

Abstract
Background:
Despite significantly reduced acute myocardial infarction (MI) mortality in recent years, ischemic heart failure (HF) continues to escalate. Therapeutic interventions effectively reversing pathologic remodeling are an urgent unmet medical need. We recently demonstrated that adiponectin receptor 1 (AdipoR1) phosphorylation by G protein-coupled receptor kinase 2 (GRK2) contributes to maladaptive remodeling in the ischemic heart. The current study clarified the underlying mechanisms leading to AdipoR1 phosphorylative desensitization, and investigated whether blocking AdipoR1 phosphorylation may restore its protective signaling, reversing post-MI remodeling.
Methods and Results:
Specific site(s) and underlying molecular mechanisms responsible for AdipoR1 phosphorylative desensitization were investigated in vitro (neonatal and adult cardiomyocytes). The effects of AdipoR1 phosphorylation inhibition upon adiponectin post-MI remodeling and HF progression were investigated in vivo. Among 4 previously identified sites sensitive to GRK2 phosphorylation, alanine substitution of Ser205 (AdipoR1S205A), but not other 3 sites, rescued GRK2-suppressed AdipoR1 functions, restoring adiponectin-induced cell salvage kinase activation and reducing oxidative cell death. The molecular investigation followed by functional determination demonstrated that AdipoR1 phosphorylation promoted clathrin-dependent (not caveolae) endocytosis and lysosomal-mediated (not proteasome) degradation, reducing AdipoR1 protein level and suppressing AdipoR1-mediated cytoprotective action. GRK2-induced AdipoR1 endocytosis and degradation were blocked by AdipoR1S205A overexpression. Moreover, AdipoR1S205E (pseudo-phosphorylation) phenocopied GRK2 effects, promoted AdipoR1 endocytosis and degradation, and inhibited AdipoR1 biological function. Most importantly, AdipoR1 function was preserved during HF development in AdipoR1-KO mice re-expressing hAdipoR1S205A. Adiponectin administration in failing heart reversed post-MI remodeling and improved cardiac function. However, re-expressing hAdipoR1WT in AdipoR1-KO mice failed to restore adiponectin cardioprotection.
Conclusion:
Ser205 is responsible for AdipoR1 phosphorylative desensitization in the failing heart. Blockade of AdipoR1 phosphorylation followed by pharmacologic adiponectin administration is a novel therapy effective in reversing post-MI remodeling and mitigating HF progression.
Keywords: Adipokines, Ischemic Heart Failure, Pathologic Remodeling, Receptor Desensitization
Subject Terms: Heart Failure, Myocardial Infarction
Graphical Abstract
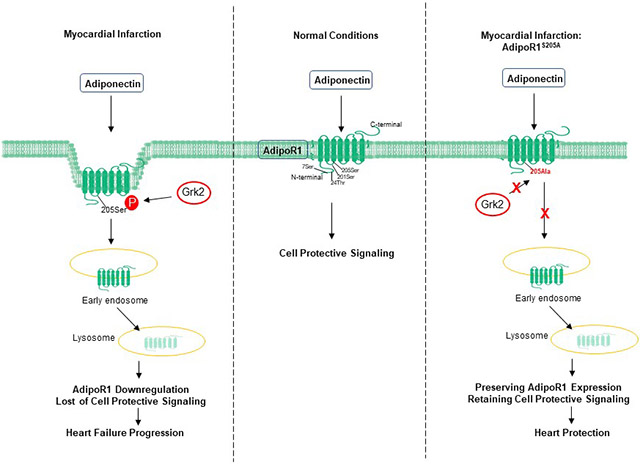
1. Introduction
The overall mortality of AMI (acute myocardial infarction) has significantly reduced (from >20% to 12.4%) in the past two decades. However, over time, many survivors of AMI will progress towards ischemic heart failure (HF), thereby increasing the HF population1. Extensive experimental and clinical studies in recent years identified many protective interventions capable of mitigating ischemic HF when initiated before or immediately after ischemia2. However, incomplete understanding of the molecular mechanisms responsible for the transition from adaptive to maladaptive remodeling in the ischemic heart has hamstrung the development of therapies effective in reversing post-MI pathologic remodeling or blocking HF progression3.
Adiponectin (APN) is an adipocyte-specific protein with strong cardiovascular regulatory function4. After more than a decade’s work, the functions of APN and its signaling pathways have largely been identified. Two specific APN receptors (AdipoR1 and AdipoR2) have been cloned5. They belong to a new family of membrane receptors (progestin and adipoQ receptor, PAQR) predicted to contain seven transmembrane domains, similar to but structurally and topologically distinct from G protein-coupled receptors (GPCRs)6. Activation of AdipoR1 (primarily expressed in muscular cells) and AdipoR2 (primarily expressed in hepatocytes) increases glucose and free fatty acid utilization, stimulates mitochondrial biogenesis and inhibits inflammatory response7. In addition to its essential role in APN signaling, AdipoR1 is critically involved in transmembrane signaling of other cardiovascular protective molecules (notably CTRP68, CTRP99, 10, tectorigenin11, and resveratrol12), as well as ischemic preconditioning13. Moreover, AdipoR1 plays a critical role in maintaining membrane fluidity in most human cell types independent of APN14. Clarifying the pathological alteration of AdipoR1 and resultant contribution to disease development may assist in identifying novel effective therapies against ischemic cardiac injury.
Basic and clinical studies demonstrate that reduced APN levels are correlated with increased AMI risk, as well as inferior cardiac functional recovery after myocardial infarction (MI) with reperfusion7. However, the role of APN in chronic HF is controversial15. Despite clear experimental evidence demonstrating APN deficiency significantly exacerbates HF progression16, several clinical observations demonstrate hyperadiponectinemia is associated with poor cardiac function and increased mortality in these patient populations17-19. Our recent study20 demonstrated for the first time that AdipoR1 is phosphorylated and desensitized by GPCR kinase 2 (GRK2) in failing cardiomyocytes. Phosphorylated AdipoR1 does not carry transmembrane protective signaling function, contributing to post-MI remodeling and HF progression. Moreover, mass spectrometry identified 4 sites (Ser7, Thr24, Ser201, and Ser205) sensitive to GRK2 phosphorylation. These results provide a likely explanation for the paradoxical relationship between elevated plasma APN and poor HF outcome. However, several important questions remain. The specific site(s) whose phosphorylation is (are) responsible for GRK2-induced AdipoR1 desensitization remains unidentified. The mechanisms underlying phosphorylative desensitization are unknown. Most importantly, whether blocking AdipoR1 phosphorylation may restore APN cardioprotective action during the chronic HF period, thereby reversing post-MI remodeling, has not been previously investigated.
Combining in vitro molecular mechanistic investigation and in vivo concept demonstration, we demonstrated that GRK2-induced AdipoR1 phosphorylation at serine205 (Ser205) is responsible for AdipoR1 desensitization in the failing heart. Genetic interventions blocking AdipoR1 phosphorylation restores APN cardiac protection during the chronic HF condition, mitigating HF progression.
2. METHODS
Data Availability.
The data that support the findings of this study are available from the corresponding author upon reasonable request.
Detailed methods for cell isolation and culture, plasmids constructions and transfections, adeno-associated virus 9 vector production and infection, cell viability and apoptosis, Western and Co-immunoprecipitation, echocardiography and strain analysis, immunofluorescent cellular and Masson’s trichrome staining, and quantitative PCR methods are provided in the Supplemental Material. Key research materials are listed in the Major Resources Table in the Supplemental Material. Representative images for cellular and animal experiments and representative western blot images were chosen to closely match the mean for the parameters assessed.
Major Resources Table.
In order to allow validation and replication of experiments, all essential research materials listed in the Methods should be included in the Major Resources Table below. Authors are encouraged to use public repositories for protocols, data, code, and other materials and provide persistent identifiers and/or links to repositories when available. Authors may add or delete rows as needed.
| Animals (in vivo studies) | ||||
|---|---|---|---|---|
| Species | Vendor or Source |
Background Strain | Sex | Persistent ID / URL |
| Mus musculus | MMRRC | B6.129P2-Adipor1tm1Dgen, C57BL/6J | F and M | MMRRC Strain #11599, https://www.mmrrc.org/catalog/sds.php?mmrrc_id=11599 |
| Genetically Modified Animals | |||||
|---|---|---|---|---|---|
| Species | Vendor or Source |
Background Strain |
Other Information | Persistent ID / URL | |
| Parent - Male | |||||
| Parent - Female | |||||
| Antibodies | |||||
|---|---|---|---|---|---|
| Target antigen | Vendor or Source |
Catalog # | Working concentration |
Lot # (preferred but not required) |
Persistent ID / URL |
| Anti-phospho-ERK1/2 rabbit monoclonal antibody | Cell Signaling Technology | 4370 | 1:2000 | https://www.cellsignal.com/product/productDetail.jsp?productId=4370 | |
| Anti-ERK1/2 rabbit monoclonal antibody | Cell Signaling Technology | 4695 | 1:1000 | https://www.cellsignal.com/product/productDetail.jsp?productId=4695 | |
| Anti-phospho-AMPKα rabbit polyclonal antibody | Cell Signaling Technology | 2531 | 1:1000 | https://www.cellsignal.com/product/productDetail.jsp?productId=2531 | |
| Anti-AMPKα rabbit monoclonal antibody | Cell Signaling Technology | 2603 | 1:1000 | https://www.cellsignal.com/product/productDetail.jsp?productId=2603 | |
| Anti-phospho-ACC rabbit monoclonal antibody | Cell Signaling Technology | 11818 | 1:1000 | https://www.cellsignal.com/product/productDetail.jsp?productId=11818 | |
| Anti-ACC rabbit polyclonal antibody | Cell Signaling Technology | 3662 | 1:1000 | https://www.cellsignal.com/product/productDetail.jsp?productId=3662 | |
| Anti-cleaved caspase-3 rabbit polyclonal antibody | Cell Signaling Technology | 9661 | 1:1000 | https://www.cellsignal.com/product/productDetail.jsp?productId=9661 | |
| Anti-caspase-3 rabbit polyclonal antibody | Cell Signaling Technology | 9662 | 1:1000 | https://www.cellsignal.com/product/productDetail.jsp?productId=9662 | |
| Anti-phospho-AP2M1 rabbit monoclonal antibody | Cell Signaling Technology | 7399 | 1:1000 | https://www.cellsignal.com/product/productDetail.jsp?productId=7399 | |
| Anti-AP2M1 rabbit monoclonal antibody | Abcam | ab75995 | 1:2000 | https://www.abcam.com/ap2m1-antibody-ep2695y-ab75995.html | |
| Anti-GRK2 rabbit polyclonal antibody | Cell Signaling Technology | 3982s | 1:1000 | https://www.cellsignal.com/product/productDetail.jsp?productId=3982s | |
| Anti-adipoR1 mouse monoclonal antibody | Santa Cruz Biotechnolog y | sc-518030 | 1:500 | https://www.scbt.com/p/adipor1-antibody-d-9?productCanUrl=adipor1-antibody-d-9&_requestid=5867389 | |
| Anti-β-arrestin1/2 rabbit monoclonal antibody | Cell Signaling Technology | 4674 | 1:1000 | https://www.cellsignal.com/product/productDetail.jsp?productId=4674 | |
| Anti-GFP rabbit polyclonal antibody | Genscript | A01704 | 1:500 | https://www.genscript.com/antibody/A01704-THE_GFP_Antibody_pAb_Rabbit.html | |
| Anti-β-actin mouse monoclonal antibody | Santa Cruz Biotechnolog y | sc-47778 | 1:200 | https://www.scbt.com/p/beta-actin-antibody-c4?requestFrom=search | |
| Anti-GAPDH rabbit monoclonal antibody | Cell Signaling Technology | 2118 | 1:1000 | https://www.cellsignal.com/product/productDetail.jsp?productId=2118 | |
| Anti-flag M2 rabbit monoclonal antibody | Sigma-Aldrich | F1804 | 1:2000 | https://www.sigmaaldrich.com/US/en/product/sigma/f1804 | |
| Anti-Rab5 rabbit monoclonal antibody | Cell Signaling Technology | 3547 | 1:200 | https://www.cellsignal.com/product/productDetail.jsp?productId=3547 | |
| Anti-LAMP2 rabbit polyclonal Antibody | Bioss | BS-2379R | 1:100 | https://www.biossusa.com/products/bs-2379r | |
| Anti-p-Ser/Phosphoserine mouse monoclonal antibody | Santa Cruz Biotechnology | sc-81514 | 1:1000 | https://www.scbt.com/p/p-ser-antibody-16b4 | |
| Secondary HRP-conjugated anti-mouse antibody | Cell Signaling Technology | 7076 | 1:1000 | https://www.cellsignal.com/product/productDetail.jsp?productId=7076 | |
| Secondary HRP-conjugated anti-rabbit antibody | Cell Signaling Technology | 7074 | 1:1000 | https://www.cellsignal.com/product/productDetail.jsp?productId=7074 | |
| Goat anti-Mouse IgG (H+L), Alexa Fluor 546 | ThermoFisher Scientific | A-11003 | 1:500 | https://www.thermofisher.com/antibody/product/Goat-anti-Mouse-IgG-HL-Cross-Adsorbed-Secondary-AntibodyPolyclonal/A-11003 | |
| Goat Anti-Rabbit IgG H&L (Cy5) | Abcam | Ab6564 | 1:2000 | https://www.abcam.com/goat-rabbit-igg-hl-cy5--preadsorbed-ab6564.html#minReactivity | |
| DNA/cDNA Clones | |||
|---|---|---|---|
| Clone Name | Sequence | Source / Repository | Persistent ID / URL |
| 3xFlag-hAdipoR1 | Human AdipoR1 cDNA | This paper/In house | |
| AAV9-cTNT-eGFP plasmid | Available at URL | A gift from Dazhi Wang | https://www.addgene.org/105543/ |
| AAV helper plasmid pAdDeltaF6 | Available at URL | A gift from Dazhi Wang | https://www.addgene.org/112867/ |
| AAV packaging plasmid pAAV2/9n | Available at URL | A gift from Dazhi Wang | https://www.addgene.org/112865/ |
| pSNAPf vector | Available at URL | New England Biolabs | https://www.neb.com/products/n9183-psnapf-vector#Product%20Information |
| Cultured Cells | |||
|---|---|---|---|
| Name | Vendor or Source | Sex (F, M, or unknown) | Persistent ID / URL |
| Primary adult cardiomyocyte | Primary cells | F and M | |
| Primary neonatal mouse ventricular myocyte | Primary cells | F and M | |
| Data & Code Availability | ||
|---|---|---|
| Description | Source / Repository | Persistent ID / URL |
2.1. Animal study protocol
All animal study experiments were performed in adherence to the National Institutes of Health Guidelines on the Use of Laboratory Animals, and were approved by the Thomas Jefferson University Committee on Animal Care. Both male and female adiponectin receptor 1 knockout (AdipoR1-KO) mice were utilized in the study.
To generate a cardiomyocyte-specific AdipoR1 mutation mouse line, a previously reported neonatal mouse AAV administration method21 was utilized. In brief, 1x1011 viral genome particles/mouse of AAV9-cTNT-eGFP, AAV9-cTNT-hAdipoR1WT or AAV9-cTNT-hAdipoR1S205A, or AAV9-cTNT-hAdipoR1S205E were injected subcutaneously in the nape of 1-2 day-old AdipoR1-KO neonatal mice. AdipoR1-KO mice injected with AAV9-cTNT-eGFP revealed cardiac-specific eGFP expression for at least 20 weeks (Figures S1A-D).
Permanent MI surgery was performed by ligating the left anterior descending coronary artery of mice with the age of 8 to 10 weeks as previously described22. One week after MI, mice were randomized to receive vehicle or APN treatment (0.25 μg/g/d) via intraperitoneal mini-osmotic pumps (ALZET, DURECT Corp Cupertino, CA). Cardiac function was determined every week after MI. Mice were sacrificed. Body, heart, and lung weight were measured.
2.2. Assessment of endocytosis via the SNAP-Surface method
hAdipoR1WT and hAdipoR1S205A were cloned into the pSNAPf vector (NEB, #E9120S) for expression of fusion proteins with a C-terminal SNAP tag. Briefly, hAdipoR1WT and hAdipoR1S205A were amplified by PCR using custom designed primers. The PCR products and pSNAPf vector were ligated via Takara ligation kit after digestion by NheI and AscI enzymes to form pSNAPf-hAdipoR1WT and pSNAPf-hAdipoR1S205A plasmids. The plasmids were then transfected into neonatal mouse ventricular myocytes (NMVMs) isolated from AdipoR1-KO mice. After 72 hours, SNAP fluorescence labeling was utilized, per manufacturer (New England Biolabs) protocol. Briefly, the NMVMs were incubated with 5 μM cell-impermeable SNAP-Surface 549 fluorescent substrate at 37°C for 30 minutes to make fusion proteins visible. Cells were washed 3 times with PBS. The images were acquired by Olympus BX51 Fluorescence Microscope. All measurements were determined by a single-blinded research fellow.
2.3. Proximity Ligation Assay (PLA)
The proximity ligation assay was performed to study hAdipoR1 and clathrin proximity via Duolink detection kit as described previously23, 24. NMVMs expressing hAdipoR1WT or hAdipoR1S205A were fixed with 2% formaldehyde for 20 minutes. Cells were then incubated with mouse anti-AdipoR1 and rabbit anti-clathrin at 4 °C overnight in a humidifying chamber. Cells were washed and incubated with secondary anti-mouse/rabbit antibodies conjugated PLA probes at 37°C for 1 hour. Slides were washed again and incubated with ligation-ligase solution, followed by incubation with amplification-polymerase solution. Cells were then mounted with a minimal volume of mounting medium containing DAPI for 30 minutes. PLA signals were detected at room temperature by Olympus BX51 Fluorescent Microscope. All measurements were determined by a single-blinded research fellow.
2.4. Statistical Analysis
The statistical analyses were performed using GraphPad Prism version 9.0.2. Shapiro-Wilk test was used to determine normality of data. For normally distributed data with three or more groups, statistical analyses were performed using a one-way ANOVA with post-hoc Tukey multiple comparison tests. Statistical analyses of the influence of two categorical independent variables on one continuous dependent variable, or multiple groups over time, a two-way ANOVA was performed with post-hoc Tukey multiple comparison tests. Group pairs underwent post hoc pairwise tests were labeled with adjusted P values to determine statistical significance in the figures. Normally distributed data are presented as mean with a standard error of the mean. For non-normally distributed data or if the sample size was less than 6, statistical analyses were performed by nonparametric analysis using a Kruskal-Wallis test with post-hoc Dunn multiple comparison tests for three or more groups. Non-normally distributed data are shown as median with 95% confidence interval. The Kaplan-Meier survival curves were analyzed by a log-rank test. No experiment-wide/across-test multiple test correction was applied; only withintest corrections were made.
The sample size represents the number of independent experimental units per group. Sample sizes were determined based on our previous experience20 performing power analysis based on a two-tailed Student’s t-test to provide adequate power to detect a 20% difference, assuming power of 80% (β=0.80) and an α of 0.05 for these studies. No animals or samples were excluded from the study. Two-sided P values were used and P values less than 0.05 were considered significant.
Detailed information about statistical tests performed for each figure are shown in Supplemental table 5.
3. Results
3.1. Identifying the site(s) responsible for AdipoR1 phosphorylative desensitization
We previously demonstrated that MI-induced AdipoR1 phosphorylation is positively correlated with GRK2 expression in WT heart, and is abolished in cardiomyocyte-specific GRK2KO mice20. Mass spectrometry identified 4 sites (Ser7,201,205 and Thr24) that are sensitive to GRK2 phosphorylation20. To identify the specific site(s) responsible for GRK2-induced AdipoR1 phosphorylative desensitization, NMVMs from AdipoR1-KO mice were transfected with vectors expressing wild type (WT) or mutated human AdipoR1 (hAdipoR1) followed by Ad-GRK2 infection. Comparable overexpression of WT or S205A constructs of AdipoR1 was confirmed 24 hours after infection (Figure S2). In AdipoR1-KO/hAdipoR1WT NMVMs, GRK2 overexpression blocked APN-induced ERK1/2, AMPK, and ACC phosphorylation (Figure 1A/B). Re-expressing a tetra-mutated AdipoR1 (all 4 sites were mutated to alanine) in AdipoR1-KO NMVMs (AdipoR1-KO/hAdipoR1S7/T24/S201/S205A) blocked GRK2 inhibitory effect after APN signaling, resulting in ERK1/2, AMPK, and ACC activation (Figures 1C/D). Re-expressing hAdipoR1S7A, hAdipoR1T24A, or hAdipoR1S201A in AdipoR1-KO NMVMs failed to block GRK2 inhibitory effect upon APN signaling (Figures S3A-F). However, re-expressing hAdipoR1S205A in AdipoR1-KO (AdipoR1-KO/hAdipoR1S205A) NMVMs completely blocked GRK2 effect, restoring APN-induced ERK1/2, AMPK and ACC phosphorylation (Figure 1E/F). To obtain more evidence supporting the critical role of Ser205 in GRK2 phosphorylative suppression of APN signaling, AdipoR1-KO NMVMs were transfected with a pseudo-phosphorylation mutation vector (Ser205 was mutated to glutamic acid, hAdipoR1S205E). APN failed to activate ERK1/2, AMPK, or ACC in these cells, even without GRK2 overexpression (Figures 1G/H).
Figure 1.
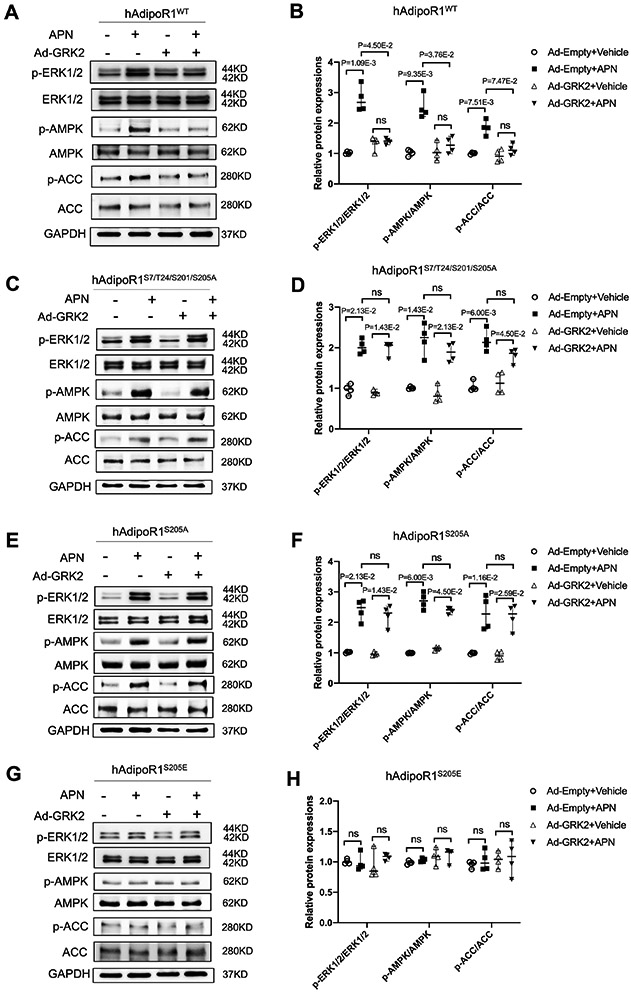
Ser205 phosphorylation is responsible for AdipoR1 desensitization and blocking AdipoR1-mediated transmembrane signaling. NMVMs isolated from adipoR1 knock out mice were transfected with 3xFlag-hAdipoR1WT, hAdipoR1S7/T24/S201/S205A, hAdipoR1S205A or hAdipoR1S205E. 24 hours after transfection, cells were infected with either an Ad-empty or Ad-GRK2 vector. 48 hours later, cells were treated with APN (2 μg/mL) for 30 minutes. A/C/E/G: Effect of APN on cell salvage kinase activation in AdipoR1-KO/hAdipoR1WT, AdipoR1-KO/hAdipoR1S7/T24/S201/S205A, AdipoR1-KO/hAdipoR1S205A or AdipoR1-KO/hAdipoR1S205E cardiomyocytes. B/D/F/H: Quantification of the Western blot results normalized to GAPDH (n=4). Statistical significance was evaluated by a Kruskal-Wallis test. Post hoc pairwise tests for indicated group pairs were performed after Dunn correction. Ns indicates not significant.
Having demonstrated that Ser205 is the responsible site blocking APN signaling when phosphorylated, we next determined whether preventing Ser205 phosphorylation may rescue APN cytoprotective effect. Cell viability, cell injury, and cell apoptosis were determined. In H2O2 treated AdipoR1-KO/hAdipoR1WT NMVMs without GRK2 overexpression, APN increased cell viability (Figure 2A), decreased LDH release (Figure 2C), and decreased cleaved caspase 3 expression (Figures 2 E/G). All these protective effects were significantly decreased when GRK2 was overexpressed (Figures 2A/C/E/G). However, APN cytoprotective effects against H2O2 were largely preserved in GRK2-overexpressing AdipoR1-KO/hAdipoR1S205A NMVMs (Figure 2B/D/F/H). Taken together, we identified Ser205 phosphorylation as responsible for AdipoR1 desensitization, blocking AdipoR1-mediated transmembrane signaling and cytoprotection.
Figure 2.
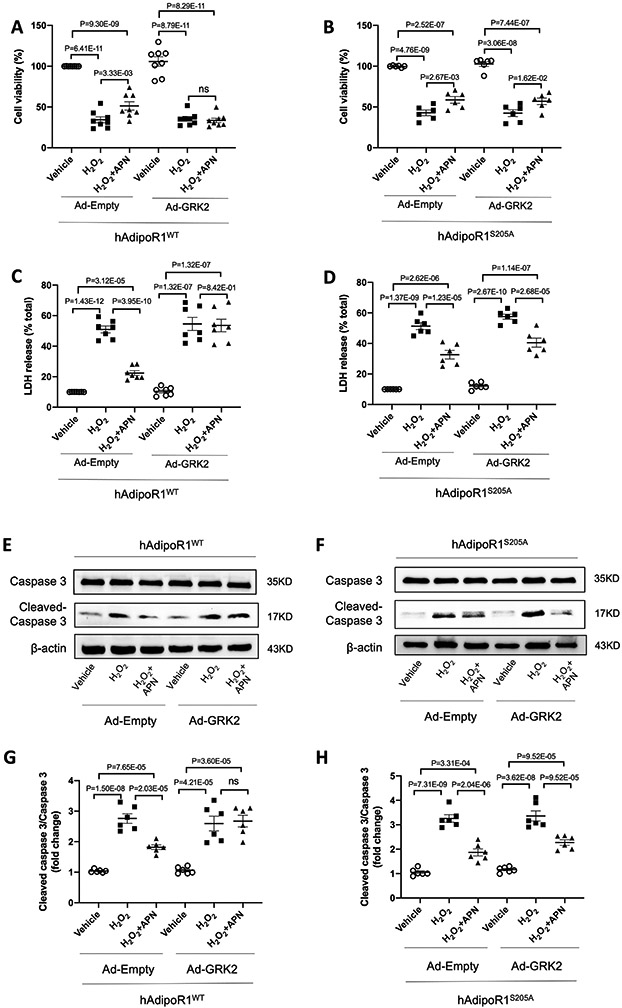
Preventing Ser205 phosphorylation rescues APN cytoprotective effect. 24 hours after being transfected with 3xFlag-hAdipoR1WT or hAdipoR1S205A, NMVMs isolated from adipoR1 knockout mice were infected with either Ad-empty or Ad-GRK2 vector. 48 hours later, cells were treated with APN (2 μg/mL) for 24h followed by H2O2 for 2h. A/B: NMVMs viability was evaluated by MTT assay (n=8 for A and n=6 for B). C/D: NMVMs injuries were determined by LDH assay (n=7 for C and n=6 for D). E-H: Western blot was performed to analyze protein expression of cleaved caspase 3 and caspase 3 in NMVMs (n=6). Statistical significance was evaluated by a one-way ANOVA. All pairwise comparisons were made. Tukey tests were used to correct for multiple comparisons. Ns indicates not significant.
3.2. Clathrin-dependent endocytosis accounts for GRK2-induced AdipoR1 desensitization
Two mechanisms are largely responsible for GRK2-induced β-adrenergic receptor desensitization, i.e., β-arrestin recruitment and receptor endocytosis/degradation25. Given the structural similarities between AdipoR1 and β-adrenergic receptors, we tested whether these mechanisms are responsible for GRK2-induced AdipoR1 desensitization. AdipoR1-KO NMVMs were transfected with 3XFlag-hAdipoR1WT and infected with Ad-GRK2. AdipoR1/β-arrestin interaction was determined by Co-IP after APN stimulation. Somewhat to our surprise, GRK2 overexpression failed to stimulate AdipoR1/β-arrestin interaction (Figure S4). To determine whether GRK2 may stimulate AdipoR1 endocytosis, two experiments were performed. First, AdipoR1-KO NMVMs were transfected with pSNAPf-hAdipoR1WT or pSNAPf-hAdipoR1S205A, followed by Ad-GRK2 infection. Cells were stained with cell-impermeable substrates to visualize protein trafficking. In pSNAPf-hAdipoR1WT expressing NMVMs, Ad-GRK2 caused significant AdipoR1 membrane to cytosol redistribution (Figure 3A). This Ad-GRK2-induced AdipoR1 redistribution was significantly attenuated in cells expressing pSNAPf-hAdipoR1S205A (Figure 3B) and mimicked by cells expressing pSNAPf-hAdipoR1S205E (Figure 3C). Second, Rab5 is a marker of the early endosome and plays an important role in endocytosis26. Adult cardiomyocytes were isolated from AdipoR1-KO mice harboring AAV9-hAdipoR1WT (AdipoR1-KO/AAV9-hAdipoR1WT) or AAV9-hAdipoR1S205A (AdipoR1-KO/AAV9-hAdipoR1S205A). GRK2-induced AdipoR1/Rab5 interaction was determined by immunofluorescent image analysis and Co-immunoprecipitation. GRK2 significantly enhanced AdipoR1/Rab5 colocalization in AdipoR1-KO/AAV9-hAdipoR1WT cardiomyocytes (Figures 3D/E), and increased AdipoR1/Rab5 interaction (Figure 3F). This effect was significantly attenuated in cardiomyocytes from AdipoR1-KO/AAV9-hAdipoR1S205A expressing mice (Figure 3D-F). Third, GRK2 reduced membrane AdipoR1 and significantly increased cytosolic AdipoR1 in cells expressing WT AdipoR1 (Figure 3G). This effect was blocked in cells expressing AdipoR1S205A (Figure 3G).
Figure 3.

Grk2 induces adipoR1 endocytosis. A/B/C: Visualization of pSNAPf-hAdipoR1WT, pSNAPf-hAdipoR1S205A or pSNAPf-hAdipoR1S205E endocytosis in NMVMs infected with Ad-empty or Ad-GRK2 24 hours after infection. D: Adult cardiomyocytes isolated from AAV9-hAdipoR1WT or AAV9-hAdipoR1S205A mice (AdipoR1-KO background) were infected with Ad-Empty or Ad-GRK2, and immune-stained against AdipoR1 (red), Rab5 (green) 24 hours after infection. Nuclei were labeled with 4’,6-diamidino-2-phenylindole (DAPI, blue). E: Pearson’s correlation coefficient quantification of red and green channels from C (n=8). F: AdipoR1/Rab5 Co-immunoprecipitation (IP: AdipoR1, IB: Rab5). Results demonstrated that GRK2OE increased AdipoR1/Rab5 Co-immunoprecipitation in cells expressing AdipoR1WT, an effect attenuated by AdipoR1S205A mutation. G: Cell fractionation followed by Western analysis: results demonstrated that GRK2OE significantly increased cytosol AdipoR1 in cells expressing WT AdipoR1, an effect attenuated by AdipoR1 S205A mutation (n=6). Statistical significance was evaluated by a two-way ANOVA. Post hoc pairwise tests for indicated group pairs were performed after Tukey correction. Ns indicates not significant.
Clathrin and caveolin are the two most important molecules mediating molecular endocytosis27. To determine which endocytic pathway is responsible for GRK2-induced AdipoR1 endocytosis and desensitization, we investigated the effects of chlorpromazine (an inhibitor of clathrin-mediated endocytosis28) and genistein (an inhibitor of caveolae-mediated endocytosis29). Treatment with chlorpromazine, but not genistein, significantly suppressed GRK2-induced AdipoR1 endocytosis (Figure 4A), suggesting clathrin (not caveolae) mediates GRK2-induced AdipoR1 endocytosis. To obtain more evidence supporting this notion, two additional experiments were performed. First, AdipoR1/clathrin interaction was visualized by Duolink in situ proximity ligation assay (PLA). In adult cardiomyocytes isolated from AdipoR1-KO/AAV9-hAdipoR1WT expressing mice, GRK2 significantly increased the number of PLA blobs. However, PLA blobs were significantly reduced in cardiomyocytes isolated from AdipoR1-KO/AAV9-hAdipoR1S205A expressing mice (Figure 4B-C). Second, clathrin-mediated endocytosis is regulated by multiple factors, particularly adaptor protein-2 (AP2) phosphorylation30. In AdipoR1-KO NMVMs expressing hAdipoR1WT, Ad-GRK2 expression (Figure S5) and AP2 phosphorylation (Figure 4D) followed a similar temporal pattern. Collectively, these results reveal that clathrin mediates GRK2-induced AdipoR1 endocytosis.
Figure 4.

Clathrin-dependent endocytosis is responsible for GRK2-induced desensitization of adipoR1. A. Visualization of pSNAPf-hAdipoR1WT endocytosis in NMVMs infected with adenovirus and treated with chlorpromazine or genistein. 24h after being transfected with pSNAPf-hAdipoR1WT, NMVMs from AdipoR1 knockout mice were infected with either Ad-empty or Ad-GRK2 vector with or without chlorpromazine (10 μmol/L) or genistein (10 μmol/L) for 48h. B. In situ PLA-based detection of AipoR1 and clathrin interaction (red particles) in adult cardiomyocytes transfected with Ad-Empty or Ad-GRK2. C. PLA analysis was quantified as PLA blobs per cell (n=8). Statistical significance was evaluated by a two-way ANOVA. Post hoc pairwise tests for indicated group pairs were performed after Tukey correction. D. Western blot was performed to analyze AP2 phosphorylation in hAdipoR1WT over-expressed NMVMs infected with adenovirus (n=5). E/F: Western blot was performed to analyze APN (2 μg/ml, 30 min)-induced ERK1/2 phosphorylation in NMVMs after being treated with chlorpromazine or genistein (n=4). For D-F, statistical significance was evaluated by a non-parametric teste (Kruskal-Wallis test). Post hoc pairwise tests for indicated group pairs were performed after Dunn correction. Ns indicates not significant. G: NMVMs viability was evaluated by MTT assay after being treated with chlorpromazine or genistein (n=6). Statistical significance was evaluated by a one-way ANOVA. All pairwise comparisons were made, and Tukey tests were used to correct for multiple comparisons. Ns indicates not significant.
Next, we determined whether blocking clathrin-mediated AdipoR1 endocytosis may restore GRK2-suppressed AdipoR1 signaling and cytoprotection. In AdipoR1-KO NMVMs expressing hAdipoR1WT, chlorpromazine (but not genistein) rescued both APN ERK1/2 activation (Figures 4E/F) and cytoprotective effect (Figure 4G) suppressed by GRK2.
3.3. GRK2 promoted AdipoR1 degradation via the lysosomal pathway
In NMVMs, GRK2 overexpression time-dependently downregulated AdipoR1 protein expression, becoming statistically significant 48 hours after Ad-GRK2 infection (Figure 5A). However, GRK2 overexpression had no significant effect on AdipoR1 mRNA expression (Figure 5B). These results suggest that protein degradation is likely involved in AdipoR1 downregulation, impairing AdipoR1-mediated signaling and cytoprotection. To test this hypothesis, AdipoR1-KO/hAdipoR1WT NMVMs were transfected with Ad-GRK2. The effect of MG-132 (a proteasome inhibitor)31 or chloroquine (a lysosome inhibitor)32 upon GRK2-induced AdipoR1 downregulation was determined. Chloroquine, not MG-132, significantly (although not completely) inhibited AdipoR1 degradation in GRK2 expressing cardiomyocytes (Figure 5C/D), suggesting lysosome-mediated protein degradation is responsible for AdipoR1 degradation after their endocytosis.
Figure 5.
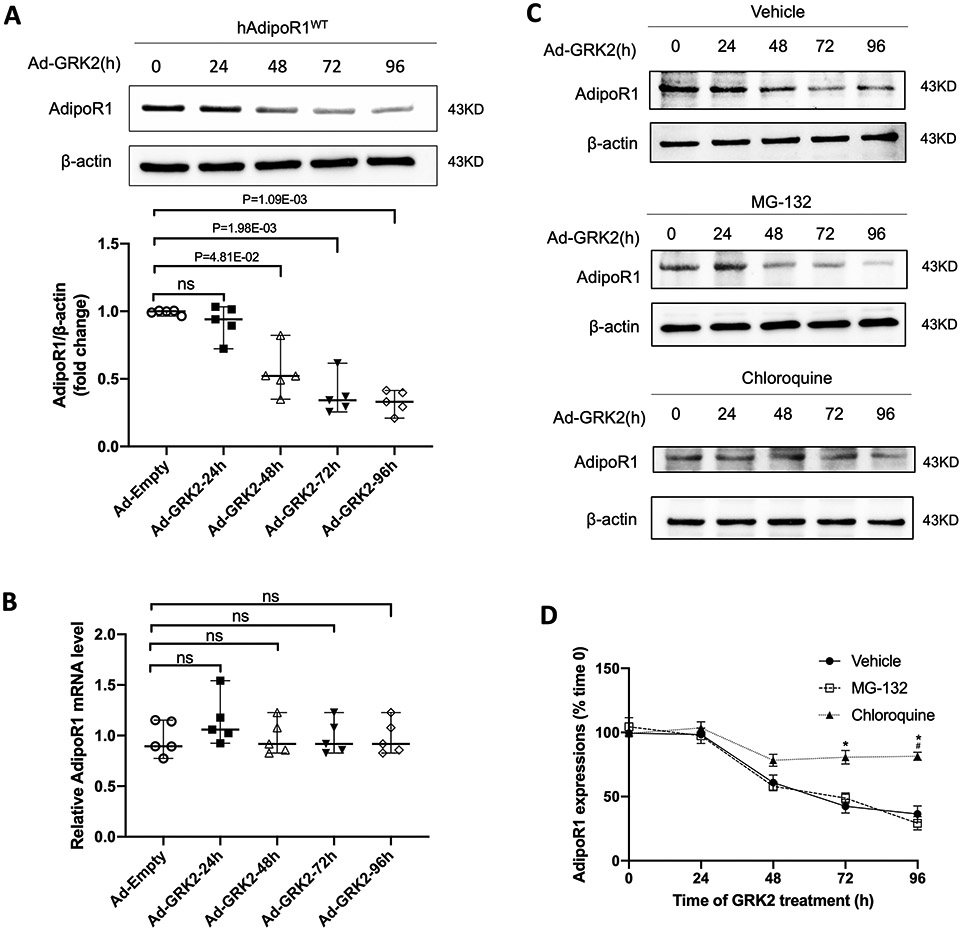
GRK2 promoted AdipoR1 degradation via lysosomal degradation in a time-dependent manner. A: Western blot was performed to analyze AdipoR1 protein expressions in AdipoR1-KO/hAdipoR1WT NMVMs infected with Ad-GRK2 for a different time (n=5). B: qPCR was used to determine AdipoR1 mRNA levels in AdipoR1-KO/hAdipoR1WT NMVMs infected with Ad-GRK2 for a different time (n=5). For A and B, statistical significance was evaluated by a Kruskal-Wallis test. Post hoc pairwise tests for indicated group pairs were performed after Dunn correction. Ns indicates not significant. C: The expressions of AdipoR1 by Western blot in AdipoR1-KO/hAdipoR1WT cardiomyocytes infected with Ad-GRK2 for different time and treated with vehicle, MG-132 (5 μg/ml) or chloroquine (200 μM). D: Quantification of Western blot (n=5 for Vehicle, n=4 for MG-132, n=5 for Chloroquine group). Statistical significance was evaluated by a Kruskal-Wallis test within each time point, and all pairwise comparisons were made. Dunn tests were used to correct for multiple comparisons. *p<0.05 Chloroquine vs. Vehicle; #p<0.05 Chloroquine vs. MG-132.
To obtain more evidence supporting this conclusion, LAMP2 (a lysosome marker) and AdipoR1 colocalization and interaction were determined by immunofluorescent staining and co-immunoprecipitation. GRK2 significantly increased LAMP2/AdipoR1 colocalization and interaction in adult cardiomyocytes from AdipoR1KO/AAV9-hAdipoR1WTmice (Figure 6A/B/C), a pathologic effect significantly attenuated in cells from AdipoR1KO/AAV9-hAdipoR1S205A mice (Figure 6A/B/C). Consistently, GRK2 time-dependently reduced AdipoR1 protein expression in adult cardiomyocytes from AdipoR1KO/AAV9-hAdipoR1WT mice, an effect blocked in cells isolated from AdipoR1KO/AAV9-hAdipoR1S205A mice (Figure 6D/E/F). Finally, expressing an AdipoR1 pseudo-phosphorylation mutation (hAdipoR1S205E) in AdipoR1-KO cardiomyocytes (AdipoR1KO/AAV9-hAdipoR1S205E) significantly reduced AdipoR1 expression without GRK2 infection (time 0). GRK2 overexpression failed to further downregulate AdipoR1 expressions in these cells (Figure 6D/E/F).
Figure 6.

AdipoR1S205A mutation blocks GRK2-induced lysosomal degradation of AdipoR1. A: Adult cardiomyocytes isolated from AAV9-hAdipoR1WT or AAV9-hAdipoR1S205A mice (AdipoR1-KO background) were infected with Ad-Empty or Ad-GRK2, and immune-stained against AdipoR1 (red), LAMP2 (green). Nuclei were labeled with DAPI (blue). B: Pearson’s correlation coefficient quantification of red and green channels from A (n=8). Statistical significance was evaluated by a two-way ANOVA. Post hoc pairwise tests for indicated group pairs were performed after Tukey correction. C: AdipoR1/LAMP2 Co-immunoprecipitation: results demonstrated that GRK2OE increased AdipoR1/LAMP2 co-immunoprecipitation in NMVMs expressing WT AdipoR1, an effect attenuated by AdipoR1 S205A mutation. D/E: The expressions of AdipoR1 by Western blot in hAdipoR1S205A or hAdipoR1S205E over-expressed cardiomyocytes infected with Ad-GRK2 for a different time. F: Quantification of Western blots (n=5 for hAdipoR1WT, n=4 for hAdipoR1S205A, n=4 for hAdipoR1WTS205E group). Statistical significance was evaluated by a Kruskal-Wallis test within each time point, and all pairwise comparisons were made. Dunn tests were used to correct for multiple comparisons. *p<0.05 hAdipoR1S205A vs. hAdipoR1WT; #p<0.05 hAdipoR1S205E vs. hAdipoR1WT.
3.4. Blocking AdipoR1 Ser205 phosphorylation restored cardioprotective signaling, reversing pathological remodeling and attenuating HF progression
We and others have previously demonstrated that APN is highly effective in heart failure prevention (APN administration during acute myocardial ischemia). However, our recent study demonstrated that due to AdipoR1 phosphorylation and desensitization, APN is not effective in heart failure therapy (APN administrated 1 week after MI, a time point significant cardiac dysfunction has developed20). To clarify whether blocking AdipoR1 phosphorylation may restore APN cardioprotective signaling, reversing pathological remodeling and blocking heart failure progression, adult AdipoR1-KO/AAV9-hAdipoR1WT and AdipoR1KO/AAV9-hAdipoR1S205A mice were subjected to permanent coronary ligation and treated with APN beginning 1 week after MI. Consistent with our previous finding, administration APN 1 week after MI failed to improve global cardiac function (M mode echocardiography) in AdipoR1-KO/AAV9-hAdipoR1WT mice (Figure 7A-C). However, in AdipoR1KO/AAV9-hAdipoR1S205A mice, APN treatment reversed cardiac function decline (Figure 7A/D/E. Tables S1-4). To better determine the effect of AdipoR1KO/AAV9-hAdipoR1S205A upon heart failure progression, speckle-tracking strain analysis (B mode echocardiography) was performed. Representative 3-dimensional wall velocity diagrams for 3 consecutive cardiac cycles revealed a dramatic reduction in wall velocity across the endocardium in MI mice (Figure 7F). APN treatment improved wall velocity in AdipoR1KO/AAV9-hAdipoR1S205A mice, an effect absent in AdipoR1KO/AAV9-hAdipoR1WT mice (Figure 7F). Strain analysis demonstrated that MI significantly reduced radial and longitudinal strain and strain rate. APN failed to protect in AdipoR1KO/AAV9-hAdipoR1WT mice (Figure 7G/H). However, administration of APN beginning 1 week after MI significantly improved radial and longitudinal strain and strain rate in AdipoR1KO/AAV9-hAdipoR1S205A mice (Figure 7G/I). APN treatment (administrated after HF development) had no significant effect upon cardiac fibrosis (Figure 8A/B), heart/body or lung/body weight ratio (Figure 8C/D), and survival rate (Figure S6) in AdipoR1-KO/AAV9-hAdipoR1WT. In contrast, APN significantly decreased cardiac fibrosis (Figure 8A/B) and heart/body and lung/body weight ratios (Figure 8C/D), and improved survival rate (Figure S6) in AdipoR1-KO/AAV9-hAdipoR1S205A mice. Finally, in WT mice (Figure S7) or AAV9-AdipoR1WT mice (Figure 8E), MI increased AdipoR1 phosphorylation and reduced total AdipoR1 expression (determined 4 weeks after MI). Treatment with paroxetine (a GRK2 inhibitor33, Figure S7) or re-expression of AAV9-AdipoR1S205A (Figure 8E) significantly attenuated MI-induced AdipoR1 phosphorylation and preserved total AdipoR1 expression. Basal AdipoR1 expression (sham) was significantly reduced in AAV9-AdipoR1S205E mice. However, MI had no significant additional effect (Figure 8E).
Figure 7.

AAV9-AdipoR1S205A restores APN’s protective effect on cardiac function following MI). A: Representative echocardiographic images (M-mode tracing) 4 weeks after MI. B-E: Quantitative analysis of cardiac function derived from echocardiography. F: 3D regional wall velocity diagrams (B-mode tracing) of left ventricular endomyocardial strain showing contraction (orange) and relaxation (blue) of 3 consecutive cardiac cycles. G: Vector diagrams showing direction and magnitude of endocardial contraction at midsystole using the parasternal long-axis. H/I: Quantitative analysis of strain and strain rate measured across the left ventricular endocardium. For B/C and H, n=6 mice for Sham, n=7 mice for MI, n=8 mice for MI+APN group. For D/E and I, n=5 mice for Sham, n=9 mice for MI, n=8 mice for MI+APN group. For B-E and H/I, statistical significance was evaluated by a two-way ANOVA with a repeated measures ANOVA (based on GLM). Post hoc all pairwise comparisons were performed after Tukey correction. AW indicates anterior wall; PW, posterior wall. **(yellow)p<0.01, *(yellow)p<0.05 MI+APN vs. Sham; **(blue)p<0.01, *(blue)p<0.05 MI vs. Sham, ##(yellow)p<0.01, #(yellow)p<0.05 MI+APN vs. MI.
Figure 8.

AAV9-AdipoR1S205A enables APN to reduce cardiac fibrosis and exert cardioprotection following MI. A: Representative images of Masson’s trichrome staining of the coronal plane. B: Quantification of fibrotic area (n = 6 mice per group). C: Heart weight–to–body weight ratio (n=8 mice for Sham, n=10 mice for MI, n=10 mice for MI+APN group). D. Lung weight–to–body weight ratio (n=8 mice for Sham, n=10 mice for MI, n=10 mice for MI+APN group). For B-D, statistical significance was evaluated by a two-way ANOVA. Post hoc pairwise tests for indicated group pairs were performed after Tukey correction. Ns indicates not significant. E. AdipoR1/p-Ser Co-immunoprecipitation showed that in AAV9-AdipoR1WT mice, MI increased AdipoR1 phosphorylation and reduced total AdipoR1 expression (determined 4 weeks after MI). Re-expression of AAV9-AdipoR1S205A significantly attenuated MI-induced AdipoR1 phosphorylation and preserved total AdipoR1 expression. Basal AdipoR1 expression (sham) was significantly reduced in AAV9-AdipoR1S205E mice. However, MI had no significant additional effect. F. Graphic illustration. AdipoR1 is phosphorylated at serine 205 and desensitized after MI, followed by endocytosis and lysosomal degradation. AdipoR1S205A mutation restores its function and exerts cardioprotection after MI.
4. Discussion
Incomplete understanding of the molecular mechanisms responsible for the transition from adaptive to pathologic remodeling after MI has hindered the development of effective therapies reversing heart failure. We recently demonstrated that AdipoR1 is phosphorylatively modified by GRK2 in the failing heart, losing its transmembrane signaling function. Importantly, significant AdipoR1 phosphorylative desensitization develops 1 week after MI, a critical time period when adaptive/pathological remodeling transition occurs20. In addition to its essential role in APN signaling, AdipoR1 is critically involved in the transmembrane signaling of other cardiovascular protective molecules8-13, 34. Therefore, the loss of AdipoR1 biological function contributes to the development of pathological remodeling and HF progression.
In this study, we made three novel observations. First, we identified Ser205 as the specific GRK2 phosphorylation site responsible for AdipoR1 desensitization. Whereas GPCRs have an intracellularly localized C-terminal domain, the N-terminal domain of AdipoR1 is intracellular6. The extracellular C-terminal domain of AdipoR1 interacts with APN, and the cytoplasmic N-terminus interacts with multiple intracellular molecules, such as APPL1, APPL2, and neutral ceramidase (nCDase), transferring APN signaling. Our previously published study identified four sites sensitive to GRK2 phosphorylation. Of these, Ser7 and Thr24 are located in the intracellular N-terminal domain. Ser201 is located in the first intracellular loop region, and Ser205 is located at the turning point from the first intracellular loop to helix III of AdipoR135. It is well-recognized that GRK2 phosphorylates β-AR at its C-terminal intracellular domain, desensitizing β-AR and blocking signaling. However, our study failed to demonstrate the two N-terminal located phosphorylation sites (Ser7 and Thr24) as being responsible for GRK2 desensitization of AdipoR1. Similarly, Ser201 mutation had no protective effect upon GRK2 suppression of AdipoR1 signaling. In contrast, alanine substitution of Ser205 blocked GRK2-induced AdipoR1 desensitization, and rescued AdipoR1 signaling function and AdipoR1-mediated cytoprotection. Moreover, Ser205 pseudo-phosphorylation phenocopied GRK2 effect, blocking APN signaling and cytoprotection. Our identification of this crucial amino acid site not only enhances understanding of AdipoR1-mediated signaling, but may also aid future precision treatment development.
The structural mechanisms responsible for Ser205 mediated phosphorylative desensitization of AdipoR1 cannot be answered by the current study. A recent study demonstrate that the N-terminal domain of AdipoR1 is dispensable in its transmembrane signaling function35, likely explaining why Ser7 and Thr24 are not the sites responsible for GRK2 desensitization of AdipoR1. In both the AdipoR1 and AdipoR2 structures, the seven transmembrane helices surround a large internal cavity, extending from the cytoplasmic surface to the middle of the outer lipid layer of the membrane. This structure is predicted to play critical role in receptor interaction with its intracellular partners, initiating transmembrane signaling35. In AdipoR1, the cavity is formed between helices VII-I-II-III. It is plausible that phosphorylation of Ser205, a residue located at the turning point of the first intracellular loop and the helix III of AdipoR1, may alter the cavity structure, therefore blocking signaling transduction. This important question will be directly addressed in our future investigation.
Second, we demonstrated that clathrin-dependent endocytosis followed by lysosomal-mediated degradation is responsible for AdipoR1 phosphorylative desensitization. It is well-recognized that β-AR phosphorylation alters its affinity, reducing G protein interaction and increasing β-arrestin interaction36-38. Previous studies suggest that G protein might be involved in AdipoR1 signaling39-47. However, we failed to demonstrate β-arrestin recruitment after AdipoR1 phosphorylation, suggesting that affinity-switch (seen in β-AR) may not be the responsible mechanism for AdipoR1 phosphorylative desensitization. Endocytosis is another major mechanism for membrane receptor desensitization. Taking several approaches, we provide evidence that receptor internalization followed by degradation is responsible for AdipoR1 phosphorylative desensitization. First, the SNAP-Surface system is an established method utilized in membrane protein internalization investigation48. Utilizing this method, we demonstrated that GRK2 overexpression significantly promoted AdipoR1 internalization in NMVMs, an effect significantly attenuated by SerS205A mutation. Second, in adult cardiomyocytes from AdipoR1-KO/AAV9-hAdipoR1WT mice, GRK2 significantly increased AdipoR1/Rab5 interaction. This effect was markedly reduced in adult cardiomyocytes from AdipoR1-KO/AAV9-hAdipoR1S205A mice. Third, clathrin-dependent and caveolae-dependent receptor internalization are two primary pathways mediating receptor internalization. We demonstrate that treatment with chlorpromazine (a clathrin-dependent endocytosis inhibitor) significantly inhibited GRK2-induced AdipoR1 endocytosis. Fourth, utilizing Duolink in situ proximity ligation assay, we observed that GRK2 significantly increased AdipoR1/clathrin interaction in adult cardiomyocytes isolated from AdipoR1-KO/AAV9-hAdipoR1WT mice. This pathologic effect was significantly reduced in adult cardiomyocytes from AdipoR1-KO/AAV9-hAdipoR1S205A mice. Fifth, proteasome and lysosome pathways are critically important in mediating protein degradation. We demonstrated that a lysosome inhibitor blocked GRK2-induced AdipoR1 degradation and upregulated AdipoR1 protein expression. Finally, re-expressing a pseudo-phosphorylation hAdipoR1S205E in AdipoR1-KO cardiomyocytes significantly reduced AdipoR1 protein expression without GRK2 overexpression. These results fill a knowledge gap in our understanding of AdipoR1 malfunction in HF progression. Additionally, these results suggest that interventions blocking AdipoR1 endocytosis and degradation may have therapeutic value against ischemic HF.
Finally, and most importantly, re-expressing AdipoR1S205A in AdipoR1-KO mice generated a mouse line resistant to GRK2-induced AdipoR1 phosphorylation, restoring APN cardioprotection. Consistent with our previous report, APN administration 1 week after MI failed to protect in AdipoR1-KO mice re-expressing WT AdipoR1. However, in cardiomyocyte-specific AdipoR1S205A re-expressing mice, APN administration reversed post-MI pathological remodeling, as evidenced by significantly reduced cardiac fibrosis, reduced weight ratios of heart/body or lung/body, and improved global and regional cardiac function. These results additionally support the role of AdipoR1 phosphorylation in post-MI pathological remodeling and HF progression. Moreover, these results call for integrative therapy, such as genetic/pharmacological inhibition of GRK2 in combination with APN administration for the most efficacious treatment of post-ischemic HF.
It should be noted the AdipoR1S205A mutation effectively blocked AdipoR1 endocytosis and subsequent degradation. However, re-expressing hAdipoR1S205A in AdipoR1-KO mice itself (without APN administration) failed to significantly mitigate HF progression. At least two possibilities exist. First, permanent coronary artery ligation results in very severe ischemic damage, making modest protective intervention ineffective. Whether re-expressing hAdipoR1S205A in AdipoR1-KO mice may protect against myocardial ischemia/reperfusion injury warrants further investigation. Second, experimental studies and clinical observations demonstrate that adipocyte APN production is significantly suppressed, and hypoadiponectinemia develops after MI49, 50. Therefore, the endogenous ligand for AdipoR1 is globally reduced in ischemic HF, losing signaling despite a functional receptor.
In summary, we provide the first direct evidence that Ser205 is responsible for AdipoR1 phosphorylative desensitization in the failing heart, contributing to post-MI maladaptive transition. Integrative interventions blocking AdipoR1 phosphorylation followed by pharmacologic APN administration may be effective novel avenues reversing post-MI remodeling and attenuating HF progression (Figure 8F).
Supplementary Material
Novelty and Significance.
What is known?
Adiponectin receptor 1 (AdipoR1) is phosphorylated and desensitized by G protein-coupled receptor kinase 2 (GRK2) in failing cardiomyocytes.
Phosphorylated AdipoR1 does not have a transmembrane protective signaling function, potentially contributing to post-MI remodeling and heart failure (HF) progression.
There are 4 sites (Ser7, Thr24, Ser201, and Ser205) in AdipoR1 sensitive to GRK2 phosphorylation.
What new information does this article contribute?
GRK2-induced AdipoR1 phosphorylation at serine205 is responsible for AdipoR1 desensitization in the failing heart.
AdipoR1 phosphorylation promoted clathrin-dependent endocytosis followed by lysosomal-mediated degradation.
Re-expressing AdipoR1S205A in AdipoR1KO cardiomyocytes blocked GRK2-induced AdipoR1 endocytosis/degradation and preserved adiponectin protective signaling, reversing MI-induced pathological remodeling.
The present study provides evidence that Ser205 is responsible for AdipoR1 phosphorylative desensitization in the failing heart, contributing to a post-MI maladaptive transition. Blocking AdipoR1 phosphorylation followed by pharmacologic adiponectin administration may be an effective and new target for reversing post-MI remodeling and attenuating HF progression.
Funding Sources
This work was supported by awards from the National Institutes of Health (HL-96686, X. Ma/Y. Wang, MPI; HL-123404, X. Ma; HL158612, Y. Wang) and the American Heart Association (20TPA35490095, Y. Wang).
Non-standard Abbreviations and Acronyms
- AdipoR
adiponectin receptor
- AdipoR1-KO
adiponectin receptor 1 knockout
- AMI
acute myocardial infarction
- APN
adiponectin
- AP2
adaptor protein-2
- GPCRs
G protein-coupled receptors
- GRK2
G protein-coupled receptor kinase 2
- hAdipoR1
human adiponectin receptor 1
- HF
heart failure
- LVEF
left ventricular ejection fraction
- LVFS
left ventricular fraction shortening
- MI
myocardial infarction
- nCDase
neutral ceramidase
- NMVMs
neonatal mouse ventricular myocytes
- PAQR
progestin and adipoQ receptor
- PLA
proximity ligation assay
- WT
wild type
Footnotes
Disclosures
The authors declare no competing interests, financial or otherwise.
References
- 1.Virani SS, Alonso A, Benjamin EJ, Bittencourt MS, Callaway CW, Carson AP, Chamberlain AM, Chang AR, Cheng S, Delling FN, et al. Heart Disease and Stroke Statistics-2020 Update: A Report From the American Heart Association. Circulation. 2020;141:e139–e596. [DOI] [PubMed] [Google Scholar]
- 2.Davidson SM, Ferdinandy P, Andreadou I, Botker HE, Heusch G, Ibanez B, Ovize M, Schulz R, Yellon DM, Hausenloy DJ, et al. Multitarget Strategies to Reduce Myocardial Ischemia/Reperfusion Injury: JACC Review Topic of the Week. J Am Coll Cardiol. 2019;73:89–99. [DOI] [PubMed] [Google Scholar]
- 3.Heusch G and Gersh BJ. Is Cardioprotection Salvageable? Circulation. 2020;141:415–417. [DOI] [PubMed] [Google Scholar]
- 4.Zhao S, Kusminski CM and Scherer PE. Adiponectin, Leptin and Cardiovascular Disorders. Circ Res. 2021;128:136–149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Yamauchi T, Kamon J, Ito Y, Tsuchida A, Yokomizo T, Kita S, Sugiyama T, Miyagishi M, Hara K, Tsunoda M, et al. Cloning of adiponectin receptors that mediate antidiabetic metabolic effects. Nature. 2003;423:762–769. [DOI] [PubMed] [Google Scholar]
- 6.Kadowaki T and Yamauchi T. Adiponectin and Adiponectin Receptors. Endocrine Reviews. 2005;26:439–451. [DOI] [PubMed] [Google Scholar]
- 7.Goldstein BJ, Scalia RG and Ma XL. Protective vascular and myocardial effects of adiponectin. Nat Clin Pract Cardiovasc Med. 2009;6:27–35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Wu W, Zhang J, Zhao C, Sun Y, Pang W and Yang G. CTRP6 Regulates Porcine Adipocyte Proliferation and Differentiation by the AdipoR1/MAPK Signaling Pathway. J Agric Food Chem. 2017;65:5512–5522. [DOI] [PubMed] [Google Scholar]
- 9.Kambara T, Shibata R, Ohashi K, Matsuo K, Hiramatsu-Ito M, Enomoto T, Yuasa D, Ito M, Hayakawa S, Ogawa H, et al. C1q/Tumor Necrosis Factor-Related Protein 9 Protects against Acute Myocardial Injury through an Adiponectin Receptor I-AMPK-Dependent Mechanism. Mol Cell Biol. 2015;35:2173–2185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Zheng Q, Yuan Y, Yi W, Lau WB, Wang Y, Wang X, Sun Y, Lopez BL, Christopher TA, Peterson JM, et al. C1q/TNF-Related Proteins, A Family of Novel Adipokines, Induce Vascular Relaxation Through the Adiponectin Receptor-1/AMPK/eNOS/Nitric Oxide Signaling Pathway. Arterioscler Thromb Vasc Biol. 2011;31:2616–2623. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Yang S, Ma C, Wu H, Zhang H, Yuan F, Yang G, Yang Q, Jia L, Liang Z and Kang L. Tectorigenin attenuates diabetic nephropathy by improving vascular endothelium dysfunction through activating AdipoR1/2 pathway. Pharmacol Res. 2020;153:104678. [DOI] [PubMed] [Google Scholar]
- 12.Ji H, Wu L, Ma X, Ma X and Qin G. The effect of resveratrol on the expression of AdipoR1 in kidneys of diabetic nephropathy. Mol Biol Rep. 2014;41:2151–9. [DOI] [PubMed] [Google Scholar]
- 13.Li H, Yao W, Liu Z, Xu A, Huang Y, Ma XL, Irwin MG and Xia Z. Hyperglycemia Abrogates Ischemic Postconditioning Cardioprotection by Impairing AdipoR1/Caveolin-3/STAT3 Signaling in Diabetic Rats. Diabetes. 2016;65:942–55. [DOI] [PubMed] [Google Scholar]
- 14.Ruiz M, Stahlman M, Boren J and Pilon M. AdipoR1 and AdipoR2 maintain membrane fluidity in most human cell types and independently of adiponectin. J Lipid Res. 2019;60:995–1004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Mado H, Szczurek W, Gasior M and Szygula-Jurkiewicz B. Adiponectin in heart failure. Future Cardiol. 2020. [DOI] [PubMed] [Google Scholar]
- 16.Shimano M, Ouchi N, Shibata R, Ohashi K, Pimentel DR, Murohara T and Walsh K. Adiponectin deficiency exacerbates cardiac dysfunction following pressure overload through disruption of an AMPK-dependent angiogenic response. J Mol Cell Cardiol. 2010;49:210–220. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.George J, Patal S, Wexler D, Sharabi Y, Peleg E, Kamari Y, Grossman E, Sheps D, Keren G and Roth A. Circulating adiponectin concentrations in patients with congestive heart failure. Heart. 2006;92:1420–1424. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Van Berendoncks AM, Garnier A, Beckers P, Hoymans VY, Possemiers N, Fortin D, Van Hoof V, Dewilde S, Vrints CJ, Ventura-Clapier Re and Conraads VM. Exercise training reverses adiponectin resistance in skeletal muscle of patients with chronic heart failure. Heart. 2011;97:1403–1409. [DOI] [PubMed] [Google Scholar]
- 19.Nakamura T, Funayama H, Kubo N, Yasu T, Kawakami M, Saito M, Momomura S and Ishikawa SE. Association of hyperadiponectinemia with severity of ventricular dysfunction in congestive heart failure. Circ J. 2006;70:1557–1562. [DOI] [PubMed] [Google Scholar]
- 20.Wang Y, Gao E, Lau WB, Wang Y, Liu G, Li JJ, Wang X, Yuan Y, Koch WJ and Ma XL. G-protein-coupled receptor kinase 2-mediated desensitization of adiponectin receptor 1 in failing heart. Circulation. 2015;131:1392–1404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Huang ZP, Kataoka M, Chen J, Wu G, Ding J, Nie M, Lin Z, Liu J, Hu X, Ma L, et al. Cardiomyocyte-enriched protein CIP protects against pathophysiological stresses and regulates cardiac homeostasis. J Clin Invest. 2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Yan W, Guo Y, Tao L, Lau WB, Gan L, Yan Z, Guo R, Gao E, Wong GW, Koch WL, et al. C1q/Tumor Necrosis Factor-Related Protein-9 Regulates the Fate of Implanted Mesenchymal Stem Cells and Mobilizes Their Protective Effects Against Ischemic Heart Injury via Multiple Novel Signaling Pathways. Circulation. 2017;136:2162–2177. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Wang Y, Wang X, Lau WB, Yuan Y, Booth D, Li JJ, Scalia R, Preston K, Gao E, Koch W et al. Adiponectin inhibits tumor necrosis factor-alpha-induced vascular inflammatory response via caveolin-mediated ceramidase recruitment and activation. Circ Res. 2014;114:792–805. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Lobysheva I, Rath G, Sekkali B, Bouzin C, Feron O, Gallez B, Dessy C and Balligand JL. Moderate caveolin-1 downregulation prevents NADPH oxidase-dependent endothelial nitric oxide synthase uncoupling by angiotensin II in endothelial cells. Arterioscler Thromb Vasc Biol. 2011;31:2098–2105. [DOI] [PubMed] [Google Scholar]
- 25.Huang ZM, Gold JI and Koch WJ. G protein-coupled receptor kinases in normal and failing myocardium. Front Biosci (Landmark Ed). 2011;16:3047–60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Ding Q, Wang Z and Chen Y. Endocytosis of adiponectin receptor 1 through a clathrin- and Rab5-dependent pathway. Cell Res. 2009;19:317–327. [DOI] [PubMed] [Google Scholar]
- 27.Chaudhary N, Gomez GA, Howes MT, Lo HP, McMahon KA, Rae JA, Schieber NL, Hill MM, Gaus K, Yap AS, et al. Endocytic crosstalk: cavins, caveolins, and caveolae regulate clathrin-independent endocytosis. PLoS Biol. 2014;12:e1001832. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Eguchi S, Takefuji M, Sakaguchi T, Ishihama S, Mori Y, Tsuda T, Takikawa T, Yoshida T, Ohashi K, Shimizu Y, et al. Cardiomyocytes capture stem cell-derived, anti-apoptotic microRNA-214 via clathrin-mediated endocytosis in acute myocardial infarction. J Biol Chem. 2019;294:11665–11674. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Matsuura S, Katsumi H, Suzuki H, Hirai N, Hayashi H, Koshino K, Higuchi T, Yagi Y, Kimura H, Sakane T, et al. l-Serine-modified polyamidoamine dendrimer as a highly potent renal targeting drug carrier. Proc Natl Acad Sci U S A. 2018;115:10511–10516. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Wrobel AG, Kadlecova Z, Kamenicky J, Yang JC, Herrmann T, Kelly BT, McCoy AJ, Evans PR, Martin S, Muller S, et al. Temporal Ordering in Endocytic Clathrin-Coated Vesicle Formation via AP2 Phosphorylation. Dev Cell. 2019;50:494–508 e11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Harhouri K, Navarro C, Depetris D, Mattei MG, Nissan X, Cau P, De Sandre-Giovannoli A and Levy N. MG132-induced progerin clearance is mediated by autophagy activation and splicing regulation. EMBO Mol Med. 2017;9:1294–1313. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Ahsan A, Zheng Y, Ma S, Liu M, Cao M, Li Y, Zheng W, Zhou X, Xin M, Hu WW, et al. Tomatidine protects against ischemic neuronal injury by improving lysosomal function. Eur J Pharmacol. 2020;882:173280. [DOI] [PubMed] [Google Scholar]
- 33.Schumacher SM, Gao E, Zhu W, Chen X, Chuprun JK, Feldman AM, JJ GT and Koch WJ. Paroxetine-mediated GRK2 inhibition reverses cardiac dysfunction and remodeling after myocardial infarction. Sci Transl Med. 2015;7:277ra31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Zhao L, Zhang JH, Sherchan P, Krafft PR, Zhao W, Wang S, Chen S, Guo Z and Tang J. Administration of rCTRP9 Attenuates Neuronal Apoptosis Through AdipoR1/PI3K/Akt Signaling Pathway after ICH in Mice. Cell Transplant. 2019;28:756–766. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Tanabe H, Fujii Y, Okada-Iwabu M, Iwabu M, Nakamura Y, Hosaka T, Motoyama K, Ikeda M, Wakiyama M, Terada T, et al. Crystal structures of the human adiponectin receptors. Nature. 2015;520:312–316. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Shenoy SK, Drake MT, Nelson CD, Houtz DA, Xiao K, Madabushi S, Reiter E, Premont RT, Lichtarge O and Lefkowitz RJ. +¦-Arrestin-dependent, G Protein-independent ERK1/2 Activation by the +¦2 Adrenergic Receptor. Journal of Biological Chemistry. 2006;281:1261–1273. [DOI] [PubMed] [Google Scholar]
- 37.Gesty-Palmer D, Chen M, Reiter E, Ahn S, Nelson CD, Wang S, Eckhardt AE, Cowan CL, Spurney RF, Luttrell LM, et al. Distinct +¦-Arrestin- and G Protein-dependent Pathways for Parathyroid Hormone Receptor-stimulated ERK1/2 Activation. Journal of Biological Chemistry. 2006;281:10856–10864. [DOI] [PubMed] [Google Scholar]
- 38.Ahn S, Nelson CD, Garrison TR, Miller WE and Lefkowitz RJ. Desensitization, internalization, and signaling functions of +¦-arrestins demonstrated by RNA interference. Proceedings of the National Academy of Sciences. 2003;100:1740–1744. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Ouchi N, Kihara S, Arita Y, Okamoto Y, Maeda K, Kuriyama H, Hotta K, Nishida M, Takahashi M, Muraguchi M, et al. Adiponectin, an adipocyte-derived plasma protein, inhibits endothelial NF-kappaB signaling through a cAMP-dependent pathway. Circulation. 2000;102:1296–1301. [DOI] [PubMed] [Google Scholar]
- 40.Ouedraogo R, Wu X, Xu SQ, Fuchsel L, Motoshima H, Mahadev K, Hough K, Scalia R and Goldstein BJ. Adiponectin suppression of high-glucose-induced reactive oxygen species in vascular endothelial cells: evidence for involvement of a cAMP signaling pathway. Diabetes. 2006;55:1840–1846. [DOI] [PubMed] [Google Scholar]
- 41.Xu SQ, Mahadev K, Wu X, Fuchsel L, Donnelly S, Scalia RG and Goldstein BJ. Adiponectin protects against angiotensin II or tumor necrosis factor alpha-induced endothelial cell monolayer hyperpermeability: role of cAMP/PKA signaling. Arterioscler Thromb Vasc Biol. 2008;28:899–905. [DOI] [PubMed] [Google Scholar]
- 42.Zhao T, Hou M, Xia M, Wang Q, Zhu H, Xiao Y, Tang Z, Ma J and Ling W. Globular adiponectin decreases leptin-induced tumor necrosis factor-alpha expression by murine macrophages: Involvement of cAMP-PKA and MAPK pathways. Cell Immunol. 2006;238:19–30. [DOI] [PubMed] [Google Scholar]
- 43.Tang YT, Hu T, Arterburn M, Boyle B, Bright JM, Emtage PC and Funk WD. PAQR proteins: a novel membrane receptor family defined by an ancient 7-transmembrane pass motif. J Mol Evol. 2005;61:372–380. [DOI] [PubMed] [Google Scholar]
- 44.Pang Y, Dong J and Thomas P. Characterization, neurosteroid binding and brain distribution of human membrane progesterone receptors delta and {epsilon} (mPRdelta and mPR{epsilon}) and mPRdelta involvement in neurosteroid inhibition of apoptosis. Endocrinology. 2013;154:283–295. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Gonzalez-Velazquez W, Gonzalez-Mendez R and Rodriguez-del VN. Characterization and ligand identification of a membrane progesterone receptor in fungi: existence of a novel PAQR in Sporothrix schenckii. BMC Microbiol. 2012;12:194. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Thomas P and Pang Y. Membrane progesterone receptors: evidence for neuroprotective, neurosteroid signaling and neuroendocrine functions in neuronal cells. Neuroendocrinology. 2012;96:162–171. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Thomas P, Pang Y, Dong J, Groenen P, Kelder J, de VJ, Zhu Y and Tubbs C. Steroid and G protein binding characteristics of the seatrout and human progestin membrane receptor alpha subtypes and their evolutionary origins. Endocrinology. 2007;148:705–718. [DOI] [PubMed] [Google Scholar]
- 48.Bitsikas V, Correa IR Jr. and Nichols BJ. Clathrin-independent pathways do not contribute significantly to endocytic flux. Elife. 2014;3:e03970. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Shibata R, Numaguchi Y, Matsushita K, Sone T, Kubota R, Ohashi T, Ishii M, Kihara S, Walsh K, Ouchi N, et al. Usefulness of adiponectin to predict myocardial salvage following successful reperfusion in patients with acute myocardial infarction. Am J Cardiol. 2008;101:1712–1715. [DOI] [PubMed] [Google Scholar]
- 50.Wang Y, Zhao J, Zhang Y, Lau WB, Jiao LY, Liu B, Yuan Y, Wang X, Tao L, Gao E, et al. Differential regulation of TNF receptor 1 and receptor 2 in adiponectin expression following myocardial ischemia. Int J Cardiol. 2013;168:2201–2206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Duran JM, Makarewich CA, Sharp TE, Starosta T, Zhu F, Hoffman NE, Chiba Y, Madesh M, Berretta RM, Kubo H, et al. Bone-derived stem cells repair the heart after myocardial infarction through transdifferentiation and paracrine signaling mechanisms. Circ Res. 2013;113:539–52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Yap L, Wang JW, Moreno-Moral A, Chong LY, Sun Y, Harmston N, Wang X, Chong SY, Vanezis K, Ohman MK, et al. In Vivo Generation of Post-infarct Human Cardiac Muscle by Laminin-Promoted Cardiovascular Progenitors. Cell Rep. 2019;26:3231–3245 e9. [DOI] [PubMed] [Google Scholar]
- 53.Wang WE, Yang D, Li L, Wang W, Peng Y, Chen C, Chen P, Xia X, Wang H, Jiang J, et al. Prolyl hydroxylase domain protein 2 silencing enhances the survival and paracrine function of transplanted adipose-derived stem cells in infarcted myocardium. Circ Res. 2013;113:288–300. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Ackers-Johnson M, Li PY, Holmes AP, O'Brien SM, Pavlovic D and Foo RS. A Simplified, Langendorff-Free Method for Concomitant Isolation of Viable Cardiac Myocytes and Nonmyocytes From the Adult Mouse Heart. Circ Res. 2016;119:909–20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Reddy GR, West TM, Jian Z, Jaradeh M, Shi Q, Wang Y, Chen-Izu Y and Xiang YK. Illuminating cell signaling with genetically encoded FRET biosensors in adult mouse cardiomyocytes. J Gen Physiol. 2018;150:1567–1582. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Kabaeva Z, Zhao M and Michele DE. Blebbistatin extends culture life of adult mouse cardiac myocytes and allows efficient and stable transgene expression. Am J Physiol Heart Circ Physiol. 2008;294:H1667–74. [DOI] [PubMed] [Google Scholar]
- 57.Wang Y, Lau WB, Gao E, Tao L, Yuan Y, Li R, Wang X, Koch WJ and Ma XL. Cardiomyocyte-derived adiponectin is biologically active in protecting against myocardial ischemia-reperfusion injury. Am J Physiol Endocrinol Metab. 2010;298:E663–70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Zinchuk V, Wu Y and Grossenbacher-Zinchuk O. Bridging the gap between qualitative and quantitative colocalization results in fluorescence microscopy studies. Sci Rep. 2013;3:1365. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Zhang F, Xia Y, Yan W, Zhang H, Zhou F, Zhao S, Wang W, Zhu D, Xin C, Lee Y, et al. Sphingosine 1-phosphate signaling contributes to cardiac inflammation, dysfunction, and remodeling following myocardial infarction. Am J Physiol Heart Circ Physiol. 2016;310:H250–61. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The data that support the findings of this study are available from the corresponding author upon reasonable request.
Detailed methods for cell isolation and culture, plasmids constructions and transfections, adeno-associated virus 9 vector production and infection, cell viability and apoptosis, Western and Co-immunoprecipitation, echocardiography and strain analysis, immunofluorescent cellular and Masson’s trichrome staining, and quantitative PCR methods are provided in the Supplemental Material. Key research materials are listed in the Major Resources Table in the Supplemental Material. Representative images for cellular and animal experiments and representative western blot images were chosen to closely match the mean for the parameters assessed.
Major Resources Table.
In order to allow validation and replication of experiments, all essential research materials listed in the Methods should be included in the Major Resources Table below. Authors are encouraged to use public repositories for protocols, data, code, and other materials and provide persistent identifiers and/or links to repositories when available. Authors may add or delete rows as needed.
| Animals (in vivo studies) | ||||
|---|---|---|---|---|
| Species | Vendor or Source |
Background Strain | Sex | Persistent ID / URL |
| Mus musculus | MMRRC | B6.129P2-Adipor1tm1Dgen, C57BL/6J | F and M | MMRRC Strain #11599, https://www.mmrrc.org/catalog/sds.php?mmrrc_id=11599 |
| Genetically Modified Animals | |||||
|---|---|---|---|---|---|
| Species | Vendor or Source |
Background Strain |
Other Information | Persistent ID / URL | |
| Parent - Male | |||||
| Parent - Female | |||||
| Antibodies | |||||
|---|---|---|---|---|---|
| Target antigen | Vendor or Source |
Catalog # | Working concentration |
Lot # (preferred but not required) |
Persistent ID / URL |
| Anti-phospho-ERK1/2 rabbit monoclonal antibody | Cell Signaling Technology | 4370 | 1:2000 | https://www.cellsignal.com/product/productDetail.jsp?productId=4370 | |
| Anti-ERK1/2 rabbit monoclonal antibody | Cell Signaling Technology | 4695 | 1:1000 | https://www.cellsignal.com/product/productDetail.jsp?productId=4695 | |
| Anti-phospho-AMPKα rabbit polyclonal antibody | Cell Signaling Technology | 2531 | 1:1000 | https://www.cellsignal.com/product/productDetail.jsp?productId=2531 | |
| Anti-AMPKα rabbit monoclonal antibody | Cell Signaling Technology | 2603 | 1:1000 | https://www.cellsignal.com/product/productDetail.jsp?productId=2603 | |
| Anti-phospho-ACC rabbit monoclonal antibody | Cell Signaling Technology | 11818 | 1:1000 | https://www.cellsignal.com/product/productDetail.jsp?productId=11818 | |
| Anti-ACC rabbit polyclonal antibody | Cell Signaling Technology | 3662 | 1:1000 | https://www.cellsignal.com/product/productDetail.jsp?productId=3662 | |
| Anti-cleaved caspase-3 rabbit polyclonal antibody | Cell Signaling Technology | 9661 | 1:1000 | https://www.cellsignal.com/product/productDetail.jsp?productId=9661 | |
| Anti-caspase-3 rabbit polyclonal antibody | Cell Signaling Technology | 9662 | 1:1000 | https://www.cellsignal.com/product/productDetail.jsp?productId=9662 | |
| Anti-phospho-AP2M1 rabbit monoclonal antibody | Cell Signaling Technology | 7399 | 1:1000 | https://www.cellsignal.com/product/productDetail.jsp?productId=7399 | |
| Anti-AP2M1 rabbit monoclonal antibody | Abcam | ab75995 | 1:2000 | https://www.abcam.com/ap2m1-antibody-ep2695y-ab75995.html | |
| Anti-GRK2 rabbit polyclonal antibody | Cell Signaling Technology | 3982s | 1:1000 | https://www.cellsignal.com/product/productDetail.jsp?productId=3982s | |
| Anti-adipoR1 mouse monoclonal antibody | Santa Cruz Biotechnolog y | sc-518030 | 1:500 | https://www.scbt.com/p/adipor1-antibody-d-9?productCanUrl=adipor1-antibody-d-9&_requestid=5867389 | |
| Anti-β-arrestin1/2 rabbit monoclonal antibody | Cell Signaling Technology | 4674 | 1:1000 | https://www.cellsignal.com/product/productDetail.jsp?productId=4674 | |
| Anti-GFP rabbit polyclonal antibody | Genscript | A01704 | 1:500 | https://www.genscript.com/antibody/A01704-THE_GFP_Antibody_pAb_Rabbit.html | |
| Anti-β-actin mouse monoclonal antibody | Santa Cruz Biotechnolog y | sc-47778 | 1:200 | https://www.scbt.com/p/beta-actin-antibody-c4?requestFrom=search | |
| Anti-GAPDH rabbit monoclonal antibody | Cell Signaling Technology | 2118 | 1:1000 | https://www.cellsignal.com/product/productDetail.jsp?productId=2118 | |
| Anti-flag M2 rabbit monoclonal antibody | Sigma-Aldrich | F1804 | 1:2000 | https://www.sigmaaldrich.com/US/en/product/sigma/f1804 | |
| Anti-Rab5 rabbit monoclonal antibody | Cell Signaling Technology | 3547 | 1:200 | https://www.cellsignal.com/product/productDetail.jsp?productId=3547 | |
| Anti-LAMP2 rabbit polyclonal Antibody | Bioss | BS-2379R | 1:100 | https://www.biossusa.com/products/bs-2379r | |
| Anti-p-Ser/Phosphoserine mouse monoclonal antibody | Santa Cruz Biotechnology | sc-81514 | 1:1000 | https://www.scbt.com/p/p-ser-antibody-16b4 | |
| Secondary HRP-conjugated anti-mouse antibody | Cell Signaling Technology | 7076 | 1:1000 | https://www.cellsignal.com/product/productDetail.jsp?productId=7076 | |
| Secondary HRP-conjugated anti-rabbit antibody | Cell Signaling Technology | 7074 | 1:1000 | https://www.cellsignal.com/product/productDetail.jsp?productId=7074 | |
| Goat anti-Mouse IgG (H+L), Alexa Fluor 546 | ThermoFisher Scientific | A-11003 | 1:500 | https://www.thermofisher.com/antibody/product/Goat-anti-Mouse-IgG-HL-Cross-Adsorbed-Secondary-AntibodyPolyclonal/A-11003 | |
| Goat Anti-Rabbit IgG H&L (Cy5) | Abcam | Ab6564 | 1:2000 | https://www.abcam.com/goat-rabbit-igg-hl-cy5--preadsorbed-ab6564.html#minReactivity | |
| DNA/cDNA Clones | |||
|---|---|---|---|
| Clone Name | Sequence | Source / Repository | Persistent ID / URL |
| 3xFlag-hAdipoR1 | Human AdipoR1 cDNA | This paper/In house | |
| AAV9-cTNT-eGFP plasmid | Available at URL | A gift from Dazhi Wang | https://www.addgene.org/105543/ |
| AAV helper plasmid pAdDeltaF6 | Available at URL | A gift from Dazhi Wang | https://www.addgene.org/112867/ |
| AAV packaging plasmid pAAV2/9n | Available at URL | A gift from Dazhi Wang | https://www.addgene.org/112865/ |
| pSNAPf vector | Available at URL | New England Biolabs | https://www.neb.com/products/n9183-psnapf-vector#Product%20Information |
| Cultured Cells | |||
|---|---|---|---|
| Name | Vendor or Source | Sex (F, M, or unknown) | Persistent ID / URL |
| Primary adult cardiomyocyte | Primary cells | F and M | |
| Primary neonatal mouse ventricular myocyte | Primary cells | F and M | |
| Data & Code Availability | ||
|---|---|---|
| Description | Source / Repository | Persistent ID / URL |