

Abstract
Effectively delivering therapeutics for treating brain tumors has been hindered by the physical and biological barriers in the brain. Even with the compromised blood-brain barrier and highly angiogenic blood-tumor barrier seen in glioblastoma (GBM), most drugs, including nanomaterial-based formulations, hardly reach intracranial tumors. This work investigated sub-5 nm ultrafine iron oxide nanoparticles (uIONP) with 3.5 nm core diameter as a carrier for delivering DNA topoisomerase inhibitor 7-ethyl-10-hydroxyl camptothecin (SN38) to treat GBM. Given a higher surface-to-volume ratio, uIONP shows 1 or 3-folds higher SN38 loading efficiency (48.3 ± 6.1%, SN38/Fe, wt%) than those with core sizes of 10 or 20 nm. SN38 encapsulated in the coating polymer exhibits pH sensitive release with <10% over 48 hours at pH 7.4, but 86% at pH 5, thus being protected from converting to inactive glucuronide by UDP-glucuronosyltransferase 1A1. Conjugating αvβ3-integrin-targeted cyclo(Arg-Gly-Asp-D-Phe-Cys) (RGD) as ligands, RGD-uIONP/SN38 demonstrated targeted cytotoxicity to αvβ3 integrin overexpressed U87MG GBM cells with a IC50 of 30.9 ± 2.2 nM. The in vivo study using an orthotopic mouse model of GBM reveals tumor-specific delivery of 11.5% injected RGD-uIONP/SN38 (10 mg Fe/kg), significantly prolonging the survival in mice by ~41%, comparing to those treated with SN38 alone (p < 0.001).
Keywords: Iron oxide nanoparticle, SN38, Glioblastoma, Drug delivery, Brain tumor, Tumor integrin, Theranostics
Graphical Abstract
Using ultrafine iron oxide nanoparticles (uIONP) with 3.5 nm core diameter as a carrier to deliver insoluble chemotherapy agent, 7-ethyl-10-hydroxyl camptothecin (SN38), to intracranial tumors is reported. With cyclic peptide RGD as the ligand targeting the tumor integrin, RGD-uIONP/SN38 exerts tumor specific cytotoxicity to U87MG glioblastoma cells, prolonging survival of mice with glioblastoma.
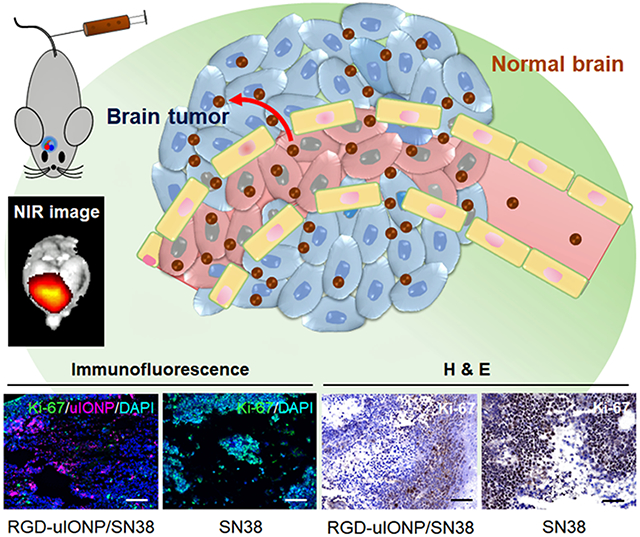
1. Introduction
Glioblastoma (GBM) is the most common malignant primary brain tumors in the central nerve system (CNS) characterized by its extremely poor prognosis with a mean 5-year survival rate of 5.8% and medium survival of less than 2 years.[1] One of the major challenges in treating GBM is the lack of effective delivery of therapeutic agents to the intracranial tumors that are protected by the blood brain barrier (BBB),[2] which represents the unique characteristics of restrictive physical and biological barriers in the CNS cancers.[3] Although the aggressive progression and angiogenesis of GBM due to the upregulations of epidermal growth factor receptor (EGFR) and vascular endothelial growth factor (VEGF) have been shown to manifest highly permeable tumor blood vessels and compromised BBB with the widened fenestration, referred as the blood-tumor barrier (BTB),[4] the intravenously (i.v.) administered therapeutic agents were still largely restricted to reach the intracranial tumors.[5]
In the past decades, nanomaterial-based drug delivery systems have been extensively developed and investigated for cancer treatment due to their advantages in improving drug solubility and pharmacokinetics, allowing for sustained release and enabling active targeting to specific biomarkers of tumors.[6] However, most of these nano-delivery systems were investigated for non-CNS cancers.[7] Efforts in developing nanomaterial-based therapeutics for GBM have not resulted in efficacious clinical outcomes, largely due to the limited and heterogeneous delivery of therapeutics to the intracranial tumors.[1a, 8] Among many nanocarriers, iron oxide nanoparticles (IONP) have been used in various clinical applications, due to the demonstrated biocompatibility, facile functionalization, and contrast enhancing effect in magnetic resonanc imaging (MRI).[6b, 9] For instance, ferumoxytol, one of the Food and Drug Administration (FDA) approved IONP formulations with core diameters ranging from 3.25 to 9 nm and hydrodynamic sizes of 17 to 30 nm,[10] has been investigated for detecting intracranial tumors in patients by MRI, taking advantage of the macrophage uptake and subsequent trafficking to the tumors.[11]
To overcome the barrier in delivering therapeutics to the brain tumor, a number of strategies have been investigated, such as receptor-mediated BBB crossing through transcytosis by targeting transferrin receptors,[12] convection enhanced delivery (CED),[13] macrophage related trafficking.[11] In addition to these strategies, nanomaterials are also know to passively target tumors through the enhanced permeability and retention (EPR) effect.[14] Particularly in the sub-5 nm size range, ultrafine IONP (uIONP) with the diameter of 3.5 nm have been shown to improve intratumoral distribution and retention comparing with larger IONP, e.g., 20 nm core size.[15] When conjugated with transferrin as the targeting ligand, uIONP exhibited a size-dependent improvement of the ligand-mediated tumor targeting and accumulation over IONP with 30 nm core by 6-fold in the mouse model of 4T1 breast tumor.[16] Importantly, high grade GBM tumors are typically highly vascularized due to angiogenesis, which is similar to breast tumors demonstrated in the 4T1 breast tumor model. Therefore, chaotic and porous tumor blood vessels presented in these tumors provide the opportunity and rationale for the EPR driven delivery of drug-loaded nanoparticles to the intracranial tumors. Although the EPR driven drug delivery depends on the unique properties of the tumor vasculature, it is not limited by the level of transmembrane receptors in the receptor-mediated transcytosis or requirement of the invasive procedure of CED as well as the time and population of macrophages needed for up taking and transporting the drug.
Here, we report the study of using uIONP coated with amphiphilic ploy(ethylene glycol)-block-allyl glycidyl ether (PEG-b-AGE) polymer as a carrier to deliver water-insoluble DNA topoisomerase I inhibitor, 7-ethyl-10-hydroxyl camptothecin (SN38), to treat intracranial tumors in the orthortopic mouse model of GBM. Cancer cells are known to develop de novo or aquired resistance to the alkylating agents, such as the standard chemotherapy agent temozolomide (TMZ) in clinic, with 90% of patients with recurrent glioma not responding to new cycle of TMZ chemotherapy.[1a, 17] In addition, pharmacokinetics and delivery of TMZ are limited by its low solubility in water (~4-5 mg/mL). Clinical administration of TMZ in patients is either done using oral formulations or through intraveneous infusion. In this study, SN38, the active metabolite of FDA approved anti-cancer drug Irinotecan®, was selected as the therapeutic agent, because (1) the drug resistance to the topoisomerase I inhibitors can be alleviated with improved delivery efficiency,[18] (2) SN38 is much more potent than Irinotecan®, and 3) its poor solubility in water and high systemic toxicity can be mitigated by using tumor targeted and polymer coated nanocarriers.[19] We applied the PEG-b-AGE polymer coating to provide a hydrophobic layer of AGE to encapsulate and protect SN38 before it is released in the tumor. To facilitate tumor specific delivery, we used cyclo(-RGDfC) peptide as the targeting ligand specific to tumor integrin αvβ3 overexpressed on angiogenic endothelial cells in the tumor vasculture and U87MG GBM cells. RGD-conjugated uIONP were labeled with near infrared (NIR) dye NIR830 for tracking the distribution in organs and the GBM tumor. The tumor targeting, delivery and treatment efficacy of RGD-conjugated uIONP/SN38 (RGD-uIONP/SN38) were investigated using an orthotopic mouse model of GBM (Scheme 1) with the results demonstrating tumor-specific delivery of 11.5% injected SN38 loaded uIONP RGD-uIONP/SN38 into the intracranial tumors and subsequent inhibition of the tumor growth and ~41% improvement in survival.
Scheme 1.

Illustration of the preparation of RGD-uIONP/SN38 labeled with near infrared dye NIR830. The prepared NIR830-RGD-uIONP/SN38 with a 3.5 nm core diameter can readily reach the intracranial tumor in the orthotopic mouse model of GBM after extravasating from the leaky tumor blood vasculatures with loosened tight junctions to accumulate in the tumor tissue, but not in the healthy brain tissue with intact BBB.
2. Results
2.1. Characterizations of Ligand Conjugated and SN38 Loaded uIONP
We prepared uIONP by thermo-decomposition and subsequently coated the oleic acid stabilized uIONP with amphiphilic PEG-b-AGE polymer following the methods in our previous reports.[20] Fourier transform infrared (FTIR) spectroscopy was performed for the polymer coated uIONP (Figure 1A) to verify the successful coating process. The bands at 3380, 2871 and 1091 cm−1 can be ascribed to the characteristic PEG peaks of O-H, C-H and C-O stretching, respectively.[21] The bands at 947 and 584 cm−1 are consistent with the reported Fe-O bending and Fe-O stretching vibrations,[22] which can be contributed by the iron oxide nanoparticle and the bonding between PEG-b-AGE coating polymer and Fe. Hydrophobic SN38 then was readily encapsulated in the amphiphilic PEG-b-AGE polymer coated on uIONP (uIONP/SN38). Encapsulation of SN38 in uIONP/SN38 did not change the solubility and stability of the overall nano-constructs in the aqueous medium. uIONP/SN38 remained highly mono-dispersed in water as observed in images (Figure 1B and C) from transmission electron microscopy (TEM). After loading SN38, the hydrodynamic diameter of PEG-b-AGE coated uIONP/SN38, measured by dynamic light scattering (DLS), slightly increased from 8.8 ± 1.3 nm (polydispersity index, PDI, = 0.02) without SN38 to 9.9 ± 1.8 nm (PDI = 0.03) (Figure 1D) with no statistical significance (p > 0.05), suggesting the limited effect of SN38 encapsulation to the overall hydrodynamic diameter. Similarly, there was no significant difference between the zeta potentials of uIONP (1.01 ± 0.17 mV) and uIONP/SN38 (0.99 ± 0.20 mV). However, the hydrodynamic diameter was increased to 11.8 ± 2.1 (PDI = 0.03) and 12.4 ± 2.6 nm (PDI = 0.04) after conjugation of RGD and subsequently near infrared (NIR) dye NIR830 on the uIONP surface (Figure 1D). The surface zeta potentials were substantially changed from the positively charged uIONP (1.01 ± 0.18 mV) and uIONP/SN38 (0.99 ± 0.22 mV) to the negatively charged RGD-uIONP/SN38 (−1.84 ± 0.24 mV) and NIR830-RGD-uIONP/SN38 (−1.64 ± 0.21 mV) (Figure 1E), which can be attributed to the neutralization of amine groups on PEG-b-AGE coated uIONP after conjugation of RGD and NIR830.[20b] The average number of amine groups on the uIONP surface was 91 ± 25 as determined by the ninhydrin assay,[20b] in accordance with the positively charged surface of uIONP. After surface functionalization with RGD and NIR830, we estimated that there were 52 ± 18 RGD and 47 ± 21 NIR830 molecules per uIONP respectively.
Figure 1.

An FTIR spectrum of PEG-b-AGE coated uIONP (A). TEM images of PEG-b-AGE polymer coated uIONP (averaged core diameter of 3.5 nm) before (B) and after (C) SN38 encapsulation (uIONP/SN38) showed the sustained mono-dispersion of uIONP/SN38. The averaged changes (N = 3) of hydrodynamic diameters (D) and zeta-potentials (E) of uIONP before and after the step-wised modifications, i.e., encapsulation of SN38 (red), conjugation of RGD ligands and labeling with NIR830 (blue and green). Incubating uIONP/SN38 in the buffer solutions of pH 5.0 and 7.4, mimicking the lysosomal and physiological pH, for 2, 4, 12, 24 and 48 hours showed a pH responsive SN38 release at pH 5 (F).
The loading efficiency of SN38 was measured at 48.3 ± 6.1% (mg SN38/mg Fe) based on the UV absorbance of SN38 at 380 nm.[23] This loading efficiency was approximately 1- and 3-folds higher than those of IONP with core diameters of 10 (22.8 ± 4.5%) and 20 nm (11.1 ± 2.8%), respectively (Supporting Information, Figure S2). The improved drug loading efficiency for uIONP over conventional IONP with larger diameters was likely due to the quadratic increase of the surface-to-volume ratio resulted from the decrease in the diameter of spherical nanoparticles.[24] To evaluate the stability of SN38 loaded on uIONP and its release profile, we incubated uIONP/SN38 in PBS (pH 7.4) and acetate buffer (pH 5.0) to mimic the physiological and lysosomal environment. Results revealed that ~86% of loaded SN38 was released from uIONP/SN38 at pH 5.0 over 48 hours, whereas only ~10% was released at the physiological pH of 7.4 (Figure 1F). The release of SN38 in the acidic environment may be attributed to the swelling of coating polymers at low pH as reported previously,[25] which led to the unfolding of hydrophobic AGE moiety and the subsequent drug release.
2.2. Specificity of Tumor Integrin Targeting by RGD Functionalized uIONP
To evaluate the targeting specificity of RGD-uIONP to U87MG human GBM cells with overexpression of αvβ3 integrin, we incubated U87MG cells with NIR830 labeled RGD-uIONP at a Fe concentration of 0.1 mg/mL for three hours at 37 °C. Human breast cancer MCF-7 and mouse macrophage Raw264.7 cells with no or low expression of αvβ3 integrin were used as the controls (Supporting Information, Figure S3).[26] Confocal fluorescence imaging of the cells showed strong NIR signal from NIR830-RGD-uIONP accumulated in U87MG cells (Figure 2A, F and K), indicating intensive uptake of NIR830-RGD-uIONP by U87MG cells with a high level of overexpression of the tumor integrin αvβ3. Meanwhile, MCF-7 cells with low αvβ3 integrin expression exhibited a low level of NIR830-RGD-uIONP uptake, evidenced by weak NIR830 signals (Figure 2C, H and M). Although macrophages were known for its non-specific phagocytosis of nanomaterials, uptake of NIR830-RGD-uIONP by Raw264.7 cells was not observed (Figure 2D, I and N) due to the substantially attenuated non-specific phagocytosis by the anti-biofouling effect from PEG-b-AGE coating on NIR830-RGD-uIONP as reported in our previous work.[20b, 20c] We further verified the ligand-mediated uptake of NIR830-RGD-uIONP by U87MG cells using the blocking assay, in which U87MG cells were co-cultured with an excess amount of RGD peptide (0.05 mM) and NIR830-RGD-uIONP (0.1 mg Fe/mL). As the result, uptake of NIR830-RGD-uIONP was markedly reduced in the presence of free RGD that competed with NIR830-RGD-uIONP in binding to targeted tumor integrin αvβ3 (Figure 2B, G and L). Furthermore, when we used non-targeted peptide cyclo(-RADfC) (RAD) to functionalized uIONP as the additional control for the above experiments, no uptake of NIR830-RAD-uIONP was observed in U87MG (Figure 2E, J and O), MCF-7 and Raw264.7 cells (Supporting Information, Figure S4). These results were further validated with Prussian blue staining for Fe and flow cytometry for U87MG and MCF-7 and Raw264.7 cells treated with RGD-uIONP and RAD-uIONP (Supporting Information, Figure S5 and S6).
Figure 2.

Fluorescence images of αvβ3 integrin targeted NIR830-RGD-uIONP incubated with U87MG GBM cells with over-expression of αvβ3 integrin (A, F and K), U87MG cells co-cultured with RGD (B, G and L), MCF-7 breast cancer cells with low expression of αvβ3 integrin (C, H and M), and Raw264.7 macrophages with low expression of αvβ3 integrin (D, I and N). NIR830-RAD-uIONP, the non-targeted control, exhibited no cellular uptake by U87MG cells (E, J and O). The scale bar is 50 μm.
2.3. Improved Bioavailability of SN38 after Encapsulation in uIONP
To examine the GBM-specific cytotoxicity of RGD-uIONP/SN38, we incubated RGD-uIONP/SN38 with U87MG GBM cells at 37 °C for 72 hours with the SN38 concentrations varying from 1 to 104 nM. The cytotoxicity analysis based on the half-maximal inhibitory concentration (IC50) showed a high potency from RGD-uIONP/SN38 with IC50 estimated at 30.9 ± 2.2 nM (Figure 3A), significantly lower than that of SN38 alone at 1770 ± 148 nM (p < 0.0001). In contrast, non-targeted RAD-uIONP/SN38 with no ligand-mediated intracellular delivery did not exhibit cytotoxicity to U87MG cells (Figure 3A). Given the roles of iron in various biological and biochemical functions and processes,[27] there have been increasing interests in whether IONPs can serve as an iron source to perturb iron homeostasis causing cytotoxicity to cells.[28] To rule out the possible effect of intracellular Fe accumulation on the cell death, we incubated U87MG cells with RGD-uIONP at the Fe concentrations equivalent to those used for RGD-uIONP/SN38, and found no obvious cytotoxicity of the RGD-uIONP vehicle (Figure 3A). When using MCF-7 breast cancer cells with low expression of αvβ3 integrin[26a, 26b] as the control, less than 10% reduction of cell viability was observed at all dosages of RGD-uIONP/SN38 comparing to the cells without treatment (Figure 3B). Similarly, RGD-uIONP/SN38 did not exhibit significant inhibitory effect on the growth of Raw264.7 macrophages (Figure 3C) or human embryonic kidney HEK293 cells (Figure 3D) with low expression of αvβ3 integrin[26b, 26c] over the same SN38 concentration range.
Figure 3.

The cytotoxicity of RGD-uIONP/SN38 (red) to U87MG GBM cells (A), MCF-7 breast cancer cells (B), Raw264.7 macrophages (C) and HEK293 embryonic kidney cells (D) measured by AlamarBlue assays in comparison to RAD-uIONP/SN38 (blue), SN38 without encapsulation (black) and vehicle RGD-uIONP (cyan) at 37 °C with SN38 concentrations ranging from 1 to 104 nM or equivalent Fe concentrations for RGD-uIONP. Each measurement was repeated and averaged ((N = 3). HPLC elution profiles of human liver microsomes treated with RGD-uIONP/SN38 and SN38 without encapsulation (E).
SN38 is known to be converted by the UDP-glucuronosyltransferase 1A1 (UGT1A1) to the non-toxic form of SN38 glucuronide (SN38G) mostly in the liver.[29] While this enzymatic conversion is important in de-toxifying the drug, it also reduces the bioavailability and activity of SN38 to treat tumors. Furthermore, inefficient detoxification of SN38 due to UGT1A1*28 polymorphism may cause a strong side-effect to patients.[30] Thus, using a tumor targeted nanocarrier for tumor-specific delivery of SN38 without releasing drugs in the circulation and normal organs may mitigate the systemic toxicity of SN38. To investigate whether encapsulation of SN38 in uIONP may protect it from UGT1A1 in the liver, we treated RGD-uIONP/SN38 with human liver microsomes containing UGT1A1 at 37 °C for one hour using SN38 as the control. The high-performance liquid chromatography (HPLC) analysis of lysates treated with SN38 alone revealed a ratio of SN38/SN38G at ~1/4, indicating that 80% of SN38 was converted to SN38G (Figure 3E). However, SN38G was not detectable in the sample of RGD-uIONP/SN38, suggesting that encapsulation of SN38 in the coating polymer of uIONP effectively protected SN38 from forming SN38G catalyzed by UGT1A1. These results are consistent with the liquid chromatography-mass spectrometry (LC-MS) of the lysates (Supporting Information, Figure S7), in which the identities of peaks were verified by the exact molecular mass of SN38 and SN38G. The conversion of SN38 to SN38G in the presence of mouse liver microsomes was also examined since the subsequent in vivo experiments were carried out in mice. Similarly, results showed no SN38 encapsulated in RGD-uIONP/SN38 being converted to SN38G, while ~50% of free SN38 was found being converted to SN38G (Supporting Information, Figure S8).
2.4. αvβ3 Integrin Targeted Delivery of RGD-uIONP/SN38 in Orthotopic Model of GBM
To investigate the delivery of RGD-uIONP/SN38 to the intracranial tumors, we performed a biodistribution analysis along with the measurement of blood half-life of RGD-uIONP/SN38 using the orthotropic mouse model of GBM derived from intracranial implantation of U87MG GBM cells. Ex vivo NIR imaging of organs and fluorescence staining of brain tumor tissues were performed to confirm the αvβ3 integrin targeted accumulation and intratumoral distribution of RGD-uIONP/SN38. NIR imaging of the tumor-bearing brains taken at 2 and 12 hours after i.v. injection confirmed the delivery of the NIR830-labeled RGD-uIONP/SN38 in the intracranial tumors (Figure 4A), with a substantially increased accumulation of NIR830-uIONP/SN38 in the tumors at 12 hours after injection comparing to that at 2-hour time point. Chemical analysis of Fe concentrations in collected tumor bearing brains at 24 hours after i.v. injection (Figure 4B) suggested the maximum accumulation of 11.5% of injected dose (ID) of RGD-uIONP (10 mg Fe/kg body weight), mostly in the tumors as shown in the NIR imaging and histological validation, comparing to the control receiving no RGD-uIONP with statistical significance (p < 0.01). In addition, uptake of NIR830-uIONP/SN38 in the liver and spleen was obvious as typically found in i.v. injected exogenous nanomaterials. Notably, a high level of NIR830 signal was observed in the kidney at both 2 and 12 hours after the injection, which was consistent with the previous reports on the partial renal clearance of sub-5 nm nanoparticles.[15, 20a, 31] However, the increased NIR830 signal at 12 hours after injection suggested that some of injected RGD-uIONP/SN38 remained in the circulation after 12 hours, avoiding the rapid clearance that is commonly seen in low molecular weight drugs.
Figure 4.

Ex vivo NIR imaging of the brain, kidney, spleen and liver of tumor bearing mice at 2 and 12 hours after i.v. injection of NIR830-RGD-uIONP/SN38 (A). Quantification of RGD-uIONP/SN38 accumulating in tumor-bearing brains (N = 3) based on Fe concentrations after subtraction of the Fe concentration in control mouse brains receiving no RGD-uIONP/SN38 (B). The statistical significance indicated the results of comparison with the control brains. Confocal fluorescence images of tumor tissues collected at 2 and 12 hours showing DAPI stained nuclei (C, G), immunofluorescence-stained tumor blood vessels (D, H) and NIR830-RGD-uIONP/SN38 (E, I), and their overlay (F, J). Pixel counts of NIR830-RGD-uIONP/SN38 over the distance from the CD31 stained tumor blood vessels (K). Immunofluorescence staining of mouse brain tumor tissues collected at 12 hours after i.v. injection showing the nuclei (L, Q), tumor blood vessels (M, R), NIR830-RGD-uIONP/SN38 (N, S), αvβ3 integrin (O), tumor associated macrophages (T) and the overlay (P, U). The scale bar is 100 μm. * p < 0.05, ** p < 0.01, ns: no significance.
To investigate whether the accumulation of NIR830-RGD-uIONP/SN38 in the tumor was resulted from their permeating leaky tumor blood vessels followed by RGD-mediated active targeting to the αvβ3 integrin, we performed a confocal microscopic analysis of the collected tumor tissue using immunofluorescence staining of blood vessels by rat antibodies against CD31, a marker for vascular endothelial cells, and rabbit anti-αvβ3 integrin antibodies for αvβ3 integrin. The fluorescence images showed that NIR830-RGD-uIONP/SN38 was mostly found within CD31-stained tumor blood vessels at 2 hours after i.v. administration (Figure 4D and E). At 12 hours after injection, a significant amount of NIR830 signals was seen in the tumor tissues outside blood vessels (Figure 4H and I), suggesting NIR830-RGD-uIONP/SN38 extravasating from the tumor blood vessels before diffusing further in the tumor. In addition, quantitative analysis of pixel counts for NIR830-RGD-uIONP/SN38 (Figure 4K) revealed a gradient of NIR830-RGD-uIONP/SN38 starting from blood vessels to the nearby tumor tissues, specifically more intense NIR830 signals overlapping with the blood vessels but less intense signals scattering in the tumor tissue near blood vessels (Figure 4G and H). The quantitative analysis of NIR830-RGD-uIONP/SN38 signals (Figure 4K) also revealed that at 12 hours after injection 32.9 ± 3.5% and 50.3 ± 6.6% of NIR830-RGD-uIONP/SN38 distributed within 20 and 50 μm from blood vessels, respectively, while some of NIR830-RGD-uIONP/SN38 diffused as far as 223 μm from blood vessels. In comparison, all NIR830-RGD-uIONP/SN38 was found in the area less than 15 μm away from blood vessels at 2-hour time point. Taking together, the results suggested that the delivery of NIR830-RGD-uIONP/SN38 to intracranial brain tumors is driven by the EPR effect. Importantly, NIR830-RGD-uIONP/SN38 appear only in the tumor tissues within tumor blood vessels immunofluorescence stained by CD31, but not in the normal brain, as no NIR830-RGD-uIONP/SN38 was found in the normal brain tissue (Supporting Information, Figure S9).
To verify whether RGD-uIONP/SN38 could specifically target U87MG cancer cells after extravasating from the tumor blood vessels, we examined the distribution of NIR830-RGD-uIONP/SN38 in the tumor tissues with immunofluorescence staining for αvβ3 integrin and CD31. Confocal fluorescence imaging of tumor tissue revealed a highly overlapped NIR830-RGD-uIONP/SN38 with αvβ3 integrin (Figure 4N and O) near tumor blood vessels (Figure 4M). These results suggested the effective targeting of U87MG GBM cells by RGD-uIONP/SN38 once they diffuse into the tumor. Notably, IONP, e.g., ferumoxytol (Feraheme®) off-label used for contrast enhanced MRI of tumors in GBM patients, has been shown to be taken up by tumor associated macrophages as they infiltrated into the tumors.[11a, 32] Hence, we further examined whether mononuclear phagocytes engulfing and trafficking of uIONP played a role in delivery of RGD-uIONP/SN38 to the GBM tumors. We used rat anti-CD68 antibody to stain CD68, a scavenger receptor expressed in the tumor associated macrophages, and anti-CD31 antibody for staining tumor vessels in the tumor tissue sections collected at 12 hours after injection. The confocal fluorescence images showed that only scattered NIR830-RGD-uIONP/SN38 were co-localized with the CD68-stained macrophages (Figure 4S and T), while most NIR830-RGD-uIONP/SN38 were spread in the tumor tissue distant from blood vessels where no CD68 stained macrophages were found. The results indicated that the macrophage-associated trafficking of nanoparticle only contributed partially, if not at all, to the time-dependent accumulation of NIR830-RGD-uIONP/SN38 in intracranial tumors comparing to the dominant EPR effect.
In the biodistribution experiment, we measured Fe content of collected organs (Supporting Information, Figure S10A) after i.v. injection of RGD-uIONP/SN38 at ID of 10 mg Fe/kg body weight. The results showed a peak accumulation (11.5% ID) in the tumor bearing brain at 24 hours, followed by the gradual decrease of Fe content at 48 (7.7% ID) and 72 hours (5.2% ID) as un-bound RGD-uIONP were slowly cleared.[15-16, 33] The blood half-time of RGD-uIONP was 7.77 hours (Supporting Information, Figure S10B) with the majority of injected RGD-uIONP (15.3 to 63.8% ID) found to accumulate in the liver within 72 hours. We also observed a steady increase of the Fe content in the kidney from 3.3 to 5.3% ID within 24 hours, followed by decreasing at 48 and 72 hours. This is likely due to the renal clearance of RGD-uIONP as reported in our previous work on the uIONP with oligosaccharide coating as well as other nanomaterials in the sub-5 nm core size range.[15, 20a, 31] The accumulation of RGD-uIONP in the spleen increased continuously within 48 hours after the injection but decreased after 72 hours. The Fe content in the lung was found to be increased from 1 to 24 hours after injection of RGD-uIONP, but went back to the normal level afterwards.
2.5. Efficacy of RGD-uIONP/SN38 in Treating Mice Bearing U87MG GBM Tumors
We performed a survival study to investigate the efficacy of RGD-uIONP/SN38 in treating mice with orthotopic U87MG GBM tumors following the treatment scheme shown in Figure 5A. Mice bearing orthotopic GBM tumors were randomly divided into five groups (N = 6/group), including: RGD-uIONP/SN38 at the high dosage (HD_NP) of 5 mg/kg body weight and low dosage (LD_NP) of 2.5 mg SN38/kg body weight, RAD-uIONP/SN38 as a non-targeted control (RAD_NP) and SN38 formulated with Cremophor EL as the drug only control (CrEL_SN38) at the dosage of 5 mg SN38/kg body weight, and PBS as no treatment control (NT). The treatment began from day 8 after tumor implantation when tumors were typically grown to the size of 20-25 mm3, mimicking a late-stage disease as GBM usually were diagnosed late in patients. RGD-uIONP/SN38 was administered through the tail vein injection every four days. The treatment dosage was determined based on the considerations of (1) the dosage of NK012, a SN38-incorporating polymeric micelles, reported in an early study of treating mice with U87MG GBM (30 mg/kg, three times every four days),[34] and (2) the estimated 11.5% ID of RGD-uIONP reaching intracranial tumors as shown in the biodistribution study.
Figure 5.

The scheme of the survival study for evaluating the efficacy of treatment using an intracranial mouse model of U87MG GBM (A). Contrast enhanced T1-weighted brain MRI of mice from the HD_NP and CrEL_SN38 groups post treatment with the enhanced tumor region segmented (colored) using a segmentation algorithm (B). Kaplan-Meier survival curve of mice bearing U87MG GBM tumors (N = 6) in the treatment groups (C). Averaged survivals of different treatment groups (D). * p < 0.05 and *** p < 0.001.
We monitored tumor growth longitudinally by Gd-DTPA contrast enhanced T1-weighted MRI of selected mice from the HD_NP and CrEL_SN38 groups every four days. After the tumor regions in slices showing enhanced tumors were segmented, mice in the CrEL_SN38 group showed an increase tumor volume on day 4 post treatment before reaching the endpoint with the second dose of treatment (Figure 5B). In contrast, the tumor growth inhibition was observed in the HD_NP group on the day 4 of the treatment, evidenced by observing a smaller tumor than that in the CrEL_SN38 group, suggesting the improved efficacy when delivering SN38 by RGD-uIONP.
The results of the survival study are summarized by the Kaplan-Meier survival curve (Figure 5C). The comparison between different treatment groups using a log-rank test revealed that HD_NP group exhibited significantly improved efficacy than the non-targeted RAD_NP (p < 0.05), CrEL_SN38 (p < 0.05) and NT group (p < 0.001), while the LD_NP group only showed higher efficacy than the NT group with statistical significance (p < 0.01). Comparing to an averaged survival of 10.5 ± 0.5 days in the NT group, mice treated with high dose of RGD-uIONP/SN38 had an averaged survival of 19.5 ± 3.1 days, which was significantly longer than the other groups (Figure 5D), i.e., 17.0 ± 4.6 days for LD_NP (p < 0.05), 14.3 ± 1.9 days for NT (p < 0.001) and 13.8 ± 2.5 days for CrEL_SN38 (p < 0.001). Notably, RGD-uIONP/SN38 exhibited greater effect on prolonging the survival of mice bearing orthotopic U87MG GBM tumor (i.e., 86% vs controls without the treatment) at a relatively lower dosage (5 mg SN38/kg/4 days), comparing to NK012, a polymeric micelle formulation of SN38 currently under a clinical trial, which showed 66% prolonged survival at a much higher dosage of 30 mg SN38/kg/4 days.[34] RGD-uIONP/SN38 treatment also exhibited a dose-dependent efficacy, as mice treated with high dose RGD-uIONP/SN38 survived significant longer than those treated with low dose (p < 0.05). Interestingly, the non-targeted RAD-uIONP/SN38 also exhibited therapeutic effect, likely through the passive tumor targeting and delivery of RAD-uIONP/SN38 to the brain tumors. The body weights of the mice were also monitored daily. Mice in all five groups exhibited stable weights before reaching the endpoint with a rapid weight loss (Supporting Information, Figure S11).
2.6. Histopathological Validation of Treatment Efficacy of RGD-uIONP/SN38
Immunohistochemistry (IHC) analysis of the tumor tissue samples collected from the animals in the groups of HD_NP, CrEL_SN38 and NT was performed using two common markers for proliferation of glioma cells, Ki-67 and EGFR, to further validate the effectiveness of RGD-uIONP/SN38 in inhibiting tumor growth. Expression levels of both EGFR (Figure 6A) and Ki-67 (Figure 6D) in the tumors were substantially reduced in mice treated with the high dose RGD-uIONP/SN38, suggesting a stronger effect of tumor growth inhibition of high dose RGD-uIONP/SN38 comparing to the animals treated with PBS, in which levels of EGFR (Figure 6C) and Ki-67 (Figure 6F) were high in the tumor based on observed intense IHC stain. Although tumors from animals receiving CrEL formulated SN38 showed a certain level of treatment induced necrosis (Figure 6B and E), the density of EGFR and Ki-67 appeared to be much higher than those treated with high dose RGD-uIONP/SN38, but similar to the PBS control without SN38 treatment.
Figure 6.

IHC analysis of EGFR and Ki-67 in the mouse brain tumor tissues collected from mice treated with high dose of RGD-uIONP/SN38 (A, D), CrEL_SN38 (B, E) and PBS (C, F). The scale bar is 100 μm.
To further verify whether the inhibited cancer cell growth and tumor progression were the results of targeted delivery of RGD-uIONP/SN38, we performed confocal fluorescence microscopic analysis to examine the spatial distribution of NIR830 labeled RGD-uIONP/SN38 in the tissue in relation to the cancer cells with expressions of EGFR and Ki-67 based on immunofluorescence staining of collected tissue samples. Using rabbit anti-EGFR and anti-Ki-67 antibodies with goat anti-rabbit IgG488 secondary antibody labeled with FITC for fluorescence imaging, the results of confocal fluorescence imaging analysis were consistent with those from IHC analysis, i.e., expressions of EGFR (Figure 7A) and Ki-67 (Figure 7I) for the high dose RGD-uIONP/SN38 treated mice were much lowered comparing to the non-treatment control (Figure 7D and L). More importantly, the majority of delivered RGD-uIONP/SN38 was found not in the regions with dense presentations of EGFR and Ki-67 (Figure 7A, B, F, I, J and N), suggesting the reduced expression of EGFR and Ki-67 and alleviated cell proliferation in the tumor regions where RGD-uIONP/SN38 were delivered. In contrast, tumor tissues of mice treated with CrEL_SN38 showed similar expression levels of EGFR and Ki-67 to those from non-treatment control (Figure 7C and K). Immunofluorescence staining for αvβ3 integrin revealed no obvious difference of αvβ3 integrin expression in the tumors receiving CrEL_SN38 and PBS, while the RGD-uIONP/SN38 treated tumors exhibited significantly reduced αvβ3 integrin level. Furthermore, fluorescent signals from stained αvβ3 integrin appeared to be not co-localized with those from RGD-uIONP/SN38 (Supporting Information Figure S12). Taking together, these results confirmed the inhibitory effect of RGD-uIONP/SN38 on the tumor growth in the delivered area. RGD-uIONP/SN38 exerts cancer cell death more effectively, comparing to treatment with CrEL_SN38 and PBS.
Figure 7.

Confocal fluorescence images of EGFR (A, C and D) and Ki-67 (I, K and L) after immunofluorescence staining and NIR830-RGD-uIONP/SN38 (B and J) in tumor-bearing brain tissues of mice treated with high dose RGD-uIONP/SN38 (A to B and I to J), CrEL_SN38 (C and K) and PBS (D and L). The overlay of fluorescence images of NIR830-RGD-uIONP/SN38, EGFR and Ki-67 with DAPI stained nuclei (E to H and M to P). The scale bar is 100 μm.
3. Discussion
Current clinical management of GBM, which is consisted of surgical resection of the tumor followed by radiation and chemotherapy, has limited success in improving patient survival. Impossible to completely resect the infiltrative GBM, the normal tissue damage associated with aggressive radiation therapy of infiltrating GBM, and the ineffective delivery of drugs to the intracranial tumors as well as tumor developing drug resistance are all responsible to the recurrence of GBM.[1a] In general, delivery of small molecule chemotherapy agents with effective dosage to brain tumors has long been challenging due to the fast clearance and efflux pump by receptors on blood vessels.[3] While nanomaterial-based delivery systems have shown to have advantages in changing pharmacokinetics of the drugs to improve drug availability, enabling biomarker targeted delivery and escaping the clearance by the efflux pump in solid tumors, most conventional nanomaterials used for drug delivery are in the core size of tens to hundreds nm, not effective in crossing the BTB in brain tumors, which have shown size-dependent permeability for nanomaterials, in both rodent models and spontaneous canine models.[6a, 6b, 7, 35] On the other hand, uIONP with sub-5 nm core size have been shown to improve both EPR driven passive targeting and ligand-mediated active targeting to the tumors than IONP with larger size.[15-16] The current study further supported this notion with the additional evidence from the intracranial tumor targeted delivery to demonstrate the advantages of sub-5 nm uIONP platform in improving the delivery efficiency of therapeutics. Our results showed RGD-uIONP/SN38 extravasated through leaky blood vessels in GBM tumors and subsequently diffused further in the tumor tissue, thus rendering an improved prognosis and outcomes for brain tumor treatment. Noteworthy, despite the observed time-dependent extravasation from blood vessesls and deeper tumor diffusion of RGD-uIONP/SN38, an exclusive conclusion cannot be made at this point that the RGD-uIONP/SN38 entering the brain tumor is solely beneifited from the EPR effect and the sub-5 nm ultrafine size. As the expression of αvβ3 integrin is also up-regulated for the endothelial cells of tumor blood vessels,[36] which can also be targeted by the RGD-uIONP/SN38, the contribution of tumor blood vessel targeting to the brain tumor delivery of RGD-uIONP/SN38 remains to be illucidated in the future systematic study. Although we have shown that the uIONP with the sub-5 nm core size exhibited significant increase of accumulation in the 4T breast tumors comparing the same formulation of IONP with a larger core size (20 or 30 nm)15,16, further investigations of the ultrafine size-enhanced nanoparticle delivery to intracranial brain tumors by comparing uIONP and RGD-uIONP/SN38 with larger IONP and drug-loaded IONP with different sizes would further validate this mechanism.
DNA topoisomerase I inhibitor SN38 is the active metabolite of chemotherapy agent Irinotecan®, but 100- to 1000-fold more potent. While this class of chemotherapy agents is a part of the standard care for many cancers,[18b] it is not used for treating GMB successfully, in parts because of its poor solubility, fast clearance and high systemic toxicity, especially when escalating the dosage to achieve the therapeutic effect.[19] We successfully encapsulated the water-insoluble SN38 molecules in the hydrophobic AGE moieties on the surface of the sub-5 nm nanocarrier for the more efficient delivery in GBM treatment. Encapsulation of SN38 on RGD-uIONP not only prevented the premature release of SN38 in the circulation and normal organs to cause systemic toxicity, but also protected its therapeutic effect before being delivered to the targeted tumor sites. Furthermore, there is a concern that patients carrying the homozygous and heterozygous UGT1A1*28 allele with reduced UGT1A1 expression may be exposed to the severe side effect of Irinotecan® and SN38 due to inefficient conversion or detoxification of SN38 by UGT1A1. For those patients, a reduced initial dose was recommended by the Food and Drug Administration (FDA).[37] The stable encapsulation of SN38 on the uIONP and subsequent tumor targeted delivery without premature release of SN38 may thus alleviate the SN38-associated side effect in these patients, while allowing for administering the proper dosage to patients carrying homozygous and heterozygous UGT1A1*28 allele.
The fluorescence imaging investigation of the intratumoral distribution of RGD-uIONP/SN38 revealed that RGD-uIONP/SN38 accumulated in the brain tumors are mostly αvβ3 integrin targeted with small amount found to be co-localized with tumor associated macrophages. While IONP, for instance ferumoxytol, have been reported to inhibit tumor growth in vivo by polarizing the tumor-associated macrophages to the pro-inflammatory subtype after phagocytosis,[38] the non-specific uptake by monocytes limits the efficient delivery of IONPs to tumors via systemic administration,[39] thus hindering their applications as targeted delivery system for therapeutics in cancer management. This study also showed evidence of RGD-uIONP/SN38 escaping from non-specific phagocytosis in certain extent, which is worthy of more systematical investigations to elucidate its interaction with nuclear monocytes in greater detail, as such interaction plays critical roles in modulating the tumor immune.[40] The developed RGD-uIONP/SN38 showed a targeted delivery of 11.5% ID to tumors in the orthotopic mouse model, ~15-folds higher than the medium delivery efficiency of nanomaterials (0.7% of ID) to solid tumors as reported in the literature,[39] even though GBM is one of the most challenging tumors for nanomaterials to be delivered.[7] Thus, the developed RGD-uIONP/SN38 demonstrates the potential as a more efficient drug delivery system than other IONP formulations.
Our survival study showed that high dose RGD-uIONP/SN38 demonstrated significantly improved efficacy and extended survival of mice over all the other groups. The pathological characterizations for EGFR and Ki-67 exhibited substantially reduced expressions in the tumor regions with delivery of RGD-uIONP/SN38, suggesting the more effective inhibition of cell proliferation in the tumors by high dose RGD-uIONP/SN38. This is supported by the observations of time-dependent diffusion of RGD-uIONP/SN38 in the intracranial tumors. Worth noting, the non-targeted RAD-uIONP/SN38 also demonstrated capability in prolonging the mouse survival comparing to the control without any treatment, which can be ascribed to the accumulation of RAD-uIONP/SN38 in the tumor via passive targeting.[15] However, similar to the results from previous studies,[16] tumor targeting of RAD-uIONP/SN38 through merely the EPR effect is not as effective as that coupled with ligand-mediated active targeting for tumor retention, evidenced by the significantly improved survival of mice receiving tumor integrin RGD-uIONP/SN38. Taking together, the results of this preclinical investigation indicate that RGD-uIONP carrier renders SN38 an improved pharmacokinetics and effective diffusion in the tumors to inhibit the cancer cell proliferation and eventually induce cancer cell death and to prolong the survival. For future development and translation of this drug delivery platform for treating brain tumor, the systematical comparison of delivery efficiency and treatment efficacy with the current standard care chemotherapy agent, TMZ, is granted to determine the proper regime and clinical value to patients.
4. Conclusion
In this study, we showed that highly potent yet hydrophobic DNA Topoisomerase I inhibitor SN38 can be encapsulated in the PEG-b-AGE co-polymer coating of sub-5 nm uIONP, rendering SN38 with improved solubility in water and high stability against detoxification by UGT1A1. After conjugating with RGD as the ligand, the RGD-uIONP/SN38 exhibited highly specific targeting and cytotoxicity to αvβ3 integrin overexpressed U87MG GBM cancer cells. In an orthotopic mouse model of U87MG GBM, the time-dependent accumulation of RGD-uIONP/SN38 in the intracranial tumors was found driven by the EPR effect followed by αvβ3 integrin targeting, instead of macrophage uptake and related trafficking to GBM tumors. Treating mice bearing intracranial GBM with the cancer cell targeted RGD-uIONP/SN38 significantly improved the survival of animals and inhibition of cancer cell proliferation.
5. Experimental Section
5.1. Materials
Ferric nitrite, sodium oleate, sodium hydroxide, sodium hydride (dry, 95%), sodium azide, sodium sulfate (anhydrous), PEG1000, allylglycidyl ether, methanesulfonyl chloride (99.5%), triphenylphosphine, 2,2-azobis(isobutyronitrile) (AIBN), (3-mercaptopropyl)trimethoxysilane, IR-783, 4-mercaptobenzoic acid, N,N’-disuccinimidyl carbonate, nickel perchlorate hexahydrate, 1,10-phenanthroline, triethylamine, trifluoroacetic acid (TFA), ninhydrin, Cremophor® EL, 3,3’-diaminobenzidine (DAB) enhanced liquid substrate system tetrahydrochloride, dimethylformamide (DMF), dimethylsulfoxide (DMSO), hexane, dichloromethane, chloroform, toluene, tetrahydrofuran (THF) ethanol (200 proof), acetonitrile and acetone were purchased from Sigma-Aldrich (St. Louis, MO, USA). SN38 was purchased from Selleck Chemicals (Houston, TX, USA). Sulfosuccinimidyl-4-(N-maleimidomethyl)cyclohexane-1-carboxylate (sulfo-SMCC), optimal cutting temperature compound (OCT), 4’,6-diamidio-2-phenylindole (DAPI), ProLong™ Gold Antifade Mountant with DAPI, AlamarBlue™ cell viability reagent, K3 EDTA blood tubes, human and mouse liver microsomes were obtained from Thermo Fisher Scientific (Rockford, IL, USA). Uridine diphosphate glucuronic acid (UDPGA) was purchased from Cayman Chemical Company (Ann Arbor, MI, USA). The MRI contrast agent Magnevist® was obtained from the pharmacy of Emory University Hospital. Phosphate-buffered saline (PBS), fetal bovine serum (FBS), Dulbecco's modification of Eagle's medium (DMEM), Roswell park memorial institute (RPMI) 1640 medium, Eagle’s minimum essential medium (EMEM), normal goat serum, penicillin-streptomycin solution and trypsin-EDTA solution were obtained from Mediatech Inc. (Herndon, VA, USA). Cyclo-(RGDfC) and cyclo-(RADfC) were purchased from Peptide International, Inc. (Louisville, KY, USA). U87MG human glioblastoma cells, Raw264.7 murine macrophage cells, HEK293 human embryonic kidney and MCF-7 human breast cancer cells were purchased from ATCC (Manassas, VA, USA). Rabbit monoclonal anti-integrin αvβ3 antibody (catalog number: ZRB1190) was purchased from Sigma-Aldrich (St. Louis, MO, USA). Rabbit polyclonal Ki-67 antibody (catalog number: PA5-19462), rabbit polyclonal anti-EGFR antibody (catalog number: PA1-1110) and rat monoclonal anti-CD31 (PECAM-1) antibody (390) (catalog number: 14-0311-82) were purchased from Thermo Fisher Scientific (Rockford, IL, USA). Rat polyclonal anti-CD68 antibody was purchased from Bio-Rad Laboratories Inc. (Hercules, CA, USA). Goat anti-rabbit IgG 488 (catalog number: ab150077), goat anti-rat IgG 555 (catalog number: ab150158) and goat anti-rat IgG 488 (catalog number: ab150157) were purchased from Abcam (Cambridge, UK). Fluoro-Gel mounting medium was purchased from Electron Microscopy Sciences (Hatfield, PA, USA).
5.2. Preparation and Characterization of SN38 loaded uIONP with PEG-b-AGE Coating
SN38 was loaded on uIONP through encapsulation in the hydrophobic layer of the amphiphilic uIONP coated with PEG-b-AGE polymer. First, oleic acid coated uIONP were prepared by thermo-decomposition from iron(III) oleate using our previous method,[20a] followed by transformation and stabilization in water through ligand exchange of oleic acids with the PEG-b-AGE polymer synthesized according to our published procedure.[20b] In a typical coating procedure, oleic acid coated uIONP (10 mg Fe) were dispersed in THF (10 mL), and mixed with PEG-b-AGE solution in THF (60 mg/mL, 5mL), with triethylamine added (final concentration of 0.1 mM) to neutralize the exchanged oleic acids. The THF solution was magnetically stirred for 48 hours to allow the optimal replacement of oleic acids by the PEG-b-AGE polymer. The encapsulation of SN38 was carried out immediately after the coating process. SN38 (10 mg) was directly added to the THF coating solution. The solution in a centrifuge tube was kept inverting on a shaker at room temperature for additional 24 hours to allow the SN38 molecules getting incorporated or absorbed in the hydrophobic AGE moiety of the coating polymer. Afterwards, the THF solution was added dropwise to vigorously stirred deionized (DI) water (45 mL) to allow the collapse of AGE hydrophobic layer onto the surface of uIONP with the SN38 encapsulated. The THF/water mixed solution was then dialyzed to remove organic solvent, triethylamine and uncoated polymers, followed by centrifugation at 3000 rpm for 5 min and filtration through a 0.22 μm filter to remove large aggregates and unloaded SN38 molecules. The hydrodynamic size and zeta potential of the SN38 loaded and PEG-b-AGE coated uIONP were measured on a Malvern Zetasizer NanoZS90 (Malvern, United Kingdom). The Fourier transform infrared (FTIR) spectrum of PEG-b-AGE coated uIONP was recorded on a Nicolet Magna-IR Fourier-Transform 560 spectrometer (ThermoFisher Scientific, Waltham, MA, USA). The Fe concentration of uIONP solution was measured by the 1,10-phenanthroline colorimetric assay as described in the literature.[20b] In a representative procedure, uIONP solution (20 μL) in concentrated nitric acid (400 μL) was heated up to 70 °C for three hours to dissassociate uIONP into Fe ions. In a sodium citrate solution (25 g/L, 95 μL), the resultant nitric acid solution (5 μL) was added, follow by the addition of hydroquinone (10 g/L in water, 40 μL), 1,10-phenanthroline (1 g/L, 40 μL) and acetate buffer (pH 3.5, 20 μL). The absorance at 508 nm was measured for the solution to quantify the Fe concentration according to the simultaneously built standard curve. The concentration of uIONP with a core diameter of 3.5 nm was calculated by assuming that the uIONP were spherical with a bulk magnetite density of 5.18 g cm−3. The number of amine groups (−NH2) from PEG-b-AGE coating on uIONP was measured by a ninhydrin assay as described previously.[20b] Briefly, ethanol/DI water (50/50, v/v) was used to dissolve ninhydrin (0.1 M) and sodium acetate trihydrate (1 M) to make the ninhydrin stock solution. Solution of PEG-b-AGE polymer coated uIONP (200 μL) was mixed with the ninhydrin stock solution (500 μL) and heated up to 80 °C for 15 min. After the solution was cooled down to room temperature, the absorbance at 570 nm was then measured for the solution to determine the concentration of −NH2 groups. The number of −NH2 groups on each uIONP was estimated by the molar ratio of −NH2 over uIONP in a given solution.
The loading efficiency of SN38 was determined by Equation (1):
| Equation (1) |
where [SN38] and [uIONP] are the concentrations of SN38 and Fe (in mg/mL) of a given uIONP/SN38 solution in water. [SN38] was determined by the absorbance at 380 nm using a Genesys 50 UV-Vis spectrophotometer (Thermo Scientific, Waltham, MA, USA) after subtracting the absorbance contribution from uIONP.[23] [uIONP] was measured by the 1,10-phenanthroline colorimetric assay as described above.
5.3. Functionalization of uIONP/SN38 with RGD and NIR830 Dye
The conjugation of RGD or RAD to the uIONP was performed by reacting the amine groups on the surface of uIONP with sulfo-SMCC following the manufacturer’s instruction to introduce maleimide groups, which readily reacted with the cysteines of RGD to covalently attach the RGD peptides to the surface of uIONP as targeting ligands. Briefly, sulfo-SMCC was mix with uIONP in the molar ratio of 20:1 (sulffo-SMCC/uIONP). The mixture was kept inverting on a shaker for two hours at room temperature, before purified using a PD-10 desalting column to remove unreacted sulfo-SMCC. The collected SMCC-uIONP was then mixed with RGD or RAD with the molar ratio of peptide/SMCC-uIONP = 20:1. The mixture was allowed to react at room temperature for one hour before un-conjugated peptide was removed by PD-10 column. The number of conjugated RGD or RAD per uIONP was estimated by the numbers of −NH2 groups on uIONP before and after conjugation. The labeling of uIONP with the near infrared dye NIR830 (excitation/emission at 791/810 nm) was carried out by mixing uIONP with NHS-NIR830 in PBS at room temperature for two hours. The hydrodynamic diameters and zeta potentials of uIONP before and after labeling with RGD peptides and NIR830 were measured using a Zetasizer (NanoZS90, Malvern Panalytical, Malvern, United Kingdom) to validate the conjugations. The number of RGD and NIR830 dye molecules conjugated to the uIONP was determined by measuring the number of −NH2 groups on uIONP before and after conjugation using Ninhydrin assays as described previously.[20b] The synthesis of NHS-NIR830 was conducted following the method in the literature using IR-783 as the starting material.[41]
5.4. Release of SN38 from uIONP
The SN38 release profile at pH 5.0 and 7.4 was determined by monitoring the changes in the amounts of SN38 loaded on unit weight of uIONP (in mg SN38/100 mg Fe) after incubating in buffered solutions over 48 hours. Since most nanomaterial-based drug complexes are internalized by targeted cancer cells via receptor-mediated endocytosis, in which the nanocarriers and cargo therapeutics have shown substantial accumulation in the early endosomes and eventually lysosomes along the trafficking pathway, the release of SN38 from the uIONP under lysosomal environment was investigated in buffered solution with pH 5, which mimics the lysosomal acidity.[42] The possibility of unintentional release of SN38 during blood circulation or clearance by healthy organs was explored in PBS mimicking the physiological condition of pH 7.4. Briefly, uIONP/SN38 was added to acetate buffer (0.1M, pH 5.0) and PBS (1X, pH 7.4) in a 2 mL centrifuge tube to reach the final volume of 1.5 mL with the Fe concentration of 1 mg/mL. The tubes were kept inverting on a shaker for 48 hours at room temperature. A portion of the solution (300 μL) was taken at 2, 4, 12, 24 and 48 hours for centrifugation at 3000 rpm for 10 min to precipitate the released SN38. Afterwards, the supernatant was taken for the measurement of SN38 loading efficiency by a UV-Vis spectrophotometer (Genesys 50, Thermo Scientific, Waltham, MA, USA) as described above. The experiment was repeated independently for three times.
5.5. Ligand-Mediated Cell Targeting of RGD-uIONP/SN38
The specificity of cell targeting of RGD-uIONP/SN38 mediated by the αvβ3 integrin was investigated in vitro using U87MG human glioblastoma cells with overexpression of αvβ3 integrin,[43] while MCF-7 human breast cancer cells[26a, 26b] and unpolarized Raw264.7 murine macrophage cells[26c] with low or no expression of αvβ3 integrin were used as the controls. Cells were maintained in culture media supplemented with 1% penicillin-streptomycin and 10% FBS in a humidified incubator with 5% CO2 at 37 °C. The U87MG and MCF-7 cells were cultured in DMEM, and Raw264.7 cells were kept in RPMI1640 medium. In a typical experiment, 5 × 104 cells were seeded into an 8-well chamber slide and cultured for 24 hours to allow cell attachment. Afterwards, the cells were cultured in the media containing NIR830-RGD-uIONP/SN38 at the Fe concentration of 0.1 mg/mL for three hours at 37 °C. For the blocking assay, RGD was added to reach the concentration of 0.05 mM in the media to co-culture with NIR830-RGD-uIONP/SN38 (0.1 mg Fe/mL) and cells. The cells were then washed with PBS for three times, followed by fixation using 4% paraformaldehyde in PBS solution for 20 min. The cells were washed again with PBS for three times before the slide was mounted with ProLong™ Gold Antifade Mountant with DAPI, and kept in 4 °C protecting from light for fluorescence imaging using a fluorescent microscope (BZ-X710, Keyence, Osaka, Japan) with a customized filter for NIR830. To analyze the cell targeting by RGD-uIONP/SN38 using flow cytometry, 105 cells were seeded in a 6-well plate and cultured in 1 mL DMEM supplemented with 1% penicillin-streptomycin and 10% FBS for 24 hours at 37 °C to allow the cells to attach. After treating cells with medium containing FITC-RGD-uIONP/SN38 at the Fe concentration of 0.02 mg/mL for three hours, the cells were harvested and washed three times with PBS. The cells were analyzed using a BD FACSymphony A3 flow cytometer. Measurements were taken three times for 1×105 cells each time. The experiment was repeated three times separately. The data was processed by Tree Star FlowJo software (FlowJo, LLC, Ashland, OR, USA).
5.6. Assay for Determining the Conversion of SN38 to SN38G
The SN38 to SN38G conversion in different forms of SN38 was investigated by treating RGD-uIONP/SN38 or SN38 alone with human or mouse liver microsomes containing UGT1A1, following the previously published methods.[29a, 44] In a typical experiment, liver microsomes (0.4 mg) and SN38 (0.4 μg) were dissolved in 0.2 mL Tris-HCl buffer with the presence of MgCl2 (10 mM) and UDPGA (5 mM) as the substrates for the glucuronidation of SN38. The solution was incubated at 37 °C for one hour, followed by addition of methanol (0.2 mL) with 1% HCl to stop the reaction. The solution was centrifuged at 2500 rpm for 30 min at 4 °C to collect the supernatant. SN38 and SN38G in the supernatant was detected by a Thermo Fisher UltiMate 3000 high performance liquid chromatography system (Waltham, MA, USA) equipped with a Acclaim™ 120 C18 columns (Thermo Fisher Scientific, Waltham, MA, USA) using the conditions adopted from the literature. Briefly, the samples (20 μL) were manually injected to the system, and eluted using a gradient of mobile phase consisting acetonitrile with 0.1% TFA (phase A) and DI water with 0.1% TFA (phase B) at a flow rate of 1 mL/min. The gradient of the mobile phase was set as: an initial eluent of 5% phase A for 5 min, followed with increased phase A to 65% by 35 min and rapid decrease of phase A to 5% in 2 min. The SN38 and SN38G in the sample were detected by the UV absorbance at 360 nm. The retention time of SN38 was determined by running the SN38 pure compound under the same HPLC condition. SN38 and SN38G were quantified based on the integrated area of corresponding peaks. To verify the molecular weights of the detected peaks, the samples were also examined by liquid chromatography-mass spectrometry (LC-MS) on a Thermo LTQ-FTMS instrument (Thermo Scientific, Waltham, MA, USA) using the same mobile phase condition.
5.7. Cytotoxicity of RGD-uIONP/SN38 to Targeted Cancer Cells
To investigate whether the cytotoxicity of RGD-uIONP/SN38 is specific to the targeted U87MG GBM cells with overexpression of αvβ3 integrins, the viability of U87MG cells after treating with RGD-uIONP/SN38 was determined by the AlamarBlue assay following our published protocol.[45] MCF-7 human breast cancer cells with low expression of αvβ3 integrins were used as a negative control. Raw264.7 murine macrophage and HEK293 human embryonic kidney cells were used as the control to assess the potential cytotoxicity of RGD-uIONP/SN38 to mononuclear phagocyte system and healthy organ cells in vivo. Briefly, 2000 cells/well were cultured in a 96-well plate for 24 hours to allow the cells to attach. Integrin-targeted RGD-uIONP/SN38, non-targeted RAD-uIONP/SN38, SN38 alone (20 mM dissolved in DMSO) and vehicle RGD-uIONP with equivalent Fe concentrations to RGD-uIONP/SN38 were incubated with the U87MG, MCF-7, Raw264.7 and HEK293 cells, respectively, at 37 °C for 72 hours with the SN38 concentrations of 0, 1, 5, 10, 50, 100, 500, 1000, 5000 and 10000 nM. The cells were then washed three times with cold PBS, before the determination of viability using AlamarBlue assay following the manufacturer’s instruction. Cell viabilities were normalized to that of cells treated with no SN38. Results were shown as the mean value of six replicates. The IC50 of RGD-uIONP/SN38 and SN38 alone were calculated by Origin 8 (OriginLab Corporation, Northampton, MA, USA) using the dose response fitting.
5.8. Preparation of Orthotopic Mouse Model of GBM
All animal experiments were carried out following a protocol approved by the Institutional Animal Care and Use Committee (IACUC) at Emory University (protocol # PROTO201700423). An intracranial U87 GBM mouse model was used to provide an orthotopic tumor microenvironment in the brain for evaluating RGD-uIONP/SN38. The mouse model was established on female nude mice (6-7 weeks old) by implanting U87MG GBM cells into the brains of mice following our established protocol.[46] Briefly, 1×105 U87MG cells were suspended in 3 μL of PBS and implanted to the bregma 2.5 mm to the right of sagittal suture and 3 mm below the skull via stereotactic injection. The tumors were allowed to grow for 10 days for use, before which the presence of the brain tumors was confirmed by the contrast enhanced T1-weighted MRI.
5.9. Delivery of RGD-uIONP/SN38 to Intracranial GBM in Mice
To investigate the targeted delivery of RGD-uIONP/SN38 to the U87MG GBM cells in the intracranial tumors, mice bearing intracranial U87 GBM were administered with a single dose of NIR830-labeled RGD-uIONP/SN38 through tail vein injection at the SN38 dosage of 5 mg/kg body weight. Mice were euthanized at 2 and 12 hours after injection (N = 3 for each time point). The brain, kidneys, spleen and liver were then collected for an ex vivo NIR imaging using an IVIS® imaging system (PerkinElmer, Waltham, MA, USA). Afterwards, the organs were embedded in OCT and frozen in liquid nitrogen. Frozen tissue sections with 6 μm thickness were prepared on a cryostat. Prussian blue staining for Fe and H&E staining were carried out for all collected organs. The brain tissue sections were also immunofluorescence stained for CD31, CD68 and αvβ3 integrins to examine the distribution of NIR830-RGD-uIONP/SN38 with respect to the blood vasculatures (CD31), microglia/macrophages (CD68) and U87MG GBM cells (αvβ3 integrin).
The quantitative analysis of RGD-uIONP/SN38 extravasating from tumor blood vessels and diffusing in the tumors was carried out by counting the pixels of NIR830-RGD-uIONP/SN38 and correlating with their distance to the nearest CD31-stained blood vessels. Four images from each mouse (12 for 2 h, 12 for 12 h) were randomly selected. Single channel images of NIR830-RGD-uIONP/SN38 and CD31 of the same tumor regions were segmented as described previously.[47] Each pixel in the segmented CD31 image was calculated for its Euclidean distance to the nearest CD31 pixel using ArcGIS pro (Esri, West Redlands, CA, USA). The resulted image was then overlaid with segmented image of NIR830-RGD-uIONP/SN38 to grant each pixel of NIR830-RGD-uIONP/SN38 the corresponding Euclidean distance. The NIR830 pixel counts with different distance to tumor blood vessels were then plotted after taking the average of 12 images for each time point.
5.10. Immunohistochemistry Analysis
The IHC staining was carried out using a DAB enhanced liquid substrate system following the manufacturer’s protocol. The immunofluorescence staining was performed following our protocols published previously.[47] Ki-67 and EGFR on collected brain tissue sections were immunofluorescence stained by rabbit Ki-67 and rabbit anti-EGFR antibodies, respectively, as the primary antibody followed with the application of goat anti-rabbit IgG 488 as the secondary antibody to validate the aggressiveness of the U87 GBM cells. To investigate the penetration of RGD-uIONP/SN38 through the BBB/BTB on the GBM mouse model, RGD-uIONP/SN38 was labeled with NIR830 tagging, and the tumor vasculature was visualized by the immunofluorescence staining of CD31 using rat monoclonal anti-CD31 (PECAM-1) antibody and goat anti-rat IgG 555. To examine whether RGD-uIONP/SN38 was capable of targeting αvβ3 integrin overexpressed by the U87MG GBM cells other than taking up by monocytes in the brain tissue, i.e., macroglia, the immunofluorescence staining of αvβ3 integrin and CD68 was conducted using rabbit anti-integrin αvβ3 antibody and rat polyclonal anti-CD68 antibody, respectively, with corresponding goat anti-rabbit IgG 488 or goat anti-rat IgG 555 antibodies as the secondary antibody. Fluorescence imaging was performed on a BZ-X710 fluorescent microscope (Keyence, Osaka, Japan) with a customized filter for NIR830. Prussian blue staining of the brain tissue sections with nuclear fast red counterstaining for cells was conducted for the brain tissue sections to visualize the distribution of uIONP following our established protocol.[20b] H&E staining was also performed using our protocol published previously.[45, 47]
5.11. Monitoring and Measurement of Tumor Growth by MRI
Contrast enhanced T1-weight MRI was performed to assess the tumor growth in the brain. MRI of mice was performed on a 3T scanner (Prisma, Siemens, Erlangen, Germany) using a 3T dedicated 1H transmit-receive quadrature volume coil. Mice were i.v. injected with Magnevist® A fat-suppressed contrast enhanced T1-weight and T2-weighted fast spin echo sequences were applied to the mouse MRI after i.v. injection of Magnevist® as the T1 contrast agent. The key parameters for MRI were as follow: TR = 3600 ms, TE = 86 ms, flip angle = 150, image matrix = 154 × 320, field of view (FOV) = 40 × 120 mm2, slice thickness = 1 mm with the number of averages = 3.
The segmentation of tumor areas in all related MRI slices was performed following our method published previously.[47] Briefly, the tumor region on each T1-weight MRI slice was extracted using FLICM algorithm integrated with local Markov random field (MRF) and Markov chain Monte Carlo (MCMC), the combination of which estimated a probability distribution over the MR images with possible labeling.
5.12. Efficacy and Survival Study of Different Treatments
To evaluate the efficacy of RGD-uIONP/SN38 in delivering therapeutics to the targeted U87MG GBM cells and thus inhibiting the growth of intracranial GBM in vivo, a mouse survival study after treating with RGD-uIONP/SN38 was performed, using non-targeted RAD-uIONP/SN38 and CrEL_SN38 as the treatment control. 30 female nude mice bearing intracranial U87MG GBM were randomly divided into five groups (N = 6 in each group) to receive the treatments of αvβ3 integrin targeted NIR830-RGD-uIONP/SN38 with high (5 mg SN38/kg body weight, HD_NP group) and low (2.5 mg SN38/kg body weight, LD_NP group) dosages, NIR830-RAD-uIONP/SN38 (RAD_NP group) and CrEL_SN38 at the dosage of 5 mg SN38/kg body weight, and PBS as the non-treatment control (NT). CrEL_SN38 was prepared following a published method by diluting the SN38 solution in ethanol/Cremophor EL using DI water to reach the SN38 final concentration of 0.5 mg/mL.[48] Treatment of mice was started from the day 8 after implantation and given twice/week. Mice from the treatment groups of HD_NP and CrEL_SN38 were selected to monitor the GBM growth by contrast enhanced T1-weight MRI every four days after treatment began. Mice were monitored daily for the body weights and euthanized once the ending points were reached following the NIH guidelines. The mouse brain, kidney, liver and spleen were collected at euthanasia and embedded in OCT to prepare tissue sections for further staining and validation. The was determined by equation (2):
| Equation (2) |
where Survival_1 and Survival_2 are the averaged survivals of mice in the two compared treatment groups with longer and shorter survival, respectively.
5.13. Biodistribution and Blood Half-Life of RGD-uIONP/SN38
To examine the pharmacokinetic profile of the RGD-uIONP/SN38 for better understanding its delivery efficiency to the intracranial GBM, a biodistribution study was performed using the U87 GBM mouse model to investigate the accumulation and clearance of RGD-uIONP/SN38 in major organs and its half-life in blood circulation, following the method we described previously.[49] Briefly, 27 female nude mice bearing intracranial U87 GBM were i.v. injected with RGD-uIONP/SN38 at the dosage of 10 mg Fe/kg body weight via tail veins. The mice were euthanized at 5 and 15 min, then 1, 2, 4, 8, 24, 48 and 72 hours after injection (N = 3 for each time point) to collect the blood via a cardiac puncture and major organs including the brain, liver, spleen, lung and kidney. Mice (N = 3) received PBS were used as the baseline. Blood samples were stored in K3 EDTA blood tubes and centrifuged at 1500 rpm for 5 min to spin-down the cells. 200 to 400 μL plasma was then collected to measure the Fe concentration using the 1,10-phenanthroline colorimetric assay as described above. The decade of Fe concentrations in mouse blood at different time points was plotted to determine the half-life of RGD-uIONP/SN38 using a pseudo-first-order kinetics. The collected organs were weighed and lyophilized before the quantification of Fe content using 1,10-phenanthroline colorimetric assay. The Fe content in organs was presented as mg Fe/g weight of organs, after subtracting the baseline Fe content obtained from the mice receiving PBS.
5.14. Statistical Analysis
Data was presented in the format of mean ± standard deviation. A two-tailed unpaired t test was used to determine the statistically significant differences when comparing the hydrodynamic diameters and zeta potentials of uIONP with and without SN38 loading (N = 3), the IC50 of RGD-uIONP/SN38 and free SN38 for U87MG cancer cells (N = 3), the inhibited cell growth of U87MG, MCF-7 and Raw264.7 cells with and without treatment of RGD-uIONP/SN38 (N = 3), and survival of animals treated with different agents or formulations (N = 6). A log-rank test was used to determine the statistical significance in the comparison of treatment efficacy in different groups of mice (N = 6) using GraphPad Prism software (GraphPad, San Diego, CA, USA). The level of significance was set at p < 0.05.
Supplementary Material
Acknowledgements
This study is supported by NIH grants 1U01CA198913, R01CA202846, R01CA154846 to H.M. and 1R41CA261544 to H.M. and L.Y. The authors thank Mr. Jie Zhang (M.S.) for the quantitative analysis of RGD-uIONP/SN38 distribution with respect to tumor blood vessels.
Footnotes
Conflict of interest
The authors declare no conflict of interest.
Supporting Information
Supporting Information is available from the Wiley Online Library or from the author.
Contributor Information
Yuancheng Li, Department of Radiology and Imaging Sciences, Emory University, Atlanta, Georgia 30329, United States of America; 5M Biomed, LLC, Atlanta, Georgia 30303, United States of America.
Manman Xie, Department of Radiology and Imaging Sciences, Emory University, Atlanta, Georgia 30329, United States of America.
Joshua B. Jones, Department of Radiology and Imaging Sciences, Emory University, Atlanta, Georgia 30329, United States of America
Zhaobin Zhang, Department of Neurosurgery, Emory University, Atlanta, Georgia 30329, United States of America.
Zi Wang, Department of Radiology and Imaging Sciences, Emory University, Atlanta, Georgia 30329, United States of America.
Tu Dang, Division of Research, Philadelphia College of Osteopathic Medicine – Georgia Campus, Suwanee, Georgia 30024, United States of America.
Xinyu Wang, Department of Pharmaceutical Sciences, Philadelphia College of Osteopathic Medicine – Georgia Campus, Suwanee, Georgia 30024, United States of America.
Malgorzata Lipowska, Department of Radiology and Imaging Sciences, Emory University, Atlanta, Georgia 30329, United States of America.
Hui Mao, Department of Radiology and Imaging Sciences, Emory University, Atlanta, Georgia 30329, United States of America.
Data Availability
The data that support the findings of this study are available from the corresponding author upon reasonable request.
References
- [1].a) Tan AC, Ashley DM, Lopez GY, Malinzak M, Friedman HS, Khasraw M, CA: A Cancer Journal for Clinicians 2020, 70, 299; [DOI] [PubMed] [Google Scholar]; b) Ostrom QT, Gittleman H, Truitt G, Boscia A, Kruchko C, Barnholtz-Sloan JS, Neuro-Oncology 2018, 20, iv1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [2].a) Daneman R, Prat A, Cold Spring Harbor Perspectives in Biology 2015, 7, a020412; [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Weller M, Wick W, Aldape K, Brada M, Berger M, Pfister SM, Nishikawa R, Rosenthal M, Wen PY, Stupp R, Reifenberger G, Nature Reviews Disease Primers 2015, 1, 15017. [DOI] [PubMed] [Google Scholar]
- [3].Arvanitis CD, Ferraro GB, Jain RK, Nature Reviews Cancer 2020, 20, 26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [4].a) Hobbs SK, Monsky WL, Yuan F, Roberts WG, Griffith L, Torchilin VP, Jain RK, Proceedings of the National Academy of Sciences of the United States of America 1998, 95, 4607; [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Monsky WL, Carreira CM, Tsuzuki Y, Gohongi T, Fukumura D, Jain RK, Clinical Cancer Research 2002, 8, 1008; [PubMed] [Google Scholar]; c) Pitz MW, Desai A, Grossman SA, Blakeley JO, Journal of Neuo-oncology 2011, 104, 629. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [5].Banks WA, Nature Reviews Drug Discovery 2016, 15, 275. [DOI] [PubMed] [Google Scholar]
- [6].a) Mitchell MJ, Billingsley MM, Haley RM, Wechsler ME, Peppas NA, Langer R, Nature Reviews Drug Discovery 2021, 20, 101; [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Huang J, Li Y, Orza A, Lu Q, Guo P, Wang L, Yang L, Mao H, Advanced Functional Materials 2016, 26, 3818; [DOI] [PMC free article] [PubMed] [Google Scholar]; c) Zheng M, Tao W, Zou Y, Farokhzad OC, Shi B, Trends in Biotechnology 2018, 36, 562; [DOI] [PubMed] [Google Scholar]; d) Jiang T, Qiao Y, Ruan M, Zhang D, Yang Q, Wang G, Chen Q, Zhu F, Yin J, Zou Y, Qian R, Zheng M, Shi B, Advanced Materials 2021, 33, 2104779; [DOI] [PubMed] [Google Scholar]; e) Liu Y, Zou Y, Feng C, Lee A, Yin J, Chung R, Park JB, Rizos H, Tao W, Zheng M, Farokhzad OC, Shi B, Nano Letters 2020, 20, 1637. [DOI] [PubMed] [Google Scholar]
- [7].Silva GA, Nature Reviews Neuroscience 2006, 7, 65. [DOI] [PubMed] [Google Scholar]
- [8].a) Yeini E, Ofek P, Albeck N, Ajamil DR, Neufeld L, Eldar-Boock A, Kleiner R, Vaskovich D, Koshrovski-Michael S, Dangoor SI, Krivitsky A, Luna CB, Shenbach-Koltin G, Goldenfeld M, Hadad O, Tiram G, Satchi-Fainaro R, Advanced Therapeutics 2021, 4, 2000124; [Google Scholar]; b) Sarkaria JN, Hu LS, Parney IF, Pafundi DH, Brinkmann DH, Laack NN, Giannini C, Burns TC, Kizilbash SH, Laramy JK, Swanson KR, Kaufmann TJ, Brown PD, Agar NYR, Galanis E, Buckner JC, Elmquist WF, Neuro-Oncology 2018, 20, 184. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [9].a) Dadfar SM, Roemhild K, Drude NI, von Stillfried S, Knuchel R, Kiessling F, Lammers T, Advanced Drug Delivery Reviews 2019, 138, 302; [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Jeon M, Halbert MV, Stephen ZR, Zhang M, Advanced Materials 2021, 33, 1906539; [DOI] [PMC free article] [PubMed] [Google Scholar]; c) Shen Z, Wu A, Chen X, Molecular Pharmaceutics 2017, 14, 1352. [DOI] [PubMed] [Google Scholar]
- [10].a) Bullivant JP, Zhao S, Willenberg BJ, Kozissnik B, Batich CD, Dobson J, International Journal of Molecular Sciences 2013, 14, 17501; [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Jahn MR, Andreasen HB, Futterer S, Nawroth T, Schunemann V, Kolb U, Hofmeister W, Munoz M, Bock K, Meldal M, Langguth P, European Journal of Pharmaceutics and Biopharmaceutics 2011, 78, 480; [DOI] [PubMed] [Google Scholar]; c) Graczyk H, Bryan LC, Lewinski N, Suarez G, Coullerez G, Bowen P, Riediker M, Journal of Aerosol Medicine and Pulmonary Drug Delivery 2015, 28, 43; [DOI] [PMC free article] [PubMed] [Google Scholar]; d) Liu Y, Naha PC, Hwang G, Kim D, Huang Y, Simon-Soro A, Jung H-I, Ren Z, Li Y, Gubara S, Alawi F, Zero D, Hara AT, Cormode DP, Koo H, Nature Communications 2018, 9, 2920. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [11].a) Barajas RF Jr., Schwartz D, McConnell HL, Kersch CN, Li X, Hamilton BE, Starkey J, Pettersson DR, Nickerson JP, Pollock JM, Fu RF, Horvath A, Szidonya L, Varallyay CG, Jaboin JJ, Raslan AM, Dogan A, Cetas JS, Ciporen J, Han SJ, Ambady P, Muldoon LL, Woltjer R, Rooney WD, Neuwelt EA, American Journal of Neuroradiology 2020, 41, 1193; [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Iv M, Samghabadi P, holdsworth S, Gentles A, Rezaii P, Harsh G, Li G, Thomas R, Moseley M, Daldrup-Link HE, Vogel H, Wintermark M, Cheshier S, Yeom KW, Radiology 2019, 290, 198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [12].Pulgar V, Frontiers in Neuroscience 2019, 12, 1019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [13].a) Jahangiri A, Chin AT, Flanigan PM, Chen R, Bankiewicz K, Aghi MK, Journal of Neurosurgery 2017, 126, 191; [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Hadjipanayis CG, Machaidze R, Kaluzova M, Wang L, Schuette AJ, Chen H, Wu X, Mao H, Cancer Research 2010, 70, 6303; [DOI] [PMC free article] [PubMed] [Google Scholar]; c) Platt S, Nduom E, Kent M, Freeman C, Machaidze R, Kaluzova M, Wang L, Mao H, Hadjipanayis CG, Clinical Neurosurgery 2012, 59, 107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [14].a) Bazak R, Houri M, Achy S. e., Hussein W, Refaat T, Molecular and Clinical Oncology 2014, 2, 904; [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Rosenblum D, Joshi N, Tao W, Karp JM, Dan P, Nature Communications 2018, 9, 1410. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [15].Wang L, Huang J, Chen H, Wu H, Xu Y, Li Y, Yi H, Wang YA, Yang L, Mao H, ACS Nano 2017, 11, 4582. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [16].Xu Y, Wu H, Huang J, Qian W, Martinson DE, Ji B, Li Y, Wang YA, Yang L, Mao H, Theranostics 2020, 10, 2479. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [17].a) Sarkaria JN, Kitange GJ, James CD, Plummer R, Calvert H, Weller M, Wick W, Clinical Cancer Research 2008, 14, 2900; [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Oliva CR, Nozell SE, Diers A, McClugage SG 3rd, Sarkaria JN, Markert JM, Darley-Usmar VM, Bailey SM, Gillespie GY, Landar A, Griguer CE, The Journal of Biological Chemistry 2010, 285, 39759. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [18].a) Rasheed ZA, Rubin EH, Oncogene 2003, 22, 7296; [DOI] [PubMed] [Google Scholar]; b) Xu Y, Villalona-Calero MA, Annals of Oncology 2002, 13, 1841. [DOI] [PubMed] [Google Scholar]
- [19].a) Vredenburgh JJ, Desjardins A, Reardon DA, Friedman HS, Neuro-Oncology 2009, 11, 80; [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Mesti T, Moltara ME, Boc M, Rebersek M, Ocvirk J, Radiology and Oncology 2015, 49, 80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [20].a) Huang J, Wang L, Zhong X, Li Y, Yang L, Mao H, Journal of Materials Chemsitry B 2014, 2, 5344; [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Li Y, Lin R, Wang L, Huang J, Wu H, Cheng G, Zhou Z, MacDonald T, Yang L, Mao H, Journal of Materials Chemsitry B 2015, 3, 3591; [DOI] [PMC free article] [PubMed] [Google Scholar]; c) Lin R, Li Y, MacDonald T, Wu H, Provenzale J, Peng X, Huang J, Wang L, Wang AY, Yang J, Mao H, Colloids and Surfaces B: Biointerfaces 2017, 150, 261. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [21].Chieng BW, Ibrahim NA, Yunus WMZW, Hussein MZ, Polymers 2014, 6, 93. [Google Scholar]
- [22].Zhang T, Zhao N, Li J, Guo H, An T, Zhao F, Ma H, RSC Advances 2017, 7, 23583. [Google Scholar]
- [23].Liu Z, Robinson JT, Sun X, Dai H, Journal of the American Chemical Society 2008, 130, 10876. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [24].Xie M, Xu Y, Huang J, Li Y, Wang L, Yang L, Mao H, WIREs Nanomedicine and Nanobiotechnology 2020, 12, e1644. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [25].a) Fu Y, Kao WJ, Expert Opinion on Drug Delivery 2010, 7, 429; [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Fan H, Jin Z, Macromolecules 2014, 47, 2674. [Google Scholar]
- [26].a) Ye Y, Chen X, Theranostics 2011, 1, 102; [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Taherian A, Li X, Liu Y, Haas TA, BMC Cancer 2011, 11, 293; [DOI] [PMC free article] [PubMed] [Google Scholar]; c) Brilha S, Wysoczanski R, Whittington AM, Friedland JS, Porter JC, Journal of Immunology 2017, 199, 982. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [27].a) Ponka P, Kidney International 1999, 55, S2; [DOI] [PubMed] [Google Scholar]; b) Wang J, Pantopoulos K, Biochemical Journal 2011, 434, 365; [DOI] [PMC free article] [PubMed] [Google Scholar]; c) Muckenthaler MU, Rivella S, Hentze MW, Galy B, Cell 2017, 168, 344. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [28].a) Bogdan AR, Miyazawa M, Hashimoto K, Tsuji Y, Trends in Biochemical Sciences 2016, 41, 274; [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Li J, Cao F, Yin H.-l., Huang Z.-j., Li Z.-t., Mao N, Sun B, Wang G, Cell Death & Disease 2020, 11, 88; [DOI] [PMC free article] [PubMed] [Google Scholar]; c) Jiang X, Stockwell BR, Conrad M, Nature Reviews Molecular Cell Biology 2021, 22, 266. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [29].a) Chen S, Yueh M-F, Bigo C, Barbier O, Wang K, Karin M, Nguyen N, Tukey RH, Proceedings of the National Academy of Sciences of the United States of America 2013, 110, 19143; [DOI] [PMC free article] [PubMed] [Google Scholar]; b) de Man FM, Goey AKL, van Schaik RHN, Mathijssen RHJ, Bins S, Clinical Pharmacokinetics 2018, 57, 1229. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [30].Iyer L, Das S, Janisch L, Wen M, Ramirez J, Karrison T, Flemming GF, Vokes EE, Schilsky RL, Ratain MJ, The Pharmacogenomics Journal 2002, 2, 43. [DOI] [PubMed] [Google Scholar]
- [31].a) Wu X, Yang H, Yang W, Chen X, Gao J, Gong X, Wang H, Duan Y, Wei D, Chang J, Journal of Materials Chemsitry B 2019, 7, 4734; [DOI] [PubMed] [Google Scholar]; b) Zhou C, Long M, Qin Y, Sun X, Zheng J, Angewandte Chemie International Edition 2011, 50, 3168. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [32].a) Hamilton BE, Nesbit GM, Dosa E, Gahramanov S, Rooney B, Nesbit EG, Raines J, Neuwelt EA, American Journal of Roentgenology 2011, 197, 981; [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Daldrup-Link HE, Golovko D, Ruffell B, Denardo DG, Castaneda R, Ansari C, Rao J, Tikhomirov GA, Wendland MF, Corot C, Coussens LM, Clinical Cancer Research 2011, 17, 5695; [DOI] [PMC free article] [PubMed] [Google Scholar]; c) Smith BR, Gambhir SS, Chemical Reviews 2017, 117, 901; [DOI] [PubMed] [Google Scholar]; d) Chen G, Roy I, Yang C, Prasad PN, Chemical Reviews 2016, 116, 2826; [DOI] [PubMed] [Google Scholar]; e) Shi J, Kantoff PW, Wooster R, Farokhzad OC, Nature Reviews Cancer 2017, 17, 20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [33].a) Wang Y, Yu L, Ding J, Chen Y, International Journal of Molecular Sciences 2019, 20, 95; [Google Scholar]; b) Chen Y, Fan Z, Yang Y, Gu C, International Journal of Oncology 2019, 54, 1143. [DOI] [PubMed] [Google Scholar]
- [34].Kuroda J.-i., Kuratsu J.-i., Yasunaga M, Koga Y, Kenmotsu H, Sugino T, Matsumura Y, Clinical Cancer Research 2010, 16, 521. [DOI] [PubMed] [Google Scholar]
- [35].a) Ohta S, Kikuchi E, Ishijima A, Azuma T, Sakuma I, Ito T, Scientific Reports 2020, 10, 18220; [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Arami H, Patel CB, Madsen SJ, Dickinson PJ, Davis RM, Zeng Y, Sturges BK, Woolard KD, Habte FG, Akin D, Sinclair R, Gambhir SS, ACS Nano 2019, 13, 2858. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [36].a) Weis SM, Cheresh DA, Cold Spring Harbor Perspectives in Medicine 2011, 1, a006478; [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Seguin J, Nicolazzi C, Mignet N, Scherman D, Chabot G, Tumour Biology 2012, 33, 1709. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [37].a) Kim TW, Innocenti F, Therapeutic Drug Monitoring 2007, 29, 265; [DOI] [PubMed] [Google Scholar]; b) Fujiwara R, Nguyen N, Chen S, Tukey RH, Proceedings of the National Academy of Sciences of the United States of America 2010, 107, 5024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [38].Zanganeh S, Hutter G, Spitler R, Lenkov O, Mahmoudi M, Shaw A, Pajarinen JS, Nejadnik H, Goodman S, Moseley M, Coussens LM, Daldrup-Link HE, Nature Nanotechnology 2016, 11, 986. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [39].Wilhelm S, Tavares AJ, Dai Q, Ohta S, Audet J, Dvorak HF, Chan WCW, Nature Reviews Materials 2016, 1, 16014. [Google Scholar]
- [40].Soetaert F, Korangath P, Serantes D, Fiering S, Ivkov R, Advanced Drug Delivery Reviews 2020, 163–164, 65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [41].a) Strekowski L, Mason CJ, Lee H, Gupta R, Sowell J, Patonay G, Journal of Heterocyclic Chemistry 2009, 40, 913; [Google Scholar]; b) Zhou Z, Chen H, Lipowska M, Wang L, Yu Q, Yang X, Tiwari D, Yang L, Mao H, Journal of Biomaterials Applications 2013, 28, 100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [42].a) Rennick JJ, Johnston APR, Parton RG, Nature Nanotechnology 2021, 16, 266; [DOI] [PubMed] [Google Scholar]; b) Cooper GM, The Cell, 2nd edition, Sinauer Associates, Sunderland (MA) 2000. [Google Scholar]
- [43].Franovic A, Elliott KC, Seguin L, Camargo MF, Weis SM, Cheresh DA, Cancer Research 2015, 75, 4466. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [44].a) Gagnon J-F, Bernard O, Villeneuve L, Tetu B, Guillemette C, Clinical Cancer Research 2006, 12, 1850; [DOI] [PubMed] [Google Scholar]; b) Chen X, Peer CJ, Alfaro R, Tian T, Spencer SD, Figg WD, Journal of Pharmaceutical and Biomedical Analysis 2012, 62, 140; [DOI] [PMC free article] [PubMed] [Google Scholar]; c) Poujol S, Pinguet F, Malosse F, Astre C, Ychou M, Culine S, Bressolle F, Clinical Chemistry 2003, 49, 1900; [DOI] [PubMed] [Google Scholar]; d) Nittayachrn P, Manaspon C, Hongeng S, Nasongkla N, Experimental Biology and Medicine 2014, 239, 1619. [DOI] [PubMed] [Google Scholar]
- [45].Zhou H, Qian W, Uckun FM, Wang L, Wang AY, Chen H, Kooby D, Yu Q, Lipowska M, Staley C, Mao H, Yang L, ACS Nano 2015, 9, 7976. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [46].Qi Q, He K, Yoo M-H, Chan C-B, Liu X, Zhang Z, Olson JJ, Xiao G, Wang L, Mao H, Fu H, Tao H, Ramalingam SS, Sun S-Y, Mischel PS, Ye K, Journal of Biological Chemistry 2012, 287, 6113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [47].Norouzi M, Yathindranath V, Thliveris JA, Kopec BM, Siahaan TJ, Miller DW, Scientific Reports 2020, 10, 11292. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [48].a) Zeng J, Li C, Duan X, Liu F, Li A, Luo C, Jia L, Gan Y, Yan L, Zheng Y, Drug Delivery 2019, 26, 354; [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Lin H-C, Chuang C-H, Cheng M-H, Lin Y-C, Fang Y-P, Pharmaceutics 2019, 11, 569. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [49].Chen H, Yeh J, Wang L, Wu X, Cao Z, Wang YA, ZHang M, Yang L, Mao H, Biomaterials 2010, 31, 5397. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The data that support the findings of this study are available from the corresponding author upon reasonable request.