1, 215–220. [DOI] [PubMed] [Google Scholar]
- 60.Keck TM; Free RB; Day MM; Brown SL; Maddaluna MS; Fountain G; Cooper C; Fallon B; Holmes M; Stang CT; Burkhardt R; Bonifazi A; Ellenberger MP; Newman AH; Sibley DR; Wu C; Boateng CA, Dopamine D4 receptor-selective compounds reveal structure-activity relationships that engender agonist efficacy. J. Med. Chem 2019, 62, 3722–3740. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Volpe DA; McMahon Tobin GA; Mellon RD; Katki AG; Parker RJ; Colatsky T; Kropp TJ; Verbois SL, Uniform assessment and ranking of opioid mu receptor binding constants for selected opioid drugs. Regul. Toxicol. Pharmacol 2011, 59, 385–390. [DOI] [PubMed] [Google Scholar]
- 62.Cai NS; Quiroz C; Bonaventura J; Bonifazi A; Cole TO; Purks J; Billing AS; Massey E; Wagner M; Wish ED; Guitart X; Rea W; Lam S; Moreno E; Casado-Anguera V; Greenblatt AD; Jacobson AE; Rice KC; Casado V; Newman AH; Winkelman JW; Michaelides M; Weintraub E; Volkow ND; Belcher AM; Ferre S, Opioid-galanin receptor heteromers mediate the dopaminergic effects of opioids. J. Clin. Invest 2019, 129, 2730–2744. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Cheng Y; Prusoff WH, Relationship between the inhibition constant (K1) and the concentration of inhibitor which causes 50 per cent inhibition (I50) of an enzymatic reaction. Biochem. Pharmacol 1973, 22, 3099–3108. [DOI] [PubMed] [Google Scholar]
- 64.Sounier R; Mas C; Steyaert J; Laeremans T; Manglik A; Huang W; Kobilka BK; Demene H; Granier S, Propagation of conformational changes during mu-opioid receptor activation. Nature 2015, 524, 375–378. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Wager TT; Hou X; Verhoest PR; Villalobos A, Moving beyond rules: the development of a central nervous system multiparameter optimization (CNS MPO) approach to enable alignment of druglike properties. ACS Chem. Neurosci 2010, 1, 435–449. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Wager TT; Hou X; Verhoest PR; Villalobos A, Central nervous system multiparameter optimization desirability: application in drug discovery. ACS Chem. Neurosci 2016, 7, 767–775. [DOI] [PubMed] [Google Scholar]
- 67.Shaik AB; Kumar V; Bonifazi A; Guerrero AM; Cemaj SL; Gadiano A; Lam J; Xi ZX; Rais R; Slusher BS; Newman AH, Investigation of novel primary and secondary pharmacophores and 3-substitution in the linking chain of a series of highly selective and bitopic dopamine D3 receptor antagonists and partial agonists. J. Med. Chem 2019, 62, 9061–9077. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Klein Herenbrink C; Sykes DA; Donthamsetti P; Canals M; Coudrat T; Shonberg J; Scammells PJ; Capuano B; Sexton PM; Charlton SJ; Javitch JA; Christopoulos A; Lane JR, The role of kinetic context in apparent biased agonism at GPCRs. Nat. Commun 2016, 7, 10842. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Hollins B; Kuravi S; Digby GJ; Lambert NA, The c-terminus of GRK3 indicates rapid dissociation of G protein heterotrimers. Cell. Signal 2009, 21, 1015–1021. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.