

Abstract
The extracellular matrix plays a critical role in mechanosensing and thereby influences the secretory properties of bone-marrow-derived mesenchymal stem/stromal cells (MSCs). As a result, interest has grown in the development of biomaterials with tunable properties for the expansion and delivery of MSCs that are used in cell-based therapies. Herein, stress-relaxing hydrogels are synthesized as hybrid networks containing both biopolymer and synthetic macromer components. Hyaluronic acid is functionalized with either aldehyde or hydrazide groups to form covalent adaptable hydrazone networks, which are stabilized by poly(ethylene glycol) functionalized with bicyclononyne and heterobifunctional small molecule crosslinkers containing azide and benzaldehyde moieties. Tuning the composition of these gels allows for controlled variation in the characteristic timescale for stress relaxation and the amount of stress relaxed. Over this compositional space, MSCs are observed to spread in formulations with higher degrees of adaptability, with aspect ratios of 1.60 ± 0.18, and YAP nuclear:cytoplasm ratios of 6.5 ± 1.3. Finally, a maximum MSC pericellular protein thickness of 1.45 ± 0.38 μm occurred in highly stress-relaxing gels, compared to 1.05 ± 0.25 μm in non-adaptable controls. Collectively, this study contributes a new understanding of the role of compositionally defined stress relaxation on MSCs mechanosensing and secretion.
Keywords: hydrogels, mesenchymal stromal cells, stress relaxation
Graphical Abstract
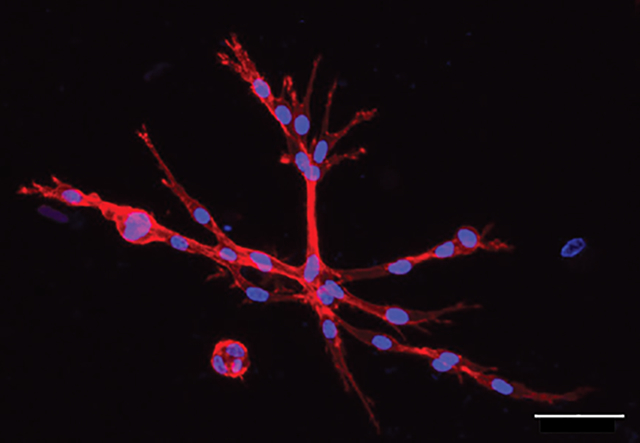
In this work, a stress-relaxing hydrogel was tuned to investigate the impact of controlled variations in stress relaxation timescales on mesenchymal stem/stromal cells (MSCs). Specifically, a hybrid network, containing both biopolymer and synthetic macromer components were synthesized to examine MSCs mechanosensing and secretory properties as a function of stress relaxation. Scale Bar = 50 μm.
1. Introduction
Mesenchymal stem/stromal cells (MSCs) are broadly used in tissue engineering and regenerative medicine applications due to their capacity for multi-lineage differentiation and potent secretory properties.[1,2] As anchorage-dependent cells, MSCs directly interact with the extracellular matrix (ECM), so the composition and mechanical properties of the biomaterials designed to deliver them can be used to tune their mechanosensing, fate, and secretory profiles. However, it is important to note that there is a dynamic reciprocity as MSCs also remodel their local surroundings via matrix degradation and deposition of nascent pericellular proteins.[3,4] For example, Loebel et al. demonstrated that MSCs secrete fibronectin, collagen, and laminin, which influence cell-matrix interactions, cell spreading and morphology, and differentiation.[5] While the effects of matrix mechanics on MSC mechanosensing, cytokine secretion, and fate specification have been well characterized, less is known regarding the impact of hydrogel dynamics, specifically that of stress relaxation, on nascent protein secretion.[5–8] In an effort to elucidate these contributions, we designed a covalent adaptable hydrogel chemistry with varying ratios of stress relaxing biopolymer to elastic synthetic polymer to test how time-dependent material behaviors influence MSC secretory properties.[5]
Synthetic hydrogels have been developed to act as 3D mimics of native tissues, and used in vitro to better understand cell-matrix and cell-cell interactions in these environments.[9,10] In other examples, synthetic hydrogels have been designed as material platforms for tissue engineering with both physical and biological properties tuned to direct various MSC behaviors.[11,12] With respect to viscoelastic properties, prior work has demonstrated both reversible addition and exchange-based reactions for 3D MSC culture, including boronic acid,[13] thioester,[14] hydrazone,[15] and allyl sulfide[16,17] based crosslinking chemistries.[13–17] Here, we employed the hydrazone chemistry, which can span a broad range of stress relaxation time scales (≈3000 s to ≈4 months), and is suitable for the culture of several cell types (e.g., C2C12 myoblasts, chondrocytes, and 3T3 fibroblasts).[15,18,19] The stress relaxation timescales of hydrazone hydrogels have been shown to directly influence the secretory properties and deposition of collagen by encapsulated chondrocytes, highlighting the utility of this chemistry in culture platforms for tissue engineering and as a means to study protein secretion.[18]
The stress relaxation afforded by the hydrazone chemistry can be fine-tuned by the experimenter’s choice of reactant structure (alkyl vs aromatic),[18] presence of catalysts,[20] and/or the stoichiometry of reactive groups[21] to impart stress relaxation time scales spanning several orders of magnitude.[18,20,21] Slower relaxation timescales (months) of aromatic hydrazones increase hydrogel matrix stability,[18] whereas faster relaxation timescales (hours), often with the use of catalysts or alkyl hydrazones, allow for stamp templating of fully formed hydrogels,[15] extrusion-based printing,[19] and injection.[15,18,19,22] However, one limitation when using fast-relaxing hydrogels for many tissue engineering and cell culture applications is the rapid erosion of cell-laden samples, leading to the dissolution of the bulk hydrogel.[13,19] Methods to remedy this issue include the introduction of thermo- and photo-responsive chemistries that lead to a small fraction of more stable crosslinks (e.g., thiol-ene addition,[19] poly(N-isopropylacrylamide)[23]).[19,23] Here, we stabilized hydrazone-based hydrogels by using a slow-gelling bioorthogonal strain-promoted azide-alkyne cycloaddition (SPAAC) reaction that retains the key characteristics of the hydrazone chemistry while mechanically stabilizing the hydrogel for long-term cell culture applications.
In this manuscript, we formulate a poly(ethylene glycol) (PEG) and hyaluronic acid (HA) (PEG-HA) hydrogel and control stoichiometry and mole fraction of each component to create materials with varying degrees of stress relaxation. We characterized the rheological properties of the HA-PEG hydrogels and found that by varying the crosslinker chemistry, the stress relaxation could be tuned across a broad range of timescales from hours to months (e.g., increased alkyl hydrazone/HA content led to faster stress relaxation). Rat MSCs (rMSCs) were encapsulated in these materials, and the viscoelastic properties led to varying degrees of response of the cells to the microenvironment via mechanosensing. Cellular changes were observed via measurements of the aspect ratios, cell volume, and Yes-associated protein 1 (YAP) localization over a wide range of stress relaxation timescales (≈2 h to 6 months). At faster timescales (≈2 h), cellular aspect ratios increased drastically to 1.60, and YAP nuclear localization (nuclear: cytoplasmic ratio of 6.5, on average) was observed, as compared to the elastic control with cellular aspect ratios of 1.2, and YAP nuclear localization of 2.8. Finally, pericellular deposition of nascent proteins was quantified in response to the effects of matrix composition, with greater deposition observed in faster-relaxing hydrogels.
2. Results and Discussion
2.1. Engineered Stress Relaxing HA-PEG Hydrogels
HA was functionalized with either an aldehyde (HA-Ald) through reduction of the HA backbone or a hydrazide (HA-Hyd) via the addition of a pendant group, (Figure 1a) which comprised the base hydrazone-crosslinked hydrogel formulation. However, significant mass loss of these hydrogels occurs during extended cell culture times due to the kinetics of the network adaptability, along with hydrolysis of the hydrazone bond.[24] To remedy this problem, irreversible crosslinks were introduced into the hydrogel.[13] Specifically, a small commercially available crosslinker, azide-PEG3-phenyl aldehyde (benzaldehyde-PEG-azide) (Figure 1b), was dosed in with the HA-Hyd groups (ranging from 12% to 100% of the functional HA-Hyd arms) to form a more stable aromatic hydrazone bond between the hydrazide and benzaldehyde groups. The hydrazone bond formed between the benzaldehyde and HA-Hyd is a significantly slower stress relaxing bond, on the order of months.[18] This reaction effectively converts hydrazide functional groups along the HA backbone into azide moieties for subsequent participation in strain-promoted azide-alkyne cycloaddition (SPAAC) reactions with a bicyclononyne (BCN) functionalized 8-arm PEG macromer (Figure 1b).[25] This SPAAC reaction forms a stable triazole bond that influences the overall viscoelastic properties of the resulting hydrogel and significantly enhances the stability of the formulation. A stable material can be achieved by incorporating this more stable crosslink into the material above the percolation threshold, where the predicted Flory–Stockmayer percolation threshold of the hydrogel was calculated to be 1.26% using Equation (1) based on an 81 arm HA-hydrazide and an 88 arm HA-aldehyde.[18,26] Changing the proportion of alkyl to benzyl hydrazone bonds allows the experimenter to tune the material to a range of compositionally-defined viscoelastic responses.
| (1) |
It is worthwhile to note that a viscoelastic hydrogel forms when reacting the HA-Ald and HA-Hyd macromers alone; however, without further stabilization, these materials are not stable for the longer timescales required for MSC encapsulation and culture (Figure 1c). Upon addition of the benzaldehyde-PEG-azide crosslinker, the hydrazide functionality was effectively converted into an azide functionality capable of participating in the SPAAC reaction to stabilize the aforementioned viscoelastic hydrogel, which dramatically changed the stress relaxation behavior (Figure 1d). Therefore, by altering the crosslinker composition, a broad range of viscoelastic properties could be achieved within the hydrogel, and stress relaxation was characterized for each formulation. For comparison, a more elastic and stable hydrogel was synthesized by reacting the HA-Hyd, functionalized with the benzaldehyde-PEG-azide crosslinker, and PEG-BCN to form a SPAAC network (Figure 1e).
Figure 1.
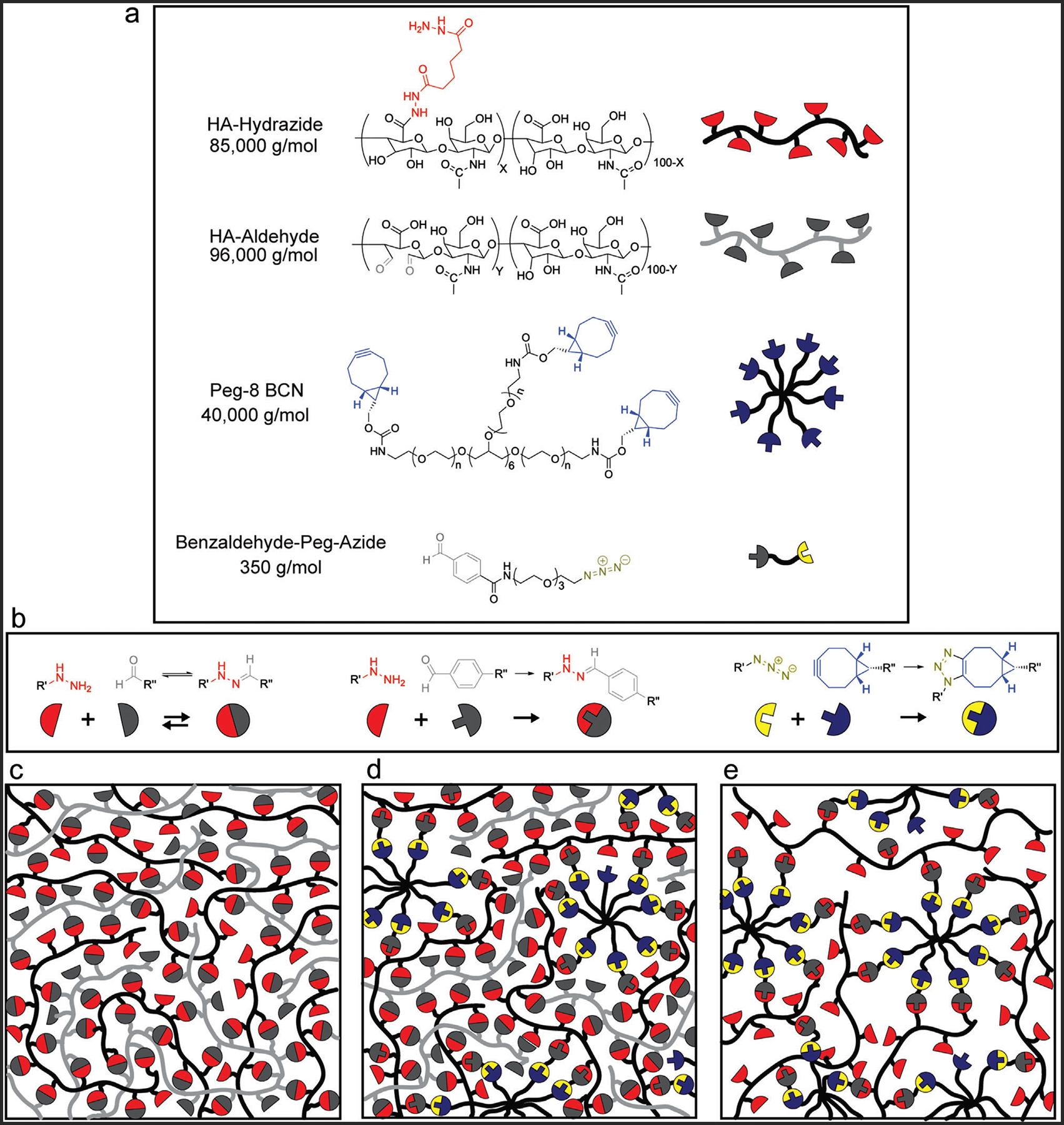
Network structure of hybrid network, stress relaxing hydrogels. a) Schematic of the chemical structures of the 85 kDa HA-hydrazide, 96 kDa HA-aldehyde, 40 kDa 8-arm PEG-BCN, and the 350 Da benzaldehyde-PEG-azide. b) The reactant groups which form the various bonds within the hydrogel including, the hydrazide and aldehyde groups which form the dynamic hydrazone bond, the hydrazide and benzaldehyde group which react to form a more stable hydrazone bond, and the azide and BCN groups which react to form the permanent triazole bond. c) Upon mixing of the HA-hydrazide and HA-aldehyde a stress relaxing hydrogel is formed. d) Upon mixing the HA-hydrazide with the benzaldehyde-PEG-azide, some of the hydrazide groups can be functionalized with an azide, enabling the reaction between the PEG-BCN and azide groups through a strain promoted cycloaddition. Upon mixing the pre-reacted HA-hydrazide and benzaldehyde-PEG-azide with the HA-aldehyde and PEG-BCN a hybrid network can form between the SPAAC bond and the dynamic hydrazone bond, forming a hydrogel. e) Through the SPAAC formation alone, an elastic hydrogel is formed.
Subsequently, hydrogels were formed at 3 wt.% total polymer in solution using stoichiometric ratios for all complementary reactive groups to achieve a range of viscoelastic properties. Specifically, hydrogels with 100%, 88%, 75%, 50%, 25%, and 0% of crosslinks comprised of fast-relaxing alkyl hydrazone bonds were fabricated. The gelation time, final storage modulus, and stress relaxation time of the hydrogels were characterized in situ using a rheometer. As shown in Figure 2, the hydrazone bonds form rapidly at physiological pH, faster than can be captured when loading the sample, as observed by the immediate gelation of the hydrogel (G′ > G″) for the initial viscoelastic condition (HA-Ald and HA-Hyd only). With the addition of the slower-gelling SPAAC crosslinks, the crossover point of G′ and G″ still occurs relatively rapidly (Figure 2a,b), reaching steady state network formation after ≈9000 s (2.5 h). The final shear storage modulus of the various hydrogel formulations ranged from 1600 to 2400 Pa (Figure 2a).
Figure 2.
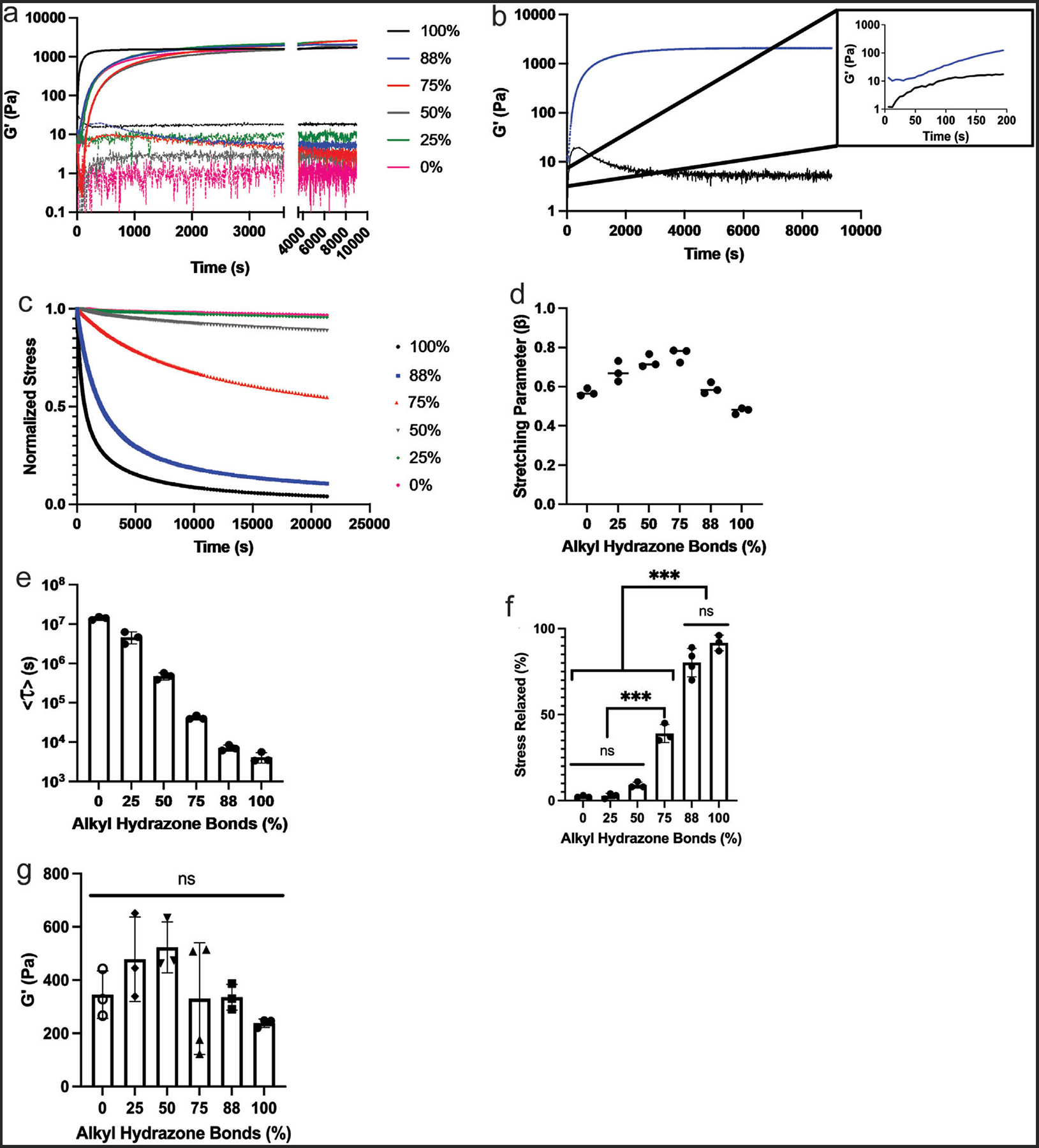
The effect of stress relaxation properties on hydrogel mechanics. a) Hydrogel in situ formation, showing the storage and loss modulus, reaching a final modulus ranging from 1600 to 2400 Pa. b) The stress relaxation behavior of the hydrogels, over 6 h, after in situ formation, with the largest amount of stress relaxed occurring in the completely viscoelastic gel, and the least stress relaxed occurring in the completely elastic condition. c) Average relaxation times calculated as a function of the percentage of hydrazone crosslinks (stress relaxation) in the hydrogel, calculated by fitting to a stretched exponential function in MATLAB. d) The fit stretching parameter (β) for each condition. e) Represents the average time constant for stress relaxation <τ >. f) The percentage of stress relaxed for each alkyl hydrazone bond condition from the 6-h stress relaxation rheological experiments was quantified. g) After 24 h swelling in PBS the hydrogels softened to a final modulus of ≈400 Pa across all conditions. Based on the composition of the hydrogel, the HA-Hydrazide functionalized with the benzaldehyde-PEG-azide to PEG-BCN ratio, dictates stress relaxation within the dual network. Values are plotted as mean ± standard deviation. ***p<0.001, n.s.: p>0.05, based on one-way ANOVA. Three replicates (n = 3) were analyzed per condition.
Following complete network formation, the stress relaxation properties of the hydrogels were tested. A 10% strain was applied over 1 s and held constant for a period of 6 h while the relaxation of the applied shear stress was monitored for each hydrogel condition (Figure 2b). The amount of stress relaxation varied with the percent of alkyl hydrazone bonds present within the hydrogel, ranging from ≈90% to only 2% of the stress relaxed for the 100% versus 0% alkyl hydrazone formulations, respectively (Figure 2b,c). As the functionalization of the HA-Hyd with the azide crosslinker was increased from 0–100%, fewer alkyl hydrazone crosslinks were present, changing the stress relaxation properties of the hydrogel. After 4 days in culture, the 88% alkyl hydrazone bonds condition maintained a stress relaxation profile which continued to relax >80% of the applied stress (Figure S1, Supporting Information). As alkyl hydrazone content further decreased, major differences in the stress relaxation properties were observed. Specifically, each sample containing 75% alkyl hydrazones or less relaxed <50% of the applied stress, with the 50%, 25%, and 0% formulations relaxing 10% or less of the applied stress (Figure 2c). These results suggest a sharp decline in the stress relaxing capabilities of the hydrogel, as fewer dynamic bonds are present in the network and the molar ratio of PEG to HA significantly exceeds 1 (Figure S2, Supporting Information).
All stress relaxation curves were normalized to their initial values and the data were fit to a Kohlrausch–Williams–Watts (KWW) function (Equation (2)):[18]
| (2) |
In this equation, the normalized stress, σ/σ0, was modeled as an exponential decay with the time constant, τk, that exists in a distribution described by the stretching parameter, β.[27] As reported in the literature, hydrogel stress relaxation encompasses a broad range of relaxation time constants due to compositional and topological heterogeneities (i.e., multiple covalent adaptable chemistries, polymer chain entanglements and loops, and differing polymer network backbones) that exist within a hydrogel network.[14,18] KWW functions are commonly used for many different materials, as an empirical relationship to describe the relaxation behavior of samples that exhibit broadly heterogenous relaxation timescales.[28,29] The heterogeneity of the relaxation time constants is qualitatively characterized through β, where a value of 1 represents a singular relaxation time constant and a broadening of the relaxation distribution as β approaches 0 (0 < β < 1).[27,30] The fitted model parameters for each hydrogel composition are shown in Figure 2d, where β > 0.5 for most samples, indicating a single relaxation mode is dominant for the time scale of stress relaxation.
Using this data, an average relaxation time constant, (<τ>), was calculated by integrating the model over the entire time domain (t = 0 to t = ∞):
| (3) |
A characteristic relaxation time for each hydrogel composition was calculated using Equation (3) and parameters fit to the stress relaxation data. These characteristic relaxation times were found to span four orders of magnitude (<τ88%> = 4.2 × 103 s to <τ0%> = 1.4 × 107 s) representing precise control over the timescale of relaxation within the hydrogels that can range from roughly one and a half hours to 6 months (Figure 2e). Further analysis of the stress relaxation data indicated that the 100% and 88% alkyl hydrazone bonds conditions showed no significant difference in percent stress relaxed, with both relaxing > 80% of the applied stress over a 6-h time course. In contrast, the 75% or less alkyl hydrazone bond conditions relaxed <50% of the applied stress over a 6-h time course (Figure 2f).
To determine the equilibrium swollen shear storage modulus of each hydrogel formulation, the samples were swollen in PBS for 24 h and subsequently analyzed with rheology. The final equilibrium storage modulus (G′) was found to be ≈400±150 Pa across all conditions, with no significant difference between any of the conditions (Figure 2g). Along with this, the swelling ratios of the extreme conditions, 88% and 0% alkyl hydrazone conditions, were analyzed in cell culture media. Over the course of 4 days, the swelling ratio achieved an equilibrium as no significant change was observed between days 2 to 4, indicating a stabilization of the hydrazone material (Figure S3, Supporting Information). Therefore, for cell culture experiments using these formulations, the primary difference between hydrogel mechanical properties was stress relaxation behavior.
2.2. Mesenchymal Stem Cell Behavior as a Function of Hydrogel Stress Relaxation
Bone marrow derived rMSCs were encapsulated at a density of 3 million cells mL−1 into the same HA-PEG hydrogel formulations supplemented with 1 mm KRGDS (RGD). RGD promotes rMSC survival by enabling cell-matrix interactions and was conjugated to the HA-Hyd via a benzaldehyde group. Viability of encapsulated rMSCs was measured using Calcein AM and ethidium homodimer live/dead stains. After 24 h, >85% viability was measured across all samples (Figure 3a). Next, rMSC morphology was investigated as a function of matrix stress relaxation, as Chaudhuri et al. found that MSC spreading was significantly influenced via stress relaxation in alginate hydrogels, with a fast-relaxing hydrogel leading to greater spreading.[31] After 4 days of culture, rMSC-laden hydrogels were fixed and stained to visualize the cytoskeletal morphology (F-actin, red) and nuclear shape (DAPI, blue) with a laser scanning confocal microscope (Figure 3b–d). Over the course of the experiment, the 100% alkyl hydrazone formulation completely degraded, which was attributed to a combination of the hydrolytic degradation of the hydrazone bond and potential reaction of the aldehydes with serum proteins in the medium.[32,33] This observation further emphasizes the need to stabilize hydrazone materials for longer-term cell studies.
Figure 3.
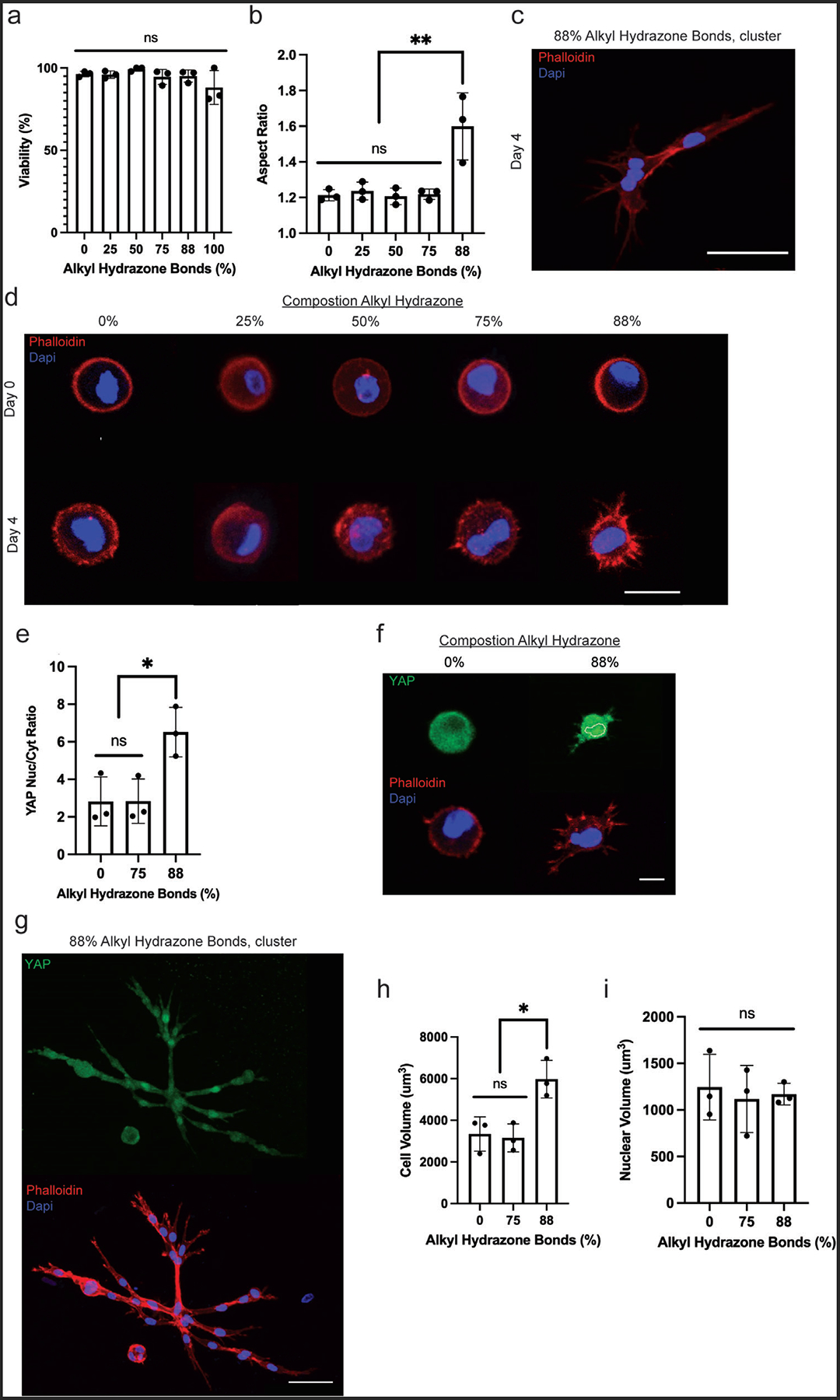
High degrees of stress relaxation led to rMSC spreading and cluster formation. a) To ensure viability post-encapsulation, cells were analyzed after 24 h in a growth medium. b) To study morphological changes as a function of stress relaxation, rMSCs were encapsulated on Day 0 and placed in a growth medium for 4 days. After 4 days, the rMSCs were fixed and immunostained. c) Representative image showing the formation of small clusters in the 88% alkyl hydrazone bonds condition by day 4, scale bar = 50 μm. d) Top: representative cell shape on Day 0 post-encapsulation across all conditions. Bottom: representative cell shape on Day 4 across all conditions, scale bar = 20 μm. e) Yap analysis of the cells after 4 days post-encapsulation, with the 88% alkyl hydrazone bonds condition showing significant Yap nuclearization. f) Top: YAP cell signal on Day 4 post-encapsulation in the 0% and 88% alkyl hydrazone bond experimental conditions. Bottom: representative cell shape on Day 4 within the 0% and 88% alkyl hydrazone bonds experimental conditions, scale bar = 10 μm. g) Top: YAP cell signal of a small cluster on Day 4 post-encapsulation in the 88% alkyl hydrazone bonds experimental condition. Bottom: representative cell shape on Day 4 of a small cluster within the 88% alkyl hydrazone bonds experimental condition. After 4 days of growth, the 88% alkyl hydrazone bonds condition showed significant cell spreading, as the cells began to become elongated within the hydrogels, scale bar = 50 μm. h) The average cellular volume of rMSCs after 4 days in the 88%, 75%, or 0% alkyl hydrazone bonds conditions. i) The average nuclear volume of rMSC nuclei after 4 days in the 88%, 75%, and 0% alkyl hydrazone bonds conditions. The Day 4 timepoint could not be acquired for the 100% alkyl hydrazone bonds condition as it had completely degraded by day 4. Values are plotted as mean ± standard deviation. *p<0.05,**p<0.01,: n.s. p>0.05, based on one-way ANOVAs. Three biological replicates (n = 3) with at least 30 MSCs analyzed per biological replicate.
Across all conditions, only the rMSCs encapsulated in 88% alkyl hydrazone crosslinks (i.e., highly adaptable, but stable for longer-term culture), showed significant spreading by Day 4, as compared to the other conditions (Figure 3b). Over the time course of culture from day 0 to day 4, the cellular aspect ratio increased from roughly 1.0 to greater than 1.60 ± 0.18 and was indicative of physical remodeling of the surrounding network. rMSCs encapsulated in the 0, 25, 50, and 75% alkyl hydrazone containing hydrogels remained largely rounded, with aspect ratios of 1.20 ± 0.038 or less, and did not show statistically significant spreading by day 4 (Figure 3b,d). In contrast, small clusters of rMSCs were observed in the 88% alkyl hydrazone; this phenotype was not seen in any other condition (Figure 3c). To determine the MSCs phenotype after 4 days in culture, the MSCs were stained for CD105, an integrin which is expressed on un-differentiated MSCs. In the 88% alkyl hydrazone condition 90 ± 9% of the MSCs expressed CD105 and in the 0% alkyl hydrazone condition 80 ± 5% of the MSCs expressed CD105 (Figures S4 and S5, Supporting Information).[34] Next, the mechanism behind cell morphology and cluster formation was investigated via actomyosin contractility. Blebbistatin was incorporated into the cell culture media (30 μm) as it inhibits the formation of myosin II, a key component of the actin-myosin construct. Upon inhibition in the 88% alkyl hydrazone condition, there was not a significant change in aspect ratio between the inhibited and uninhibited conditions (Figure S6, Supporting Information). However, the cell protrusions were seen to take on a unique morphology, and the formation of clusters was no longer readily observed, indicating a dependence upon actomyosin contractility (Figure S7, Supporting Information).
Previously, network adaptability in protease degradable HA hydrogels was shown to induce MSC spreading and YAP nuclear localization.[35] With this in mind, mechanical sensing as a function of the network adaptability was measured via analysis of the nuclear to cytoplasmic YAP ratio. By day 4, the 88% alkyl hydrazone bond condition showed greater nuclear localization of YAP, with an average nuclear to cytoplasmic ratio of 6.5 ± 1.3, as compared to the slower stress relaxing conditions of 75% and 0%, which had average nuclear to cytoplasmic ratios of 2.8 ± 1.2 and 2.8 ± 1.3, respectively (Figure 3e,f). Along with this, small clusters of cells were again observed by day 4 in the 88% alkyl hydrazone bond condition and stained for YAP; however, the clusters were not included in the analysis, as differentiating them proved challenging due to the multinuclear structures (Figure 3g).
Further, Lee et al. showed MSC volume expansion occurred in faster-relaxing hydrogels, leading to increased MSC spreading.[36] Therefore, the cellular and nuclear volumes were analyzed on day 4 for the 88%, 75%, and 0% alkyl hydrazone bond conditions to better understand the effects of YAP localization. Noticeably, there was a modest increase in cell volume in the 88% alkyl hydrazone condition as compared to the 75% and 0% alkyl hydrazone conditions, with an average cell volume of ≈6100 ± 4500 μm3, as compared to ≈3200 ± 1700 and 3500 ± 1600 μm3, respectively (Figure 3h). In contrast, the nuclear volumes remained consistent across all conditions, with an average volume of ≈1200 μm3 (Figure 3i).
With the statistically significant increase in cellular volume, the YAP nuclear to cytoplasmic ratios are better understood. As YAP is translocated to the nucleus the nuclear volume remains relatively consistent across conditions, therefore, YAP density in the nucleus increases. In contrast, the intensity in the cytoplasm decreases as the cytoplasmic volume increases (with increasing alkyl hydrazone bonds and HA content). The change in cellular morphology and YAP in the 88% alkyl hydrazone condition suggests a critical proportion of adaptable crosslinks in the pericellular region allow for viscoelasticity-induced cell spreading and morphological changes. Specifically, rMSCs respond to networks that have fast stress relaxation timescales (≈ 2 h), which allow for microenvironmental rearrangements that lead to changes in cell morphology and clustering.[13] Interestingly, the 88% alkyl hydrazone bond condition was the only condition that showed significant cell spreading or YAP nuclear localization, which may be partly explained by the molar ratios of HA to PEG, as the 88% alkyl hydrazone bond condition was the only condition with a HA to PEG ratio >1.
Cell spreading and morphology in covalent adaptable networks require coordinated relaxation of multiple bonds over the size scale of microns. However, rMSCs are metabolically active and can deposit ECM that impacts cell signaling, even after short culture periods. With this in mind, we characterized matrix deposition as a function of the hydrogel stress relaxation properties by measuring nascent protein deposition in rMSC-laden gels. Nascent protein was visualized in the hydrogels via the incorporation of a noncanonical amino acid L-Homopropargylglycine (HPG). The HPG was incorporated into newly synthesized proteins and enabled the visualization of the deposited proteins via a copper-catalyzed click-reaction of an azide-functionalized fluorophore onto the HPG (Figure 4a). In addition, the cell membrane was visualized using an HCS cell mask blue stain (Figure 4a). rMSCs were encapsulated in all hydrogel conditions. The 88% alkyl hydrazone condition showed extensive protein deposition throughout the pericellular region, especially in comparison to the elastic control (Figure 4b,c). The 88% alkyl hydrazone condition had a mean protein thickness of 1.45 ± 0.38 μm, and an average maximum protein thickness of 2.31 ± 0.77 μm (Figure 4d). In contrast, the 75% alkyl hydrazone condition had a mean protein thickness of 1.21 ± 0.30 μm and an average maximum protein thickness of about 1.88 ± 0.59 μm, with the 50, 25, and 0% alkyl hydrazone bond hydrogels having a mean protein thickness of 1.05 ± 0.25 μm or less, and an average maximum protein thickness of about 1.7 ± 0.57 μm or less. The total area of deposited nascent proteins indicated that the fastest-relaxing hydrogel (88% alkyl hydrazone) had the greatest amount of total nascent protein deposition (Figure 4e).
Figure 4.
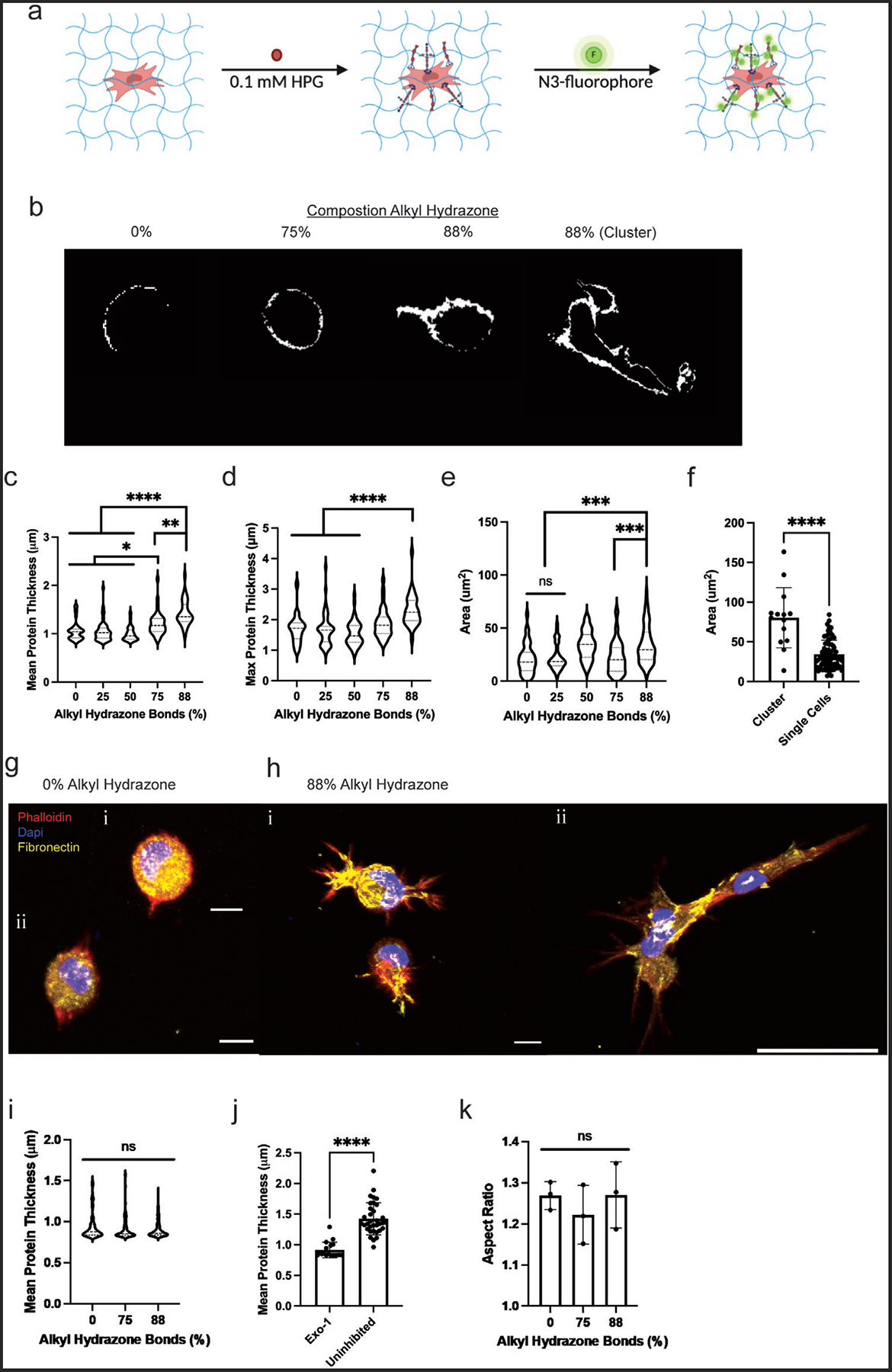
Nascent protein deposition increases with increasing stress relaxation. a) HPG was incorporated into the secreted proteins as a methionine homologue, and then fluorescently labeled with an azide-fluorophore, which enabled the visualization of the secreted proteins. b) Shows the visualization of the secreted proteins in the significantly different populations for protein deposition, 88%, 75%, and 0% alkyl hydrazone bonds conditions and includes both single cell and clustered nascent protein deposition from the 88% alkyl hydrazone bonds condition. c) Graphical representation of the mean secreted protein thickness, with the 88% alkyl hydrazone bonds condition secreting, on average, a protein thickness of 1.5 μm, and the 0% alkyl hydrazone bonds condition secreting, on average, a protein thickness of 1 μm. d) Represents the maximum secreted protein with 88% alkyl hydrazone bonds condition secreting roughly 2.5 μm, and the 0% alkyl hydrazone bonds condition secreting 1.8 μm. e) The average area of the deposited proteins surrounding the rMSCs in each condition, with the 88% alkyl hydrazone bonds condition depositing the greatest area of secreted protein. f) The average area of deposited protein between rMSCs clustered versus single cell rMSCs in the 88% viscoelastic condition. g) (i,ii) Represents the deposition of fibronectin in the 0% alkyl hydrazone bonds condition, where it is localized around the cell, scale bar = 10 μm. h) (i,ii) Represents the deposition of fibronectin in the 88% alkyl hydrazone bonds condition, with highly localized deposition in the spread portions of the cell, shown in both single cells and clustered rMSCs, scale bar = 10 μm and 50 μm, respectively. i) The mean secreted protein thickness upon inhibition with Exo-1. j) Compares the mean secreted protein thickness within the 88% alkyl hydrazone bonds condition, for both the Exo-1 inhibited and uninhibited conditions. k) The aspect ratios for the 0, 75, and 88% alkyl hydrazone conditions upon inhibition of Exo-1, showing the reduction in MSC spreading. Values are plotted as mean ± standard deviation, ****p< 0.0001, ***p<0.001, **p<0.01, *p<0.05, n.s. p>0.05, based on one-way ANOVAs and Student’s t-tests. Three biological replicates (n = 3) with at least 30 MSCs and 5 slices per single cell were analyzed per biological replicate.
Furthermore, the rMSCs in the 88% alkyl hydrazone condition could be separated into two distinct phenotypes: those which clustered together and those that remained as single cells. Cells within these two groups were analyzed for the total amount of nascent protein deposition and rMSCs residing in clusters secreted greater amounts of nascent proteins as compared to single cells (Figure 4f). To extend on this analysis, the fibronectin content within the nascent proteins deposited was quantified. Fibronectin was highly secreted and shown to localize in the branches extending from the rMSCs in the 88% alkyl hydrazone condition. In the 0% alkyl hydrazone condition, fibronectin deposition was seen uniformly around the cell (Figure 4g,h).
Taken together, this data suggests that rMSCs secrete a significant pericellular matrix, around the cellular niche in fast-relaxing hydrogels. Previously, Loebel et al. found an increase in protein deposition over time within a dynamic hydrogel. In this study, we sought to better understand the role of stress relaxation of compositionally defined hydrogels on the composition and deposition of the pericellular matrix. Since stress relaxation can affect mechanosensing over time, we completed Exo-1 inhibition studies, blocking exocytosis. (Figure 4i). With Exo-1 inhibition, nascent protein deposition was significantly reduced in both the 88% and 75% alkyl hydrazone bond conditions, with respective deposited pericellular protein thicknesses of 0.91 ± 0.11 and 0.91 ± 0.14 μm, which was comparable to the 0% alkyl hydrazone controls (0.94 ± 0.16 μm) (Figure 4i,j). Further analysis of exocytosis inhibition revealed a reduction in rMSC spreading with Exo-1 treatment, where the 88% and 75% alkyl hydrazone bond conditions had aspect ratios of 1.27 ± 0.08 and 1.22 ± 0.07 μm, respectively. These values are similar to that of the 0% alkyl hydrazone control of 1.26 ± 0.03 μm (Figure 4k). This illustrates not only the importance of cell-matrix interactions, but of pericellular protein deposition, as both are necessary for mechanosensing, which leads to cellular spreading. Effectively, this work showed a dependence of rMSC spreading and clustering upon the degree of relaxation-dependent pericellular protein deposition, suggesting a critical amount of protein deposition for certain phenotypes to occur on viscoelastic substrates.
In summary, this work highlights the use of tunable material platforms to influence the mechanosensing and secretory profiles of rMSCs, in response to the extent of compositionally defined stress relaxation. It would be of interest to investigate the cytokine secretory profiles of MSCs from various tissue sources in differing stress relaxing environments to better understand their dependence on the matrix microenvironments, in the context of compositionally independent stress relaxation rates.[35] This information could further instruct the development of platforms for expanding MSCs and delivering them in vivo for clinical application. Ultimately, we show that short-term stress relaxation dependence influences rMSC spreading, cluster formation, and the resulting protein secretion.
3. Conclusion
A stress relaxing biomaterial platform was engineered with controlled material properties by utilizing a covalent adaptable network formed with dynamic hydrazone bonds and a stabilizing SPAAC network formed via an elastic triazole bond. This hybrid network enabled control over the degree of stress relaxation by varying the amount of hydrazone-functionalized HA with an azide crosslinker and PEG-alkyne; this led to control over the ratio of the alkyl hydrazone to SPAAC crosslinks present. Hydrogel formulations were varied from 100%, 88%, 75%, 50%, 25%, and 0% alkyl hydrazone bonds, leading to stress relaxation properties ranging from >80% to <10%. The degree of stress relaxation impacted rMSC cell spreading and mechanosensing. Specifically, cell aspect ratios of 1.60 ± 0.18 and YAP nuclear to cytoplasmic ratios of 6.5 ± 1.3 were measured in the 88% alkyl hydrazone bond condition. In contrast, the 0% alkyl hydrazone bond condition had aspect ratios of 1.21 ± 0.03 and YAP ratios of 2.8 ± 1.3, on average. Finally, the influence of stress relaxation of rMSC ECM deposition was investigated; increased stress relaxation led to enhanced pericellular protein deposition. The 88% alkyl hydrazone bond condition had a mean protein thickness of 1.45 ± 0.38 μm, whereas the 75% alkyl hydrazone bond condition had a mean protein thickness of 1.21 ± 0.30 μm and the 50%, 25%, and 0% alkyl hydrazone bond conditions thicknesses were all 1.05 ± 0.25 μm or less. In summary, this novel biomaterial system permitted systematic control over the degree of stress relaxation using covalently adaptable linkages in hybrid gels containing both biopolymer and synthetic polymer components, and the corresponding influence of matrix relaxation on rMSCs mechanosensing and ECM deposition was revealed.
4. Experimental Section
Synthesis of HA-Aldehyde and Characterization:
HA-Ald was synthesized following previously established protocols.[19] Briefly, Ha (500 kDa, 500 mg) was dissolved in dH2O (50 mL), then, sodium periodate (267.5 mg, 1:1 periodate/HA molar ratio) was added into the reaction mixture. The reaction was stirred in the dark for 2 h and successively quenched with the addition of ethylene glycol (70μL). The reaction was then dialyzed for 3 days against dH2O (8000 MWCO) and lyophilized for 3 days to yield a white powder (471 mg, 94% yield). HA-Ald was flash frozen in liquid nitrogen and stored at −20 °C until further use. The functionalization of the HA-Ald macromer was quantified using a 2,4,6-Trinitrobenzene Sulfonic Acid (TNBS) assay as previously described.[37,38] Briefly, the HA-Ald was dissolved at 2 wt.% (w/v) and then reacted with tert-Butyl carbazate (t-BC, in 1% trichloroacetic acid) in dH2O. After 24 h, the HA-Ald/t-BC and t-BC standards were reacted with 0.5 mL TNBS (6 mm in 0.1 sodium tetraborate at pH 8) for 1 h. Samples were then reacted with 0.5 N hydrochloric acid and measured at an absorbance of 340 nm on a microplate reader (≈35% functionalization).
Synthesis of HA-Hydrazide and Characterization:
HA-Hyd was synthesized following previously established protocols.[19] Briefly, Ha (60 kDa, 500 mg) was dissolved in dH2O (100 mL) before adipic acid dihy-drazide (≈6.5 mg, >60x molar excess) was added in large molar excess to the reaction and the pH was adjusted to 6.8. 1-Ethyl-3-(3-dimethylaminopropyl)carbodiimide (EDC, 776 mg, 4 mmol) and hydroxybenzotriazole (HOBt, 765 mg, ≈6 mmol) were separately dissolved in a DMSO/dH2O mixture (1:1) and added dropwise to the reaction. The pH was adjusted to 6.8 every 30 min for 4 h and then allowed to react for 24 h. The solution was dialyzed against dH2O (8000 MWCO) for 3 days and then lyophilized for 3 days. Once dried, the products were weighed, dissolved in a 5 wt.% NaCl/dH2O solution, and precipitated into pure ethanol and dialyzed for another 3 days. Products were then lyophilized for 3 days to yield a white powder (430 mg, 86% yield), and flash frozen in liquid nitrogen and stored at −20 °C until use. HA-Hyd was characterized using 1H NMR (≈40% functionalization).
Synthesis of PEG-BCN and Characterization:
PEG-BCN was synthesized following previously established protocols.[25] Briefly, an 8-arm 40kDa PEG-amine (1.0 g, 0.2 mmol amine) and BCN-oSu (0.1 g, 0.343 mmol, 1.7x) were added to a 50 mL round bottom (RB) flask (dried overnight at 80 °C). A minimal amount of anhydrous DMF was added to the RB flask to dissolve the contents (10 mL). The flask was then placed under argon and stirred at room temperature. N,N-Diisopropylethyleamine (0.8mmol, 4x) was added to the solution and the reaction was left to proceed overnight. The following day, the reaction mixture was diluted with dH2O and dialyzed for 3 days (8000 MWCO). The dialyzed solution was lyophilized for 3 days, resulting in a dried white powder as the PEG-BCN product (0.985 g, 98% yield). End group functionalization was confirmed using 1H NMR (>95% functionalization).
Peptide Synthesis:
Benzaldehyde-KGRGDS was synthesized using standard Fmoc chemistry and Rink Amide MBHA resin on a Protein Technologies Tribute Peptide Synthesizer (0.5 mmol scale), as previously described.[15] After deprotection of the N-terminus, the resin was transferred to a peptide synthesis glass vessel and washed with DCM (3×10 mL). HATU (760.5 mg, 2.0 mmol), 4-Formylbenzoic acid (mg, 2.0 mmol), DIPEA (700 μL), DMF (10 mL), and DCM (3 mL) were added to the vessel, and the mixture was stirred at room temperature for 3 h. The solution was drained from the vessel under argon pressure and the resin was washed with DCM (5×10mL). Synthesized peptides were cleaved using, a peptide cleavage solution formed by dissolving dithiothreitol (DTT) and phenol (1:1) in a solution of 95% trifluoroacetic acid (TFA), 2.5% tri-isopropylsilane (TIPS), and 2.5% deionized water. The synthesized peptides were cleaved at room temperature for 2 h. Cleaved peptides were precipitated in cold diethyl ether, recovered by centrifugation, and desiccated overnight. The cleaved peptides were then purified by reverse-phase HPLC (Waters Delta Prep 4000) purification on a C18 column using a linear acetonitrile:water gradient. The collected fractions of purified peptides were identified by matrix-assisted laser desorption/ionization-time-of-flight (MALDI-TOF) mass spectrometry.
Hydrogel Formation and Rheological Characterization:
Functionalized HAs were dissolved in phosphate buffered saline (PBS, pH 7.4) at 3 wt.%. Functionalized PEG-BCN was dissolved in PBS at 10 wt.% and the azide-PEG3-phenolaldehyde (BroadPharm) crosslinker was dissolved at a concentration of 20 mm. Hydrogels were formed with 3 w/v% final polymer content based on stoichiometry. In situ rheology measurements were performed using a TA Instruments DHR-3 rheometer equipped with an 8 mm parallel plate geometry. Hydrogel formation was evaluated by time sweeps (1.0 Hz; 0.5% strain). For stress relaxation experiments, 6 h of measurement followed a 10% strain applied over 1 s, as dictated by the amount of time for the 100% viscoelastic condition to relax the majority of the applied stress (>90%). Formulations were defined by varying the percentage of alkyl hydrazone bonds present within the hydrogel by modifying the HA-hydrazide with a small molecule, benzaldehyde-PEG-azide, which effectively converts the HA-hydrazide to an azide. The formulations consisted of 100% (purely alkyl hydrazone), 88% alkyl hydrazone (i.e., 12% of the functional HA-hydrazide arms were functionalized with benzaldehyde-PEG-azide), 75%, 50%, 25%, and 0% (purely SPAAC, i.e., 100% of the functional HA-hydrazide arms were functionalized with benzaldehyde-PEG-azide), incorporating the necessary proportions of PEG-BCN and HA to achieve these functionalities. Mineral oil was applied to the hydrogel perimeter to prevent evaporation during the experiment. Theoretical models were fit using the curve fit application in MATLAB.
Hydrogel Swelling Ratio:
Hydrogels were made to be 100 μL gels at 3 wt.%, for the 88% and 0% alkyl hydrazone conditions. The theoretical mass was then calculated to be 3 mg and used as the dry mass of the hydrogels in Equation S1, Supporting Information. Every 24 h the mass of the hydrogels was recorded, and the swelling ratios were calculated, based on the swollen mass and the theoretical dry mass of the hydrogels. These swelling ratios were then normalized to the equilibrium swollen ratio.
Cell Culture and Encapsulation:
Sprague–Dawley rMSCs were purchased from Cyagen and were expanded and cultured following the manufacturer’s protocol. Briefly, the cells were obtained at passage 2 and expanded to passage 5 in growth medium specifically, Dulbecco’s Modified Eagle Medium (ThermoFisher) of low glucose (1 ng mL−1 glucose) supplemented with 10% FBS (Gibco), 50 μg mL−1 streptomycin (Gibco), and 0.5 μg mL−1 of Amphotericin B (Gibco). The media was changed after 24 h, and then every three days until 80–90% cell confluency was reached. When the desired cell confluency was reached, the cells were stored at passage 5 in liquid nitrogen. Hydrogels were fabricated by pre-reacting the HA-Hyd with the benzaldehyde-PEG-azide crosslinker, and KGRGDS based on stoichiometry for the desired 3 wt.% hydrogel formulation, of 100% (purely alkyl hydrazone), 88%, 75%, 50%, 25%, and 0% (purely SPAAC), and allowing them to react overnight. The following day, the HA-Ald, PEG-BCN, and PBS were mixed at stoichiometric ratios in a modified syringe barrel. rMSCs at passage 5 were thawed and placed in growth medium. The rMSC suspension was centrifuged (200 rcf, 5 min) and the resulting pellet was resuspended in the HA-Hyd, benzaldehyde-PEG-azide, KRGDS mixture for a cell density of 1–3 million cells mL−1. The cell suspension was then mixed into the syringe barrel with the HA-Ald, PEG-BCN, and PBS. The hydrogels were allowed to react until a soft gel formed (5 min or less) and then placed onto either benzaldehyde functionalized coverslips (for the 100, 88, and 75% conditions) or azide functionalized coverslips (for the 50, 25, and 0% conditions), and allowed to react for an additional 2 min before 1 mL growth medium was added.
Cell Viability, Morphology, and YAP Staining:
For viability assays, cells were encapasulated at a concentration of 1 million cells mL−1 and cultured for 24 hours. The cells were stained with Calcein AM (Life Technologies)/ethidium homodimer (Thermo Fisher Scientific) Live/Dead solution following the manufacturer’s protocol. To analyze morphological changes and YAP, rMSCs grown in the HA-PEG hydrogels, at a concentration of 3 million cells mL−1, were fixed with formalin (30 min, room temperature) following 4 days of growth and then rinsed with PBS. After fixation, the hydrogels were permeabilized with 0.1% Triton X-100 in PBS (1h, room temperature) and blocked using 5% BSA (Fisher Scientific) in PBS (1h, room temperature). The samples were incubated with DAPI (Sigma-Aldrich, 1:1000) and Alexa Flour 647 Phalloidin (Invitrogen,1:300) in blocking buffer (overnight, 4 °C) (morphology) or incubated with DAPI (Sigma-Aldrich, 1:1000), Alexa Flour 647 Phalloidin (Invitrogen,1:300) and YAP (Santa Cruz Biotechnology, 1:500) in blocking buffer. After washing with PBS with 0.05% Tween 20 (PBST) three times to remove excess stain, the fluorescently labeled rMSCs were imaged using confocal microscopy (Zeiss LSM 710) with a Plan-Aprochromat 20x (NA −1.0) water objective. ImageJ (NIH) was used to visualize and quantify morphological changes using the analyze particles plugin. Imaris was used to visualize and quantify YAP localization and nuclear: cytoplasm ratios following previously published protocols.[13] Briefly, 3D views were reconstructed from z-stack images (step size 1.0 μm). The cell and nucleus were determined from the Alexa Fluor 647 Phalloidin and DAPI channels, respectively. Using the YAP channel, the YAP nuclear to cytosolic ratio was calculated based on the following formula,
| (4) |
where Inuc and Icell are the total intensities of the YAP signal inside the nucleus and cell, respectively, and Vnuc and Vcell are the volumes of the nucleus and the cell, respectively.
Phenotypic Characterization and Blebbistatin Inhibition of MSCs:
Cells were encapsulated at a concentration of 3 million cells mL−1 and cultured in cell culture media, as explained above. The media was changed every 3 days, and on day 4 the cells were fixed, permeabilized, blocked, and stained for DAPI (Sigma-Aldrich, 1:1000), Rhodamine Phalloidin (Cytoskeleton, 1:300), and Anti-CD105 (Abcam, 1:250) as described above. For the Blebbistatin inhibition study, cell culture media containing the additional Blebbistatin (Sigma-Aldrich) at a concentration of 30 μm was replenished every 3 days. After 4 days, the cells were again fixed, permeabilized, blocked, and stained for DAPI (Sigma-Aldrich, 1:1000), Rhodamine Phalloidin (Cytoskeleton, 1:300), Anti-CD29 (BD, 1:250), and Anti-CD44 (Abcam, 1:250). The labeled rMSCs were imaged using confocal microscopy (Zeiss LSM 710) with a Plan-Aprochromat 20x (NA −1.0) water objective. ImageJ was used to visualize and quantify CD105, CD29, and CD44 positive cells.
Nascent Protein Deposition:
Cells were encapsulated at a concentration of 3 million cells mL−1 and cultured in glutamine-, methionine- and cystine-free high glucose DMEM (Thermo Fisher Scientific) supplemented with 0.201 mm cystine (Sigma-Aldrich), 100 μg mL−1 sodium pyruvate (Sigma-Aldrich), 50 μg mL−1 2-Phospho-L-ascorbate trisodium salt (Sigma-Aldrich), 10% FBS (Gibco), 50 μg mL−1 streptomycin (Gibco), 0.5 μg mL−1 of Amphotericin B (Gibco) and 0.1 mm L-Homopropargylglycine (HPG). Media was changed every 2 days. After 4 days, the hydrogels were fixed with formalin (30 min, room temperature) and then washed with PBS three times (5 min, room temperature). Then, the cells were stained with HCS Cell Mask Blue Stain (Invitrogen, 1:1000, 30 min, room temperature) in PBS. The hydrogels were then further washed with 1% BSA (10 min, room temperature) three times. Upon completion of the washes, a monofunctional azide was added in PBS at a dilution of 1:500 (45 min, room temperature). Again, the hydrogels were washed three times with 1% BSA in PBS and then an Alexa Flour 647 azide dye in a copper sulfate solution was added to the hydrogels (overnight, 4 °C). The following day, the hydrogels were washed three times with PBS (10 min, room temperature) and then imaged. The deposited protein was imaged using confocal microscopy (Zeiss LSM 710) and the deposited protein thickness was quantified following previously published protocols.[5] Briefly, the maximum intensity projection of a z-slice encompassing a 1 μm section of the cell was projected for each channel and the resulting projection was binarized utilizing Otsu’s thresholding. The HCS Cell Mask channel was subtracted from the nascent protein channel to yield a mask showing only nascent protein that was secreted on the cell exterior for the given 1 μm section of the cell being analyzed. The features of the extracellular secreted protein were quantified using the “BoneJ” plugin (ImageJ) and the mean thickness, maximum thickness, and area of each slice was reported. A total of 5 slices were analyzed per cell, and a total of 30 cells were analyzed per condition to yield significance.
Statistical Analysis:
All data were collected using 3 hydrogel replicates per condition. For each hydrogel at least 30 cells were analyzed. With the nascent protein deposition, 30 cells were analyzed per hydrogel, with 5 z-slices per cell analyzed. Data were compared using one-way ANOVAs with Tukey post-hoc comparisons or Student’s t-test in Prism 8 (GraphPad Software, Inc). Data were presented as mean ± standard deviation.
Supplementary Material
Acknowledgements
The authors acknowledge support from the National Institutes of Health, United States (Grant: R01DE016523). This work was supported in part by a grant from the United States Defense Advanced Research Projects Agency (DARPA, Grant: W911NF-19-2-0024) and by a grant from the National Science Foundation (NSF, Grant: 2033723). A.N.B. was supported by a Department of Education Graduate Assistance in Areas of National Need (DoEd GAANN) fellowship. M.W.Y. was supported by the NSF GRFP. Figure 1 schematics were created with Adobe Illustrator, Figure 4a was created with Biorender.com and Adobe Illustrator.
Footnotes
Supporting Information
Supporting Information is available from the Wiley Online Library or from the author.
Conflict of Interest
The authors declare a competing financial conflict of interest, a provisional patent has been filed (PCT/US22/14729). A.N.B, M.W.Y, B.V.S., and K.S.A. are named as inventors on the patent and have the potential to receive royalties from this patent.
Contributor Information
Alexandra N. Borelli, Department of Chemical and Biological Engineering, University of Colorado Boulder, Boulder, CO 80303, USA The BioFrontiers Institute, University of Colorado Boulder, Boulder, CO 80303, USA.
Mark W. Young, Department of Chemical and Biological Engineering, University of Colorado Boulder, Boulder, CO 80303, USA The BioFrontiers Institute, University of Colorado Boulder, Boulder, CO 80303, USA.
Bruce E. Kirkpatrick, Department of Chemical and Biological Engineering, University of Colorado Boulder, Boulder, CO 80303, USA The BioFrontiers Institute, University of Colorado Boulder, Boulder, CO 80303, USA; Medical Scientist Training Program, University of Colorado Anschutz Medical Campus, Aurora, CO 80045, USA.
Matthew W. Jaeschke, Department of Chemical and Biological Engineering, University of Colorado Boulder, Boulder, CO 80303, USA The BioFrontiers Institute, University of Colorado Boulder, Boulder, CO 80303, USA.
Sarah Mellett, Department of Chemical and Biological Engineering, University of Colorado Boulder, Boulder, CO 80303, USA.
Seth Porter, Department of Chemical and Biological Engineering, University of Colorado Boulder, Boulder, CO 80303, USA.
Michael R. Blatchley, Department of Chemical and Biological Engineering, University of Colorado Boulder, Boulder, CO 80303, USA The BioFrontiers Institute, University of Colorado Boulder, Boulder, CO 80303, USA.
Varsha V. Rao, Department of Chemical and Biological Engineering, University of Colorado Boulder, Boulder, CO 80303, USA The BioFrontiers Institute, University of Colorado Boulder, Boulder, CO 80303, USA.
Balaji V. Sridhar, Department of Physical Medicine and Rehabilitation, University of Colorado, Aurora, CO 80231, USA
Kristi S. Anseth, Department of Chemical and Biological Engineering, University of Colorado Boulder, Boulder, CO 80303, USA The BioFrontiers Institute, University of Colorado Boulder, Boulder, CO 80303, USA.
Data Availability Statement
The data that support the findings of this study are available from the corresponding author upon reasonable request.
References
- [1].da Silva Meirelles L, Fontes AM, Covas DT, Caplan AI, Cytokine Growth Factor Rev. 2009, 20, 419. [DOI] [PubMed] [Google Scholar]
- [2].Caplan AI, Correa D, Cell Stem Cell 2011, 9, 11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [3].Salzlechner C, Walther AR, Schell S, Merrild NG, Haghighi T, Huebscher I, Undt G, Fan K, Bergholt MS, Hedegaard MAB, Gentleman E, Mater. Adv 2020, 1, 2888. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [4].Huebsch N, Arany PR, Mao AS, Shvartsman D, Ali OA, Bencherif SA, Rivera-Feliciano J, Mooney DJ, Nat. Mater 2010, 9, 518. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [5].Loebel C, Mauck RL, Burdick JA, Nat. Mater 2019, 18, 883. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [6].Seib FP, Prewitz M, Werner C, Bornhäuser M, Biochem. Biophys. Res. Commun 2009, 389, 663. [DOI] [PubMed] [Google Scholar]
- [7].Maia FR, Fonseca KB, Rodrigues G, Granja PL, Barrias CC, Acta Biomater. 2014, 10, 3197. [DOI] [PubMed] [Google Scholar]
- [8].Chaudhuri O, Gu L, Darnell M, Klumpers D, Bencherif SA, Weaver JC, Huebsch N, Mooney DJ, Nat. Commun 2015, 6, 6365. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [9].Guvendiren M, Burdick JA, Curr. Opin. Biotechnol 2013, 24, 841. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [10].Tibbitt MW, Anseth KS, Biotechnol. Bioeng 2009, 103, 655. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [11].Foyt DA, Norman MDA, Yu TTL, Gentleman E, Adv. Healthcare Mater 2018, 7, 1700939. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [12].Drury JL, Mooney DJ, Biomaterials 2003, 24, 4337. [DOI] [PubMed] [Google Scholar]
- [13].Tang S, Ma H, Tu HC, Wang HR, Lin PC, Anseth KS, Adv. Sci 2018, 5, 1800638. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [14].Brown TE, Carberry BJ, Worrell BT, Dudaryeva OY, McBride MK, Bowman CN, Anseth KS, Biomaterials 2018, 178, 496. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [15].McKinnon DD, Domaille DW, Cha JN, Anseth KS, Adv. Mater 2014, 26, 865. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [16].Brown TE, Marozas IA, Anseth KS, Adv. Mater 2017, 29, 1605001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [17].Marozas IA, Cooper-White JJ, Anseth KS, New J. Phys 2019, 21, 045004. [Google Scholar]
- [18].Richardson BM, Wilcox DG, Randolph MA, Anseth KS, Acta Biomater. 2019, 83, 71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [19].Wang LL, Highley CB, Yeh YC, Galarraga JH, Uman S, Burdick JA, J. Biomed. Mater. Res., Part A 2018, 106, 865. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [20].Lou J, Liu F, Lindsay CD, Chaudhuri O, Heilshorn SC, Xia Y, Adv. Mater 2018, 30, 1705215. [DOI] [PubMed] [Google Scholar]
- [21].McKinnon DD, Domaille DW, Brown TE, Kyburz KA, Kiyotake E, Cha JN, Anseth KS, Soft Matter 2014, 10, 9230. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [22].Marquardt LM, Doulames VM, Wang AT, Dubbin K, Suhar RA, Kratochvil MJ, Medress ZA, Plant GW, Heilshorn SC, Sci. Adv 2020, 6, eaaz1039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [23].Wang H, Heilshorn SC, Adv. Mater 2015, 27, 3717. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [24].Kalia J, Raines RT, Angew. Chem., Int. Ed. Engl 2008, 47, 7523. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [25].Deforest CA, Tirrell DA, Nat. Mater 2015, 14, 523. [DOI] [PubMed] [Google Scholar]
- [26].Rice MA, Anseth KS, J. Biomed. Mater. Res., Part A 2004, 70, 560. [DOI] [PubMed] [Google Scholar]
- [27].Johnston DC, Phys. Rev. B 2006, 74, 184430. [Google Scholar]
- [28].Walker M, Godin M, Harden JL, Pelling AE, APL Bioeng. 2020, 4, 036107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [29].Taherunnisa S, Rami Reddy M, Piasecki M, al -, Saha D, Joshi YM, Bandyopadhyay R, Rep. Prog. Phys 1996, 59, 1133. [Google Scholar]
- [30].Wu JH, Jia Q, Sci. Rep 2016, 6, 20506. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [31].Chaudhuri O, Gu L, Klumpers D, Darnell M, Bencherif SA, Weaver JC, Huebsch N, Lee HP, Lippens E, Duda GN, Mooney DJ, Nat. Mater 2016, 15, 326. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [32].Fritz KS, Petersen DR, Free Radical Biol. Med 2013, 59, 85. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [33].Lee KY, Bouhadir KH, Mooney DJ, Macromolecules 2000, 33, 97. [Google Scholar]
- [34].Lin CS, Xin ZC, Dai J, Lue TF, Histol. Histopathol 2013, 28, 1109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [35].Caliari SR, Vega SL, Kwon M, Soulas EM, Burdick JA, Biomaterials 2016, 103, 314. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [36].pyo Lee H, Stowers R, Chaudhuri O, Nat. Commun 2019, 10, 529. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [37].Purcell BAJ, Brendan P, David L, Manoj CB, Shauna DM, Ryan WJ, Kia ZN, Heather D, Sarah P, Christine LB, James S, Parker FD, Joseph GH, Robert GC, Francis SG, Nat. Mater 2014, 13, 653.24681647 [Google Scholar]
- [38].Su WY, Chen YC, Lin FH, Acta Biomater. 2010, 6, 3044. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The data that support the findings of this study are available from the corresponding author upon reasonable request.