

Abstract
Gut commensals with the capacity to translocate across the intestinal barrier can drive the development of diverse immune-mediated diseases1–4. However, the key factors that dictate bacterial translocation remain unclear. Recent studies have revealed that gut microbiota strains can adapt and evolve throughout the lifetime of the host5–9, raising the possibility that changes in individual commensals themselves over time may impact their propensity to elicit inflammatory disease. Here we show that within-host evolution of the model gut pathobiont Enterococcus gallinarum facilitates bacterial translocation and initiation of inflammation. Using a combination of in vivo experimental evolution and comparative genomics, we found that E. gallinarum diverges into independent lineages adapted to colonise either luminal or mucosal niches in the gut. Compared to ancestral and luminal E. gallinarum, mucosally-adapted strains evade detection and clearance by the immune system, exhibit increased translocation to and survival within the mesenteric lymph nodes and liver, and induce increased intestinal and hepatic inflammation. Mechanistically, these changes in bacterial behaviour are associated with non-synonymous mutations or indels in defined regulatory genes in E. gallinarum, altered microbial gene expression programs, and remodelled cell wall structures. Lactobacillus reuteri also exhibited broadly similar patterns of divergent evolution and enhanced immune evasion in a monocolonisation-based model of within-host evolution. Overall, these studies define within-host evolution as a critical regulator of commensal pathogenicity that provides a unique source of stochasticity in the development and progression of microbiota-driven disease.
Gut commensals with pathogenic potential, referred to as pathobionts3, are believed to play causal roles in a diverse array of inflammation-associated diseases4, ranging from inflammatory bowel disease to metabolic syndrome. However, putative disease-driving pathobionts are also detected in ostensibly healthy humans10 and pathobiont colonisation often precedes overt disease development by years to decades. Genetic and environmental factors clearly contribute to this stochasticity3,4, as host genetic predisposition, microbiome composition, and environmental exposures can all impact disease susceptibility. However, the full concert of factors that contribute to stochasticity in pathobiont-driven inflammatory disease remains to be defined.
Although commensal strains are often conceptualized as static functional units that collectively comprise dynamic host-associated microbial communities, it is now clear that discrete microbial strains can adapt and evolve over the lifespan of an individual through within-host evolution5,6. Billions of de novo mutations are generated daily within the gut microbiome6 and enable commensal diversification and adaptation. We hypothesized that within-host evolution may contribute to the acquisition or enhancement of pathological behaviours by commensal pathobionts, and thus provide a novel microbial source of stochasticity in microbiota-driven inflammatory diseases. To test this hypothesis, we focused on the model pathobiont Enterococcus gallinarum.
Strain divergence in lupus-prone mice
E. gallinarum is a gram-positive facultative anaerobe found in more than 6% of human gut microbiomes11. E. gallinarum translocation to the mesenteric lymph nodes (mLNs) and liver accelerates the development of mouse models of lupus, autoimmune hepatitis and primary sclerosing cholangitis and has been detected in liver biopsies from autoimmune patients1,2. To examine the role of within-host evolution in bacterial translocation, we first isolated E. gallinarum strains from the liver and faeces of autoimmune-prone (NZW x BXSB) F1 mice (Fig. 1a), which were previously reported to exhibit E. gallinarum translocation1. We then performed whole-genome sequencing (WGS) on five faecal isolates and six liver isolates from these mice, along with seven additional E. gallinarum strains from diverse sources. Phylogenetic comparisons of the core genomes and pangenomes of these strains revealed a tight clustering of all (NZW x BXSB) F1-derived isolates (Fig. 1b, Extended Data Fig. 1a), indicating that these isolates were derived from the same lineage.
Fig 1|. Divergent evolution of liver and faecal E. gallinarum isolates in specific-pathogen free (NZW x BXSB) F1 mice and monocolonised C57BL/6 mice.
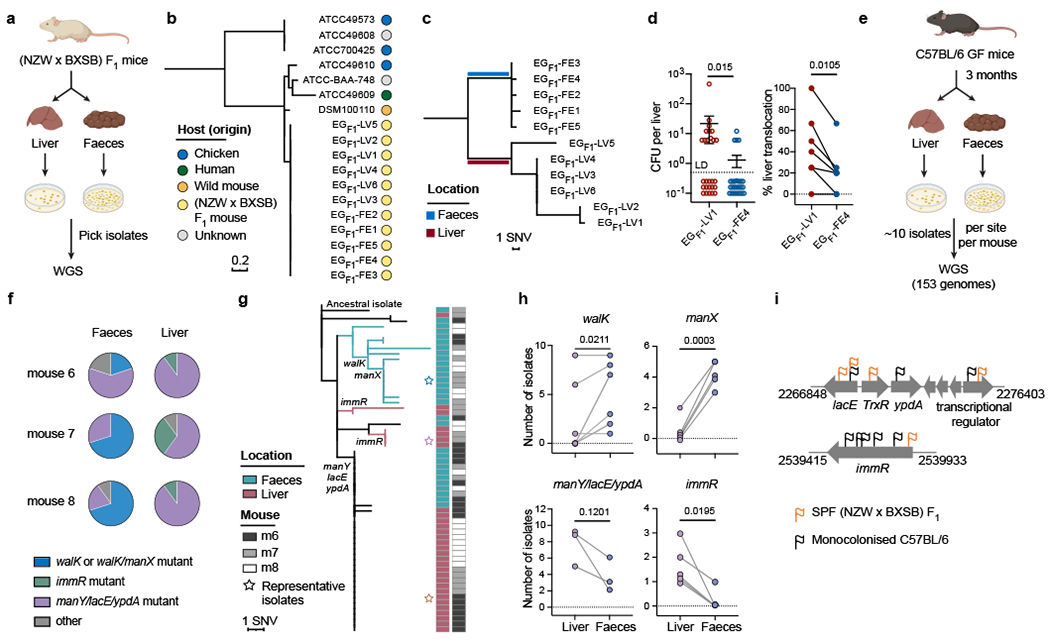
a, Schematic of E. gallinarum isolation from SPF (NZW x BXSB) F1 mice. b, Phylogenetic tree of E. gallinarum strains based on core-genome alignments. c, Phylogenetic tree of liver and faecal E. gallinarum isolates from (NZW x BXSB) F1 mice. d, E. gallinarum liver translocation in monocolonised mice. Bacterial load in liver (left) and percentage of mice showing liver translocation (right). n = 27 mice across 6 independent experiments. Data represent mean ± SEM. e, Schematic of experimental evolution in E. gallinarum monocolonised C57BL/6 mice. f, Distribution of select mutations in liver and faecal populations. Representative data from mice 6, 7, and 8 (co-housed) are presented. g, Phylogenetic tree of E. gallinarum isolates sampled from mice 6-8. h, Frequency of E. gallinarum isolates from faeces and liver harbouring mutations in the indicated genes. Each circle represents one mouse. walK (n = 6), manX (n = 5), manY/lacE/ypdA (n = 3), immR (n = 5). i, Mutations detected in liver E. gallinarum isolates from (NZW x BXSB) F1 and monocolonised mice. Each flag represents one unique mutation. c, g, Reference-based alignments. Unpaired two-tailed t-test (d, left) or paired two-tailed t-test (d, right, h). SNV, single-nucleotide variant. LD, limit of detection.
To enable high-resolution comparisons between (NZW x BXSB) F1-derived isolate genomes, we established a closed reference genome for a single faecal E. gallinarum isolate (EGF1-FE1) (Extended Data Fig. 1b). Using reference-based alignments, we identified 26 single-nucleotide variants (SNVs) and 10 indels (small insertions or deletions) across the eleven isolates as compared to the reference isolate (Extended Data Fig. 1c, Supplementary Table 1). Thirteen genes exhibited non-synonymous mutations and indels across at least two strains; many of these genes were predicted to encode transcriptional regulators, or were related to nutrient utilization (e.g., phosphotransferase system (PTS), ATP-binding cassette (ABC) transporters) and environmental sensing (two-component systems) (Extended Data Fig. 1d). Intriguingly, liver and faecal isolates segregated into two site-specific lineages, suggesting potential divergence of E. gallinarum into two populations with differential capacity to translocate to the liver (Fig. 1c). To directly test this possibility, we monocolonised groups of germ-free (GF) C57BL/6 mice with a representative liver or faecal isolate (EGF1-LV1 and EG F1-FE4, see Supplementary Table 7 for a summary of all strains) and examined bacterial translocation. Compared to the faecal isolate, the liver E. gallinarum isolate exhibited significantly higher levels of liver translocation (Fig. 1d). By contrast, the faecal E. gallinarum isolate predominated in the faeces (Extended Data Fig. 1e) after bi-colonisation of specific-pathogen-free (SPF) C57BL/6 mice with equal doses of these two strains. These results suggest that faecal isolates show enhanced competitive fitness in the gut lumen, whereas liver isolates exhibit an enhanced capacity to translocate to the liver.
Divergent evolution in gnotobiotic mice
Our data suggest that within-host evolution of E. gallinarum may drive divergence into at least two lineages that occupy distinct host-associated niches and exhibit differential translocation. However, because the mice from which these isolates were collected were naturally colonised by E. gallinarum, we cannot formally exclude the possibility that this divergence was due to the acquisition of distinct strains at birth. To definitively test the role of within-host evolution in E. gallinarum strain divergence and to map the evolutionary paths of E. gallinarum adaptation within individual hosts, we next established a simplified in vivo experimental evolution model using monocolonised C57BL/6 mice.
We inoculated eight GF mice with an individual faecal isolate from (NZW x BXSB) F1 mice (EGF1-FE4; hereafter referred to as the ancestral strain) and housed these mice in four separate cage-level gnotobiotic isolators (isocages) to enable within-host bacterial evolution and potential translocation to the liver (Extended Data Fig. 2a). After three months, we collected and whole-genome sequenced six to ten isolates from the liver and ten isolates from the faeces of each animal (153 isolates total; Fig. 1e). We detected 159 mutations across all isolates as compared to the ancestral strain, including 97 SNVs and 62 indels (Supplementary Table 2). All isolates harboured de novo mutations as compared to the ancestral strain, indicating that the colonising strain was either displaced during a selective sweep or reduced to a low abundance. In coding regions, 77/84 SNVs were non-synonymous and 37/39 indels resulted in disruptive in-frame deletions or frameshift mutations (Supplementary Table 2). While most mutations were unique to a single isolate, 57 mutations located in 30 genes were detected in multiple isolates (Extended Data Fig. 2b, c). Like liver isolates from (NZW x BXSB) F1 mice, mutations in these experimentally evolved isolates were enriched in genes encoding transcriptional regulators or involved in nutrient transport and environmental sensing (Extended Data Fig. 2d).
To examine whether mutations in specific genes were associated with site-specific colonisation across animals and cages, we compared the frequency of all detected mutations between liver and faecal populations. We found that mutations in walK and manX were enriched in faecal isolates, while mutations in manY, lacE, ypdA and immR were more common among liver isolates (Fig. 1f–h, Extended Data Fig. 3a–d). Mutations in these genes were detected in mice across independent experiments (i.e., across isocages), indicating parallel adaptive evolution. Similar to the SPF liver and faecal isolates, a representative faecal walK/manX mutant exhibited enhanced competitive fitness in the colon in bi-colonised mice as compared to representative liver isolates with mutations in manY, lacE, ypdA or immR or the ancestral isolate (Extended Data Fig. 3e). Altogether, these data suggest that within-host evolution drives the divergence of E. gallinarum into multiple lineages with distinct niche preferences.
Although their specific functions in E. gallinarum are poorly-characterized, manX, manY and lacE encode components of mannose-specific enzyme II12 and lactose-specific enzyme II13 of the PTS system, which mediate mannose and lactose transport respectively; walK14 and ypdA15 encode sensor histidine kinases from two-component systems and walK regulates cell wall metabolism16; and, immR encodes a helix-turn-helix transcriptional regulator that controls the expression of mobile genetic elements and potentially mediates adaptation to oxidative stresses17 (see Supplementary Table 8 for a summary of genes). Mutations in immR, lacE, and a downstream gene encoding a transcriptional regulator were also detected in liver isolates from SPF (NZW x BXSB) F1 mice (Fig. 1i). This shared mutational pattern across liver isolates from monocolonised and naturally-colonised autoimmune-prone SPF mice suggests that E. gallinarum may face similar selective pressures and acquire shared features in these two independent settings.
Liver strains seed from mucosal niches
We next sought to identify the niche(s) where liver isolates were selected and maintained. We reconstructed the evolutionary relationships between liver and faecal strains in each monocolonised mouse and noted that multiple mice (particularly those with high levels of liver translocation) harboured distinct lineages of E. gallinarum in the liver, some of which were connected to a faecal lineage and others that were detected only in the liver (Fig. 2a, Extended Data Fig. 4a–d). For example, in mouse seven, manY/lacE/ypdA triple mutants were detected in both liver and faeces, while immR mutants were only detected in the liver (Fig. 2a). This observation could result from either long-term colonisation and survival of this lineage in the liver, or incomplete sampling of an intestinal E. gallinarum population that exhibits enhanced liver translocation. To distinguish between these two possibilities, we first examined whether E. gallinarum liver isolates can persistently colonise the liver by intravenously injecting SPF mice with representative manY/lacE/ypdA and immR mutant liver strains and tracking their survival in the liver over time. Although both strains were detected at high levels in all animals after three days, most animals harboured no detectable E. gallinarum in their livers after two weeks (Fig. 2b, c). These results suggest that liver E. gallinarum strains cannot colonise the liver long-term and, instead, bacteria must continually re-seed the liver by translocating from the gut. Since many liver-associated lineages are poorly represented in faecal samples, we speculated that liver-translocating microbes may drain from a mucosal niche within the intestine. We thus assessed the distribution of lineage-defining mutations in E. gallinarum in the mucosa and lumen of the small and large intestine, as well as in the liver itself, via shotgun metagenomics (Fig. 2d). We found that mutations enriched in faecal populations were dominant in the intestinal lumen, particularly the colonic lumen. By contrast, liver-associated mutations were highly enriched in populations associated with the intestinal mucosa (Fig. 2e). Together, these data imply that liver isolates of E. gallinarum are derived primarily from subpopulations of intestinal E. gallinarum that colonise mucosa-associated niches.
Fig 2 |. E. gallinarum liver isolates are primarily derived from lineages associated with the intestinal mucosa.
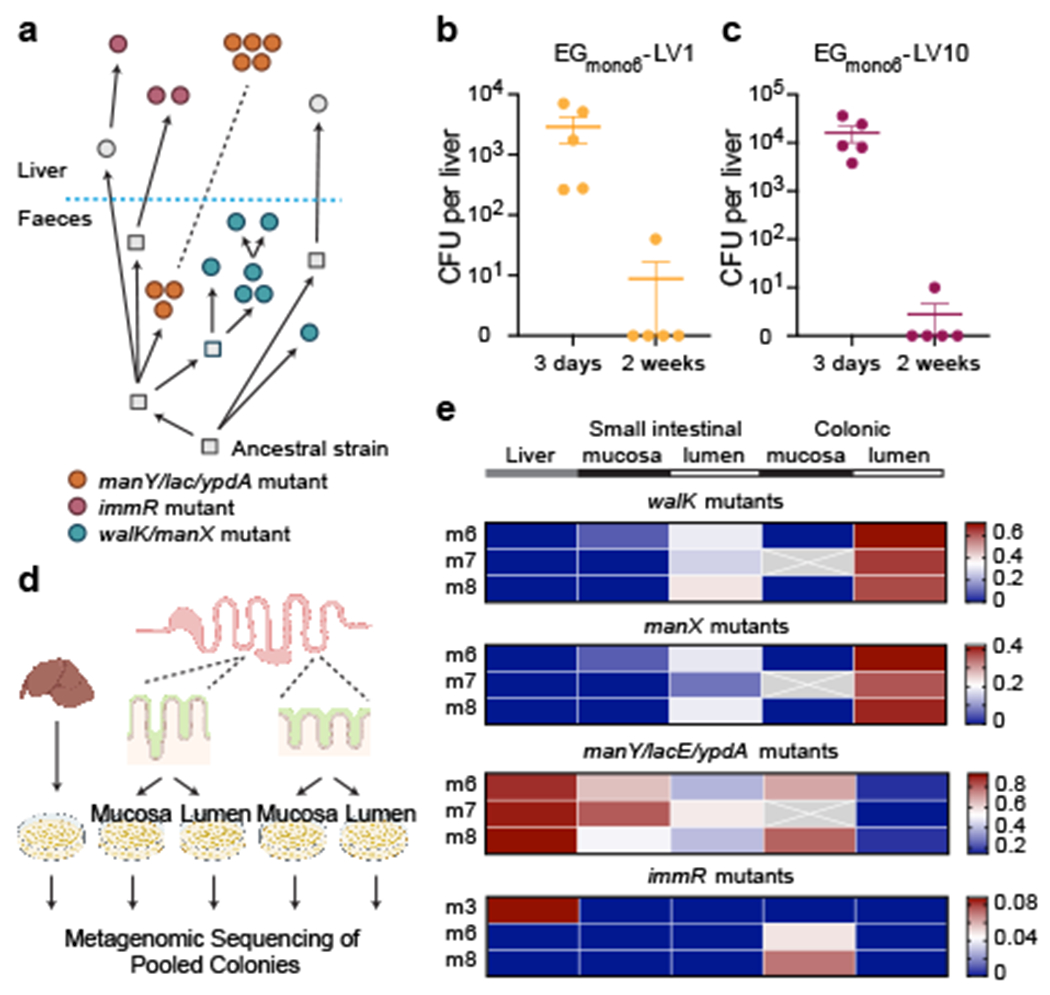
a, Reconstruction of the phylogenetic history of E. gallinarum isolates sampled from mouse 7. Circles represent sequenced isolates and squares represent hypothetical intermediate genotypes. Arrows connect related genotypes and dashed lines connect genetically identical isolates. Liver and faecal populations are separated by a blue dashed line. b, c, Liver clearance of intravenously injected E. gallinarum liver isolates EGmono6-LV1 (manY/lacE/ypdA mutant) and EGmono6-LV10 (immR mutant). See Supplementary Table 7 for a summary of all mutants. Bacterial load of EGmono6-LV1 (b) and EGmono6-LV10 (c) recovered from livers 3 days or 2 weeks post-injection. n = 5 mice. Representative of three independent experiments. Data represent mean ± SEM. d, Schematic of metagenomic sequencing of E. gallinarum populations across tissue sites in monocolonised mice. e, Allele frequency for select mutations across tissue sites. Rows represent mice and columns represent tissue sites. Crosses represent missing values.
Liver strains evade immune clearance
To definitively test whether monocolonisation-derived liver and faecal E. gallinarum isolates exhibit differential niche preference, we bi-colonised GF mice with representative liver (manY/lacE/ypdA or immR mutant) and faecal isolates (walK/manX mutant). We found that liver isolates were enriched in the small intestinal mucosa (SIM) and exhibited the lowest abundance in the colonic lumen (Fig. 3a). Host-associated niches, such as the colonic inner mucus layer and small intestinal crypts, are generally hostile to microbes due to robust host defences18. We speculated that liver isolates may originate from gut populations that have acquired increased resistance to immune clearance, which allows them to occupy mucosal niches. To test this hypothesis, we measured the sensitivity of liver and faecal isolates to killing by antimicrobial peptides and enzymes, or engulfment by phagocytes. As predicted, liver isolates were significantly more resistant to killing by cathelicidin-related antimicrobial peptide (mCRAMP) (Extended data Fig. 5a), lysozyme-mediated growth inhibition (Fig. 3b), and phagocytosis by bone marrow-derived macrophages (BMDMs) as compared to the faecal isolate (Fig. 3c).
Fig 3 |. E. gallinarum liver isolates exhibit increased resistance to immune clearance and altered cell wall structures.
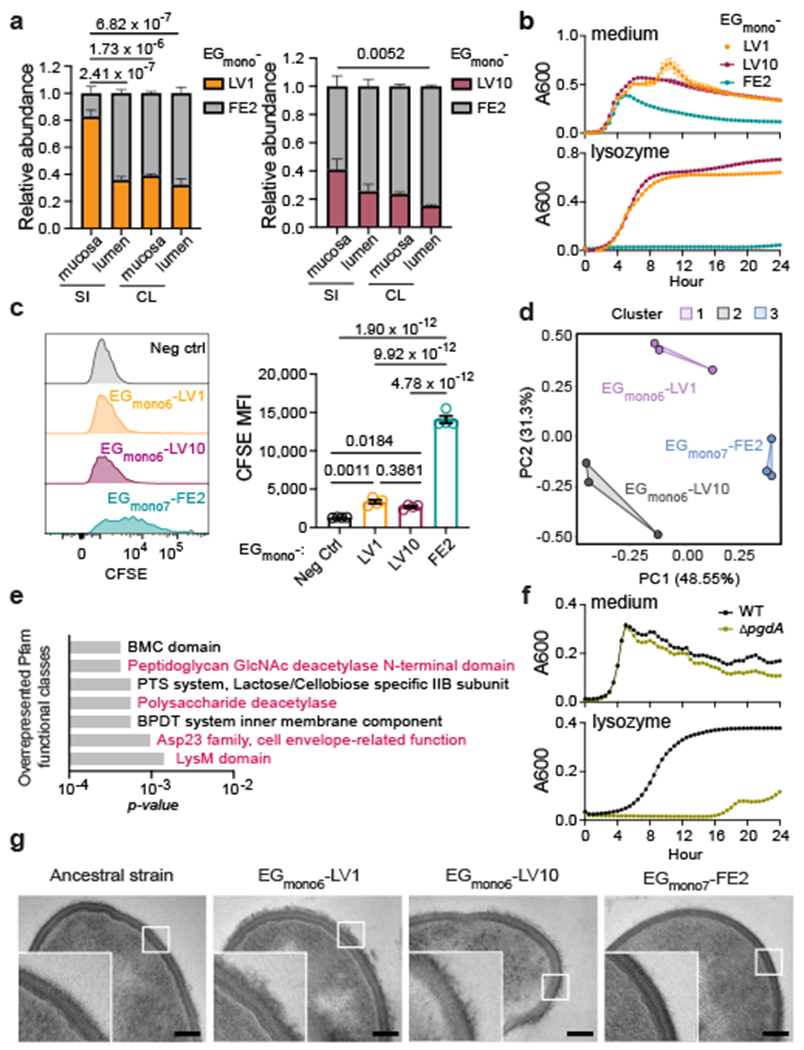
a, Relative abundance of E. gallinarum isolates in C57BL/6 mice bi-colonised with representative faecal (EGmono7-FE2) and liver EGmono6-LV1 (left) or EGmono6-LV10 (right) isolates. n = 3 mice. b, Growth curves of E. gallinarum isolates with or without 5 mg/ml lysozyme. n = 5 biological replicates. c, Phagocytosis of CFSE-labelled E. gallinarum by BMDMs as detected by flow cytometry. Representative histograms and quantification of MFI of CFSE signals in live F4/80+ cells. Neg ctrl, BMDM cells only. n = 4 biological replicates. d, e, Transcriptomic analyses of cultured E. gallinarum isolates. d, Principal component analysis. Clusters were generated by K-means. n = 3 independent cultures. e, Overrepresented Pfam functional classes (EGmono6-LV1 vs. EGmono7-FE2). f, Growth curves of WT versus pgdA mutant strains with or without 10 mg/ml lysozyme. n = 3 biological replicates. g, TEM images of cultured E. gallinarum isolates. Scale bars, 100 nm. Results are representative of n = 2 (a, f, g) or 3 (b-e) independent experiments. Data in (a-c, f) represent mean ± SEM. Some error bars in (b, f) were too small to show. Two-way (a) or one-way (c) ANOVA with Tukey’s post hoc test. SI, small intestine. CL, colon. Neg ctrl, negative control. CFSE, carboxyfluorescein succinimidyl ester. MFI, mean fluorescence intensity. BPDT, binding-protein-dependent transport.
To identify potential mechanisms of immune evasion, we assessed microbial gene expression for two representative liver isolates and one faecal isolate via RNA-sequencing (RNA-seq). The global transcriptomes of these three isolates clustered separately (Fig. 3d) and liver and faecal isolates exhibited approximately 1000 differentially expressed genes, indicating that the observed mutations in regulatory genes had widespread effects on bacterial gene expression (Extended data Fig. 5c, d). Both faecal and liver isolates also differed from the ancestral isolate (Extended Data Fig. 5e–g). Many genes with increased expression in liver isolates were related to bacterial cell wall structures (Fig. 3e, Extended Data Fig. 5h, i). For instance, homologs of pgdA, which encodes a peptidoglycan N-acetylglucosamine (GlcNAc) deacetylase, contribute to lysozyme resistance in other species19,20. Accordingly, a pgdA insertion mutant of E. gallinarum was significantly more sensitive to lysozyme-mediated inhibition (Fig. 3f) and liver clearance after intravenous injection (Extended Data Fig. 5j) as compared to the wild-type control. Liver isolates also exhibited a thick layer of capsular polysaccharides as measured by transmission electron microscopy (TEM), which was largely absent in the ancestral strain and faecal isolate (Fig. 3g). Notably, bacterial capsules can act as virulence factors for Enterococcus species by facilitating evasion of the innate immune response21. Overall, the bacterial cell wall modifications observed in liver isolates may explain their increased resistance to immune clearance.
Immunogenicity of divergent strains
We next examined whether liver and faecal lineages of E. gallinarum induced distinct host responses in the intestine via RNA-seq analysis of ileal epithelial cells (IECs) and whole ileal tissues. Eight hours after microbial colonisation, faecal isolate-colonised mice displayed increased expression of immune response genes in IECs, including genes involved in coordinating enhanced barrier defence, as compared to liver isolate-colonised mice (Extended Data Fig. 6a, b). In particular, genes involved in antigen processing and presentation (e.g., H2-DMb1, Tapbp), autophagy (e.g., Atg13, Atg14), and mucin production (e.g., Muc2, Muc3) were upregulated in IECs from faecal isolate-colonised mice (Extended Data Fig. 6b). At this early time point, whole ileal tissues exhibited equivalent responses to both liver and faecal isolates. The enhanced immune responses in IECs elicited by the faecal isolate persisted two weeks after colonisation (Fig. 4a, Extended Data Fig. 6c–e). However, at this later time point, liver isolate-colonised mice exhibited increased inflammatory signatures in ileal tissues (Extended Data Fig. 6c, d), including increases in markers of mononuclear phagocytes (e.g., Cd36, Cd44, Cd11c, Ly6c1, Cx3cr1) and phagocyte chemoattractants (e.g., Cxcl12, Cxcl14, Ccl11) (Fig. 4a). Notably, liver and faecal isolate-colonised mice exhibited roughly equivalent ileal bacterial loads (Extended Data Fig. 6f), indicating that the induction of distinct intestinal immune responses was not merely due to differential bacterial densities.
Fig 4 |. E. gallinarum liver isolates elicit limited barrier defences, exhibit increased bacterial translocation, and induce gut and liver inflammation.
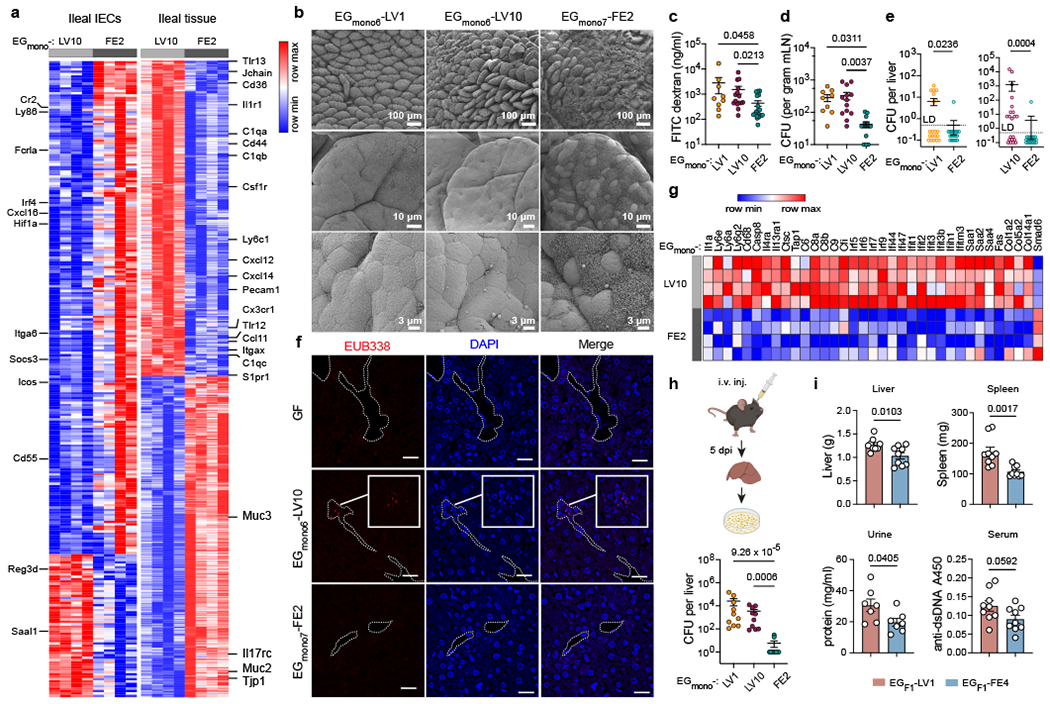
a, Differentially expressed genes in ileal epithelia or whole ileal tissues of C57BL/6 mice monocolonised with E. gallinarum for 2 weeks. Left and right heatmaps display distinct gene sets. n = 4 mice. b, Scanning electron microscope (SEM) images of distal ilea from monocolonised mice. c, Gut permeability of mice monocolonised with EGmono6-LV1 (n = 9), EGmono6-LV10 (n = 15), or EGmono7-FE2 (n = 18) for two weeks. d, e, Bacterial translocation in monocolonised mice. d, Bacterial load in mLNs. EGmono6-LV1 (n = 9), EGmono6-LV10 (n = 13), EGmono7-FE2 (n = 14). e, Bacterial load in liver. Data were combined from five independent experiments. Left panel, EGmono6-LV1 (n = 20), EGmono7-FE2 (n = 18). Right panel, n = 21 mice. f, Fluorescence in situ hybridization imaging of liver translocation in E. gallinarum colonised mice. EUB338 probe detects bacteria. Dashed lines highlight vessel edges. Scale bars, 20 μm. g, Transcriptomic profiles of livers from monocolonised mice. n = 4 mice. h, Liver clearance of intravenously injected E. gallinarum isolates. Bacterial load in livers 5 dpi. n = 10 mice. i, Imiquimod-induced autoimmune phenotypes in mice monocolonised with EGF1-LV1 or EGF1-FE4. Liver and spleen weights, urine protein concentrations, and serum anti-dsDNA autoantibodies. n = 9 mice. Results are representative of n = 2 (a-c, f, g, i) or 3 (h) or 5 (d) independent experiments. Data in (c-e, h, i) represent mean ± SEM. Two-tailed Wald test with Benjamini-Hochberg correction (FDR = 0.05) (a, g), Kruskal-Wallis test with Benjamini-Hochberg correction (FDR = 0.05) (c, h), one-way ANOVA with Tukey’s post hoc test (d), two-tailed Mann-Whitney test (e), unpaired two-tailed t-test (i). LD, limit of detection. dpi, days post-injection.
Consistent with RNA-seq results, the faecal isolate induced more mucus production (Fig. 4b) and increased goblet cell hyperplasia (Extended Data Fig. 6g) in the terminal ileum. Finally, faecal isolate-colonised mice exhibited increased expression of intraepithelial lymphocytes (IELs) markers, including Cd3e, Cd8a, Trdc and Itgae (Extended Data Fig. 6b) and displayed increased CD3+ IELs by immunohistochemistry (IHC) (Extended Data Fig. 6h). Altogether, these data support a model whereby faecal E. gallinarum elicits an enhanced epithelial cell response that leads to reinforcement of the epithelial barrier through increased mucus production and IEL recruitment22–24, while liver isolates evade initial detection at the epithelial surface and instead initiate inflammatory responses in the lamina propria.
Translocation elicits liver inflammation
As mice colonised with E. gallinarum liver isolates exhibited reduced intestinal mucus, IEL recruitment, and tight junction protein expression (Tjp1) (Fig. 4a), we speculated that they may also display impaired gut integrity. Indeed, liver isolate-colonised mice showed increased intestinal permeability as compared to faecal isolate-colonised mice (Fig. 4c). The enhanced barrier integrity in faecal isolate-colonised mice was independent of Toll-like receptor signalling but dependent on lymphocytes as Rag1−/− mice exhibited equivalent barrier integrity regardless of colonisation state (Extended Data Fig. 6i).
Based on the “leaky gut” model25, we next sought to examine whether liver isolates also exhibit increased translocation across the intestinal barrier into systemic sites such as the liver. In monocolonised mice, both representative liver isolates showed enhanced bacterial translocation to the mLNs (Fig. 4d) and liver (Fig. 4e, Extended Data Fig. 6j) as compared to a faecal isolate. Furthermore, mice monocolonised with representative liver isolates for two weeks exhibited increased hepatic inflammatory gene expression as compared to faecal isolate-colonised mice, including upregulation of genes encoding pro-inflammatory cytokines, interferon-stimulated genes, serum amyloid A proteins and collagen (Fig. 4g, Extended Data Fig. 6k).
As liver isolates were more resistant to mCRAMP, lysozyme, and macrophage phagocytosis in vitro, we also speculated that liver isolates may persist longer in the liver in vivo. To test this possibility, we intravenously injected mice with liver or faecal isolates and assessed the bacterial load in the liver after five days. While the faecal isolate was almost completely cleared by this time point, both liver isolates persisted at high levels (Fig. 4h). These results suggest that within-host evolution of E. gallinarum facilitates both enhanced translocation to and survival in the liver for at least 5 days but fewer than two weeks (Fig. 2b, c).
SPF liver strains aggravate autoimmunity
Given the enhanced liver-translocating capacity and shared mutational patterns across both naturally and experimentally evolved liver E. gallinarum isolates, we speculated that liver isolates derived from SPF (NZW x BXSB) F1 mice may exhibit broadly similar phenotypes to liver isolates from monocolonised mice. Like monocolonisation-derived liver isolates, a representative liver isolate from (NZW x BXSB) F1 mice had a thicker polysaccharide capsule and was more resistant to mCRAMP and liver clearance as compared to a representative faecal isolate (Extended Data Fig. 7a–c). Moreover, gnotobiotic mice monocolonised with the F1 liver isolate exhibited increased gut permeability and ileal inflammation as compared to mice colonised with the F1 faecal isolate (Extended Data Fig. 7d, e). Since E. gallinarum translocation can exacerbate systemic autoimmunity in (NZW x BXSB) F1 mice1, we next tested whether the F1 liver isolate may elicit enhanced autoimmune-associated phenotypes as compared to the F1 faecal isolate by treating monocolonised C57BL/6 mice with the TLR7 agonist imiquimod to induce autoimmunity26 (Extended Data Fig. 7f). We found that liver isolate-colonised mice showed more bacterial translocation (Extended Data Fig. 7g–i) and exhibited exacerbated lupus-like manifestations (Fig. 4i), including hepatosplenomegaly, proteinuria, and increased anti-dsDNA autoantibodies in sera. These data suggest that within-host evolution of E. gallinarum leads to broadly similar phenotypic and functional consequences in both simplified experimental models of in vivo evolution (i.e., monocolonisations) and naturally colonised hosts harbouring complex microbial communities. Furthermore, they show that a liver translocating strain of E. gallinarum exacerbates chemically-induced autoimmune-associated phenotypes.
Evolution in diverse microbial settings
To examine whether divergent evolution of E. gallinarum also occurs in WT mice in the presence of a complex gut microbiota, we colonised GF mice with the F1 faecal isolate plus a nine-species mock microbial community (hereinafter referred to as MC mice) or gavaged SPF C57BL/6 mice with the same faecal isolate (Fig. 5a). Seven weeks later, half of the MC mice exhibited E. gallinarum liver translocation (Fig. 5a, top). We picked colonies from the liver and faeces of one mouse with liver translocation and reconstructed their evolutionary histories via WGS. Like monocolonised mice and select (NZW x BXSB) F1 mice, liver E. gallinarum isolates from the MC mouse also acquired mutations at the immR gene. By contrast, faecal isolates largely consisted of an mga mutant lineage (Fig. 5b, Supplementary Table 3). Unlike MC mice, WT SPF mice colonised with E. gallinarum for one and a half years had no detectable liver translocation (Fig 5a, centre), potentially due to the small population size of E. gallinarum present in this setting (undetectable by 16S rRNA gene sequencing) combined with the robust barrier integrity present in healthy SPF WT mice. However, E. gallinarum isolates from the SIM or faeces of these mice were genetically divergent (Fig. 5c, Supplementary Table 4)—spxA or yxdM mutants predominated in the SIM, while norD/yocH double mutants were enriched in the faeces (Supplementary Table 8). The different mutational patterns observed across disparate experimental models may be due to differences in microbiota composition, host genetics, or inflammatory states. Despite their genetic differences, a representative liver isolate from MC mice and SIM isolates from SPF mice displayed enhanced resistance to liver clearance after intravenous injection as compared to matched faecal isolates (Fig. 5d, e). These data demonstrate that E. gallinarum can diverge into mucosal versus luminal lineages under multiple settings, including in healthy WT SPF animals; however, liver translocation was only detectable under specific genetic or environmental conditions (e.g., in autoimmune-prone SPF animals or in monocolonised or MC mice).
Fig 5 |. Within-host evolution across additional microbial contexts and bacterial species.
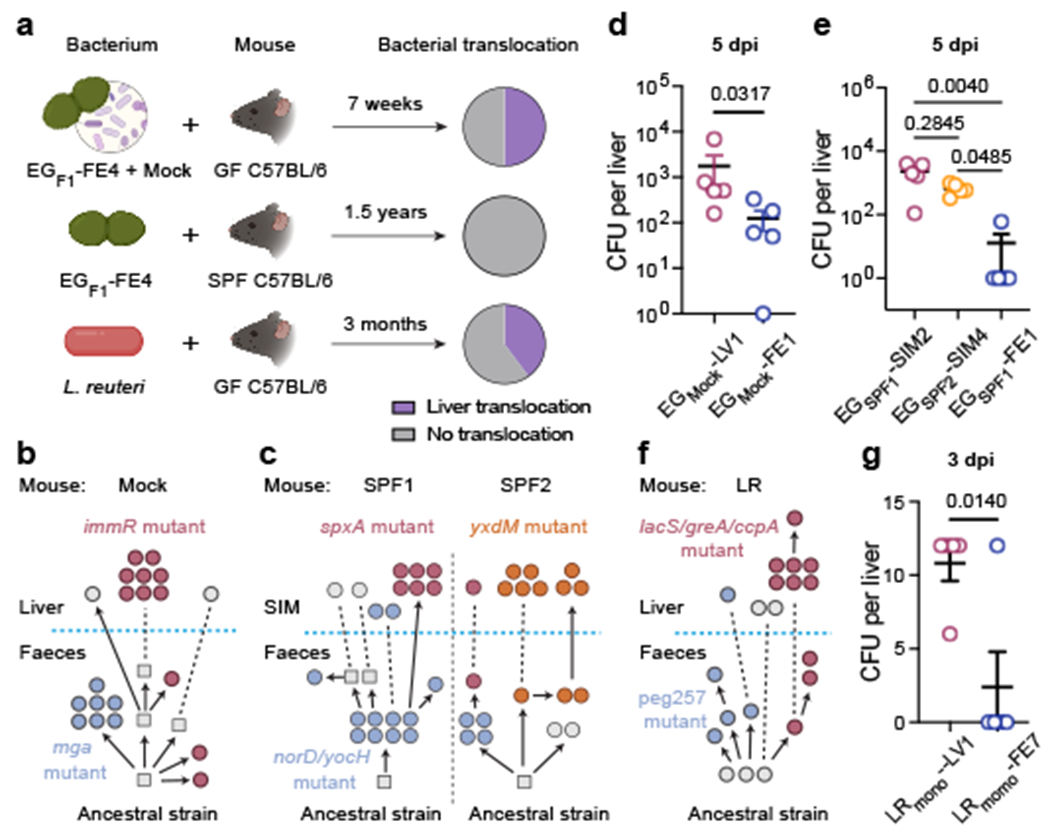
a, Schematic of experimental evolution of E. gallinarum in a mock (top, n = 6 mice) and SPF (centre, n = 3 mice) community or L. reuteri in monocolonisation (bottom, n = 5 mice). Pie charts showing proportions of mice with liver translocation. b, c, Reconstructed phylogenetic histories of E. gallinarum isolates sampled from an MC mouse (b) or two co-housed SPF mice (c). d, e, Liver clearance of intravenously injected E. gallinarum. Representative liver (EGmock-LV1) and faecal (EGmock-FE1) isolates from an MC mouse (d). Representative SIM (EGSPF1-SIM2, EGSPF2-SIM4) and faecal (EGSPF1-FE1) isolates from SPF mice (e). Liver bacterial load 5 dpi. n = 5 mice. f, Reconstructed phylogenetic history of L. reuteri isolates. g, Liver clearance of intravenously injected liver (LRmono-LV1) and faecal (LRmono-FE7) L. reuteri isolates. Liver bacterial load 3 dpi. n = 5 mice. Schematics in (b, c, f) are organized as described in Fig 2a. Genotypes of bacterial isolates examined in (d, e, g) are described in Supplementary Table 7. Results are representative of two (d, e, g) independent experiments. Data in (d, e, g) represent mean ± SEM. Two-tailed Mann-Whitney test (d), Kruskal-Wallis test with Benjamini-Hochberg correction (FDR = 0.05) (e), unpaired two-tailed t-test (g). SIM, small intestinal mucosa. dpi, days post-injection.
Evolution of distinct gut commensals
Finally, we wondered whether divergent evolution into unique lineages adapted to mucosal versus luminal niches is unique to E. gallinarum or would also occur in other gut commensals. We addressed this question using two phylogenetically and phenotypically distinct species: Lactobacillus reuteri, which can translocate to the liver in a mouse model of lupus26, and Bacteroides fragilis which can colonise mucosal crypts27 but is not thought to translocate to the liver. We monocolonised mice with either L. reuteri or B. fragilis for three months before assessing liver translocation. A subset of L. reuteri colonised mice exhibited liver translocation (Fig. 5a, bottom), whereas B. fragilis-colonised mice had no detectable bacterial translocation (Extended Data Fig. 8a), indicating that the capacity to translocate to the liver in this setting is not generic to all microbes. Liver and faecal isolates of L. reuteri were genotypically divergent: a lacS/greA/ccpA triple mutant was enriched in the liver population while peg257 mutants modestly predominated in the faeces (Fig. 5f, Supplementary Table 5, 8). A representative L. reuteri liver isolate also exhibited enhanced liver persistence in vivo as compared to a faecal isolate (Fig. 5g). While B. fragilis failed to translocate to the liver in this model, we found that B. fragilis isolates from the SIM and faeces were also genetically divergent (Extended Data Fig. 8b, Supplementary Table 6), which suggests potential adaption to mucosa-associated versus luminal niches. Although intravenously injected B. fragilis strains were rapidly cleared by the liver, SIM isolates still showed significantly increased liver persistence at very early timepoints as compared to a faecal isolate (Extended Data Fig. 8c). Taken together, these results suggest that microbial adaptations to mucosal versus luminal niches may occur in multiple commensal species, but the ability to evolve the capacity for liver translocation is potentially taxon-dependent.
Discussion
We show here that the gut pathobiont E. gallinarum diverges into independent lineages with differential niche-preferences and pathogenicity through within-host evolution (Extended Data Fig. 9). One lineage of E. gallinarum is adapted to colonise the intestinal lumen and is highly susceptible to immune clearance, whereas the second lineage is adapted for mucosal colonisation and evades immune detection and elimination. This mucosally-adapted lineage also exhibits enhanced translocation across the epithelium into the mLNs and liver and drives gut and liver inflammation in susceptible mice. Mechanistically, this enhanced translocation capacity is associated with reduced barrier enhancement upon microbial colonisation and increased survival within the liver, implying that adaptations that enable mucosal colonisation may also ‘pre-adapt’28 E. gallinarum to persist within internal organs.
Given the predominance of non-translocating strains in the faeces, we speculate that a trade-off between mucosal colonisation and bacterial transmissibility may limit the transmission of more pathogenic strains to new hosts29. Indeed, we observed that co-housed mice commonly shared faecal strains but often harboured genetically unique liver-translocating strains (e.g., Fig. 5c, Extended Data Fig. 8b). Preferential horizontal or vertical transmission of faecal strains may thus reset the microbial evolutionary clock for each new recipient or generation. These dynamics may partially explain the incomplete penetrance of pathobiont-driven disease as most individuals would initially inherit non-pathogenic strains and may also contribute to the age-related onset of many putative microbiota-driven pathologies.
We focused largely on E. gallinarum as it was previously reported to translocate to the mesenteric lymph nodes and liver in multiple mouse models of inflammatory disease1,2. However, not all commensal microbes will be equally capable of evolving this capacity during the limited lifespan of an individual host—indeed, while both E. gallinarum and L. reuteri exhibited liver translocation in monocolonised mice, B. fragilis failed to translocate. Our data suggest that one predictor of ‘nascent pathogenicity’ may be the ability to remodel the cell wall through the modulation of broad transcriptional programs that are co-ordinately regulated by transcriptional regulators such as immR or sensor histidine kinases such as ypdA and walK; these master regulators may act as ideal platforms for microbial adaptation via within-host evolution by enabling mutations in a single gene to co-ordinately re-program multiple features of bacterial behaviour.
While these studies relied heavily on simplified gnotobiotic mouse models, we also observed broadly similar patterns of divergent evolution in the presence of complex microbial communities; nonetheless, additional studies will be necessary to establish the impacts of various genetic, environmental, and microbial contexts on the pace and trajectory of within-host evolution. Notably, analogous instances of within-host evolution may also occur in humans. For example, Clostridium innocuum strains recovered from the intestinal mucosa versus mesenteric adipose tissue of Crohn’s disease patients were genetically divergent30; bloodborne isolates of the Enterococcus faecalis acquired resistance to immune clearance via mutations in the transcriptional regulator gntR31; and, compared to nose-colonising strains, infecting variants of Staphylococcus aureus accumulated adaptive mutations in rsp or agr genes, which regulate bacterial quorum sensing and toxin production32.
In summary, our results demonstrate that within-host evolution of a gut pathobiont can facilitate bacterial translocation and thus reveal a previously unappreciated microbial source of stochasticity in the development and progression of diverse microbiota-driven pathologies. Understanding the determinants and dynamics of within-host pathobiont evolution may eventually enable improved predictions of microbial impacts on human (patho)physiology and point towards novel therapeutic strategies to mitigate the diverse array of inflammatory disorders that are aided and abetted by invasive pathobionts.
Methods
Data reporting
No statistical methods were used to predetermine sample size. The experiments were not randomized, and the investigators were not blinded to allocation during experiments and outcome assessment.
Mice
SPF C57BL/6 mice were purchased from Jackson Laboratory (Bar Harbor, Maine, USA). (NZW x BXSB) F1 mice used in this study were generously provided by Martin Kriegel1 and maintained in a specific pathogen-free environment at Yale University. GF wild-type and Rag1−/− C57BL/6 mice were originally from University of Chicago Animal Resources Centre and University of Michigan Gnotobiotics respectively. GF MyD88−/−TRIF−/− C57BL/6 were kindly provided by Kathy McCoy at the International Microbiome Centre, University of Calgary and were originally generated by Bruce Beutler at the Scripps Research Institute. GF mice were bred and maintained under sterile conditions in CBC’s (Class Biologically Clean, Madison, WI, USA) Quad flexible film isolators in the Palm Lab Gnotobiotic Facility at Yale School of Medicine. All gnotobiotic experiments were conducted by transferring mice to positive pressure ventilated microisolator cages (Techniplast, ISO72P). Mice were on a dark/light cycle of 12 hours/12 hours with temperature and humidity controlled. Both male and female mice were used for these studies and mice were age and sex-matched within each experiment. All mouse experiments were approved by the Yale University Institutional Animal Care and Use Committee.
Bacteria
Lactobacillus reuteri was cultured in MRS broth (BD Difco), and other bacterial strains were cultured in Gifu Anaerobic Media (GAM, HiMedia Laboratories) broth at 37°C in an anaerobic chamber (Coy Laboratory Products Inc.) unless otherwise specified.
Isolation of E. gallinarum from (NZW x BXSB) F1 mice
E. gallinarum isolates were sampled from different 18-week-old (NZW x BXSB) F1 male mice. Liver isolates were collected from the livers as previously described1. To sample faecal isolates, faecal contents were eluted with GAM broth and diluted to 5 × 103 and 5 × 102 cells/ml. 100 μl of the diluted faecal material was plated on GAM agar plates and incubated for 24 or 48 h. Approximately 384 colonies were picked from each faecal sample and the bacterial species was assessed by next-generation sequencing of the v4 region of the 16S rRNA gene followed by further Sanger sequencing of the full-length 16S rRNA gene for putative E. gallinarum strains.
Experimental evolution in mice
Monocolonisation
For E. gallinarum, eight 8–12 week old GF C57BL/6 mice (four wild-type and four Rag1−/−) were gavaged with approximately 6 × 107 CFU of EGF1-FE4 and housed in four isocages. As our data did not reveal any apparent impacts of host genetics on bacterial evolution (Supplementary Table 2), we have presented all experimental evolution data together here. In addition, another two groups of GF C57BL/6 mice were inoculated with approximately 6 × 107 CFU of either L. reuteri or B. fragilis. After three months, mice were euthanized and the liver, small intestine and colon were aseptically dissected. The entire liver was mashed through a 70 µm cell strainer with a plunger and washed with 1 ml sterile PBS. For the small intestine and colon, tissues were opened longitudinally, and luminal contents were collected and resuspended in sterile PBS. Intestinal tissue was then washed thoroughly in cold sterile PBS by vigorous shaking. The washed tissue was then carefully checked to ensure no luminal particles remained. Intestinal tissue was cut into small pieces and transferred to a 2 ml sterile screw-cap tube containing 1 ml PBS and 15–20 BigPrep™ Lysing Matrix D beads (MP Biomedicals). Tubes were secured and vigorously shaken in a bead-beater (Biospec) for 30 sec. The tissue was then filtered through a 70 µm cell strainer, and the flowthrough was collected and saved as the “mucosa” sample. To avoid cross-contaminations, all the samples were processed mouse by mouse. 200 µl of liver homogenate, 100 µl of diluted lumen content (10−5 for small intestine; 10−6 for colon) and 100 µl of diluted mucosal sample (10−4-10−3) were spread on GAM agar plates and cultured anaerobically for 24 h. Up to 12 colonies were randomly picked from each plate and inoculated into fresh GAM broth to make glycerol stocks and used for further study. The unused liver homogenates, lumen and mucosal samples were mixed with an equal volume of 40% glycerol and saved as glycerol stocks for long-term storage in −80 ºC.
Mice colonised with a mock microbial or SPF consortium
Eight-week-old GF mice were inoculated with EGF1-FE4 and a nine-species microbial community (IgA− consortium)33 and housed in isocages for 7 weeks. SPF C57BL/6 mice were gavaged with EGF1-FE4 and housed for 1.5 years before assessing liver translocation. The isolation of E. gallinarum isolates from these mice were largely identical to isolation from monocolonised mice except the faecal or mucosal homogenates were plated on GAM agar containing 8 mg/L vancomycin and cultured aerobically.
Whole-genome sequencing of single bacterial isolates
Single colonies were picked from streaked plates and inoculated into GAM broth. After overnight culture, the bacterial DNA was extracted using DNeasy UltraClean Microbial Kit (Qiagen, 12224–250) following manufacturer’s protocol. Sequencing libraries were prepared using Illumina Nextera XT library preparation kit (FC-131–1024) and barcoded using Nextera XT Index Kit v2 Sets. The quality of libraries was analysed by Agilent Bioanalyzer before pooling. E. gallinarum isolates derived from (NZW x BXSB) F1 mice were sequenced on a MiSeq sequencer using a 2 × 250 bp v2 kit (Illumina); all other strains were sequenced on a NovaSeq S4 2 × 150 bp at Yale Centre for Genome Analysis (YCGA). All the sequenced genomes generated reads at a coverage of more than 100X.
Oxford Nanopore sequencing
A single colony of EGF1-FE1 was picked from a streaked plate and inoculated into GAM broth to grow to mid-log phase. The high molecular weight DNA was gently extracted using Quick-DNA HMW MagBead Kit (Zymo Research Cat # D6060). The quality of DNA was roughly assessed by Nanodrop ™ 8000 (Thermo Fisher Scientific), and the concentration was measured by Qubit 3.0 (Invitrogen). Approximately 400 ng of HMW genomic DNA was used for library construction using Rapid Barcoding Sequencing Kit (Oxford Nanopore Technologies, SQK-RBK004). The indexed library was loaded on a flow cell and sequenced with a MinION for 48 h. Data acquisition and basecalling was performed by MinKNOW in real-time.
Metagenomic sequencing of E. gallinarum population
Tissue stocks of liver, mucosa and luminal content of the small intestine and colon from the experimental evolution model were plated on GAM agar plates and cultured for 24 h. All the bacterial colonies on the plate were scraped, pooled, and resuspended in 1ml sterile PBS followed by centrifugation at 8,000 g for 5 min to pellet cells. Bacterial DNA extraction, library preparation and quality control (Q.C.) were performed as described above for bacterial WGS. All the metagenomic libraries were pooled and sequenced on a NovaSeq S4 2 × 150 bp at YCGA.
Hybrid assembly of complete reference genome
To close the genome of EGF1-FE1, hybrid assembly of Illumina short reads and Oxford Nanopore long reads was performed by using Unicycler34 (v. 0.4.8). The Illumina short reads were Q.C. checked with FastQC35 (v. 0.11.9), trimmed, and filtered by using Trimmomatic36 (v. 0.36) to remove Nextera adapters and low-quality reads. For ONT reads, the quality of raw reads was checked by using NanoStat37 (v. 1.1.2), and then filtered by Filtlong (v. 0.2.0) to only extract best reads up to total bases of 185M (50X ONT coverage). Default settings were used in Unicycler for hybrid assembly. The closed genome of E. gallinarum was annotated in RAST server38,39 (v. 2.0) and Prokka40 (v. 1.14.5).
Variant calling and allele frequency inference
The closed genome of EGF1-FE1 was used as a reference sequence for all (NZW x BXSB) F1-derived isolates. For experimentally evolved E. gallinarum isolates or populations, EGF1-FE4 was used as the reference genome. The draft genomes of L. reuteri and B. fragilis parental strains were used as the reference sequences for evolved L. reuteri and B. fragilis isolates, respectively. Before mapping, the raw Illumina reads were Q.C. checked, trimmed, and filtered by using FastQC and Trimmomatic as described above. Reference-based alignment was performed by using Snippy41 (v. 4.4.3) and Breseq42 (v. 0.35.5) for the detection of SNVs, indels, and structural variants. For single isolate genotyping, the consensus mode of Breseq and the “snippy-multi” of Snippy was applied. For metagenomes, the polymorphism mode of Breseq was used. Each metagenomic sample had reads of more than 300X coverage depth. The genome of EGF1-FE4 was generated by using the “gdtools APPLY” function of Breseq after reads alignment.
Phylogenetic tree construction
A phylogenetic tree of E. gallinarum ATCC, DSM, and (NZW x BXSB) F1-derived strains was constructed based on core-genome alignments by Parsnp in Harvest suite43 (v. 1.1.2). Trimmed raw reads were assembled as scaffolds by SPAdes44 (v. 3.13.1). Scaffolds were then re-ordered by Mauve45 (v. 2.3.1) based on a reference genome EGF1-FE1. Ordered draft genomes were used as input for core-genome alignments by Parsnp. For other phylogenetic trees in this study, reference-based alignment with maximum-likelihood tree building method was used. SNVs called by Snippy and Breseq were used for tree generation in UGENE46 (v. 1.32.0). The substitution model was set as GTR with all the other setting as default. All the trees were visualized in iTOL47.
Pangenome analysis of Enterococcus strains
Scaffolds of Enterococcus strains longer than 500 bp were annotated by Prokka, and the pangenome was computed by roary48 (v. 3.11.2) and visualized by anvio49 (v. 6.2).
Tissue RNA sequencing
Six-week-old GF C57BL/6 mice were gavaged with approximately 6 × 107 CFU of E. gallinarum. Two-weeks after monocolonisation, mice were euthanized, a small piece of liver and 2 cm of distal ileum was dissected and immediately flushed with RNAprotect Tissue Reagent (Qiagen, 76104). Samples were individually saved in tubes containing 1 ml of RNAprotect at 4 ºC overnight and processed the following day. To isolate ileal epithelial cells, another 6 cm of ileum was dissected and immediately left in cold PBS on ice. Fat and visible Peyer’s patches were carefully removed, and the intestine was opened longitudinally and washed in cold PBS to remove luminal contents. Ileal tissues were then cut into small pieces and put into 10 ml of 2 mM EDTA/PBS in 15 ml conical tubes followed by shaking at 180 rpm for 30 min at 37 ºC. After a 30-min digestion, epithelial cells were collected by filtering through 70 µm cell strainers. Cells were washed once with cold PBS and pelleted by centrifugation at 1500 rpm for 5 min at 4 ºC. The pelleted ileal epithelial cells were resuspended and lysed in 600 ul of RLT buffer (RNeasy Mini Kit, Qiagen, 74106) with 1% 2-mercaptoethanol (Sigma-Aldrich). Lysed cells were frozen at −80 ºC until RNA extraction. RNA was isolated using the RNAeasy mini kit with on-column DNase digestion (Qiagen) according to the manufacturer’s instructions.
Libraries were generated by using the Illumina Stranded Total RNA Prep with Ribo-Zero Plus kit (20040529) following manufacturer’s protocol. The concentration and quality of libraries were assessed by Agilent Bioanalyzer. Libraries were equally pooled and sequenced on a NovaSeq S4 2 × 100 bp at YCGA. Each library generated more than 5 million reads.
The first “T” nucleotide introduced by Illumina Stranded Total RNA prep workflows was trimmed by cutadapt50 (v. 3.2). Trimmed reads were aligned to mouse genome (GRCm38, Ensembl51) by using STAR52 (v. 2.7.7a) with default settings. The mapping results were used to calculate read counts over each gene by HTSeq53 (v. 0.13.5). The read counts for each gene were used to analyse differential gene expression using the DESeq254 (v. 1.30.1) package in R. Pathway analysis was performed in GSEA55 (v. 4.1.0) software.
Bacterial transcriptomics
Single colonies of E. gallinarum were inoculated into GAM broth for overnight culture. The bacterial density was assessed by measuring OD600, and then diluted to OD600 = 0.05 in 3 ml of GAM broth. Three independent cultures were included for each isolate. Bacteria were collected at mid-log phase after 3 h culture. To preserve RNA, one volume of bacteria culture was immediately mixed with two volumes of RNAprotect Bacteria Reagent (Qiagen, 76506) by 5 sec vortex followed by 5 min incubation at room temperature. Bacterial pellets were collected after centrifugation and processed for RNA extraction. Pellets were resuspended with 120 μl of TE/protease K/lysozyme mixtures (70 μl 1x TE buffer + 30 μl 100 mg/ml lysozyme + 20 μl 20 mg/ml Qiagen proteinase K) and incubated at room temperature for 15 min on a rotor. 700 μl of RLT with 1% 2-mercaptoethanol was then added and vortexed vigorously for 10 sec. The suspension was transferred into a 2 ml screw-cap tube containing 25–50 mg lysing matrix B beads (MP Biomedicals) for cell disruption in the bead-beater for 10 min. After 10 sec centrifugation at 13,000 g, supernatant was transferred into a new tube and added with 590 μl of 80% ethanol. Mixed lysate was transferred to RNeasy mini spin column for RNA extraction. From this step, RNA was isolated according to the manufacturer’s instructions with on-column DNase digestion (Qiagen). The procedures of library preparation and sequencing were largely the same as ileal transcriptomics except 15 cycles were used for library amplification as suggested.
The complete genome of E. gallinarum EGM181 (ASM1484419v1) was downloaded from GenBank database and used as the reference sequence for functional analyses. Trimmed reads were aligned to the reference genome by using Bowtie256 (v. 2.4.2), and the calculation of read counts over each gene was performed by BEDTools57 (v. 2.30.0). Overrepresented functional classes were identified by GSEA-pro v3 (http://gseapro.molgenrug.nl/).
Intestinal permeability assay with FITC-dextran
Mice were fasted for 4 h prior to FITC-Dextran (4 kDa, Sigma-Aldrich) administration. 200 μl of 40 mg/ml FITC-dextran was orally administrated via gavage in each mouse. Blood samples were collected through retro-orbital vein after 3 h. Serum was diluted 1:5 with PBS and placed in a fluorescence plate reader to determine the concentration by fluorescence excitation at 485 nm/535 nm reference. Duplicates of serum samples, blank serum samples (without FITC-dextran administration) as well as a concentration series of FITC-dextran to generate a standard curve were included.
Assessment of bacterial translocation
Mice were euthanized and the liver, spleen and mLNs were aseptically dissected. The tissues were mashed through 70 µm cell strainers with a plunger and washed with 1 ml sterile PBS. 200 μl of tissue homogenate was spread and cultured on GAM agar plates. CFU numbers were counted after 48 h culture. The species of bacterial colonies were validated by Sanger sequencing of the full-length 16S rRNA gene.
Intravenous injection of bacteria
SPF C57BL/6 mice were injected with approximately 4 × 106 CFU of bacteria at late-log growth phase through retro-orbital vein. To count bacterial load in liver, mice were euthanized and the liver was aseptically processed as described above. 100 µl of liver homogenate was plated on GAM agar plates and cultured anaerobically for 24 h before CFU counting.
Determination of Minimum Inhibitory Concentrations (MIC) of mCRAMP
The MIC was determined as described previously58 with minor modifications. Briefly, bacteria were anaerobically cultured overnight and inoculated into 96-well plates at a starting A600 of 0.01 in a total volume of 100 μl medium supplemented with select concentrations of mCRAMP (Anaspec). Duplicates were included. The 96-well plates were incubated at 37°C and the A600 was monitored continuously using a microplate reader (BioTek) in an anaerobic chamber. Positive controls containing no mCRAMP were performed with every isolate and the determination of MIC was made when the positive controls reached late log phase. The MIC was determined as the lowest concentration of mCRAMP that reduced growth (A600) by more than 50% when compared to the positive controls.
In vitro growth inhibition by lysozyme
Bacteria were anaerobically cultured overnight and inoculated into 96-well plates at a starting A600 of 0.01, in a total volume of 200 μl medium supplemented with 5 or 10 mg/ml lysozyme (Thermo Fisher Scientific). The A600 was monitored as described above.
BMDM phagocytosis
Bacteria were anaerobically cultured for 24 h. To label cells with carboxyfluorescein succinimidyl ester (CFSE), cultured bacteria were washed and resuspended in 10 μM CFSE (Biolegend) in PBS. Cells were incubated at room temperature for 1 h on a rotor and washed thoroughly with sterile PBS three times. CFSE-labelled bacterial cells were resuspended in sterile PBS and corrected to a concentration of a OD600 of 1. Bone marrow was isolated from C57BL/6 mice and grown in DMEM medium supplemented with 10% FBS, 1% Pen-Strep and 20 ng/ml M-CSF (Biolegend) for 7 d. The matured BMDMs were detached with Trypsin-EDTA and plated in 24-well plates. After 8 h, re-attached BMDMs were treated with CFSE-labelled bacteria at a multiplicity of infection (MOI) ~ 15. BMDMs without bacteria treatment were included as negative controls. Cells were collected after 2 h coculture and stained for F4/80 (BM8, eBiosceince) and live/dead (Invitrogen) markers on ice for 30 min. Phagocytosis was analysed by LSRII (BD Biosciences) using FlowJo (v. 10.4).
In vivo competition in SPF mice
EGF1-LV1 and EG F1-FE4 were cultured to late-log phase and equally mixed for gavage. The number of bacteria was estimated by OD600. Six-week-old SPF C57BL/6 mice were gavaged with approximately 6 × 107 CFU of bacterial mixture. Faeces were collected a week post-gavage. Diluted faecal material was plated on GAM agar plates containing 8 mg/L vancomycin and aerobically cultured for 24 h. At least twelve E. gallinarum colonies were randomly picked from each plate for genotyping. To distinguish EGF1-LV1 and EG F1-FE4 isolates, rpoN gene was selected based on whole-genome sequences due to a small insertion in EGF1-LV1. Primers used to amplify rpoN gene were forward: 5’-AGCAGCGAGGAGATACCATC-3’; reverse: 5’- TTTCCCATTGACGGCACGA-3’. The PCR program was set as follows: 98 ºC for 1 min followed by 30 cycles of 98 ºC for 10 sec, 55 ºC for 20 sec and 72 ºC for 3 min. Afterwards, samples were kept at 72 ºC for 5 min. The PCR product was purified by using AMPure XP beads (Beckman Coulter) and sequenced by Sanger sequencing. EGF1-LV1 contains “CTTTTTTTTT” (9Ts) and EGF1-FE4 contains “CTTTTTTTT” (8Ts).
Bi-colonisation of E. gallinarum isolates in GF mice
E. gallinarum liver and faecal isolates were cultured to late-log phase, and equally mixed for gavage. GF C57BL/6 mice were gavaged with E. gallinarum mixture and euthanized four weeks (EGmono6-LV1 vs. EGmono7-FE2) or seven days (EGmono6-LV10 vs. EGmono7-FE2) post-gavage. The processing of small intestine and colon to isolate lumenal versus mucosal samples were performed as described in previous sections. Samples were cultured on GAM agar plates under aerobic conditions. After 48 h culture, the colony of EGmono7-FE2 isolate was more transparent than EGmono6-LV1 and EGmono6-LV10 (Extended Data Fig. 3f). The relative abundance of liver and faecal isolates was measured by colony morphology-based counting. To ensure the accuracy of this method, a subset of colonies was selected from each plate and analysed by sanger sequencing to validate genotypes. To distinguish liver and faecal isolates, PCR was performed to amplify the walK gene using the following primers: forward 5’- CTCTCCCACATGACCGATGG-3’; reverse 5’- TCTCGGCTTTCTGCCCAAAT-3’. The same PCR program was used as described in previous section and the PCR product was purified and analysed by Sanger sequencing. Sequences containing “TGCGAAT” represent EGmono7-FE2 isolate, while sequences containing “TGCGAAC” represent liver isolates.
TEM of E. gallinarum
Bacteria were fixed in 4% paraformaldehyde overnight then rinsed once, and further fixed in 2.5% glutaraldehyde, 2% paraformaldehyde in 0.1M sodium cacodylate buffer pH 7.4 for 1 h. Cells were rinsed well and were spun down in 2% agar in 0.1M sodium cacodylate. Chilled blocks were trimmed and post fixed with reduced osmium tetroxide (1% osmium tetroxide and 1% potassium ferrocyanide in 0.1M cacodylate) for 1 h, rinsed and then en-bloc stained in aqueous 2% uranyl acetate for 1 h. Cells were rinsed in distilled water, dehydrated in an ethanol series from 50% to 100% and two changes of 100% propylene oxide, infiltrated with Embed 812 (Electron Microscopy Sciences) epoxy resin and baked overnight at 60 ºC. Hardened blocks were cut using a Leica UltraCut UC7. 60 nm sections were collected on formvar/carbon coated grids and contrast stained using 2% uranyl acetate and lead citrate. Sections on grids were viewed using FEI Tencai Biotwin TEM at 80 Kv. Images were taken using Morada CCD and iTEM (Olympus) v. 2.4.2.
SEM of mouse ileum
Dissected tissue was fixed with 4% paraformaldehyde overnight at 4 ºC followed by further fixation with 2.5% glutaraldehyde in 0.1M sodium cacodylate buffer pH 7.4 for 1 h once sample was pinned open. Samples were rinsed in 0.1M sodium cacodylate buffer and post fixed in 2% osmium tetroxide in 0.1M sodium cacodylate buffer pH 7.4. Samples were then rinsed in buffer and dehydrated through an ethanol series to 100%. The samples were dried using a Leica 300 critical point dryer with liquid carbon dioxide as transitional fluid, glued to aluminium stubs using a carbon adhesive, and sputter coated with 4 nm platinum/palladium using a Cressington 208HR. Digital images were acquired in Zeiss CrossBeam 550 between 1.5–2 kV at a working distance of 8–12 mm.
Histology and immunochemistry
Mouse ileum was immersion-fixed in carnoy’s fixative (60% ethanol, 30% chloroform, 10% glacial acetic acid) on ice for 3 h. Sections of paraffin-embedded samples were stained with periodic acid–Schiff (Sigma-Aldrich) or anti-CD3 antibodies (1:300 dilutions, polyclonal, Abcam, ab5690) and analysed using light microscopy. Digital light microscopic images were recorded with a AmScope MU300 camera and AmScope imaging software. Representative images were captured under 200X magnification. Villi in five randomly selected windows under 100X magnification were counted for statistical analysis.
Liver 16S rRNA FISH
Paraffin-embedded liver slides were heated at 50 ºC for 1 h and paraffin was removed by xylene followed by absolute ethanol twice at room temperature for 5 min each. Slides were dried at 50 ºC for 10 min and stained with EUB-338 probe in hybridization buffer (0.9 M NaCl, 0.02 M Tris pH 7.5, 20% Formamide, 0.05% SDS) at 46 ºC for 2 h. After washing twice with post-hybridization buffer (0.215 M NaCl, 0.02 M Tris pH 7.5, 0.025 M EDTA, 0.05% SDS) at 46 ºC for 5 min and rinsed with deionized water, air dried slides were cured by ProLong™ Diamond Antifade Mountant with DAPI (Thermo Fisher Scientific) and sealed with nail polish. Images were captured by Zeiss LSM 880 airyscan.
Imiquimod-induced lupus model
Eight-week-old GF mice were monocolonised with EGF1-LV1 or EGF1-FE4 for 8 weeks and received topical administration of 1.25 mg of 5% imiquimod cream on the ear every other day. Urine and serum were collected at the end of experiment. Proteinuria was quantified by BCA Protein Assay kit (Thermo Fisher Scientific) and the level of anti-dsDNA autoantibodies was assessed by ELISA as described in previous studies1. The liver, spleen and mLNs were dissected, weighed, and analysed for bacterial translocation.
Generation of E. gallinarum pgdA insertion mutant
pORI28-pgdA non-replicative plasmid was constructed in the pORI28 vector backbone59 using standard cloning techniques. Briefly, an internal 648bp fragment of pgdA gene was amplified by using forward primer: 5’- AGCTCTTAGACGAGCTTGCTCA −3’ and reverse primer: 5’- GATCCACCATCTGTCCTAACATG −3’; and cloned into pORI28 vector using blunt cloning with forward primer: 5’- ACGCGTCGACGTCATATGGA −3’ and reverse primer: 5’- CTGCAGAAGCTTCGAATTCG −3’. The construct was transformed and propagated in E. coli EC1000 by electroporation. Sequence was confirmed by sanger sequencing. The plasmid was then extracted from 100 ml culture using QIAprep Spin Miniprep Kit and concentrated using Amicon ultra 30KDa 0.5ml filter unit. The pgdA gene insertion mutant was created by using the method described in Lauté-Caly. et al.60 with minor modifications. A single colony of E. gallinarum was grown overnight statically in BHI at 37 ºC. The overnight culture was diluted in fresh BGYT (BHI+yeast extract+glucose+Tris) supplemented with 0.5M sucrose, and a range of glycine (0% to 5%) and grown for 18 h. The glycine concentration that gave rise to ~60% growth when compared to growth without glycine was chosen for making electrocompetent cells (2% glycine). The cells were chilled on ice for 10–15 min and washed 3 times with the chilled electroporation buffer (0.5M sucrose + 10% glycerol). 50 ul of the electrocompetent cells were treated with 10 μg/ml lysozyme and 10 U/ml mutanolysin for 30 min at 37 °C. Cells were then transformed by electroporation with 5–10 μg of pORI28-pgdA plasmid DNA and recovered in BHI broth for 4 h before plating on BHI agar supplemented with 0.25M sucrose and 20 ug/ml erythromycin. Plasmid insertion was confirmed for successful transformants by PCR amplification and sanger sequencing.
Cartoon schematics
Figure schematics in Fig. 1a, e; 2d; 4h; 5a and Extended Data Fig. 2a; 4a–d; 7f; 8a; 9 are created with BioRender.com.
Statistical analysis
Statistical analyses were performed with Graphpad Prism (v. 9.1.0) or R (v. 4.0.4). Fisher’s exact tests were performed in R and were used to identify differential enrichments of mutated bacterial genes in liver or faecal population. Principle component analysis and K-means clustering of bacterial transcriptomics was performed in R. Differential gene expression analysis was performed by DESeq2 (v. 1.30.1) in R. Data plotted in figures were analysed by Graphpad Prism and were assessed for normal distribution. For data that passed normality tests, unpaired two-tailed t-tests were used to compare two groups; paired two-tailed t-tests were used to compare rates of liver translocation; one-way ANOVA with Tukey’s post hoc tests were used to compare multiple groups; two-way ANOVA with Tukey’s post hoc tests or two-way ANOVA with Sidak’s post hoc tests were used in bi-colonisation experiments. For data that failed to pass normality tests, two-tailed Mann-Whitney tests were used to compare two groups; Kruskal-Wallis tests with Benjamini-Hochberg correction (FDR = 0.05) were used to compare multiple groups. Samples sizes are indicated in each figure legend and p values are indicated in the figures. p values for Kruskall-Wallis are approximated based on the chi-square distribution and account for rank ties.
Extended Data
Extended Data Fig. 1 |. Mutations detected in E. gallinarum isolates from SPF (NZW x BXSB) F1 mice.
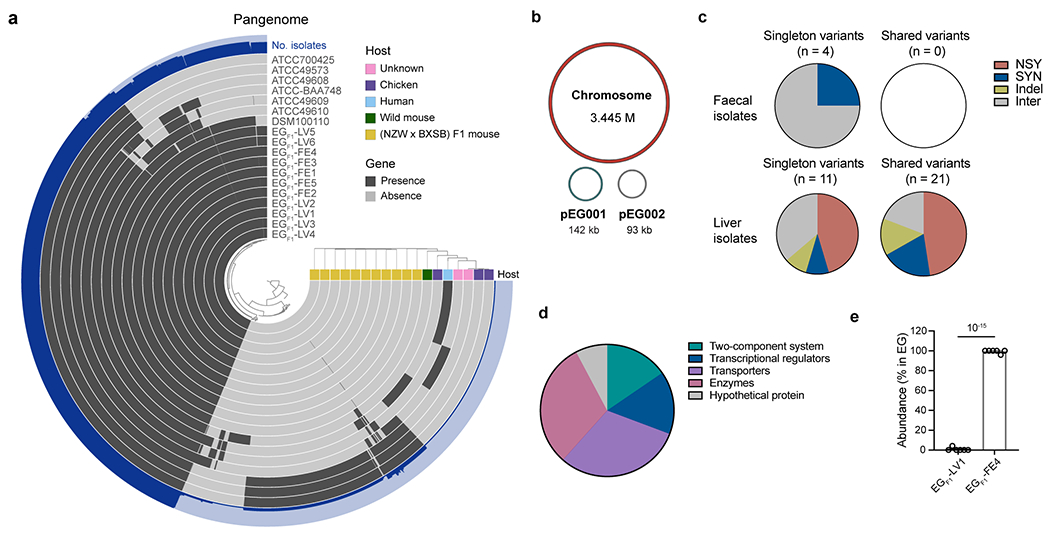
a, Pangenome of eighteen E. gallinarum strains. b, Schematic of the complete genome of E. gallinarum strain EGF1-FE1. c, Number of singleton or shared variants detected in E. gallinarum faecal or liver isolates. d, Functional classes of genes with nonsynonymous mutations or indels. n = 13 genes. e, Relative abundance of liver and faecal E. gallinarum isolates in the faeces of co-colonised SPF C57BL/6 mice. Mice were gavaged with equal doses of EGF1-LV1 and EGF1-FE4 and faecal samples were analysed one week after colonisation. n = 6 mice across two independent experiments. Data represent mean ± SEM. Unpaired two-tailed t-test (e). NSY, nonsynonymous SNVs; SYN, synonymous SNVs; Indel, small insertion/deletion variant; Inter, intergenic variant. EG, E. gallinarum.
Extended Data Fig. 2 |. Mutations detected in E. gallinarum isolates from monocolonised C57BL/6 mice.
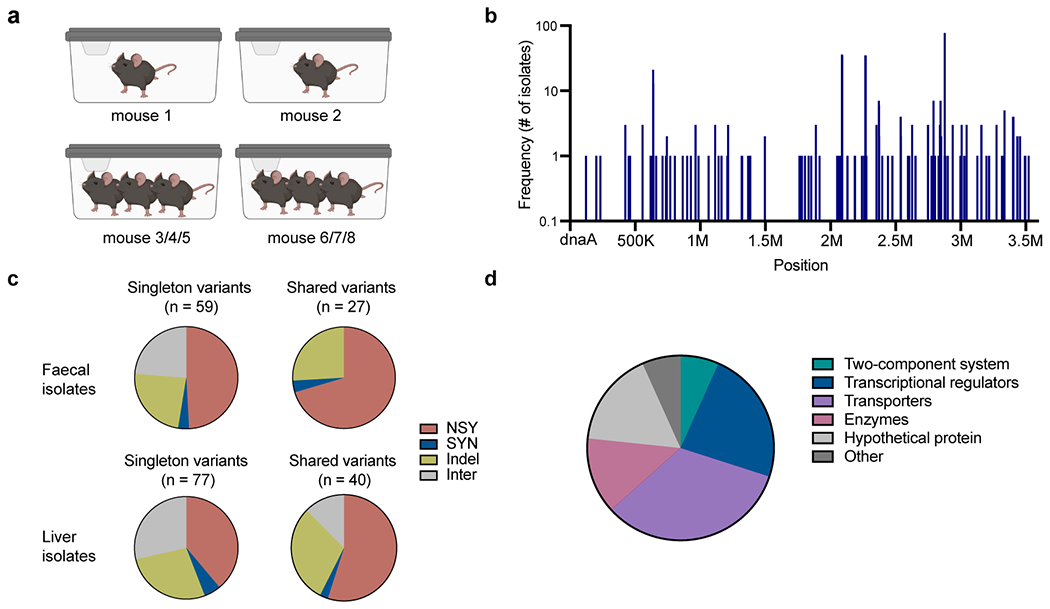
a, Schematic of cage setup of the experimental evolution in monocolonised mice. b, Distribution of detected mutations across the E. gallinarum genome. n = 159 unique mutations detected in 153 isolates. c, Number of singleton or shared variants. d, Functional classes of genes with nonsynonymous mutations or indels. n = 30 genes. NSY, nonsynonymous SNVs; SYN, synonymous SNVs; Indel, small insertion/deletion variant; Inter, intergenic variant.
Extended Data Fig. 3 |. Within-host evolution of E. gallinarum in monocolonised mice.
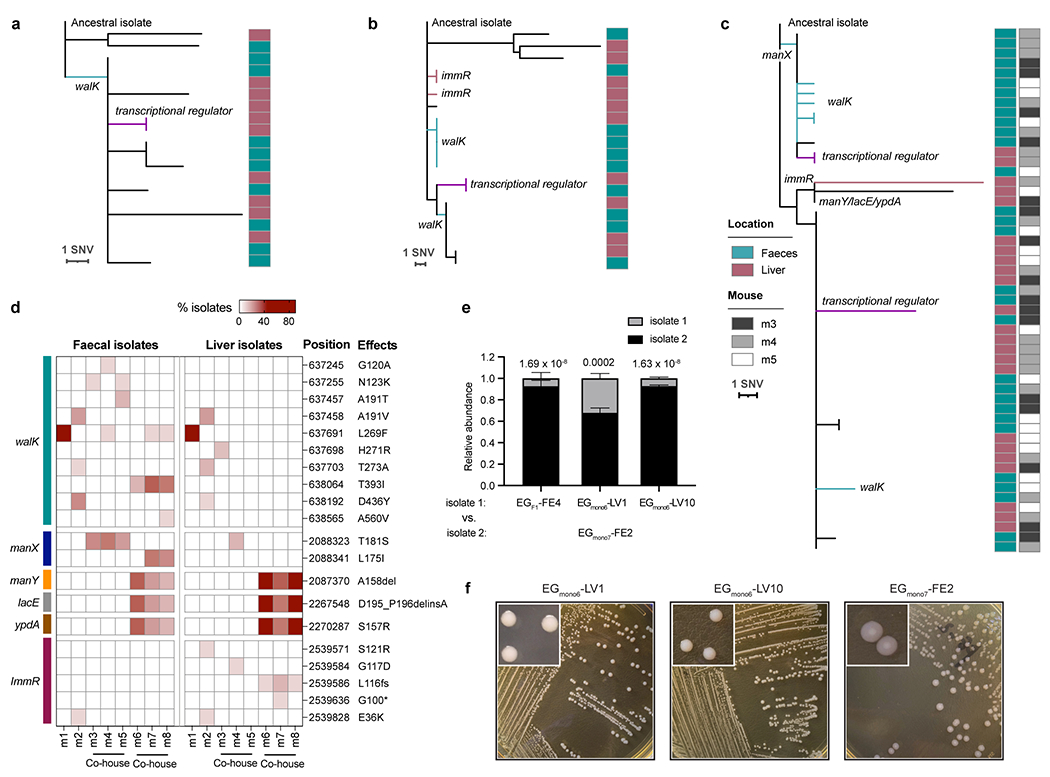
a-c, Phylogenetic tree of liver and faecal E. gallinarum isolates sampled from mouse 1 (a), mouse 2 (b) or mice 3-5 (c). Reference-based alignments. d, Position, effects, and frequency of mutations at walK, manX, manY, lacE, ypdA, and immR in liver or faecal isolates sampled from each individual mouse. Rows represent mutations and columns represent individual mice. e, Relative abundance of E. gallinarum isolates in the faeces of bi-colonised mice. EGF1-FE4, EGmono6-LV1, or EGmono6-LV10 was co-gavaged with an equal dose of EGmono7-FE2 and samples were collected 4 weeks after co-colonisation. See Supplementary Table 7 for a summary of all mutants. n = 3 mice. Representative of two independent experiments. Data represent mean ± SEM. Two-way ANOVA with Sidak’s post hoc test. f, Colony morphologies of EGmono6-LV1, EGmono6-LV10, and EGmono7-FE2 after 48-hour aerobic culture on GAM agar plates.
Extended Data Fig. 4 |. Reconstructed evolutionary paths of E. gallinarum within individual hosts.
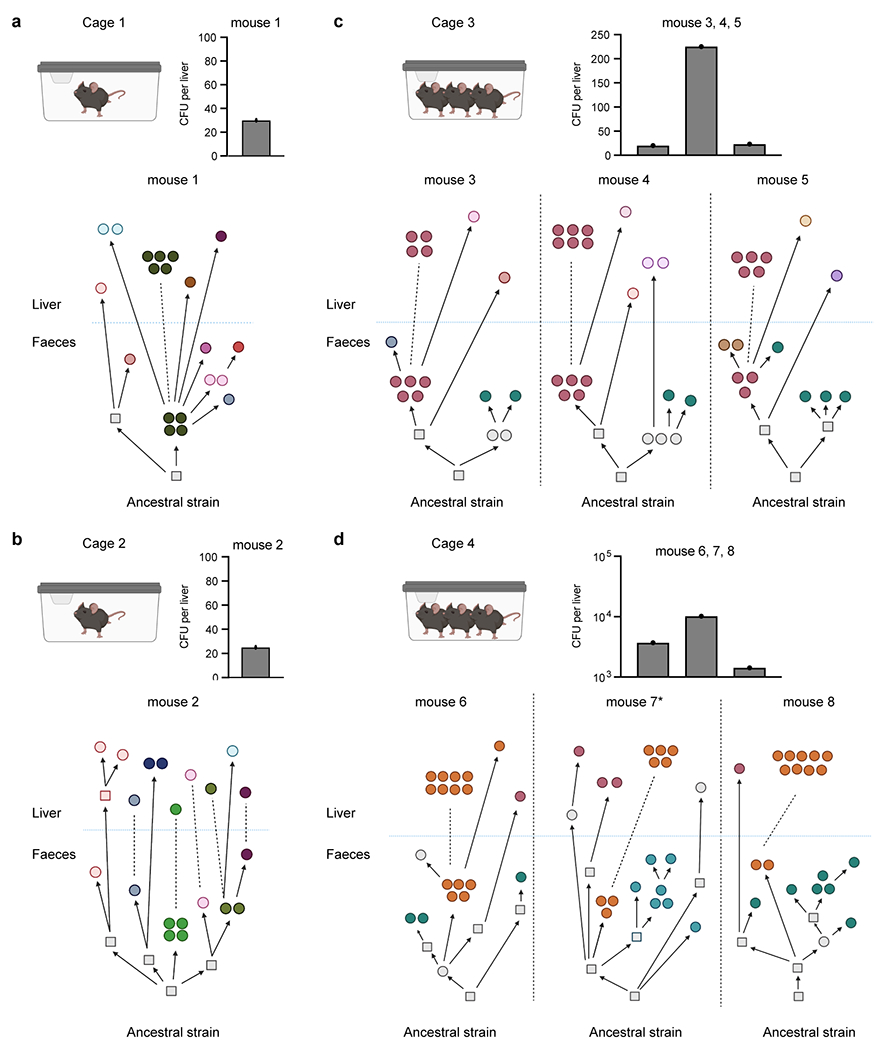
Reconstructed phylogenetic histories of E. gallinarum isolates from mouse 1 (a), mouse 2 (b), mice 3-5 (c), and mice 6-8 (d). The mouse 7 is also presented in Fig. 2a. Bar plots show bacterial load recovered from the liver of each monocolonised mouse. n = 1 mouse. Circles represent sequenced isolates and squares represent hypothetical intermediate genotypes. Different colours indicate distinct genotypes. Arrows connect related genotypes and dashed lines connect genetically identical isolates. Liver and faecal populations are separated by a blue dashed line.
Extended Data Fig. 5 |. Transcriptomic comparisons of liver and faecal E. gallinarum isolates, and susceptibility of E. gallinarum pgdA mutant to liver clearance.
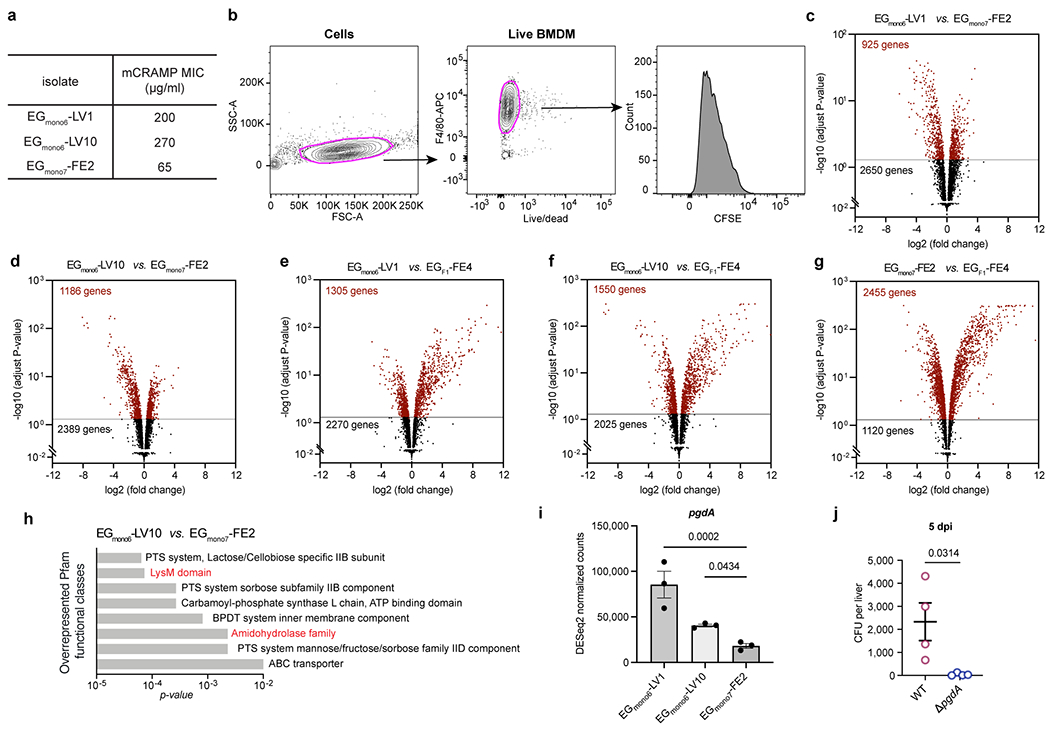
a, The minimum inhibitory concentrations (MIC) of mCRAMP against E. gallinarum isolates. b, Gating strategy used to select live macrophages and quantify CFSE signal in Fig. 3c. c-g, Volcano plots showing adjusted p-value versus fold change for 3575 genes expressed in EGmono6-LV1 (c), EGmono6-LV10 (d) as compared to EGmono7-FE2, or in EGmono6-LV1 (e), EGmono6-LV10 (f) and EGmono7-FE2 (g) isolate as compared to the ancestral strain EGF1-FE4. Horizontal lines show the p-value cut-off for significance after correction for multiple testing (adjust p-value < 0.05). Differentially expressed genes are coloured as red. n = 3 independent cultures. h, Overrepresented Pfam functional classes (EGmono6-LV10 vs. EGmono7-FE2). i, Expressions of pgdA gene in cultured E. gallinarum isolates. n = 3 independent cultures. j, Liver clearance of intravenously injected wild-type or pgdA mutant strains of E. gallinarum. Liver bacterial load 5 days post-injection. n = 4 mice. Representative of two independent experiments. Data in (i, j) represent mean ± SEM. Two-tailed Wald test with Benjamini-Hochberg correction (FDR = 0.05) (c-g), one-way ANOVA with Benjamini-Hochberg correction (FDR = 0.05) (i), unpaired two-tailed t-test (j).
Extended Data Fig. 6 |. Liver and faecal E. gallinarum isolates elicit distinct immune responses in the intestine and liver.
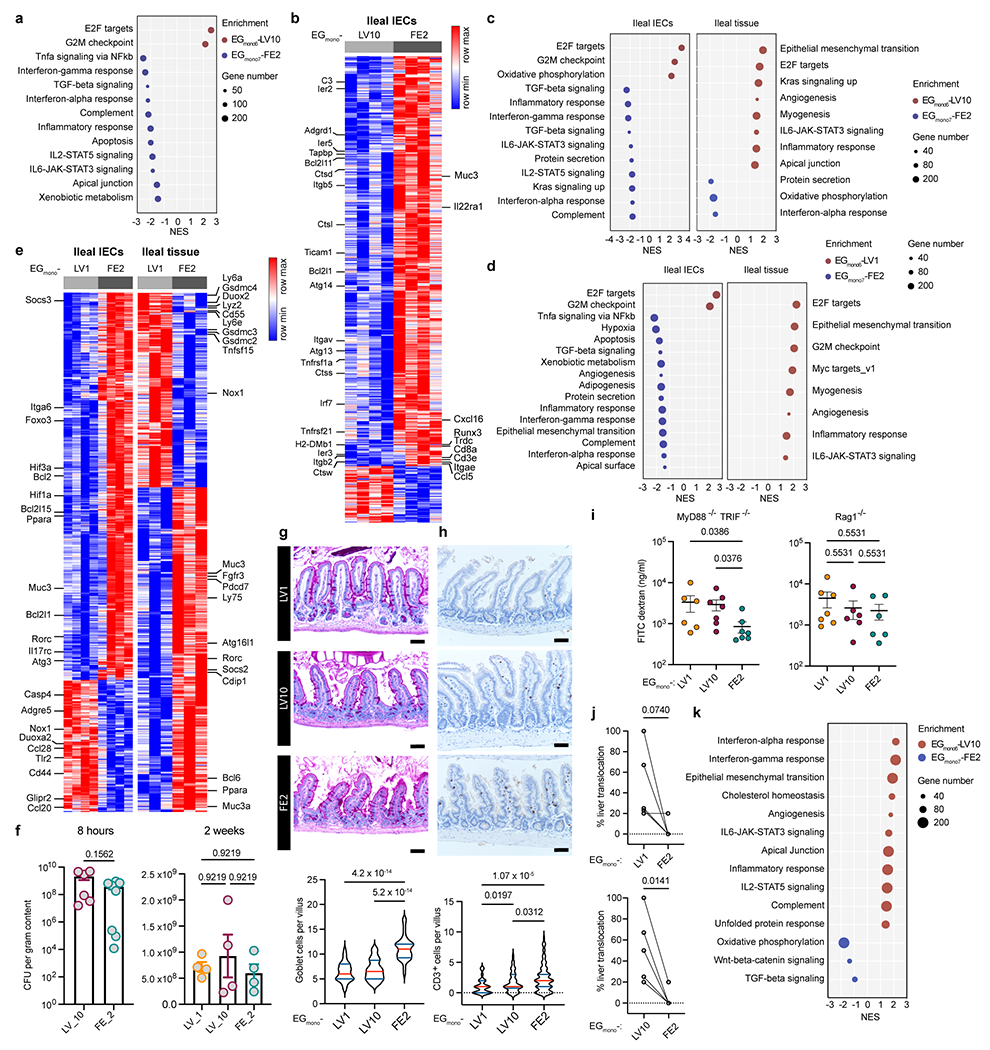
a-e, Transcriptomics of ileal epithelia and ileal tissues. a, b, Eight-hour monocolonisation. Enriched pathways (a) and differentially expressed genes (b) in IECs between mice colonised by EGmono6-LV10 or EGmono7-FE2. n = 4 mice. c-e, Two-week monocolonisation. c, Pathways enriched in IECs or ileal tissues induced by EGmono6-LV10 or EGmono7-FE2 colonisation. d, e, Enriched pathways (d) and differentially expressed genes (e) in IECs or ileal tissues between mice colonised by EGmono6-LV1 or EGmono7-FE2. IECs (n = 4), ileal tissues (n = 3). Left and right heatmaps display distinct gene sets. f, Density of E. gallinarum in distal ileum after 8-hour or 2-week monocolonisation. 8 hours: EGmono6-LV10 (n = 6), EGmono7-FE2 (n = 8). 2 weeks: n = 4 mice. g, h, PAS staining (g) or CD3 immunohistochemistry staining (h) showing goblet cells (g, n = 40 villi) or IELs (h, n = 90 villi) in distal ileum of monocolonised mice 2 weeks (g) or 18 hours (h) post-gavage. Scale bars, 50 μm. i, Gut permeability of monocolonised mice. MyD88−/−TRIF−/−: EGmono6-LV1 (n = 6), EGmono6-LV10 and EGmono7-FE2 (n = 7). Rag1−/−: EGmono6-LV1 (n = 7), EGmono6-LV10 and EGmono7-FE2 (n = 6). j, Percentage of monocolonised mice showing liver translocation. k, Pathways enriched in livers induced by EGmono6-LV10 or EGmono7-FE2 monocolonisation. Results are representative of n =2 (a-i) or 5 (j) independent experiments. Data represent mean ± SEM (f, i), violin plots (g, h) showing median (red) and quartiles (blue). Two-tailed Wald test with Benjamini-Hochberg correction (FDR = 0.05) (b, e), two-tailed Wilcoxon test (f, left panel), Kruskal-Wallis test with Benjamini-Hochberg correction (FDR = 0.05) (f, right panel; h, i), one-way ANOVA with Tukey’s post hoc test (g), paired two-tailed t-test (j). NES, normalized enrichment score.
Extended Data Fig. 7 |. Liver E. gallinarum isolates from SPF (NZW x BXSB) F1 mice exhibit similar features as experimentally evolved liver isolates from monocolonisations.
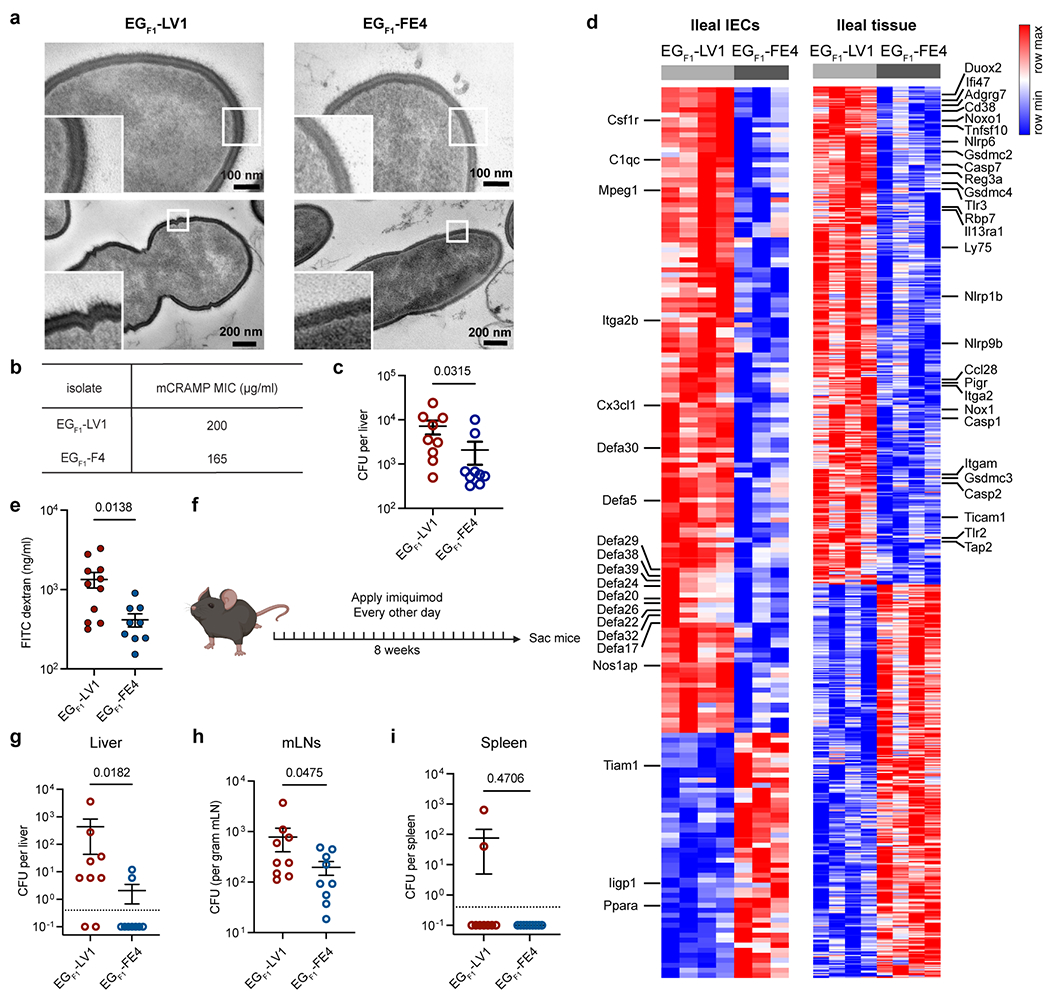
a, TEM images of cultured E. gallinarum isolates from (NZW x BXSB) F1 mice. Two bacterial cells of each isolate were shown. b, Minimum inhibitory concentration for mCRAMP against (NZW x BXSB) F1 E. gallinarum isolates. c, Liver clearance of intravenously injected (NZW x BXSB) F1 E. gallinarum isolates. Liver bacterial load 5 days post-injection. n = 9 mice. d, Differentially expressed genes in IECs (EGF1-LV1: n = 4, EGF1-FE4: n = 3) or ileal tissues (n = 4) between mice monocolonised with EGF1-LV or EGF1-FE4 for 2 weeks. Left and right heatmaps display distinct gene sets. e, Gut permeability of mice monocolonised with EGF1-LV1 (n = 11) or EGF1-FE4 (n = 9) for 2 weeks. f-i, Imiquimod-induced autoimmune phenotypes in mice monocolonised with EGF1-LV1 or EGF1-FE4. f, Schematic of imiquimod-induced autoimmunity. E. gallinarum translocation to the liver (g), mLNs (h) and spleen (i). n = 9 mice. Results in (a-i) are representative of two independent experiments. Data in (c, e, g-i) represent mean ± SEM. Two-tailed Mann-Whitney test (c, g-i), unpaired two-tailed t-test (e), two-tailed Wald test with Benjamini-Hochberg correction (FDR = 0.05) (d).
Extended Data Fig. 8 |. Divergent evolution of B. fragilis in monocolonised mice.
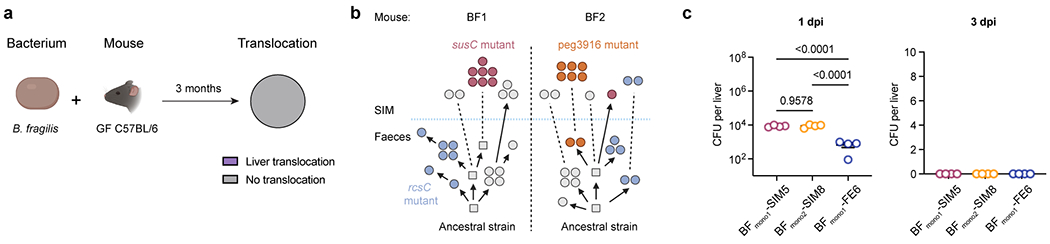
a, Schematic of experimental evolution of B. fragilis in monocolonised mice. Pie charts showing proportions of mice with liver translocation. n = 5 mice. b, Reconstructed phylogenetic histories of B. fragilis isolates sampled from two co-housed mice (BF1 and BF2). Circles represent sequenced isolates and squares represent hypothetical intermediate genotypes. Arrows connect related genotypes and dashed lines connect genetically identical isolates. SIM and faecal populations are separated by a blue dashed line. c, Liver clearance of intravenously injected B. fragilis. Representative SIM (BFmono1-LV5, BFmono2-LV8) and faecal (BFmono1-LV6) isolates. Liver bacterial load 1 or 3 dpi. n = 4 mice. Representative of two independent experiments. Data represent mean ± SEM. One-way ANOVA with Tukey’s post hoc test (c). SIM, small intestinal mucosa. dpi, days post-injection.
Extended Data Fig. 9 |. Impacts of within-host evolution of E. gallinarum on bacterial translocation and initiation of inflammation in the intestine and liver.
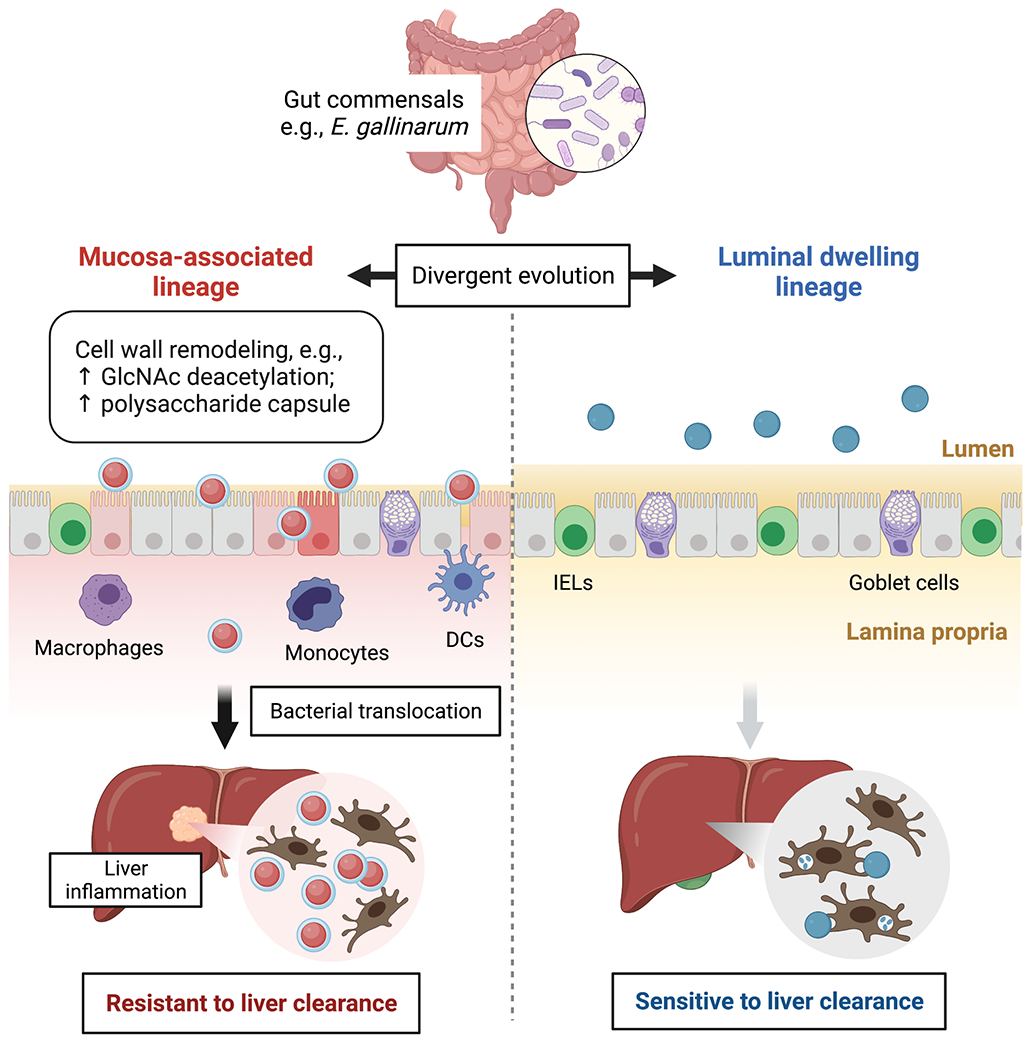
Within-host evolution of E. gallinarum facilitates divergence into independent lineages with distinct niche preferences and capacities for translocation. Faecal lineages preferentially colonise the intestinal lumen and are highly susceptible to immune recognition and clearance. By contrast, mucosa-associated lineages are adapted to colonise mucosal niches and exhibit enhanced resistance to immune recognition and clearance via remodelling of the bacterial cell wall. Functionally, the faecal lineage elicits robust immune responses at the intestinal epithelium, including enhanced mucus production and increased IEL recruitment; these responses reinforce the integrity of the gut barrier and limit bacterial translocation. By contrast, mucosally-adapted E. gallinarum lineages evade initial detection at the epithelial surface, translocate across the ‘leaky’ gut barrier, exhibit increased survival after translocation, and trigger inflammation in the intestinal lamina propria and liver.
Supplementary Material
Acknowledgements
We thank R. Medzhitov, A. Wang, and members of the Palm Lab for useful discussions and comments, and Y. Cao, T. A. Rice, and S. R. Leopold for experimental assistance. We thank Centre for Cellular and Molecular Imaging (CCMI) Electron Microscopy Facility at Yale University for electron micrograph acquisition, the Yale Research Histology Core for histological staining and Yale Centre for Genome Analysis for next-generation sequencing.
This work was supported by the National Institute on Aging of the National Institutes of Health under award number R01AG068863 (to N.W.P.). Approximately 50% of the funding for this research project (~$300,000) was financed with NIH funds; the remainder was financed by nongovernmental sources. N.W.P. also gratefully acknowledges support from the Common Fund of the National Institutes of Health (DP2DK125119), Leona M. and Henry B. Helmsley Charitable Trust (3083), Chan Zuckerberg Initiative, Michael J. Fox Foundation for Parkinson’s Research, Ludwig Family, Mathers Foundation, Pew Charitable Trust, NIGMS (RM1GM141649), and F. Hoffmann-La Roche Ltd. M.A.K. acknowledges support from the Lupus Research Alliance. The funders of this work had no role in study design, data collection and analysis, decision to publish, or preparation of the manuscript. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health. Schematic figures were created with BioRender.com
Competing interests
N.W.P. is a co-founder of Artizan Biosciences, Inc. and Design Pharmaceuticals, Inc., and has received research funding from Artizan Biosciences, Inc. and F. Hoffmann-La Roche AG. M.A.K. holds a patent on the use of microbiota manipulations to treat immune-mediated diseases (Patent No. PCT/US18/14368) and consults for Eligo Biosciences, Inc. All other authors have no competing interests.
Footnotes
Supplementary information is available for this paper.
Data availability
The sequencing data generated in this study is available at NCBI Sequence Read Archive (SRA) database. Genomic sequences are under the accession PRJNA743649 and RNA-seq data is under the accession PRJNA743979. Source data are provided with this paper.
References
- 1.Manfredo Vieira S et al. Translocation of a gut pathobiont drives autoimmunity in mice and humans. Science 359, 1156–1161, doi: 10.1126/science.aar7201 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Nakamoto N et al. Gut pathobionts underlie intestinal barrier dysfunction and liver T helper 17 cell immune response in primary sclerosing cholangitis. Nat Microbiol 4, 492–503, doi: 10.1038/s41564-018-0333-1 (2019). [DOI] [PubMed] [Google Scholar]
- 3.Chow J, Tang H & Mazmanian SK Pathobionts of the gastrointestinal microbiota and inflammatory disease. Curr Opin Immunol 23, 473–480, doi: 10.1016/j.coi.2011.07.010 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Ruff WE, Greiling TM & Kriegel MA Host-microbiota interactions in immune-mediated diseases. Nat Rev Microbiol 18, 521–538, doi: 10.1038/s41579-020-0367-2 (2020). [DOI] [PubMed] [Google Scholar]
- 5.Schloissnig S et al. Genomic variation landscape of the human gut microbiome. Nature 493, 45–50, doi: 10.1038/nature11711 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Zhao S et al. Adaptive evolution within gut microbiomes of healthy people. Cell Host Microbe 25, 656–667 e658, doi: 10.1016/j.chom.2019.03.007 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Elhenawy W, Tsai CN & Coombes BK Host-specific adaptive diversification of Crohn’s disease-associated adherent-invasive Escherichia coli. Cell Host Microbe 25, 301–312 e305, doi: 10.1016/j.chom.2018.12.010 (2019). [DOI] [PubMed] [Google Scholar]
- 8.Barroso-Batista J et al. Specific eco-evolutionary contexts in the mouse gut reveal Escherichia coli metabolic versatility. Curr Biol 30, 1049–1062 e1047, doi: 10.1016/j.cub.2020.01.050 (2020). [DOI] [PubMed] [Google Scholar]
- 9.Yilmaz B et al. Long-term evolution and short-term adaptation of microbiota strains and sub-strains in mice. Cell Host Microbe 29, 650–663 e659, doi: 10.1016/j.chom.2021.02.001 (2021). [DOI] [PubMed] [Google Scholar]
- 10.Group, N. H. W. et al. The NIH Human Microbiome Project. Genome Res 19, 2317–2323, doi: 10.1101/gr.096651.109 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Integrative, H. M. P. R. N. C. The Integrative Human Microbiome Project. Nature 569, 641–648, doi: 10.1038/s41586-019-1238-8 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Plumbridge J Control of the expression of the manXYZ operon in Escherichia coli: Mlc is a negative regulator of the mannose PTS. Mol Microbiol 27, 369–380, doi: 10.1046/j.1365-2958.1998.00685.x (1998). [DOI] [PubMed] [Google Scholar]
- 13.Honeyman AL & Curtiss R 3rd. Isolation, characterization and nucleotide sequence of the Streptococcus mutans lactose-specific enzyme II (lacE) gene of the PTS and the phospho-beta-galactosidase (lacG) gene. J Gen Microbiol 139, 2685–2694, doi: 10.1099/00221287-139-11-2685 (1993). [DOI] [PubMed] [Google Scholar]
- 14.Dubrac S & Msadek T Tearing down the wall: peptidoglycan metabolism and the WalK/WalR (YycG/YycF) essential two-component system. Adv Exp Med Biol 631, 214–228, doi: 10.1007/978-0-387-78885-2_15 (2008). [DOI] [PubMed] [Google Scholar]
- 15.Fried L, Behr S & Jung K Identification of a target gene and activating stimulus for the YpdA/YpdB histidine kinase/response regulator system in Escherichia coli. J Bacteriol 195, 807–815, doi: 10.1128/JB.02051-12 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Dobihal GS, Brunet YR, Flores-Kim J & Rudner DZ Homeostatic control of cell wall hydrolysis by the WalRK two-component signaling pathway in Bacillus subtilis. Elife 8, doi: 10.7554/eLife.52088 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Auchtung JM, Lee CA, Garrison KL & Grossman AD Identification and characterization of the immunity repressor (ImmR) that controls the mobile genetic element ICEBs1 of Bacillus subtilis. Mol Microbiol 64, 1515–1528, doi: 10.1111/j.1365-2958.2007.05748.x (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Donaldson GP, Lee SM & Mazmanian SK Gut biogeography of the bacterial microbiota. Nat Rev Microbiol 14, 20–32, doi: 10.1038/nrmicro3552 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Boneca IG et al. A critical role for peptidoglycan N-deacetylation in Listeria evasion from the host innate immune system. Proc Natl Acad Sci U S A 104, 997–1002, doi: 10.1073/pnas.0609672104 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Benachour A et al. The lysozyme-induced peptidoglycan N-acetylglucosamine deacetylase PgdA (EF1843) is required for Enterococcus faecalis virulence. J Bacteriol 194, 6066–6073, doi: 10.1128/JB.00981-12 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Thurlow LR, Thomas VC, Fleming SD & Hancock LE Enterococcus faecalis capsular polysaccharide serotypes C and D and their contributions to host innate immune evasion. Infect Immun 77, 5551–5557, doi: 10.1128/IAI.00576-09 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Dalton JE et al. Intraepithelial gammadelta+ lymphocytes maintain the integrity of intestinal epithelial tight junctions in response to infection. Gastroenterology 131, 818–829, doi: 10.1053/j.gastro.2006.06.003 (2006). [DOI] [PubMed] [Google Scholar]
- 23.Hoytema van Konijnenburg DP et al. Intestinal epithelial and intraepithelial T cell crosstalk mediates a dynamic response to infection. Cell 171, 783–794 e713, doi: 10.1016/j.cell.2017.08.046 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Olivares-Villagomez D & Van Kaer L Intestinal intraepithelial lymphocytes: sentinels of the mucosal barrier. Trends Immunol 39, 264–275, doi: 10.1016/j.it.2017.11.003 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.McPherson AC, Pandey SP, Bender MJ & Meisel M Systemic immunoregulatory consequences of gut commensal translocation. Trends Immunol 42, 137–150, doi: 10.1016/j.it.2020.12.005 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Zegarra-Ruiz DF et al. A Diet-Sensitive Commensal Lactobacillus Strain Mediates TLR7-Dependent Systemic Autoimmunity. Cell Host Microbe 25, 113–127 e116, doi: 10.1016/j.chom.2018.11.009 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Lee SM et al. Bacterial colonization factors control specificity and stability of the gut microbiota. Nature 501, 426–429, doi: 10.1038/nature12447 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Brown SP, Cornforth DM & Mideo N Evolution of virulence in opportunistic pathogens: generalism, plasticity, and control. Trends Microbiol 20, 336–342, doi: 10.1016/j.tim.2012.04.005 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Culyba MJ & Van Tyne D Bacterial evolution during human infection: Adapt and live or adapt and die. PLoS Pathog 17, e1009872, doi: 10.1371/journal.ppat.1009872 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Ha CWY et al. Translocation of viable gut microbiota to mesenteric adipose drives formation of creeping fat in humans. Cell 183, 666–683 e617, doi: 10.1016/j.cell.2020.09.009 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Van Tyne D et al. Impact of antibiotic treatment and host innate immune pressure on enterococcal adaptation in the human bloodstream. Sci Transl Med 11, doi: 10.1126/scitranslmed.aat8418 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Young BC et al. Severe infections emerge from commensal bacteria by adaptive evolution. Elife 6, doi: 10.7554/eLife.30637 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Palm NW et al. Immunoglobulin A coating identifies colitogenic bacteria in inflammatory bowel disease. Cell 158, 1000–1010, doi: 10.1016/j.cell.2014.08.006 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Wick RR, Judd LM, Gorrie CL & Holt KE Unicycler: Resolving bacterial genome assemblies from short and long sequencing reads. PLoS Comput Biol 13, e1005595, doi: 10.1371/journal.pcbi.1005595 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Andrews S FastQC: A quality control tool for high throughput sequence data [Online]. Available online at: http://www.bioinformatics.babraham.ac.uk/projects/fastqc/. (2010).
- 36.Bolger AM, Lohse M & Usadel B Trimmomatic: a flexible trimmer for Illumina sequence data. Bioinformatics 30, 2114–2120, doi: 10.1093/bioinformatics/btu170 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.De Coster W, D’Hert S, Schultz DT, Cruts M & Van Broeckhoven C NanoPack: visualizing and processing long-read sequencing data. Bioinformatics 34, 2666–2669, doi: 10.1093/bioinformatics/bty149 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Aziz RK et al. The RAST Server: rapid annotations using subsystems technology. BMC Genomics 9, 75, doi: 10.1186/1471-2164-9-75 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Overbeek R et al. The SEED and the Rapid Annotation of microbial genomes using Subsystems Technology (RAST). Nucleic Acids Res 42, D206–214, doi: 10.1093/nar/gkt1226 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Seemann T Prokka: rapid prokaryotic genome annotation. Bioinformatics 30, 2068–2069, doi: 10.1093/bioinformatics/btu153 (2014). [DOI] [PubMed] [Google Scholar]
- 41.T S. snippy: fast bacterial variant calling from NGS reads, <https://github.com/tseemann/snippy> (2015).
- 42.Deatherage DE & Barrick JE Identification of mutations in laboratory-evolved microbes from next-generation sequencing data using breseq. Methods Mol Biol 1151, 165–188, doi: 10.1007/978-1-4939-0554-6_12 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Treangen TJ, Ondov BD, Koren S & Phillippy AM The Harvest suite for rapid core-genome alignment and visualization of thousands of intraspecific microbial genomes. Genome Biol 15, 524, doi: 10.1186/s13059-014-0524-x (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Bankevich A et al. SPAdes: a new genome assembly algorithm and its applications to single-cell sequencing. J Comput Biol 19, 455–477, doi: 10.1089/cmb.2012.0021 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Darling AC, Mau B, Blattner FR & Perna NT Mauve: multiple alignment of conserved genomic sequence with rearrangements. Genome Res 14, 1394–1403, doi: 10.1101/gr.2289704 (2004). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Okonechnikov K, Golosova O, Fursov M & team U Unipro UGENE: a unified bioinformatics toolkit. Bioinformatics 28, 1166–1167, doi: 10.1093/bioinformatics/bts091 (2012). [DOI] [PubMed] [Google Scholar]
- 47.Letunic I & Bork P Interactive Tree Of Life (iTOL) v5: an online tool for phylogenetic tree display and annotation. Nucleic Acids Res 49, W293–W296, doi: 10.1093/nar/gkab301 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Page AJ et al. Roary: rapid large-scale prokaryote pan genome analysis. Bioinformatics 31, 3691–3693, doi: 10.1093/bioinformatics/btv421 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Eren AM et al. Community-led, integrated, reproducible multi-omics with anvi’o. Nat Microbiol 6, 3–6, doi: 10.1038/s41564-020-00834-3 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Martin M Cutadapt removes adapter sequences from high-throughput sequencing reads. EMBnet. journal 17, 10–12 (2011). [Google Scholar]
- 51.Howe KL et al. Ensembl 2021. Nucleic Acids Res 49, D884–D891, doi: 10.1093/nar/gkaa942 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Dobin A et al. STAR: ultrafast universal RNA-seq aligner. Bioinformatics 29, 15–21, doi: 10.1093/bioinformatics/bts635 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Anders S, Pyl PT & Huber W HTSeq—a Python framework to work with high-throughput sequencing data. Bioinformatics 31, 166–169, doi: 10.1093/bioinformatics/btu638 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Love MI, Huber W & Anders S Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol 15, 550, doi: 10.1186/s13059-014-0550-8 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Subramanian A et al. Gene set enrichment analysis: a knowledge-based approach for interpreting genome-wide expression profiles. Proc Natl Acad Sci U S A 102, 15545–15550, doi: 10.1073/pnas.0506580102 (2005). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Langmead B & Salzberg SL Fast gapped-read alignment with Bowtie 2. Nat Methods 9, 357–359, doi: 10.1038/nmeth.1923 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Quinlan AR & Hall IM BEDTools: a flexible suite of utilities for comparing genomic features. Bioinformatics 26, 841–842, doi: 10.1093/bioinformatics/btq033 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Cullen TW et al. Antimicrobial peptide resistance mediates resilience of prominent gut commensals during inflammation. Science 347, 170–175, doi: 10.1126/science.1260580 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Leenhouts K et al. A general system for generating unlabelled gene replacements in bacterial chromosomes. Mol Gen Genet 253, 217–224, doi: 10.1007/s004380050315 (1996). [DOI] [PubMed] [Google Scholar]
- 60.Laute-Caly DL et al. The flagellin of candidate live biotherapeutic Enterococcus gallinarum MRx0518 is a potent immunostimulant. Sci Rep 9, 801, doi: 10.1038/s41598-018-36926-8 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The sequencing data generated in this study is available at NCBI Sequence Read Archive (SRA) database. Genomic sequences are under the accession PRJNA743649 and RNA-seq data is under the accession PRJNA743979. Source data are provided with this paper.