

Abstract
Background & Aims
Beyond the classical description of eosinophil functions in parasite infections and allergic diseases, emerging evidence supports a critical role of eosinophils in resolving inflammation and promoting tissue remodeling. However, the role of eosinophils in liver injury and the underlying mechanism of their recruitment into the liver remain unclear.
Methods
Hepatic eosinophils were detected and quantified using flow cytometry and immunohistochemical staining. Eosinophil-deficient (ΔdblGata1) mice were used to investigate the role of eosinophils in three models of acute liver injury. In vivo experiments using IL-33−/− mice and macrophage-depleted mice, as well as in vitro cultures of eosinophils and macrophages, were performed to interrogate the mechanism of eotaxin-2 (CCL24) production.
Results
Hepatic accumulation of eosinophils was observed in patients with acetaminophen (APAP)-induced liver failure, whereas few eosinophils were detectable in healthy liver tissues. In mice treated with APAP, CCl4 or concanavalin A, eosinophils were recruited into the liver and played a profound protective role. Mice deficient of macrophages or IL-33 exhibited impaired hepatic eosinophil recruitment during acute liver injury. CCL24, but not CCL11, was increased after treatment of each hepatotoxin in an IL-33 and macrophage-dependent manner. In vitro experiments demonstrated that IL-33, through stimulating IL-4 release from eosinophils, promoted macrophages to produce CCL24.
Conclusions
This is the first study to demonstrate that hepatic recruitment of and protection by eosinophils occur commonly in various models of acute liver injury. The data further provide insights into the mechanism of hepatic eosinophil recruitment. The findings support further exploration of eosinophils as a therapeutic target to treat APAP-induced acute liver injury.
Keywords: Eosinophils, acute liver injury, IL-33, macrophages, CCL24
Lay summary
The current study unveils that eosinophils are recruited into the liver and play a protective function during acute liver injury caused by APAP overdose. The data demonstrate that IL-33-activated eosinophils trigger macrophages to release high amount of CCL24, which promotes hepatic eosinophil recruitment. The findings implicate the potential of using eosinophils as an effective cell-based therapy for treatment of APAP-induced acute liver injury.
Introduction
Acute liver injury caused by ischemia/reperfusion (IR), hepatitis virus, autoimmunity, or adverse drug reactions can lead to liver failure (1), for which the only treatment at present is liver transplantation. To expand on therapeutic options, it is important to better understand the underlying mechanisms of acute liver injury, especially endogenous protective pathways that can be harnessed to limit tissue damage.
Eosinophils are traditionally thought to be involved in host response against parasite infection and the pathogenesis of allergic diseases (2, 3). However, emerging evidence has ascribed eosinophils to functions of promoting tissue repair and resolving inflammation. It is reported that eosinophils protect against acute lung injury (4), promote resolution of airway inflammation (5), and airway remodeling (6). The understanding of eosinophils’ involvement in liver injury is in its infancy. There are very few published studies and they report contradictory results (7–10). We recently reported that eosinophils were recruited into the liver after IR injury and played a profound hepato-protective function (7). However, it remains unknown how eosinophils are recruited into the liver and what role they play in other acute liver injury conditions.
Although very few eosinophils can be detected in the liver under homeostatic conditions (11), their accumulation in the liver has been observed in several pathological conditions, such as viral hepatitis, hepatic allograft rejection, and drug-induced liver injury (12–14). In addition, the molecular and cellular mechanism accounting for hepatic eosinophil recruitment has not been studied. Eotaxin-1 (CCL11) and eotaxin-2 (CCL24), through acting on CCR3, are the most potent eosinophil chemoattractants. For example, it is reported that the interaction between CCL24 and CCR3 plays a dominant role in eosinophil recruitment to the lung in OVA-induced experimental asthma model (15), whereas CCL11 is more important for basal recruitment of eosinophils to tissues (16). Moreover, both CCL11 and CCL24 contribute to eosinophil infiltration into various types of tumors (17). Interestingly, it has been shown that injection of exogenous IL-33 can significantly increase the number of eosinophils in the blood and various tissues (18, 19). The underlying mechanism, however, is not clear. One study showed that exogenous IL-33 administration could induce CCL24 up-regulation (20). Since IL-33 is an alarmin released during tissue injury, it is a plausible upstream signal in eosinophil recruitment.
In the current study using in vitro and mouse models, we uncovered the critical role of an eosinophil/macrophage cross-talk, through the IL-33/IL-4/CCL24 axis, in eosinophil recruitment into injured livers. Moreover, our data demonstrate a hepato-protective function of infiltrated eosinophils in APAP-induced acute liver injury.
Materials and methods
Human samples
Paraffin-embedded human liver biopsies of explants were obtained from patients diagnosed with APAP-induced liver failure and enrolled in the Acute Liver Failure Study Group (ALFSG). The study was approved by all the study site institutional review boards, including UT Southwestern (STU 062010–126). The bio-repository is maintained by the National Institute of Diabetes, Digestive and Kidney Diseases (NIDDK). All patients and their legally authorized representatives had given informed consent for the release of information and use of available tissue samples. Some of the clinical data are shown in supplemental Table 1. Paraffin-embedded human liver biopsies from healthy individuals were obtained through the Liver Tissue Cell Distribution System at the University of Minnesota, which is funded by National Institutes of Health (NIH contract #HHSN276201200017C).
Animals
Breeders of IL-33fl/fl-eGFP reporter (cat#030619), Balb/c (cat#000651), and ΔdblGata1 mice (cat#005653) were purchased from the Jackson laboratory. Breeders of IL-33−/− mice (on C57BL/6N genetic background) were obtained from J. Brown (University of Colorado Anschutz Medical Campus). C57BL/6N from Taconic Biosciences were used as WT control mice. All colonies were established and maintained in the UTHealth animal facility. All experiments were performed according to the guidelines of the Institutional Animal Care and Use Committee at UTHealth.
Experimental Method
Eight- to ten-weeks old mice were injected with acetaminophen (APAP, i.p.), carbon tetrachloride (CCl4, i.p.), or concanavalin A (ConA, i.v.) to induce acute liver injury. Flow cytometric and immunohistochemistry (IHC) analyses were performed to detect hepatic eosinophils. The degrees of liver injuries were determined by measurements of serum ALT levels and evaluation of liver histopathology. Adoptive transfer of bone marrow-derived eosinophils to ΔdblGata1 mice was performed to investigate the hepato-protective function of these cells. The underlying molecular mechanism of hepatic recruitment of eosinophils was explored by multiple in vivo and in vitro approaches. More detailed methods can be found in the Supplemental Information.
Results
Eosinophils are recruited to the liver after acute injury
Overdose of APAP is a major cause of acute liver failure in the clinic. Although N-acetylcysteine (NAC) can reduce APAP-induced liver injury (AILI), its effectiveness drops sharply around 8h after APAP overdose. Thus, identifying new therapeutic strategies harnessing endogenous protective mechanisms remains an active area of research. To examine if eosinophils are recruited to the liver in patients with APAP-induced liver failure, we obtained liver explant samples and performed IHC staining with an antibody specific for eosinophil peroxidase (EPX). Compared with liver biopsies from healthy individuals, those from patients with AILI displayed a dramatic increase in the number of eosinophils (Fig. 1A, B). Similarly, in a mouse model of AILI, both the percentage and the absolute number of eosinophils in the liver were increased after APAP challenge and peaked at 48h (Fig. 1C). Eosinophils were identified as CD45+SSChiSiglecF+CCR3+ cells by flow cytometric analyses, and as cells expressing major basic protein (MBP) by IHC. (Fig. 1D, E). We observed similar recruitment of eosinophils to the liver after APAP treatment in male and female mice (Fig. 1 and supplemental Fig. 1), suggesting that this phenomenon was not sex-dependent. Moreover, this phenomenon was not limited to AILI, as our data showed that after CCl4- or ConA-induced acute liver injury, eosinophils were also recruited to the liver (supplemental Fig. 2). These data are the first to reveal that eosinophils are recruited into the liver during AILI and in other animal models of acute liver injury.
Fig.1. Eosinophils accumulate in the liver after APAP treatment.
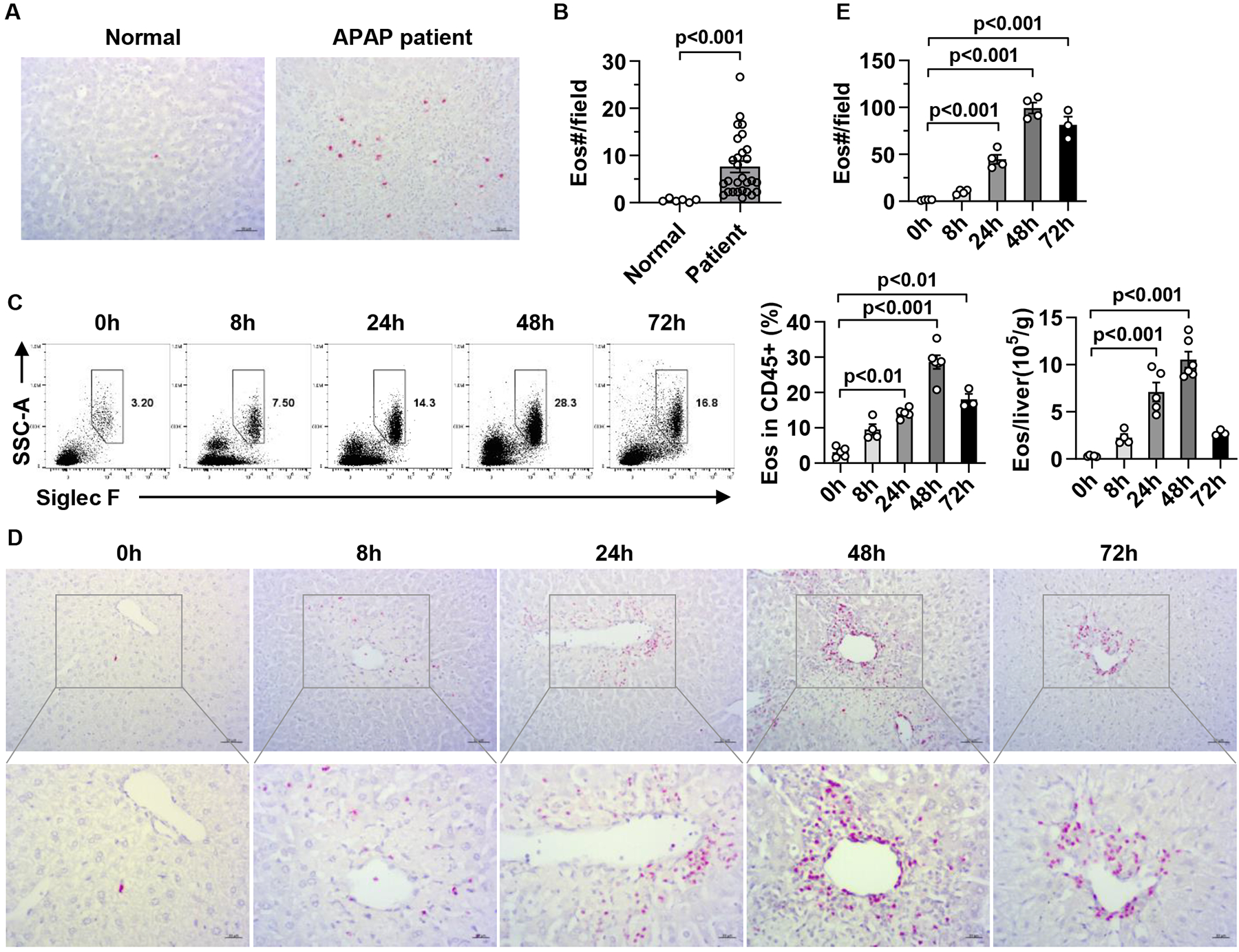
(A, B) Eosinophils in liver samples from individuals with normal livers (Normal, n=6) and patients with APAP-induced liver failure (APAP patient, n=28) were detected by IHC staining using anti-human EPX antibody (scale bar, 50μm). The numbers of eosinophils per field (776×582 pixels) were quantified. (C-E) Male C57BL/6 mice were injected i.p. with APAP. (C) The percentage and absolute number of eosinophils in the liver were measured after APAP treatment by flow cytometry (n=3–6/group). Eosinophils were identified as CD45+CD11b+SSChiSiglec F+CCR3+ cell. (D, E) Eosinophils were detected by IHC staining using anti-mouse MBP antibody and quantified (n= 3–4/group, scale bar, 50μm). Two-tailed unpaired Student’s t test was performed in B. One-way ANOVA with Tukey post hoc test was performed in C and E.
Haptic eosinophils protect against acute liver injury
To investigate the role of recruited eosinophils in acute liver injury, we treated WT and eosinophil-deficient mice (ΔdblGata1) with APAP, CCl4 or ConA. In all three cases, ΔdblGata1 mice developed worsened liver injury at 24h compared with WT mice, as demonstrated by increased serum levels of alanine aminotransferase (ALT) and areas of hepatocyte necrosis (Fig. 2A–C and supplemental Fig. 3). We focused our study on AILI, as it is the most clinically relevant among the three models. To further substantiate the protective function of eosinophils, we adoptively transferred WT bone marrow-derived eosinophils (bmEos) to ΔdblGATA1 mice immediately prior to APAP treatment. The data showed a dramatic reduction of serum ALT levels and the extents of liver necrosis (Fig. 2A–C). Even when the bmEos transfer was delayed to 8h or 12h after APAP treatment, significant attenuation of AILI in ΔdblGATA1 recipient mice was still evident (Fig. 2D–F). Repeating the adoptive transfer experiments with varying numbers of bmEos revealed that 5 million cells were sufficient to achieve a significant protection against liver injury (supplemental Fig.4). These data demonstrated a profound hepato-protective effect of eosinophils during acute liver injury.
Fig. 2. Eosinophils protect against APAP-induced liver injury.
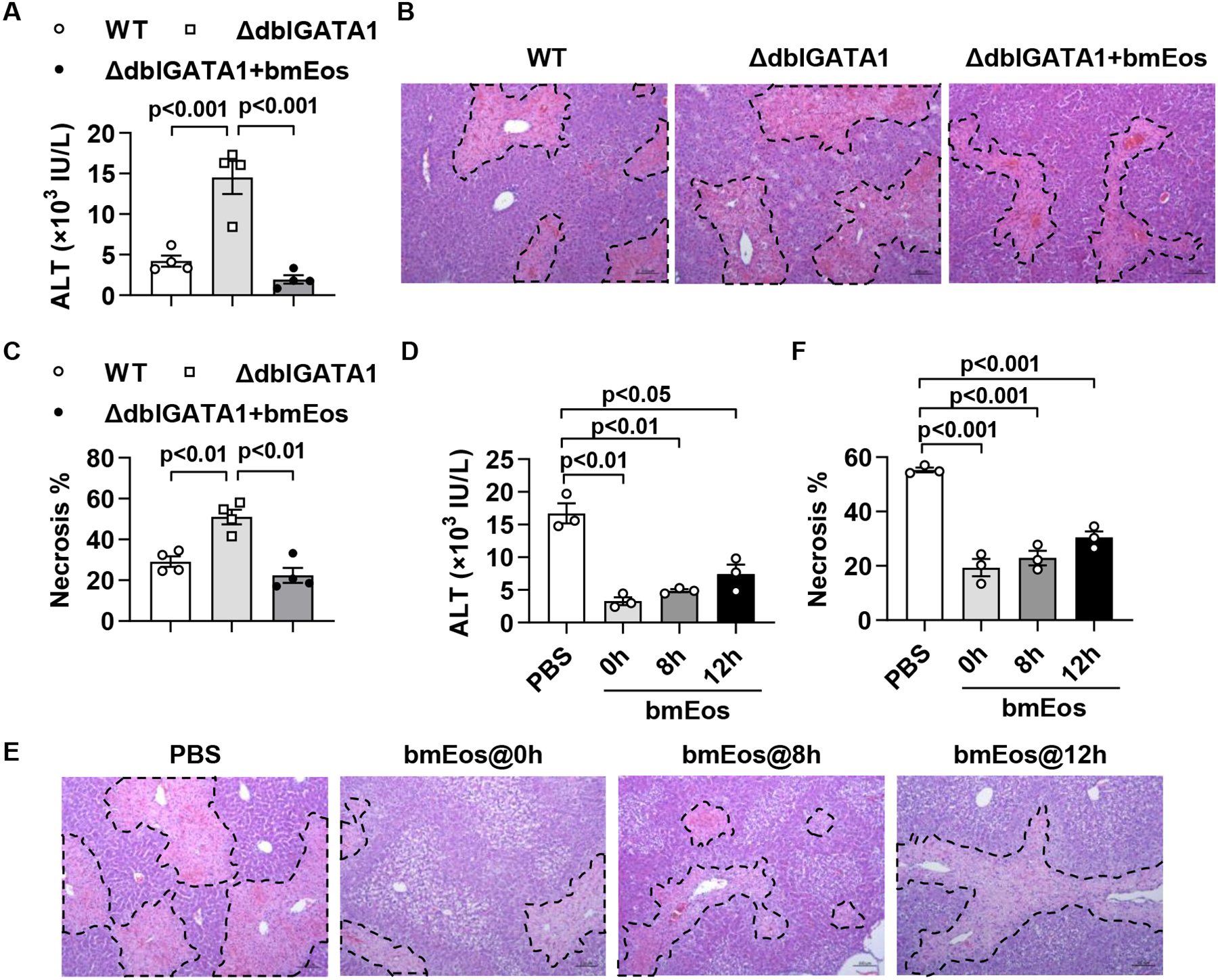
(A to C) WT Balb/c and ΔdblGATA1 mice were i.v. injected with PBS or WT bmEos (7.5×106) immediately before treatment with APAP (n=4/group). Serum ALT levels were measured, and liver necrosis (scale bars, 100μm) was evaluated and quantified at 24h after APAP treatment. (D to F) ΔdblGATA1 mice were i.v. injected with PBS or WT bmEos (7.5×106) at 0h, 8h, or 12h after APAP treatment (n=3/group). Serum levels of ALT were measured and liver necrosis (scale bars, 100μm) was examined at 24h after APAP treatment. One-way ANOVA with Tukey post hoc test was performed in A, C, D, and F.
Macrophages play an essential role in eosinophil recruitment after acute liver injury
To understand how eosinophils are recruited into the liver after injury, we set out to determine which cell type(s) are important in this process. It has been reported that platelets are essential for leukocyte recruitment into inflamed tissues (21). However, when we depleted platelets using an anti-CD41 antibody at 24h prior to APAP challenge, we did not observe any effect on the number of eosinophils recruited to the liver (supplemental Fig. 5A, B). Studies of acute peritonitis induced by zymosan or thioglycollate have demonstrated that neutrophils are recruited first at around 4–6h, which is then followed by eosinophil infiltration (22, 23). In many models of tissue inflammation, including AILI, neutrophils and monocytes are among the first leukocytes recruited (24, 25). To investigate whether neutrophils or macrophages are required for eosinophil recruitment, we depleted neutrophils using anti-Ly6G Ab or macrophages using liposome-entrapped clodronate (CLD) at 24h before APAP treatment. Neutrophil depletion did not affect the numbers of eosinophils recruited into the liver (supplemental Fig. 5C, D). However, compared with empty liposome-treated control mice, CLD-treated, macrophage-depleted mice had much fewer eosinophils in the liver after APAP treatment (Supplemental Fig. 6A, B). Likewise, macrophage-depletion also resulted in impaired eosinophil recruitment to the liver in mice treated with CCl4 or ConA (Supplemental Fig. 6C–F). These data demonstrated that macrophages play a crucial role in eosinophil recruitment during acute liver injury.
IL-33 facilitates hepatic eosinophil recruitment in a macrophage-dependent manner
To understand the mechanism by which macrophages promote eosinophil recruitment, we hypothesized that damage associated molecular patterns (DAMPs) might be important in activating macrophages to recruit eosinophils. High mobility group box 1(HMGB1) and IL-1α are widely reported as DAMPs in models of sterile tissue injury, including AILI. More recently, IL-33 has been identified as another DAMP or alarmin released during tissue injury (26–28). To examine if any of these DAMPs triggers eosinophil recruitment to the liver, we injected i.v. HMGB1, IL-1α, and IL-33 to naive WT mice and measured the number of eosinophils in the liver after 24h. As shown in Fig. 3A, B, exogenous IL-33, but not IL-1α or HMGB1 induced eosinophil recruitment to the liver. We next hypothesized that if IL-33 played a role in eosinophil recruitment during AILI, it should be released after APAP challenge. Indeed, our data showed a significant increase in serum levels of IL-33 after APAP treatment (Fig. 3C). To further confirm that IL-33 was necessary for eosinophil recruitment, we subjected IL-33−/− mice to APAP-, CCl4- and ConA-induced acute liver injury. Compared with WT mice, IL-33−/− mice displayed dramatically reduced eosinophil recruitment into the liver after injury (Fig. 3D–F and supplemental Fig. 7). These data provide strong evidence that IL-33 is essential in inducing eosinophil recruitment after acute liver injury.
Fig. 3. IL-33 promotes eosinophil recruitment to the liver.
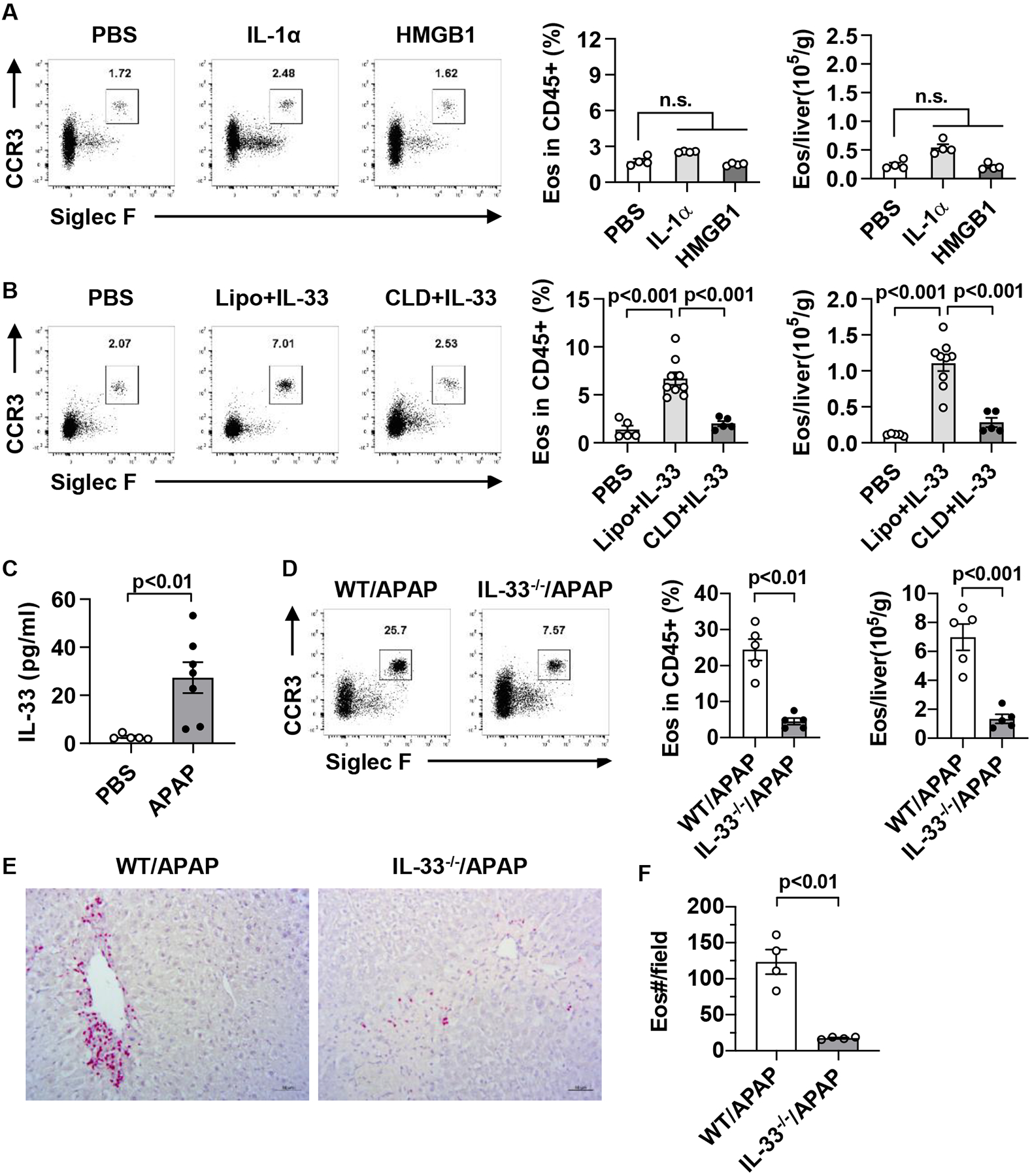
(A) C57BL/6 mice were injected i.v. with PBS, IL-1α, or HMGB1 twice on one day spaced out by 8h (0.5 μg/mouse each dose). The percentage and number of hepatic eosinophils were measured by flow cytometry at 24h after first dose (n=4/group). (B) C57BL/6 mice were injected i.v. with PBS (n=5) or IL-33 (n=9) twice on one day spaced out by 8h (0.5 μg/mouse each dose). Some IL-33-treated mice were injected with CLD (n=5) to deplete macrophages or empty liposomes (Lipo) as control (n=9) at 24h prior to the initial IL-33 treatment. All mice were sacrificed at 24h after the first dose of IL-33 for the measurement of the percentage and number of hepatic eosinophils. (C) C57BL/6 mice were injected i.p. with PBS (n=5) or APAP (n=7). After 24h, serum levels of IL-33 were measured by ELISA. (D, E) WT and IL-33−/− mice were injected i.p. with APAP. After 48h, eosinophils in the liver were measured using flow cytometry (D, n=5/group) and IHC staining (E-F, n=4/group, scale bar, 50μm). Two-tailed unpaired Student’s t test was performed in A to D, and F. n.s. indicates no statistical significance.
To determine if IL-33-mediated eosinophil recruitment is dependent on macrophages, we depleted macrophages in naive mice at 24h before IL-33 treatment. As shown in Fig. 4B, IL-33-induced hepatic recruitment of eosinophils was abrogated when macrophages were depleted. Furthermore, we found that injecting exogenous IL-33 could restore hepatic eosinophil recruitment in IL-33−/− mice, but not when macrophages were depleted (supplemental Fig. 8).
Fig. 4. Liver sinusoidal endothelial cells are the main source of IL-33 release during AILI.
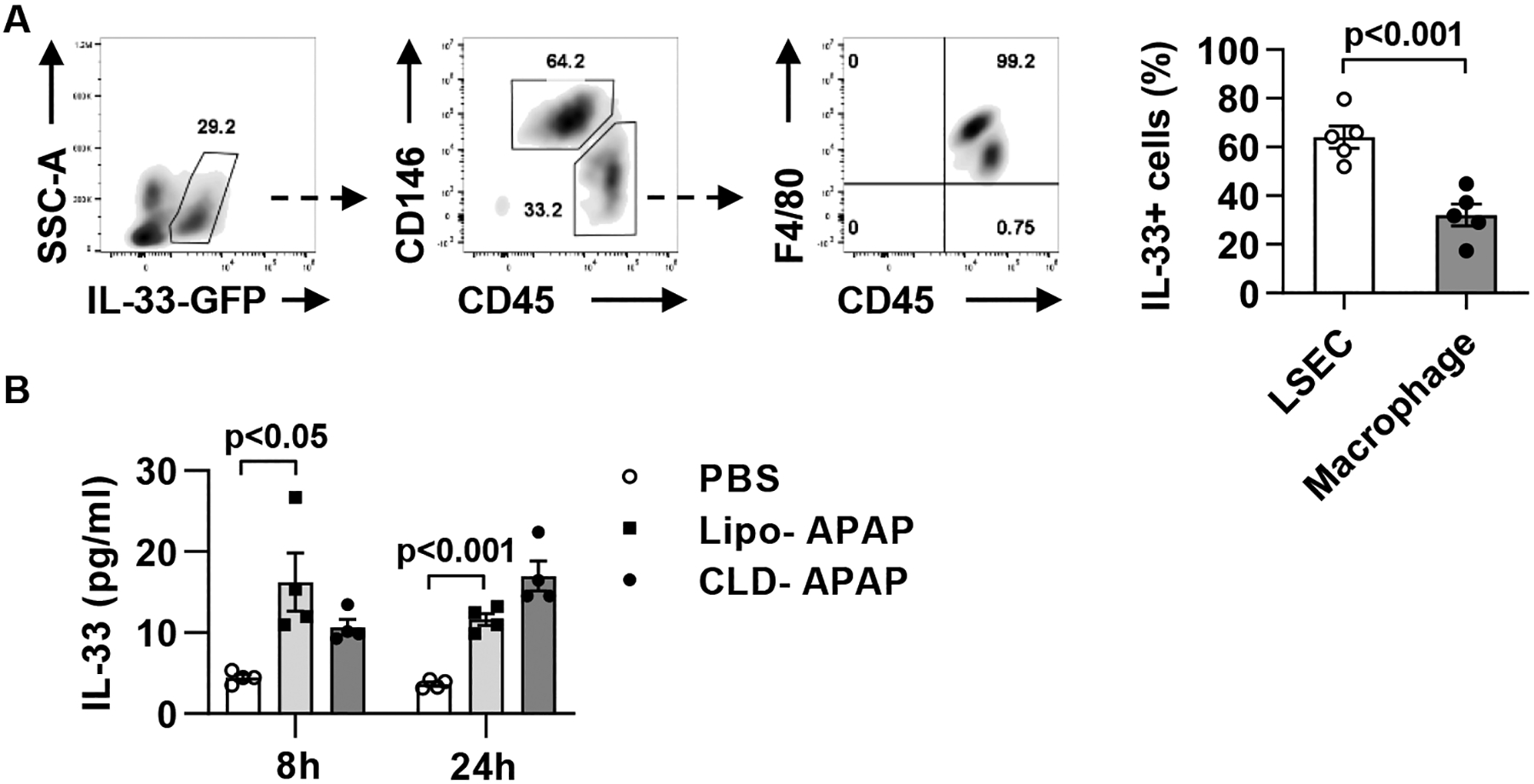
(A) IL-33-GFP reporter mice were injected i.p. with APAP (n=5). After 24h, liver non-parenchymal cell were isolated and analyzed for GFP+ cells by flow cytometry. (B) WT C57BL/6 mice were injected i.p. with PBS (n=3) or APAP (n=8). The APAP-treated mice were divided into two groups (n=4/group) treated with empty liposomes (Lipo) or CLD to deplete macrophages 24h prior to APAP challenge. All mice were sacrificed at 24h after APAP treatment and serum levels of IL-33 were measured by ELISA. Two-tailed unpaired Student’s t test was performed in A and B.
Although the release of IL-33 has been reported in animal models of liver injury and in patients with liver diseases (29, 30), the cellular source of IL-33 release has not been identified. To determine which cells release IL-33 during AILI, we treated IL-33-GFP reporter mice with APAP and isolated hepatocytes and liver non-parenchymal cells (NPCs) after 24h. Flow cytometric analyses identified two IL-33-positive cell types that could potentially release IL-33. Liver sinusoidal endothelial cells (LSECs, CD45−CD146+) accounted for about 65% of IL-33+ cells, and the remaining were CD45+F4/80+ macrophages (Fig. 4A). To further determine which of the two cell types contributed to IL-33 release, we depleted macrophages and found no effect on the serum levels of IL-33 (Fig. 4B). The data implied that LSECs are more likely the cellular source of IL-33 release.
IL-33 plays a role in macrophage production of CCL24
To address the question of what macrophages produce to recruit eosinophils, we investigated CCL11 and CCL24, which are widely reported eosinophil-specific chemoattractants (31). We observed an increase in mRNA but not protein levels of CCL11 after CCl4 treatment; however, neither mRNA nor protein levels of CCL11 were altered after APAP- or ConA-treatment (Fig. 5A, B and supplemental Fig. 9A–D). In contrast, both hepatic mRNA and serum protein levels of CCL24 were markedly increased after treatment of each hepatotoxin (Fig. 5C, D and supplemental Fig. 9E–H). To determine if CCL24 was produced by macrophages, we measured the serum level of CCL24 in control and macrophage-depleted mice. The data showed that macrophage-depletion dramatically reduced level of CCL24 in all three models of acute liver injury (Fig. 5D, and supplemental Fig. 9F, H), indicating an important role of macrophages in CCL24 production.
Fig. 5. CCL24, but not CCL11, is up-regulated in a macrophage-dependent manner during AILI.
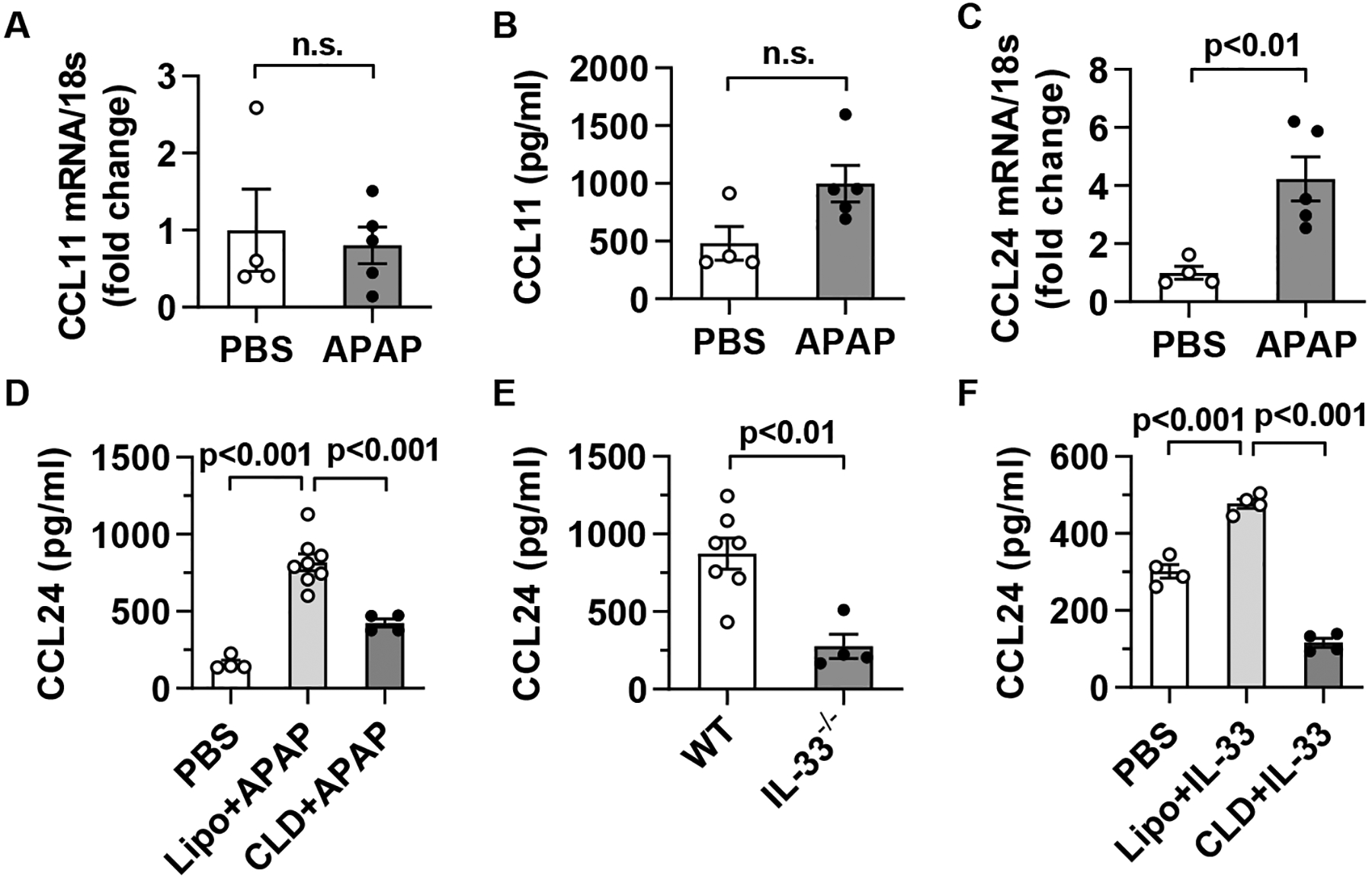
(A-D) C57BL/6 mice were injected i.p. with PBS or APAP (n=4–5/group). After 24h, CCL11 and CCL24 mRNA expression in liver tissues (A, C) and protein levels in sera (B, D) were measured by qPCR and ELISA, respectively. Some mice were treated with CLD to deplete macrophages at 24h prior to APAP challenge. (E) WT (n=7) and IL-33−/− (n=4) mice were treated with APAP, and after 24h serum levels of CCL24 were measured. (F) C57BL/6 mice were treated with PBS or IL-33 as described in Fig. 3B. Some IL-33-treated mice were injected with CLD to deplete macrophages following the same protocol described in Fig. 3B. All mice were sacrificed at 24h after the first dose of IL-33 and serum levels of CCL24 were measured (n=4/group). Two-tailed unpaired Student’s t test was performed in A to F. n.s. indicates no statistical significance.
Since our data suggested that IL-33-induced macrophage activation plays a critical role in eosinophil recruitment, we tested if IL-33 was important in triggering CCL24 expression by macrophages. We found that the serum levels of CCL24 were significantly lower in APAP-treated IL-33−/− mice than WT mice (Fig. 5E). Moreover, exogenous IL-33 induced CCL24 production in naive mice, but not in macrophage-depleted mice (Fig. 5F). These data suggested that IL-33, released during acute liver injury, plays an important role in promoting CCL24 production by macrophages, thereby mediating eosinophil recruitment into the liver.
IL-33, through activating eosinophils, indirectly promotes CCL24 production by macrophages.
To investigate whether IL-33 directly activates macrophages to produce CCL24, we treated bone marrow-derived macrophages (BMDM) in vitro with IL-33. As shown in Fig. 6A, IL-33 did not directly induce CCL24 release, suggesting that another cell type may be involved. Because IL-33 functions through its receptor, suppression of tumorigenicity 2 (ST2), we set out to identify ST2-expressing cells in the liver. The data revealed that almost all ST2+ cells were eosinophils (Fig. 6B). To examine if eosinophils are involved in inducing CCL24 production by macrophages, we isolated eosinophils from the livers of APAP-treated mice by using magnetic-activated cell sorting (MACS). We then cultured purified eosinophils or the remaining cell mixture (not containing eosinophils) with BMDM in the presence of IL-33. The data showed that eosinophils were required for IL-33-induced CCL24 expression by macrophages (Fig. 6C).
Fig. 6. IL-33-stimulated eosinophils, through release of IL-4, induce CCL24 production by macrophages.
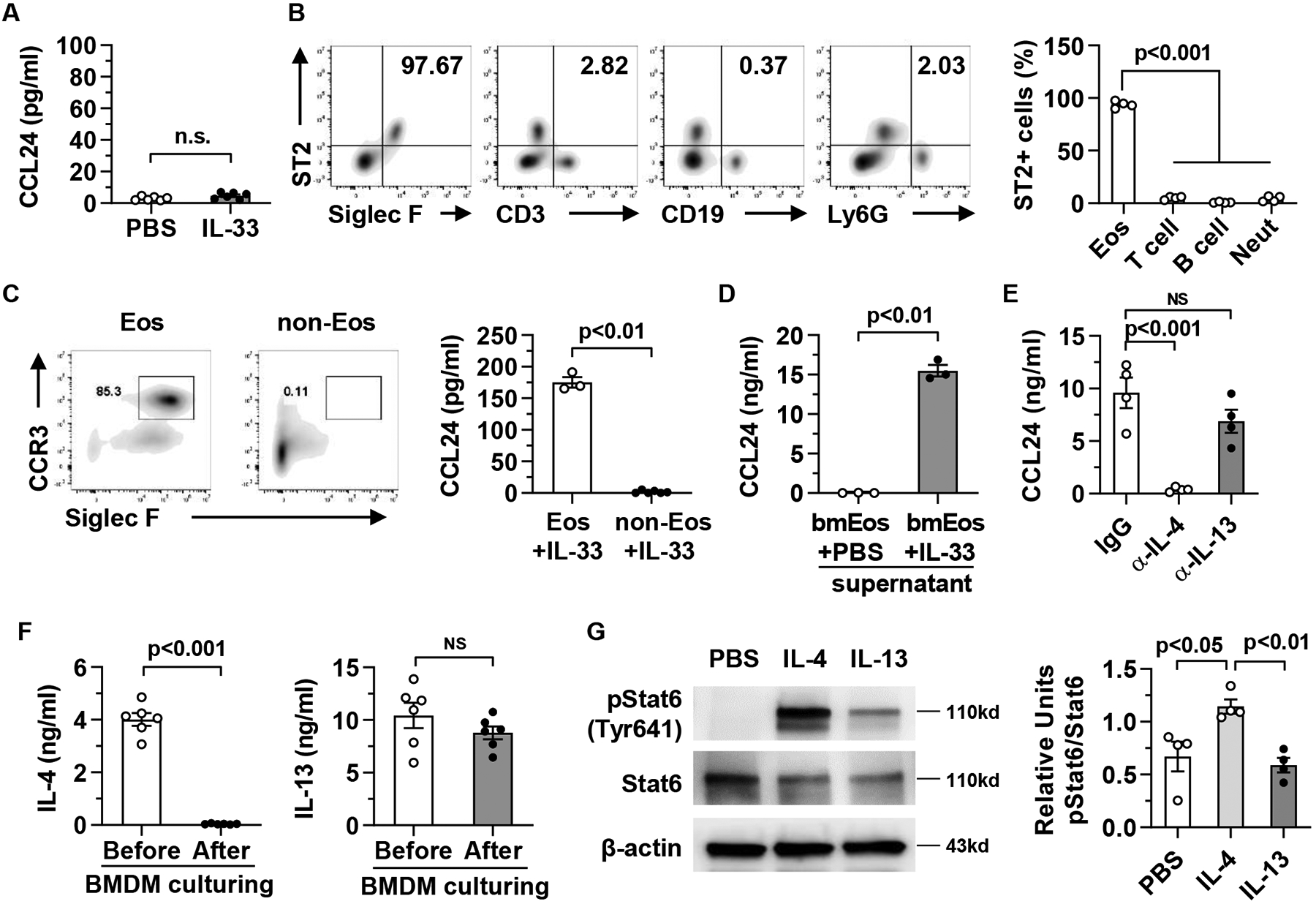
(A) BMDMs (4×106 cells/ml per well), generated from C57BL/6 mice, were stimulated with mouse recombinant IL-33 (20 ng/ml). After 24h, CCL24 protein levels in the supernatants were measured by ELISA. (B) Liver mononuclear cells (MNCs) were isolated from WT mice (n=4) at 24h after APAP treatment and analyzed for ST2 expression by flow cytometry. (C) From liver MNCs, isolated at 24h after APAP treatment, eosinophils (Eos) were purified by Siglec F-positive selection using MACS and the purity is around 85%. The Siglec F-negative portion of cell mixture (non-Eos) was also collected. Eos and non-Eos (1×106 cells/ml per well) were cultured together with BMDMs (4×106 cells/well) in presence of IL-33. After 24h co-culturing, CCL24 protein levels in the supernatants were measured by ELISA. (D) Bone marrow-derived eosinophils (bmEos, 4×106 cells/ml per well) were stimulated with PBS or IL-33 (20 ng/ml). After 24h, the conditioned media (CM) were collected and used to culture BMDMs (4×106 cells/ml per well) for another 24h. CCL24 protein levels in the BMDM culture supernatants were measured by ELISA. (E) The culturing system was set up as described in D with the addition of anti-IL-4 antibody or anti-IL-13 antibody in the CM collected from IL-33-stimulated bmEos. (F) The protein levels of IL-4 and IL-13 in the CM from IL-33-stimulated bmEos were measured before and after culturing of BMDMs. (G). BMDMs was treated with PBS, mouse recombinant IL-4 or IL-13 for 1h. Expression levels of phosphorylated-Stat6, Stat6, and β-actin were measured and quantified (n=4/group). Two-tailed unpaired Student’s t test was performed in A to D, and F. One-way ANOVA with Tukey post hoc test was performed in E and G. n.s. indicates no statistical significance.
To further elucidate the mechanism, we cultured BMDM with conditional media (CM) collected from IL-33-stimulated eosinophils and found induction of CCL24 (Fig. 6D). The data suggested that soluble factor(s) released from IL-33-activated eosinophils were important. We and others have shown that IL-33 stimulates eosinophils to release IL-4 and IL-13 (7, 32). To determine whether IL-4 and/or IL-13 played a critical role in CCL24 production by macrophages, we added anti-IL-4 or anti-IL-13 neutralizing antibody to the CM. As shown in Fig. 6E, neutralizing IL-4, but not IL-13, abrogated CCL24 production by macrophages. Interestingly, we found that IL-4, but not IL-13, was depleted from the CM after culturing BMDM for 24h (Fig. 6F), suggesting that IL-4 was consumed by macrophages. Furthermore, treatment of BMDM with the same concentration IL-4 or IL-13 showed that IL-4 induced a much stronger response than IL-13 in terms of Stat6 activation (Fig. 6G). Taken together, these data demonstrated that IL-4, but not IL-13, released by eosinophils after IL-33 stimulation played a key role in CCL24 production by macrophages.
Discussion
The current study demonstrates that eosinophils are recruited to the liver during acute injury and play a protective role in multiple modes of liver injury. IL-33, which is up-regulated and released by LSEC during acute liver injury, acts on eosinophils and initiates the positive feedback loop involving a cross-talk between eosinophils and macrophages. IL-33 activates eosinophils to release IL-4. IL-4 plays a critical role in inducing macrophages to produce CCL24, which in turn recruits more eosinophils. Furthermore, our data also show that the recruited eosinophils confer hepato-protection against liver injury caused by APAP overdose.
IL-33 expression in injured livers has been investigated by IHC staining. It was found to be mainly expressed in endothelial cells in CCl4-induced liver injury, but predominantly in hepatocytes in ConA-induced hepatitis (33). In an LPS-induced inflammation model, IL-33-positive staining demarcates the liver sinusoids, implying endothelial cell expression of IL-33 (34). It is known that IL-33 is released as an alarmin during tissue injury (28). We observed a significant elevation of IL-33 in the serum of mice after APAP treatment, similar to our previous finding of IL-33 release during hepatic IR injury (7). However, the cellular source of IL-33 release was unknown. The current study using IL-33-GFP reporter mouse and macrophage depletion approach identified LSEC as a main source of IL-33 release during APAP-induced liver injury.
Two recent genome-wide association studies have highlighted a correlation between IL-33 and the number of circulating eosinophils (35, 36). One study discovered certain sequence variants in IL-33 strongly correlating with blood eosinophil numbers and the risk of developing asthma (36). Another study found that a loss of function mutation of IL-33 was associated with reduced blood eosinophil numbers (35). Studies in mice reported that multiple injections of IL-33 over a 7-day period could significantly induce eosinophil accumulation in various tissues, such as the spleen and bone marrow (18, 19). IL-33-induced eosinophilia was also found in the lung when IL-33 was administered intranasally for 3 days (37). These studies provide direct evidence that IL-33 induces eosinophilia. However, because the IL-33 treatments were extended for several days in these studies, it was unclear whether IL-33-induced eosinophilia was due to eosinophil recruitment into the tissues or enhanced eosinophil differentiation from progenitor cells in the bone marrow. Our finding that eosinophils accumulate within 24h after IL-33 injections strongly suggests that IL-33 drives eosinophil recruitment to the liver.
The notion that IL-33 might act directly as an eosinophil chemoattractant has been negated by an in vitro study of eosinophil migration in response to IL-33 (38). Our data are consistent with an indirect action of IL-33 on eosinophil recruitment, such that IL-33-activated eosinophils, through releasing IL-4, promote CCL24 production by macrophages to recruit more eosinophils in a feed-forward loop. Although the role of IL-4 in promoting macrophages to produce CCL24 has been reported (39–41), our study is the first to link IL-33-activated eosinophils with IL-4 release and CCL24 production by macrophages. Interestingly, a previous study showed that although IL-33 could not induce eosinophil recruitment into the lung of ST2−/− mice, adoptive transfer of WT eosinophils to ST2−/− mice actually triggered the infiltration of ST2−/− eosinophils into the lung. Although the study did not define the mechanism, our findings would suggest that the transferred WT eosinophils responded to IL-33, thereby promoting macrophages to release CCL24 and recruiting eosinophils regardless of ST2 expression (37).
Our study highlights a cross-talk interaction between eosinophils and macrophages that plays a key role in eosinophil recruitment to sterile inflamed liver. This interaction is important and applies to several distinct biological conditions. For example, eosinophil-derived IL-4 or IL-13 promotes alternative activation of macrophages (M2) in adipose tissues, thereby maintaining glucose homeostasis (42). IL-4-activated macrophages promote eosinophilia, important in anti-helminth infection in mice (43). A more recent study documented a cooperative interaction between eosinophils and tissue-resident macrophages in cutaneous leishmaniasis, in which eosinophils were required to maintain a M2-like phenotype of macrophages and that the macrophages promoted eosinophil recruitment to the dermis (39).
There have been very few studies of the role of eosinophils in acute liver injury and the results are contradictory (7–10, 44). For example, one study of ConA-induced hepatitis showed that eosinophil-depletion by anti-CCR3 antibody reduced ConA-induced injury, implicating a pathological role of eosinophils (10). This study used repeated high doses of anti-CCR3 antibody, raising the question whether in addition to eosinophils, other CCR3+ cells that may be affected. Another study showed that ST2−/− mice developed more severe ConA-induced hepatitis and pretreatment with IL-33 prevented liver injury in WT mice (44). Our finding that IL-33 induces hepatic eosinophil recruitment suggests that the protective effect of IL-33 may be attributed, in part, to eosinophils. Moreover, we found that eosinophil-deficient mice developed exacerbated liver injury caused by APAP, CCl4, or ConA treatment. These data together with our previous observation in hepatic IR injury strongly support a protective role of eosinophils in acute liver injury (7).
In summary, the present study for the first time demonstrates hepatic recruitment of eosinophils after APAP overdose and a profound hepato-protective function of these cells against AILI. The data also provide novel insights into the molecular and cellular mechanisms involved in hepatic eosinophil accumulation. The findings suggest that strategies to promote eosinophil recruitment and/or adoptive transfer of eosinophils may represent a novel cell-based therapeutic approach to treat patients with APAP overdose-induced liver failure.
Supplementary Material
Highlights.
Eosinophils are recruited to the liver in multiple models of acute injury and play a protective role.
A cross-talk interaction between eosinophils and macrophages plays an important role in eosinophil recruitment to the injured liver in a feed-forward loop.
IL-33 is selectively released from liver sinusoidal endothelial cells during APAP-induced liver injury.
IL-33, through stimulating IL-4 release from eosinophils, promotes macrophages to produce CCL24.
Financial support:
This work was supported by the NIH DK121330, DK122708, DK122796, DK109574 and the University of Texas System Translational STARs award to C.J.; the NSFC 81873570 to L.X.; the AST Research Network/CSL Behring Fellowship Basic Science Research grant to Y.Y.; the NIAAA R00AA026648 to K.L.P.; the NIH AI132840-01A, AI145108 and the Mayo Foundation grant to E.A.J. ALFSG was funded by U-01 DK 058369 from NIDDK to UT Southwestern Medical Center.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Competing interests: The authors declare that they have no competing interests.
Data Availability Statement:
The data that support the findings of this study are available on request from the corresponding author.
References
- 1.Stravitz RT, Lee WM. Acute liver failure. Lancet. 2019;394(10201):869–81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Klion AD, Nutman TB. The role of eosinophils in host defense against helminth parasites. The Journal of allergy and clinical immunology. 2004;113(1):30–7. [DOI] [PubMed] [Google Scholar]
- 3.Gleich GJ. Mechanisms of eosinophil-associated inflammation. The Journal of allergy and clinical immunology. 2000;105(4):651–63. [DOI] [PubMed] [Google Scholar]
- 4.Krishack PA, Hollinger MK, Kuzel TG, Decker TS, Louviere TJ, Hrusch CL, et al. IL-33-mediated Eosinophilia Protects against Acute Lung Injury. American journal of respiratory cell and molecular biology. 2021;64(5):569–78. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Takeda K, Shiraishi Y, Ashino S, Han J, Jia Y, Wang M, et al. Eosinophils contribute to the resolution of lung-allergic responses following repeated allergen challenge. The Journal of allergy and clinical immunology. 2015;135(2):451–60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Humbles AA, Lloyd CM, McMillan SJ, Friend DS, Xanthou G, McKenna EE, et al. A critical role for eosinophils in allergic airways remodeling. Science. 2004;305(5691):1776–9. [DOI] [PubMed] [Google Scholar]
- 7.Wang Y, Yang Y, Wang M, Wang S, Jeong JM, Xu L, et al. Eosinophils attenuate hepatic ischemia-reperfusion injury in mice through ST2-dependent IL-13 production. Science translational medicine. 2021;13(579). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Goh YP, Henderson NC, Heredia JE, Red Eagle A, Odegaard JI, Lehwald N, et al. Eosinophils secrete IL-4 to facilitate liver regeneration. Proc Natl Acad Sci U S A. 2013;110(24):9914–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Proctor WR, Chakraborty M, Chea LS, Morrison JC, Berkson JD, Semple K, et al. Eosinophils mediate the pathogenesis of halothane-induced liver injury in mice. Hepatology. 2013;57(5):2026–36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Louis H, Le Moine A, Flamand V, Nagy N, Quertinmont E, Paulart F, et al. Critical role of interleukin 5 and eosinophils in concanavalin A-induced hepatitis in mice. Gastroenterology. 2002;122(7):2001–10. [DOI] [PubMed] [Google Scholar]
- 11.Yu YR, O’Koren EG, Hotten DF, Kan MJ, Kopin D, Nelson ER, et al. A Protocol for the Comprehensive Flow Cytometric Analysis of Immune Cells in Normal and Inflamed Murine Non-Lymphoid Tissues. PloS one. 2016;11(3):e0150606. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Tarantino G, Cabibi D, Camma C, Alessi N, Donatelli M, Petta S, et al. Liver eosinophilic infiltrate is a significant finding in patients with chronic hepatitis C. Journal of viral hepatitis. 2008;15(7):523–30. [DOI] [PubMed] [Google Scholar]
- 13.Bjornsson E, Kalaitzakis E, Olsson R. The impact of eosinophilia and hepatic necrosis on prognosis in patients with drug-induced liver injury. Alimentary pharmacology & therapeutics. 2007;25(12):1411–21. [DOI] [PubMed] [Google Scholar]
- 14.Nagral A, Ben-Ari Z, Dhillon AP, Burroughs AK. Eosinophils in acute cellular rejection in liver allografts. Liver transplantation and surgery : official publication of the American Association for the Study of Liver Diseases and the International Liver Transplantation Society. 1998;4(5):355–62. [DOI] [PubMed] [Google Scholar]
- 15.Pope SM, Zimmermann N, Stringer KF, Karow ML, Rothenberg ME. The eotaxin chemokines and CCR3 are fundamental regulators of allergen-induced pulmonary eosinophilia. Journal of immunology. 2005;175(8):5341–50. [DOI] [PubMed] [Google Scholar]
- 16.Marichal T, Mesnil C, Bureau F. Homeostatic Eosinophils: Characteristics and Functions. Frontiers in medicine. 2017;4:101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Korbecki J, Kojder K, Siminska D, Bohatyrewicz R, Gutowska I, Chlubek D, et al. CC Chemokines in a Tumor: A Review of Pro-Cancer and Anti-Cancer Properties of the Ligands of Receptors CCR1, CCR2, CCR3, and CCR4. International journal of molecular sciences. 2020;21(21). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Johnston LK, Hsu CL, Krier-Burris RA, Chhiba KD, Chien KB, McKenzie A, et al. IL-33 Precedes IL-5 in Regulating Eosinophil Commitment and Is Required for Eosinophil Homeostasis. Journal of immunology. 2016;197(9):3445–53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Schmitz J, Owyang A, Oldham E, Song Y, Murphy E, McClanahan TK, et al. IL-33, an interleukin-1-like cytokine that signals via the IL-1 receptor-related protein ST2 and induces T helper type 2-associated cytokines. Immunity. 2005;23(5):479–90. [DOI] [PubMed] [Google Scholar]
- 20.Kienzl M, Hasenoehrl C, Valadez-Cosmes P, Maitz K, Sarsembayeva A, Sturm E, et al. IL-33 reduces tumor growth in models of colorectal cancer with the help of eosinophils. Oncoimmunology. 2020;9(1):1776059. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Rossaint J, Margraf A, Zarbock A. Role of Platelets in Leukocyte Recruitment and Resolution of Inflammation. Frontiers in immunology. 2018;9:2712. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Lastrucci C, Baillif V, Behar A, Al Saati T, Dubourdeau M, Maridonneau-Parini I, et al. Molecular and cellular profiles of the resolution phase in a damage-associated molecular pattern (DAMP)-mediated peritonitis model and revelation of leukocyte persistence in peritoneal tissues. FASEB journal : official publication of the Federation of American Societies for Experimental Biology. 2015;29(5):1914–29. [DOI] [PubMed] [Google Scholar]
- 23.Yamada T, Tani Y, Nakanishi H, Taguchi R, Arita M, Arai H. Eosinophils promote resolution of acute peritonitis by producing proresolving mediators in mice. FASEB journal : official publication of the Federation of American Societies for Experimental Biology. 2011;25(2):561–8. [DOI] [PubMed] [Google Scholar]
- 24.Graubardt N, Vugman M, Mouhadeb O, Caliari G, Pasmanik-Chor M, Reuveni D, et al. Ly6C(hi) Monocytes and Their Macrophage Descendants Regulate Neutrophil Function and Clearance in Acetaminophen-Induced Liver Injury. Frontiers in immunology. 2017;8:626. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Holt MP, Cheng L, Ju C. Identification and characterization of infiltrating macrophages in acetaminophen-induced liver injury. J Leukoc Biol. 2008;84(6):1410–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Zhang C, Feng J, Du J, Zhuo Z, Yang S, Zhang W, et al. Macrophage-derived IL-1alpha promotes sterile inflammation in a mouse model of acetaminophen hepatotoxicity. Cellular & molecular immunology. 2018;15(11):973–82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Bertheloot D, Latz E. HMGB1, IL-1alpha, IL-33 and S100 proteins: dual-function alarmins. Cellular & molecular immunology. 2017;14(1):43–64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Martin NT, Martin MU. Interleukin 33 is a guardian of barriers and a local alarmin. Nature immunology. 2016;17(2):122–31. [DOI] [PubMed] [Google Scholar]
- 29.Sun Z, Chang B, Gao M, Zhang J, Zou Z. IL-33-ST2 Axis in Liver Disease: Progression and Challenge. Mediators of inflammation. 2017;2017:5314213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.McHedlidze T, Waldner M, Zopf S, Walker J, Rankin AL, Schuchmann M, et al. Interleukin-33-dependent innate lymphoid cells mediate hepatic fibrosis. Immunity. 2013;39(2):357–71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Rothenberg ME, Hogan SP. The eosinophil. Annual review of immunology. 2006;24:147–74. [DOI] [PubMed] [Google Scholar]
- 32.Qiu Y, Nguyen KD, Odegaard JI, Cui X, Tian X, Locksley RM, et al. Eosinophils and type 2 cytokine signaling in macrophages orchestrate development of functional beige fat. Cell. 2014;157(6):1292–308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Arshad MI, Rauch M, L’Helgoualc’h A, Julia V, Leite-de-Moraes MC, Lucas-Clerc C, et al. NKT cells are required to induce high IL-33 expression in hepatocytes during ConA-induced acute hepatitis. European journal of immunology. 2011;41(8):2341–8. [DOI] [PubMed] [Google Scholar]
- 34.Pichery M, Mirey E, Mercier P, Lefrancais E, Dujardin A, Ortega N, et al. Endogenous IL-33 is highly expressed in mouse epithelial barrier tissues, lymphoid organs, brain, embryos, and inflamed tissues: in situ analysis using a novel Il-33-LacZ gene trap reporter strain. Journal of immunology. 2012;188(7):3488–95. [DOI] [PubMed] [Google Scholar]
- 35.Smith D, Helgason H, Sulem P, Bjornsdottir US, Lim AC, Sveinbjornsson G, et al. A rare IL33 loss-of-function mutation reduces blood eosinophil counts and protects from asthma. PLoS genetics. 2017;13(3):e1006659. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Gudbjartsson DF, Bjornsdottir US, Halapi E, Helgadottir A, Sulem P, Jonsdottir GM, et al. Sequence variants affecting eosinophil numbers associate with asthma and myocardial infarction. Nature genetics. 2009;41(3):342–7. [DOI] [PubMed] [Google Scholar]
- 37.Stolarski B, Kurowska-Stolarska M, Kewin P, Xu D, Liew FY. IL-33 exacerbates eosinophil-mediated airway inflammation. Journal of immunology. 2010;185(6):3472–80. [DOI] [PubMed] [Google Scholar]
- 38.Suzukawa M, Koketsu R, Iikura M, Nakae S, Matsumoto K, Nagase H, et al. Interleukin-33 enhances adhesion, CD11b expression and survival in human eosinophils. Laboratory investigation; a journal of technical methods and pathology. 2008;88(11):1245–53. [DOI] [PubMed] [Google Scholar]
- 39.Lee SH, Chaves MM, Kamenyeva O, Gazzinelli-Guimaraes PH, Kang B, Pessenda G, et al. M2-like, dermal macrophages are maintained via IL-4/CCL24-mediated cooperative interaction with eosinophils in cutaneous leishmaniasis. Science immunology. 2020;5(46). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Makita N, Hizukuri Y, Yamashiro K, Murakawa M, Hayashi Y. IL-10 enhances the phenotype of M2 macrophages induced by IL-4 and confers the ability to increase eosinophil migration. International immunology. 2015;27(3):131–41. [DOI] [PubMed] [Google Scholar]
- 41.Kurowska-Stolarska M, Stolarski B, Kewin P, Murphy G, Corrigan CJ, Ying S, et al. IL-33 amplifies the polarization of alternatively activated macrophages that contribute to airway inflammation. Journal of immunology. 2009;183(10):6469–77. [DOI] [PubMed] [Google Scholar]
- 42.Wu D, Molofsky AB, Liang HE, Ricardo-Gonzalez RR, Jouihan HA, Bando JK, et al. Eosinophils sustain adipose alternatively activated macrophages associated with glucose homeostasis. Science. 2011;332(6026):243–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Turner JD, Pionnier N, Furlong-Silva J, Sjoberg H, Cross S, Halliday A, et al. Interleukin-4 activated macrophages mediate immunity to filarial helminth infection by sustaining CCR3-dependent eosinophilia. PLoS pathogens. 2018;14(3):e1006949. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Volarevic V, Mitrovic M, Milovanovic M, Zelen I, Nikolic I, Mitrovic S, et al. Protective role of IL-33/ST2 axis in Con A-induced hepatitis. Journal of hepatology. 2012;56(1):26–33. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The data that support the findings of this study are available on request from the corresponding author.