

Abstract
Introduction/Aims:
With current and anticipated disease-modifying treatments, including gene therapy, an early diagnosis for Duchenne muscular dystrophy (DMD) is crucial to assure maximum benefit. In 2009, a study from the Muscular Dystrophy Surveillance, Tracking, and Research Network (MD STARnet) showed an average diagnosis age of 5 years among males with DMD born from January 1, 1982 to December 31, 2000. Initiatives were implemented by the US Centers for Disease Control and Prevention (CDC) and patient organizations to reduce time to diagnosis. We conducted a follow-up study in a surveillance cohort born after January 1, 2000 to determine whether there has been an improvement in time to diagnosis.
Methods:
We assessed the age of diagnosis among males with DMD born from January 1, 2000 to December 31, 2015 using data collected by six US MD STARnet surveillance sites (Colorado, Iowa, western New York State, the Piedmont region of North Carolina, South Carolina, and Utah). The analytic cohort included 221 males with definite or probable DMD diagnosis without a documented family history. We computed frequency count and percentage for categorical variables, and mean, median, and standard deviation (SD) for continuous variables.
Results:
The mean [median] ages in years of diagnostic milestones were: first signs, 2.7 [2.0]; first creatine kinase (CK), 4.6 [4.6]; DNA/muscle biopsy testing, 4.9 [4.8]; and time from first signs to diagnostic confirmation, 2.2 [1.4].
Discussion:
The time interval between first signs of DMD and diagnosis remains unchanged at 2.2 years. This results in lost opportunities for timely genetic counseling, implementation of standards of care, initiation of glucocorticoids, and participation in clinical trials.
Keywords: delay, diagnostic criteria, Duchenne muscular dystrophy, MD STARnet, muscle dystrophy, surveillance
INTRODUCTION
Studies of individuals with Duchenne muscular dystrophy (DMD) have shown an average age at diagnosis of approximately 5 years, which has not changed over time.1–4 Creatine kinase (CK) level is almost always elevated in patients with DMD and is therefore a useful, although underutilized, screening test in patients suspected of having DMD.
Achieving an early diagnosis is important. Implementation of established care guidelines for patients with DMD that focus on proactive monitoring of disease progression and the use of corticosteroids have proven critical in guiding clinicians to establish a standard of treatment.5–7 New disease-modifying treatments, such as exon-skipping drugs, have recently been approved, and promising gene replacement therapies are under development.8–12 As a result, there is continued interest from clinical and patient communities to diagnose DMD as early in life as possible.
Using data from four sites of the Muscular Dystrophy Surveillance, Tracking, and Research Network (MD STARnet), we found an interval of 2.5 years between recognition of the first signs and symptoms of DMD and a confirmatory diagnosis (mean age, 4.9 years) among individuals born from January 1, 1982 through December 12, 2000.1 This finding represented a lack of change from the previous two decades.2,3 We conducted a follow-up study using a surveillance cohort born after January 1, 2000 to determine whether there has been an improvement in time to diagnosis.
METHODS
Individuals with DMD were identified through the MD STARnet; methods for surveillance activities have been described previously.13 Briefly, MD STARnet is a population-based surveillance system for childhood-onset dystrophinopathy among individuals born between January 1, 2000 and December 31, 2015 and residing in one of six sites (Colorado [CO], Iowa [IA], 21 counties in western New York State [wNY], 33 counties in the Piedmont region of North Carolina [NC], South Carolina [SC], and Utah/Nevada [UT]).
Trained personnel identified cases and obtained data from medical records (ie, neuromuscular clinics, physical medicine and rehabilitation clinics, hospitals, outpatient clinics) and administrative sources (ie, birth defects registries, birth and death certificates, state hospital discharge databases). A clinical review committee reviewed and classified each of the cases using published case classification definitions (definite, probable, possible, asymptomatic, female).14 All sites obtained institutional review board approval or exemption. Most sites (CO, IA, NC, wNY, SC) also had public health authority to conduct surveillance for muscular dystrophy (MD).
Figure S1 shows the analytic cohort with study inclusion and exclusion criteria. Individuals from Nevada were excluded due to incomplete data on early diagnostic milestones. The final analytic cohort included 221 individuals with DMD.
We computed frequency count and percentage for categorical variables, and mean, median (Md), and standard deviation (SD) for continuous variables. Due to violations of variance homogeneity across race/ethnicity, the Kruskal-Wallis test was used to determine overall significance, and the Dwass-Steel-Critchlow-Fligner method was used to evaluate all pairwise comparisons. For analysis of mean changes by year of birth, analysis of variance with planned comparisons (2000 vs each subsequent year through 2005) was used. Data analysis was performed using SAS version 9.4 software (SAS Institute).
RESULTS
Family/primary caregivers were most commonly the first to note concerns, followed by the child’s primary care provider. Initial medical evaluation was performed most frequently by a primary care provider, followed by a neurologist or neuromuscular specialist (Table 1). For the total sample, the mean age at first signs and symptoms reported by the caregiver was 2.7 (SD = 1.8, Md = 2.0) years, followed by first CK at 4.6 (SD = 2.3, Md = 4.6) years and DNA/muscle biopsy confirmatory testing at 4.9 (SD = 2.3, Md = 4.8) years. The average time from first symptoms to diagnostic confirmation was 2.2 (SD = 2.5, Md = 1.4) years. The ages at first concern, and subsequent CK testing and initial visit to a neurology or neuromuscular specialist, were significantly later among non-Hispanic black individuals compared with non-Hispanic white individuals (Figure 1). In addition, the age at CK testing was significantly later among Hispanics compared with non-Hispanic whites. No other differences in ages by race/ethnicity were found. Additional details are available in Table S1.
TABLE 1.
Demographic and characteristics of individuals with Duchenne muscular dystrophy in the Muscular Dystrophy Surveillance, Tracking, and Research Network, 2000–2015
| Characteristics | n(%) |
|---|---|
| Total | 221 |
| Race/ethnicity | |
| Hispanic | 37 (16.7) |
| Non-Hispanic black | 15 (6.8) |
| Non-Hispanic white | 150 (67.9) |
| Unknown | 19 (8.6) |
| First concern noted by: | |
| Family or primary caregiver | 117 (52.9) |
| Child's primary care provider | 50 (22.6) |
| School (teacher, RN, PT, PE) | 25 (11.3) |
| Therapist (PT, OP) | 9 (4.1) |
| Other specialitiesa | 5 (2.3) |
| Orthopedist/podiatrist | 2 (0.1) |
| Unknown | 13 (15.9) |
| First professional evaluating concerns | |
| Primary care providera | 93 (42.1) |
| Neurologist/neuromuscular specialist | 32 (14.5) |
| First professional evaluating concerns | |
| Therapist (early intervention, PT, ST) | 23 (10.4) |
| Orthopedist/podiatrist | 23 (10.4) |
| Gastroenterology/hepatology | 20 (9.0) |
| Developmental pediatrician/rehabilitation medicine | 11 (5.0) |
| Other specialtiesb | 11 (5.0) |
| Unknown | 8 (3.6) |
Abbreviations: OP, occupational therapy; PE, physical education; PT, physical therapist; RN, registered nurse; ST, speech therapy.
Emergency physician, gastroenterologist, hospital physician, or hematologist/oncologist.
Family practice, nurse practitioner, pediatrician, or unknown specialty.
FIGURE 1.
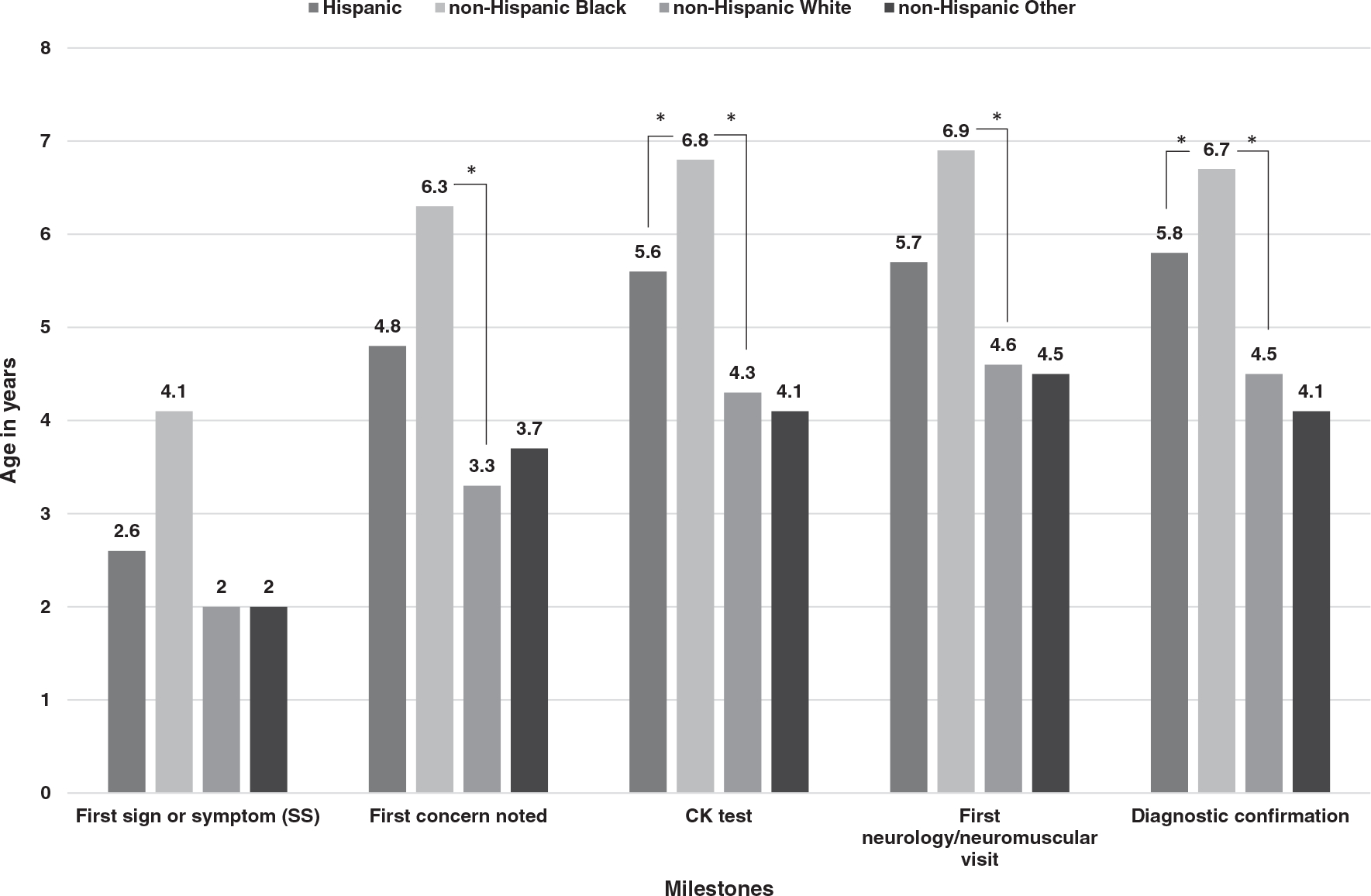
Histogram of age in years at important clinical and diagnostic milestones by race/ethnicity among males with Duchenne muscular dystrophy in the Muscular Dystrophy Surveillance, Tracking and Research Network, 2000–2015. Statistically significant differences (P < .05) were determined by Dwass-Steel-Critchlow-Fligner method pairwise analysis and denoted by asterisk
The diagnosis was genetically confirmed by DNA testing in 96.4% of all individuals, whereas muscle biopsy was performed in only a small number (n = 27). Definitive diagnosis (DNA testing or muscular biopsy) occurred at earlier mean ages for non-Hispanic whites and showed statistically significant later ages among Hispanics and non-Hispanic blacks compared with non-Hispanic whites (Table S1).
Of the individuals in the final analytic cohort, 39 were diagnosed incidentally after CK testing ordered for other medical reasons revealed elevated CK. The time to definitive diagnosis was 2.6 (SD = 2.4, Md = 1.9) years among those not diagnosed due to an incidental CK test result, whereas this interval was 0.5 (SD = 2.1, Md = 0.9) year in those suspected of muscular dystrophy due to incidental CK testing.
We found no significant differences over time in mean ages for diagnostic milestones, including time from earliest symptom to confirmatory testing, for males born from 2000 to 2005 (Figure S2 A–F).
DISCUSSION
In this study we have demonstrated a persistent 2.2-year time interval between first signs and symptoms of DMD and diagnostic confirmation and an average age of 4.9 years at diagnosis among males without a family history of DMD in selected geographical regions in the United States. This interval between symptom onset and diagnosis has not changed in the past three decades1–4 and is persistently later in minority groups, as reported elsewhere.15
Our study also showed that nearly all patients born and diagnosed after January 1, 2000 were genetically confirmed. Knowing a patient’s specific genetic mutation is very important because there are now four US Food and Drug Administration (FDA)-approved exon-skipping drugs that are indicated for specific subgroups of DMD patients with mutations amenable to exon 45, 51, or 53 skipping. Precise genetic confirmation is also a requirement for participation in the ongoing gene therapy and mutation-specific treatment clinical trials.
With partial support from the CDC, a National Task Force for Early Identification of Childhood Neuromuscular Disorders was convened. This group was comprised of representatives from a range of professional organizations involved in the care of children and from several pediatric neuromuscular disease advocacy groups. The task force developed a tool to assist health-care providers in early identification and evaluation of children with motor delay with the goal of decreasing the age at pediatric neuromuscular disease diagnosis, including DMD.16 This resource includes a modified algorithm emphasizing early CK testing in children with motor delays. In addition, the American Academy of Pediatrics published a complementary algorithm for the surveillance and screening of children for motor delays for pediatric care providers17 and developed a website, Physical Developmental Delay: What to Look For, to provide a resource for parents who may be worried about the motor development of their child.18
Although such ongoing educational efforts may improve early recognition of DMD, newborn screening (NBS) for DMD could ensure early diagnosis and help to mitigate racial disparities that currently exist. The unchanged time interval to diagnosis, recent FDA approval of four exon-skipping drugs, and preliminary results of successful gene transfer in children with DMD age 4 to 7 years have underscored the potential value of NBS for DMD.8–10,19 Since 1976, 10 DMD NBS programs have screened more than 1.8 million newborns20 and current programs are underway in New York, North Carolina, and Boston.21,22 Many ethical, legal, and social issues have been identified and are important to consider in the design of DMD NBS programs before widespread implementation including: (1) whether both males and females should be screened for an X-linked disorder; (2) genetic treatment availability for a subset of the DMD population; and (3) identification and follow-up of newborns with other conditions in which CK is elevated.20,23,24
The time to diagnosis of DMD among males without family history remains unchanged and results in lost opportunities for timely genetic counseling, implementation of standards of care, access to newly approved disease-modifying medications, and participation in clinical trials. Educating pediatric providers to identify children with DMD and other conditions causing muscle weakness or motor delay has been one approach to reducing the time to diagnosis. More studies can inform whether early treatment improves the outcomes of children with DMD and whether newborn screening is a feasible approach to achieving early and equitable diagnosis.
Supplementary Material
ACKNOWLEDGMENTS
This project was supported by the Cooperative Agreement Nos. DD001126, DD001119, DD001123, DD001116, DD001117, DD001108, DD001120, DD001054 funded by the US Centers for Disease Control and Prevention (CDC). The findings and conclusions in this report are those of the authors and do not necessarily represent the official position of the CDC.
Funding information
US Centers for Disease Control and Prevention, Cooperative Agreement Numbers: DD001126, DD001119, DD001123, DD001116, DD001117, DD001108, DD001120, DD001054; National Institutes of Health, Grant Numbers: P50 NS053672 (NIAMS) and U24 NS-107181 (NINDS), Friedreich’s Ataxia Research Alliance; Italfarmaco; PTC; Sarepta; Pfizer; Retrotope; Reata; AMO (all to K.D.M.).
Abbreviations:
- CDC
US Centers for Disease Control and Prevention
- CK
creatine kinase
- DMD
Duchenne muscular dystrophy
- FDA
US Food and Drug Administration
- MD
muscular dystrophy
- Md
median
- MD STARnet
Muscular Dystrophy Surveillance, Tracking, and Research Network
- NBS
newborn screening
Footnotes
CONFLICT OF INTEREST
Dr. Emma Ciafaloni has received personal compensation for serving on advisory boards and/or as a consultant for Viela Bio, Avexis, Biogen, Medscape, Amicus, PTC Therapeutics, Sarepta Therapeutics, Ra Pharma, Wave, and Strongbridge Biopharma plc. Dr. Ciafaloni has received personal compensation for serving on a speaker’s bureau for Biogen as well as research and/or grant support from the CDC, CureSMA, Muscular Dystrophy Association, National Institutes of Health, Orphazyme, the Patient-Centered Outcomes Research Institute, Parent Project Muscular Dystrophy, PTC Therapeutics, Santhera, Sarepta Therapeutics, Orphazyme, and the US Food and Drug Administration. Dr. Ciafaloni has also received royalties from Oxford University Press and compensation from Medlink for editorial duties.
Dr. Katherine Mathews receives research funding from NIH (NIAMS) P50 NS053672, NIH (NINDS) U24 NS-107181, and the Friedreich’s Ataxia Research Alliance, and serves as a site PI for clinical research sponsored by Italfarmaco, PTC, Sarepta, Pfizer, Retrotope, Reata and AMO.
The other authors declare no conflicts of interest.
ETHICAL PUBLICATION STATEMENT
We confirm that we have read the Journal’s position on issues involved in ethical publication and affirm that this report is consistent with those guidelines.
SUPPORTING INFORMATION
Additional supporting information may be found in the online version of the article at the publisher’s website.
DATA AVAILABILITY STATEMENT
Due to privacy concerns (detailed personal information was obtained from a small number of individuals living in a defined surveillance area), data from the MD STARnet is not publicly available. Data used for this analysis are maintained at the Centers for Disease Control and Prevention. Researchers interested in MD STARnet should contact MD STARnet at MDSTARnet@CDC.gov.
REFERENCES
- 1.Ciafaloni E, Fox DJ, Pandya S, et al. Delayed diagnosis in Duchenne muscular dystrophy: data from the Muscular Dystrophy Surveillance, Tracking, and Research Network (MD STARnet). J Pediatr. 2009;155:380–385. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Bushby KM, Hill A, Steele JG. Failure of early diagnosis in symptomatic Duchenne muscular dystrophy. Lancet. 1999;353:557–558. [DOI] [PubMed] [Google Scholar]
- 3.Mohamed K, Appleton R, Nicolaides P. Delayed diagnosis of Duchenne muscular dystrophy. Eur J Paediatr Neurol. 2000;4:219–223. [DOI] [PubMed] [Google Scholar]
- 4.van Ruiten HJ, Straub V, Bushby K, et al. Improving recognition of Duchenne muscular dystrophy: a retrospective case note review. Arch Dis Child. 2014;99:1074–1077. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Birnkrant DJ, Bushby K, Bann CM, et al. Diagnosis and management of Duchenne muscular dystrophy, part 1: diagnosis, and neuromuscular, rehabilitation, endocrine, and gastrointestinal and nutritional management. Lancet Neurol. 2018;17:251–267. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Birnkrant DJ, Bushby K, Bann CM, et al. Diagnosis and management of Duchenne muscular dystrophy, part 2: respiratory, cardiac, bone health, and orthopaedic management. Lancet Neurol. 2018;17:347–361. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Birnkrant DJ, Bushby K, Bann CM, et al. Diagnosis and management of Duchenne muscular dystrophy, part 3: primary care, emergency management, psychosocial care, and transitions of care across the lifespan. Lancet Neurol. 2018;17:445–455. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Frank DE, Schnell FJ, Akana C, et al. Increased dystrophin production with golodirsen in patients with Duchenne muscular dystrophy. Neurology. 2020;94:e2270–e2282. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Clemens PR, Rao VK, Connolly AM, et al. Safety, tolerability, and efficacy of Viltolarsen in boys with Duchenne muscular dystrophy amenable to exon 53 skipping: a phase 2 randomized clinical trial. JAMA Neurol. 2020;77:982–991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Mendell JR, Goemans N, Lowes LP, et al. Longitudinal effect of eteplirsen versus historical control on ambulation in Duchenne muscular dystrophy. Ann Neurol. 2016;79:257–271. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Ho PP, Lahey LJ, Mourkioti F, et al. Engineered DNA plasmid reduces immunity to dystrophin while improving muscle force in a model of gene therapy of Duchenne dystrophy. Proc Natl Acad Sci USA. 2018;115:E9182–E9191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Mendell JR, Sahenk Z, Lehman K, et al. Assessment of systemic delivery of rAAVrh74.MHCK7. Micro-dystrophin in children with Duchenne muscular dystrophy: a nonrandomized controlled trial. JAMA Neurol. 2020;77:1122–1131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.lMiller LA, Romitti PA, Cunniff C, et al. The muscular dystrophy surveillance tracking and research network (MD STARnet): surveillance methodology. Birth Defects Res A Clin Mol Teratol. 2006;76:793–797. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Mathews KD, Cunniff C, Kantamneni JR, et al. Muscular Dystrophy Surveillance Tracking and Research Network (MD STARnet): case definition in surveillance for childhood-onset Duchenne/Becker muscular dystrophy. J Child Neurol. 2010;25:1098–1102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Holtzer C, Meaney FJ, Andrews J, et al. Disparities in the diagnostic process of Duchenne and Becker muscular dystrophy. Genet Med. 2011;13:942–947. [DOI] [PubMed] [Google Scholar]
- 16.American Academy of Pediatrics. Does my child have physical developmental delays? Updated 2022. https://www.healthychildren.org/English/MotorDelay/Pages/default.aspx. Accessed January 21, 2022.
- 17.Noritz GH, Murphy NA. Motor delays: early identification and evaluation. Pediatrics. 2013;131:e2016–e2027. [DOI] [PubMed] [Google Scholar]
- 18.American Academy of Pediatrics. Does my child have physical developmental delays? Updated 2022. https://www.healthychildren.org/english/motordelay/pages/default.aspx. Accessed January 21, 2022.
- 19.Mendell JR, Al-Zaidy S, Shell R, et al. Single-dose gene-replacement therapy for spinal muscular atrophy. N Engl J Med. 2017;377:1713–1722. [DOI] [PubMed] [Google Scholar]
- 20.Gatheridge MA, Kwon JM, Mendell JM, et al. Identifying non-Duchenne muscular dystrophy-positive and false negative results in prior Duchenne muscular dystrophy newborn screening programs: a review. JAMA Neurol. 2016;73:111–116. [DOI] [PubMed] [Google Scholar]
- 21.Al-Zaidy SA, Lloyd-Puryear M, Kennedy A, et al. A roadmap to newborn screening for Duchenne muscular dystrophy. Int J Neonatal Screen. 2017;3:8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Parad RB, Sheldon Y, Bhattacharjee A. Implementation of hospital-based supplemental Duchenne muscular dystrophy newborn screening (sDMDNBS): a pathway to broadening adoption. Int J Neonatal Screen. 2021;7:77. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Lloyd-Puryear MA, Crawford TO, Brower A, et al. Duchenne muscular dystrophy newborn screening, a case study for examining ethical and legal issues for pilots for emerging disorders: considerations and recommendations. Int J Neonatal Screen. 2018;4:6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Ross LF, Clarke AJ. A historical and current review of newborn screening for neuromuscular disorders from around the world: lessons for the United States. Pediatr Neurol. 2017;77:12–22. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
Due to privacy concerns (detailed personal information was obtained from a small number of individuals living in a defined surveillance area), data from the MD STARnet is not publicly available. Data used for this analysis are maintained at the Centers for Disease Control and Prevention. Researchers interested in MD STARnet should contact MD STARnet at MDSTARnet@CDC.gov.