

Hypoplastic left heart syndrome (HLHS) is a severe form of single ventricle congenital heart disease (CHD) characterized by the underdevelopment of the left ventricle, mitral valve, aortic valve, and ascending aorta. Coronary arterial abnormalities such as thickened wall, kinking arteries, and coronary arterial fistulous communications have been revealed by postmortem examinations1, which may impact ventricular development and intra-cardiac hemodynamics, leading to a poor prognosis after surgical palliation1. However, the intrinsic defect in coronary vessels and its genetic basis remain unclear. Through single-cell RNA sequencing (scRNA-seq) analysis of human fetal heart with an underdeveloped left ventricle (ULV) and ECs differentiated from induced pluripotent stem cells (iPSCs) with HLHS, we uncovered an abnormal population of coronary arterial ECs with loss of arterial features and decreased proliferation, which were attributed to HLHS de novo mutation (DNM) KMT2D mediated NOTCH signaling defect.
To reveal the transcriptomic changes in HLHS coronary vessels, heart ECs (CDH5+) were sorted from dissociated human fetal heart (healthy control vs. ULV) (A) and iPSC-derived ECs (iPSC-ECs, healthy control vs. HLHS)2 and subjected to scRNA-seq. We first excluded the endocardial population (NPR3+) by selecting CDH5+/NPR3− vascular ECs2 for downstream analysis. Out of the six sub-clusters in fetal heart vascular ECs, we focused on four clusters (Cluster 0-3) containing predominate vascular ECs and excluded those in transitional states. EC subtypes were annotated by multiple cell-type-specific markers, such as vein (NR2F2+, Cluster 0&2), artery (MECOM+, Cluster 1&3), and late artery (GJA5+, Cluster 1)3 (A, left panel). Gene ontology (GO) analysis was performed based on differentially expressed genes (DEGs) between control and ULV vascular ECs (A, right panel). Compared with control, ULV showed defects in general EC functions such as cell junction organization and EC migration. Notably, C1 late arterial cluster exhibited cell-type specific defects in endothelium development, EC proliferation, artery morphogenesis, and Notch signaling, which were not observed in other EC subclusters. These results intrigued us to further focus on understanding the coronary arterial ECs (AECs) abnormalities in HLHS.
Figure.
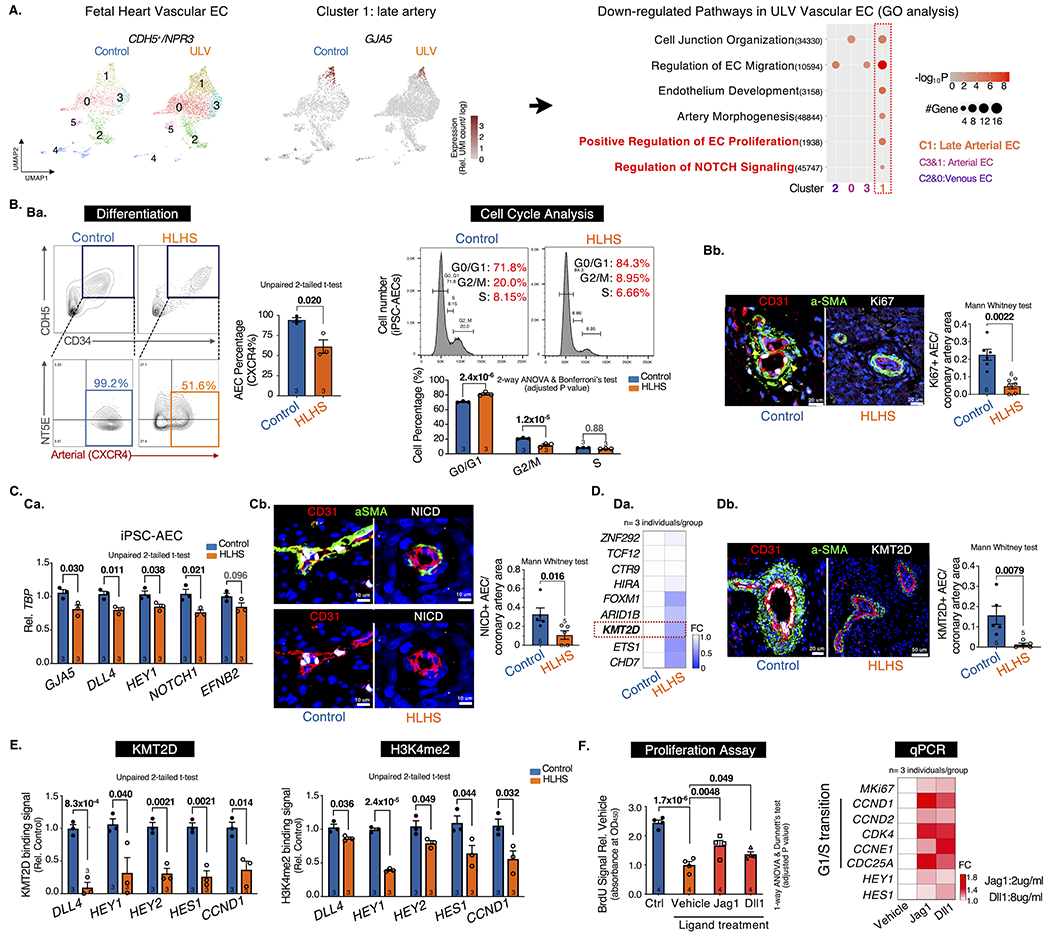
A. scRNA-seq analysis of vascular ECs (CDH5+/NPR3−). UMAP projection of Control vs. ULV human fetal left heart vascular ECs were shown on the left. Cluster1: GJA5+ late artery. Cluster4: venous-to-endocardial transitional population. N=1 per group. Then Gene Ontology analysis were performed on each cluster and showed downregulated pathways in ULV vascular ECs vs. control. GO terms: adjusted p-value < 0.05. UMI: Unique Molecular Identifier.
B. Artery differentiation and proliferation were impaired in HLHS. Ba, flow cytometry analysis of iPSC-AEC progenitors percentage (left) and their cell-cycle statuses (right). Bb, Immunostaining and quantification of Ki67+ arteries in human heart tissue. aSMA: smooth muscle actin.
C. NOTCH pathway was suppressed in HLHS AECs. Ca, qPCR of NOTCH genes in iPSC-AECs. Cb, Immunostaining and quantification of NICD+ arteries in human heart tissue.
D. KMT2D expression was decreased in HLHS AECs. Da, qPCR of HLHS DNM gene expression in iPSC-ECs. Db, Immunostaining and quantification of KMT2D+ arteries in human heart tissue.
E. KMT2D binding capacity and H3K4me2 signal were reduced in HLHS iPSC-AECs. (Evaluated via ChIP-qPCR)
F. NOTCH ligands treatment improved the proliferation of HLHS iPSC-AECs. Both BrdU proliferation assay and qPCR showed improved proliferation in HLHS iPSC-AECs. N=4 technical repeats. FC: fold change normalized to control.
Statistics (GraphPad Prism 9.3.1): Based on additional literature support from similar studies, our samples fit normal distribution. Parametric test: unpaired 2-tailed t-test (2 groups), ANOVA (>2 groups) with post hoc tests as indicated; non-parametric test: Mann-Whitney (2 groups). Mean±SEM; n= biological replicates as indicated.
Next, we generated pure AECs from three HLHS and age-matched control iPSC lines2 using a published protocol4. HLHS iPSCs exhibited impaired AEC differentiation as evidenced by the reduced CXCR4+NT5E−/low AEC progenitors compared to the control (Ba, left panel). Mature AECs were further enriched by CXCR4+ cell sorting. HLHS AECs demonstrated impaired proliferation with increased G0/G1 and decreased S/G2/M cell percentage (Ba, right panel), accompanied by the downregulation of Ki67 and G1/S transition genes (CCND1/2), and the upregulation of G1/S transition inhibitor CDKN2A/P16 (data not shown). This was further validated in HLHS human fetal heart tissue showing reduced proliferative AECs labeled by Ki67 (Bb). Consistent with GO analysis showing NOTCH defect in ULV AECs (A), HLHS iPSC-AECs exhibited decreased expression of NOTCH targeted genes (e.g., DLL4, HEY1, NOTCH1) (Ca). Reduced NOTCH intracellular domain (NICD) was also observed in HLHS fetal heart tissue (Cb).
Previously, we revealed that the majority of the HLHS DNMs encoded genes were highly enriched in endocardial and endothelial populations in human fetal heart2. To further understand the genetic underpinnings in the HLHS cases we studied, we first examined the expression levels of several key chromatin remodelers harboring HLHS DNMs. We identified five genes (e.g., FOXM1, KMT2D) that significantly reduced in HLHS iPSC-ECs (Da). Among them, KMT2D, a lysine methyltransferase, favors NICD-RBPJ complex mediated gene activation by maintaining a permissive chromatin status via catalyzing H3K4me1, me2 and me35. KMT2D protein level was also reduced in HLHS coronary arteries (Db). Additionally, ChIP-qPCR revealed reduced KMT2D binding capacity and H3K4me2 signals to the promoter loci of several NOTCH targeted genes in mature HLHS AECs (E), which are critical in maintaining arterial characteristics (DLL4, HEY1/2, HES1) and cell proliferation (CCND1). Intriguingly, the treatment of NOTCH ligand Jag1 and Dll1 improved cell proliferation of HLHS AECs and upregulated G1/S transition genes downstream of the NOTCH pathway (F).
In summary, our study revealed that KMT2D-mediated NOTCH defect contributed to the coronary AECs abnormalities in HLHS. The NOTCH-related defects were more pronounced in coronary AECs compared with other cardiac cell types. Disruption of NOTCH signaling led to impaired proliferation and maintenance of arterial features in HLHS AECs, which may partially explain the decreased vascular density and coronary artery malformation in HLHS fetal heart1. Notably, NOTCH ligands rescued HLHS associated gene expression abnormalities and cellular phenotypes, providing a potential therapeutic target.
Acknowledgments
We thank Drs. Kyle Loh, Nanhua Zhang for providing intellectual consultant to the project. We also thank ReGen Theranostics, Inc Rochester, MN as the manufacturer for the iPSC lines.
Sources of Funding
This work was supported by single ventricle gift fund from Stanford University, Todd and Karen Wanek Family Program for Hypoplastic Left Heart Syndrome from Mayo Clinic, NIH 2R24HD000836-52 from the Eunice Kennedy Shriver National Institute of Child Health and Human Development (The University of Washington Birth Defects Research Laboratory). Z.Y. received support from American Heart Association (AHA) predoctoral fellowship (Jan/2022-Dec/2023). X.Z. received support from the Stanford Aging and Ethnogeriatrics (SAGE) Research Center under NIH/NIA grant P30AG059307. S.M. is funded by the Heart and Stroke Foundation of Canada Chair, Canadian Institutes of Health Research (CIHR) and ERA PerMed, and the Ted Rogers Centre for Heart Research. J.C.W is funded by R01 HL141371 & R01 HL126527.
Footnotes
Consent for iPSC generation was obtained from both control and patients under approved IRBs: Mayo Clinic: 10-006845; Stanford: IRB 5443. Tissue collection and use in the research were approved by the University of Washington: IRB STUDY00000380. Human tissue sections were obtained under approved IRBs: Hospital for Sick Children IRB 1000011284; Mount Sinai Hospital REB# 08-0009-E.
Disclosures
SM serves on the Advisory Boards of Bristol Myers Squibb, and Tenaya Therapeutics.
Data Availability
Raw data and complete methods can be made available upon request from the corresponding author. Single-cell RNA sequencing Seurat object have been deposited in the GEO database under accession number GSE138979. Data not shown in the main figure can be accessed via: https://www.biorxiv.org/content/10.1101/2021.08.30.457716v3
References
- 1.Cole CR and Eghtesady P. The myocardial and coronary histopathology and pathogenesis of hypoplastic left heart syndrome. Cardiology in the Young. 2016;26:19–29. [DOI] [PubMed] [Google Scholar]
- 2.Miao Y, Tian L, Martin M, Paige SL, Galdos FX, Li J, Klein A, Zhang H, Ma N, Wei Y, et al. Intrinsic Endocardial Defects Contribute to Hypoplastic Left Heart Syndrome. Cell Stem Cell. 2020;27:574–589.e8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Su T, Stanley G, Sinha R, D’Amato G, Das S, Rhee S, Chang AH, Poduri A, Raftrey B, Dinh TT, et al. Single-cell analysis of early progenitor cells that build coronary arteries. Nature. 2018;559:356–362. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Gage BK, Liu JC, Innes BT, Macparland SA, McGilvray ID, Bader GD and Keller GM. Generation of Functional Liver Sinusoidal Endothelial Cells from Human Pluripotent Stem-Cell-Derived Venous Angioblasts. Cell Stem Cell. 2020;27:254–269.e9. [DOI] [PubMed] [Google Scholar]
- 5.Oswald F, Rodriguez P, Giaimo BD, Antonello ZA, Mira L, Mittler G, Thiel VN, Collins KJ, Tabaja N, Cizelsky W, et al. A phospho-dependent mechanism involving NCoR and KMT2D controls a permissive chromatin state at Notch target genes. Nucleic Acids Research. 2016;44:4703–4720. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
Raw data and complete methods can be made available upon request from the corresponding author. Single-cell RNA sequencing Seurat object have been deposited in the GEO database under accession number GSE138979. Data not shown in the main figure can be accessed via: https://www.biorxiv.org/content/10.1101/2021.08.30.457716v3